

Abstract
Despite numerous research efforts, drug delivery through the oral route remains a major challenge to formulation scientists. The oral delivery of drugs poses a significant challenge because more than 40% of new chemical entities are practically insoluble in water. Low aqueous solubility is the main problem encountered during the formulation development of new actives and for generic development. A complexation approach has been widely investigated to address this issue, which subsequently improves the bioavailability of these drugs. This review discusses the various types of complexes such as metal complex (drug-metal ion), organic molecules (drug-caffeine or drug-hydrophilic polymer), inclusion complex (drug-cyclodextrin), and pharmacosomes (drug-phospholipids) that improves the aqueous solubility, dissolution, and permeability of the drug along with the numerous case studies reported in the literature. Besides improving solubility, drug-complexation provides versatile functions like improving stability, reducing the toxicity of drugs, increasing or decreasing the dissolution rate, and enhancing bioavailability and biodistribution. Apart, various methods to predict the stoichiometric ratio of reactants and the stability of the developed complex are discussed.
Graphical Abstract

Keywords: complexation, cyclodextrins, ion-resin complexation, organic molecular complexes, pharmacosomes, phase solubility
Introduction
The drug development stage of new and generic products is painstaking and tedious. Developing a new pharmaceutical active ingredient or product costs around 2.6 billion dollars [1]. Therefore, formulation scientists are looking to develop novel formulations with better therapeutic outcomes for the existing drugs instead of developing a new drug. In order to develop an excellent pharmaceutical formulation with the intended high therapeutic efficacy, the minimum effective concentration of the drug should be achieved at the target site of action with a rapid onset. However, more than 40% of new chemical entities are insoluble in water [2]. Therefore, the drug must be in the solution at the absorption site to get absorbed and exert its intended action [2]. Low aqueous solubility is the main problem encountered during the formulation development of new actives and for generic development [2]. Adequate drug solubility in aqueous media is the prerequisite for successful drug delivery. Most biological membranes are enclosed by aqueous coats such as mucus, and only the dissolved active molecules can penetrate these membranes. An inadequate/poor aqueous solubility can affect the rate and extent of drug absorption after oral administration and slow the product development process for other routes of administration, including parenteral, nasal, ocular, dermal, and transdermal routes. Many approaches were adopted to enhance the aqueous solubility and dissolution of hydrophobic actives, such as particle size reduction, the addition of cosolvents or surfactants, salt formation, pH adjustment (for drugs with pH-dependent solubility), amorphous solid dispersions, and development of water-soluble drug complexes [3–5]. Formulation scientists screen many solubilizing approaches during the preformulation stage in the product development process to achieve adequate drug solubility or dissolution of the active. Every so often, adequate aqueous solubility can only be achieved by combining more than one solubilizing approach [6].
The drug solubility is often intrinsically related to particle size; the surface area to volume ratio increases with smaller particle size. The larger surface area permits more significant interaction with the solvent molecules, thus, increasing the drug's solubility [2]. Particle size reduction achieves an efficient and economic solubility enhancement [2]. However, the mechanical stress inherent to milling and grinding could impart significant physical stress on the drug inducing degradation in the finished dosage form [2]. The drug may experience thermal stress during comminution and raise concerns during the processing of thermosensitive pharmaceuticals [2]. In addition, the small particle size possesses high cohesive forces leading to poor flow property.
The Co-solvency approach means the addition of water and drug-miscible solvents to increase the solubility of poorly soluble drugs. Because of the low toxicity and the ability to solubilize hydrophobic pharmaceuticals, the co-solvency approach has been widely used to prepare parenteral dosage forms. However, parenteral formulations might require dilution with water to decrease the solvent content before administration, thus, reducing the solvation capacity [7]. In addition, Co-solvency has been adopted only as an approach for preparing liquid formulations. However, liquid dosage forms are more susceptible to chemical degradation when compared to solid dosage forms.
pH adjustment can combine with co-solvents to increase the solubility of the poorly water-soluble drug. However, the risk for drug precipitation upon dilution of the formulation with aqueous media having a pH at which the drug is less soluble could be a possible drawback. In addition, local or systemic tolerability and toxicity issues related to using a non-physiological pH and extreme pH values could occur. Furthermore, solubilized drugs in liquid formulations are often chemically less stable when compared to solid formulations. Moreover, the adjusted pH could accelerate hydrolysis or help in the catalysis of other possible degradation mechanisms [8].
The primary, fundamental, and oldest approach is using surfactants to help improve the dissolution performance of poorly water-soluble drugs. Surfactants increase the wetting of lipophilic drug particles by decreasing the interfacial tension between drug particles and the aqueous media. However, conventional micellar solutions have several disadvantages, including system instability over long storage periods and gastric irritation when used in excess quantities [9].
Salt formation is the most popular and effective method for improving solubility and dissolution rates of weakly acidic and basic actives [10]. However, organic compounds frequently undergo self-association in aqueous solutions due to their amphiphilic nature [11, 12]. Bile salts are good examples of how these organic compounds show surface activity and undergo self-association in their aqueous solutions due to their amphiphilic character [13]. Many published investigations reported similar aggregation for the salt forms of many drug molecules in solution [14–17]. Due to this self-aggregation phenomenon, salts and non-salt forms of active moieties in saturated solutions are lower than the measured concentrations in the solution, thus, leading to non-ideal pH-solubility behavior [10].
Amorphous solid dispersions are utilized frequently to improve oral bioavailability by improving the dissolution rate of poorly water-soluble drug molecules during the drug product development process [18]. However, the improved solubility using amorphous solid dispersions is due to the higher energy of the amorphous form; however, the high energy state could pose crucial challenges in manufacture, storage, shipping, or dissolution [19]. Moreover, understanding the molecular interactions between actives and the polymeric carrier in the solid state of the amorphous solid dispersions and the supersaturated solution still needs further elucidation to boost the predictability of physicochemical stability and in vivo performance of these systems [20].
Complexation is a widely used approach in the pharmaceutical field to improve the solubility of various drugs that are poorly water-soluble and, thus, their bioavailability. Unlike other solubility enhancement techniques, which require special precautions to mask the taste (e.g., taste masking polymers), complexing agents have the inherent ability to taste mask the drug. Occasionally, the enhanced solubility of the drugs with disagreeable taste may worsen the taste even more. Aside from increasing solubility, complexation can improve drug permeation, bioavailability, and biodistribution. They can also improve the drug's stability (for example, thermal stability) [21].
Complexes are intermolecular associations of ligands and substrates [6]. Complexes are classified into two types based on whether the acceptor component is a metal ion or an organic molecule; metal-ion complexes and organic molecular complexes. For instance, metal complexes or coordination compounds result from a Lewis acid–base reaction between two or more different chemical entities or donor–acceptor mechanisms [22]. In non-metallic complexes, the drug is bound to a non-metallic atom in a neutral molecule or ion in an ionic compound [22]. Another class is the inclusion complexes, wherein one compound gets entrapped in the molecular framework of another. Many intermolecular forces are involved in forming different complexes; all are non-covalent bonds, including van der Waals forces of dispersion, ionic bonds, and dipolar and induced dipolar forces. Hydrogen bonding also provides a major force in some molecular complexes, while coordinate covalence is significant for metal complexes [6, 22]. All categories of complexation discussed in this review with their applications in the pharmaceutical field and a few of the marketed products, recent patents, and research are listed in Tables I, II, and III, respectively.
Table I.
FDA Approved Products Developed Using Complexation Technique
| Complexing agent | Dosage Form | Active Ingredient | Marketed product | Approval Date | Uses | Ref |
|---|---|---|---|---|---|---|
| Inclusion Complexation | ||||||
| Cyclodextrin | Oral chewable | Cetirizine HCl | CHILDREN'S ZYRTEC ALLERGY | 2007 | Taste Masking | [23] |
| Oral | Drospirenone + Ethinyl estradiol | YAZ | 2001 | Stability of Ethinyl estradiol | [24] | |
| Topical gel | Metronidazole | METROGEL | 1988 | Solubility | [25] | |
| HPβCD | Injection | Telavancin | VIBATIV | 2009 | [26] | |
| Diclofenac sodium | DYLOJECT | 2014 | [27] | |||
| Letermovir | PREVYMIS | 2017 | [28] | |||
| Tecovirimat | TPOXX | 2018 | [29] | |||
| Oral solution | Larotrectinib | VITRAKVI | 2018 | [30] | ||
| Oral solid | Cladribine | MAVENCLAD | 2019 | [31] | ||
| SBEβCD | Injection | Remdesivir | VEKLURY® | 2020 | [32] | |
| Voriconazole | VFEND® | 2002 | [33] | |||
| Delafloxacin | BAXDELA | 2017 | [34] | |||
| Posaconazole F | Noxafil | 2006 | [35] | |||
| Brexanolone | ZULRESSO | 2019 | [36] | |||
| Fosphenytoin sodium | CEREBYX® | 1996 | stability | [37] | ||
| Ion-resin Complexation | ||||||
| Sodium polystyrene sulfonate | Extended-release chewable tablet | Methylphenidate hydrochloride | QuilliChew ER. | 2015 | Prolong dissolution | [38] |
| Extended-release oral suspension | Methylphenidate hydrochloride | QUILLIVANTTM XR | 2012 | [39] | ||
| Extended-release oral suspension | Chlorpheniramine polistirex; codeine polistirex | TUZISTRA XR | 2015 | [40] | ||
| Extended-release oral suspension | Chlorpheniramine polistirex; hydrocodone polistirex | TUSSIONEX PENNKINETIC | 2012 | |||
| Extended-release oral suspension | Dextromethorphan polistirex | DELSYM | 2017 | [41] | ||
| Pharmacosomes | ||||||
| Phospholipid [1-α-dimyristoylphosphatidylcholine (DMPC) and l-α-dimyristoylphosphatidylglycerol (DMPG)] | Injection | Amphotericin B | ABELCET | 1995 | Biodistribution and reduce toxicity | [42, 43] |
| Metal Complexes | ||||||
| Silver | Topical cream | Silver sulfadiazine | Silvadene | 1982 | Pharamcodynamics propertics | [44] |
| Zinc | Ophthalmic ointment | Bacitracin zinc | Lumi-sporyn | 1982 | stability | [45] |
Table II.
Recent Patents on Pharmaceutical Dosage Forms Developed Using Complexation Technique
| Patent Number | Api | Title | Complexing Agent | Purpose/Use | Current Assignee/Inventors | Granted Year | Reference |
|---|---|---|---|---|---|---|---|
| US11452729B2 | Fulvestrant | Fulvestrant compositions and method of use | sulfobutylether beta-cyclodextrin | Solubility enhancement | Shimoda Biotech Pty Ltd | 2022 | [46] |
| AU2018200444B2 | Peptide proteasome inhibitors | Cyclodextrin complexation methods for formulating peptide proteasome inhibitors | sulfobutylether beta-cyclodextrin | Solubility and stability enhancement | Onyx Therapeutics Inc | 2019 | [47] |
| US11369684B2 | Meloxicam | Pharmaceutical compositions comprising meloxicam | sulfobutylether beta-cyclodextrin | improve the bioavailability or pharmacokinetics | Axsome Therapeutics Inc | 2022 | [48] |
| EP2785352B1 | Alfaxalone | Stable injectable pharmaceutical compositions comprising 2-hydroxypropyl-beta-cyclodextrin and alfaxalone | 2-hydroxypropyl-beta-cyclodextrin | Solubility enhancement | Jurox Pty Ltd | 2020 | [49] |
| EP2616046B1 | Aspirin, Naproxen sodium, Acetaminophen, or Ibuprofen | Aqueous drug delivery system comprising off-flavor masking agent | alpha-, beta- or gamma-cyclodextrin, or their derivative | Taste Masking | Bev-RX Inc | 2016 | [50] |
| AU2018214110B2 | Opiates, opioids, tranquillizers, stimulants, or narcotics | Abuse resistant capsule | polacrilex resin, sodium polystyrene sulfonate, potassium polyacrilin, or colestyramine resin; | ABUSE RESISTANT | RP Scherer Technologies LLC | 2019 | [51] |
| US10744176B2 | Acidic active pharmaceutical ingredient, particularly ketoprofen | Edible oral strip or wafer dosage form containing ion exchange resin for taste masking | anion exchange resin | Taste Masking | LTS Lohmann Therapie Systeme GmbH and Co KG | 2020 | [52] |
| WO2007101551A2 | Curcumin | Curcumin phospholipid complexes that have improved bioavailability | soy phospholipids | Improve bioavailability | Indena SpA | 2019 | [53] |
| US10729792B2 | Texaphyrin | Texaphyrin-phospholipid conjugates and methods of preparing same | phosphatidylcholine, phosphatidylethanoloamine, phosphatidylserine or phosphatidylinositol | Improve bioavailability | University Health Network | 2020 | [54] |
Table III.
Recent Research on Complexation-based Pharmaceutical Dosage Forms
| Drug | Type of Complexation | Method of Preparation | Outcome | Reference |
|---|---|---|---|---|
| Galangin | Inclusion Complex (β-CD) | Nanoprecipitation | Improved solubility, dissolution, and pharmacokinetics | [55] |
| Carbamazepine | Inclusion Complex (β-CD and HPβ-CD) | Hot Liquid Extrusion | Improved solubility, dissolution, and pharmacokinetics | [56] |
| Naringenin | Ternary Inclusion Complex (HPβ-CD and hydrophilic polymers) | Hot Melt Extrusion | Improved solubility and dissolution | [57] |
| Sorafenib tosylate | Inclusion Complex (β-CD) | Kneading method | Improved solubility and dissolution | [58] |
| Atomoxetine hydrochloride | Inclusion Complex (β-CD) | Kneading method | Taste masking | [59] |
| Azithromycin | Ion-exchange resin complexaion (Kyron T-135 and Doshion-P542 AB) | Batch process | Taste masking | [60] |
| Quinine hydrochloride dihydrate | Ion-exchange resin complexaion (Amberlite™ IRP88 and Amberlite™ IRP64 | Hot Melt Extrusion | Taste masking | [61] |
| Ursolic acid | Pharmacosome (hydrogenated soybean phophatidylcholine) | Reflux method | Bioavailability and hepatoprotectivity | [62] |
| Heparin | Pharmacosome (Soy lecithin) | Solvent evaporation method | Bioavailability | [63] |
| Flurbiprofen | Organic molecular complexes (Lidocaine) | Solvent evaporation method | Enhancing solubility and permeability of Flurbiprofen | [64] |
Inclusion Complexes
An inclusion complex forms between a host (ligand) and guest (substrate), where the guest molecules (drug) entrap within the framework of the host molecule [65]. In this complex, no covalent bonds are created nor broken within or between the ligand and the substrate. The driving forces for the complex formation are usually hydrogen bonds, electrostatic, van der Waals forces, conformational strain, and charge transfer interactions [6]. The inclusion complexes include clathrates, channel lattice complexes, and conventional drug complexes with hosts containing cavities (e.g., cyclodextrin). In clathrates, the host molecules are usually the polymers, which cover the guest molecules like a cage [66]. In channel lattice complexes, the guest molecule is usually a long-chain compound enveloped by host molecules forming channel-like crystal structures. For example, Thakral et al. (2008) developed the channel lattice complex using urea to improve the solubility and stability of 13-cis retinoic acid [67]. However, the conventional drug complexes with hosts containing cavities are formed between the guest drug and host molecules. In addition, the host molecule possesses a cavity to accommodate the guest molecule. Various supramolecular cyclic host molecules possess cavities in their molecular structure, like cyclodextrins, cucurbiturils, calixarenes, and crown ethers (Fig. 1). Of the various supramolecular cyclic host molecules, only inclusion complexes with cyclodextrin have been transformed into marketed pharmaceutical products.
Fig. 1.

ɑ-cyclodextrin (A), β-cyclodextrin (B), γ-cyclodextrin (C), Hydroxypropyl-β-cyclodextrin (D), Cucurbit(7)uril (E), Cucurbit(8)uril (F), sulfonatocalix(4)arene (G) & sulfonatocalix(8)arene (H).
Cyclodextrins
Cyclodextrins (CDs) are cyclic oligosaccharides composed of α-D-glucopyranoside subunits linked together by α-1,4 glycosidic bonds. Their structure resembles a truncated cone with a hydrophobic central cavity and hydrophilic outer surface [68]. The most common CDs are natural αCD, βCD, and γCD consisting of 6, 7, and 8 α-D-glucopyranoside subunits, respectively. The increase in subunits proportionates to the increase in central cavity size, making them selectively accommodate a diversity of guests based on molecular size [69]. In addition, these natural CDs can be chemically modified to obtain CDs of pharmaceutical interest; these include hydroxypropylated CD derivatives (e.g., HPβCD and HPγCD), randomly methylated CDs (RMβCD), and sulfobutylether CDs (e.g., SBEβCD and SBEγCD).
Drug-CD Complexes
The Drug-CD inclusion complexes can be prepared by various techniques like solvent evaporation, co-precipitation, freeze drying, precipitation with compressed anti-solvents, microwave irradiation method, supercritical antisolvent technology, hot liquid extrusion, etc. [56, 70–73]. CDs can accommodate the drug molecule in its cavity, forming drug-CD complexes and changing the included drug's physicochemical properties, like enhancing aqueous solubility, stability, and permeability. The most common application of CD complexation in the pharmaceutical field is to improve the solubility of the included drug molecules [69]. The drug, upon complexation, gets hosted in the central cavity of the CD; when this drug-CD complex is added to aqueous media, the hydroxy groups on the outer surface of the drug-CD complex form hydrogen bonds with water molecules, allowing the drug to be dissolved. [74]. In addition to inclusion complex formation, CDs and drug-CD complexes at higher concentrations can form aggregates in aqueous media, and aggregate formation is proportional to CD and drug-CD complex concentration. These aggregates are formed through non-inclusion complexation or micellar effects. Numerous studies reported that aggregates could also enhance the solubility of lipophilic drugs by acting as drug reservoirs [75].
Various types of CDs have been explored to enhance the solubility and dissolution of lipophilic drugs (Fig. 2). For example, Das et al. used SBβCD to form Ibuprofen (IBU) inclusion complexes to improve their solubility. The phase solubility studies showed the formation of complexes in the stoichiometric ratio of 1:1. The results revealed the amorphous state of the drug within the complex and 9.98 and 21-fold improvements in solubility when IBU-SBβCD inclusion complexes prepared in the weight ratio of 1:1 and 1:5 (IBU:SBβCD), respectively [76]. In another study, HPβCD was utilized by Yan et al. to enhance the solubility of a flavonoid called Baicalein (BAI). The complex was formed in a stoichiometric ratio of 1:1 based on phase solubility studies. Analytical methods confirmed the amorphous state of BAI in the inclusion complex. The solubility of the BAI improved from 1.37 µg/mL (Pure BAI) to 147.38 µg/mL (BAI- HPβCD)[70].
Fig. 2.

Inclusion complexation of pomalidomide (POM) with sulfobutylether-β-cyclodextrin.
Reproduced with permission from ref [77].
Apart from improving solubility, the CD complexation can be utilized for taste masking. For instance, Liu et al. developed an orodispersible film of donepezil to facilitate swallowing in Alzheimer’s patients. The unacceptable bitter taste of the drug was masked by using the CD complexation technique. The authors utilized HPβCD to produce inclusion complexes; subsequently, these complexes were formulated into an orodispersible film. The taste masking effect was studied using the electronic tongue method; the results showed that inclusion complexes and the films prepared from the inclusion complex were palatable compared to the drug, physical mixture, and films prepared with the drug and sucralose [78]. In addition, the cyclodextrin complexation can improve the drug's stability (e.g., photolytic, thermal). In a study by Yildiz et al., the carvacrol-CD complex thermal gravimetric analysis showed higher degradation temperature than pure carvacrol [79]. Similarly, with the FDA-approved product Yaz (drospirenone/Ethinyl estradiol oral tablets), the stability of Ethinyl estradiol was improved using CD complexation [24].
The selection of cyclodextrin for complexation is determined by several factors, including the route of administration and the physical properties of the drug and cyclodextrins. Due to the toxicity associated with cyclodextrins, they are not suitable for administration through all the routes. Except for the ɑ-CD, all other natural CD’s are prohibited in parenteral formulations. In contrast, the HPβCD and SBEβCD are commonly used in parenteral formulations. RMβCD, on the other hand, is only approved for use in ocular and nasal applications [80].
The size of the cyclodextrin and drug molecule can influence the complex formation i.e., the CD’s cavity size should perfectly accommodate the drug molecule. For instance, the ɑ-CD is suitable to form the complex with small-sized molecules or side chains of large molecules. Whereas the β-CD is used for molecules with benzene or phenyl rings, which is prominent in most drug molecules. The γ-CD is used for the complexation of large molecules [81]. The suitability of cyclodextrin’s size to the selected drug molecule can be evaluated either by using in-silico (molecular docking or molecular modeling studies) or in-vitro analytical techniques (phase solubility studies or Job’s plot). The drug's solubility also influences complex formation; drugs with lower intrinsic solubility have shown a greater relative increase in solubility. The ionic nature of the drug and cyclodextrin also influences complexation efficiency and stability. Ionized drug complexes typically have higher stability constants than unionized drug complexes. For instance, both the unionized and cationic forms of chlorpromazine form a 1:1 complex with β-cyclodextrin; however, the unionized form has a four-fold higher stability constant than the cationic form. Furthermore, cyclodextrin with the opposite charge of the drug will produce complexes with high stability constants [82].
Ternary Inclusion Complexes
Even though cyclodextrin complexation is a promising technique to improve solubility, it has poor complexation efficacy (CE), resulting in the requirement of large quantities of cyclodextrin to attain the required solubility of the drug. Using large quantities of cyclodextrin increases the bulkiness of the final formulation. The correlation between the final bulk (weight) of formulation and cyclodextrin can be explained by the following equation [83]:
| 1 |
where MWCD is the molecular weight of CD, MWDrug is the drug’s molecular weight.
It can be observed from Eq. 1 that the formulation bulkiness is directly proportional to the molecular weight of cyclodextrin and indirectly proportional to CE. However, from equation-7, it can be inferred that the CE is the product of the intrinsic solubility of the drug (S0) and the equilibrium constant or stability constant (Km:n). Thus, increasing S0 or Km:n, can improve the CE. The S0 of a drug in aqueous media can be improved by converting the drug either into an amorphous form, ionizing the drug, creating salt formation, or adding co-solvents. While the methods to improve Km:n include adding auxiliary substances or counter-ionic drugs. [75]. Of the currently investigated methods, the formation of ternary complexes by adding auxiliary substances to the drug:CD inclusion complex is widely employed to increase CE. Various water-soluble auxiliary substances, such as polymers, hydroxyl acids, amino acids, can prepare ternary complexes [84]. In these systems, the CDs and auxiliary substances act synergistically to improve the drug’s CE, Km:n & solubility in aqueous media. The increase in CE permits the use of less CD and reduces the final formulation’s bulkiness without compromising the drug’s solubility [85].
Many studies have been reported to improve the solubility of various drugs through the ternary complexation approach. For example, Zafar et al. adopted the ternary inclusion complex approach to improve genistein’s solubility. The binary complex was prepared using βCD in a stoichiometric ratio of 1:1. The D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) was added to the drug-CD inclusion complex at a concentration of 0.05% w/w to form a ternary complex. Km:n and CE values increased from 419.67 M−1 and 0.31% of the drug-CD inclusion complex to 654.98 M−1 and 0.48% of the ternary complex. In addition, the ternary complex exhibited higher solubility than the binary complex, which was 48.09-fold and 23.36-fold compared to pure genistein [86]. In another study, Suvarna et al. developed the ternary inclusion complex of Irbesartan to improve its aqueous solubility. The ternary complexes were formed in a stoichiometric ratio of 1:1:1 (Drug:HPβCD:L-Arginine). The value of Km:n improved from 19.57 M−1 (drug-CD) to 316.04 M−1 (ternary), while CE increased from 0.137 (drug-CD) to 2.212% (ternary). The solubility of the Irbesartan improved to 6.22 mg/mL by ternary complex compared to 0.68 mg/mL and 0.007 mg/mL of drug-CD complex and pure Irbesartan, respectively [87].
Selecting CD and auxiliary substances is essential to develop a suitable formulation. Both components can be selected by applying phase solubility studies, where improving CE and Km:n from binary to ternary inclusion complex act as the critical factor. In general, the CD can be selected based on the molecular size of the drug and the size of the interior cavity of the CD. The cavity size of the CD should be neither too loose for a drug to escape from the complex nor insufficient for the drug to get hosted in the CD [88]. The auxiliary substance can be selected based on the physicochemical properties of the drug. For example, the primary auxiliary substance could significantly enhance the solubility of an acidic drug [89]. L-arginine is widely employed among all amino acids as it shows a synergetic effect with CD to solubilize the drug [68].
Cucurbiturils (CBs)
CBs are barrel-shaped macrocyclic compounds made up of glycoluril units. Like CDs, these compounds can be obtained in various sizes based on the presence of glycoluril subunits (n = 5–8, 10). The most used CBs in drug delivery are CB (6), CB (7) & CB (8) [90]. The CBs have poor solubility in water, with a maximum solubility of 20–30 mM for CB (5) and CB (7). In contrast, the solubility of CB (6) and CB (8) is very low at 0.018 mM and < 0.01 mM, respectively [91]. However, their solubility in body fluids is comparatively higher; for example, the CB (6) has a solubility of 33–37 mM in blood, 1–4 mM in gastric fluid, and 5–7 mM in intestinal fluid [91]. The surface of the CBs can be modified to improve the aqueous solubility like CDs. The CB’s hydrophobic cavity facilitates the host–guest complexation between the drug and CBs. The complex is stabilized by forming H-bonds or ion–dipole interactions between the drug and CB’s cavity [92]. The formed complex improves drug solubility, physio-chemical stability, and controlled drug release. In comparison to drug-CD complexes, the complexes of CBs possess high stability constants of about 1015 M−1 [92]. The investigation of complexation with CBs is similar to CDs, focusing on the size of host moiety, stoichiometry, and stability of the complex.
Liu et al. improved the solubility of thiabendazole through complexation with CBs. The CB (7), symmetrical tetra-methylcucurbituril (6) {TMeQ (6)} and meta-hexamethyl-substituted cucurbituril (6) {HMeQ (6)} were utilized in the complexation. The complexation with HMeQ (6) showed the greatest improvement in solubility, with a 40-fold improvement compared to pure thiabendazole’s aqueous solubility [93]. Similarly, Huang et al. utilized the CBs to improve the solubility of kinetin. The solubility and stability of complexes were in the order of HMeQ (6) (43-fold) > TMeQ (6) (37-fold) > CB (7) (35-fold) [94].
Calixarenes (CAs)
Similar to the molecules mentioned above, CAs also belong to macrocyclic molecules with phenol subunits [95]. Unlike CDs and CBs, these molecules are conical shaped, with a lower narrow rim and a wide upper rim for accommodating the guest molecules. The CAs usually have an even number of subunits, e.g., 4, 6, and 8, as they can be prepared easily [96]. Various hydrophilic groups like sulfonato, carboxy, phosphonato, and amino groups can be attached at the para-positions of CAs to make the compounds more water soluble [95]. The central cavity is comparatively smaller than CDs. Similar to other inclusion complex-forming molecules, these molecules form host–guest complexes, thereby improving the solubility of poorly soluble drugs.
Yang et al. studied the solubility enhancement efficiency of 4-sulphonic CAs (n = 4, 6, and 8) on nifedipine. The solubility studies revealed that the 0.008 M 4-sulphonic calix(8)arene showed the most significant improvement in the solubility of nifedipine (1.11 * 10–5 M to 3.28 * 10–5 M at pH.5) followed by 0.008 M 4-sulphonic calix(4)arene. In the case of 0.008 M 4-sulphonic calix(6)arene, the solubility of nifedipine decreased compared to the absence of the complexing agent. However, the solubility was improved slightly at a high concentration of 4-sulphonic calix(6)arene and pH 5 [97].
In another study by Menon et al., the para-sulphonato CAs were utilized for the solubility enhancement of carvedilol. The authors utilized para-sulphonato CAs (n = 4 and 6). Based on the phase solubility studies, the inclusion complexes were formed in the ratio of drug: para-sulphonato calix(4)arene (1:2) and drug: para-sulphonato calix(6)arene (1:1). The stability constants were 3.07*104 M−1 and 6.0*105 M−1 for a drug:para-sulphonato calix(4)arene and drug:para-sulphonato calix(6)arene respectively. The in vitro dissolution studies revealed the dissolution enhancement ability of the complexing agents with 100% release in less than 45 min compared to 4 h for the pure drug [98].
Pharmacosome (PHC)
PHC is a nonvesicular association of drug and phospholipid. The complexing agent in PHC is an amphiphilic and zwitterionic molecule called phospholipids, an essential constituent of the cell plasma membrane. The amphiphilic nature of the phospholipids is due to the presence of a hydrophilic head (phosphate group – negatively charged) and a hydrophobic tail (long-chain fatty acid) [99]. The PHC is not suitable for all the drugs, since the phospholipids form complexes with polar functional groups (––COOH, ––OH, and ––NH2) containing active hydrogen. Therefore, only the drug containing these functional groups are suitable for PHC formation. The drug and phospholipids are bonded in complexes by forming either a non-covalent or an ionic bond. For example, the non-covalent bond is formed between the oxygen atom of the phosphate group in the phospholipid and the active hydrogen of the drug.
In contrast, the ionic bond is formed between the quaternary amine (positive charge) of the phospholipid and the ionized carboxylic group (negatively charged) of the acidic drug [100, 101]. Moreover, reports of hydrophobic interactions between the drug and phospholipid to form complexes have been found. It is demonstrated that molecules with conjugated π-electron systems can form various types of complexes with phospholipids [102]. In the case of covalent bond formation between the drug and phospholipids, the resulting moieties should be termed prodrugs rather than PHC [6]. The amphiphilic nature of PHC helps improve the drug’s solubility in both aqueous and non-aqueous media. Though the improvement of aqueous solubility is minute, they can significantly improve oral bioavailability and biodistribution. In addition, PHC can improve the metabolic stability of the drug and improves bioavailability. For instance, upon parenteral administration, the gemcitabine gets metabolized to form 2′-deoxy-2′, 2′- diflurouridine (dFdU). The in-vivo studies of the gemcitabine-phospholipid complex revealed the inhibition of gemcitabine metabolism and conversion into dFdU, thus enhancing the bioavailability [103]. This technique is more favorable to API’s that needs enhancement of bioavailability either by permeation enhancement and/or evasion from metabolism.
For example, FDA approved Amphotericin B injectable lipid complex (Abelcet) is complex 1:1 complex of Amphotericin B and 1- α-dimyristoylphosphatidylcholine (DMPC) and 1- α-dimyristoylphosphatidylglycerol (DMPG) (7:3), wherein the lipid complex improved the biodistribution of Amphotericin B compared to its pure form [42]. The improvement in the oral bioavailability by PHC is due to its ability to envelope the drug in aqueous media and thus protect the drug from enzymatic or pH-dependent degradation, parallelly helping reduce the irritability of GIT from the drug. In addition, the absorption of PHC is like the endogenous absorption of phospholipids, which occurs via enterocytes, therefore, protects the drug from P-gp efflux [104]. During the formation of PHC, the stoichiometric ratio of drug and phospholipid is an essential factor; in most of the reported studies, the drug to phospholipid was in the ratio of 1:1. Several therapeutic moieties have been complexed with phospholipids to improve either of the solubility, bioavailability, or both.
Amirinejab et al. utilized a phospholipid complexation approach to improve the dissolution profile of IBU, as shown in Fig. 3. The complex was formed using phosphatidylcholine in the stoichiometric ratio of 1:1, 1:0.5, and 1:0.25 (Drug:Phospholipid). The IBU crystals were observed in the preparation containing IBU: Phospholipid in the stoichiometric ratio of 1:0.25. The solubility measurements showed that 1:0.5 (IBU: Phospholipid) had the most significant improvement by ~ 2.5 fold compared to the pure drug in phosphate buffer (pH 7.2). At the same time, the complexes exhibited less solubility than the pure drug in pH 1.2. The dissolution profiles of pure drugs and complexes are in-line with solubility studies, wherein the dissolution in pH 7.2 is in the order of 1:0.5 > 1:1 > pure IBU, and at pH 1.2, the order is 1:0.5 < 1:1 < pure IBU. The decrease in solubility and dissolution rate in the acidic pH (1.2) is due to the non-ionization of IBU leading to its entrapment in PHC, thus decreasing the free drug in the acidic media [101].
Fig. 3.

Pharmacosomes of Ibuprofen.
Reproduced with permission from ref [101].
In another study by Zhang et al., the pharmacokinetic and bioavailability of kaempferol (bioavailability; 2%) was improved by using phospholipid complexation. The drug was complexed with phosphatidylcholine in a stoichiometric ratio of 1:1. The aqueous solubility of the formed complexes was improved by ~ 216 fold, while ~ threefold improved the n-Octanol solubility. The dissolution of complexes was superior to the pure drug in both pH 1.2 and pH 6.8 dissolution media after 2 h, with the release of 60.13% and 56.13% compared to 32.76% and 34.67%, respectively. The Cmax of the drugs from the complex was increased by ~ 2.75-fold in comparison with pure drug after oral administration to male SD rats [105].
Similarly, Biswas et al. improved the bioavailability of piperine using phospholipid complexation. Wherein Piperine (PIP) was complexed with hydrogenated phosphatidylcholine (HPTC) in a stoichiometric ratio of 0.5:1 to 1.5:1 (HPTC: PIP). The solubility of the complex in water and n-Octanol was improved by ~ 29.5-fold and ~ 3.2-fold, respectively, compared to pure PIP. Furthermore, the Cmax of the drug from complex (9.75 ± 2.17 μg/ml) was significantly improved compared to the Cmax of PIP from the pure drug (8 ± 0.21 μg/ml) [106]. These studies demonstrated phospholipid complexation’s ability to improve the drug’s bioavilability. The preparation method in most of the studies was found to be stirring or refluxing the drug and phospholipid in an organic solvent, followed by solvent evaporation [101, 105]. In addition, co-grinding, mechanical dispersion, and anti-solvent precipitation methods were also employed [107–109].
Drug-Resin Complexes (DRC)
Ion-Exchange Resins (IER) are high molecular weight, water-insoluble, highly cross-linked polymeric structures with ionizable cationic or anionic functional groups. During the DRC formation, the ionizable functional groups on IER can exchange their ions with similarly charged strong affinitive drug ions in a solution [110]. The interaction between the drug and IER in a suitable solution can be described in the following equation-2.
| 2 |
The strength of exchanging ions depends on the affinity of functional groups, which could divide the IERs into strongly acidic cation [sulphonic acid groups (–SO3H)], weakly acidic cation [carboxylic acid groups (–COOH)], strongly basic anion [quaternary ammonium groups(–NR3OH)], and weakly basic anion [amine groups (–NH2, –NHR, and –NR2)] exchange resins. The DRC is limited to the drugs in the salt form, and most of the FDA-approved drugs have either hydrochloride or hydrobromide salt forms. The selection of resin should be based on the type of API; for an anionic API, a cation exchange resin should be used and vice-versa. The drug’s in vitro or in vivo release from the DRC requires higher affinity ions to replace the drug [111]. The DRC reacts with high-affinity ions like K+ in the gastric environment, releasing the drug according to the following equation-3.
| 3 |
The drug cannot be extracted from the DRC without these charged ions; therefore, these systems can be used to develop abuse and tamper-resistant dosage forms [112, 113]. In addition, the FDA approved various drug-resin complexes, and all are for extended-release dosage form design, wherein the extended-release property is obtained by coating the DRC particles with extended-release polymers. Apart from this, the DRC can be used for improving solubility, dissolution, and taste masking. Therefore, this complexation is mostly used to develop abuse-deterrent formulations (particularly for opioids) and extended-release dosage forms, especially the extended-release suspensions (currently approved by FDA).
Kulthe et al. enhanced the solubility of Atorvastatin Calcium using ion-resin complexation. Cholestyramine was used as a complexing agent in the stoichiometric ratio of 1:1 (drug: resin). The saturation solubility studies showed an improvement in the solubility by ~ 3.25 fold following the amorphous conversion of the drug [114]. Panraksa et al. utilized the ion-exchange resin complexation for taste masking of nizatidine. The authors used strong anion exchange resins the Amberlite IRP-69 (sodium polystyrene sulphonate) and Dowex-50 (hydrogen form—polystyrene sulphonic acid); the resins were protonated by washing with acid (0.1N HCl). The nizatidine was added to the resin in water to form complexes. The nizatidine showed high complexation efficiency in a stoichiometric ratio of 1:3 (drug:resin) with Dowex-50 because of like-charged ions, the H+ compared to Amberlite IRP-69 with Na+ ions. The dissolution studies exhibited the complete release within 1 h. The in vivo taste masking studies reported a pleasant taste for the DRC [115].
Xin et al. improved the dissolution of IBU by preparing DRC with poly (ethylene–g–styrene–trimethyl–ammonium–chloride; strong anionic resin). The resin was activated in alkaline media (1 M NaOH); the activated resin and IBU were added to water and stirred to form a complex. As a result, the dissolution of IBU from DRC was higher than pure IBU in pH 7.0 phosphate buffer. Also, the in vivo studies revealed that the DRC was ~ 3.5-fold and less likely to form ulcers than pure IBU [116]. Despite having various pharmaceutical applications, preparing these complexes requires large quantities of solvent compared to other pharmaceutical complexation techniques. Modern technologies like hot-melt extrusion (HME) have been studied but not widely employed in preparing these complexes [61].
Metal Ion Complexes
Metal complexes, also known as coordination complexes, are formed between the metal ions (Lewis acids) and Ligands (API – Lewis bases). During the formation of coordination complex, the ligands (H2O:, H3N:, Cl−:) donate pair of electrons to metal ions (e.g., Fe2+, Cu2+, Zn2+, and Pt4+) [6]. Cisplatin, a water-soluble anticancer drug, is the best example of a metal complex in the pharmaceutical market, with the platinum ion complexed with two chloride atoms and two ammonia moieties [117]. Similarly, there are various metal-based FDA-approved drugs like carboplatin (platinum), oxaliplatin (platinum), silver sulfadiazine (silver), bacitracin zinc (Zinc), etc. Although the presence of these complexes in pharmaceuticals is negligible, it is recently gaining more attention, especially in nutraceuticals. Most of the metal complexes studied to date are mainly used to improve either the stability or the pharmacodynamic properties of the drug molecules, rather than improving the solubility. During the formation of metal-drug complexes, the electronegativity of the metal drives the formation of stable metal complexes. Typically, the more electronegatively charged the metal ion is, the more stable the formed complex, which is difficult to dissociate in the aqueous media, resulting in poor aqueous solubility. As a result, the metal-ion used in complex formation should be carefully chosen to balance the solubility and stability of metal-drug complexes [118].
Similarly, chelates are also known as coordination complexes because the same substrate binds to more than one ligand site (chelating agent). Chelating agents, such as EDTA, are frequently used to remove metal ion impurities in pharmaceutical formulations that have the potential to degrade the pharmaceutical product. Pharmaceutical drug-metal ion chelates exist, but chelate formation is not always necessary to improve drug solubility. However, some metal complexes of water-insoluble drugs may form water-soluble drug-metal complexes [119].
Few studies have reported improving the solubility of various water-insoluble APIs upon complexation with metal ions. For example, Higuchi et al. demonstrated that the solubility of oxytetracycline increased upon forming chelates with Ca2+, and the stoichiometry of complexation was in higher order for Ca2+ (1:2) [119]. Similarly, Ross et al. studied the effect of various metal-ion complexes on the aqueous solubility of fluoroquinolones antimicrobials. The authors complexed the lomefloxacin with various metal ions. The stoichiometry (drug:metal ion) was varied with a different metal ion, for example, Ca2+ (1:1), Mg2+ (2:1), Bi3+ (2:1), Fe3+ (2:1) and Al3+ (3:1). The solubility of the lomefloxacin was increased upon complexation with all the metal ions following AL-type phase solubility curve, except for Bi3+ (B-type phase solubility curve) attributed to the limited aqueous solubility of Bi3+ salts [118].
In another study conducted by Sareen et al., the solubility of curcumin was improved upon complexation with Zinc. The stoichiometry of complexation was 1:1(Curcumin:Zinc). The solubility of the curcumin-zinc complex was increased by twofold compared to pure curcumin throughout various pHs of 1.2, 6.8, and 7.4 [120]. Similarly, Hieu et al. studied the effect of various metal ions complexation on the solubility of curcumin. The metals used are zinc (Zn2+), calcium (Ca2+), and iron (Fe3+) which have an ionic electronegativity of 1.65, 1.00, and 1.9 respectively [121]. The complexes were prepared with calcium and Zinc in the stoichiometry of 1:1 (curcumin:metal-ion), whereas 3:1 with Fe3+. The complexation efficiency was in the order of iron > zinc > calcium. The solubility studies conducted in water with various concentrations of Tween 80 (0.1 to 2.5% w/v) revealed an improvement in the solubility of complexes with an increase in Tween 80 concentrations. The solubility of the curcumin complex was in the order of calcium > zinc > iron, while the stability of the complexes is in the order of calcium < zinc < iron. Therefore, the complexes of metal-ions with high electronegativity produce strong complexes, which cannot be cleaved easily in the aqueous media resulting in a low solubility [122]. Despite various studies demonstrating their ability in solubility enhancement, the metal complexes are limited to a few API molecules. Apart from this, the metal complexes exert heavy metal-ion toxicity and possess less bioavailability due to their trans-chelation with amino-acid, and peptides in the gastrointestinal tract [123].
Organic Molecular Complexes (OMCs)
Organic molecular complexes (OMCs) are non-covalently bound substrates and ligands [6]. OMCs are formed due to weak non-covalent interactions, including hydrogen bonding, charge-transfer interactions, hydrophobic interactions, electrostatic interactions, and dispersion forces between substrates and ligands [6]. In aqueous solutions, the bound molecules of the substrate are frequently in dynamic equilibrium with their corresponding unbound substrate molecules [6]. OMCs could form as a result of non-covalent interaction between small substrates and small ligands (e.g., drug-drug), between small substrates and large ligands (e.g., drug-polymer or drug-protein), and between large substrates and large ligands (e.g., protein-carbohydrate) [6]. OMCs are prepared mainly as water-soluble complexes of poorly water-soluble drugs [6].
Small Substrate and Small Ligand OMCs/Stacking Complexes
Stacking complexes are formed due to the overlap of the planar regions of aromatic compounds [124]. The hydrocarbon moieties of nonpolar molecules tend to aggregate and be squeezed out of the water environment due to the strong hydrogen bonding between water molecules [124]. The aggregation of the hydrocarbon moieties is due to the large planar nonpolar regions in the nonpolar molecule. The stacked arrangement reduces the exposure of the hydrophobic regions of nonpolar moieties to water [125]. Therefore, the complex is formed due to the interaction between the planer hydrophobic regions of the ligand and the substrate (drug). The drug and the ligand may not have an affinity towards each other but interact to minimize the exposure of their molecules to water [125].
Stacked complexes are either homogeneous or mixed so stacking may occur between the same molecules (self-association) or different molecules (co-association) [125]. Various models and theories have been suggested to explain the dependence of the stacking phenomenon on the properties of the active and the ligand. Based on the maximum aromatic overlap model, the size of the pi-electron system of the ligand is the most crucial factor in determining the strength of the interaction in the complexation process [125, 126]. In addition, the electrostatic force of the donor–acceptor type plays an essential role in the complexation process [125, 127]. Moreover, the role of hydrogen bonding in stacking complexation has also been investigated; however, a clear relationship has not been established [125, 128, 129]. It was also postulated that high log P drugs have a more vital driving force towards complexation. However, this theory considers the whole non-polarity of the substrate molecules. Therefore, only a part of the substrate may be complexing; therefore, the total non-polarity theory may not be correct [128, 129]. Some ligands that are well known to form stacking OMCs are Nicotinamide, Pyrene, Anthracene, Salicylic acid, Methylene blue, Ferulic acid, Benzoic acid, Gentisic acid, Theobromine, Purine, Naphthalene, and Caffeine [124].
Caffeine Based OMCs
Caffeine (CAF) is a methylxanthine alkaloid that occurs naturally in more than 60 plants, including tea leaves, guarana, coffee beans, cocoa, and kola nuts [130–132]. CAF has central nervous system stimulant, vasodilating, and diuretic pharmacological activities [130]. In addition, some recent investigations have reported potential anticancer activity for CAF [133, 134]. OMCs between poorly water-soluble drugs and CAF have been successfully used to improve the solubility, in vitro dissolution rate, photostability, and, thus, in vivo therapeutic efficacy of many actives. Therefore, CAF has been well-established as an excellent complexing ligand for improving the therapeutic outcomes of many poorly water-soluble drugs. CAF interacts mainly with acidic drugs that have organic acid anions. This interaction could occur by either a dipole–dipole interaction or a hydrogen bond between the polarized carbonyl groups of CAF and the hydrogen atom of the acidic drug, as illustrated in Fig. 4 [132]. However, sometimes less water-soluble OMCs of drugs such as gentisic acid are formed. CAF forms hydrogen-bonded OMCs with drugs that have a proton donor functional group, including phenols, phenol derivatives, and aliphatic alcohols [132, 135].
Fig. 4.

Organic molecular complexes of caffeine with p-aminobenzoic acid by a hydrogen bond (a) and dipole–dipole force (b).
Reproduced with permission from ref [132].
Higuchi et al. prepared many OMCs of caffeine and various drugs, including the antimicrobial sulfathiazole, in the early 1950s. Adding CAF to the aqueous medium containing sulfathiazole resulted in ~ threefold improvement in the drug solubility due to the formation of 1:1 sulfathiazole-caffeine OMC [136]. Goto et al. investigated the effect of complexation between CAF with salicylic acid, aspirin, dehydroacetic acid, and ethyl p-hydroxybenzoate on oral absorption in a rabbit animal model. The tested drugs associated with CAF form water-soluble complexes. The in vivo experimental results showed a proportional relationship between the absorption rate constants and complex ratios in administered solutions [137].
Shakeel et al. prepared CAF-celecoxib (1:1) OMC to enhance celecoxib's solubility and in vitro dissolution rate. In vitro dissolution testing for pure celecoxib, complex, and marketed capsule (Celebrex®) showed the highest % release (90.5) celecoxib for the caffeine–celecoxib complex, suggesting that CAF is a promising complexing ligand for solubility and dissolution enhancement of poorly water-soluble drugs such as the non-steroidal anti-inflammatory drug celecoxib [130]. Butt et al. prepared CAF and Soluplus OMCs containing the anti-hyperlipidemic drug rosuvastatin calcium to improve the solubility and dissolution rate of the drug. The prepared OMCs significantly improved the rate and the extent of drug release from tablets in pH 1.2, 6.6, and 6.8 buffers. They provided a rosuvastatin calcium concentration higher than the saturation solubility of the drug [138].
Ahmed et al. studied the effect of CAF complexation with the vitamin B2 riboflavin on the photolysis kinetics of the vitamin at different pH (2.0–10.5). CAF inhibited vitamin photolysis due to the complex formation between CAF and riboflavin. Moreover, the photochemical interaction of riboflavin with CAF indicated that a pH of ~ 6 is the most proper for stabilizing the riboflavin due to the formation of a complex with the highest stability constant at this pH [139].
Nicotinamide Based OMCs
In addition to stacking, at least two more theories have been suggested to explain how nicotinamide (NCT) solubilizes drugs. First, it has been proposed that NCT acts like a chaotrope that breaks the strongly associated structure of water and, thus, has higher drug solubility [125]. Some researchers postulated that NCT develops micellar aggregates in water, followed by the inclusion of the substrate into these micellar aggregates. The critical hydrotrope concentration is the concentration at which the aggregation starts [140]. The is still a debate about whether these aggregates are higher-order complexes or micelles. In either case, it was demonstrated that the production of the aggregates was a crucial step in the solubilization of the substrate.
Sanghvi et al. investigated the solubility enhancement of eleven poorly water-soluble drugs (phenobarbitone, griseofulvin, phenytoin, ketoprofen, estrone, amiodarone, carbendazim, 2-phenoxy propionic acid, XK-469, benzoyl phenyl urea derivative, and PG-300995) after complexation with NCT [125]. The solubilization efficiency of NCT was compared to that of inclusion complexation with HPβCD and SBβCD. At low NCT concentrations, a 1:1 complex was formed, while at higher NCT concentrations 1:2 (drug: NCT) complex was formed. The NCT solution (20% w/v) showed solubility improvements up to 4000-fold for the benzoyl phenyl urea derivative. Moreover, NCT was more effective than cyclodextrins for solubilizing carbendazim, griseofulvin, and PG-300995. The authors also studied the mechanism of drug solubilization by studying the effects of NCT concentration on water's conductivity and surface tension. A slight break in both tested characteristics at 10% NCT concentration, indicating self-association at higher NCT concentrations [125].
Agrawal et al. investigated the effect of different hydrotropes (NCT, piperazine, sodium ascorbate, sodium salicylate, and sodium benzoate) complexation on the solubility of the non-steroidal anti-inflammatory drug nimesulide [141]. The solubility augmentation power of the tested hydrotropes was ranked in this increasing order–NCT < sodium benzoate < sodium salicylate < sodium ascorbate < piperazine–with a solubility enhancement ratio of 12 < 58 < 68 < 156 < 3248, respectively for 2 M hydrotrope concentration [141].
Small Substrate and Large Ligand OMCs
Many hydrophilic polymers interact with poorly water-soluble drugs to form water-soluble complexes. For example, many commercial drug products use polyvinylpyrrolidone (Povidone; PVP) as a solubilizer. According to the FDA’s inactive ingredient database, PVP, PVP K12, PVP K15, PVP K17, PVP K25, PVP K25/30, PVP K30, and PVP K90 are used in many commercial formulations for auricular, intramuscular, ophthalmic, oral, subcutaneous, sublingual, topical, transdermal, vaginal, intravenous, and buccal routes. PVP forms hydrogen bonding with many poorly water-soluble drugs, including acetaminophen [142], amphotericin B [143], diflunisal [144], hydrocortisone [145], metronidazole [146], and sulindac [147]. Water-soluble cellulose derivatives such as hypromellose (HPMC; hydroxypropyl methylcellulose) have also been investigated for enhancing the aqueous solubility of poorly water-soluble drugs, including acetazolamide, prazepam, hydrocortisone, and sulfamethoxazole [145]. In addition, Polyhydroxy oxyethylene phosphate has been used as a solubilizer for paclitaxel through complex formation [148]. Some proteins, such as serum albumin, form water-soluble complexes of poorly soluble drugs, including paclitaxel [149] and rifapentine [150].
Afrasiabi et al. reported that the aqueous solubility of acetaminophen increased from 14.3 mg/mL to 26.7 mg/mL after complexation with PVP (8% w/v) polymer. Dialysis studies revealed the weak, physical, and reversible interaction between PVP and the drug. In addition, Infrared spectroscopy revealed a hydrogen bonding interaction between PVP and acetaminophen; therefore, the increase in drug solubility is probably attributed to forming a water-soluble PVP complex [142]. Rodrigues et al. investigated the complex formation between the poorly water-soluble non-steroidal anti-inflammatory drug diflunisal with PVP. X-ray diffraction analysis revealed the transformation of diflunisal from crystalline to amorphous state in solid dispersions depending on the PVP content. DSC thermograms of solid dispersion systems revealed a solid-state interaction. The increased release rate of the drug from solid dispersion compared to the physical mixture was attributed to the formation of a water-soluble PVP complex [144].
Loftsson et al. studied the effect of water-soluble polymers (HPMC and PVP) on the aqueous solubility of acetazolamide, prazepam, hydrocortisone, and sulfamethoxazole. The tested polymers (0.10–0.25% w/v significantly improved the aqueous solubility of the tested actives. In addition, the solubility studies revealed that a significant fraction (30–50%) of tested drug molecules are bound to the polymers in dilute aqueous polymer solutions [145].
Ostrovskii et al. used human serum albumin, succinylated gelatin, and sodium caseinate to produce water-soluble forms of the antituberculosis drug rifapentine by precipitation or homogenization. The ultrasonic homogenization generated stable colloidal dispersions with a 100-fold improvement in the aqueous solubility of the drug (9– 10 mg/mL). Furthermore, the dilution of the suspensions led to the dissociation of the aggregates formed during and formation of an apparent drug solution. The particle size was 10– 20 nm, which would not lead to embolization upon infusion of that water-soluble drug complex. The results showed that the selected approach could be a promising platform for preparing parenteral formulations for tuberculosis [150].
Large Substrate and Small Ligand OMCs
Most macromolecules, such as protein drugs, are water-soluble. However, proteins could precipitate from aqueous solutions and form protein aggregates. Carbohydrates (Polyols) can form complexes with these macromolecules in aqueous solutions and prevent this irreversible phenomenon [151].
Determination of Stoichiometry Ratio of Reactants (Drug and Complexing Agent)
The stoichiometry of complexation and stability of the complexes are the main factors to be considered during the formation of drug complexes. The most common stoichiometric ratio between the drug and complexing agents is 1:1, and sometimes this ratio can be higher, like one molecule of drug can form a complex with more than one molecule of complexing agent and vice-versa [83]. There are various methods to ensure complex formation, determine the stoichiometry of complexation and evaluate the stability of the formed complex. Some of the widely adopted techniques are described below.
In silico Molecular Modeling
The in silico molecular modeling can give an insight into the interaction between the drug and the complexing agent, such as whether the drug and complexing agent are interacting to form a complex, the type of interaction, and the interaction ratio. These studies can reduce the time and cost of the developmental process of drug complexes. Various in silico methods like molecular docking, molecular dynamics, artificial intelligence, and artificial neural networks have been used to determine the interactions for the formation of complexes [152]. Among all these methods, molecular docking has been extensively used to determine the interaction between different CDs and guest molecules [153]. For instance, Mahipal et al. used AutoDock Vian software to investigate the interaction between the sorafenib tosylate and CDs (α-CD, β-CD, and γ-CD) (Fig. 5A), wherein the authors found that the interaction between the sorafenib tosylate and β-CD is strongest among the other natural CDs with the binding affinity of -6.1 kcal/mol. The studies also revealed the stoichiometry of sorafenib tosylate and β-CD in the ratio of 1:1 [58]. However, this molecular docking technique has been confined to the application for CD complexes because molecular docking requires the target (i.e., the CD) with docking site-like proteins. On the other hand, the complexation interaction can be studied by molecular dynamics, which enables the study of the interaction between various molecules and stimulates various process parameters like temperature and pressure conditions [152].
Fig. 5.

A) Molecular docking studies and the structures of SFNT (a), β-CD (b), top view of SFNT/β-CD inclusion complex (c), bottom view of SFNT/β-CD inclusion complex (d), side view of SFNT/β-CD inclusion complex (e), and hydrogen bond (yellow dotted line) representation in SFNT/β-CD inclusion complex (f).
Reproduced with permission from ref [58]. B) Molecular structure simulation of IPA association produced using Gauss View package. Reproduced with permission from ref [101]. C) The conformation of the system of methylphenidate hydrochloride and ion exchange resins. Reproduced with permission from ref [154].
Different types of complexes have also been studied using molecular dynamic simulations. For example, Sherje et al. used the Maestro module in Schrodinger software to study the interaction between etodolac, HPβCD, and L-arginine in ternary inclusion complex formation [155]. Apart from the application to CD complexes, molecular dynamic modeling can be used to study the interaction between the drug and other complexing agents like resin and phospholipids.
Similarly, Amirinejad et al. used Gaussian 16 software to study the phospholipid complexes interaction between the IBU and 1,2-dilinoyl-sn-glycero-3- phosphocholine (Fig. 5B) [101]. Li et al. studied the interaction between methylphenidate hydrochloride and resin (sodium polystyrene sulfonate) using Gaussview 5 software (Fig. 5C) [154]. In summary, the application of molecular simulation to determine the various characteristics of complexes has been rapidly increasing with the progress in the improvement of in-silico modeling.
Phase Solubility Studies
Higuchi and Connors developed a phase solubility method to examine the effect of the host in solubilizing the guest molecule [156]. The phase solubility diagrams are plotted between the concentration of host (X-axis) vs. the concentration of dissolved guest (drug) (Y-axis). Based on the obtained linearity of the curve, the phase solubility diagrams are divided into A and B types, as illustrated in Fig. 6. The A-type curve indicates the formation of soluble drug-CD complexes. The A-type curves are divided into three subtypes, AL, AP, and AN. The AL-type curve indicates the solubility of the drug increases linearly with an increase in CD concentration, thus inferring the formation of monomolecular drug-CD complexes (1:1). In these curves, the slope values are usually equal to or less than unity and the Km:n can be determined using equation-5. The AP-type curve represents the positive deviation of the curve from linearity, which occurs when the solubilization of drug increases without the further addition of CD. At the same time, the AP-type curve may indicate the formation of higher-order complexes concerning CD (one molecule of drug reacts with more than one CD). The two-CD molecules and a molecule of the drug need not be complexed, as it is essential to remember that this method indicates the influence of CD on the solubilization of the drug rather than complex formation. The solubilization increase can also be attributed to the formation of either non-inclusion complexes, CD aggregates, or both. In AP-type curves, the slope values is usually greater than unity and the Km:n can be determined using equation-6. The AN-type represents the negative deviation from the linearity, which occurs when the solubilization of the drug decreases with the further addition of CD. The AN-type can be obtained when the CDs or their complexes self-associate at higher CD concentrations or due to the changes in the complexation media, like viscosity and surface tension, due to higher concentrations of CDs. In AN-type curves, the slope values is usually lesser than unity and the Km:n can be determined using equation-5 [74, 75, 83, 157].
Fig. 6.
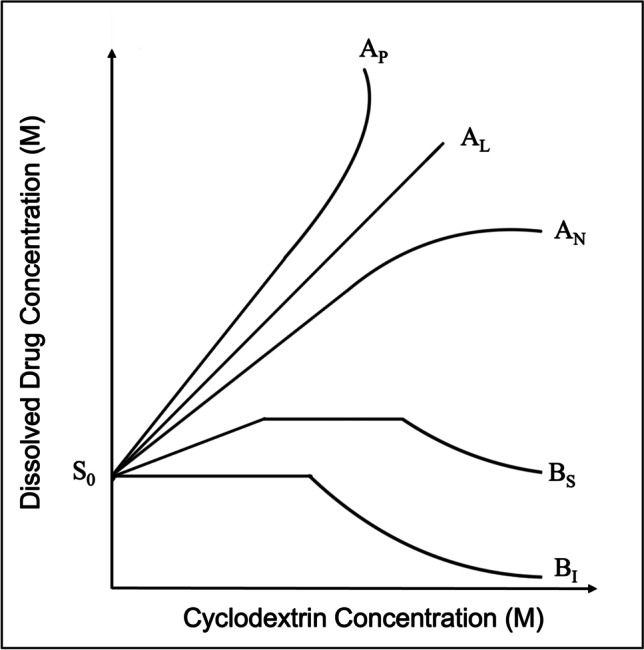
Phase Solubility Curve and its classification.
In contrast, B-type curves imply the formation of complexes with limited solubility and are commonly obtained using natural CDs. The BS-type curves have three regions the linear, plateau, and descending region. The linear region can be mathematically considered as AL-type. The plateau region indicates the solubilization of all the available drugs in the complexation media or the inefficiency of the complexing agent to form complexes. The further addition of CD results in the formation of insoluble complexes that precipitate depletion of solubilized drug concentrations, thus forming the descending region in the BS-type curve. The BI-type curves represent the formation of insoluble complexes and are like BS-type, except they do not have initial linear and plateau regions [157]. The following equation-4 defines the equilibrium or stability constant (Km:n) in a drug complex.
| 4 |
m*D and n*C represent the number of molecules of the drug and the complexing agent, respectively [83].
The value of Km:n for cyclodextrin:drug in 1:1 and 2:1 ratio can be calculated using the following equation-5&6 respectively.
| 5 |
| 6 |
where S0 is the intrinsic solubility of the drug (intercept) and the slope is the slope obtained from the drug-complexing agent phase solubility diagram as represented in Fig. 0.6 [83]. The value of Km:n changes with the ratio of reacting species; for instance, the value of K1:1 ranges from 102 to 103 M−1 and essentially not more than 104 M−1, while for K1:2 the value ranges from 10 to 500 M−1 [75]. The value Km:n is highly vulnerable to the S0 of the drug, and sometimes it is complicated to obtain the value of S0 for poorly soluble drugs. Further, the lipophilic drugs tend to self-associate in aqueous media leading to errors in calculating Km:n. Under these circumstances, it would be more accurate to calculate CE as in equation-7 rather than Km:n [74].
| 7 |
The slope for the above equations can be determined from the phase solubility diagram.
Job’s Plot
The job plot is often referred to as a method of continuous variation and is used to determine the stoichiometry ratio of reactants (drug and complexing agent) in chemical equilibrium.
m*D and n*C represent the number of molecules of the drug and the complexing agent, respectively. While DmCn represents the molecular complex. Unlike the phase solubility studies, the job’s plot is plotted between the mole fraction of the drug or complexing agent (x-axis) vs. dependent variable like analytical measurement (y-axis) that correlates linearly with concentrations of drug or complexing agent [158]. Most often, the analytical measurement employed in this method is UV–VIS absorbance; other measurements may include but are not limited to conductivity, circular dichroism, and melting point depression.
In this study, concentrations of the drug (0-1 M) and complexing agent (1-0 M) are varied simultaneously, but the total concentration of the drug and complexing agent remains constant. Plotting the analytical measurements (y-axis) against the x-axis gives a curve. The curve’s maximum gives a qualitative insight into the stoichiometry ratio of reactants. For instance, the maximum of the curve at a 0.5-mol fraction of drug or complexing agent of the X-axis (Fig. 7A) indicates the stoichiometry ratio of reactants is 1:1. Whereas if the maximum (0.33 on X-axis) is inclined towards the left side of the curve (Fig. 7B), it indicates the stoichiometry ratio of reactants in 1:2. Similarly, if the maximum of the curve is inclined towards the right side of X-axis, it indicates the stoichiometry ratio of reactants in 2:1. The sharpness of the curve indicates the rough estimation of equilibrium constant Km:n. The sharp curve indicates a higher Km:n, while the bell shape indicates a lower Km:n (Fig. 7C) [159].
Fig. 7.

Different curves of Job’s Plot, where P – Measured physical or analytical property, XA – Mole fraction of Drug. While figure A – represents the stoichiometry of complexation in 1:1 (Maximum of curve at XA is 0.5), figure B – represents the stoichiometry of complexation in 1:2 (Maximum of curve at XA is 0.33) and figure C – represents the various shapes of job’s plot in 1:1 stoichiometric complexation indicating equilibrium constant (km:n)
Reproduced with permission from ref [159].
Thermodynamics of Drug-Complex Interaction
The thermodynamic parameters i.e., Gibbs free energy, enthalpy, and entropy of a complexation can be obtained from the stability constant of CD-complex against temperature. As mentioned earlier the complexation involves electrostatic interaction, confirmational changes, van der Waals forces, hydrogen bonding, etc. Compared to all, the hydrophobic interactions (van der Waals forces) are important in the complex formation. These van der Waals forces are always associated with slightly positive enthalpy and large positive entropy. However, the water molecules present in the CD’s hydrophobic cavity must be liberated to accommodate the guest API molecule. The water molecules present in the CD’s hydrophobic core are devoid of any hydrogen bonding, thus they possess great flexibility to release from the CD’s core. The free existence (lack of hydrogen bonding) of water molecules in the CD’s hydrophobic core makes them enthalpically rich compared to water molecules present in the bulk media surrounding the CD’s surface. Therefore, the release of these water molecules is always associated with positive enthalpy and large negative entropy values. This entropy and enthalpy compensation can be observed upon adding up all the energies from the hydrophobic interaction and liberation of water from the CD’s core, resulting in slight positive enthalpy. Also, the formation of the CD complex is independent of the chemical nature of the guest molecule, rather it depends on the size of the guest molecule. Thus, the CD complexation can generally be categorized as an enthalpically driven reaction [157].
While most other complexation mechanisms involve either formation of an H-bond or an ionic bond, both bonds are enthalpically driven. The formation of an ionic bond is a strong interaction between positive and negative ions. Whereas the formation of an H-bond is a weak interaction between the partially positive and negatively charged atoms. This opposite change interaction always favors large negative enthalpy change in a system, since the interaction between the oppositely charged molecules is usually spontaneous and releases energy i.e., exothermic [160].
The phospholipid's interaction with drugs is more predictable and depends on the functional groups as mentioned in section-3. The formation of pharmacosomes is enthalpically driven because of the high enthalpically active interaction between the phospholipid and drug, which is usually by H-bond formation or ionic bond formation [161]. Similarly, ionic interactions are typically used to form drug-resin complexes and metal complexes. While organic molecular complexes are typically formed via the H-bond formation [6]. Therefore, all the drug complexes in pharmaceuticals are enthalpically driven.
Conclusions
It is clear from the facts provided in this review that drug complexes, by diverse molecular structures and physicochemical properties, can be utilized as a practical approach to overcoming many drug delivery issues of poorly water-soluble drug molecules. Drug complexes can fulfill the requirement as effective delivery vehicles because they can increase the aqueous solubility, dissolution rate, and bioavailability of poorly water-soluble drugs. Among all complexation approaches, inclusion complexation with cyclodextrins has been prominently utilized as an effective solubility and dissolution enhancement approach. In addition to solubility, the permeability of the drug moieties can be improved through lipid complexes. Considering various reports, it is crucial to thoroughly understand the preformulation aspects to predict the stoichiometry ratio of reactants and stability of the complex. Various in-silico and in vitro models can effectively predict the various preformulation aspects and assist in developing a stable drug complex. Since different regulatory authorities currently approve many pharmaceutical products based on complexation approach, this could open the possibility for other applications to prepare successful drug delivery platforms. The applications of the complexation approach are not limited to the topics reviewed above but also have been expanded to the food, cosmetics, and agriculture industries.
Author contribution
Siva Ram Munnangi, Ahmed Adel Ali Youssef, and Preethi Lakkala: Conceptualization, Writing – original draft, Writing – review & editing. Nagarjuna Narala and Sagar Narala: Writing – review & editing. Sateesh Kumar Vemula, Michael Repka: Supervision.
Data Availability
No data was used for the research described in the article.
Declaration
Competing Interest
The authors have no conflicts of interest to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jangid AK, Agraval H, Gupta N, Yadav UCS, Sistla R, Pooja D, et al. Designing of fatty acid-surfactant conjugate based nanomicelles of morin hydrate for simultaneously enhancing anticancer activity and oral bioavailability. Colloids Surf B Biointerfaces. 2019;175:202–211. doi: 10.1016/j.colsurfb.2018.11.073. [DOI] [PubMed] [Google Scholar]
- 2.Savjani KT, Gajjar AK, Savjani JK. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012;2012:1–10. doi: 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alzahrani A, Nyavanandi D, Mandati P, Youssef AAA, Narala S, Bandari S, et al. A systematic and robust assessment of hot-melt extrusion-based amorphous solid dispersions: Theoretical prediction to practical implementation. Int J Pharm. 2022;624:121951. doi: 10.1016/j.ijpharm.2022.121951. [DOI] [PubMed] [Google Scholar]
- 4.Narala S, Nyavanandi D, Alzahrani A, Bandari S, Zhang F, Repka MA. Creation of Hydrochlorothiazide Pharmaceutical Cocrystals Via Hot-Melt Extrusion for Enhanced Solubility and Permeability. AAPS PharmSciTech. 2022;23:56. doi: 10.1208/s12249-021-02202-8. [DOI] [PubMed] [Google Scholar]
- 5.Narala S, Nyavanandi D, Srinivasan P, Mandati P, Bandari S, Repka MA. Pharmaceutical Co-crystals, Salts, and Co-amorphous Systems: A novel opportunity of hot-melt extrusion. J Drug Deliv Sci Technol. 2021;61:102209. doi: 10.1016/j.jddst.2020.102209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loftsson T. Drug solubilization by complexation. Int J Pharm Elsevier. 2017;531:276–280. doi: 10.1016/j.ijpharm.2017.08.087. [DOI] [PubMed] [Google Scholar]
- 7.Kale AR, Kakade S, Bhosale A. A Review on: Solubility Enhancement Techniques. J Curr Pharma Res. Current Pharma Research; 2020;10:3630–47.
- 8.Jagtap S, Magdum C, Jadge D, Jagtap R. Solubility Enhancement Technique: A Review. J Pharm Sci Res. Cuddalore, India: Journal of Pharmaceutical Sciences and Research 2018;10:2205–11.
- 9.Trivedi R, Kompella UB. Nanomicellar formulations for sustained drug delivery: strategies and underlying principles. Nanomed. 2010;5:485–505. doi: 10.2217/nnm.10.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serajuddin ATM. Salt formation to improve drug solubility. Adv Drug Deliv Rev. 2007;59:603–616. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Mukerjee P. Micellar properties of drugs: micellar and nonmicellar patterns of self-association of hydrophobic solutes of different molecular structures—monomer fraction, availability, and misuses of micellar hypothesis. J Pharm Sci Elsevier. 1974;63:972–981. doi: 10.1002/jps.2600630647. [DOI] [PubMed] [Google Scholar]
- 12.Attwood D, Mosquera V, Villar VP. Thermodynamic properties of amphiphilic drugs in aqueous solution. J Chem Soc Faraday Trans. 1989;85:3011–7. doi: 10.1039/f19898503011. [DOI] [Google Scholar]
- 13.Carey MC, Small DM. Micelle formation by bile salts: physical-chemical and thermodynamic considerations. Arch Intern Med American Medical Association. 1972;130:506–527. doi: 10.1001/archinte.1972.03650040040005. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Morris KR, Chu B. Aggregation behavior of fosinopril sodium—A new angiotensin-converting enzyme inhibitor. J Pharm Sci Wiley Online Library. 1995;84:609–613. doi: 10.1002/jps.2600840516. [DOI] [PubMed] [Google Scholar]
- 15.Rades T, Müller-Goymann CC. Investigations on the micellisation behaviour of fenoprofen sodium. Int J Pharm Elsevier. 1997;159:215–222. doi: 10.1016/S0378-5173(97)00288-3. [DOI] [Google Scholar]
- 16.Fini A, Fazio G, Gonzalez-Rodriguez M, Cavallari C, Passerini N, Rodriguez L. Formation of ion-pairs in aqueous solutions of diclofenac salts. Int J Pharm Elsevier. 1999;187:163–173. doi: 10.1016/S0378-5173(99)00180-5. [DOI] [PubMed] [Google Scholar]
- 17.Peresypkin A, Kwei G, Ellison M, Lynn K, Zhang D, Rhodes T, et al. Supramolecular Behavior of the Amphiphilic Drug (2R)-2-Ethylchromane-2-Carboxylic Acid Arginine Salt (a Novel PPARα/γ Dual Agonist) Pharm Res Springer. 2005;22:1438–1444. doi: 10.1007/s11095-005-5883-2. [DOI] [PubMed] [Google Scholar]
- 18.Pandi P, Bulusu R, Kommineni N, Khan W, Singh M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int J Pharm. 2020;586:119560. doi: 10.1016/j.ijpharm.2020.119560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Y, Ho C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J Pharm Sci. 2015;104:3237–3258. doi: 10.1002/jps.24541. [DOI] [PubMed] [Google Scholar]
- 20.Butreddy A, Sarabu S, Almutairi M, Ajjarapu S, Kolimi P, Bandari S, et al. Hot-melt extruded hydroxypropyl methylcellulose acetate succinate based amorphous solid dispersions: Impact of polymeric combinations on supersaturation kinetics and dissolution performance. Int J Pharm. 2022;615:121471. doi: 10.1016/j.ijpharm.2022.121471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arima H, Higashi T, Motoyama K. Improvement of the bitter taste of drugs by complexation with cyclodextrins: applications, evaluations and mechanisms. Ther Deliv Future Science. 2012;3:633–644. doi: 10.4155/tde.12.28. [DOI] [PubMed] [Google Scholar]
- 22.Sinko PJ, editor. Martin’s Physical Pharmacy and Pharmaceutical Sciences. 6. Baltimore, MD: Lippincott Williams & Wilkins; 2010. [Google Scholar]
- 23.Havlir T, Kasraian K. Palatable chewable tablet [Internet]. 2009 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US7482022B2/en?q=Palatable+chewable+tablet&oq=Palatable+chewable+tablet
- 24.Backensfeld T, Heil W, Lipp R. Compositions of estrogen-cyclodextrin complexes [Internet]. 2007 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US7163931B2/en
- 25.Chang Y, Dow GJ. Aqueous compositions containing metronidazole [Internet]. 2010 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/CA2470492C/en
- 26.Patel JH, Churchwell MD, Seroogy JD, Barriere SL, Grio M, Mueller B. Telavancin and hydroxy propyl-beta-cyclodextrin clearance during continuous renal replacement therapy: an in vitro study. Int J Artif Organs. 2009;32:745–751. doi: 10.1177/039139880903201006. [DOI] [PubMed] [Google Scholar]
- 27.Wright C, Carr DB, Mermelstein FH. Formulations of low dose diclofenac and beta-cyclodextrin [Internet]. 2011 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US20110218247/en
- 28.Paulus K, Schwab W, Grunder D, Hoogevest P van. Pharmaceutical composition containing an antivirally active dihydroquinazoline derivative [Internet]. 2020 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US10603384B2/en
- 29.Tyavanagimatt SR, Stone MACL, Weimers WC, Kasi GK, Samuel PNK, Bolken T, et al. ST-246 liquid formulations [Internet]. 2016 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US9233097B2/en
- 30.Reynolds M, Smith SA. Liquid formulations of (S)-N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-A]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide [Internet]. 2018 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US10137127B2/en
- 31.Bodor NS, Dandiker Y. Oral formulations of cladribine [Internet]. 2014 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US8785415B2/en
- 32.Larson N. Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections [Internet]. 2020 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US10675296B2/en
- 33.Celador IB, Canudo VB, Navarro JA, Valle MIT del, Rosés MP. Pharmaceutical compositions comprising voriconazole [Internet]. 2013 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/EP2561863A1/en
- 34.Zhāngguìmín, Xún míng jīn Wángmènghuá. Delafloxacin meglumine freeze-dried preparation for injection and preparation method thereof [Internet]. 2021 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/CN113018268A/en
- 35.Heimbecher SK, Monteith D, Pipkin JD. Posaconazole intravenous solution formulations stabilized by substituted β-cyclodextrin [Internet]. 2016 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US9358297B2/en
- 36.COLQUHOUN H, KANES SJ. Neuroactive steroids, compositions, and uses thereof [Internet]. 2017 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/WO2017156103A1/en
- 37.Mǎ tāo, Lǐ xù, Mèng shūjuān. Stable fosphenytoin composition of sodium and preparation thereof [Internet]. 2017 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/CN106265501A/en
- 38.Tu Y-H, Perumal A, Kathala K. Methylphenidate extended release chewable tablet [Internet]. 2017 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US9545399B2/en
- 39.Mehta K, Tu Y-H, Perumal A. Orally effective methylphenidate extended release powder and aqueous suspension product [Internet]. 2015 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US8956649B2/en
- 40.McMahen R, Tengler M, Sloane M, Lockhart D. Methods of Formulating and Designing Liquid Drug Suspensions Containing Ion Exchange Resin Particles [Internet]. 2014 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US20140033806A1/en
- 41.Fischer FX, Khanna SC. Resinate sustained release dextromethorphan composition [Internet]. 1988 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/US4788055A/en
- 42.Cavassin FB, Baú-Carneiro JL, Vilas-Boas RR, Queiroz-Telles F. Sixty years of Amphotericin B: An Overview of the Main Antifungal Agent Used to Treat Invasive Fungal Infections. Infect Dis Ther. 2021;10:115–147. doi: 10.1007/s40121-020-00382-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonner DP, Clark JM. Amphotericin B lipid complex [Internet]. 1995 [cited 2022 Nov 25]. Available from: https://patents.google.com/patent/EP0421733B1/en
- 44.Frei A. Metal Complexes, an Untapped Source of Antibiotic Potential? Antibiotics. Multidisciplinary Digital Publishing Institute; 2020;9:90. [DOI] [PMC free article] [PubMed]
- 45.Ming L-J, Epperson JD. Metal binding and structure–activity relationship of the metalloantibiotic peptide bacitracin. J Inorg Biochem. 2002;91:46–58. doi: 10.1016/S0162-0134(02)00464-6. [DOI] [PubMed] [Google Scholar]
- 46.Swart H. Fulvestrant compositions and method of use [Internet]. 2022 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/US11452729B2/en?q=cyclodextrin+complexation+solubility+enhancement&oq=cyclodextrin+complexation+for+solubility+enhancement
- 47.Dalziel S, Jumaa M, Lewis E, Shwonek P. Cyclodextrin complexation methods for formulating peptide proteasome inhibitors [Internet]. 2019 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/AU2018200444B2/en?q=cyclodextrin+complexation+solubility+enhancement&oq=cyclodextrin+complexation+for+solubility+enhancement&page=1
- 48.Tabuteau H. Pharmaceutical compositions comprising meloxicam [Internet]. 2022 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/US11369684B2/en?q=cyclodextrin+complexation+solubility+enhancement&oq=cyclodextrin+complexation+for+solubility+enhancement&page=4
- 49.Pasloske KS, Lau K, Richardson SJ, WILLIS AA. Stable injectable pharmaceutical compositions comprising 2-hydroxypropyl-beta-cyclodextrin and alfaxalone [Internet]. 2020 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/EP2785352B1/en?q=hydroxypropyl+betadex+complexation+solubility+enhancement&oq=hydroxypropyl+betadex+complexation+for+solubility+enhancement&page=1
- 50.Reed KA. Aqueous drug delivery system comprising off-flavor masking agent [Internet]. 2016 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/EP2616046B1/en?q=cyclodextrin+complexation+taste+masking&oq=cyclodextrin+complexation+for+taste+masking
- 51.WENGNER S. Abuse resistant capsule [Internet]. 2019 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/AU2018214110B2/en?q=sodium+polystyrene+sulfonate+%2b+solubility+enhancement&oq=sodium+polystyrene+sulfonate++%2b+solubility+enhancement
- 52.Li M, Krumme M. Edible oral strip or wafer dosage form containing ion exchange resin for taste masking [Internet]. 2020 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/US10744176B2/en?q=ion+exchange+resin+%2b+taste+masking&oq=ion+exchange+resin++%2b+taste+masking&page=1
- 53.Giori A, Franceschi F. Phospholipid complexes of curcumin having improved bioavailability [Internet]. 2019 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/EP1991244B1/en?q=phospholipid+complexes&oq=phospholipid+complexes
- 54.Zheng G, KECA J. Texaphyrin-phospholipid conjugates and methods of preparing same [Internet]. 2020 [cited 2023 Jan 23]. Available from: https://patents.google.com/patent/US10729792B2/en?q=phospholipid+complexes&oq=phospholipid+complexes
- 55.Abbas ZS, Sulaiman GM, Jabir MS, Mohammed SAA, Khan RA, Mohammed HA, et al. Galangin/β-Cyclodextrin Inclusion Complex as a Drug-Delivery System for Improved Solubility and Biocompatibility in Breast Cancer Treatment. Molecules. Multidisciplinary Digital Publishing Institute; 2022;27:4521. [DOI] [PMC free article] [PubMed]
- 56.Manne ASN, Hegde AR, Raut SY, Rao RR, Kulkarni VI, Mutalik S. Hot liquid extrusion assisted drug-cyclodextrin complexation: a novel continuous manufacturing method for solubility and bioavailability enhancement of drugs. Drug Deliv Transl Res. 2021;11:1273–1287. doi: 10.1007/s13346-020-00854-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Granados PA, Pinho LAG, Sa-Barreto LL, Gratieri T, Gelfuso GM, Cunha-Filho M. Application of hot-melt extrusion in the complexation of naringenin with cyclodextrin using hydrophilic polymers. Adv Powder Technol. 2022;33:103380. doi: 10.1016/j.apt.2021.11.032. [DOI] [Google Scholar]
- 58.Donthi MR, Munnangi SR, Krishna KV, Marathe SA, Saha RN, Singhvi G, et al. Formulating Ternary Inclusion Complex of Sorafenib Tosylate Using β-Cyclodextrin and Hydrophilic Polymers: Physicochemical Characterization and In Vitro Assessment. AAPS PharmSciTech. 2022;23:254. doi: 10.1208/s12249-022-02406-6. [DOI] [PubMed] [Google Scholar]
- 59.Özyılmaz ED, Comoglu T. Development of pediatric orally disintegrating mini-tablets containing atomoxetine hydrochloride-β-cyclodextrin inclusion complex using experimental design. Drug Dev Ind Pharm. Taylor & Francis; 2022;48:667–81. [DOI] [PubMed]
- 60.Siddiqui F, Shoaib MH, Ahmed FR, Qazi F, Yousuf RI, Usmani MT, et al. Formulation development and optimization of taste-masked azithromycin oral suspension with ion exchange resins: Bioanalytical method development and validation, in vivo bioequivalence study, and in-silico PBPK modeling for the paediatric population. J Drug Deliv Sci Technol. 2023;79:104048. doi: 10.1016/j.jddst.2022.104048. [DOI] [Google Scholar]
- 61.Tan DCT, Ong JJ, Gokhale R, Heng PWS. Hot melt extrusion of ion-exchange resin for taste masking. Int J Pharm Int J Pharm. 2018;547:385–394. doi: 10.1016/j.ijpharm.2018.05.068. [DOI] [PubMed] [Google Scholar]
- 62.Biswas S, Mukherjee PK, Harwansh RK, Bannerjee S, Bhattacharjee P. Enhanced bioavailability and hepatoprotectivity of optimized ursolic acid–phospholipid complex. Drug Dev Ind Pharm. Taylor & Francis; 2019;45:946–58. [DOI] [PubMed]
- 63.Qiu X-L, Fan Z-R, Liu Y-Y, Wang D-F, Wang S-X, Li C-X. Preparation and Evaluation of a Self-Nanoemulsifying Drug Delivery System Loaded with Heparin Phospholipid Complex. Int J Mol Sci. Multidisciplinary Digital Publishing Institute; 2021;22:4077. [DOI] [PMC free article] [PubMed]
- 64.Xu Q, Furuishi T, Fukuzawa K, Yonemochi E. Physicochemical Properties and Transdermal Absorption of a Flurbiprofen and Lidocaine Complex in the Non-Crystalline Form. Pharmaceutics. Multidisciplinary Digital Publishing Institute; 2023;15:318. [DOI] [PMC free article] [PubMed]
- 65.Dhall M, Madan AK. Comparison of cyclodextrins and urea as hosts for inclusion of drugs. J Incl Phenom Macrocycl Chem. 2017;89:207–227. doi: 10.1007/s10847-017-0748-y. [DOI] [Google Scholar]
- 66.Powell HM. 15. The structure of molecular compounds. Part IV. Clathrate compounds. J Chem Soc 1948;61–73. [DOI] [PubMed]
- 67.Thakral S, Madan AK. Urea co-inclusion compounds of 13 cis-retinoic acid for simultaneous improvement of dissolution profile, photostability and safe handling characteristics. J Pharm Pharmacol. 2008;60:823–832. doi: 10.1211/jpp.60.7.0003. [DOI] [PubMed] [Google Scholar]
- 68.Aiassa V, Garnero C, Longhi MR, Zoppi A. Cyclodextrin Multicomponent Complexes: Pharmaceutical Applications. Pharmaceutics [Internet]. Pharmaceutics; 2021 [cited 2022 May 6];13. Available from: https://pubmed.ncbi.nlm.nih.gov/34371790/ [DOI] [PMC free article] [PubMed]
- 69.Rincón-López J, Almanza-Arjona YC, Riascos AP, Rojas-Aguirre Y. Technological evolution of cyclodextrins in the pharmaceutical field. J Drug Deliv Sci Technol. 2021;61:102156. doi: 10.1016/j.jddst.2020.102156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan T, Ji M, Sun Y, Yan T, Zhao J, Zhang H, et al. Preparation and characterization of baicalein/hydroxypropyl-β-cyclodextrin inclusion complex for enhancement of solubility, antioxidant activity and antibacterial activity using supercritical antisolvent technology. J Incl Phenom Macrocycl Chem. 2020;96:285–295. doi: 10.1007/s10847-019-00970-2. [DOI] [Google Scholar]
- 71.Das S, Mohanty S, Maharana J, Jena SR, Nayak J, Subuddhi U. Microwave-assisted β-cyclodextrin/chrysin inclusion complexation: An economical and green strategy for enhanced hemocompatibility and chemosensitivity in vitro. J Mol Liq. 2020;310:113257. doi: 10.1016/j.molliq.2020.113257. [DOI] [Google Scholar]
- 72.Ghosh A, Biswas S, Ghosh T. Preparation and Evaluation of Silymarin β-cyclodextrin Molecular Inclusion Complexes. J Young Pharm JYP. 2011;3:205–210. doi: 10.4103/0975-1483.83759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shukla SK, Chan A, Parvathaneni V, Kanabar DD, Patel K, Ayehunie S, et al. Enhanced solubility, stability, permeation and anti-cancer efficacy of Celastrol-β-cyclodextrin inclusion complex. J Mol Liq. 2020;318:113936. doi: 10.1016/j.molliq.2020.113936. [DOI] [Google Scholar]
- 74.Saokham P, Muankaew C, Jansook P, Loftsson T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules. 2018. [DOI] [PMC free article] [PubMed]
- 75.Jansook P, Ogawa N, Loftsson T. Cyclodextrins: structure, physicochemical properties and pharmaceutical applications. Int J Pharm. 2018;535:272–284. doi: 10.1016/j.ijpharm.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 76.Das SK, Kahali N, Bose A, Khanam J. Physicochemical characterization and in vitro dissolution performance of ibuprofen-Captisol® (sulfobutylether sodium salt of β-CD) inclusion complexes. J Mol Liq. 2018;261:239–249. doi: 10.1016/j.molliq.2018.04.007. [DOI] [Google Scholar]
- 77.Szabó Z-I, Orbán G, Borbás E, Csicsák D, Kádár S, Fiser B, et al. Inclusion complexation of the anticancer drug pomalidomide with cyclodextrins: fast dissolution and improved solubility. Heliyon. 2021;7:e07581. doi: 10.1016/j.heliyon.2021.e07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu T, Wan X, Luo Z, Liu C, Quan P, Cun D, et al. A donepezil/cyclodextrin complexation orodispersible film: Effect of cyclodextrin on taste-masking based on dynamic process and in vivo drug absorption. Asian J Pharm Sci. 2019;14:183–192. doi: 10.1016/j.ajps.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yildiz ZI, Celebioglu A, Kilic ME, Durgun E, Uyar T. Fast-dissolving carvacrol/cyclodextrin inclusion complex electrospun fibers with enhanced thermal stability, water solubility, and antioxidant activity. J Mater Sci. 2018;53:15837–15849. doi: 10.1007/s10853-018-2750-1. [DOI] [Google Scholar]
- 80.Shimpi S, Chauhan B, Shimpi P. Cyclodextrins: application in different routes of drug administration. Acta Pharm Zagreb Croat. 2005;55:139–156. [PubMed] [Google Scholar]
- 81.Jambhekar SS, Breen P. Cyclodextrins in pharmaceutical formulations II: solubilization, binding constant, and complexation efficiency. Drug Discov Today. 2016;21:363–368. doi: 10.1016/j.drudis.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 82.Boudeville P, Burgot J. A New pH-metric Methodology for the Determination of Thermodynamic Inclusion Constants of Guest/Cyclodextrin Complexes. J Pharm Sci. 1995;84:1083–1089. doi: 10.1002/jps.2600840910. [DOI] [PubMed] [Google Scholar]
- 83.Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: basic science and product development. J Pharm Pharmacol. John Wiley & Sons, Ltd; 2010;62:1607–21. [DOI] [PubMed]
- 84.Jadhav P, Petkar B, Pore Y, Kulkarni A, Burade K. Physicochemical and molecular modeling studies of cefixime-l-arginine- cyclodextrin ternary inclusion compounds. Carbohydr Polym. 2013. [DOI] [PubMed]
- 85.Kurkov SV, Loftsson T. Cyclodextrins Int J Pharm Elsevier. 2013;453:167–180. doi: 10.1016/j.ijpharm.2012.06.055. [DOI] [PubMed] [Google Scholar]
- 86.Zafar A, Alruwaili NK, Imam SS, Alsaidan OA, Alharbi KS, Mostafa EM, et al. Formulation of ternary genistein β-cyclodextrin inclusion complex: In vitro characterization and cytotoxicity assessment using breast cancer cell line. J Drug Deliv Sci Technol. 2022;67:102932. doi: 10.1016/j.jddst.2021.102932. [DOI] [Google Scholar]
- 87.Suvarna V, Gujar P, Murahari M, Sharma D, Chamariya R. Supramolecular ternary inclusion complexes of Irbesartan with hydroxypropyl-beta-cyclodextrin. J Drug Deliv Sci Technol. 2022;67:102964. doi: 10.1016/j.jddst.2021.102964. [DOI] [Google Scholar]
- 88.Szente L, Singhal A, Domokos A, Song B. Cyclodextrins: Assessing the Impact of Cavity Size, Occupancy, and Substitutions on Cytotoxicity and Cholesterol Homeostasis. Mol J Synth Chem Nat Prod Chem. 2018;23:1228. doi: 10.3390/molecules23051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gladys G, Claudia G, Marcela L. The effect of pH and triethanolamine on sulfisoxazole complexation with hydroxypropyl-beta-cyclodextrin. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2003;20:285–293. doi: 10.1016/s0928-0987(03)00202-1. [DOI] [PubMed] [Google Scholar]
- 90.Walker S, Oun R, McInnes FJ, Wheate NJ. The Potential of Cucurbit[n]urils in Drug Delivery. Isr J Chem. 2011;51:616–624. doi: 10.1002/ijch.201100033. [DOI] [Google Scholar]
- 91.Wheate NJ, Limantoro C. Cucurbit[n]urils as excipients in pharmaceutical dosage forms. Supramol Chem Taylor & Francis. 2016;28:849–856. doi: 10.1080/10610278.2016.1178746. [DOI] [Google Scholar]
- 92.Macartney DH. Encapsulation of Drug Molecules by Cucurbiturils: Effects on their Chemical Properties in Aqueous Solution. Isr J Chem. 2011;51:600–615. doi: 10.1002/ijch.201100040. [DOI] [Google Scholar]
- 93.Liu Q, Tang Q, Xi Y-Y, Huang Y, Xiao X, Tao Z, et al. Host–guest interactions of thiabendazole with normal and modified cucurbituril: 1H NMR, phase solubility and antifungal activity studies. Supramol Chem Taylor & Francis. 2015;27:386–392. doi: 10.1080/10610278.2014.999768. [DOI] [Google Scholar]
- 94.Huang Y, Xue S-F, Tao Z, Zhu Q-J, Zhang H, Lin J-X, et al. Solubility enhancement of kinetin through host–guest interactions with cucurbiturils. J Incl Phenom Macrocycl Chem. 2008;61:171–177. doi: 10.1007/s10847-008-9410-z. [DOI] [Google Scholar]
- 95.Da Silva E, Lazar AN, Coleman AW. Biopharmaceutical applications of calixarenes. J Drug Deliv Sci Technol. 2004;14:3–20. doi: 10.1016/S1773-2247(04)50001-1. [DOI] [Google Scholar]
- 96.Ukhatskaya EV, Kurkov SV, Matthews SE, Loftsson T. Encapsulation of drug molecules into calix[n]arene nanobaskets role of aminocalix[n]arenes in biopharmaceutical field. J Pharm Sci. 2013;102:3485–512. doi: 10.1002/jps.23681. [DOI] [PubMed] [Google Scholar]
- 97.Yang W, de Villiers MM. The solubilization of the poorly water soluble drug nifedipine by water soluble 4-sulphonic calix[n]arenes. Eur J Pharm Biopharm Off J Arbeitsgemeinschaft Pharm Verfahrenstechnik EV. 2004;58:629–636. doi: 10.1016/j.ejpb.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 98.Menon SK, Mistry BR, Joshi KV, Modi NR, Shashtri D. Evaluation and solubility improvement of Carvedilol: PSC[n]arene inclusion complexes with acute oral toxicity studies. J Incl Phenom Macrocycl Chem. 2012;73:295–303. doi: 10.1007/s10847-011-0056-x. [DOI] [Google Scholar]
- 99.Singh RP, Gangadharappa HV, Mruthunjaya K. Phospholipids: Unique carriers for drug delivery systems. J Drug Deliv Sci Technol. 2017;39:166–179. doi: 10.1016/j.jddst.2017.03.027. [DOI] [Google Scholar]
- 100.Hüsch J, Dutagaci B, Glaubitz C, Geppert T, Schneider G, Harms M, et al. Structural properties of so-called NSAID–phospholipid-complexes. Eur J Pharm Sci. 2011;44:103–116. doi: 10.1016/j.ejps.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 101.Amirinejad M, Davoodi J, Abbaspour MR, Akhgari A, Hadizadeh F, Badiee A. Preparation, characterization and improved release profile of ibuprofen-phospholipid association. J Drug Deliv Sci Technol. Elsevier; 2020;60:101951.
- 102.Afanas’eva YuG, Fakhretdinova ER, Spirikhin LV, Nasibullin RS. Mechanism of interaction of certain flavonoids with phosphatidylcholine of cellular membranes. Pharm Chem J. 2007;41:354–6.
- 103.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Controlled Release. 2004;100:331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 104.Kuche K, Bhargavi N, Dora CP, Jain S. Drug-Phospholipid Complex-a Go Through Strategy for Enhanced Oral Bioavailability. AAPS PharmSciTech [Internet]. AAPS PharmSciTech; 2019 [cited 2022 Jul 21];20. Available from: https://pubmed.ncbi.nlm.nih.gov/30610392/ [DOI] [PubMed]
- 105.Zhang K, Gu L, Chen J, Zhang Y, Jiang Y, Zhao L, et al. Preparation and evaluation of kaempferol-phospholipid complex for pharmacokinetics and bioavailability in SD rats. J Pharm Biomed Anal. J Pharm Biomed Anal; 2015;114:168–75. [DOI] [PubMed]
- 106.Biswas S, Mukherjee PK, Kar A, Bannerjee S, Charoensub R, Duangyod T. Optimized piperine-phospholipid complex with enhanced bioavailability and hepatoprotective activity. Pharm Dev Technol Pharm Dev Technol. 2021;26:69–80. doi: 10.1080/10837450.2020.1835956. [DOI] [PubMed] [Google Scholar]
- 107.Murugan V, Mukherjee K, Maiti K, Mukherjee PK. Enhanced Oral Bioavailability and Antioxidant Profile of Ellagic Acid by Phospholipids. J Agric Food Chem. American Chemical Society; 2009;57:4559–65. [DOI] [PubMed]
- 108.Sikarwar MS, Sharma S, Jain AK, Parial SD. Preparation, characterization and evaluation of Marsupsin-phospholipid complex. AAPS PharmSciTech. 2008;9:129–137. doi: 10.1208/s12249-007-9020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo B, Liu H, Li Y, Zhao J, Yang D, Wang X, et al. Application of phospholipid complex technique to improve the dissolution and pharmacokinetic of probucol by solvent-evaporation and co-grinding methods. Int J Pharm. 2014;474:50–56. doi: 10.1016/j.ijpharm.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 110.Zhang TY, Du RF, Wang YJ, Hu JL, Wu F, Feng Y. Research Progress of Preparation Technology of Ion-Exchange Resin Complexes. AAPS PharmSciTech [Internet]. AAPS PharmSciTech; 2022 [cited 2022 Jul 29];23. Available from: https://pubmed.ncbi.nlm.nih.gov/35381945/ [DOI] [PubMed]
- 111.Anand V, Kandarapu R, Garg S. Ion-exchange resins: carrying drug delivery forward. Drug Discov Today. 2001;6:905–914. doi: 10.1016/S1359-6446(01)01922-5. [DOI] [PubMed] [Google Scholar]
- 112.Mehta K, Tu Y-H, Chaudhuri A, Perumal A. Abuse resistant opioid drug - ion exchange resin complexes having hybrid coatings [Internet]. 2013 [cited 2022 Sep 18]. Available from: https://patents.google.com/patent/WO2013119231A1/en
- 113.Ghebre-Sellassie I, Terefe H. Tamper-resistant pharmaceutical dosage forms and process for making same [Internet]. 2018 [cited 2022 Sep 18]. Available from: https://patents.google.com/patent/US10010620B2/en?q=ion-resin+complexation+%2b+tamper+deterrent&oq=ion-resin+complexation+%2b+tamper+deterrent
- 114.Kulthe VV, Chaudhari PD. Drug Resinates an Attractive Approach of Solubility Enhancement of Atorvastatin Calcium. Indian J Pharm Sci. 2013;75:523–532. [PMC free article] [PubMed] [Google Scholar]
- 115.Panraksa P, Boonsermsukcharoen K, Hwang K-M, Park E-S, Jantrawut P. Taste Masking of Nizatidine Using Ion-Exchange Resins. Processes. Multidisciplinary Digital Publishing Institute; 2019;7:779.
- 116.Che X, Wang LH, Yang Y, Yuan Y, Wang QF, Wang Y, et al. Ibuprofen ion-exchange fiber complex: improved dissolution and gastric tolerance based on ion exchange. Drug Dev Ind Pharm. Drug Dev Ind Pharm; 2013;39:744–51. [DOI] [PubMed]
- 117.Quintanilha JCF, Saavedra KF, Visacri MB, Moriel P, Salazar LA. Role of epigenetic mechanisms in cisplatin-induced toxicity. Crit Rev Oncol Hematol. 2019;137:131–142. doi: 10.1016/j.critrevonc.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 118.Ross DL, Riley CM. Physicochemical properties of the fluoroquinolone antimicrobials III. Complexation of lomefloxacin with various metal ions and the effect of metal ion complexation on aqueous solubility. Int J Pharm. 1992;87:203–13. doi: 10.1016/0378-5173(92)90244-V. [DOI] [Google Scholar]
- 119.Higuchi T, Bolton S. The Solubility and Complexing Properties of Oxytetracycline and Tetracycline III: Interactions in Aqueous Solution With Model Compounds, Biochemicals, Metals, Chelates, and Hexametaphosphate. J Am Pharm Assoc Sci Ed. Elsevier; 1959;48:557–64. [DOI] [PubMed]
- 120.Sareen R, Jain N, Dhar KL. Curcumin-Zn(II) complex for enhanced solubility and stability: an approach for improved delivery and pharmacodynamic effects. Pharm Dev Technol Pharm Dev Technol. 2016;21:630–635. doi: 10.3109/10837450.2015.1041042. [DOI] [PubMed] [Google Scholar]
- 121.Duan J, Ma B, Liu F, Zhang S, Wang S, Kong Y, et al. Coordination ability determined transition metal ions substitution of Tb in Tb-Asp fluorescent nanocrystals and a facile ions-detection approach. Nanoscale Royal Soc Chem. 2018;10:7526–7535. doi: 10.1039/c7nr09267a. [DOI] [PubMed] [Google Scholar]
- 122.Hieu TQ, Thao DTT. Enhancing the Solubility of Curcumin Metal Complexes and Investigating Some of Their Biological Activities. J Chem. Hindawi Limited; 2019;2019.
- 123.Wang Z, Li J, Lin G, He Z, Wang Y. Metal complex-based liposomes: Applications and prospects in cancer diagnostics and therapeutics. J Controlled Release. 2022;348:1066–1088. doi: 10.1016/j.jconrel.2022.06.012. [DOI] [PubMed] [Google Scholar]
- 124.Patel JN, Rathod DM, Patel NA, Modasiya MK. Techniques to improve the solubility of poorly soluble drugs. Int J Pharm Life Sci. 2012;3.
- 125.Sanghvi R, Evans D, Yalkowsky SH. Stacking complexation by nicotinamide: A useful way of enhancing drug solubility. Int J Pharm Elsevier. 2007;336:35–41. doi: 10.1016/j.ijpharm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 126.Nakano NI, Igarashi SJ. Molecular interactions of pyrimidines, purines, and some other heteroaromatic compounds in aqueous media. Biochemistry ACS Publications. 1970;9:577–583. doi: 10.1021/bi00805a019. [DOI] [PubMed] [Google Scholar]
- 127.Badwan AA, El-Khordagui LK, Saleh AM, Khalil SA. The solubility of benzodiazepines in sodium salicylate solution and a proposed mechanism for hydrotropic solubilization. Int J Pharm Elsevier. 1982;13:67–74. doi: 10.1016/0378-5173(82)90143-0. [DOI] [Google Scholar]
- 128.Kenley RA, Jackson SE, Winterle JS, Shunko Y, Visor GC. Water soluble complexes of the antiviral drugs, 9-[(1, 3-dihydroxy-2-propoxy) methyl] guanine and acyclovir: the role of hydrophobicity in complex formation. J Pharm Sci Elsevier. 1986;75:648–653. doi: 10.1002/jps.2600750706. [DOI] [PubMed] [Google Scholar]
- 129.Suzuki H, Sunada H. Mechanistic studies on hydrotropic solubilization of nifedipine in nicotinamide solution. Chem Pharm Bull (Tokyo). The Pharmaceutical Society of Japan; 1998;46:125–30. [DOI] [PubMed]
- 130.Shakeel F, Faisal MS. Caffeine: A potential complexing agent for solubility and dissolution enhancement of celecoxib. Pharm Biol Taylor & Francis. 2010;48:113–115. doi: 10.3109/13880200903030074. [DOI] [PubMed] [Google Scholar]
- 131.Hampp A. The extraction of caffeine from tea: A modification of the procedure of Murray and Hansen. J Chem Educ. ACS Publications; 1996;73:1172.
- 132.Belayneh A, Kassa F. The Effect of Coffee on Pharmacokinetic Properties of Drugs : A Review. BioMed Res Int. 2020;2020:1–11. doi: 10.1155/2020/7909703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tomita K, Tsuchiya H. Caffeine enhancement of the effect of anticancer agents on human sarcoma cells. Jpn J Cancer Res. Wiley Online Library; 1989;80:83–8. [DOI] [PMC free article] [PubMed]
- 134.Sabisz M, Skladanowski A. Modulation of cellular response to anticancer treatment by caffeine: inhibition of cell cycle checkpoints, DNA repair and more. Curr Pharm Biotechnol Bentham Science Publishers. 2008;9:325–336. doi: 10.2174/138920108785161497. [DOI] [PubMed] [Google Scholar]
- 135.Escott-Stump S. Nutrition and diagnosis-related care. Lippincott Williams & Wilkins; 2008.
- 136.Higuchi T, Lach JL. Investigation of Some Complexes Formed in Solution by Caffeine*: IV. Interactions Between Caffeine and Sulfathiazole, Sulfadiazine, P-Aminobenzoic Acid, Benzocaine, Phenobarbital, and Barbital. J Am Pharm Assoc Sci Ed. Elsevier; 1954;43:349–54. [DOI] [PubMed]
- 137.GOTO S, TAKAMATSU R, SHIBAO M, IGUCHI S. Effect of combination of pharmaceuticals on gastrointestinal absorption. I. Combination of caffeine with a few absorbable drugs. Chem Pharm Bull (Tokyo). The Pharmaceutical Society of Japan; 1968;16:332–7. [DOI] [PubMed]
- 138.Butt S, Hasan SMF, Hassan MM, Alkharfy KM, Neau SH. Directly compressed rosuvastatin calcium tablets that offer hydrotropic and micellar solubilization for improved dissolution rate and extent of drug release. Saudi Pharm J. 2019;27:619–628. doi: 10.1016/j.jsps.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ahmad I, Ahmed S, Sheraz MA, Aminuddin M, Vaid FHM. Effect of caffeine complexation on the photolysis of riboflavin in aqueous solution: a kinetic study. Chem Pharm Bull (Tokyo). The Pharmaceutical Society of Japan; 2009;57:1363–70. [DOI] [PubMed]
- 140.Balasubramanian D, Srinivas V, Gaikar VG, Sharma MM. Aggregation behavior of hydrotropic compounds in aqueous solution. J Phys Chem ACS Publications. 1989;93:3865–3870. doi: 10.1021/j100346a098. [DOI] [Google Scholar]
- 141.Agrawal S, Pancholi SS, Jain NK, Agrawal GP. Hydrotropic solubilization of nimesulide for parenteral administration. Int J Pharm Elsevier. 2004;274:149–155. doi: 10.1016/j.ijpharm.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 142.Afrasiabi Garekani H, Sadeghi F, Ghazi A. Increasing the aqueous solubility of acetaminophen in the presence of polyvinylpyrrolidone and investigation of the mechanisms involved. Drug Dev Ind Pharm. Taylor & Francis; 2003;29:173–9. [DOI] [PubMed]
- 143.Charvalos E, Tzatzarakis MN, Van Bambeke F, Tulkens PM, Tsatsakis AM, Tzanakakis GN, et al. Water-soluble amphotericin B–polyvinylpyrrolidone complexes with maintained antifungal activity against Candida spp. and Aspergillus spp. and reduced haemolytic and cytotoxic effects. J Antimicrob Chemother. Oxford University Press; 2006;57:236–44. [DOI] [PubMed]
- 144.Rodriguez-Espinosa C, Martinez-Oharriz MC, Martin C, Goni MM, Velaz I, Sanchez M. Dissolution kinetics for coprecipitates of diflunisal with PVP K30. Eur J Drug Metab Pharmacokinet Springer. 1998;23:109–112. doi: 10.1007/BF03189324. [DOI] [PubMed] [Google Scholar]
- 145.Loftsson T, Fri H, Gu TK. The effect of water-soluble polymers on aqueous solubility of drugs. Int J Pharm Elsevier. 1996;127:293–296. doi: 10.1016/0378-5173(95)04207-5. [DOI] [Google Scholar]
- 146.Vakulˈskaya TI, Larina LI, Lopyrev VA. Polarographic investigation of the metronidazole complex with polyvinylpyrrolidone. Chem Heterocycl Compd Springer. 2012;47:1372–1377. doi: 10.1007/s10593-012-0923-4. [DOI] [Google Scholar]
- 147.Tros de Llarduya MC, Martín C, Goni MM, Martinez-Oharriz MC. Solubilization and interaction of sulindac with polyvinylpyrrolidone K30 in the solid state and in aqueous solution. Drug Dev Ind Pharm. Taylor & Francis; 1998;24:295–300. [DOI] [PubMed]
- 148.Mitova V, Hristova T, Cherkezova R, Koseva N, Yusa S, Troev K. Polyphosphoester-based paclitaxel complexes. J Appl Polym Sci. Wiley Online Library; 2015;132. [PubMed]
- 149.Di Costanzo F, Gasperoni S, Rotella V, Di Costanzo F. Targeted delivery of albumin bound paclitaxel in the treatment of advanced breast cancer. OncoTargets Ther. Dove Press; 2009;2:179. [DOI] [PMC free article] [PubMed]
- 150.Ostrovskii KP, Osipova NS, Vanchugova LV, Shipulo EV, Pereverzeva É, Treshchalin ID, et al. Use of proteins to increase the aqueous solubility of rifapentine. Pharm Chem J Springer. 2016;50:407–412. doi: 10.1007/s11094-016-1460-8. [DOI] [Google Scholar]
- 151.Jana M, Bandyopadhyay S. Conformational flexibility of a protein–carbohydrate complex and the structure and ordering of surrounding water. Phys Chem Chem Phys. Royal Society of Chemistry; 2012;14:6628–38. [DOI] [PubMed]
- 152.Mazurek AH, Szeleszczuk Ł, Gubica T. Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. Int J Mol Sci. Multidisciplinary Digital Publishing Institute; 2021;22:9422. [DOI] [PMC free article] [PubMed]
- 153.Quevedo MA, Zoppi A. Current trends in molecular modeling methods applied to the study of cyclodextrin complexes. J Incl Phenom Macrocycl Chem. 2018;90:1–14. doi: 10.1007/s10847-017-0763-z. [DOI] [Google Scholar]
- 154.Li C, Han X, Hong X, Li X, Zhang H, Wang Z, et al. Study on the Complexation and Release Mechanism of Methylphenidate Hydrochloride Ion Exchange Resin Complex. Polymers. 2021;13:4394. doi: 10.3390/polym13244394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sherje AP, Kulkarni V, Murahari M, Nayak UY, Bhat P, Suvarna V, et al. Inclusion Complexation of Etodolac with Hydroxypropyl-beta-cyclodextrin and Auxiliary Agents: Formulation Characterization and Molecular Modeling Studies. Mol Pharm. 2017;14:1231–1242. doi: 10.1021/acs.molpharmaceut.6b01115. [DOI] [PubMed] [Google Scholar]
- 156.HIGUCHI, T. A phase solubility technique. Adv Anal Chem Instrum. 1965;4:117–211.
- 157.Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007; [DOI] [PubMed]
- 158.Olson EJ, Bühlmann P. Getting More out of a Job Plot: Determination of Reactant to Product Stoichiometry in Cases of Displacement Reactions and n:n Complex Formation. J Org Chem American Chemical Society. 2011;76:8406–8412. doi: 10.1021/jo201624p. [DOI] [PubMed] [Google Scholar]
- 159.Renny JS, Tomasevich LL, Tallmadge EH, Collum DB. Method of Continuous Variations: Applications of Job Plots to the Study of Molecular Associations in Organometallic Chemistry. Angew Chem Int Ed. 2013;52:11998–12013. doi: 10.1002/anie.201304157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Laidler KJ. A glossary of terms used in chemical kinetics, including reaction dynamics (IUPAC Recommendations 1996) Pure Appl Chem De Gruyter. 1996;68:149–192. doi: 10.1351/pac199668010149. [DOI] [Google Scholar]
- 161.Gautschi N, Van Hoogevest P, Kuentz M. Amorphous drug dispersions with mono- and diacyl lecithin: On molecular categorization of their feasibility and UV dissolution imaging. Int J Pharm. 2015;491:218–230. doi: 10.1016/j.ijpharm.2015.06.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.