

Abstract
Numerous omics studies, primarily genomics analyses, have been conducted to fully understand the molecular biological characteristics of cancer. In recent years, the depth of proteomic analysis, which comprehensively analyzes proteins and molecules that function directly in vivo, has increased dramatically. Proteomics using mass spectrometry (MS) is a promising technology to directly examine proteoforms, including post‐translational modifications and variants originating from genomic aberrations. Recent advances in MS‐based proteomics have enabled direct, in depth, and quantitative analysis of the expression levels of various cancer‐related proteins, as well as their cancer‐specific proteoforms, and proteins that fluctuate with cancer initiation and progression in cell lines and tissue samples. Additionally, the integration of proteomic data with genomic, epigenomic, and transcriptomic data has formed the growing field of proteogenomics, which is already yielding new biological and diagnostic knowledge. Deep proteomic profiling provides clinically useful information in various aspects, including understanding the mechanisms of cancer development and progression and discovering targets for diagnosis and drug development. Furthermore, it is expected to make a significant contribution to the promotion of personalized medicine. In this review, recent advances and impacts in MS‐based clinical proteomics are highlighted with a focus on oncology.
Keywords: biomarker, cancer, glycoproteomics, mass spectrometry, proteogenomics
Clinical proteomics has become increasingly important in recent years. In this review, we provide an overview of state‐of‐the‐art mass spectrometry techniques in cancer research and describe our targeted glycoform analysis for the development of specific cancer biomarkers and our immunopeptidomics studies for the identification of tumor‐specific immunopeptides.
Abbreviations
- DIA
data‐independent acquisition
- ESI‐MS
electrospray ionization mass spectrometry
- FDA
Food and Drug Administration
- HLA
human leukocyte antigen
- LacdiNAc
GalNAcβ1‐4GlcNAc
- LC
liquid chromatography
- MS
mass spectrometry
- PEA
proximity extension assay
- PSA
prostate‐specific antigen
- PTM
post‐translational modification
- WES
whole‐exome sequencing
1. INTRODUCTION
Cancer biomarkers are proteins and other substances that are characteristically produced by different types of cancer, and their detection using enzymes or antibodies has played a major role in determining diagnosis and treatment strategies. In recent years, however, technological advances in genomics have led to the application of liquid biopsy to genomic medicine for cancer. Liquid biopsy is used to search for traces of cancer in body fluids (blood, urine, etc.). The widespread use of liquid biopsy is largely due to innovative advances in genomic analysis technology that enable single‐cell analysis. This modality has received considerable attention given its noninvasive nature and the low risks associated with prospective sampling. 1 As a result of the development of highly sensitive and specific genetic analysis technologies, several tests, including FoundationOne Liquid CDx and Guardant 360 CDx, have already been approved by the US Food and Drug Administration (FDA) as liquid biopsy tests for companion diagnostics. 2 However, the recent significant progress in various omics analytical technologies as well as data science has been remarkable. Although there have been clinical applications of genetic diagnosis, the integration of data from multiple modalities to transcend single‐omics is expected to improve in the future.
Proteins are responsible for most of the biochemical functions of the cell and are physically or functionally associated with other proteins and biomolecules. To understand the function of individual proteins and their spatiotemporal dynamics and fluctuations in complex biological systems, it is necessary to measure the variation and abundance of proteins in those systems. Furthermore, proteins are the targets of most cancer therapeutics, including in the growing field of immunotherapy. Therefore, clinical proteomics is becoming increasingly important. 3 However, the proteome in clinical samples is highly complex and is difficult to analyze in detail. The dynamic range of expressed proteins depends on the type of matrix, ranging from 8 orders of magnitude in tissue specimens 4 to approximately 12 orders of magnitude in body fluids such as plasma. 5 Although proteomics has lagged far behind genomic technology, mass spectrometry (MS)‐based proteomics has improved significantly over the past years and is increasingly used in various biological research. 6 , 7 In recent clinical proteomics, the exploration for novel biomarkers and therapeutic targets has been conducted by covering the entire proteome or by in‐depth examination of post‐translational modifications (PTMs) or cancer‐specific amino acid alterations. In this review, the latest mass spectrometric techniques in cancer research are outlined, as well as our targeted glycoform analyses for the development of specific cancer biomarkers and the immunopeptidomics for the identification of the tumor‐specific immunopeptides.
2. APPROACHES TO THE DEVELOPMENT OF DIAGNOSTIC AND THERAPEUTIC AGENTS BY APPLYING STATE‐OF‐THE‐ART PROTEOMICS
2.1. Overview of mass spectrometric platforms for proteomics
Mass spectrometric techniques have become a prominent method for the global and individual characterization of proteins. The typical approach of MS‐based protein analysis is the bottom‐up proteomics 6 (Figure 1). First, plasma proteins or extracted proteins from cells or tissue specimens are enzymatically digested into peptides. Peptides are usually separated by liquid chromatography (LC) and subsequently analyzed by electrospray ionization MS (ESI‐MS). LC‐ESI tandem MS (LC‐ESI‐MS/MS) data provide information on both the abundances and sequences of peptides in the sample.
FIGURE 1.
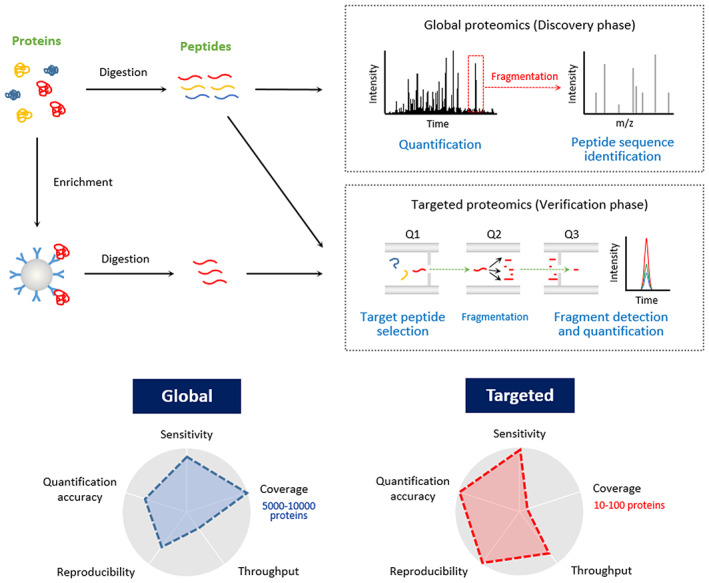
Liquid chromatography tandem mass spectrometry (LC‐MS/MS) workflow for proteomics. Proteins are extracted and digested into peptides by enzymes such as trypsin. Prior to digestion, enrichment steps, such as immunoprecipitation, can be performed. Peptides are separated by LC and then introduced and detected at the mass analyzer. In the global method, a full spectrum of peptide ions is obtained, followed by fragmentation to identify the peptide sequence. In targeted proteomics, peptides with a known mass‐to‐charge ratio (m/z) are selected at the first quadrupole (Q1) and then fragmented and monitored
Global proteomics is used to obtain an overall picture of the protein profile in a sample. These methods are used in the discovery phase to provide complete coverage of the proteome without bias. 6 Thousands of proteins can be identified and quantified in a single experiment. 8
In targeted proteomics, on the other hand, a relatively small number of proteins (typically <10) are subject to analysis. 9 For this reason, targeted proteomics is often used for candidate verification. This method requires information on the amino acid sequences of target proteins to set the optimal mass parameters specific to each of these proteins. 10 For example, the multiple reaction monitoring (MRM)/selected reaction monitoring (SRM) method specifically detects and quantifies specific peptides by combining a first quadrupole (Q1) filter, which allows the target peptide ion to pass, with a Q3 filter, which allows fragment ions after fragmentation (Figure 1). Absolute quantification of target proteins is also possible by using stable isotope–labeled peptides 11 or recombinant protein libraries 12 as internal standards. The main advantages of the targeted method over global proteomics are reproducibility, sensitivity, and throughput. Targeted methods can also be combined with enrichment methods, such as immunoprecipitation, which helps in the in‐depth characterization of target proteins.
Recently, a number of studies using alternative MS acquisition strategies, such as data‐independent acquisition (DIA), have been reported. DIA, also known as sequential window acquisition of all theoretical mass spectra (SWATH‐MS), 13 is a global proteomics approach that is expected to play an increasing role in the future as a method that enables comprehensive, quantitative, and reproducible analysis. 14 , 15 In DIA, theoretically all peptides in a sample are sequentially fragmented within a specified range of mass‐to‐charge ratio (m/z) values, and MS/MS spectra are acquired. These spectra are then matched to a predefined spectral library to identify proteins. Although the complexity of the DIA dataset made computational processing difficult, recent technological innovations in data science have made it more practical, and it is now possible to identify 10,000 proteins in a single shot. 16
The data obtained by the MS methods mentioned above are complex and of large size. Recent advances in informatics platforms support robust and precise interpretation of proteomics datasets. 17 , 18 , 19
2.2. Exploration of biomarkers in plasma by MS‐based proteomics
Cancer biomarkers are useful for diagnosis, prognosis, staging, monitoring therapeutic response or progression, and follow‐up. Compared with tissue biopsy, fluid‐based testing has many advantages: It is minimally invasive, has low risks associated with longitudinal sampling, is less labor intensive, and reflects the patient's tumor status in real time. 1 However, there are only approximately 30 blood‐ or body fluid–based biomarkers approved by the FDA which are clinically used. 20
The definitive goal of biomarker development is to establish reliable, accurate, and clinically applicable methods for the diagnosis of diseases. MS‐based proteomics has long been a powerful tool for cancer biomarker profiling in the context of various body fluids. 21 Although blood is the most widely used body fluid in clinical testing and a promising source of potential biomarkers, studying the plasma proteome has been challenging because very high concentrations of proteins, such as albumin, often mask potential disease biomarkers at lower concentrations. However, advances in MS‐based proteomic detection systems and sample preparation methods, such as removal of highly abundant proteins or fractionation of proteins or peptides, are partially overcoming these problems. 5 Recently, Blume et al achieved over seven orders of magnitude coverage of plasma proteome analysis by using unique nanoparticles. They successfully detected over 1000 proteins in a single pool of plasma with high reproducibility, including 53 FDA‐approved protein biomarkers. 22
Despite the tremendous efforts of researchers, discovering new serological biomarkers is not an easy task. One solution to this problem is to combine multiple markers to increase detection performance. A successful example of biomarker discovery by plasma/serum proteomics is the OVA1 test. OVA1 is an FDA‐cleared blood test that evaluates the levels of five ovarian cancer–associated markers (i.e., CA‐125, beta‐2 microglobulin, transferrin, apolipoprotein A1, and transthyretin). 23 , 24 , 25 The challenge of improving the performance of cancer markers by combining multiple proteins continues to be ongoing. 22 , 26 Given the rapid development of computer science, a new trend in the future may be scores that combine parameters from different modalities. For example, the CancerSEEK test analyzes mutations in 16 genes commonly found in cancer (e.g., TP53 and KRAS) and multiple cancer‐related proteins (e.g., CEA and AFP) and scores them using a logistic regression algorithm. 27 , 28
3. POST‐TRANSLATIONAL MODIFICATION ANALYSIS FOR PRECISE BIOMARKERS OF CANCER
3.1. Mass spectrometric analysis of proteoforms
Proteoforms are distinct protein forms resulting from genetic variations, splicing isoforms, and PTMs (e.g., phosphorylation and glycosylation). 29 In several important clinical areas, proteoforms have already been identified and reported to be associated with the progression of disease, including cancer. 30 Currently, MS is used to analyze proteoforms, providing deep, quantitative information on target proteins. Although recent improvements in MS have enabled large‐scale identification of proteins, analyzing proteoforms in complex samples is still very challenging. This is because the abundance of these variants is usually only a small fraction of the sample. Therefore, an appropriate enrichment step is necessary to analyze proteoform variants in depth. 7 , 31
Defining PTMs that are specifically expressed by cancer cells can greatly improve the diagnostic performance of known cancer biomarkers. In recent years, The Cancer Genome Atlas (TCGA) has identified many alterations in the expression of glycosylation‐associated genes that correlate with cancers. 32 Aberrant glycosylation is well defined as a hallmark of cancer and represents a promising source of potential biomarkers. 33 , 34 New technologies and new methods for glycan analysis support quantitative and qualitative characterization of cancer‐specific glycoforms in combination with the protein backbone with sufficient sensitivity and specificity for clinical applications. 34 , 35 As an example, we summarize key research on prostate‐specific antigen (PSA) glycoform analysis for improving the diagnostic accuracy of PSA.
3.2. PSA glycoform as a prostate cancer biomarker
Prostate cancer is the most frequently diagnosed cancer in men worldwide, 36 accounting for 27% of all newly diagnosed cancers in the United States. 37 PSA is detected in the blood of patients with prostate cancer and is utilized for diagnosis, to follow‐up the response to therapy and to monitor the clinical course of the disease. 38 , 39 However, the role of PSA in screening asymptomatic men for prostate cancer is controversial due to its low specificity. 40 , 41 In fact, only 25% of cases with PSA levels within the gray zone (4.0‐10.0 ng/mL) were pathologically confirmed as prostate cancer. 42 , 43 In recent years, several new biomarkers, such as the prostate health index (PHI) and prostate cancer antigen 3 (PCA3), have become available to improve the accuracy of prostate cancer diagnosis. 44
One recent approach to improve the specificity of the serum PSA test is to examine cancer‐specific glycoforms on PSA. With recent advances in glycan structure analysis technology, promising glycan structures have been identified as biomarkers. 45 , 46 Among them, α2,3‐sialylation, 47 , 48 , 49 core fucosylation, 50 or GalNAcβ1‐4GlcNAc (LacdiNAc) structure 51 , 52 , 53 have received much attention as promising candidates (Figure 2), and platforms for corresponding clinical application are being developed.
FIGURE 2.
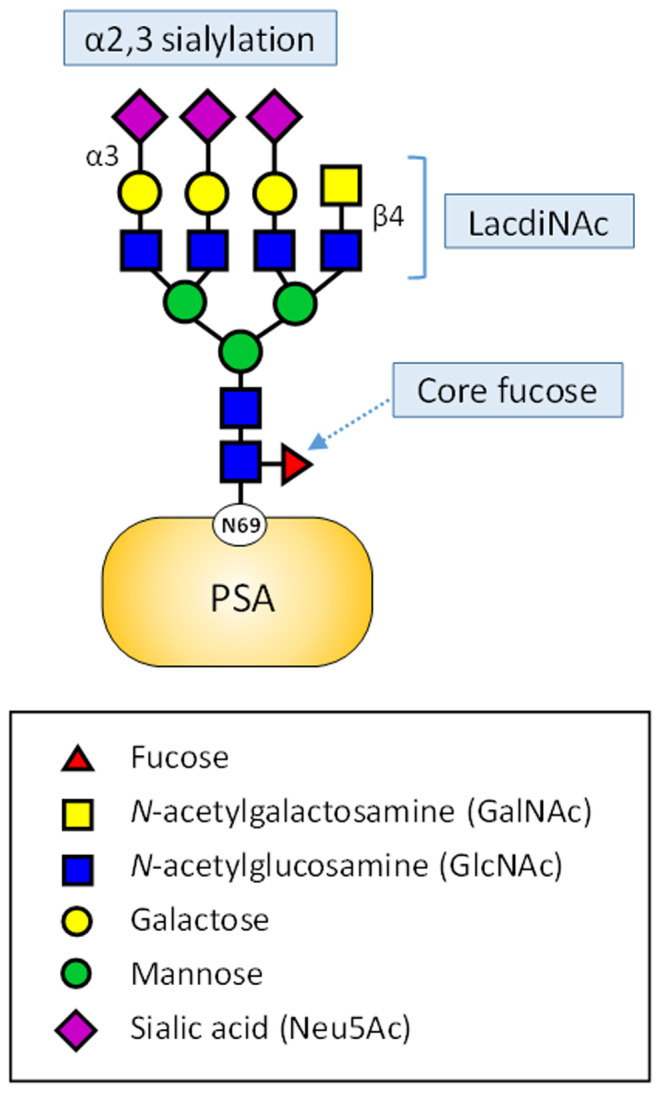
Prostate cancer–specific glycoforms on serum prostate‐specific antigen (PSA). Characteristic N‐glycan structures of PSA whose blood level increases with the progression of prostate cancer. Human PSA has a single N‐glycan at Asn69. Glycan nomenclature follows the Symbol Nomenclature for Glycans guidelines 71
Analysis of glycopeptides by MS allows identification of the glycosylation sites and the complete repertoire of glycan structures of a target protein. By using in‐depth quantitative MS, we found that the abundance of multisialylated LacdiNAc structures was significantly elevated in prostate cancer patients compared with benign prostate hyperplasia (BPH) patients 53 (Figure 3). The LacdiNAc structure is poorly expressed in humans, whereas its increased expression has been reported in prostate, ovarian, and pancreatic cancers. 51 , 54 , 55 In particular, increased expression of β‐N‐acetylgalactosaminyltransferase 4 (B4GALNT4), a glycosyltransferase responsible for the LacdiNAc structure, has been observed in prostate cancer patients. 51 A new diagnostic model, the PSA G‐index, was constructed by logistic regression analysis, and the AUC was 1.0 (both sensitivity and specificity were 100%, n = 30).
FIGURE 3.
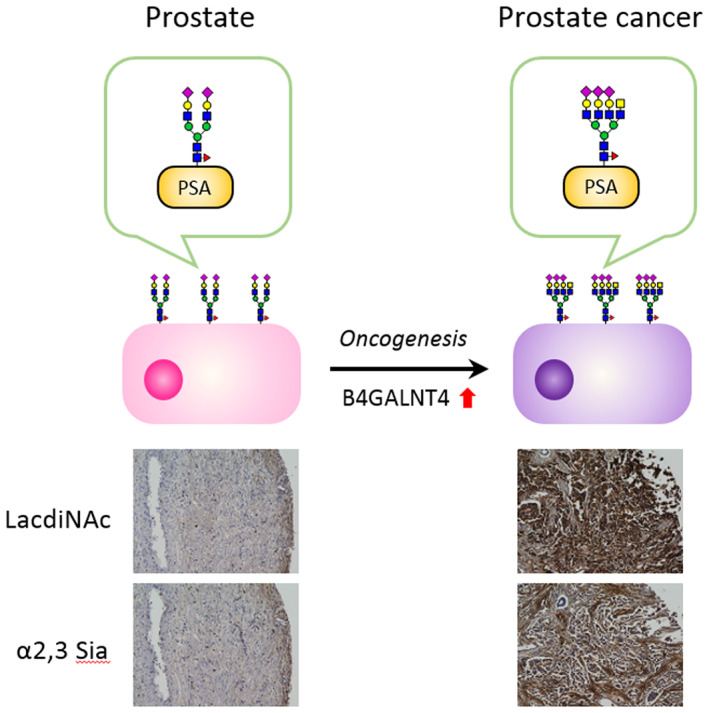
Lectin histochemical staining of prostate tissue specimens and proposed model for the production of prostate cancer–specific glycoforms on prostate‐specific antigen (PSA). LacdiNAc structures are rarely expressed in normal prostate tissue, while prostate cancer cells showed a characteristic tendency to express glycoproteins containing LacdiNAc and α2,3‐sialylated structures, resulting in increased multisialylated LacdiNAc structures on PSA of prostate cancer patients. These findings were consistent with the results of glycoform analysis of PSA by mass spectrometry 53
These studies indicate that cancer‐specific glycan structures could be useful targets for cancer diagnosis. Recent enhancements in glycoinformatics platforms have also enabled global proteomic studies of glycoproteins. 56 With improved analytical techniques and more efficient sample analysis, glycoproteomics is expected to become a promising tool for biomarker discovery.
4. CANCER PROTEOGENOMICS
With the advances of technologies such as next‐generation DNA sequencing and RNA sequencing (RNA‐seq), genome analysis in cancer has shown tremendous impacts, including the identification of driver mutations and the development of targeted therapies. Deep sequencing analyses such as whole‐genome sequencing (WGS), whole‐exome sequencing (WES), and RNA‐seq from clinical samples, including cancer tissues, can be used to construct personalized proteome databases. With these personalized databases, the mutant proteins that carry cancer‐specific somatic mutations can be identified by MS. This integrated field of genomics and proteomics is called proteogenomics. All genomic, epigenomic, and transcriptomic data are available for proteogenomics according to the research purpose and the sample types, such as preclinical cancer models and tumor specimens. 7 , 57 , 58 More recently, proteogenomic detection of amino acid alterations caused by somatic mutations in cancer cells has flourished as the sensitivity and depth of MS have improved.
One example of cancer proteogenomics is immunopeptidomics, a type of peptide genomics that retrieves immunopeptide sequences from a personalized proteome database (Figure 4). The presentation of tumor antigens by human leukocyte antigen (HLA) molecules on the surface of cancer cells is important as a trigger for the recognition of cancer cells by tumor‐killing T cells. The efficacy of T‐cell–based cancer immunotherapy depends on the recognition of HLA‐binding peptides presented on the surface of cancer cells. Therefore, for the development of cancer vaccines and T‐cell–based adaptive immunotherapy, it is crucial to characterize and classify immunogenic epitopes derived from endogenously expressed source proteins. Cancer‐specific amino acid alterations due to somatic mutations in cancer cells are known to be one of the sources of tumor‐specific immunopeptides (neoantigens) that play important roles in immune cell recognition of cancer cells as nonself cells. 59 To date, MS is the only analytical technique that can directly identify the amino acid sequences of the actual presented immunopeptides 60 , 61 , 62 (Figure 4).
FIGURE 4.
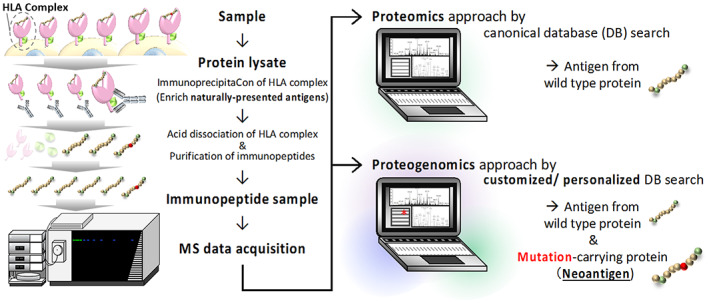
Schematic workflow for immunopeptidomics by a proteogenomics approach. General sample preparation of immunopeptides (left panel) and neoantigen identification by proteogenomics approach (right panel) are shown. The central mediators of cancer immunotherapy are immunopeptides that are specifically presented only by cancer cells. The tumor‐specific immunopeptides are called neoantigens. The immunopeptide is presented extracellularly by forming a human leukocyte antigen (HLA) complex. After isolating the HLA complex by immunoprecipitation, an enriched immunopeptide sample can be obtained through the dissociation of the HLA complex and the immunopeptide purification process (left panel). In the discovery phase, the general proteomics approach, which refers to the canonical sequence of the human proteome, can only identify antigens that can be presented by both nontumor and normal cells (upper right panel). Therefore, careful verification of the cancer specificity of the antigen (e.g., whether the source protein of the antigen is highly expressed in cancer cells) is critical for practical application. The proteogenomics approach identifies neoantigens by reference to the amino acid sequence of cancer‐specific mutations constructed from genomic and transcriptomic information (lower right panel). Identification of neoantigens by mass spectrometry (MS) generally ensures natural and actual presentation and cancer specificity of the antigen of interest, contributing to more effective antigen selection
Advances in proteogenomics methodology have expanded the exploration of neoantigens not only to canonical tumor antigens derived from coding regions but also to noncanonical peptides (i.e., antigens originating from aberrant translation of non–protein‐coding transcripts and aberrant or regulated transcription of noncoding genomic regions) 63 , 64 (Figure 5). The identification of a large number of immunopeptides with such a diverse landscape from clinical samples by sophisticated global immunopeptidomic analysis will enable profiling of cancer‐specific immunopeptides. The profiling of cancer‐specific immunopeptides may also contribute to the construction of advanced predictive algorithms for precision medicine in cancer. In contrast to this broadening of the immunopeptide landscape, more focused targeted MS is also gaining attention. This methodology is useful for gaining insight into more specific targets, for example, to determine if a desired neoantigen is present in a sample. As most somatic mutations found in cancer cells are personal and unique, the overlap of neoantigens between patients is highly limited. From a health‐economic perspective, designing immunotherapies based on these mutations is not currently appropriate. On the other hand, immunotherapies targeting cancer driver mutations are expected to become more common as off‐the‐shelf immunotherapies due to their overlap among cancer patients and pancancer status. By means of a global and targeted immunopeptidomic approach, we have identified neoantigens, including oncogenic KRAS (G12V), from cell lines and colorectal cancer tissue. 62 Given that the MS‐based approach is an ideal method for the direct and robust analysis of immunopeptides in clinical samples, it will contribute to neoantigen selection for the successful development of more effective cancer immunotherapy.
FIGURE 5.
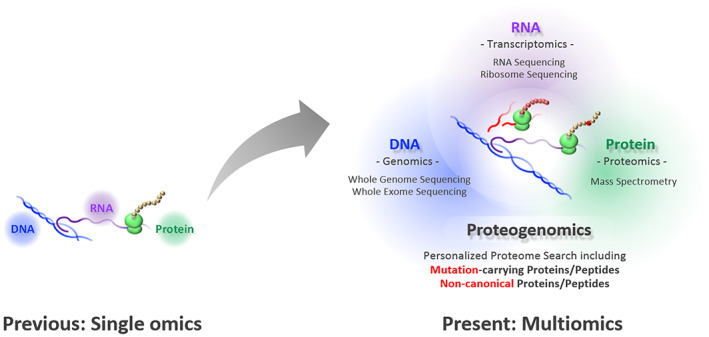
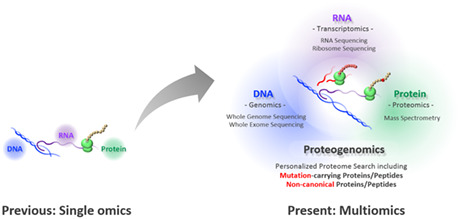
Transcending single omics to integrated multiomics. A few decades ago, genomics, transcriptomics, and proteomics were established independently as academic disciplines. Following the massive advances in deep sequencing technology, new frontiers in the proteome landscape are emerging with the development of mass spectrometry (MS). MS‐based proteomics now bridges an individual's genomic blueprint with clinically relevant physiology as proteogenomics and, beyond that, as multiomics
In this way, MS is playing a growing role in multiomics studies that are essential for precision medicine.
5. CONCLUSION
In this review, the recent impact and applications of mass spectrometric technologies in cancer research are introduced. In‐depth analysis of tumor‐derived proteins/peptides may provide important and practical insights into cancer biology. This information would also be useful in the development of novel cancer biomarkers and drugs that take into account the characteristics and clinical needs in oncology.
Just as single‐cell WES and single‐cell RNA‐seq were major breakthroughs in the field of genetic analysis, very recently, more sensitive and innovative technologies have begun to be reported in the field of proteomics, enabling single‐cell analysis. 65 , 66 , 67 , 68 These single‐cell proteomics are expected to contribute to cancer research by identifying and characterizing rare cells, such as cancer stem cells, and detecting the early stages of drug resistance.
Although mass spectrometric techniques are currently difficult to routinely apply in clinical use, further improvements in sample processing and MS instruments are expected to enable clinical proteomics at reasonable throughput. On the other hand, while MS is still the most efficient method to search for target molecules, a number of innovative non–MS‐based technologies have been developed in recent years, enabling highly sensitive and multiplexed analysis of proteins. 21 For example, the proximity extension assay (PEA), which is commercialized by Olink, uses matched pairs of antibodies attached to unique DNA oligonucleotides for sensitive DNA‐readout methodologies. 69 , 70 Compared with MS, PEA is less comprehensive but confers advantages in sensitivity and specificity and requires less sample volume. The latest PEA can simultaneously measure approximately 3000 validated proteins from a few microliters of blood sample. The impact of these novel proteomics will be enormous, both academically and technologically. Clinical proteomics will be one of the promising solutions for the application of proteomics to integrate multiparametric data to build new diagnostics or to understand cancer biology.
FUNDING INFORMATION
This work was partly supported by the Japan Agency for Medical Research and Development (AMED) under grant number JP22ama221409 to Y.H.
CONFLICT OF INTEREST
K.U. has consulted for LSI Medience Corporation and received compensation. K.U. is also an editorial board member of Cancer Science. Y.H. and Y.M. have no conflicts of interest.
ACKNOWLEDGMENTS
We thank all members of our laboratory for scientific discussion.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board. N/A.
Informed Consent. N/A.
Registry and the Registration No. of the study/trial. N/A.
Animal Studies. N/A.
Haga Y, Minegishi Y, Ueda K. Frontiers in mass spectrometry–based clinical proteomics for cancer diagnosis and treatment. Cancer Sci. 2023;114:1783‐1791. doi: 10.1111/cas.15731
REFERENCES
- 1. Alix‐Panabières C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479‐491. [DOI] [PubMed] [Google Scholar]
- 2. Chelakkot C, Yang H, Shin YK. Relevance of circulating tumor cells as predictive markers for cancer incidence and relapse. Pharmaceuticals. 2022;15:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macklin A, Khan S, Kislinger T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin Proteomics. 2020;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang D, Eraslan B, Wieland T, et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol Syst Biol. 2019;15:e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paul J, Veenstra TD. Separation of serum and plasma proteins for In‐depth proteomic analysis. Separations. 2022;9:89. [Google Scholar]
- 6. Aebersold R, Mann M. Mass‐spectrometric exploration of proteome structure and function. Nature. 2016;537:347‐355. [DOI] [PubMed] [Google Scholar]
- 7. Mani DR, Krug K, Zhang B, et al. Cancer proteogenomics: current impact and future prospects. Nat Rev Cancer. 2022;22:298‐313. [DOI] [PubMed] [Google Scholar]
- 8. Muntel J, Gandhi T, Verbeke L, et al. Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC‐MS instrumentation and data analysis strategy. Mol Omics. 2019;15:348‐360. [DOI] [PubMed] [Google Scholar]
- 9. Geyer PE, Holdt LM, Teupser D, Mann M. Revisiting biomarker discovery by plasma proteomics. Mol Syst Biol. 2017;13:942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vidova V, Spacil Z. A review on mass spectrometry‐based quantitative proteomics: targeted and data independent acquisition. Anal Chim Acta. 2017;964:7‐23. [DOI] [PubMed] [Google Scholar]
- 11. Ludwig C, Claassen M, Schmidt A, Aebersold R. Estimation of absolute protein quantities of unlabeled samples by selected reaction monitoring mass spectrometry. Mol Cell Proteomics. 2012;11:M111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsumoto M, Matsuzaki F, Oshikawa K, et al. A large‐scale targeted proteomics assay resource based on an in vitro human proteome. Nat Methods. 2017;14:251‐258. [DOI] [PubMed] [Google Scholar]
- 13. Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R. Data‐independent acquisition‐based SWATH‐MS for quantitative proteomics: a tutorial. Mol Syst Biol. 2018;14:e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collins BC, Hunter CL, Liu Y, et al. Multi‐laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH‐mass spectrometry. Nat Commun. 2017;8:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Messner CB, Demichev V, Bloomfield N, et al. Ultra‐fast proteomics with scanning SWATH. Nat Biotechnol. 2021;39:846‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawashima Y, Nagai H, Konno R, et al. Single‐shot 10K proteome approach: over 10,000 protein identifications by data‐independent acquisition‐based single‐shot proteomics with ion mobility spectrometry. J Proteome Res. 2022;21:1418‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol. 2008;26:1367‐1372. [DOI] [PubMed] [Google Scholar]
- 18. Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry‐based proteomics. Nat Methods. 2017;14:513‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M. DIA‐NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods. 2020;17:41‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romano G. Tumor markers currently utilized in cancer care. Mater Methods. 2015;5:1456 (last modified: 2022‐10‐21). [Google Scholar]
- 21. Ding Z, Wang N, Ji N, Chen ZS. Proteomics technologies for cancer liquid biopsies. Mol Cancer. 2022;21:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blume JE, Manning WC, Troiano G, et al. Rapid, deep and precise profiling of the plasma proteome with multi‐nanoparticle protein corona. Nat Commun. 2020;11:3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rai AJ, Zhang Z, Rosenzweig J, et al. Proteomic approaches to tumor marker discovery. Arch Pathol Lab Med. 2002;126:1518‐1526. [DOI] [PubMed] [Google Scholar]
- 24. Zhang Z, Bast RC Jr, Yu Y, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004;64:5882‐5890. [DOI] [PubMed] [Google Scholar]
- 25. Fung ET. A recipe for proteomics diagnostic test development: the OVA1 test, from biomarker discovery to FDA clearance. Clin Chem. 2010;56:327‐329. [DOI] [PubMed] [Google Scholar]
- 26. Sun Y, Guo Z, Liu X, et al. Noninvasive urinary protein signatures associated with colorectal cancer diagnosis and metastasis. Nat Commun. 2022;13:2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science. 2018;359:926‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lennon AM, Buchanan AH, Kinde I, et al. Feasibility of blood testing combined with PET‐CT to screen for cancer and guide intervention. Science. 2020;369:eabb9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith LM, Kelleher NL. Proteoforms as the next proteomics currency. Science. 2018;359:1106‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith LM, Agar JN, Chamot‐Rooke J, et al. The human proteoform project: defining the human proteome. Sci Adv. 2021;7:eabk0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ongay S, Boichenko A, Govorukhina N, Bischoff R. Glycopeptide enrichment and separation for protein glycosylation analysis. J Sep Sci. 2012;35:2341‐2372. [DOI] [PubMed] [Google Scholar]
- 32. Bellis SL, Reis CA, Varki A, Kannagi R, Stanley P. Glycosylation changes in cancer. In: Varki A, Cummings RD, Esko JD, et al., eds. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; 2022:631‐644. [Google Scholar]
- 33. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540‐555. [DOI] [PubMed] [Google Scholar]
- 34. Haga Y, Ueda K. Glycosylation in cancer: its application as a biomarker and recent advances of analytical techniques. Glycoconj J. 2022;39:303‐313. [DOI] [PubMed] [Google Scholar]
- 35. Haga Y, Yamada M, Fujii R, et al. Fast and ultrasensitive Glycoform analysis by supercritical fluid chromatography‐tandem mass spectrometry. Anal Chem. 2022;94:15948‐15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209‐249. [DOI] [PubMed] [Google Scholar]
- 37. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7‐33. [DOI] [PubMed] [Google Scholar]
- 38. Wallner LP, Jacobsen SJ. Prostate‐specific antigen and prostate cancer mortality: a systematic review. Am J Prev Med. 2013;45:318‐326. [DOI] [PubMed] [Google Scholar]
- 39. Stephan C, Ralla B, Jung K. Prostate‐specific antigen and other serum and urine markers in prostate cancer. Biochim Biophys Acta. 2014;1846:99‐112. [DOI] [PubMed] [Google Scholar]
- 40. Thompson IM, Ankerst DP, Chi C, et al. Operating characteristics of prostate‐specific antigen in men with an initial PSA level of 3.0 ng/ml or lower. Jama. 2005;294:66‐70. [DOI] [PubMed] [Google Scholar]
- 41. Harvey P, Basuita A, Endersby D, Curtis B, Iacovidou A, Walker M. A systematic review of the diagnostic accuracy of prostate specific antigen. BMC Urol. 2009;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Catalona WJ, Partin AW, Slawin KM, et al. Use of the percentage of free prostate‐specific antigen to enhance differentiation of prostate cancer from benign prostatic disease: a prospective multicenter clinical trial. Jama. 1998;279:1542‐1547. [DOI] [PubMed] [Google Scholar]
- 43. Mistry K, Cable G. Meta‐analysis of prostate‐specific antigen and digital rectal examination as screening tests for prostate carcinoma. J Am Board Fam Pract. 2003;16:95‐101. [DOI] [PubMed] [Google Scholar]
- 44. Duffy MJ. Biomarkers for prostate cancer: prostate‐specific antigen and beyond. Clin Chem Lab Med. 2020;58:326‐339. [DOI] [PubMed] [Google Scholar]
- 45. Gilgunn S, Conroy PJ, Saldova R, Rudd PM, O'Kennedy RJ. Aberrant PSA glycosylation–a sweet predictor of prostate cancer. Nat Rev Urol. 2013;10:99‐107. [DOI] [PubMed] [Google Scholar]
- 46. Scott E, Munkley J. Glycans as biomarkers in prostate cancer. Int J Mol Sci. 2019;20:1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ishikawa T, Yoneyama T, Tobisawa Y, et al. An automated micro‐Total immunoassay system for measuring cancer‐associated alpha2,3‐linked Sialyl N‐glycan‐carrying prostate‐specific antigen may improve the accuracy of prostate cancer diagnosis. Int J Mol Sci. 2017;18:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoneyama T, Ohyama C, Hatakeyama S, et al. Measurement of aberrant glycosylation of prostate specific antigen can improve specificity in early detection of prostate cancer. Biochem Biophys Res Commun. 2014;448:390‐396. [DOI] [PubMed] [Google Scholar]
- 49. Hatano K, Yoneyama T, Hatakeyama S, et al. Simultaneous analysis of serum alpha2,3‐linked sialylation and core‐type fucosylation of prostate‐specific antigen for the detection of high‐grade prostate cancer. Br J Cancer. 2021;126:764‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fujita K, Hatano K, Tomiyama E, et al. Serum core‐type fucosylated prostate‐specific antigen index for the detection of high‐risk prostate cancer. Int J Cancer. 2021;148:3111‐3118. [DOI] [PubMed] [Google Scholar]
- 51. Fukushima K, Satoh T, Baba S, Yamashita K. Alpha1,2‐Fucosylated and beta‐N‐acetylgalactosaminylated prostate‐specific antigen as an efficient marker of prostatic cancer. Glycobiology. 2010;20:452‐460. [DOI] [PubMed] [Google Scholar]
- 52. Kaya T, Kaneko T, Kojima S, et al. High‐sensitivity immunoassay with surface plasmon field‐enhanced fluorescence spectroscopy using a plastic sensor chip: application to quantitative analysis of total prostate‐specific antigen and GalNAcbeta1‐4GlcNAc‐linked prostate‐specific antigen for prostate cancer diagnosis. Anal Chem. 2015;87:1797‐1803. [DOI] [PubMed] [Google Scholar]
- 53. Haga Y, Uemura M, Baba S, et al. Identification of Multisialylated LacdiNAc structures as highly prostate cancer specific glycan signatures on PSA. Anal Chem. 2019;91:2247‐2254. [DOI] [PubMed] [Google Scholar]
- 54. Machado E, Kandzia S, Carilho R, Altevogt P, Conradt HS, Costa J. N‐glycosylation of total cellular glycoproteins from the human ovarian carcinoma SKOV3 cell line and of recombinantly expressed human erythropoietin. Glycobiology. 2011;21:376‐386. [DOI] [PubMed] [Google Scholar]
- 55. Peracaula R, Royle L, Tabares G, et al. Glycosylation of human pancreatic ribonuclease: differences between normal and tumor states. Glycobiology. 2003;13:227‐244. [DOI] [PubMed] [Google Scholar]
- 56. Kawahara R, Chernykh A, Alagesan K, et al. Community evaluation of glycoproteomics informatics solutions reveals high‐performance search strategies for serum glycopeptide analysis. Nat Methods. 2021;18:1304‐1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, Paulovich AG. Clinical potential of mass spectrometry‐based proteogenomics. Nat Rev Clin Oncol. 2019;16:256‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chong C, Coukos G, Bassani‐Sternberg M. Identification of tumor antigens with immunopeptidomics. Nat Biotechnol. 2022;40:175‐188. [DOI] [PubMed] [Google Scholar]
- 59. Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen‐based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1:11‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bassani‐Sternberg M, Coukos G. Mass spectrometry‐based antigen discovery for cancer immunotherapy. Curr Opin Immunol. 2016;41:9‐17. [DOI] [PubMed] [Google Scholar]
- 61. Hirama T, Tokita S, Nakatsugawa M, et al. Proteogenomic identification of an immunogenic HLA class I neoantigen in mismatch repair‐deficient colorectal cancer tissue. JCI Insight. 2021;6:e146356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Minegishi Y, Kiyotani K, Nemoto K, et al. Differential ion mobility mass spectrometry in immunopeptidomics identifies neoantigens carrying colorectal cancer driver mutations. Commun Biol. 2022;5:831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chong C, Marino F, Pak H, et al. High‐throughput and sensitive Immunopeptidomics platform reveals profound Interferongamma‐mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol Cell Proteomics. 2018;17:533‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chong C, Muller M, Pak H, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non‐canonical peptides in tumor immunopeptidomes. Nat Commun. 2020;11:1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Specht H, Emmott E, Petelski AA, et al. Single‐cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021;22:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tsai CF, Zhang P, Scholten D, et al. Surfactant‐assisted one‐pot sample preparation for label‐free single‐cell proteomics. Commun Biol. 2021;4:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Woo J, Williams SM, Markillie LM, et al. High‐throughput and high‐efficiency sample preparation for single‐cell proteomics using a nested nanowell chip. Nat Commun. 2021;12:6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gebreyesus ST, Siyal AA, Kitata RB, et al. Streamlined single‐cell proteomics by an integrated microfluidic chip and data‐independent acquisition mass spectrometry. Nat Commun. 2022;13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96‐plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9:e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wik L, Nordberg N, Broberg J, et al. Proximity extension assay in combination with next‐generation sequencing for high‐throughput proteome‐wide analysis. Mol Cell Proteomics. 2021;20:100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Neelamegham S, Aoki‐Kinoshita K, Bolton E, et al. Updates to the symbol nomenclature for glycans guidelines. Glycobiology. 2019;29:620‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]