

Abstract
Ischemic stroke is a common complication of sickle cell disease (SCD) and without intervention can affect 11% of children with SCD before the age of 20. Within the Trans-Omics for Precision Medicine (TOPMed), a genome-wide association study (GWAS) of ischemic stroke was performed on 1,333 individuals with SCD from Brazil (178 cases, 1,155 controls). Via a novel Cox proportional hazards analysis, we searched for variants associated with ischemic stroke occurring at younger ages. Variants at genome wide significance (P<5×10−8) include two near genes previously linked to non-SCD early onset stroke (<65 years): ADAMTS2 (rs147625068, P=3.70 × 10−9) and CDK18 (rs12144136, P=2.38 × 10−9). Meta-analysis which included the independent SCD cohorts Walk-PHaSST and PUSH exhibited consistent association for variants rs1209987 near gene TBC1D32 (P = 3.36×10−10), rs188599171 near CUX1 (P = 5.89×10−11), rs77900855 near BTG1 (P=4.66×10−8), and rs141674494 near VPS13C (1.68×10−9). Finding from this study support a multi-variant model of early ischemic stroke risk and possibly a shared genetic architecture between SCD individuals and non-SCD individuals <65 years.
Keywords: GWAS, sickle cell disease, Brazil, stroke, ischemic stroke
INTRODUCTION
Ischemic stroke is one of the most common comorbidities of sickle cell disease (SCD), occurring 20 times more frequently than seen in non-SCD children1,2. Without early preventative therapy, 11% of children with SCD experience at least one overt ischemic stroke before the age of 203, with recurrence ranging from 60–92%4 and mortality of roughly 5%5. Ischemic stroke is more common than hemorrhagic stroke in children, with hemorrhagic stroke more common in ages 20–35 years. The pathophysiology of ischemic stroke within SCD is complex1, and the current standard for risk assessment, transcranial doppler ultrasonography (TCD) of cerebral vessels, is difficult to perform on children under age 3. Also, some individuals with normal TCD still experience stoke3,6,7.
Genetic risk of stroke has been investigated in SCD and non-SCD populations. Within SCD, stroke risk is highest in HbSS homozygotes compared to other SCD genotypes, whereas the coinheritance of alpha-thalassemia is protective against stroke1. There is mounting evidence for the existence of other heritable genetic risk factors for stroke.8–15 A GWAS of 677 African Americans with SCD discovered two nonsynonymous variants in the genes GOLGB1 and ENPP1 associated with protection from stroke13. The same variant in ENPP1 was confirmed within a Brazilian SCD cohort, however GOLGB1 was not replicated15. Recently, APOE variants were also shown to be associated with ischemic stroke in children with SCD ages four and younger16.
Investigating genetic risk within non-SCD stroke may provide additional insight into the similarity or differences compared to SCD. Within non-SCD pediatric ischemic stroke studies, risk variants near genes ADAMTS2, ADAMTS12, and ADAMTS13 haven been discovered, and both ADAMTS2 and ADAMTS12 have been replicated17,18. In separate studies, variants near CDK1819 , HABP220, were linked to early onset ischemic stroke but have not been replicated. Gaps in our knowledge of the genetic contributors to ischemic stroke risk within SCD remain. To address this, we conducted the largest genome-wide association study of stroke in individuals with sickle cell disease (N=1,333). This was conducted within the Brazil Sickle Cell Disease Cohort Study as part of the Recipient Epidemiology Donor Evaluation Study III (REDS-III)21.
METHODS
Two novel approaches were implemented in this study. Variants were measured via whole genome sequencing, avoiding the need to impute on reference panels which may have little to no overlap in ancestry with the study cohort. In addition, a Cox proportional hazards model was used to estimate differences in time to stroke, allowing for the discovery of genetic variants associated with earlier stroke events.
Study Population
The Brazil SCD Cohort Study is part of the National Institutes of Health, National Heart Lung and Blood Institute (NHLBI) REDS-III program.22 The Brazil National Research Ethics Commission, local ethical committees at each participating center and the Institutional Review Boards at University of California, San Francisco (UCSF) and the REDS-III data coordinating center, Research Triangle Institute, International (RTI) all reviewed and approved the study.
2,793 sickle cell disease individuals were enrolled at 6 different centers in Brazil: Hemominas Belo Horizonte, Juiz de Fora, and Montes Claros, Hemorio, Rio de Janeiro, Instituto de Tratamento do Câncer Infantil, Sao Paulo, and Hemope, Recife. Enrollment included interviews, medical abstraction, and blood collection. Written informed consent was obtained from participants ≥18 years or from guardians of younger patients, and assent was obtained for children aged 7 to 17. Medical records were abstracted for clinical history using standardized definitions23. Participants were routine patients at these centers, and relatively complete medical histories were available, including acute complications. This included a history of ischemic stroke defined as an acute neurological syndrome resulting from impaired cerebral blood flow without evidence of hemorrhage. Diagnostic criteria included either a magnetic resonance image (MRI) or a computerized tomography (CT) scan showing an infarctive central nervous system event consistent with symptoms and signs or diagnosis based on examination and clinical history with neurologic symptoms/signs lasting >24 hours. Controls were defined as individuals with SCD who had no history of stroke. While MRI/CT was available to confirm cases, current guidelines for care of SCD does not include MRI/CT when neurological symptoms are absent.
DNA collection and sequencing
Whole blood was collected in ethylenediaminetetraacetic acid (EDTA), and DNA was extracted from the buffy coat via ethanol precipitation. Purified DNA was quantified with RT-PCR, and normalized to 10 ng/uL. SCD genotypes were confirmed using allele-specific pyrosequencing (Qiagen, Hilden, Germany)24 and Sanger sequencing of exons 1 and 2 of gene HBB if the pyrosequencing results conflicted with medical records. Additional sequencing of HBB exon 3, introns, and promoter was performed for samples with unresolved genotypes at the Hemoglobinopathy Reference Laboratory at UCSF Benioff Children’s Hospital Oakland.
Whole genome sequencing was conducted within the NHLBI Trans-Omics for Precision Medicine (TOPMed) program (freeze 6a)25. Average sequencing depth was 38 fold, and variants were called jointly across roughly 140,000 samples, including the N=1,333 individuals in the current study, using the GotCloud pipeline.25 The list of roughly 18 million variants were filtered to include only those at observed minor allele frequency >1%, resulting in a final list of 14.3 million variant autosomal sites.
Exclusion Criteria
Individuals were excluded from this analysis for the following reasons: not having TOPMed DNA sequence data, being a duplicate sample, inability to link DNA to clinical records, missing age or stroke status, and not being an HbSS homozygote. Samples were also removed if they were clear outliers along principal components 1–3 of the genotype data. Finally, we also excluded individuals with no history of stroke who were either undergoing chronic transfusion therapy (CTT) or had abnormal transcranial doppler (TCD) readings (Supplemental Fig 1). This was done to ensure a more homogenous population included in the study.
Statistical Analyses
A Cox proportional hazards model was used to measure single nucleotide polymorphisms (SNP) associations. Age at stroke was used for cases, and age at enrollment was right censored for controls without a history of ischemic stroke. Principal components (PC) were calculated from LD pruned genotypes26 using the entire freeze6a TOPMed cohort and was then was subset to the N=1,333 participants in the study. Statistical tests were performed using the R package survival27 with the following model:
Where t is time in years to first stroke (or years to enrollment for controls), PC is the principal component eigenvector of the observed genotype covariance matrix, and SNP represents the genotype of the individual. Genotypes were converted to integers prior to populating the model in the following way: 0 = homozygous reference; 1 = heterozygous reference/variant; 2 = homozygous variant. Genome-wide significance was defined as P < 5 ×10−8. Conditional regression was performed on candidate 5Mb genomic regions surrounding the lead SNPs defined by lowest P-value. No independent loci within the candidate regions were observed. Code for this analysis is available online: https://github.com/earleyej/cox-gwas.
All genotype-level analyses were performed in a HIPPA compliant environment on DNAnexus (https://www.dnanexus.com/). Annotated views of genomic candidate regions were created with LocusZoom28 using the full (ALL) 1000 genomes reference for linkage disequilibrium (LD) estimates. Tests on GWAS summary statistics, including gene-level association and enrichment tests were performed using the online portal Functional Mapping and Annotation of GWAS (FUMA29). Gene-level association was performed on the full 14 million P-values using Multi-marker Analysis of GenoMic Annotation (MAGMA)30 using default settings. Gene set pathway analysis was performed on the Gene Ontology (GO) database (N=9,988). Colocalization analysis was performed with the R package coloc31 on 1Mb regions surrounding each independent genome-wide significant SNP with the lowest P-value for that region (aka ‘lead’ SNP). Additional gene set enrichment was performed on the GWAS Catalog32 of genes using hypergeometric tests and corrected for multiple hypothesis testing using Benjamini-Hochberg FDR33. Candidate genes were defined as being within 500Kb of a lead SNP, or if no genes were present within 500Kb, then the most proximal gene was used for that region. Meta-analysis of candidate SNPs with Walk-PHaSST and PUSH cohorts was performed using Fisher’s method.
Multi-variant survival analysis
Individuals were categorized reflecting the number of lead SNPs they carried. All categories were required to have at least 10 participants, for a total of 5 categories (0, 1, 2, 3, and ≥4). These were used as categorical variables in a univariate survival analysis. P-values were calculated using Gehan-Wilcoxon34, a non-parametric test which has more sensitivity in detecting differences between groups where the hazard ratio is higher at earlier timepoints.
RESULTS
Cohort characteristics
Of the 2,793 SCD individuals enrolled in the REDS-III Brazil SCD Cohort, 1,460 were excluded, with a final set of 1,333 individuals (Supplemental Figure 1). Younger age was strongly associated with ischemic stroke (OR=0.9; CI=0.89–0.92; P<0.001; Figure 1A) but not sex (male risk OR=0.9; CI, 0.65–1.24, P=0.5) or Hemocenter (OR=0.98; CI, 0.89–1.08; P=0.7). Many patients reported taking hydroxyurea (HU); however, the majority of participants initiated treatment after their first stroke and thus we did not consider HU in our analysis for time to first ischemic stroke. Genome-wide association was conducted on HbSS homozygotes only (N=1,333). Within this set were 178 (13.4%) individuals who experienced ischemic stroke (Table 1) with a median age at stroke of 9.5 years (Table 1, Fig 1A).
Figure 1.
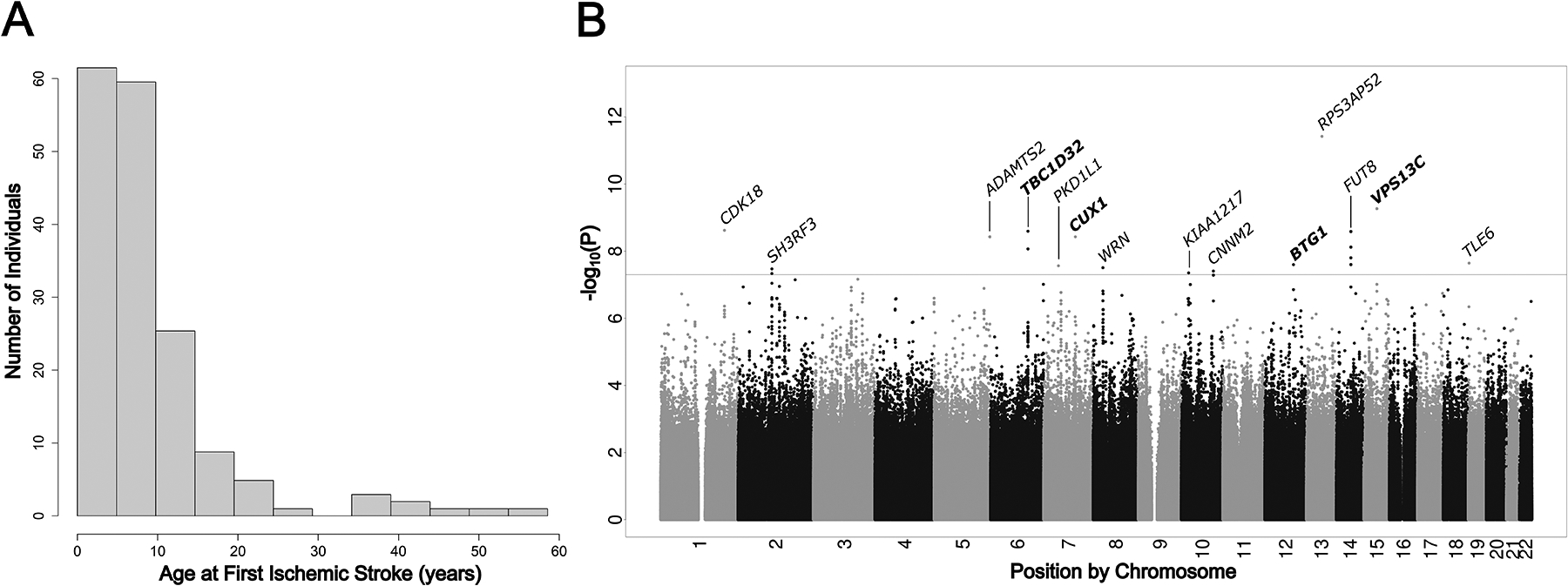
(A) Histogram of age at first ischemic stroke for the GWAS cohort. (B) Manhattan plot of results from proportional hazards GWAS P-values for ischemic stroke in HbSS SCD (N=1,333). Horizontal dotted line represents the genome-wide significance threshold of P = 5 × 10–8. The nearest gene to each lead genome-wide significant SNP is annotated.
Table 1.
Demographics of HbSS Individuals within the REDS-III Brazil SCD Cohort
| Total cohort size | N=1,333 |
|---|---|
| Sex (%) | |
| Female | 720 (54.0) |
| Male | 613 (46.0) |
| Age at enrollment, mean (±SD), y | 20.6 (±13.1) |
| Age at first stroke, mean (±SD), y | 9.5 (±9.4) |
| Stroke, no. (%) | |
| cases | 178 (13.4) |
| controls | 1155 (86.6) |
| Hemocenter, no. (%) | |
| Hemominas BH | 313 (23.5) |
| Hemominas JFO | 149 (11.2) |
| Hemominas MOC | 205 (15.4) |
| Hemope | 285 (21.4) |
| Hemorio | 332 (24.9) |
| ITACI SP | 49 (0.04) |
Genetic architecture of ischemic stroke in Brazilian Sickle Cell
We observed 28 genome-wide significant variants (Supplemental Table 1) across 14 independent regions associated with stroke occurring at younger ages (Figure 1B, Table 2). No systemic inflation of P-values was observed (Genomic Control Lambda = 1.02; Supplemental Figure 2). Most variants sites were intergenic; however, three regions were intronic to genes CUX1, KIAA1217, and CNNM2 (Supplemental Figure 3). Also Included in this list of 14 lead SNPs were three regions previously linked to stroke – ADAMTS2, CDK18, FUT818–20.
Table 2.
Cox regression results for lead genome-wide significant SNPs
| Chr | Position (b38) | Effect Allele | Nearest Protein Coding Gene | rsID | P-Value | Effect AF (%) | HR | 95% CI | Variant Type | Functional Annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 205,490,782 | G | CDK18 | rs12144136 | 2.38 × 10−9 | 5.1 | 2.7 | 2.4 – 3.0 | SNP | Intergenic |
| 2 | 109,059,503 | A | SH3RF3 | rs10181988 | 3.35 × 10−8 | 2.7 | 3.2 | 2.8 – 3.6 | SNP | Intergenic |
| 5 | 179,375,279 | A | ADAMTS2 | rs147625068 | 3.70 × 10−9 | 1.0 | 5.4 | 4.8 – 5.9 | SNP | Intergenic |
| 6 | 120,572,043 | T | TBC1D32 | rs1209987 | 2.52 × 10−9 | 10.3 | 2.3 | 2.0 – 2.6 | SNP | Intergenic |
| 7 | 47,681,969 | C | PKD1L1 | rs181930335 | 2.71 × 10−8 | 2.3 | 3.6 | 3.2 – 4.1 | SNP | Intergenic |
| 7 | 102,180,550 | A | CUX1 | rs188599171 | 3.70 × 10−9 | 1.1 | 5.6 | 5.0 – 6.2 | SNP | Intronic |
| 8 | 31,319,867 | A | WRN | rs115684945 | 3.10 × 10−8 | 1.8 | 3.4 | 3.0 – 3.8 | SNP | Intergenic |
| 10 | 23,744,565 | A | KIAA1217 | rs115292858 | 4.43 × 10−8 | 2.2 | 3.8 | 3.3 – 4.3 | SNP | Intronic |
| 10 | 102,993,950 | - | CNNM2, NT5C2 | rs367825668 | 3.90 × 10−8 | 1.7 | 3.4 | 3.0 – 3.9 | DEL | Intronic |
| 12 | 92,093,367 | G | BTG1 | rs77900855 | 2.51 × 10−8 | 1.0 | 5.0 | 4.5 – 5.6 | SNP | Intergenic |
| 13 | 68,609,784 | C | RPS3AP52 | rs73204086 | 3.78 × 10−12 | 1.1 | 5.5 | 5.0 – 6.0 | SNP | Intergenic |
| 14 | 65,317,148 | T | FUT8 | rs150572699 | 2.58 × 10−9 | 1.4 | 3.9 | 2.5 – 6.2 | SNP | Intergenic |
| 15 | 61,683,898 | G | VPS13C | rs141674494 | 5.40 × 10−10 | 1.1 | 6.3 | 5.7 – 6.9 | SNP | Intergenic |
| 19 | 2,961,825 | G | TLE6 | rs116211928 | 2.25 × 10−8 | 1.4 | 4.8 | 4.2 – 5.3 | SNP | Intergenic |
Chr = Chromosome; AF = Allele Frequency; HR = Hazard Ratio; 95% CI = 95% Confidence Interval; Genomic positions in GRCh38 build.
Out of the 14 genome-wide lead variants, 5 were only observed in heterozygous state whereas 9 were observed in both heterozygous and homozygous variant state. Individuals possessing at least one of these 14 variants exhibited increased risk for ischemic stroke at earlier ages (P < 0.001 for all 14 variants, Gehan-Wilcoxon) (Supplemental Figure 4).
Multiple variant survival analysis
To investigate the impact of possessing multiple lead risk variants within the same individual, participants were divided into categories based on the number of lead SNPs they harbored. Individuals with zero lead SNPs had a stroke rate of 4% (n=716), whereas those with at least one of the 14 lead variants had an increased rate at 14% (n=430), and this rate increased even further for individuals with two (36%; n=125), three (70%; n=50), or four or more variants (83%; n=12) (Figure 2A). The relative impact of these alleles on cohort level risk was measured by calculating the product of minor allele frequency with the hazard ratio (Figure 2B). The variant rs12144136 near CDK18 exhibited the highest relative weight of risk for early stroke within this cohort.
Figure 2.
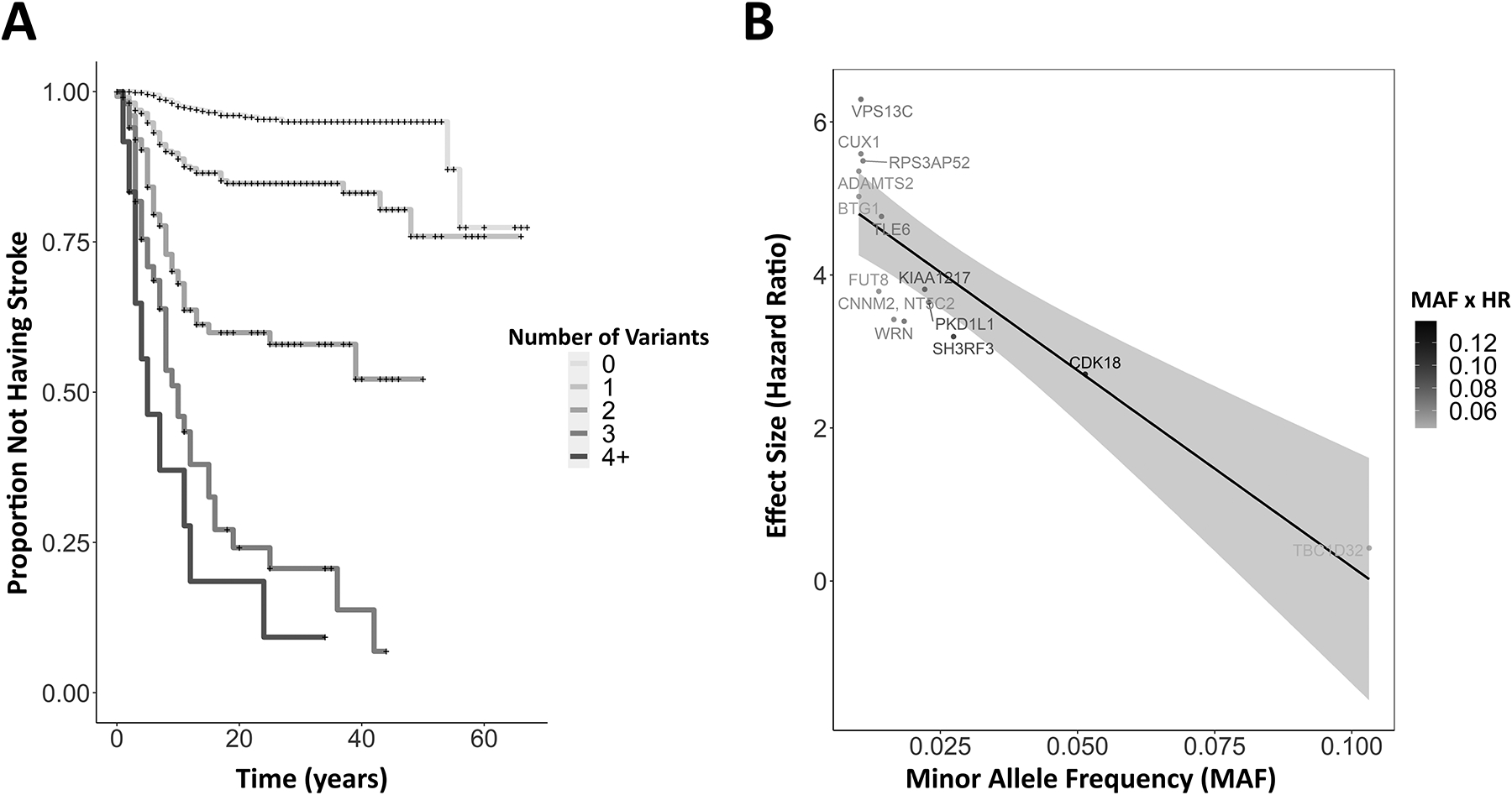
(A) Kaplan-Meier plot of individuals harboring multiple co-occurring genome-wide significant risk alleles for early stroke. Curves represent subsets of the study cohort who possessed zero (N=716), one (N=430), two (N=125), three (N=50), or four or more (N=12) stroke risk alleles in any combination from the 14 lead SNPS. (+) represents right-censored data. (B) A scatter plot of observed hazard ratios (HR) and minor allele frequencies (MAF). Points represent the 14 lead SNPs and are annotated with the nearest gene. The relative impact of each allele on the cohort-scale risk of earlier ischemic stroke is calculated as the product of MAF and HR and is color coded from gray (lower impact) to black (higher impact).
Cis-co-localization analysis of ischemic stroke using GTEx
All 14 candidate SNPs from this study occur in inter-genic or intronic regions. To investigate evidence of these SNPs influencing gene expression, the lead SNPs were searched against the GTEx database of gene expression to identify cis- and trans- expression quantitative trail loci (eQTL) variant associations across different tissue types. The lead SNP on chromosome 1, rs12144136, is a cis-eQTL for the gene CDK18 within the Skeletal Muscle tissue category (Supplemental Table 2) which appears to upregulate this gene. No other SNPs at genome-wide significance showed evidence of cis-eQTL activity.
Expanding to 1Mb regions around each lead SNP, we assessed co-localization of cis-eQTL associations with 24 different tissue types relevant to stroke. Three of the 14 candidate regions showed strong evidence (Posterior Probability, PP > 0.9) of colocalization with a cis-eQTL (Supplemental Table 2). On chromosome 12, two cis-eQTLs were found for genes LUM in the frontal cortex (rs77217583) and NUDT4 (rs117881990) in the hypothalamus. On chromosome 14, one cis-eQTL was found for RAB15 in the substantia nigra; and on chromosome 15, one cis-eQTL was found for VPS13C in cervical spinal cord. Three other moderately strong signals (P > 0.5) were detected on chromosome 8 – one cis-eQTL for WRN expression in heart left ventricle and two cis-eQTLs for PURG in the substantia nigra.
Gene-level Association
We performed a gene-level association on 19,021 protein coding genes using the 14M site summary statistics (Supplemental Figure 5; Supplementary Table 3). No genes exhibited genome-wide significant enrichment defined at the Bonferroni-corrected alpha of 2.6×10−6; however, three genes exhibited some evidence of association (P<10−4), including GPR15 (P=3.9×10-5), MMP26 (P=4.6×10−5), and GOLT1B (P=7.5×10-5).
Gene-set enrichment analysis was performed to predict impact on biological functions using the Gene Ontology (GO) database (Supplemental Table 4). While no gene set achieved the Bonferroni-corrected p-value of 5×10−6, top results at the P<0.001 threshold included the platelet dense tubular network (P = 3.63×10−5), the hemoglobin complex (P = 0.0007), gas transport (P = 0.0006), and regulation of systemic arterial blood pressure by hormone (P = 0.0008).
Replication with other SCD cohorts
Candidate SNP analysis of the 14 lead SNPs was performed within two independent SCD cohorts: Walk-PHaSST (12 cases, 300 controls)35 and PUSH (14 cases, 202 controls)36 (Supplemental Table 5). Cox regression for stroke timing, adjusting for sex and the first genetic principal component, showed increased risk for early stroke at SNPs rs1209987 near gene TBC1D32 (P = 0.01) and rs188599171 near gene CUX1 (P = 0.001) within the combined Walk-PHaSST/PUSH cohort. Meta-analysis with the REDS-III Brazil SCD cohort and the combined Walk-PHaSST/PUSH cohort showed genome-wide significance for four SNPs: rs1209987 near gene TBC1D32 (P = 3.36×10–10), rs188599171 near CUX1 (P = 5.89×10–11), rs77900855 near BTG1 (P=4.66×10–8), and rs141674494 near VPS13C (1.68×10−9), although we note these results are being driven largely by the REDS-III Brazil SCD cohort as none of the SNPs reached genome-wide significance within Walk-PHaSST or PUSH.
A comparison was also made to the results from Flanagan et al13 (122 cases, 167 controls). None of the 14 lead SNPs were genotyped in this cohort, and investigating the surrounding 1Mb regions did not uncover any loci exhibiting association with ischemic above the P=0.001 threshold (results not shown).
A third and final comparison was made to an independent cohort of SCD children with genome sequence data, comprised of 15 patients with a history of ischemic stroke, 26 patients with a history of abnormal TCD, and 140 control patients (J.M.F. and R.E.W. Unpublished data, 2022). A combined Armitage test for trend on the combined stoke/abnormal TDC (N=413) versus controls (N=140) showed modest, but not genome-wide significant, replication of rs116211928 near gene TLE6 (P = 0.022).
Candidate-gene Enrichment Analysis to GWAS Catalog
To assess overlap in genetic architecture between early ischemic stroke and other relevant cardiovascular traits, we performed an enrichment test against the GWAS Catalog of gene by trait associations. Candidate genes from the current study were defined as located within 500Kb of a lead SNP, or the most proximal gene if none were present in that region, resulting in a list of 52 genes (Supplemental Table 6). Significant overlap in gene sets was observed for mean arterial pressure (3/9 genes; P = 5.82 × 10−4), hypertension (5/98 genes; P = 1.48 × 10−3), immature fraction of reticulocytes (4/104 genes; P = 1.64 × 10−2), and myocardial infarction (3/55 genes; P = 3.53 × 10−2) (Figure 3).
Figure 3.
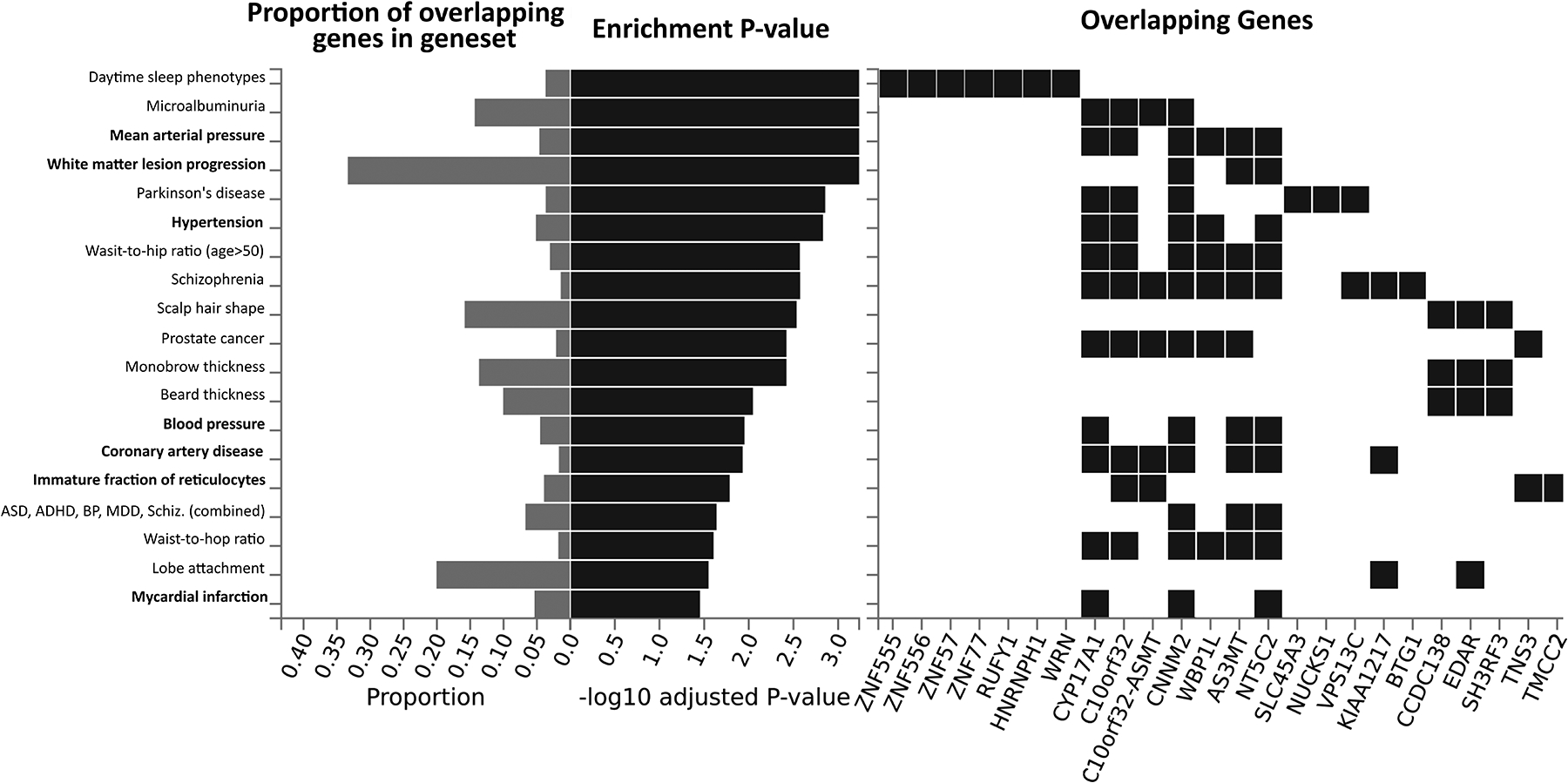
Gene enrichment test results against the GWAS Catalog of reported gene by trait associations. Candidate genes were defined as being within 500kb of a lead SNP. Left, horizontal bar plots of the proportion of genes within each trait category observed in the candidate gene list (gray) and the P values (black) from a hypergeometric enrichment test (FDR adjusted). Traits relevant to ischemic stroke are listed in bold. Right, a visual matrix showing which candidate genes overlap a given trait.
Comparison to previous ischemic stroke studies in SCD and non-SCD
Previous candidate SNP studies of stroke in SCD have evaluated 12 SNPs, as well as one additional SNP from a GWAS8–15. None of these candidates SNPs reached genome-wide significance in the current study (Supplemental Table 7). Expanding the search to surrounding 1Mb region centered on the 13 candidate SNPs uncovered the lead SNP rs147625068 (P = 3.7×10−9) near gene LTC4S on chromosome 9.
A similar search was performed for non-SCD early onset ischemic stroke candidate genes: ADAMTS2 (rs469568), ADAMTS12 (rs1364044), CDK18 (rs77571454), HABP2 (rs11196288)17–20. In the current study, the variant rs77571454 near gene CDK18 was not observed, and none of the other three variants reached genome-wide significance (Supplemental Table 8). Searching within the 1Mb region centered on these four candidate SNPs uncovered the two lead SNPs rs12144136 (P = 2.38×10−9), near CDK18, and rs147625068 (P = 3.7×10−9), near ADAMTS2, and one additional variant near ADAMTS12 (rs534089428, P = 2.43×10−5).
DISCUSSSION
We have discovered 14 genetic regions associated with ischemic stroke occurring at earlier ages in SCD individuals. This study included 1,333 HbSS individuals, the largest genome-wide evaluation of ischemic stroke within SCD to date. We implemented a Cox proportional hazards model to estimate risk at earlier timepoints. Results implicate gene candidates previously linked with non-SCD early-onset stroke – ADAMTS2, CDK18; genes linked to cardiovascular disease risk factors – BTG1, CNMM2, CUX1, FUT8, KIAA1217, LUM, NT5C2, NUDT4, PKD1L1, WRN; and eight novel genes – SH3RF3, RAB15, RPS3AP52, TBC1D32, VPS13C, and TLE6. To our knowledge this is the first independent replication of genetic risk associated with increased stroke at alleles near genes CDK18 and FUT8.
Previous candidate gene studies of stroke in SCD implicated five alleles within genes TGFBR3, TEK, ANXA2, IL4R, and ADCY910–12; however, the single published GWAS of ischemic stroke in SCD did not find evidence for alleles near these genes influencing stroke risk but instead found alleles near two novel genes – GOLGB1 and ENPP113. We did not find evidence of alleles near these genes exhibiting genome-wide significant association to stroke. The reasons for this are unclear, although we should note the previous GWAS enrolled African Americans whereas the current work enrolled Brazilians, each having distinct histories of admixture.
Results from this work implicate genes previously linked to non-SCD early onset stroke: ADAMTS2 and CDK1817–19. Alleles near genes ADAMTS2 and ADAMTS12 were previously associated with pediatric stroke in two separate U.S. populations17,18. ADAMTS2 also appears to have a role in cerebral aneurysm37 and cardiac muscle hypertrophy38 possibly through regulation of the extracellular matrix39. We observed one variant, rs534089428, within 500kb of ADAMTS12 at P = 2×10−5.
High reticulocyte count is a reliable risk factor for ischemic stroke in sickle cell individuals1. Variants affecting candidate genes from the current study, CUX1, CNNM2, and NUDT4, have been previously associated with higher reticulocyte counts40 41. CNNM2 has also been linked to numerous stroke related traits – hypertension, coronary artery disease, and mean corpuscular hemoglobin42–49. CNNM2 gene is expressed throughout the body but is particularly high in the brain, kidney, and endocrine tissue50 and appears to play a role in renal magnesium uptake51,52
This study is limited in a few ways. First, this work did not consider the influence of environmental exposures and lifestyles on ischemic stroke risk. Non-acute, unreported comorbidities may also play a role. This study is larger than previous at N=1,333, but the observed association of alleles at a frequency of 1–5% within a case size of N=178 means that relatively few individuals are driving this signal, and future studies in independent cohorts will need to validate these findings. In addition, we define candidate genes based on proximity to lead SNPs and note the possibility risk alleles may actually be linked to distant genes via, for example, trans-QTL effects.
In conclusion, we have identified risk alleles associated with earlier ischemic stroke. Findings from this work have overlapped previously identified non-SCD early onset ischemic stroke genes – ADAMTS2 and CDK18– and discovered 16 novel genes. Future studies will be needed to confirm the clinical significance of these findings and to disentangle the potentially complex interaction among these alleles.
Supplementary Material
Acknowledgements:
EJE, SK, GPP wrote the paper; GPP, CD, BC, SK, ES designed the study; EJE, FF, GPP performed the analysis; CSA, DOWR, DTSC collected data; JMF, REW, XZ, VG, MG, YZ, MN, SN provided lookup analysis for independent SCD cohorts. Special thanks to patients and research staff at each hemocenter in Brazil participating in this study. We recognize the following people for their commitment and contribution to this project: Alfredo Mendrone Jr., Cesar de Almeida Neto, Roberta Calcucci, Erivanda Bezerra, Carolina Miranda, Franciane Mendes de Oliveira, Valquıria Reis, Nayara Durte, Barbara Malta, Jose Wilson Sales, Maria Aparecida Souza, Rodrigo Ferreira, Maria do Carmo Valgueir, Regina Gomes, Airly Goes Maciel, Rebeca Talamatu Dantas, Flavia Herculano, Ana Claudia Pereira, Ana Carla Alvarenga, Adriana Grilo, Fabiana Canedo, Pedro Losco Takecian, Mina Cintho Ozahata, Rodrigo Muller de Carvalho, Christopher McClure, Simone A. Glynn.
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Genome Sequencing for “NHLBI TOPMed: REDS-III_Brazil” (phs001468.v3.p1) was performed at Baylor (HHSN268201500015C, HHSN268201600033I). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal August 2022.
Funding:
The NHLBI REDS-III program was supported by NHLBI contracts HHSN2682011-00001I, -00002I, -00003I, -00004I, -00005I, -00006I, -00007I, -00008I, -00009I, 75N2019D00033, and R21 HL135367.
Footnotes
Disclosures: None.
REFERENCES
- 1.Belisario AR, Silva CM, Velloso-Rodrigues C, Viana MB. Genetic, laboratory and clinical risk factors in the development of overt ischemic stroke in children with sickle cell disease. Hematol Transfus Cell Ther. 2018;40(2):166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Earley CJ, Kittner SJ, Feeser BR, et al. Stroke in children and sickle-cell disease: Baltimore-Washington Cooperative Young Stroke Study. Neurology. 1998;51(1):169–176. [DOI] [PubMed] [Google Scholar]
- 3.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]
- 4.Kirkham FJ, Lagunju IA. Epidemiology of Stroke in Sickle Cell Disease. J Clin Med. 2021;10(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strouse JJ, Jordan LC, Lanzkron S, Casella JF. The excess burden of stroke in hospitalized adults with sickle cell disease. Am J Hematol. 2009;84(9):548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams RJ, McKie VC, Carl EM, et al. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann Neurol. 1997;42(5):699–704. [DOI] [PubMed] [Google Scholar]
- 7.Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood. 2004;103(10):3689–3694. [DOI] [PubMed] [Google Scholar]
- 8.Hoppe C, Klitz W, Cheng S, et al. Gene interactions and stroke risk in children with sickle cell anemia. Blood. 2004;103(6):2391–2396. [DOI] [PubMed] [Google Scholar]
- 9.Taylor JGt, Tang DC, Savage SA, et al. Variants in the VCAM1 gene and risk for symptomatic stroke in sickle cell disease. Blood. 2002;100(13):4303–4309. [DOI] [PubMed] [Google Scholar]
- 10.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat Genet. 2005;37(4):435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belisario AR, Sales RR, Toledo NE, et al. Reticulocyte count is the most important predictor of acute cerebral ischemia and high-risk transcranial Doppler in a newborn cohort of 395 children with sickle cell anemia. Ann Hematol. 2016;95(11):1869–1880. [DOI] [PubMed] [Google Scholar]
- 13.Flanagan JM, Sheehan V, Linder H, et al. Genetic mapping and exome sequencing identify 2 mutations associated with stroke protection in pediatric patients with sickle cell anemia. Blood. 2013;121(16):3237–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoppe C, Klitz W, D’Harlingue K, et al. Confirmation of an association between the TNF(−308) promoter polymorphism and stroke risk in children with sickle cell anemia. Stroke. 2007;38(8):2241–2246. [DOI] [PubMed] [Google Scholar]
- 15.Belisario AR, Nogueira FL, Rodrigues RS, et al. Association of alpha-thalassemia, TNF-alpha (−308G>A) and VCAM-1 (c.1238G>C) gene polymorphisms with cerebrovascular disease in a newborn cohort of 411 children with sickle cell anemia. Blood Cells Mol Dis. 2015;54(1):44–50. [DOI] [PubMed] [Google Scholar]
- 16.Brewin JN, Smith AE, Cook R, et al. Genetic Analysis of Patients With Sickle Cell Anemia and Stroke Before 4 Years of Age Suggest an Important Role for Apoliprotein E. Circ Genom Precis Med. 2020;13(5):531–540. [DOI] [PubMed] [Google Scholar]
- 17.Witten A, Ruhle F, de Witt M, et al. ADAMTS12, a new candidate gene for pediatric stroke. PLoS One. 2020;15(8):e0237928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arning A, Hiersche M, Witten A, et al. A genome-wide association study identifies a gene network of ADAMTS genes in the predisposition to pediatric stroke. Blood. 2012;120(26):5231–5236. [DOI] [PubMed] [Google Scholar]
- 19.Yamada Y, Kato K, Oguri M, et al. Identification of nine genes as novel susceptibility loci for early-onset ischemic stroke, intracerebral hemorrhage, or subarachnoid hemorrhage. Biomed Rep. 2018;9(1):8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng YC, Stanne TM, Giese AK, et al. Genome-Wide Association Analysis of Young-Onset Stroke Identifies a Locus on Chromosome 10q25 Near HABP2. Stroke. 2016;47(2):307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carneiro-Proietti ABF, Kelly S, Miranda Teixeira C, et al. Clinical and genetic ancestry profile of a large multi-centre sickle cell disease cohort in Brazil. Br J Haematol. 2018;182(6):895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinman S, Busch MP, Murphy EL, et al. The National Heart, Lung, and Blood Institute Recipient Epidemiology and Donor Evaluation Study (REDS-III): a research program striving to improve blood donor and transfusion recipient outcomes. Transfusion. 2014;54(3 Pt 2):942–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballas SK, Lieff S, Benjamin LJ, et al. Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol. 2010;85(1):6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Martino CC, Alencar CS, Loureiro P, et al. Use of an automated pyrosequencing technique for confirmation of sickle cell disease. PLoS One. 2019;14(12):e0216020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.A Package for Suvival Analysis in R. [computer program]. 2020. [Google Scholar]
- 28.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B. 1995;57(1):289–300. [Google Scholar]
- 34.Gehan EA. A generalized Wilcoxon test for comparing arbitrarily singly-censored samples. Biometrika. 1965;52(1–2):203–224. [PubMed] [Google Scholar]
- 35.Sachdev V, Kato GJ, Gibbs JS, et al. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation. 2011;124(13):1452–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minniti CP, Sable C, Campbell A, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica. 2009;94(3):340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arning A, Jeibmann A, Kohnemann S, et al. ADAMTS genes and the risk of cerebral aneurysm. J Neurosurg. 2016;125(2):269–274. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Chen W, Zhang J, et al. Critical Role of ADAMTS2 (A Disintegrin and Metalloproteinase With Thrombospondin Motifs 2) in Cardiac Hypertrophy Induced by Pressure Overload. Hypertension. 2017;69(6):1060–1069. [DOI] [PubMed] [Google Scholar]
- 39.Mead TJ, Apte SS. ADAMTS proteins in human disorders. Matrix Biol. 2018;71–72:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Astle WJ, Elding H, Jiang T, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016;167(5):1415–1429 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vuckovic D, Bao EL, Akbari P, et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell. 2020;182(5):1214–1231 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wain LV, Vaez A, Jansen R, et al. Novel Blood Pressure Locus and Gene Discovery Using Genome-Wide Association Study and Expression Data Sets From Blood and the Kidney. Hypertension. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeuchi F, Akiyama M, Matoba N, et al. Interethnic analyses of blood pressure loci in populations of East Asian and European descent. Nat Commun. 2018;9(1):5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.German CA, Sinsheimer JS, Klimentidis YC, Zhou H, Zhou JJ. Ordered multinomial regression for genetic association analysis of ordinal phenotypes at Biobank scale. Genet Epidemiol. 2020;44(3):248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeong H, Jin HS, Kim SS, Shin D. Identifying Interactions between Dietary Sodium, Potassium, Sodium-Potassium Ratios, and FGF5 rs16998073 Variants and Their Associated Risk for Hypertension in Korean Adults. Nutrients. 2020;12(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu TH, Fann JC, Chen SL, et al. Gradient Relationship between Increased Mean Corpuscular Volume and Mortality Associated with Cerebral Ischemic Stroke and Ischemic Heart Disease: A Longitudinal Study on 66,294 Taiwanese. Sci Rep. 2018;8(1):16517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Harst P, Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122(3):433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dichgans M, Malik R, Konig IR, et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. Stroke. 2014;45(1):24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koyama S, Ito K, Terao C, et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet. 2020;52(11):1169–1177. [DOI] [PubMed] [Google Scholar]
- 50.Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 51.Arjona FJ, de Baaij JH, Schlingmann KP, et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet. 2014;10(4):e1004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartmann K, Seweryn M, Handelman SK, Rempala GA, Sadee W. Non-linear interactions between candidate genes of myocardial infarction revealed in mRNA expression profiles. BMC Genomics. 2016;17(1):738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.