

Summary
Candida auris and Candida albicans can result in invasive fungal diseases. And yet, these species can stably and asymptomatically colonize human skin and gastrointestinal tracts. To consider these disparate microbial lifestyles, we first review factors shown to influence the underlying microbiome. Structured by the damage response framework, we then consider the molecular mechanisms deployed by C. albicans to switch between commensal and pathogenic lifestyles. Next, we explore this framework with C. auris to highlight how host physiology, immunity, and/or antibiotic receipt are associated with progression from colonization to infection. While treatment with antibiotics increases the risk an individual will succumb to invasive candidiasis, the underlying mechanisms remain unclear. Here, we describe several hypotheses that may explain this phenomenon. We conclude by highlighting future directions integrating genomics with immunology to advance our understanding of invasive candidiasis and human fungal disease.
Keywords: Candida, mycobiome, antibiotics, damage response framework, commensal, pathogen, medical mycology, microbiome, antibiotic stewardship
eTOC.
In this review, Proctor and colleagues spotlight two fungal pathogens – Candida auris and Candida albicans -- named by the World Health Organization as urgent health threats. They explore the role of the microbiome, host physiology, host immunity, and antibiotics in modulating switches between commensalism, pathogenesis, and infection, in Candida species.
Introduction
We often think of the components of the human microbiota as commensals and deploy these concepts with some degree of equivalence, but these are not synonymous terms. A useful framework for considering the organisms that live on our body is the damage response framework, developed by Pirofski and Casadevall, which defines a lexicon based on a synthesis of the history of the field of microbial pathogenesis1. In this framework, microbiota consists simply of microbes that colonize the body at a given time point while commensalism or pathogenesis denote behavioral lifestyles, most easily identified by the outcome of a given interaction, between a host and a colonist. Following adherence, the outcome which denotes commensalism is colonization with the apparent absence of disease. The outcome that defines pathogenesis is colonization with “damage that affects host homeostasis”2, which may manifest as inflammation, tissue destruction, or disease. The damage response framework importantly resolves the paradox that even organisms like Malassezia species, which are widely regarded as commensals of the skin of virtually all healthy adults3, have been implicated in inflammatory responses such as atopic dermatitis4, inflammatory bowel disease (IBD)5, and invasive disease and mortality6–8. Conversely, C. auris, one of the world’s most feared fungi, asymptomatically colonizes up to 90% of humans while adopting the lifestyle of a commensal, challenging our best efforts at detection during surveillance9. Whether an organism adopts the commensal or pathogenic lifestyle depends on the immune and health status of the host, the local tissue environment (e.g., pH, salivary flow rates, oxygen concentration, etc.), the microbe’s genetic potential, its epigenetic profile, as well as microbial consortium at the site of adherence. It is even possible that individual microbes within a population will adopt different lifestyles simply because one or more of these variables differs between adherence sites.
Irrespective of behavior or lifestyle (commensal, pathogen), a microbe must adhere either to host tissue directly or to other microbial colonists to engage with the host. Initial adherence is followed by an outcome, either clearance of an individual microbe or its successful growth and colonization of a tissue, shaped by intrinsic and extrinsic microbial factors. For example, outcomes for Candida engagement with a host depend upon the intrinsic adoption of either yeast or hyphal forms, each necessary for pathogenesis10. The yeast morphology arises after budding of daughter cells following cytokinesis and nuclear division. The hyphal morphology is filamentous and includes one or more tubular cells separated by porous septa. In C. albicans, Ume6 is a master regulator of the shape-shifting yeast to hyphal switch11. Ume6 is variably expressed based on the intensity of different host cues, such as pH, and tunes production of an adhesin and secreted aspartic protease 6 (Sap6). Levels of these proteins determine whether individual cells are hyper-competitive for colonization or get rapidly cleared by activating a pro-inflammatory response, permitting yeast and hyphal forms of C. albicans to co-exist in a single host’s gut. Within a microbial population, different microbes will experience different outcomes, following adherence, depending on the interaction of each organism with the host (Table 1).
Table 1: Examples of genetically encoded microbial circuitry in bacteria (A. veronii) and fungi that determine organismal lifestyle in a wide variety of hosts ranging from invertebrates to plants and mammals.
Within the “Experimental model paradigm” column, “Infection model” represents microbe-centric studies that permit investigation of genes required for virulence, often measured by mortality, while “Commensal-pathogen switch” experimental paradigms permit examination of gene function governing transitions between commensal and pathogenic lifestyles.
| Microbial population | Host Model | Experimental model paradigm | Body site | Microbial Switch | Outcomes | Citation |
|---|---|---|---|---|---|---|
| C. albicans | Mouse | Commensal-pathogen switch | Gut | Ume6/Sap6 | Colonization vs. Pro-inflammatory clearance | 11 |
| C. albicans | In vitro / Mouse | Commensal-pathogen switch | Gut | Efg1/Wor1 | Phenotypic switch to optimize metabolism and colonization vs. clearance | 12 |
| C. albicans | In vitro / Mouse | Commensal-pathogen switch | Gut | Efg1-Ec1-dependent factor Candidapepsin | Phenotypic switch to activate IL-1β dependent pro-inflammatory program | 13 |
| C. albicans | In vitro / Mouse | Commensal-pathogen switch | Gut | Sef1/Sfu1 | Activates or represses iron uptake genes involved in virulence or colonization | 14 |
| Botrytis cinerea | Arabidopsis thaliana | Commensal-pathogen switch | Leaves | BcSpd1 | Suppression of plant defense and activation of virulence genes or clearance | 15 |
| Aeromonas veronii | Zebrafish | Commensal-pathogen switch | Gut | AimA | Colonization vs. clearance by activation of neutrophils | 16 |
| Parastagonospora nodorum | Wheat | Infection | Seedlings | PnPf2 | Activates transcriptional program that results in expression of 12 effector genes involved in necrosis | 17 |
| Blastomyces dermatitidis B. gilchristii | In vitro / Mouse | Infection | Lung | BAD1 | Inhibits TGF-α production permitting adherence and pathogenic switch. Null mutants are avirulent | 18 |
| Aspergillus fumigatus | Mouse | Infection | Lung | CpCA | Activates stress response program required for virulence | 19 |
| C. auris | Mouse | Infection | Bloodstream | IncRNA, DINOR | Tunes stress response program, modulating genome integrity, cell filamentation, and virulence | 20 |
| C. auris | Caenorhabditis elegans | Infection | C. elegans | Hog1 | Activates a stress response program that’s required for virulence | 21 |
Genetically encoded C. albicans regulatory switches also govern metabolic programs, potentially optimizing organismal fitness within the population for differential nutrient utilization regimes at spatially segregated locations along the gastrointestinal (GI) tract12. Population substructure encompassing individual cells with distinct morphologies, cell surface properties, levels of metabolic activity, and gene expression profiles have been observed in other host-associated fungi, such as Cryptococcus neoformans22. These studies suggest that microbes are genetically hardwired to fine-tune organismal behavior towards commensalism on the one hand or pathogenesis on the other, using transcriptional switches, depending on environmental cues. Rather than designating microbes that are “sometimes commensal”, “sometimes pathogen” as opportunists, the damage response framework considers that at any given moment microbes within a population may adopt different lifestyles, leading to divergent outcomes – in line with the emergent belief that single cell behavior is an important determinant of the population as a whole.
In this review, we consider two Candida species, designated by the World Health Organization (WHO) as critical priorities23, because they famously adopt both commensal and pathogenic lifestyles in humans. Given that both species must compete with and embed themselves into the microbiota, we begin by discussing factors that modulate the human microbiota, often triggering a switch in colonists to transition from commensal to pathogenic lifestyle. Next, we review the literature on molecular determinants of Candida colonization, dissecting known molecular cues that underly the switch from commensal to pathogenic lifestyle in Candida species, as well as mechanisms by which Candida colonization may shape the host’s systemic health. We then use the emerging fungal pathogen C. auris as a case study to contextualize the damage response framework, while introducing how colonization and invasive disease drive both acute outbreaks and endemic spread. Finally, we review the evidence behind multiple alternative hypotheses that explain the increased risk of C. auris infection and associated mortality following antibiotic treatment.
Factors that shape the human microbiota and can provoke microbial lifestyle switches
The human microbiota is comprised of the bacteria, fungi, archaea, viruses, and protists that colonize our bodies. Some microbial species establish stable colonization and are detected on an individual on the timescale of months to years while other species transiently colonize24. Regardless of the duration of association, microbial colonists potentially shape host health long past their physical contact with the host25. The ability of an organism to colonize successfully may depend on a host’s experience with prior colonists. This is well illustrated by the observation that C. albicans engenders Th17 producing cells that are cross-reactive against a wide variety of fungi, ranging from Malassezia to Aspergillus26. More broadly, microbial services include training the host immune system locally and at distal anatomical sites27 and serving as keystone species to organize microbial communities. On time scales ranging from days to a lifespan, the human microbiome experiences an onslaught of disturbances28, both exogenous and those associated with host physiology. These disturbances can reconfigure the community and facilitate adoption of a pathogenic lifestyle by colonists. Here, we briefly spotlight several sources of disturbance – antimicrobials, host physiology, and microbial invasion – that influence outcomes associated with microbial adherence.
Humans are exposed to various chemicals – ranging from antibiotics to antifungals to disinfectants – that can modulate the microbiota, often in unexpected ways. For example, oral administration of several antibiotics used to treat dermatologic disorders can select for antibiotic resistance genes in skin commensals such as Staphylococcus epidermidis, which may then serve as a reservoir for the horizontal transfer of resistance genes to other species, such as S. aureus29. In addition, antibiotics used to treat bacterial infections can lead to an expansion of Candida in the oral cavity30, vagina31, or the gut32, the latter of which may facilitate invasive fungal infection via bloodstream translocation from the gut33. While antifungals treat fungal infections, they also deplete the gut mycobiota which surprisingly elicits inflammation in the lungs as well as the gut34. Specifically, in a mouse model of allergic airway disease, the anti-fungal drugs fluconazole, amphotericin-B, or 5-fluorocytosine were associated with immunopathology characterized by heightened eosinophilic pulmonary infiltration, elevated serum IgE and IgG1, and increased production of Th2 cytokines (IL-4, IL-5, IL-10), presumably in response to depletion of the gut mycobiota34. Finally, the antiseptic chlorhexidine is commonly used to “decolonize skin” to prevent surgical site infections and/or to reduce the risk of bloodstream infections in hospitalized patients35, but decolonization is indiscriminate, inducing a shift in the skin microbiota36. Aside from antimicrobial agents meant to eliminate certain microbes from the host or otherwise reduce microbial load, commonly used prescription medications can modulate the composition of the bacterial component of the microbiota37 with many expected to have impacts on the fungal community38, though this merits further exploration.
Ecosystems ranging from rocky inter-tidal ecologic environments to the human microbiota are shaped by disturbance regimes, defined here as periodic processes that govern the dynamics and health of the ecosystem39. For humans, disturbance regimes arise with a full range of changes to host behavior, physiology or health status. For example, the microbiota of the human oral cavity is shaped by diurnal fluctuations in salivary flow rates40,41, which are tuned by the parasympathetic nervous system. Additionally, flow rates periodically rise or fall due to autonomic responses related to chewing, tasting acidity, or even thinking salacious or anxious thoughts. These periodic and frequent fluctuations in salivary flow rates mark a disturbance regime that maintains the oral microbiota. Medications with anti-cholinergic effect or radiation therapy to the head, nose or throat ablate these fluctuations in salivary flow, resulting in hyposalivation42,43. The chronic loss of punctuated increases and decreases in salivation (i.e., the loss of the disturbance regime) leads to a marked shift in the spatial organization of the microbiota44, predisposing the host to anterior dental caries45 and oral candidiasis46. In other words, the loss of this disturbance regime provokes the switch from a commensal to pathogenic lifestyle in Candida and other species. As a second example, at puberty, hormonal fluctuations, secondary gland maturation, and subsequent lipid availability on the skin modify the composition of the human microbiome by facilitating colonization of skin by auxotrophic and lipophilic Malassezia species47. Puberty thus marks the onset of a disturbance regime that is characterized by the glandular production of antimicrobials and sebum, as well as immunological responses. Once adults pass the seventh decade of life, however, androgen levels fall, and the sebaceous glands eventually succumb to dysfunction and hyperplasia, potentially accounting for the increased diversity of the skin microbiota of elderly individuals, and subsequent increased risk of skin infection48. Thus, host physiological processes maintain the health of the microbiota, whereas losses of periodic physiological disturbances provoke shifts in ecosystem health.
Invasion or predominance of the microbiota by microbes that tend not to colonize (or only colonize at low abundance) also represents a disturbance. This is for several reasons49. First, invasive species potentially modify existing disturbance regimes. The antibiotic-mediated loss of the commensal microbiota and ascension of Candida species in the gut, for example, is associated with the loss of tonic immunity50, a disturbance regime underlying host homeostasis. Second, invasive species can modify the biotic or abiotic properties of an ecosystem, such as the local destruction of teeth, nails, or soft tissue, which can create an environment favorable for the invasion or expansion of other species51.Third, through competition, cooperation or metabolic cross-feeding52, invasive species can moderate interactions among the community32 and facilitate the successful invasion of other species. The Rakoff-Nahoum group observed that the gut microbiota of preterm infants is initially predominated by Staphylococcus species, and that Staphylococci are often succeeded by either Klebsiella or Enterococcus53. In silico modeling and animal experiments revealed the basis for these disparate patterns of succession. When Klebsiella ascends to dominance over Staphylococcus, it inhibits Staphylococcus while promoting the ascension of C. parapsilosis. Importantly, Candida was inhibited by Staphylococcus so its displacement by Klebsiella is needed prior to its expansion. Likewise, Enterococcus appears to ascend and inhibit Klebsiella, explaining empirical patterns in humans. In the extreme, succession patterns such as these can culminate in an over-abundance in the microbiota of multiple species typically found circulating in hospital environments54, leading to community composition of older adults residing in nursing homes that differs markedly from that of similarly aged community dwellers55.
C. albicans as a model to understand the diversity of lifestyles
In the United States, Candida species are reported to cause up to 22% of bloodstream infections with patients in intensive care units accounting for 50% of cases56. The burden of invasive fungal disease is increasing due to an aging global population, as well as the increasingly sophisticated iatrogenic treatments used to manage complex patient populations, such as those undergoing transplantation or cancer treatment. While Candida is a leading cause of bloodstream infection, it silently colonizes the GI tract of the majority of adults. Candida also colonizes the skin of healthy adults, although typically at low abundance and prevalence compared to the more dominant Malassezia species57. Some have proposed that the prevalence of Candida-colonized adults may be higher in the western world, linked to high sugar and fat diets58–60 and high antibiotic utilization. An increased load of Candida within the gut can increase the risk of systemic Candida infection33, and in recent years has been associated with a multitude of diseases ranging from allergic lung inflammation and GI tract cancers to alcohol-associated liver disease61–63. Therefore, there is a clear need to improve understanding of the molecular cues that drive the switch from the commensal to the pathogenic lifestyle in Candida species.
Determinants of Candida colonization resistance
Unlike humans, mice are typically resistant to persistent Candida GI colonization by standard laboratory strains, which provides an opportunity to study the mechanisms by which C. albicans colonizes the host. Colonization resistance may be overcome by depleting the microbiota, host deficiency of key mucosal defense molecules, or by strain-specific C. albicans resistance to host defenses, as outlined below. Depleting the gut microbiota with broad-spectrum antibiotics prior to oral delivery of C. albicans breaks colonization resistance. The bacteria Bacteroides and Firmicutes are essential for promoting Candida colonization resistance in the gut, and their depletion activates a signaling axis involving hypoxia inducible factor (HIF)-1– and cathelicidin-related antimicrobial peptide (CRAMP)64. Specifically, HIF-1α activation in intestinal epithelial cells mediates downstream production of the antimicrobial peptide CRAMP (LL-37 in humans) that helps to expel Candida from the GI tract. Indeed, mice deficient in CRAMP are less resistant to Candida gut colonization and can be stably colonized with the C. albicans lab strain SC531465 without antibiotic treatment. Strains of C. albicans that can colonize the murine GI tract in the absence of antibiotics (e.g., strain 529L) are more resistant to CRAMP-mediated killing65, further underscoring the HIF-1α/CRAMP axis as a critical determinant of colonization resistance. Treatments that boost HIF-1α activation help to reduce Candida colonization in mice, which is important for preventing mortality from invasive infection64. Invasive C. albicans infections that originate from the GI tract cause a hepato-splenic pattern of infection in patients with prior expansion of commensal Candida populations and disrupted mucosal barrier and/or innate immune defenses33,66,67.
Within the gut, C. albicans typically exists as yeast but recent studies have also uncovered its presence in the filamentous (hyphal) form11. The mechanisms governing the ratio of yeast to hyphae in the gut are unclear, and the degree to which immune selection or fungal factors moderate outcomes is unknown. Of note, strains with differing capacities to overcome GI colonization resistance in mice have significantly different responses to hyphal-inducing growth conditions in vitro yet form a similar cell morphology within the gut lumen in vivo65. Colonizing yeast cells, called GI-induced transition (GUT) cells, are distinct from yeast cells grown in the lab or found within tissues during systemic Candida infection12. For example, GUT cells down-regulate genes involved in iron acquisition, adhesion, and other gene modules typically associated with infection. The master regulator of GUT morphology is the transcription factor Wor1, and over-expression of Wor1 in C. albicans results in increased fitness to colonize the murine GI tract. Furthermore, the Noble lab recently showed that the master regulator Ume6 inhibits gut colonization by modulating the expression of hypha-associated proteases and adhesion molecules, rather than by directly affecting fungal morphogenesis11. However, why some fungal virulence factors, such as adhesins, should alter GI colonization capacity, is still unknown. Another genetic determinant of colonization “fitness” within the mouse GI tract was recently uncovered by the Bennett lab68. They showed that loss of the filamentation regulator Efg1 during gut passage in mice, as well as Candida strains cultured from the gut of colonized humans, was associated with increased fitness for gut commensalism and increased virulence during systemic infection. Collectively, the expanding knowledge of molecular factors that regulate fitness within the GI tract shows promise for developing targeted strategies to modulate fungal colonization and subsequent invasive fungal infections in humans.
Candida and its crosstalk with the mammalian host
The microbiota and the mucosal immune system both exert significant influence over Candida morphological transitions. For example, IL-17 is significantly induced by C. albicans hyphae69, and Th17 and γδ T cells are critical for clearing this morphology during murine mucosal Candida infections70,71. In the gut, there is a high concentration of Th17 cells within the intestinal lamina propria, which may inhibit or quickly clear hyphae from the gut. Indeed, Candida expands within the GI tract and invades intestinal tissue when Th17 cells are depleted by broad-spectrum antibiotics72. Candida also secretes mediators to inhibit IL-17 immune responses during invasive infection. Specifically, secretion by C. albicans of the lipase Lip2, made predominantly by hyphae, suppressed IL-23 production from tissue-resident dendritic cells that in turn dampened IL-17 production by γδ T cells73. Infection of mice with a lip2-null C. albicans mutant resulted in exaggerated IL-17 production and enhanced clearance and protection from infection. Therefore, Candida can modulate the lipid microenvironment using secreted mediators, which may have significant consequences for host-fungal interactions. Whether C. albicans can directly modulate the metabolic milieu of the gut is unclear, although initial studies have indicated that Candida GI colonization of infants can affect the production of immune-stimulatory metabolites (discussed below).
Gut colonizing Candida yeasts appear to induce Th17 cells systemically. Humans colonized with Candida have increased frequency of circulating antifungal Th17 cells which are cross-reactive to unrelated fungi including Malassezia and Aspergillus26. Intestinal inflammation boosts the number of circulating antifungal Th17 cells, which may act in a pathologic manner at distant anatomical sites. For example, Th17 cells cross-reactive to Aspergillus spores were found to augment allergic-type inflammation within the lung, thus exacerbating asthma pathology and sensitization to inhaled fungal spores. In mice, colonization with Candida also induces systemic Th17 cells that exacerbated lung inflammation in allergic disease but had no impact on control of viral pneumonia74. However, Th17 cells induced by C. albicans gut colonization were protective against invasive infection caused by C. albicans or S. aureus. Protection against systemic fungal (Aspergillus) and bacterial (Staphylococcus, Pseudomonas) infections also occurred independently of adaptive immunity when mice were colonized with gut-adapted Candida strains, implicating trained immunity in this phenomenon75. Taken together, these studies show that Candida gut colonization modulates both local and systemic host immunity. While this tradeoff may afford protection against invasive infections, it could promote pathology in multiple organs along the gut-lung axis. Indeed, young babies with increased carriage of Candida had a higher risk of developing childhood asthma76. Mechanistically, early colonization with Candida changed the metabolite profile within the gut, skewing T cell polarization ex vivo. Adult T cells exposed to the metabolite cocktail derived from the guts of Candida colonized babies prevented the differentiation of regulatory T cells (Tregs) and favored the production of the Th2 cytokine IL-4. Similar functional skewing in vivo may explain the increased susceptibility to allergic pathology in the lung that is driven by IL-4 and lack of suppression by Tregs. Moreover, recent work from the Corry lab identified the peptide toxin candidalysin as the C. albicans-derived factor that drives direct type-2 allergenic responses within the lung following inhalation by C. albicans77. Continued investigations into Candida colonization and subsequent systemic pathology may thus reveal insight into molecular mediators of host homeostasis, and physiologic disturbance regimes that shape microbial communities.
In addition to driving systemic pathology, Candida can exacerbate localized GI inflammatory disease. During intestinal inflammation, Candida may produce candidalysin, which forms pores that penetrate the membrane of epithelial cells to induce damage and enable hyphal invasion across mucosal barriers78. The GI tract of patients with IBD are enriched in Candida species13, which when cultured are more likely to produce candidalysin than are strains isolated from healthy controls. Candidalysin activated the inflammasome and IL-1β production from macrophages, both of which strongly correlated with the severity of ulcerative colitis. Candidalysin-mediated immune cell damage and activation drove pathologic Th17 immunity in the gut, contributing towards the inflammatory sequalae seen in IBD. Similarly, patients with either gastric or colonic cancers were found to have an increased abundance of Candida species in their microbiome61. Moreover, Candida was detected directly within solid tumors, suggesting that the fungus may be capable of invading tumor tissues. Of interest, the presence of fungi within GI tract tumors was predictive of mortality in humans. Collectively, these studies demonstrate that Candida colonization has far-reaching consequences for health and disease, which calls for further mechanistic understanding.
C. auris as a model to understand the damage response framework
Genomic sequencing of C. auris revealed that four clades from distinct continents separated by tens of thousands of single nucleotide polymorphisms (SNPs) emerged nearly simultaneously79. Efforts to ascertain the origin of C. auris identified the first isolate in banked Candida isolate collections from 199680. However, how C. auris quickly emerged as 4 distinct clades remains an enigma. Over the last decade, outbreaks have been reported across the globe. In New York City, a highly inter-connected web of healthcare facilities, including skilled nursing homes, long-term care facilities, long-term acute care hospitals and acute care hospitals, have reported a regional outbreak detected as both clinical and surveillance cases81. Of concern, recent reports suggest the COVID-19 pandemic was associated with an apparent increase in C. auris transmission in acute care hospitals82. Due to its high propensity for spread and high frequency of resistance to antifungals and disinfectants, the U.S. Centers for Disease Control and Prevention (CDC) described C. auris as an urgent health threat in 201983, and the WHO listed it as one of the world’s most threatening fungi in 202223. Cases of colonization can escape detection as a result of its ability to colonize human skin long-term, without provoking a clinically apparent inflammatory response. Consequently, C. auris readily establishes outbreaks in congregant settings including cycles of endemic infection. Here, we explore what is known about its ability to colonize skin, the consequences of persistent colonization, and explanations for how antibiotics increase the likelihood of colonization and infection with C. auris Consideration of the impact of antibiotics on invasive C. auris infection fits within the damage response framework and highlights a role for antibiotic stewardship in the management of fungal disease.
Persistent colonization of skin
Skin and nares are the primary sites of colonization for C. auris while bloodstream infection is the major clinical risk. Patients who are colonized with C. auris can remain persistently, asymptomatically colonized for months or even as long as a year84. Colonization of individuals in healthcare facilities experiencing endemic spread tends to be multi-focal with more than one body site testing positive, but without any single site identifying all colonized patients54. Bioburden of C. auris is strikingly high with colony forming unit (CFU) counts exceeding a million in nares, axilla/groin, fingertips, and toe web samples54,85. The bioburden of C. auris on humans exceeds that observed for low abundance members of the skin mycobiome, C. parapsilosis, C. orthopsilosis, and C. tropicalis57. Ex vivo studies suggest that C. auris can form dense, multi-layer biofilms on porcine skin cultivated in sweat with population densities exceeding that of other Candida species86. C. auris colonization does not induce clinically apparent skin inflammation in humans, an observation recapitulated in animal studies which report a lack of skin erythema or histological evidence of hyperkeratosis or spongiosis87. The lack of skin inflammation cannot be attributed to superficial colonization of skin, where immune cells may not infiltrate, as C. auris has been found to reside within deeper skin tissue, including in glands and hair follicles88. Moreover, colonization with C. auris leads to enrichment of CD4+ IL-17A+ and CD4+ IL-17F+ (Th17) cells as well as CD8+ IL-17A (Tc17) cells, γδ T cells, and type-3 innate lymphoid cells within the skin compartment. Ablation of IL-17 signaling results in an increase in C. auris colonization with contributions noted for both innate and adaptive IL-17–producing lymphoid cell populations, suggesting intimate tuning of C. auris colonization bioburden by the host immune system. While recognized as a human pathogen, C. auris behaves essentially as a skin commensal.
Current work has focused on elucidating the molecular determinants of virulence in C. auris using experimental infection paradigms, rather than paradigms meant to identify commensal-pathogen switches (Table 1). Such studies have revealed genes such as Hog121 and at least one long non-coding RNA, DINOR20, that drive the regulation of stress response and virulence programs in C. auris. Moreover, passage of C. auris through a mammalian host induces atypical morphology, giving rise to subpopulations of aggregative and filamentous isolates, a phenomenon observed in some clinical studies. Importantly, aggregative strains appear to be less virulent than non-aggregative strains and exhibit divergent transcriptional profiles89,90. ACE2 and TOR3 were identified as transcriptional switches underlying the aggregating phenotype in C. auris while ELM1 was identified as a regulator of filamentation90. It is intriguing to speculate that these transcriptional switches govern the transition between the commensal to pathogenic lifestyle in C. auris, though future work is needed to elucidate the molecular determinants of C. auris colonization.
Clinical significance of persistent colonization
Skin colonization with C. auris is the largest risk factor for a subsequent bloodstream infection, highlighting the importance of controlling fungal colonization. Moreover, early clinical studies have reported patients who resolve one bout of systemic C. auris infection often experience recurrent infection84, reinforcing the need to decolonize patients. Contamination of the hospital environment (Figure 1A) by live C. auris is a reservoir for recolonization and/or infection with a required minimum contact period of 4 hours91. In the environment, a higher count of live cells was recovered from non-porous compared to porous hospital surfaces85. In the UK, C. auris isolates were detected on reusable equipment, including axillary skin-surface temperature probes, more often than in the general environment92. Investigators have postulated that bedding and clothing may serve as sources of C. auris exposure, contributing to persistent multi-focal colonization91. In addition to these environmental sources, autoinoculation from sites experiencing occult or even sub-surface colonization may play a contributing role in perpetuating colonization54,88. Together, these studies demonstrate that an integrated approach will be required to prevent and control C. auris colonization.
Figure 1: Implications of persistent skin colonization by Candida auris.
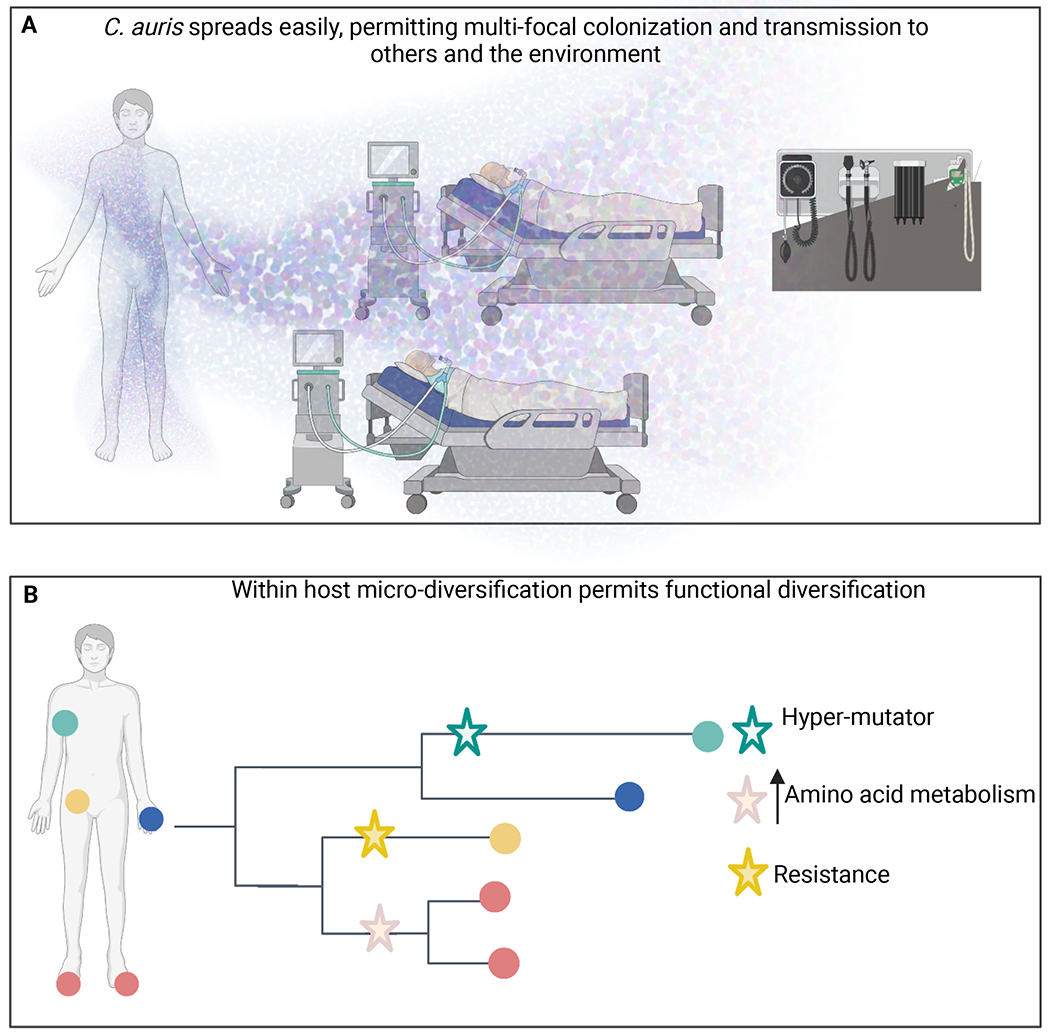
A) Skin shedding distributes a million microbes per hour into the environment, creating a reservoir for transmission. B) Persistent colonization of multiple body sites permits micro-diversification, which may prove to be adaptive. Created with BioRender.com.
In addition to the risk posed to patients, persistent colonization is clinically important as it permits the genomic diversification and possible adaptive evolution of C. auris which poses a longer-term risk (Figure 1B). An investigation of the genomic diversity of C. auris isolates revealed its micro-diversification across body sites on a single individual93. Samples from 5 body sites of a single individual yielded clones separated by 4-9 SNPs. A larger study revealed that more than 20% of patients were colonized by multiple C. auris isolates that differed from each other by more than 5 SNPs92. Population metagenomes of C. auris isolates from temperature probes matched the mixed colonization of some patients, suggesting that micro-diversity on the patient is reflected on environmental surfaces. These findings have been replicated with surveillance and clinical cultures94,95. In addition to human samples, C. auris strains can also colonize organic surfaces such as apples96. Intriguingly, C. auris isolates on individual apples were separated by as few as 1-8 or as many as 107 SNPs. This suggests that the apples were colonized more than once with distinct C. auris strains and that microevolution occurred after colonization, consistent with observations in humans.
Given that C. auris chronically colonizes individuals at high densities, and standard clinical surveillance tends to sample limited body sites, the full extent to which C. auris micro-diversification occurs is not yet known. Nor have studies yet illuminated the functional consequences of C. auris micro-diversification. However, we can gain insight into the significance of this population structure by considering other microbes that colonize patients long term. Micro-diversification of Pseudomonas aeruginosa in patients with cystic fibrosis (CF) generates functional diversity soon after initial colonization97 with adaptation to chronic infection occurring via parallel evolution within 3 years98. Specifically, isolates evolved along independent trajectories towards reduced mobility, exhibiting lineage specific evolution in terms of the elaboration of quorum-sensing molecules and secretion of proteases. Intriguingly, parallel evolution of antibiotic resistance occurred at spatially segregated sites with population variants diversifying according to sub-lineage specific mutation rates. Similar results are seen for other bacteria in which the co-existence of distinct sub-lineages in the CF lung extends over the course of years99.
Providing parallel evidence, in vitro evolution of C. auris recapitulates clinical reports in humans; that is, population diversity with the evolution of several distinct sub-lineages occurring in vitro in just a handful of passages100. Recurrent mutations were found in multiple clinical isolates in 15 genes involved in nutrient acquisition, as well as in transcriptional and cell cycle regulation genes. A hypermutator clinical isolate of C. auris was identified with a missense mutation in the C. auris homolog of the mismatch DNA repair component MLH1. Mutations in DNA mismatch repair genes have arisen during micro-evolution underlying hyper-mutator strains of A. fumigatus, Saccharomyces cerevisiae, C. albicans, C. glabrata, Cryptococcus deuterogattii, and C. neoformans101. In the C. albicans diploid genome, the rate of micro-evolution is driven in part by recombination-induced mutagenesis102. Moreover, the rate of mutation in C. albicans is roughly 100 times higher during passage through the murine oral cavity than during in vitro passage, suggesting that a multitude of host factors select for enrichment of SNPs103. In the haploid genome of C. glabrata, frameshift and nonsynonymous mutations have been associated with enrichment in cell surface genes, as well as genes associated with antifungal resistance, including ERG4 and FKS2104. Thus, micro-diversification potentially permits sub-lineages to evolve along distinct lines – generating an insurance policy that permits survival in the face of a fluctuating host environment. Just as a viral quasi-species is adaptive, so too might be the micro-diversity of fungal species.
Multiple alternative hypotheses to explain antibiotics as a risk factor for C. auris infection
Antibiotic receipt is an independent risk factor for C. auris infection in global studies from South Korea, Venezuela, India, Pakistan, and the United States80,105,106. A large-scale study examining the role of antimicrobial drug exposure found that cephalosporins, carbapenems, clindamycin, trimethoprim-sulfamethoxazole, and colistin use is associated with a significantly increased odds of colonization by fluconazole-resistant Candida species107. Our work previously reported receipt of antibiotics as a risk factor for colonization of humans with C. auris54. What factors may contribute to this outcome? We propose several alternative, non-mutually exclusive hypotheses that may explain this phenomenon including disruption of colonization resistance and tonic immune stimulation (Figure 2A1–4), sepsis induced immunosuppression (Figure 2B), and modulation of organismal fitness by the antibiotic (Figure 2C). Herein, we survey the literature supporting each hypothesis, and highlight critical gaps in our knowledge.
Figure 2: Alternative hypotheses explaining host susceptibility to infection and colonization with C. auris.
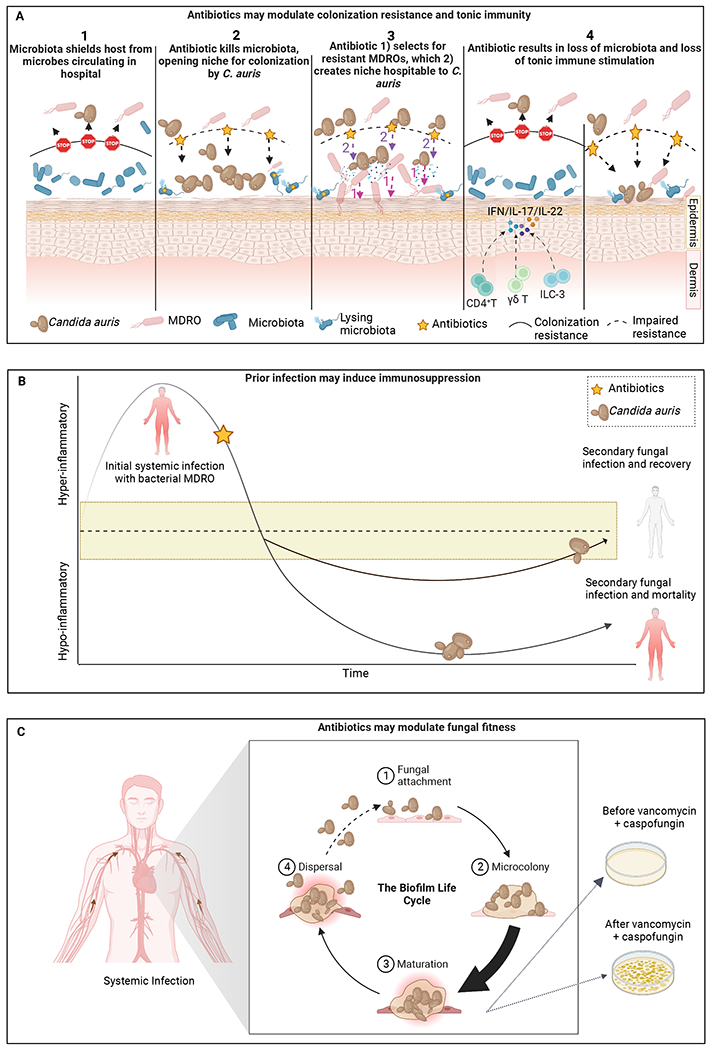
A) Antibiotics may open a niche for C. auris colonization by modulating the commensal microbiota via several potential mechanisms. B) Adapted from117, an initial bout of bacterial sepsis may induce immunosuppression, paving the way for secondary infection and mortality, with antibiotic serving as a red herring. The yellow banding indicates a range of immune-competence. C) Vancomycin and other antibiotics may directly modulate the fitness of C. auris by increasing biofilm formation and inducing a state of tolerance, or via other mechanisms. Created with BioRender.com.
Depletion of colonization resistance and/or loss of tonic immune stimulation
Antibiotic-mediated depletion of the commensal microbiota may permit C. auris to colonize a site from which it would otherwise be excluded (Figure 2A.1). Studies of humans have been observational and cannot be used to infer causality. Nonetheless, some consistent themes have emerged. In an observational study of surveillance samples from 3 nursing homes in California, Illinois, and New York, we reported that C. auris colonization is associated with an apparent enrichment of Proteobacteria commonly involved in nosocomial infection such as Acinetobacter baumannii, K. pneumoniae, and E. faecalis108. Surveillance samples, in that study, were not linked to clinical data, so it was impossible to determine the extent to which factors such as antibiotic usage or health status were associated with the apparent bloom of Proteobacteria or C. auris colonization. A larger point prevalence survey of a ventilator skilled nursing facility in Illinois revealed that the relative abundance of species that tend to be highly abundant on the skin of healthy adults – including Staphylococcus hominis, Corynebacterium striatum, Corynebacterium accolens, and Staphylococcus caprae – is negatively correlated with C. auris abundance54. Concomitantly, the relative abundance of multidrug-resistant organisms (MDROs) – including P. aeruginosa, A. baumannii, Proteus mirabilis, K. pneumoniae, and Providencia stuartii – is positively correlated with C. auris relative abundance. Where MDROs predominated, bacterial diversity was reduced. Intriguingly, a reduction of C. auris abundance over time, as measured by ITS1 sequencing, was accompanied by an increase in bacterial diversity, as well as a transition to a more ‘normal’ bacterial community, as measured by 16S sequencing. Yet, due to the observational nature of this report, it was not possible to determine whether C. auris colonization led to the depletion of commensal species and apparent enrichment of Proteobacteria, or whether the antibiotic (or some other factor) did. Thus, we still do not understand which potential mechanism (Figure 2A) explains the association between C. auris colonization and antibiotic administration in humans, highlighting a critical gap in our understanding.
A murine model of invasive candidiasis could provide insight into the modes of action of antibiotics against C. auris109. Using germ free, antibiotic treated, and control mice, the De Jesus group examined the role of the microbiome in moderating systemic infection following intravenous (IV) injection or gavage of a C. auris Clade I isolate. They reported a significant increase in uterine C. auris after IV administration of C. auris in immunocompetent mice following treatment with antibiotics compared to mice with an intact microbiome, with no other differences across other examined organ systems (i.e., heart, lungs, cecum, liver, spleen, kidney, stomach, and brain). Following gavage, immunocompetent mice experienced a significant increase in liver tissue bioburden following treatment with antibiotics, but no other organ systems carried higher burdens of C. auris in germ free or antibiotic exposure conditions, compared to controls. Differences between germ free immunocompetent mice and controls were not significant. Taken together, these data suggest that the antibiotic rather than the microbiota may have mediated the increased risk of uterine or liver infection following IV injection or gavage of C. auris. Neutropenic germ-free mice, in contrast, showed significantly higher risk of infection in the heart and lungs following IV injection of C. auris, while antibiotics appeared to increase infection of the cecum and stomach in neutropenic mice, suggesting that the microbiota may play a role in protecting against infection in the immunocompromised host. While C. auris appeared to be the dominant fungus, in that study, results from the 16S survey were not reported, so it was not possible to understand the specific bacteria involved in conferring colonization resistance, or their response to immunosuppression or to the antibiotic exposure. Additional work that aims to disambiguate the timing of C. auris colonization relative to the enrichment of Proteobacteria, the delivery of antibiotics, the specific antibiotic classes that mediate these effects, and immunological status will help elucidate these complex relationships.
While results for C. auris are inconclusive, recent work using C. albicans has begun to illuminate our understanding of the interaction between antibiotics, colonization resistance, and immunity. Huang and colleagues compared the gut microbiome of rats infected with C. albicans after antibiotic treatment, rats inoculated with fungi only, and rats without any treatment110. Strikingly, infection with fungi after antibiotic treatment is associated with a significant decrease in bacterial diversity as compared to infection with fungi alone. This suggests that treatment with the antibiotic, rather than infection with the fungus, results in reduced bacterial diversity, an important distinction considering C. albicans produces the quorum sensing compound farnesol which could act directly against a wide range of bacteria111. Consistent with this finding, a small-scale survey of the human gut microbiome following a 6-day course of antibiotics (of varying classes) revealed a significant decrease in bacterial diversity accompanied by a significant increase in fungal diversity32. In particular, Candida spp. increased 15-fold from baseline to treatment for most antibiotics. Of special interest, over 1/3 of fungi present before treatment showed significant changes in relative abundance 90 days after treatment, suggesting a long-lasting imprint of antibiotics on the mycobiome. Persistent perturbation of the mycobiome is particularly consequential considering our recent work reporting that antibiotic treatment triggers not just a net decrease in bacterial diversity and an expansion of Enterobacteriaceae/Candida in the gut, but also immune dysregulation characterized by low local levels of Th17, IL-22, and GM-CSF producing lymphoid cells72 (Figure 2A.4). The concerted changes in the composition of the microbiota and loss of IL-17A and GM-CSF, but not IL-22, enhanced Candida-associated mortality in mice, facilitated by the joint translocation of C. albicans with bacterial species (e.g., Escherichia coli, Lactobacillus murinus, and Enterococcus gallinarum) from the gut into the bloodstream. Additionally, Artis and colleagues proposed that the commensal microbiota primes the innate immune system for a robust interferon response to viral challenge. They demonstrated increased mortality and tissue pathology in the murine lung as a result of an aberrant transcriptional profile underlying dampened interferon signaling, preventing clearance of influenza following antibiotic treatment112. Gut colonization by C. albicans was sufficient to reverse this effect, promoting tonic stimulation, and conferring protection against mortality due to viral infection113. It is tempting to speculate that C. auris, which typically colonizes the skin rather than the GI tract, may also take advantage of loss of colonization resistance as well as dysregulated immunity following antibiotics. Finally, as reviewed elsewhere, it is possible that reciprocal interactions between fungi and bacteria play a pivotal role in determining the fate of fungal species following antibiotic mediated ablation of bacterial diversity114.
Collateral damage from prior systemic infection
Anecdotal data suggest that infection with a bacterial MDRO or a prior bout of candidemia is a risk factor for future colonization with C. auris80,105,115,116. Given that bacteremia or candidemia often precedes infection with C. auris it may be possible that antibiotics serve as a red herring and that immune dysregulation related to sepsis may be the underlying risk factor. While initially controversial, it is now accepted that prior sepsis induces states of immunosuppression117–119 after resolution of the cytokine storm, which may increase the permissiveness of the host for C. auris colonization or infection (Figure 2B). However, clinical studies reporting the incidence of infection with C. auris after antibiotic treatment have not reported immunological data. Additional work is thus needed to understand the extent to which immunosuppression related to sepsis underlies the association between C. auris and antibiotic use. Given that bloodstream infections involving Candida spp. and various MDROs is associated with increased mortality120, future clinical studies reporting antibiotics as a risk factor should report immunological status of the host, so that this risk factor can be disentangled.
Antibiotics may directly modulate fungal fitness
In 1929, Alexander Fleming famously reported that fungi produce antibiotics that can kill bacteria by observing the effects of fungal contamination on bacterial plates. Before they were used as pharmaceuticals to kill bacteria infecting humans, antibiotics were simply compounds used by microbes to optimize their fitness in their native environments where they had to compete against both fungal and bacterial species for space and nutrients. Indeed, a rich history of literature from the 1940s-1950s suggests that antibiotics that kill bacteria have direct effects on fungi, which may be inhibitory or stimulatory38. For example, trimethoprim is commonly used to treat urinary tract infections and is active against both gram-positive and gram-negative bacteria121. Trimethoprim also has inhibitory activity against certain fungi including Paracoccidioides brasiliensis and Pneumocystis jirovecii122. Sulfa drugs similarly target folate synthesis and are broad spectrum antagonists of bacteria. When tested against 70 Aspergillus strains, encompassing 6 species, most sulfonamides exhibited fungistatic activity, except for pentamidine, which was fungicidal for A. nidulans123. Pentamidine is also active and fungicidal against Fusarium species, particularly F. oxysporum124,125.
Intriguingly, penicillin was shown to stimulate, rather than inhibit, the growth of C. albicans in vitro, leading Foley & Winter to conclude that the antibiotic directly or indirectly (via the metabolites released by the lysis of antibiotic-sensitive Candida cells) stimulated the growth of survivors126. Other data conflict, reporting that penicillin does not stimulate growth of C. albicans127. These discrepant results are likely due to differences in growth media and the Candida species deployed in each study. More recently, work examining the impact of vancomycin, rifampin, and amoxicillin on the growth of C. auris also reported antibiotics as stimulating the growth of Candida128. Specifically, vancomycin resulted in a 28% increase in CFUs in C. auris biofilms cultivated in vitro, an effect not observed with either amoxicillin or rifampicin. Second, treatment of the C. auris biofilm with vancomycin increased the tolerance of the biofilm towards the antifungal caspofungin. Finally, using an insect model, larvae infected with C. auris in the presence of vancomycin resulted in a significant increase in mortality compared to C. auris alone. Taken together, these data suggest that antibiotics may modulate the fitness of C. auris directly via unknown mechanisms (Figure 2C). In a murine model, we did not see an increase in C. auris skin colonization following pre-treatment with tetracycline or a combination of ampicillin, metronidazole, neomycin, and vancomycin88. Therefore, additional work examining the effects of different classes of antibiotics on the fitness of C. auris is needed. It is also worth noting that certain antibiotics may suppress the host immune system129, providing another mechanism by which antibiotics may modulate fungal fitness, predisposing the host to invasive infection. Finally, as reviewed elsewhere, it is possible that specific reciprocal interactions between fungi and bacteria will play a pivotal role in determining the fate of fungal species following antibiotic mediated ablation of bacterial diversity114.
Perspectives and future directions
Given the urgent concern posed by C. auris infections, we are not accustomed to considering that C. auris behaves, at times, like a commensal. We have historically used dichotomous thinking to describe the organisms that live on our bodies -- either they are commensals, or they are pathogens. The damage response framework provides a useful lexicon1 whereby members of the microbiota adopt different behaviors, either the commensal or pathogenic lifestyle, and they can transition from one to the other depending on environmental cues, including shifts in disturbance regime, invasion of the microbiota by other organisms, or disturbance associated with chemicals, like antibiotics. Moreover, different members of a given population may exist in the commensal or pathogenic lifestyle at the same time, as evidenced by the simultaneous presence of yeast and hyphal forms of C. albicans in the gut. Fully incorporating recognition of behavioral phenotypes into our understanding of the microbiota permits us to define and target a set of switches that regulate the behavioral lifestyle of any given microbe.
A consensus has emerged that C. auris undergoes micro-diversification on its host on the timescale of months. As additional studies of C. auris outbreaks are undertaken, it is critically important investigators build on this consensus. Integrating spatial and longitudinal genomic data with clinical data will permit us to identify the ecological and evolutionary forces shaping the adaptation of C. auris to the human host and its hospital environment. In turn, we hope that this information may inspire therapeutic modalities to prevent colonization and infection with C. auris or to lock it out of the pathogenic state.
Furthermore, C. albicans remains a leading cause of nosocomial bloodstream infection with high mortality despite treatment. Leveraging this well-defined model system, future work must continue to innovate, developing experimental paradigms that elucidate fungal-derived and host immune factors that govern the balance between commensalism and pathogenesis. Such work should continue to build on our nascent understanding of how colonization at one site modulates immune responses at distal anatomical sites. At the same time, future work should begin to elucidate the role other members of the human microbiota play in promoting protective or pathogenic immune responses to C. albicans commensalism and pathogenesis.
More broadly, it is currently difficult to determine the extent to which each hypothesis proposed in Figure 2 increments the risk of invasive candidiasis following antibiotic treatment. This gap in our knowledge limits our power to intervene. Consequently, future studies should illuminate the mechanisms by which antibiotics act on fungi. Moreover, studies quantifying risk factors for invasive fungal infection should include host immunological profiles in their models to increase our understanding of the extent to which sepsis-induced immunosuppression, or loss of tonic immunity, increases the risk of invasive fungal disease post-antibiotic treatment. Moving forward, the challenge will be to develop translational strategies that integrate information from basic science, genomic, and clinical studies in order to flip the fungal switch in favor of patient outcomes.
Acknowledgements
This work was supported by the NIH Division of Intramural Research of the NHGRI and the NIAID. The authors would like to thank Jonathan Nicklas for his helpful suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Casadevall A, and Pirofski LA (2000). Host-pathogen interactions: basic concepts of microbial commensalism, colonization, infection, and disease. Infect Immun 68, 6511–6518. 10.1128/IAI.68.12.6511-6518.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casadevall A, and Pirofski LA (2015). What is a host? Incorporating the microbiota into the damage-response framework. Infect Immun 83, 2–7. 10.1128/IAI.02627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrd AL, Belkaid Y, and Segre JA (2018). The human skin microbiome. Nat Rev Microbiol 16, 143–155. 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 4.Sparber F, De Gregorio C, Steckholzer S, Ferreira FM, Dolowschiak T, Ruchti F, Kirchner FR, Mertens S, Prinz I, Joller N, et al. (2019). The Skin Commensal Yeast Malassezia Triggers a Type 17 Response that Coordinates Anti-fungal Immunity and Exacerbates Skin Inflammation. Cell Host Microbe 25, 389–403 e386. 10.1016/j.chom.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Limon JJ, Skalski JH, and Underhill DM (2017). Commensal Fungi in Health and Disease. Cell Host Microbe 22, 156–165. 10.1016/j.chom.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhimi W, Theelen B, Boekhout T, Otranto D, and Cafarchia C (2020). Malassezia spp. Yeasts of Emerging Concern in Fungemia. Front Cell Infect Microbiol 10, 370. 10.3389/fcimb.2020.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedrosa AF, Lisboa C, and Rodrigues AG (2018). Malassezia infections with systemic involvement: Figures and facts. J Dermatol 45, 1278–1282. 10.1111/1346-8138.14653. [DOI] [PubMed] [Google Scholar]
- 8.Vijaya Chandra SH, Srinivas R, Dawson TL Jr., and Common JE (2020). Cutaneous Malassezia: Commensal, Pathogen, or Protector? Front Cell Infect Microbiol 10, 614446. 10.3389/fcimb.2020.614446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubin R (2022). On the Rise, Candida auris Outwits Treatments and Travels Incognito in Health Care Settings. JAMA. 10.1001/jama.2022.17760. [DOI] [PubMed] [Google Scholar]
- 10.Chow EWL, Pang LM, and Wang Y (2021). From Jekyll to Hyde: The Yeast-Hyphal Transition of Candida albicans. Pathogens 10. 10.3390/pathogens10070859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witchley JN, Penumetcha P, Abon NV, Woolford CA, Mitchell AP, and Noble SM (2019). Candida albicans Morphogenesis Programs Control the Balance between Gut Commensalism and Invasive Infection. Cell Host Microbe 25, 432–443 e436. 10.1016/j.chom.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pande K, Chen C, and Noble SM (2013). Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet 45, 1088–1091. 10.1038/ng.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li XV, Leonardi I, Putzel GG, Semon A, Fiers WD, Kusakabe T, Lin WY, Gao IH, Doron I, Gutierrez-Guerrero A, et al. (2022). Immune regulation by fungal strain diversity in inflammatory bowel disease. Nature 603, 672–678. 10.1038/s41586-022-04502-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C, Pande K, French SD, Tuch BB, and Noble SM (2011). An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10, 118–135. 10.1016/j.chom.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, He S, Zhang S, A R, Li W, and Liu S (2022). The Necrotroph Botrytis cinerea BcSpd1 Plays a Key Role in Modulating Both Fungal Pathogenic Factors and Plant Disease Development. Front Plant Sci 13, 820767. 10.3389/fpls.2022.820767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rolig AS, Sweeney EG, Kaye LE, DeSantis MD, Perkins A, Banse AV, Hamilton MK, and Guillemin K (2018). A bacterial immunomodulatory protein with lipocalin-like domains facilitates host-bacteria mutualism in larval zebrafish. Elife 7. 10.7554/eLife.37172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DAB, John E, Rybak K, Phan HTT, Singh KB, Lin SY, Solomon PS, Oliver RP, and Tan KC (2019). A specific fungal transcription factor controls effector gene expression and orchestrates the establishment of the necrotrophic pathogen lifestyle on wheat. Sci Rep 9, 15884. 10.1038/s41598-019-52444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finkel-Jimenez B, Wuthrich M, and Klein BS (2002). BAD1, an essential virulence factor of Blastomyces dermatitidis, suppresses host TNF-alpha production through TGF-beta-dependent and -independent mechanisms. J Immunol 168, 5746–5755. 10.4049/jimmunol.168.11.5746. [DOI] [PubMed] [Google Scholar]
- 19.Krappmann S, Bignell EM, Reichard U, Rogers T, Haynes K, and Braus GH (2004). The Aspergillus fumigatus transcriptional activator CpcA contributes significantly to the virulence of this fungal pathogen. Mol Microbiol 52, 785–799. 10.1111/j.1365-2958.2004.04015.x. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Chow EWL, Wang H, Xu X, Cai C, Song Y, Wang J, and Wang Y (2021). LncRNA DINOR is a virulence factor and global regulator of stress responses in Candida auris. Nat Microbiol 6, 842–851. 10.1038/s41564-021-00915-x. [DOI] [PubMed] [Google Scholar]
- 21.Day AM, McNiff MM, da Silva Dantas A, Gow NAR, and Quinn J (2018). Hog1 Regulates Stress Tolerance and Virulence in the Emerging Fungal Pathogen Candida auris. mSphere 3. 10.1128/mSphere.00506-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alanio A, Vernel-Pauillac F, Sturny-Leclere A, and Dromer F (2015). Cryptococcus neoformans host adaptation: toward biological evidence of dormancy. mBio 6. 10.1128/mBio.02580-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.WHO fungal priority pathogens list to guide research, development and public health action. Geneva: World Health Organization; 2022. License: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- 24.Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, and Knight R (2018). Current understanding of the human microbiome. Nat Med 24, 392–400. 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derrien M, and van Hylckama Vlieg JE (2015). Fate, activity, and impact of ingested bacteria within the human gut microbiota. Trends Microbiol 23, 354–366. 10.1016/j.tim.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, Röhmel J, Eschenhagen P, Grehn C, Seidel K, et al. (2019). Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell 176, 1340–1355.e1315. 10.1016/j.cell.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 27.Runge S, and Rosshart SP (2021). The Mammalian Metaorganism: A Holistic View on How Microbes of All Kingdoms and Niches Shape Local and Systemic Immunity. Front Immunol 12, 702378. 10.3389/fimmu.2021.702378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Relman DA (2012). The human microbiome: ecosystem resilience and health. Nutr Rev 70 Suppl 1, S2–9. 10.1111/j.1753-4887.2012.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jo JH, Harkins CP, Schwardt NH, Portillo JA, Program NCS, Zimmerman MD, Carter CL, Hossen MA, Peer CJ, Polley EC, et al. (2021). Alterations of human skin microbiome and expansion of antimicrobial resistance after systemic antibiotics. Sci Transl Med 13, eabd8077. 10.1126/scitranslmed.abd8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pankhurst CL (2013). Candidiasis (oropharyngeal). BMJ Clin Evid 2013, 1304. [PMC free article] [PubMed] [Google Scholar]
- 31.Goncalves B, Ferreira C, Alves CT, Henriques M, Azeredo J, and Silva S (2016). Vulvovaginal candidiasis: Epidemiology, microbiology and risk factors. Crit Rev Microbiol 42, 905–927. 10.3109/1040841X.2015.1091805. [DOI] [PubMed] [Google Scholar]
- 32.Seelbinder B, Chen J, Brunke S, Vazquez-Uribe R, Santhaman R, Meyer AC, de Oliveira Lino FS, Chan KF, Loos D, Imamovic L, et al. (2020). Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome 8, 133. 10.1186/s40168-020-00899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhai B, Ola M, Rolling T, Tosini NL, Joshowitz S, Littmann ER, Amoretti LA, Fontana E, Wright RJ, Miranda E, et al. (2020). High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat Med 26, 59–64. 10.1038/s41591-019-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, Brown J, Funari VA, Wang HL, Crother TR, et al. (2016). Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 19, 865–873. 10.1016/j.chom.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Horo JC, Silva GL, Munoz-Price LS, and Safdar N (2012). The efficacy of daily bathing with chlorhexidine for reducing healthcare-associated bloodstream infections: a meta-analysis. Infect Control Hosp Epidemiol 33, 257–267. 10.1086/664496. [DOI] [PubMed] [Google Scholar]
- 36.SanMiguel AJ, Meisel JS, Horwinski J, Zheng Q, Bradley CW, and Grice EA (2018). Antiseptic Agents Elicit Short-Term, Personalized, and Body Site-Specific Shifts in Resident Skin Bacterial Communities. J Invest Dermatol 138, 2234–2243. 10.1016/j.jid.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vich Vila A, Collij V, Sanna S, Sinha T, Imhann F, Bourgonje AR, Mujagic Z, Jonkers D, Masclee AAM, Fu J, et al. (2020). Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat Commun 11, 362. 10.1038/s41467-019-14177-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afeltra J, and Verweij PE (2003). Antifungal Activity of Nonantifungal Drugs. European Journal of Clinical Microbiology and Infectious Diseases 22, 397–407. 10.1007/s10096-003-0947-x. [DOI] [PubMed] [Google Scholar]
- 39.Proctor DM, and Relman DA (2017). The Landscape Ecology and Microbiota of the Human Nose, Mouth, and Throat. Cell Host Microbe 21, 421–432. 10.1016/j.chom.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dawes C (1972). Circadian rhythms in human salivary flow rate and composition. J Physiol 220, 529–545. 10.1113/jphysiol.1972.sp009721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collado MC, Engen PA, Bandin C, Cabrera-Rubio R, Voigt RM, Green SJ, Naqib A, Keshavarzian A, Scheer F, and Garaulet M (2018). Timing of food intake impacts daily rhythms of human salivary microbiota: a randomized, crossover study. FASEB J 32, 2060–2072. 10.1096/fj.201700697RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prado-Mel E, Ciudad-Gutierrez P, Rodriguez-Ramallo H, Sanchez-Fidalgo S, Santos-Ramos B, and Villalba-Moreno AM (2022). Association between anticholinergic activity and xerostomia and/ or xerophthalmia in the elderly: systematic review. BMC Pharmacol Toxicol 23, 94. 10.1186/s40360-022-00637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dreizen S, Brown LR, Handler S, and Levy BM (1976). Radiation-induced xerostomia in cancer patients. Effect on salivary and serum electrolytes. Cancer 38, 273–278. [DOI] [PubMed] [Google Scholar]
- 44.Proctor DM, Fukuyama JA, Loomer PM, Armitage GC, Lee SA, Davis NM, Ryder MI, Holmes SP, and Relman DA (2018). A spatial gradient of bacterial diversity in the human oral cavity shaped by salivary flow. Nat Commun 9, 681. 10.1038/s41467-018-02900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ooshima T, Hashida T, Fuchihata H, Fujiwara T, Yoshida T, Izumitani A, Sobue S, and Hamada S (1991). Dental caries induction in hyposalivated rats. Caries Res 25, 138–142. 10.1159/000261356. [DOI] [PubMed] [Google Scholar]
- 46.Molek M, Florenly F, Lister INE, Wahab TA, Lister C, and Fioni F (2022). Xerostomia and hyposalivation in association with oral candidiasis: a systematic review and meta-analysis. Evid Based Dent. 10.1038/s41432-021-0210-2. [DOI] [PubMed] [Google Scholar]
- 47.Park J, Schwardt NH, Jo JH, Zhang Z, Pillai V, Phang S, Brady SM, Portillo JA, MacGibeny MA, Liang H, et al. (2022). Shifts in the Skin Bacterial and Fungal Communities of Healthy Children Transitioning through Puberty. J Invest Dermatol 142, 212–219. 10.1016/j.jid.2021.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russell-Goldman E, and Murphy GF (2020). The Pathobiology of Skin Aging: New Insights into an Old Dilemma. Am J Pathol 190, 1356–1369. 10.1016/j.ajpath.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Loughlin LS, and Green PT (2017). Secondary invasion: When invasion success is contingent on other invaders altering the properties of recipient ecosystems. Ecol Evol 7, 7628–7637. 10.1002/ece3.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pang IK, and Iwasaki A (2012). Control of antiviral immunity by pattern recognition and the microbiome. Immunol Rev 245, 209–226. 10.1111/j.1600-065X.2011.01073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marsh PD (2003). Are dental diseases examples of ecological catastrophes? Microbiology (Reading) 149, 279–294. 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- 52.Kurkjian HM, Akbari MJ, and Momeni B (2021). The impact of interactions on invasion and colonization resistance in microbial communities. PLoS Comput Biol 17, e1008643. 10.1371/journal.pcbi.1008643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao C, Coyte KZ, Bainter W, Geha RS, Martin CR, and Rakoff-Nahoum S (2021). Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 591, 633–638. 10.1038/s41586-021-03241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Proctor DM, Dangana T, Sexton DJ, Fukuda C, Yelin RD, Stanley M, Bell PB, Baskaran S, Deming C, Chen Q, et al. (2021). Integrated genomic, epidemiologic investigation of Candida auris skin colonization in a skilled nursing facility. Nature Medicine 27, 1401–1409. 10.1038/s41591-021-01383-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larson PJ, Zhou W, Santiago A, Driscoll S, Fleming E, Voigt AY, Chun OK, Grady JJ, Kuchel GA, Robison JT, and Oh J (2022). Associations of the skin, oral and gut microbiome with aging, frailty and infection risk reservoirs in older adults. Nat Aging 2, 941–955. 10.1038/s43587-022-00287-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, and Kullberg BJ (2018). Invasive candidiasis. Nature Reviews Disease Primers 4, 18026. 10.1038/nrdp.2018.26. [DOI] [PubMed] [Google Scholar]
- 57.Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, Program, N.I.H.I.S.C.C.S., et al. (2013). Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370. 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mishra AA, and Koh AY (2018). Adaptation of Candida albicans during gastrointestinal tract colonization. Curr Clin Microbiol Rep 5, 165–172. 10.1007/s40588-018-0096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heisel T, Montassier E, Johnson A, Al-Ghalith G, Lin YW, Wei LN, Knights D, and Gale CA (2017). High-Fat Diet Changes Fungal Microbiomes and Interkingdom Relationships in the Murine Gut. mSphere 2. 10.1128/mSphere.00351-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fajstova A, Galanova N, Coufal S, Malkova J, Kostovcik M, Cermakova M, Pelantova H, Kuzma M, Sediva B, Hudcovic T, et al. (2020). Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling. Cells 9. 10.3390/cells9122701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dohlman AB, Klug J, Mesko M, Gao IH, Lipkin SM, Shen X, and Iliev ID (2022). A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 185, 3807–3822 e3812. 10.1016/j.cell.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bacher P, Jochheim-Richter A, Mockel-Tenbrink N, Kniemeyer O, Wingenfeld E, Alex R, Ortigao A, Karpova D, Lehrnbecher T, Ullmann AJ, et al. (2015). Clinical-scale isolation of the total Aspergillus fumigatus-reactive T-helper cell repertoire for adoptive transfer. Cytotherapy 17, 1396–1405. 10.1016/j.jcyt.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 63.Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, Bluemel S, Hartmann P, Xu J, Koyama Y, et al. (2017). Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest 127, 2829–2841. 10.1172/JCI90562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, Simms-Waldrip TR, Xie Y, Hooper LV, and Koh AY (2015). Activation of HIF-1α and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat Med 21, 808–814. 10.1038/nm.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McDonough LD, Mishra AA, Tosini N, Kakade P, Penumutchu S, Liang SH, Maufrais C, Zhai B, Taur Y, Belenky P, et al. (2021). Candida albicans Isolates 529L and CHN1 Exhibit Stable Colonization of the Murine Gastrointestinal Tract. mBio 12, e0287821. 10.1128/mBio.02878-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koh AY, Köhler JR, Coggshall KT, Van Rooijen N, and Pier GB (2008). Mucosal Damage and Neutropenia Are Required for Candida albicans Dissemination. PLoS Pathog 4, e35. 10.1371/journal.ppat.0040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lionakis MS, and Levitz SM (2018). Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu Rev Immunol 36, 157–191. 10.1146/annurev-immunol-042617-053318. [DOI] [PubMed] [Google Scholar]
- 68.Liang SH, Anderson MZ, Hirakawa MP, Wang JM, Frazer C, Alaalm LM, Thomson GJ, Ene IV, and Bennett RJ (2019). Hemizygosity Enables a Mutational Transition Governing Fungal Virulence and Commensalism. Cell Host Microbe 25, 418–431 e416. 10.1016/j.chom.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng SC, van de Veerdonk FL, Lenardon M, Stoffels M, Plantinga T, Smeekens S, Rizzetto L, Mukaremera L, Preechasuth K, Cavalieri D, et al. (2011). The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J Leukoc Biol 90, 357–366. 10.1189/jlb.1210702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206, 299–311. 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Conti HR, Peterson AC, Brane L, Huppler AR, Hernandez-Santos N, Whibley N, Garg AV, Simpson-Abelson MR, Gibson GA, Mamo AJ, et al. (2014). Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J Exp Med 211, 2075–2084. 10.1084/jem.20130877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drummond RA, Desai JV, Ricotta EE, Swamydas M, Deming C, Conlan S, Quinones M, Matei-Rascu V, Sherif L, Lecky D, et al. (2022). Long-term antibiotic exposure promotes mortality after systemic fungal infection by driving lymphocyte dysfunction and systemic escape of commensal bacteria. Cell Host Microbe. 10.1016/j.chom.2022.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Basso P, Dang EV, Urisman A, Cowen LE, Madhani HD, and Noble SM (2022). Deep tissue infection by an invasive human fungal pathogen requires lipid-based suppression of the IL-17 response. Cell Host Microbe 30, 1589–1601.e1585. 10.1016/j.chom.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shao T-Y, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, Pham G, Burg AR, Ruff B, Gonzalez T, et al. (2019). Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell Host Microbe 25, 404–417.e406. 10.1016/j.chom.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tso GHW, Reales-Calderon JA, Tan ASM, Sem X, Le GTT, Tan TG, Lai GC, Srinivasan KG, Yurieva M, Liao W, et al. (2018). Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595. 10.1126/science.aat0537. [DOI] [PubMed] [Google Scholar]
- 76.Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, Panzer AR, LaMere B, Rackaityte E, Lukacs NW, et al. (2016). Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nature Medicine 22, 1187–1191. 10.1038/nm.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu Y, Zeng Z, Guo Y, Song L, Weatherhead JE, Huang X, Zeng Y, Bimler L, Chang CY, Knight JM, et al. (2021). Candida albicans elicits protective allergic responses via platelet mediated T helper 2 and T helper 17 cell polarization. Immunity 54, 2595–2610 e2597. 10.1016/j.immuni.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Höfs S, Gratacap RL, Robbins J, Runglall M, et al. (2016). Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–68. 10.1038/nature17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lockhart SR, Etienne KA, Vallabhaneni S, Farooqi J, Chowdhary A, Govender NP, Colombo AL, Calvo B, Cuomo CA, Desjardins CA, et al. (2017). Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin Infect Dis 64, 134–140. 10.1093/cid/ciw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee WG, Shin JH, Uh Y, Kang MG, Kim SH, Park KH, and Jang HC (2011). First three reported cases of nosocomial fungemia caused by Candida auris. J Clin Microbiol 49, 3139–3142. 10.1128/JCM.00319-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adams E, Quinn M, Tsay S, Poirot E, Chaturvedi S, Southwick K, Greenko J, Fernandez R, Kallen A, Vallabhaneni S, et al. (2018). Candida auris in Healthcare Facilities, New York, USA, 2013-2017. Emerg Infect Dis 24, 1816–1824. 10.3201/eid2410.180649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thoma R, Seneghini M, Seiffert SN, Vuichard Gysin D, Scanferla G, Haller S, Flury D, Boggian K, Kleger GR, Filipovic M, et al. (2022). The challenge of preventing and containing outbreaks of multidrug-resistant organisms and Candida auris during the coronavirus disease 2019 pandemic: report of a carbapenem-resistant Acinetobacter baumannii outbreak and a systematic review of the literature. Antimicrob Resist Infect Control 11, 12. 10.1186/s13756-022-01052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.CDC. Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services, CDC; 2019. 10.15620/cdc:82532. [DOI] [Google Scholar]
- 84.Vallabhaneni S, Kallen A, Tsay S, Chow N, Welsh R, Kerins J, Kemble SK, Pacilli M, Black SR, Landon E, et al. (2016). Investigation of the First Seven Reported Cases of Candida auris, a Globally Emerging Invasive, Multidrug-Resistant Fungus - United States, May 2013-August 2016. MMWR Morb Mortal Wkly Rep 65, 1234–1237. 10.15585/mmwr.mm6544e1. [DOI] [PubMed] [Google Scholar]
- 85.Zhu Y, O’Brien B, Leach L, Clarke A, Bates M, Adams E, Ostrowsky B, Quinn M, Dufort E, Southwick K, et al. (2020). Laboratory Analysis of an Outbreak of Candida auris in New York from 2016 to 2018: Impact and Lessons Learned. J Clin Microbiol 58. 10.1128/JCM.01503-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Horton MV, Johnson CJ, Kernien JF, Patel TD, Lam BC, Cheong JZA, Meudt JJ, Shanmuganayagam D, Kalan LR, and Nett JE (2020). Candida auris Forms High-Burden Biofilms in Skin Niche Conditions and on Porcine Skin. mSphere 5, e00910–00919. doi: 10.1128/mSphere.00910-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghannoum M, Herrada J, McCormick TS, and Long L (2021). A Novel Transdermal Application for Clearing Skin Colonization by Candida auris. Antimicrobial Agents and Chemotherapy 65, e02303–02320. doi: 10.1128/AAC.02303-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang X, Hurabielle C, Drummond RA, Bouladoux N, Desai JV, Sim CK, Belkaid Y, Lionakis MS, and Segre JA (2021). Murine model of colonization with fungal pathogen Candida auris to explore skin tropism, host risk factors and therapeutic strategies. Cell Host Microbe 29, 210–221 e216. 10.1016/j.chom.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Borman AM, Szekely A, and Johnson EM (2016). Comparative Pathogenicity of United Kingdom Isolates of the Emerging Pathogen Candida auris and Other Key Pathogenic Candida Species. mSphere 1. 10.1128/mSphere.00189-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Santana DJ, and O’Meara TR (2021). Forward and reverse genetic dissection of morphogenesis identifies filament-competent Candida auris strains. Nat Commun 12, 7197. 10.1038/s41467-021-27545-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schelenz S, Hagen F, Rhodes JL, Abdolrasouli A, Chowdhary A, Hall A, Ryan L, Shackleton J, Trimlett R, Meis JF, et al. (2016). First hospital outbreak of the globally emerging Candida auris in a European hospital. Antimicrobial Resistance & Infection Control 5, 35. 10.1186/s13756-016-0132-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eyre DW, Sheppard AE, Madder H, Moir I, Moroney R, Quan TP, Griffiths D, George S,Butcher L, Morgan M, et al. (2018). A Candida auris Outbreak and Its Control in an Intensive Care Setting. N Engl J Med 379, 1322–1331. 10.1056/NEJMoa1714373. [DOI] [PubMed] [Google Scholar]
- 93.Roberts SC, Zembower TR, Ozer EA, and Qi C (2021). Genetic Evaluation of Nosocomial Candida auris Transmission. J Clin Microbiol 59. 10.1128/JCM.02252-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de St Maurice A, Parti U, Anikst VE, Harper T, Mirasol R, Dayo AJ, Garner OB, Prabaker KK, and Yang S (2022). Clinical, microbiological, and genomic characteristics of clade-III Candida auris colonization and infection in southern California, 2019-2022. Infect Control Hosp Epidemiol, 1–9. 10.1017/ice.2022.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Luca DG, Alexander DC, Dingle TC, Dufresne PJ, Hoang LM, Kus JV, Schwartz IS, Mulvey MR, and Bharat A (2022). Four genomic clades of Candida auris identified in Canada, 2012-2019. Med Mycol 60. 10.1093/mmy/myab079. [DOI] [PubMed] [Google Scholar]
- 96.Yadav A, Jain K, Wang Y, Pawar K, Kaur H, Sharma KK, Tripathy V, Singh A, Xu J, and Chowdhary A (2022). Candida auris on Apples: Diversity and Clinical Significance. mBio 13, e0051822. 10.1128/mbio.00518-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Markussen T, Marvig RL, Gomez-Lozano M, Aanaes K, Burleigh AE, Hoiby N, Johansen HK, Molin S, and Jelsbak L (2014). Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 5, e01592–01514. 10.1128/mBio.01592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bartell JA, Sommer LM, Haagensen JAJ, Loch A, Espinosa R, Molin S, and Johansen HK (2019). Evolutionary highways to persistent bacterial infection. Nat Commun 10, 629. 10.1038/s41467-019-08504-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, and Kishony R (2014). Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet 46, 82–87. 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burrack LS, Todd RT, Soisangwan N, Wiederhold NP, and Selmecki A (2022). Genomic Diversity across Candida auris Clinical Isolates Shapes Rapid Development of Antifungal Resistance In Vitro and In Vivo. mBio 13, e0084222. 10.1128/mbio.00842-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gambhir N, Harris SD, and Everhart SE (2022). Evolutionary Significance of Fungal Hypermutators: Lessons Learned from Clinical Strains and Implications for Fungal Plant Pathogens. mSphere 7, e0008722. 10.1128/msphere.00087-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, and Bennett RJ (2018). Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc Natl Acad Sci U S A 115, E8688–E8697. 10.1073/pnas.1806002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Forche A, Cromie G, Gerstein AC, Solis NV, Pisithkul T, Srifa W, Jeffery E, Abbey D, Filler SG, Dudley AM, and Berman J (2018). Rapid Phenotypic and Genotypic Diversification After Exposure to the Oral Host Niche in Candida albicans. Genetics 209, 725–741. 10.1534/genetics.118.301019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Helmstetter N, Chybowska AD, Delaney C, Da Silva Dantas A, Gifford H, Wacker T, Munro C, Warris A, Jones B, Cuomo CA, et al. (2022). Population genetics and microevolution of clinical Candida glabrata reveals recombinant sequence types and hypervariation within mitochondrial genomes, virulence genes, and drug targets. Genetics 221. 10.1093/genetics/iyac031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chowdhary A, Anil Kumar V, Sharma C, Prakash A, Agarwal K, Babu R, Dinesh KR, Karim S, Singh SK, Hagen F, and Meis JF (2014). Multidrug-resistant endemic clonal strain of Candida auris in India. Eur J Clin Microbiol Infect Dis 33, 919–926. 10.1007/s10096-013-2027-1. [DOI] [PubMed] [Google Scholar]
- 106.Calvo B, Melo AS, Perozo-Mena A, Hernandez M, Francisco EC, Hagen F, Meis JF, and Colombo AL (2016). First report of Candida auris in America: Clinical and microbiological aspects of 18 episodes of candidemia. J Infect 73, 369–374. 10.1016/j.jinf.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 107.Ben-Ami R, Olshtain-Pops K, Krieger M, Oren I, Bishara J, Dan M, Wiener-Well Y, Weinberger M, Zimhony O, Chowers M, et al. (2012). Antibiotic exposure as a risk factor for fluconazole-resistant Candida bloodstream infection. Antimicrob Agents Chemother 56, 2518–2523. 10.1128/AAC.05947-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang X, Welsh RM, Deming C, Proctor DM, Thomas PJ, Gussin GM, Huang SS, Kong HH, Bentz ML, Vallabhaneni S, et al. (2021). Skin Metagenomic Sequence Analysis of Early Candida auris Outbreaks in U.S. Nursing Homes. mSphere 6, e00287–00221. doi: 10.1128/mSphere.00287-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pichowicz AM, Torres SR, Torres-Velez FJ, Longyear AD, Singh N, Lasek-Nesselquist E, and De Jesus M (2022). Depletion of the Microbiota Has a Modest but Important Impact on the Fungal Burden of the Heart and Lungs during Early Systemic Candida auris Infection in Neutropenic Mice. Microorganisms 10. 10.3390/microorganisms10020330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huang D, Li H, Lin Y, Lin J, Li C, Kuang Y, Zhou W, Huang B, and Wang P (2022). Antibiotic-induced depletion of Clostridium species increases the risk of secondary fungal infections in preterm infants. Front Cell Infect Microbiol 12, 981823. 10.3389/fcimb.2022.981823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peleg AY, Hogan DA, and Mylonakis E (2010). Medically important bacterial-fungal interactions. Nat Rev Microbiol 8, 340–349. 10.1038/nrmicro2313. [DOI] [PubMed] [Google Scholar]
- 112.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. (2012). Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170. 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang TT, Shao TY, Ang WXG, Kinder JM, Turner LH, Pham G, Whitt J, Alenghat T, and Way SS (2017). Commensal Fungi Recapitulate the Protective Benefits of Intestinal Bacteria. Cell Host Microbe 22, 809–816 e804. 10.1016/j.chom.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shirtliff ME, Peters BM, and Jabra-Rizk MA (2009). Cross-kingdom interactions: Candida albicans and bacteria. FEMS Microbiol Lett 299, 1–8. 10.1111/j.1574-6968.2009.01668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sayeed MA, Farooqi J, Jabeen K, Awan S, and Mahmood SF (2019). Clinical spectrum and factors impacting outcome of Candida auris: a single center study from Pakistan. BMC Infect Dis 19, 384. 10.1186/s12879-019-3999-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tian S, Bing J, Chu Y, Chen J, Cheng S, Wang Q, Zhang J, Ma X, Zhou B, Liu L, et al. (2021). Genomic epidemiology of Candida auris in a general hospital in Shenyang, China: a three-year surveillance study. Emerg Microbes Infect 10, 1088–1096. 10.1080/22221751.2021.1934557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Boomer JS, Green JM, and Hotchkiss RS (2014). The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence 5, 45–56. 10.4161/viru.26516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Venet F, and Monneret G (2018). Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol 14, 121–137. 10.1038/nrneph.2017.165. [DOI] [PubMed] [Google Scholar]
- 119.Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, Bauer M, and Riedemann NC (2011). The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care 15, R183. 10.1186/cc10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pittet D, Li N, and Wenzel RP (1993). Association of secondary and polymicrobial nosocomial bloodstream infections with higher mortality. Eur J Clin Microbiol Infect Dis 12, 813–819. 10.1007/BF02000400. [DOI] [PubMed] [Google Scholar]
- 121.Gleckman R, Blagg N, and Joubert DW (1981). Trimethoprim: mechanisms of action, antimicrobial activity, bacterial resistance, pharmacokinetics, adverse reactions, and therapeutic indications. Pharmacotherapy 1, 14–20. 10.1002/j.1875-9114.1981.tb03548.x. [DOI] [PubMed] [Google Scholar]
- 122.Stevens DA, and Vo PT (1982). Synergistic interaction of trimethoprim and sulfamethoxazole on Paracoccidioides brasiliensis. Antimicrobial Agents and Chemotherapy 21, 852–854. doi: 10.1128/AAC.21.5.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Afeltra J, Meis JFGM, Vitale RG, Mouton JW, and Verweij PE (2002). In Vitro Activities of Pentamidine, Pyrimethamine, Trimethoprim, and Sulfonamides against Aspergillus Species. Antimicrobial Agents and Chemotherapy 46, 2029–2031. doi: 10.1128/AAC.46.6.2029-2031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lionakis MS, Chamilos G, Lewis RE, Wiederhold NP, Raad II, Samonis G, and Kontoyiannis DP (2006). Pentamidine is active in a neutropenic murine model of acute invasive pulmonary fusariosis. Antimicrob Agents Chemother 50, 294–297. 10.1128/AAC.50.1.294-297.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lionakis MS, Lewis RE, Samonis G, and Kontoyiannis DP (2003). Pentamidine is active in vitro against Fusarium species. Antimicrob Agents Chemother 47, 3252–3259. 10.1128/AAC.47.10.3252-3259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Foley GE, and Winter WD Jr. (1949). Increased mortality following penicillin therapy of chick embryos infected with Candida albicans var. stellatoidea. J Infect Dis 85, 268–274. 10.1093/infdis/85.3.268. [DOI] [PubMed] [Google Scholar]
- 127.Huppert M, Macpherson DA, and Cazin J (1953). Pathogenesis of Candida albicans infection following antibiotic therapy. I. The effect of antibiotics on the growth of Candida albicans. J Bacteriol 65, 171–176. 10.1128/jb.65.2.171-176.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maione A, Pietra AL, Salvatore MM, Guida M, Galdiero E, and de Alteriis E (2022). Undesired Effect of Vancomycin Prolonged Treatment: Enhanced Biofilm Production of the Nosocomial Pathogen Candida auris. Antibiotics 11, 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Melenotte C, Pontarotti P, Pinault L, Mege JL, Devaux C, and Raoult D (2021). Could beta-Lactam Antibiotics Block Humoral Immunity? Front Immunol 12, 680146. 10.3389/fimmu.2021.680146. [DOI] [PMC free article] [PubMed] [Google Scholar]