

Abstract
Immune microenvironment could affect the biological progress in prostate cancer (PCa) through N6 methyl adenosine (m6A) methylation. The purpose of this study was to investigate the crosstalk between m6A methylation and immune microenvironment and explore potential biomarkers to improve the immunotherapeutic response. Firstly, according to 11 differentially expressed m6A genes between normal and tumor samples, PCa patients were divided into immune microenvironment subtype 1 (IMS1) and IMS2 based on m6A gene profiles extracted from The Cancer Genome Atlas (TCGA) database. IMS2 showed an immune “cold” phenotype with worse prognoses, and HNRNPC was identified as the biomarker of IMS2 by the protein‐protein interaction network. Furthermore, through bioinformatics analyses and in vitro experiments, we found that HNRNPC‐high patients showed a suppressive immune‐infiltrating tumor microenvironment with a higher infiltration of regulatory T (Treg) cells. Finally, we cocultured transfected PCa cells with peripheral blood mononuclear cells (PBMC) and verified that HNRNPC inhibits tumor immunity by elevating the activation of Treg cells and suppression of effector CD8 T cell. In conclusion, we identified a “cold” immune phenotype in PCa, and HNRNPC regulating the activation of Treg cells. Activation of the immune microenvironment through targeting HNRNPC may be a potential therapeutic option for advanced PCa.
Keywords: HNRNPC, M6A methylation, prostate cancer, regulatory T (Treg) cells, tumor immune microenvironment
Harboring a high expression level of m6A reader HNRNPC, progressed prostate cancer (PCa) exhibited a “cold” immune phenotype, which was mainly induced by increasing Treg abundance. Targeting HNRNPC may help activate the immune microenvironment or elevate the sensitiveness to anti‐CTLA4 therapies.
Abbreviations
- AS
aneuploidy score
- BCR
B cell receptor
- BCR
biochemical recurrence
- BH
Hochberg
- CTLA‐4
cytotoxic T lymphocyte associated protein 4
- DEMG
differentially expressed m6A gene
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- HLA
human leukocyte antigen
- hnRNA
heteronuclear RNA
- hnRNP
heterogeneous nuclear ribonucleoprotein
- HRD
homologous recombination defects
- ICD
immunogenic cell death
- ICI
immune checkpoint inhibitor
- ICP
immune checkpoints
- IMS
immune microenvironment subtype
- IRG
immune‐related genes
- ITH
intratumor heterogeneity
- iTreg
induced regulatory T cells
- m6A
N6 methyl adenosine
- mCRPC
metastatic castration‐resistant prostate cancer
- MDSC
myeloid‐derived suppressor cell
- MHC
major histocompatibility complex
- NK
natural killer cell
- NKT
natural killer T cell
- nTreg
natural regulatory T cell
- PCa
prostate cancer
- PD‐1
programmed death‐1 receptor
- PD‐L1
programmed death‐1 ligand
- PPI
protein‐protein interaction
- PRAD
prostate adenocarcinoma
- TCGA
The Cancer Genome Atlas
- TCIA
The Cancer Immunome Atlas
- TCR
T cell receptor
- Th1
T helper 1 cell
- TMB
tumor mutation burden
- TME
tumor microenvironment
- TPM
transcripts per million
- UTR
untranslated region
- WES
whole‐exome sequencing
1. INTRODUCTION
Prostate cancer (PCa) is a common malignancy in men. 1 , 2 Despite the rapid response to androgen deprivation therapy, most PCa patients eventually progress to fatal metastatic castration‐resistant PCa (mCRPC). 3 , 4 Recently, immune checkpoint inhibitors (ICIs) have become choices for progressive PCa. 5 , 6 Although immunotherapy has proven to be an effective and important new strategy for the management of PCa patients, only a few patients benefit from immunotherapy. 7 , 8 , 9 , 10 This phenomenon may be attributed to the varied heterogeneity of the immune microenvironment among individuals. 11 , 12 Therefore, it is important to further explore the regulatory mechanisms of the tumor immune microenvironment to optimize the management of immunotherapy.
N6‐methyladenosine (m6A) is the most common post‐transcriptional modification of mRNA and mediates more than 60% of RNA methylation. 13 , 14 The abnormal methylation level of m6A is closely related to stem cell differentiation and the immune response, which plays an important role in the progression of various cancers. 15 , 16 , 17 , 18 The abundance of m6A methylation modification in tumors mainly depends on the expression of methylation regulators, including methyltransferases (“writers”) and demethylases (“erasers”) in cells, while binding proteins (“readers”) perform a series of biological functions by binding to the methylation sites of m6A. 19
In a previous study, Thorsson et al identified six “immune subtypes”: wound‐healing, IFN‐γ–dominant, inflammatory, lymphocyte‐depleted, immunologically quiet, and TGF‐β–dominant, which play different characters of immune features. 20 Currently, several methods have emerged to characterize the immune tumor microenvironment (TME) for the assessment of total lymphocytic infiltrates, including immune gene expression signatures, such as immunogenic cell death (ICD) and immune checkpoint (ICP) genes, neoantigen prediction, T cell receptor (TCR) and B cell receptor (BCR) repertoire inference, and somatic DNA alterations. 21 , 22 , 23 These methods and strategies provide a new basis for our research into the immune microenvironment of PCa.
In this study, we systematically evaluated the expression profile of m6A methylation regulators in PCa to improve the risk stratification of prognosis and promote therapeutic decision‐making in PCa. The relationship between m6A regulatory factors and immune cell infiltration was analyzed in silico and verified in vitro. Our study attempted to illustrate the m6A methylation regulatory mechanism and the prostate tumor immune microenvironment status of PCa, thus providing a basis for improving immunotherapy strategies in PCa.
2. MATERIALS AND METHODS
2.1. Data processing
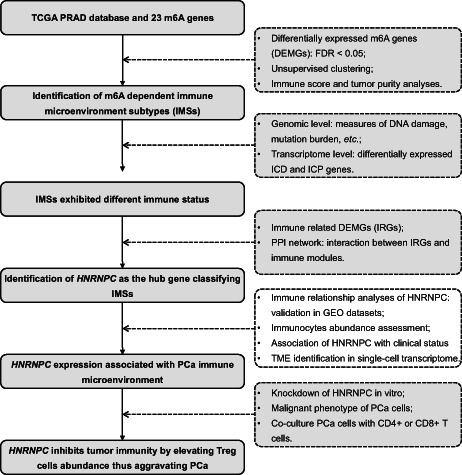
Figure 1 shows a flowchart of this study. The mRNA expression profiles and related clinical data were collected from The Cancer Genome Atlas (TCGA) data portal and Gene Expression Omnibus (GEO) datasets (GSE70768, GSE32571, GSE60329, GSE46602). The mRNA expression data were transformed to values in transcripts per million (TPM). A total of 23 m6A genes were integrated from several studies, as shown in Figure S1. The single‐cell transcriptome data were extracted from GEO dataset: GSE141445 including 13 samples, 12 primary samples, and 1 metastatic sample.
FIGURE 1.
Flow chart of this study
2.2. Differentially expressed m6A genes (DEMGs) and cluster analysis
We used Wilcoxon rank sum test to perform the differential expression analyses. M6A genes that met the criterion of false discovery rate (FDR) <0.05 between tumor and normal samples were identified as DEMGs. Based on the expression level of DEMGs, patients were then separated into subgroups using unsupervised clustering (named immune microenvironment subtypes [IMSs] subsequently).
Differences in the clinical characteristics of patients between IMSs were measured, including clinical M stage, pathological T stage, pathological N stage, Gleason score, and biochemical recurrence (BCR). In addition, we investigated the differences in tumor purity and immune scores between clusters. Next, we calculated the ssGSEA score of immunostimulatory and inhibitory states using gene lists collected from previous studies, which are provided in Table S1.
2.3. Identifying the immune landscape of IMS clusters
First, we counted the proportion of C1‐C6 immune subtypes in the two IMSs. Considering that immune infiltration was related to “measures of DNA damage,” we compared several measures of DNA damage, including fraction alteration, number of segments, aneuploidy score (AS), homologous recombination defects (HRD), and intratumor heterogeneity (ITH) between IMSs. Next, a comparison was performed of the genome changes, including silent/nonsilent mutation, indel neoantigens, and SNV neoantigen counts. Differences in BCR and TCR diversity between clusters were also measured using the Shannon index. All data used were obtained from Thorsson et al 20 Finally, we compared the expression levels of ICD and ICP genes between the two IMSs.
Furthermore, whole‐exome sequencing (WES) from TCGA database data was used to calculate the tumor mutation burden (TMB) score in PCa with exons uniformly counted as 40 M regions and to describe the mutation spectrum of two clusters to search for specific mutations.
2.4. Correlations of DEMGs with immune microenvironment
We performed correlation analyses of DEMGs with immune score and tumor purity to identify genes related to the immune microenvironment (named immune‐related genes [IRGs]). Genes with expression levels negatively correlated with immune score and positively correlated with tumor purity were determined as IRGs. To assess the outcome of immunotherapy for IMSs, we compared the expression of the following marker genes, which have been proven to affect immunotherapy in different ways: CD274 (PD‐L1), 21 CXCL9, 24 MEX3B, 25 HAVCR2 (TIM3), 22 CTLA4, and CD38. 26
2.5. Identification of hub genes
The STRING database (https://string‐db.org/) and Cytoscape software were used to establish a protein‐protein interaction (PPI) network with a minimum required interaction score of 0.40 to evaluate the interactions between proteins coded by IRGs and immune modules, including the major histocompatibility complex (MHC) module, receptor module, chemokine module, immunostimulatory module, and immunoinhibitory module. IRGs that interacted with immune modules were identified as hub genes.
2.6. Validation of hub genes through in silico analyses
First, GEO datasets (GSE70768, GSE32571, GSE60329, and GSE46602) were applied to validate the correlations between IRGs and the immune microenvironment. Second, we analyzed the correlation between the expression of hub genes and MEX3B, which can downregulate HLA‐A expression on the surface of tumor cells, thereby rendering the tumor cells unable to be recognized and killed by T cells. 25 Next, tumor patients were separated into a high‐expression group and a low‐expression group according to the median hub gene expression levels; differences in the abundance of immune cells between the two groups were measured. Finally, based on TCGA database, we compared the differences in the expression levels of hub genes between different clinical conditions. Kaplan‐Meier (K‐M) plots were developed to perform survival analysis of hub genes.
2.7. Single‐cell transcriptome data analyses
The single‐cell transcriptome data were preprocessed using the “Seurat” package. Well‐established markers for each cell type of PCa, integrated with the automatic cell annotation tool “SingleR,” 27 were applied to annotate the cell types, dividing the cells into luminal epithelial cells (AR), basal epithelial cells (TP63), T cells (CD3E), B cells (CD19), fibroblasts (MYL9), mast cells (KIT), monocytes (CD14), and endothelial cells (CD34). We calculated percentages of cell types among samples and analyzed their correlation with HNRNPC‐positive cell rate. “ProjecTIL” was used to parse human scRNA‐seq T cell data in the context of murine TIL profiles. 28 We then compared the difference of T cell subgroup composition between HNRNPC‐positive and ‐negative cells.
We used “CellChat” R package to study cell‐cell communication. 29 The samples were divided into positive and negative groups according to whether the positive rate of HNRNPC was greater than 70%. We compared the strength of interaction among different cell types.
2.8. Cell lines and cell culture
Prostate cancer cells (C4‐2B, LNCaP, DU145, and PC3) and normal human prostate epithelial cells (RWPE‐1) were purchased from the American Type Culture Collection (ATCC). All cells were tested and confirmed to be free of mycoplasma contamination before use. C4‐2B cells were cultured in DMEM/F12 (4:1). LNCaP cells were cultured in RPMI‐1640. DU145 cells were cultured in MEM. PC3 cells were cultured in the F‐12 K medium. RWPE‐1 cells were cultured in keratinocyte serum‐free medium (K‐SFM). All media were provided by Gibco and were treated with 10% FBS and 100 U/mL penicillin/streptomycin in a 5% CO2 incubator.
2.9. Cell transfection
Three specific shRNAs targeting HNRNPC and scrambled shRNA were synthesized by GenePharma. Cells were seeded in six‐well plates at a density of 8 × 106 cells/well, and the plates were placed at 37°C with 5% CO2. When cell confluence reached 70%, transfections were performed using the Lipofectamine 3000 kit (Invitrogen) according to the manufacturer's instructions.
2.10. Quantitative real‐time PCR (RT‐qPCR)
Cells were seeded in six‐well plates at a density of 1.5 × 106 cells/well and incubated for 24 hours. Total RNA was extracted from the cells using TRIzol® reagent (Invitrogen), and the extracted RNA was reverse‐transcribed into cDNA using the ReverTra Ace qPCR RT Kit (Toyobo). Quantitative PCR was performed using SYBR® qPCR Mix (Toyobo) based on the 2−ΔΔCt method. GAPDH and U6 were used as the endogenous control genes. All experiments were performed in triplicates.
2.11. Cell‐counting kit‐8 (CCK‐8) assays
Cell viability was determined using a CCK‐8 kit (Beyotime Institute of Biotechnology), following the manufacturer's instructions. The cells (2 × 104 cells/well) were seeded in a 96‐well plate. After cell growth for 12, 24, 48, or 72 hours, 10 μL of CCK‐8 solution was added to each well. Two hours later, absorbance was measured at 450 nm using a microplate reader. All experiments were performed in triplicates.
2.12. EdU assays
The cells were cultured in 24‐well plates until 70% confluence. After 10 μL EdU solution was added and 2 hours incubation, the cells were fixed with 4% paraformaldehyde. After washing, a Click‐iTR EdU kit was used to detect EdU. The nuclei were stained with DAPI and the cells were observed using a fluorescence microscope (Olympus). All experiments were performed in triplicates.
2.13. TUNEL assays
Cells were grown in 24‐well plates until 70% confluence. Cultured cells were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton‐X 100. TUNEL assays were performed according to the manufacturer's instructions (Roche). Briefly, cells were first incubated in a terminal deoxynucleotidyl transferase (TdT) reaction cocktail, followed by treatment with a Click‐iT reaction cocktail. Nuclei were stained with DAPI. The cells were observed and imaged using a fluorescence microscope (Olympus). All experiments were performed in triplicates.
2.14. Flow cytometry analysis
Cells (2 × 106) were seeded into each well of a six‐well plate and incubated for 24 hours. To assess apoptosis, cells were harvested after transfection for flow cytometry analysis. Briefly, after double‐staining with an Annexin V‐FITC/PI apoptosis kit (Multi Sciences), apoptosis was determined using a flow cytometer (BD). The apoptotic cells were gated as Annexin V‐FITC+PI+ and Annexin V‐FITC+PI−. The apoptosis rate was defined as the percentage of apoptotic cells in total cells. Flow cytometric analysis was performed to determine the percentage of Treg cells. The cocultured peripheral blood mononuclear cells (PBMCs) were digested with 0.25% trypsin and washed with PBS containing 0.5% (w/v) bovine serum albumin (BSA). Lymphocytes in PBMCs were gated by forward scattering (FSC) and lateral scattering (SSC). CD4+ T cells were gated by CD3 and CD4 staining. Isolated CD4+ T cells were then incubated with FITC‐conjugated anti‐CD25 and anti‐CD127 antibodies. 30 All experiments were performed in triplicates.
2.15. Transwell assays
After transfection, 2 × 105 cells were seeded into each well of the upper transwell chamber (8 pm pore size, Corning). A medium containing 10% FBS was added to the lower chamber. After incubation for 24 hours, cells on the upper side of the upper chamber were wiped off, and cells on the lower side of the upper chamber were fixed with 4% paraformaldehyde and stained with 5% crystal violet for 5 minutes. Finally, stained cells were counted under a microscope in five random visual fields. All experiments were performed in triplicates.
2.16. Wound‐healing assays
Cells were seeded into six‐well plates (2 × 106/well) and incubated for 24 hours. Wounds were created by passing a plastic tip across the monolayer cells. The time of wound infliction was considered to be 0 hours, and wound closure was photographed 24 hours later using a microscope. All experiments were performed in triplicates.
2.17. Coculture of PBMCs and PCa cells
Peripheral blood mononuclear cells obtained from Procell (Wuhan) were cultured for 72 hours for activation in RPMI‐1640 medium (Hyclone; GE Healthcare) with 10% FCS (Gibco; Thermo Fisher Scientific, Inc.), 2 mm l‐glutamine, 0.5% streptomycin and penicillin, 25 mm HEPES, 3 μG/mL anti‐CD28 antibody, 1 μG/mL anti‐CD3 antibody, and 100 u/mL IL‐2. PCa cells (PC3 or DU145) with or without HNRNPC interference were cocultured indirectly with activated PBMCs in six‐well transwell coculture plates (0.4 μm polyester film) for 48 hours, where PBMCs cells were planted in the upper layer and PCa cells were plated in the lower layer.
2.18. Statistical analyses
All analyses were performed using R software (version 4.0.5). Differences between groups were evaluated using Wilcoxon rank‐sum tests for continuous data and Fisher's exact tests for categorical variables. Pearson's test was used for the correlation analysis. The R package “ConsensusClusterPlus” was used for unsupervised clustering. ssGSEA scores were calculated using the “GSVA” package. K‐M plots were constructed using the K‐M “survival” package. The immune score and the tumor purity were calculated with the “estimate” package based on gene expression levels. CIBERSORT and ImmuCellAI (http://bioinfo.life.hust.edu.cn/ImmuCellAI) were used to calculate the infiltration score, assess the response to immunotherapy, and estimate the abundance of immune cells. All analyses were two‐sided, and statistical significance was set at p < 0.05.
3. RESULTS
3.1. Identification of m6A‐dependent IMSs
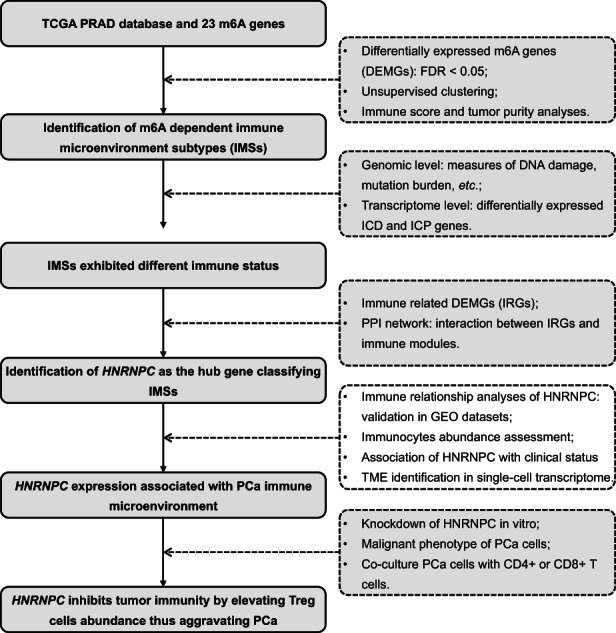
We extracted the m6A regulatory gene (shown in Figure S1) expression profile data of 52 normal and 395 tumor tissues from TCGA. Eleven m6A genes were found to be differentially distributed between PCa and normal tissues with an FDR < 0.05, including RBM15B, IGF2BP2, FMR1, HNRNPA2B1, HNRNPC, METTL3, ZC3H13, YTHDF1, FTO, YTHDF2, and ALKBH5 (Figure 2A). Using unsupervised clustering based on 11 identified enzymes, PCa patients were separated into two clusters with different molecular and immune characteristics (Figure 2B). The baseline characteristics of the 203 group1 and 292 group2 patients are shown in Table S2. After excluding individuals with missing values, the heatmap is shown in Figure 2C, which shows the distributions of tumor stage, BCR condition, Gleason score, m6A gene expression, tumor purity, and immune score between subgroups. Group2 exhibited an abundance enrichment of m6A genes. Furthermore, patients in group2 had higher tumor purity and lower immune scores (Figure 2D,E). Therefore, we named the two subgroups immune microenvironment subtype 1 (IMS1) and IMS2 in the further study. Additionally, we found that patients with IMS2 had a more advanced tumor condition and poorer prognosis than those with IMS1 (Figure 2C,G,H). More importantly, IMS1 samples showed significantly higher scores for both stimulatory and inhibitory immune responses than IMS2 samples (p all < 0.05, Figure 2F), indicating that IMS1 is an immune “hot” phenotype, while IMS2 is an immune “cold” phenotype. All above mentioned suggested that the enrichment of m6A gene may affect the reprogramming of tumor immune microenvironment, thus leading to different prognoses and survival outcomes.
FIGURE 2.
DEMGs and cluster analysis. A, The bar plot of 11 DEMGs. B, Unsupervised clustering of PCa patients from TCGA PRAD dataset. C, The heatmap plot of differences in the clinical M stage, pathologic T stage, pathologic N stage, Gleason score, incidence of biochemical recurrence (BCR), tumor purity, and immune score between two clusters. D, E, Differences in the tumor purity and the immune score between two clusters. F, Differences in the state of tumor immune microenvironment, including stimulatory (left) and inhibitory (right) immune scores, between two clusters. G, Kaplan‐Meier plot of the comparison in the overall survival between two clusters. H, The number of patients in different clusters stratified by survival years. *p < 0.05; **p < 0.01; ***p < 0.001. DEMGs, differentially expressed m6A genes; PCa, prostate cancer; PRAD, prostate adenocarcinoma; TCGA, The Cancer Genome Atlas
3.2. IMSs exhibited different immune status on genomic and transcriptome levels
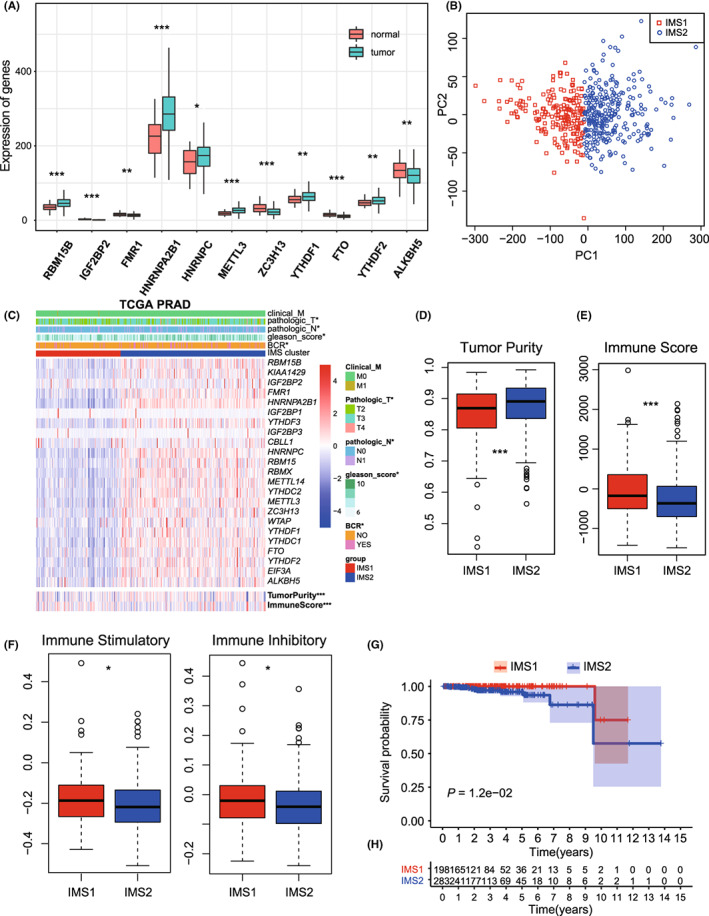
The measures of DNA damage, including altered fraction, AS, HRD, ITH, and number of segments, were all significantly stronger in IMS2 than in IMS1, which represents a poorer prognosis of tumor patients (p all < 0.05, Figure 3A–E). The mutation rates were also found to be significantly different between IMSs (p all < 0.05, Figure 3F,G). Further comparison of the mutation spectra revealed several high‐frequency mutations. For example, LRP1B and RYR2 were specifically mutated in IMS1, while SPTA1, ATM, and CSMD3 were mutated in IMS2 (Figure S2A,B). Notable difference was not found in the percentage of immune subtypes (C1‐C4), neoantigen counts (both Indel and SNV), BCR/TCR diversity, and TMB between IMSs (Figures S3 and S4A).
FIGURE 3.
Immune landscape of immune microenvironment subtype (IMS) clusters. A‐E, Measures of DNA damage, fraction altered, number of segments, aneuploidy score (AS), homologous recombination defects (HRD), intratumor heterogeneity (ITH), between clusters. F, G, silent and nonsilent mutation rate of IMS clusters. H, Correlation between the expression level of HNRNPC and immune score in TCGA database. I, Protein‐protein interaction (PPI) network of the interactions between immune‐related genes (IRGs) and major histocompatibility complex (MHC) module. J, Correlations of HNRNPC expression with MEX3B expression in TCGA database
The expression of most ICD and ICP genes varied significantly between IMSs (Figure S3D,E), indicating a difference in the tumor immune status. Most importantly, well‐established immunotherapy markers showed obvious differences. CD274 (PD‐L1), 21 T‐cell immunoglobulin mucin‐3 (TIM‐3), 22 and CTLA4, 23 which represent the most effective predictive biomarkers for checkpoint inhibitor–based immunotherapy, were expressed at higher levels in IMS2 (p all < 0.05, Figure S2C). CXCL9, the ligand of CXCR3, was also expressed at higher levels in IMS2 cells (Figure S2C, p = 1.94 × 10−9). CXCL9 has been identified as a biomarker for sensitivity to PD‐1 blockade, and augmenting the intratumoral function of this CXCR3‐CXCL9 chemokine system could improve clinical outcomes. 24 MEX3B was also significantly higher in IMS2 cells (p = 2.7 × 10−14), which can inhibit the killing effect by preventing T cells from recognizing tumor cells. 25 Furthermore, tumors treated with PD‐1/PD‐L1–blocking antibodies developed drug resistance through the upregulation of CD38, 26 which showed higher expression in IMS1 (p = 5.43 × 10–3, Figure S2C).
3.3. Identification of HNRNPC as the hub gene classifying IMSs
To investigate the crosstalk between m6A modification and the immune microenvironment, we analyzed the immune correlation of 11 DEMGs. Correlation analysis identified six IRGs, HNRNPC, HNRNPA2B1, YTHDF1, YTHDF2, METTL3, and RBM15B, whose expression levels were significantly negatively correlated with the immune score (R = −0.19, −0.18, −0.15, −0.13, −0.28, and −0.23, respectively; p = 2.4 × 10−5, 5.3 × 10−5, 7.2 × 10−4, 4.0 × 10−3, 1.8 × 10−10, and 1.6 × 10−7, respectively; Figure 3H and Figure S4B–F) and positively correlated with tumor purity (R = 0.20, 0.19, 0.17, 0.16, 0.29, and 0.23, respectively; p = 4.8 × 10−6, 2.5 × 10−5, 1.3 × 10−4, 2.6 × 10−4, 4.0 × 10−11, and 1.3 × 10−7, respectively; Figure S4G–L), suggesting that these genes may be associated with a low degree of immune invasion and tumor progression (Figure S5). In addition, PPI network analysis was performed to identify hub regulatory factors. As HNRNPC was associated with the most nodes, it was identified as a hub gene, which also showed association with the MHC module simultaneously (Figure 3I). The PPI network with the immunoinhibitory and immunostimulatory modules also confirmed the immune correlation of m6A regulatory factors (Figure S6). HNRNPC was also significantly upregulated in IMS2 (Figure 2C); therefore, we noted it as the representative classifying IMS in further studies.
3.4. HNRNPC expression associated with PCa immune microenvironment
To investigate the effect of the hub m6A regulator HNRNPC on the tumor immune microenvironment of PCa, four GEO datasets were used for validation. Consistent results were obtained for the different datasets. In the GSE46602, GSE32571, and GSE60329 datasets, HNRNPC showed a negative correlation with immune score, but a positive correlation with tumor purity (Figure S7). In dataset GSE70768 with the largest sample size, the same result was obtained with statistical significance (R = −0.20 and 0.24, respectively; p = 5.3 × 10−3 and 5.9 × 10−4, respectively; Figure S7A). Additionally, the expression level of HNRNPC was negatively correlated with the infiltration score (R = −0.34; p = 8.7 × 10−7; Figure S7C). Meanwhile, we found that the expression of HNPNPC was significantly correlated with MEX3B (R = 0.40, p < 2.2 × 10−16; Figure 3J). Finally, the correlation between HNRNPC and the immune cells was investigated. A high abundance of natural regulatory T cells (nTreg), induced regulatory T cells (iTreg), T helper 1 cells (Th1), central memory cells, and monocytes could be identified in the HNRNPC low‐expression group (p all < 0.05, Figure 4A). Overexpression of Treg cells is likely to be the reason for the tumor's immunosuppressive status. Meanwhile, the abundance of exhausted cells, natural killer T cells (NKT), and natural killer cells (NK) was significantly lower in the high‐expression group (p all < 0.05, Figure 4A). To verify the relationship between HNRNPC expression and abundant of Treg, cytotoxic T cells, we performed a correlation analysis in the TCGA‐PRAD cohort, finding that the expression of HNRNPC was significantly positively correlated with Treg (iTreg and nTreg) scores, and negatively with cytotoxic T cell scores (R = 0.1, 0.31 and 0.11, respectively, p = 2.2 × 10−2, 1.1 × 10−12 and 1.1 × 10−2, respectively; Figure 4B–D). Furthermore, HNRNPC was highly expressed in progressive PCa, such as higher pathologic T stage, higher pathologic N stage, and high Gleason score (Figure 4E–H) in the TCGA PRAD dataset. In addition, the OS of patients with high HNRNPC expression was poorer than that of patients with low expression (Figure 4I).
FIGURE 4.
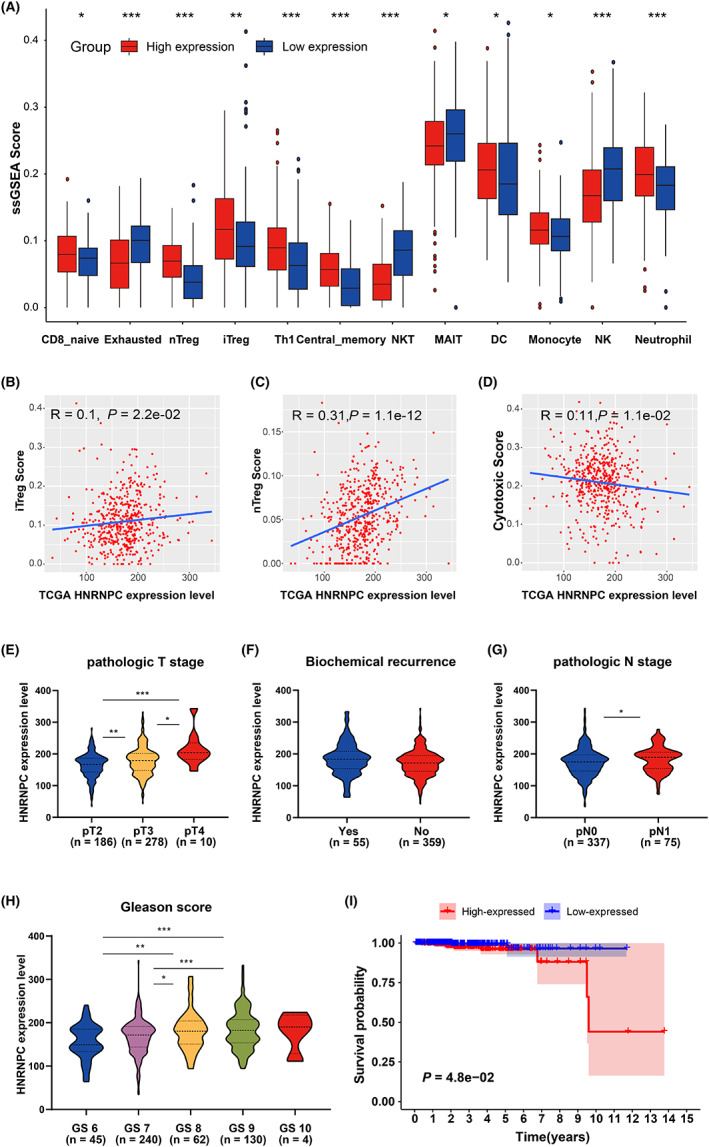
Validation of the correlation between HNRNPC and immune microenvironment. A, Differences in the abundance of immune cells between the high‐expression group and low‐expression group (separated according to the median of HNRNPC expression level in GSE70768). *p < 0.05. B‐D, Correlations between HNRNPC expression and iTreg (B), nTreg (C), and cytotoxic T cells score (D). E‐H, Expression difference of HNRNPC between tumor patients with different pathologic T stages (E), biochemical recurrence condition (F), pathologic N stage (G), and Gleason score (H). I, Kaplan‐Meier plot of the comparison in the overall survival between HNRNPC high‐expression patients and low‐expression patients. TCGA, The Cancer Genome Atlas; PRAD, prostate adenocarcinoma
3.5. Treg proportion in HNRNPC‐positive cells was significantly higher than in negative cells on the single‐cell level
To accurately map T cell status on the single‐cell level, we described the single‐cell transcriptome atlas of PCa. First, after data preprocessing, we obtained a total of 36,424 cells (Figure 5A,B and Figure S8A). We annotated nine cell types: luminal cells (AR), T cells (KRT3E), B cells (MS4A1), mast cells (KIT), monocytic cells (CD14), endothelial (CD34), fibroblast (ACAT2), basal (KRT19), and other epithelial cells (Figure S8B). Furthermore, we analyzed the correlation between the proportion of each cell type and HNRNPC‐positive cell rate, the result showed that HNRNPC‐positive cell rate was significantly positively correlated with epithelium cells, and negatively with T cells (Figure 5E). Through subanalyses on T cells, we got nine clusters including CD4_NaiveLike, CD8_EarlyActive, CD8_EffectorMemory, CD8_NaiveLike, CD8_Tex, CD8_Tpex, Tfh, Th1, and Treg. According to the expression of HNRNPC, we separated the cells into HNRNPC‐positive and ‐negative cells. The proportion of Treg cells in positive cells was much higher than that in HNRNPC‐negative cells. Furthermore, the number of naïve CD8 T cells and active CD8 T cells was lower in HNRNPC‐positive cells, which indicates that cells with high expression of HNRNPC were in a state of immunosuppression (Figure 5D–F).
FIGURE 5.
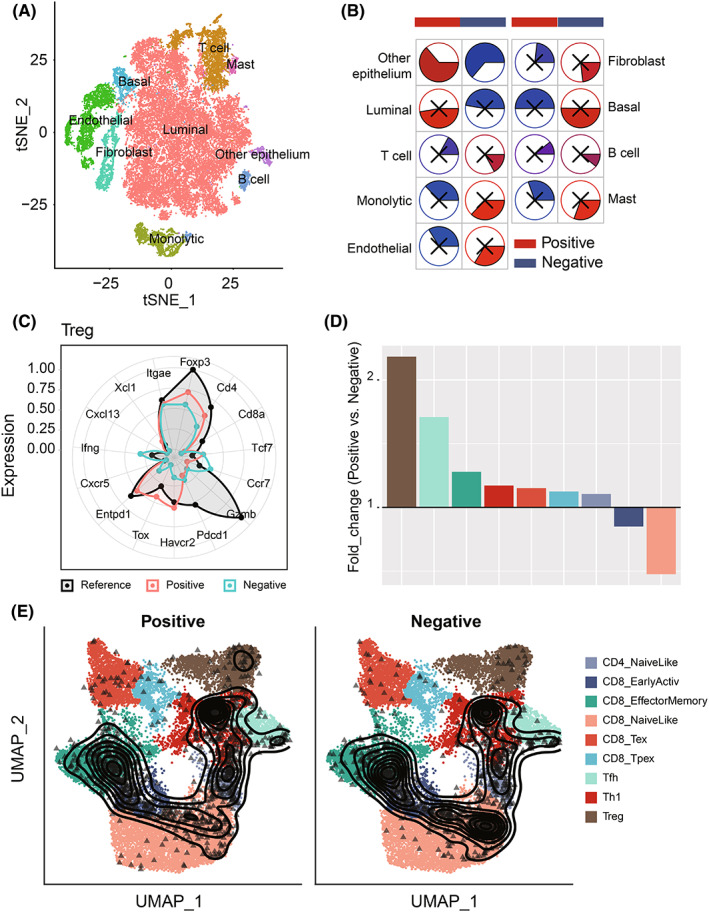
Comparison of Treg proportion in HNRNPC‐positive cells and ‐negative cells on the single‐cell level. A, The tSNE clustering visualization of single‐cell RNA sequencing of prostate cancer. B, The tSNE plots of cells from patients with cells colored based on the cell types. C, Well‐known markers of eight cell types. D, The fold change of T cell subset composition of positive and negative cells. E, The tSNE plot of T cells colored by subtype and HNRNPC expression.
Furthermore, we explored specific cell‐cell communication in positive samples. According to the cutoff value of the positive rate of 70%, we divided 13 samples into two groups: positive group (five samples) and negative group (eight samples) (Figure S9A). The differentially enriched interaction between cell types is shown in the heatmap, which indicated that the cell‐cell interaction in the positive group was stronger than that in the negative group, especially between T cells and other cells (Figure S9B). Then, different signal flow analyses implied that a large number of signal flows were activated in the positive group, including SPP1, TGF‐β, which were associated with immunosuppression (Figure S9C). Finally, we discovered ligand‐receptor interaction specifically in the HNRNPC‐positive group. The epithelial cells in the HNRNPC‐positive group interacted with other types of cells through such ligand‐receptor pairs: MIF‐(CD74+CXCR4) and MIF‐(CD74+CD44) with T cells; VEGFR‐PDGFA (a tumor angiogenesis related interaction), and NAMPT‐(ITGA5+ITGB1) (an immunosuppression‐associated interaction) with endothelial cells, and JAG1‐NOTCH3 and PDGFA‐PDGFRB (two pairs associated with pericytes and formation of tumor‐associated fibroblasts) with fibroblast (Figure S9D).
3.6. Knockdown of HNRNPC inhibited the proliferation and migration ability of PCa cell in vitro
We performed PCR analysis to confirm the expression levels of HNRNPC in PCa cell lines. Compared with the normal prostate epithelial cell line RWPE‐1, four PCa cell lines (C4‐2B, Lncap, DU145, and PC3) showed elevated HNRNPC expression (Figure 6A). To evaluate the oncogenic effect of HNRNPC in vitro, DU145 and PC3 cells with the highest HNRNPC levels were stably transfected with an shRNA targeting HNRNPC, whereas cells transfected with a scrambled vector were used as a negative control. Using shRNAs, the expression of HNRNPC was successfully knocked down in two PCa cell lines (DU145 and PC3) (Figure 6B, Figure S10A). Among the shRNAs, shRNA‐1 and shRNA‐2 were used for functional assays with the highest knockdown efficiency. We investigated the effect of HNRNPC on PCa cell proliferation. CCK‐8 assay demonstrated that knockdown of HNRNPC significantly decreased the proliferative ability of DU145 and PC3 cells (Figure 6C, Figure S10B). These results were further confirmed by EdU incorporation assays in both DU145 and PC3 cells (Figure 6D, Figure S10C). These results indicated that HNRNPC is required for PCa cell proliferation in vitro. Furthermore, we evaluated the effect of HNRNPC knockdown on apoptosis of PCa cells using TUNEL staining and flow cytometry. Our results showed that the apoptosis level of PCa cells was significantly decreased by HNRNPC downregulation (Figure 6 E‐F, Figure S10D,E). Transwell and wound‐healing assays were performed to detect the effects of HNRNPC on the migration and invasion of PCa cells. The results showed that HNRNPC could also promote malignancy in PCa cells, as evidenced by the decreased migration and invasion ability in DU145 and PC3 cells after knockdown of HNRNPC (Figure 6G, Figure S10F,G).
FIGURE 6.
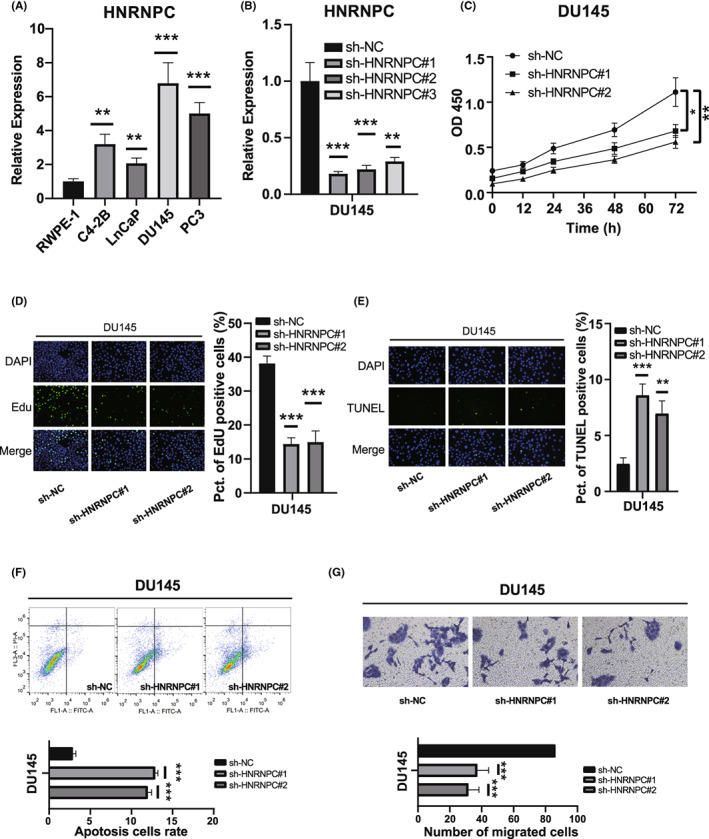
Knockdown of HNRNPC‐promoted cell apoptosis inhibited the cell proliferation and migration of prostate cancer (PCa) cells. A, Relative expression of HNRNPC in human prostate epithelial cell line RWPE‐1 and PCa cell lines. B, qRT‐PCR analyses of HNRNPC mRNA in DU145 cells treated with negative control (NC) or HNRNPC shRNAs. C, D, CCK‐8 assays and EdU incorporation assays for DU145 cells treated with negative control (NC) or HNRNPC shRNAs. E, F, TUNEL assays and flow‐cytometric analysis for DU145 cells treated with NC or HNRNPC shRNAs. G, Transwell assays and wound‐healing assays for DU145 cells
3.7. HNRNPC inhibits tumor immunity by elevating the activation of Treg cells and suppression of CD8 cells
The above immune correlation analysis suggested that HNRNPC could mediate the activation of Treg cells. To determine whether HNRNPC‐mediated PCa cells induced activation of Tregs, we cocultured transfected PCa cells with PBMC T cells (Figure 7A). The expression of the Treg markers (CD25+CD127−) was significantly decreased in PBMC cells incubated with HNRNPC‐knockdown PCa cells compared with that in the NC group (Figure 7B,C), indicating the activation of Treg cells in HNRNPC low‐expression tumors. Furthermore, the number of effector CD8 T cells (CD8+CD107a+IFN‐γ+) was significantly increased in PBMC cells incubated with HNRNPC‐knockdown PCa cells compared with that in the NC group (Figure 7D,E), representing the suppression of CD8 cells.
FIGURE 7.
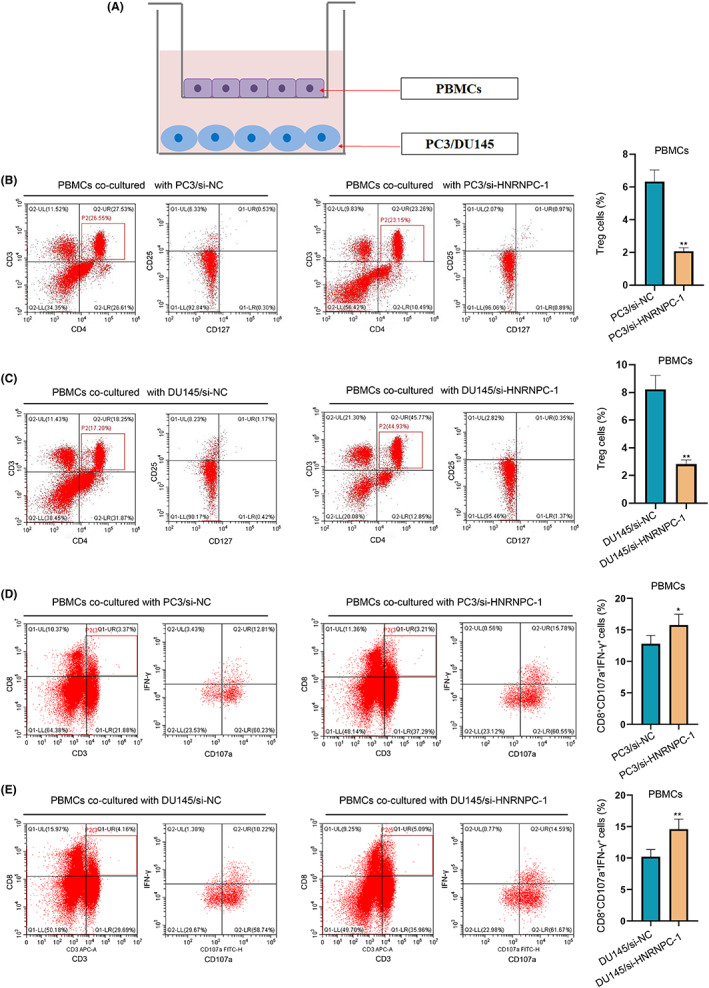
Coculture of peripheral blood mononuclear cells (PBMCs) and prostate cancer (PCa) cells. A, The sketch of the coculture system. B, C, The expression of Treg markers (CD25+CD127−) after coculture‐transfected PC3 cells and DU145 cell. D, E, The expression of effector CD8 T cell markers (CD8+CD107a+INF‐γ+) after coculture‐transfected PC3 cells and DU145 cell
4. DISCUSSION
Prostate cancer is a heterogeneous disease with different ethnic characteristics that originates from prostatic epithelial cells. 31 , 32 Targeting the immunosuppressive mechanism in the TME has revolutionized cancer treatment, while clinical trials have shown minimal efficacy in patients with PCa. M6A methylation is the most common form of mRNA modification and plays a pivotal role in post‐transcriptional regulation. Studies showed that METTL3 and YTHDF2 promote PCa migration and invasion by mediating m6A modification, 33 , 34 suggesting that m6A regulates the occurrence and development of PCa in a unique way. Furthermore, studies evaluated the m6A regulation patterns of PCa and correlated these modification patterns with the tumor immune microenvironment characteristics. 35 , 36 Although these studies coanalyzed m6A methylation and the TME of PCa, they did not discuss the underlying mechanism of tumor immune status.
In our study, the expression patterns, prognostic values, and effects on the TME of m6A regulators in PCa were demonstrated. Subsequently, we identified two subtypes of PCa (IMS1 and IMS2) by consensus clustering of m6A regulatory factors. IMS1/IMS2 subtypes have different prognoses and clinicopathological features in PCa patients, which are closely related to immune scores and immune cell infiltration. The prognosis of IMS1 patients was significantly better than that of patients with IMS2, which may be related to higher immune infiltration. This was consistent with previous findings that patients with high immune scores had superior survival compared with those with low immune scores. 37 As most m6A regulators were highly expressed in IMS2 cells, it was suggested that the abundance of m6A modification may remodel the tumor immune microenvironment, leading to an immune “cold” phenotype. At the same time, IMS2 had a higher proportion of DNA damage, such as HRD and ITH. Wang et al showed that a higher HRD score was associated with poor clinical outcomes in PRAD. 38 Several studies have found that ITH can be a prognostic factor for solid tumors, as it diminishes the immune response. 39 , 40 Consistently, the mutation spectrum showed that TP53 and ATM comutations especially occurred in IMS2, which has implications as a biomarker for guiding ICI treatment. TP53 and ATM comutation was associated with better OS than a single mutation and no mutation among patients with any cancer. 41 Most importantly, IMS2 showed higher expression of PD‐L1 and CXCL9, which are all biomarkers of sensitivity to PD‐1 blockade therapy. 42 At the same time, IMS2 cells showed lower expression of CD38, which represented low resistance to anti‐PD‐1 immunotherapy. 26 These results suggest that the IMS2 group probably responded to PD‐1 blockade therapy. However, other ICPs including TIM3 and CTLA4 were also significantly highly expressed in IMS2, which means that the inhibitory immune microenvironment of this group of patients may not be activated by pure antiPD‐1/PD‐L1 therapy. HNRNPC, a m6A reader, was identified as the hub gene interacting with HLA‐A, HLA‐B, and B2M. We found MEX3B, which was proved to mediate resistance to cancer immunotherapy by downregulating HLA‐A expression, showed higher expression in IMS2. Moreover, HNRNPC could regulate the m6A modification of HLA‐A, HLA‐B, and B2M (shown in the RM2Target and RMBase databases 43 , 44 ), and a previous study showed that B2M mutations are closely related to the abundance of Treg cells, which might lead to an immunosuppression. 45 In conclusion, a novel immunotherapeutic strategy based on the identification of IMS2 patients might greatly benefit their survival.
We identified HNRNPC as a hub gene that interacts with m6A methylation via immune processes and can effectively classify IMSs. Some studies found an association between HNRNPC and tumor progression, 46 , 47 , 48 as well as immune cell infiltration. 49 However, which immune cells can be regulated by HNRNPC, thus remodeling the TME is still unclear.
Our results suggest that HNRNPCs promote the activation of Treg cells. Immunosuppression in the TME, mediated by Tregs, is the main mechanism of tumor immune escape, which is associated with poor prognosis. 50 On the single‐cell level, we described the tumor immune microenvironment, especially the T cell subset. The rate of HNRNPC positive cells was significantly positively correlated with the proportion of epithelial cells and negatively correlated with the proportion of T cells. More importantly, the proportion of Treg cells in positive cells is higher, which represented a state of immunosuppression, consistent with our findings in the bulk RNA‐seq dataset. Observing the flow of communication signals between cells, we found that SPP1 and TGF‐β signal flows were activated in the positive group, which indicates immunosuppression in several cancers. 51 , 52 , 53 , 54 Furthermore, the epithelial cells in the positive group mainly interact with T cells through MIF‐(CD74+CXCR4) and MIF‐(CD74+CD44), which can mediate the formation of an immunosuppressive microenvironment caused by tumor hypoxia. 55 Our study indicated that HNRNPC can promote the infiltration of Tregs by coculture of PBMCs and PCa cells, which may be a potential mechanism by which HNRNPC promotes PCa progression through inhibiting T cell activity. Therefore, we suspected that HNRNPC could promote the infiltration of Tregs and T cell exhaustion to reshape the immunosuppressive microenvironment and promote the progression of PCa.
Our study has several limitations. First, our results were confirmed in both TCGA and GEO databases. Owing to the lack of sufficient in‐house data, the results were not externally verified. Therefore, it is necessary to validate these results in a multicenter cohort. In addition, our results showed that HNRNPC cells promoted Treg cell activation and suppression of effector CD8 T cells. However, the regulatory mechanism of m6A methylation needs to be further elucidated to improve precise immunotherapy of PCa.
Our findings provide new insights into the m6A‐TME interplay in PCa progression and present a potential target. We identified a “cold” immune phenotype in progressed PCa, and m6A reader HNRNPC regulating the abundance of Treg cells may be the mechanism of m6A methylation–mediated response to anti‐CTLA‐4. Multiple immunosuppressive phenotypes suggested that single checkpoint inhibitor is likely to produce drug resistance. Therefore, activation of the immune microenvironment through targeting upstream m6A regulators may be a potential therapeutic option for advanced PCa.
ACKNOWLEGMENTS
None.
FUNDING INFORMATION
This work was supported by the Medical Research Project of the Jiangsu Provincial Health and Family Planning Commission (No. H2018052) and Young Talents Program of the Jiangsu Cancer Hospital (No. 2017YQL‐04).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: N/A.
Supporting information
Supporting information S1.
Cheng Y, Li L, Wei X, et al. HNRNPC suppresses tumor immune microenvironment by activating Treg cells promoting the progression of prostate cancer. Cancer Sci. 2023;114:1830‐1845. doi: 10.1111/cas.15745
Yifei Cheng, Lu Li, and Xiyi Wei contributed equally to this article.
Contributor Information
Feng Qi, Email: qf199408@163.com.
Yanyan Zhang, Email: dabaoyan@njmu.edu.cn.
Xiao Li, Email: leex91@163.com.
DATA AVAILABILITY STATEMENT
TCGA data are openly available at https://portal.gdc.cancer.gov/.
The GEO data are openly available at https://www.ncbi.nlm.nih.gov/gds.
REFERENCES
- 1. Siegel R, Miller K, Fuchs H, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7‐33. doi: 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- 2. Bray F, Ferlay J, Soerjomataram I, Siegel R, Torre L, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 3. Siegel R, Miller K, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7‐34. doi: 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 4. Halabi S, Kelly W, Ma H, et al. Meta‐analysis evaluating the impact of site of metastasis on overall survival in men with castration‐resistant prostate cancer. J Clin Oncol. 2016;34(14):1652‐1659. doi: 10.1200/jco.2015.65.7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huinen Z, Huijbers E, van Beijnum J, Nowak‐Sliwinska P, Griffioen A. Anti‐angiogenic agents – overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol. 2021;18:527‐540. doi: 10.1038/s41571-021-00496-y [DOI] [PubMed] [Google Scholar]
- 6. Yap T, Parkes E, Peng W, Moyers J, Curran M, Tawbi H. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021;11:1368‐1397. doi: 10.1158/2159-8290.Cd-20-1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beer T, Kwon E, Drake C, et al. Randomized, double‐blind, phase III trial of Ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy‐naive castration‐resistant prostate cancer. J Clin Oncol. 2017;35(1):40‐47. doi: 10.1200/jco.2016.69.1584 [DOI] [PubMed] [Google Scholar]
- 8. Maia M, Hansen A. A comprehensive review of immunotherapies in prostate cancer. Crit Rev Oncol Hematol. 2017;113:292‐303. doi: 10.1016/j.critrevonc.2017.02.026 [DOI] [PubMed] [Google Scholar]
- 9. Graff J, Alumkal J, Drake C, et al. Early evidence of anti‐PD‐1 activity in enzalutamide‐resistant prostate cancer. Oncotarget. 2016;7(33):52810‐52817. doi: 10.18632/oncotarget.10547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kwon E, Drake C, Scher H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration‐resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184‐043): a multicentre, randomised, double‐blind, phase 3 trial. Lancet Oncol. 2014;15(7):700‐712. doi: 10.1016/s1470-2045(14)70189-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gubin M, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature. 2014;515(7528):577‐581. doi: 10.1038/nature13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang G, Zhao D, Spring D, DePinho R. Genetics and biology of prostate cancer. Genes Dev. 2018;32:1105‐1140. doi: 10.1101/gad.315739.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du K, Zhang L, Lee T, Sun T. mA RNA methylation controls neural development and is involved in human diseases. Mol Neurobiol. 2019;56(3):1596‐1606. doi: 10.1007/s12035-018-1138-1 [DOI] [PubMed] [Google Scholar]
- 14. Wang S, Sun C, Li J, et al. Roles of RNA methylation by means of N‐methyladenosine (mA) in human cancers. Cancer Lett. 2017;408:112‐120. doi: 10.1016/j.canlet.2017.08.030 [DOI] [PubMed] [Google Scholar]
- 15. Geula S, Moshitch‐Moshkovitz S, Dominissini D, et al. Stem Cells. m6A mRNA Methylation Facilitates Resolution of naïve Pluripotency toward Differentiation. Science (New York, NY). 2015;347(6225):1002‐1006. doi: 10.1126/science.1261417 [DOI] [PubMed] [Google Scholar]
- 16. Li H, Tong J, Zhu S, et al. mA mRNA methylation controls T cell homeostasis by targeting the IL‐7/STAT5/SOCS pathways. Nature. 2017;548(7667):338‐342. doi: 10.1038/nature23450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen T, Hao Y, Zhang Y, et al. M(6)a RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16(3):289‐301. doi: 10.1016/j.stem.2015.01.016 [DOI] [PubMed] [Google Scholar]
- 18. Cui Q, Shi H, Ye P, et al. mA RNA methylation regulates the self‐renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622‐2634. doi: 10.1016/j.celrep.2017.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Y, Hsu P, Chen Y, Yang Y. Dynamic transcriptomic mA decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616‐624. doi: 10.1038/s41422-018-0040-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812‐830 e14. doi: 10.1016/j.immuni.2018.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gibney G, Weiner L, Atkins M. Predictive biomarkers for checkpoint inhibitor‐based immunotherapy. Lancet Oncol. 2016;17(12):e542‐e551. doi: 10.1016/s1470-2045(16)30406-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koyama S, Akbay E, Li Y, et al. Adaptive resistance to therapeutic PD‐1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cha E, Klinger M, Hou Y, et al. Improved survival with T cell clonotype stability after anti‐CTLA‐4 treatment in cancer patients. Sci Transl Med. 2014;6(238):238ra70. doi: 10.1126/scitranslmed.3008211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chow M, Ozga A, Servis R, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity. 2019;50(6):1498‐1512.e5. doi: 10.1016/j.immuni.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang L, Malu S, McKenzie J, et al. The RNA‐binding protein MEX3B mediates resistance to cancer immunotherapy by downregulating HLA‐A expression. Clin Cancer Res. 2018;24(14):3366‐3376. doi: 10.1158/1078-0432.Ccr-17-2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen L, Diao L, Yang Y, et al. CD38‐mediated immunosuppression as a mechanism of tumor cell escape from PD‐1/PD‐L1 blockade. Cancer Discov. 2018;8(9):1156‐1175. doi: 10.1158/2159-8290.Cd-17-1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aran D, Looney AP, Liu L, et al. Reference‐based analysis of lung single‐cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163‐172. doi: 10.1038/s41590-018-0276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andreatta M, Corria‐Osorio J, Muller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single‐cell transcriptomics data using reference atlases. Nat Commun. 2021;12(1):2965. doi: 10.1038/s41467-021-23324-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin S, Guerrero‐Juarez CF, Zhang L, et al. Inference and analysis of cell‐cell communication using CellChat. Nat Commun. 2021;12(1):1088. doi: 10.1038/s41467-021-21246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giganti G, Atif M, Mohseni Y, et al. Treg cell therapy: how cell heterogeneity can make the difference. Eur J Immunol. 2021;51(1):39‐55. doi: 10.1002/eji.201948131 [DOI] [PubMed] [Google Scholar]
- 31. Bulten W, Kartasalo K, Chen P, et al. Artificial intelligence for diagnosis and Gleason grading of prostate cancer: the PANDA challenge. Nat Med. 2022;28:154‐163. doi: 10.1038/s41591-021-01620-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scelo G, Larose TL. Epidemiology and risk factors for kidney cancer. J Clin Oncol. 2018;36:3581. doi: 10.1200/jco.2018.79.1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Pan C, Wang X, et al. Silencing of METTL3 effectively hinders invasion and metastasis of prostate cancer cells. Theranostics. 2021;11(16):7640‐7657. doi: 10.7150/thno.61178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Xie H, Ying Y, et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6‐methyladenosine‐dependent way in prostate cancer. Mol Cancer. 2020;19(1):152. doi: 10.1186/s12943-020-01267-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Z, Zhong J, Zeng J, et al. Characterization of the m6A‐associated tumor immune microenvironment in prostate cancer to aid immunotherapy. Front Immunol. 2021;12:735170. doi: 10.3389/fimmu.2021.735170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Y, Sun H, Zheng J, Shao C. Analysis of RNA m(6)a methylation regulators and tumour immune cell infiltration characterization in prostate cancer. Artif Cells Nanomed Biotechnol. 2021;49(1):407‐435. doi: 10.1080/21691401.2021.1912759 [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Song L, Shen J, et al. Prognostic and predictive values of immune infiltrate in patients with head and neck squamous cell carcinoma. Hum Pathol. 2018;82:104‐112. doi: 10.1016/j.humpath.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 38. Knijnenburg T, Wang L, Zimmermann M, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23(1):239‐254.e6. doi: 10.1016/j.celrep.2018.03.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021;27(2):212‐224. doi: 10.1038/s41591-021-01233-9 [DOI] [PubMed] [Google Scholar]
- 40. Wolf Y, Bartok O, Patkar S, et al. UVB‐induced tumor heterogeneity diminishes immune response in melanoma. Cell. 2019;179(1):219‐235.e21. doi: 10.1016/j.cell.2019.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y, Chen G, Li J, et al. Association of Tumor Protein p53 and ataxia‐telangiectasia mutated Comutation with response to immune checkpoint inhibitors and mortality in patients with non‐small cell lung cancer. JAMA Netw Open. 2019;2(9):e1911895. doi: 10.1001/jamanetworkopen.2019.11895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Litchfield K, Reading J, Puttick C, et al. Meta‐analysis of tumor‐ and T cell‐intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184(3):596‐614.e14. doi: 10.1016/j.cell.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bao X, Zhang Y, Li H, et al. RM2Target: a comprehensive database for targets of writers, erasers and readers of RNA modifications. Nucleic Acids Res. 2023;51(D1):D269‐D279. doi: 10.1093/nar/gkac945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xuan JJ, Sun WJ, Lin PH, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46(D1):D327‐D334. doi: 10.1093/nar/gkx934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Echterdiek F, Janikovits J, Staffa L, et al. Low density of FOXP3‐positive T cells in normal colonic mucosa is related to the presence of beta2‐microglobulin mutations in lynch syndrome‐associated colorectal cancer. Onco Targets Ther. 2016;5(2):e1075692. doi: 10.1080/2162402X.2015.1075692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu Z, Chen Q, Shu L, Zhang C, Liu W, Wang P. Expression profiles of m6A RNA methylation regulators, PD‐L1 and immune infiltrates in gastric cancer. Front Oncol. 2022;12:970367. doi: 10.3389/fonc.2022.970367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo W, Tan F, Huai Q, et al. Comprehensive analysis of PD‐L1 expression, immune infiltrates, and m6A RNA methylation regulators in esophageal squamous cell carcinoma. Front Immunol. 2021;12:669750. doi: 10.3389/fimmu.2021.669750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gruber A, Schmidt R, Ghosh S, et al. Discovery of physiological and cancer‐related regulators of 3' UTR processing with KAPAC. Genome Biol. 2018;19(1):44. doi: 10.1186/s13059-018-1415-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S, Xu G, Chao F, Zhang C, Han D, Chen G. HNRNPC promotes proliferation, metastasis and predicts prognosis in prostate cancer. Cancer Manag Res. 2021;13:7263‐7276. doi: 10.2147/CMAR.S330713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jacobs J, Nierkens S, Figdor C, de Vries I, Adema G. Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? Lancet Oncol. 2012;13(1):e32‐e42. doi: 10.1016/s1470-2045(11)70155-3 [DOI] [PubMed] [Google Scholar]
- 51. Watermann C, Pasternack H, Idel C, et al. Recurrent HNSCC Harbor an immunosuppressive tumor immune microenvironment suggesting successful tumor immune evasion. Clin Cancer Res. 2021;27(2):632‐644. doi: 10.1158/1078-0432.CCR-20-0197 [DOI] [PubMed] [Google Scholar]
- 52. Ciummo SL, D'Antonio L, Sorrentino C, et al. The C‐X‐C motif chemokine ligand 1 sustains breast cancer stem cell self‐renewal and promotes tumor progression and immune escape programs. Front Cell Dev Biol. 2021;9:689286. doi: 10.3389/fcell.2021.689286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zeng F, Zhang Y, Han X, Zeng M, Gao Y, Weng J. Predicting non‐alcoholic fatty liver disease progression and immune deregulations by specific gene expression patterns. Front Immunol. 2020;11:609900. doi: 10.3389/fimmu.2020.609900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pawlik A, Anisiewicz A, Filip‐Psurska B, et al. Calcitriol and its analogs establish the immunosuppressive microenvironment that drives metastasis in 4T1 mouse mammary gland cancer. Int J Mol Sci. 2018;19(7):2116. doi: 10.3390/ijms19072116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y, Wang Z, Jia F, et al. CXCR4‐guided liposomes regulating hypoxic and immunosuppressive microenvironment for sorafenib‐resistant tumor treatment. Bioact Mater. 2022;17:147‐161. doi: 10.1016/j.bioactmat.2022.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information S1.
Data Availability Statement
TCGA data are openly available at https://portal.gdc.cancer.gov/.
The GEO data are openly available at https://www.ncbi.nlm.nih.gov/gds.