

Abstract
Gold nanoparticles (Au-NPs) have been designated as superior tools for biological applications owing to their characteristic surface plasmon absorption/scattering and amperometric (electron transfer) properties, in conjunction with low or no immediate toxicity towards biological systems. Many studies have shown the ease of designing application-based tools using Au-NPs but the interaction of this nanosized material with biomolecules in a physiological environment is an area requiring deeper investigation. Immune cells such as lymphocytes circulate through the blood and lymph and therefore are likely cellular components to come in contact with Au-NPs. The main aim of this study was to mechanistically determine the functional impact of Au-NPs on B-lymphocytes. Using a murine B-lymphocyte cell line (CH12.LX), treatment with citrate-stabilized 10 nm Au-NPs induced activation of an NF-kB-regulated luciferase reporter, which correlated with altered B lymphocyte function (i.e. increased antibody expression). TEM imaging demonstrated that Au-NPs can pass through the cellular membrane and therefore could interact with intracellular components of the NF-kB signaling pathway. Based on the inherent property of Au-NPs to bind to –thiol groups and the presence of cysteine residues on the NF-kB signal transduction proteins IkB kinases (IKK), proteins specifically bound to Au-NPs were extracted from CH12.LX cellular lysate exposed to 10 nm Au-NPs. Electrophoresis identified several bands, of which IKKa and IKKb were immunoreactive. Further evaluation revealed activation of the canonical NF-kB signaling pathway as evidenced by IkBa phosphorylation at serine residues 32 and 36 followed by IkBa degradation and increased nuclear RelA. Additionally, expression of an IkBa super-repressor (resistant to proteasomal degradation) reversed Au-NP-induced NF-kB activation. Altered NF-kB signaling and cellular function in B-lymphocytes suggests a potential for off-target effects with in vivo applications of gold nanomaterials and underscores the need for more studies evaluating the interactions of nanomaterials with biomolecules and cellular components.
1. Introduction
With the advent of nanomaterials came the realization of their enormous potential to many interdisciplinary sciences, especially nano-medicine. The specific properties of nanomaterials (electronic, mechanical, physicochemical) have shown to be primarily dependent on their shape and dimensions.1 Of all the metallic nanoparticles available today, gold has attracted special interest owing to its plasmonic properties in conjunction with low or no immediate toxicity towards biological systems. Moreover, the ease of synthesis and chemical stability, adds to the overall advantages of gold nanoparticles (Au-NPs). Over the years, the primary focus has been to evaluate the material properties of nanoparticles (NPs), which led to the development of state of the art synthesis and characterization techniques. However, with the ever-growing increase in the nano-bio applications of Au-NPs, there is a need to understand their effect on biological systems. Immune cells such as lymphocytes circulate through the blood and lymph and therefore are likely cellular components to come in contact with Au-NPs. Additionally, the non-inert nature of these nanomaterials has been revealed in both in vitro and in vivo studies demonstrating toxic effects or altered cellular function following Au-NP exposure.2–8 Indeed Au-NPs are chemically active towards biomolecules9–13 and have the capability to alter the fate of chemical reactions and modulate cellular signaling pathways and cellular function.
NF-κB/Rel proteins represent a family of transcription factors that not only regulate inflammatory responses but also the expression of a wide variety of genes encompassing a spectrum of cellular activities such as proliferation, differentiation, and apoptosis.14,15 NF-κB activation is mediated through at least three major pathways, ‘canonical’ (or classical), ‘non-canonical’ (or alternate), or ‘atypical’, which differ in phosphorylation and degradation patterns of IκBα, primary NF-κB dimers formed (i.e. p50/RelA, p50/cRel vs. p52/RelB), and specific signal transducing proteins.14,15 Inhibitory κB kinase (IKK) plays a pivotal role in intracellular signaling and is a point of convergence for a large number of signaling cascades including the activation of the NF-κB pathway.14 The IKK complex consists of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ). In the canonical pathway, activation of IKK by an inducing signal (e.g. lipopolysaccharide) leads to phosphorylation of IκBα at serines 32 and 36 followed by ubiquitination and proteosomal degradation of IκBα. IκBα degradation releases NF-κB dimers (i.e. p50/RelA), which translocate to the nucleus and elicit initiation of gene transcription (Fig. 10).16–19
Fig. 10.
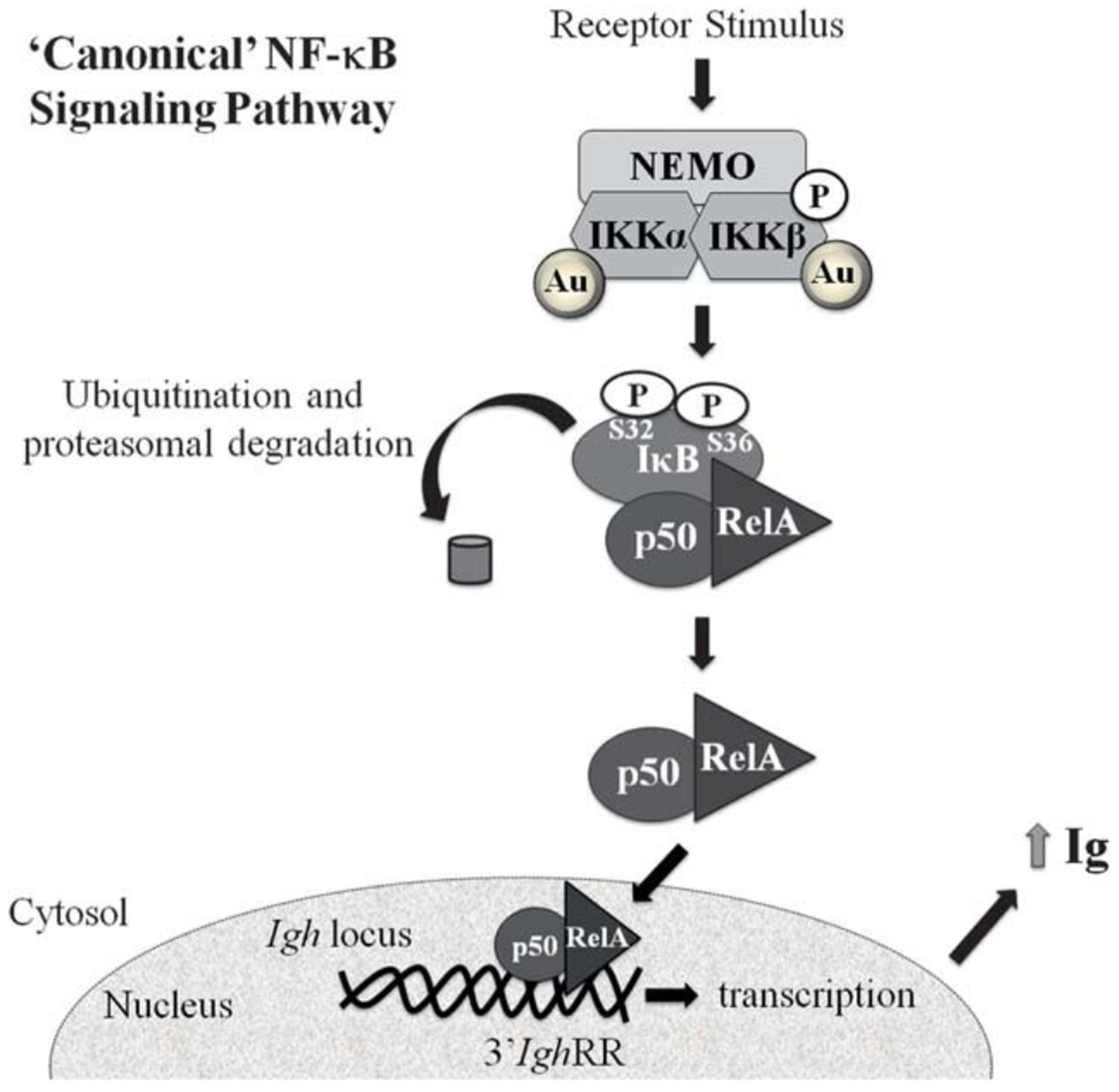
Proposed mechanism of NF-κB activation and altered Ig expression by Au-NPs. The present study supports 3’IghRR activation and increased Ig expression by 10 nm Au-NPs that is mediated through activation of the ‘canonical’ NF-κB signaling pathway. A physical interaction between 10 nm Au-NPs and IKKα and IKKβ was also demonstrated, which may play a role in Au-NP-induced NF-κB activation.
Gold compounds have long been used as anti-inflammatory agents. Although the exact mechanism is unclear, gold compounds have been shown to inhibit NF-κB and IKK activity.20–24 Immune function, including the regulation of humoral immunity, has been linked to NF-κB/Rel activity.25 B- lymphocytes, the main effector cells of humoral immunity, secrete antibodies that bind antigens or invading pathogens and non-self molecules with specificity and facilitate the clearance of these antigens from the host. Antibody production in B-lymphocytes may be partially mediated through NF-κB/Rel binding to kB motifs in enhancer regions that regulate tran- scription of the immunoglobulin (Ig) heavy and light chains (primary components of antibodies). Within the Ig heavy chain (Igh) gene, a transcriptional regulatory region 3’ of the heavy chain constant regions (referred to as 3’IghRR) mediates Igh expression and therefore overall antibody levels; furthermore, 3’IghRR activity is influenced by NF-κB.26–28
The main aim of this study was to determine the impact of Au-NPs on B-lymphocytes. Because of the inherent property of gold to bind to –thiol groups, we hypothesized that potential protein interactions with Au-NPs could alter both NF-kB signaling pathways and antibody expression. A review of the available literature suggests that proteins adsorb to the NP surface depending on the material properties such as charge, size, hydrophobicity, and conductivity.29 Utilizing a well-characterized murine B-lymphocyte cell line (CH12.LX), we identified intracellular 10 nm Au-NPs through the use of TEM, demonstrating that the Au-NPs could cross the cellular membrane and physically interact with cellular proteins. Transient transfection studies utilizing an NF-κB-regulated luciferase reporter demonstrated an increase in NF-κB activity following Au-NP treatment of the CH12.LX cells. In CH12.γ2b-3’IghRR cells (a CH12.LX variant), ELISA analysis of Ig expression showed a correlation between Au-NP-induced activation of NF-κB and an increase in both 3’IghRR activity and IgA expression suggesting a potential for Au-NP-mediated immune dysfunction.
Further mechanistic analysis demonstrated a physical interaction between Au-NPs and IKKa and IKKb in the cellular lysate, likely due to the Au-NPs binding cysteine residues in IKK.30,31 Additionally Au-NP treatment of the CH12.LX cells induced IκBα phosphorylation at serines 32 and 36 followed by IκBα degradation and nuclear translocation of RelA. These results suggest the activation of the ‘canonical’ NF-κB pathway, which was confirmed using the CH12.IκBαAA cell line (variant of CH12.LX) that expresses an inducible IκBα super-repressor. Expression of the IκBα super-repressor inhibited Au-NP-induced activation of NF-kB. Taken together, these results demonstrate that not only are B-lymphocytes a sensitive and direct target of Au-NPs, but further investigation of Au-NPs and their interactions with intracellular proteins is warranted.
2. Experimental
Cell lines and culture conditions
The CH12.LX B-cell line was derived from the murine CH12 B-cell lymphoma32 and has been extensively utilized to study a variety of cellular processes specific to B-lymphocytes. The CH12.γ2b-3’IghRR cell line is a subclone isolated from CH12.LX cells that stably express a transgene (γ2b Igh gene) regulated by the 3’IghRR.33,34 The CH12. IκBα AA B-cell line, developed and generously provided by Dr Gail Bishop,35 is a variant of the CH12.LX cell line that stably expresses an IPTG (isopropyl b-d-1-thiogalactopyranoside; Sigma-Aldrich)-inducible IκBα super repressor (IκBα AA) resistant to degradation and negative feedback regulation by NF-κB/Rel proteins.35 All cells were grown in RPMI 1640 media (MediaTech, Herndon, VA) supplemented with 10% bovine calf serum (Hyclone, Logan, UT), 13.5 mM HEPES, 100 units per mL penicillin, 100 mg mL−1 streptomycin, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, and 50 mM β-mercaptoethanol. Cells were maintained at 37 °C in an atmosphere of 5% CO2. Cell viability was determined by assaying 1.0 mL of cell suspension for Trypan Blue exclusion staining with a Beckman Coulter ViCell instrument (Beckman Coulter, Brea, CA).
Synthesis and characterization of gold nanoparticles (Au-NPs)
Tetrachloroauric(iii) acid monohydrate (~52% Au) was purchased from MP Biomedicals. Sodium citrate was purchased from Fischer Scientific. Au-NPs were synthesized by the Turkevich method36 modified to obtain the 10 nm size. Briefly, an aqueous solution of 1 mM HAuCl4 was brought to boil with vigorous stirring. The concentration of sodium citrate solution (58.2 mM) was adjusted to obtain the desired 10 nm size of Au-NPs, and then 10 mL of sodium citrate solution was added to the boiling HAuCl4 solution with stirring and boiled for 10 min. The solution was stirred rapidly for an additional 15 min without heating. The solution was then brought to boil again and 5 mL of additional sodium citrate solution was added, after which the solution was stirred vigorously for an additional 10 min without heating. The synthesized Au-NP solution was characterized for size distribution and dispersion using Ultra-Violet-Visible spectroscopy (UV-Vis, Varian Cary 300 spectrometer), Dynamic Light Scattering (DLS, Malvern Zetasizer Nano), and Transmission Electron Microscopy (TEM, Hitachi H-7600). Nanoparticle suspensions (7 mL) were dried on formvar/carbon film-coated Cu grids (Canemco Inc. Canada) for TEM imaging.
Cellular uptake of Au-NPs
Uptake of Au-NPs by the CH12.LX cells was determined as previously described.37 Briefly, CH12.LX cells were treated with 50 mg mL−1 of 10 nm Au-NPs for different intervals of time (0, 1, 3, 4 and 24 h). Cells were then centrifuged at 3000 rpm and washed with 1× PBS. The cell pellet was then fixed using a 2% solution of 1 : 1 paraformaldehyde : gluteraldehyde. After washing with 1x PBS, the pellet was stained with osmium tetroxide for visualization of the NPs. The pellet was then washed 5 times with 1× PBS. After a series of dehydration steps with ethanol (50%, 70%, 90% and 100%), the pellet was cured in LR white resin (Electron Microscopy Sciences, Hatfield, PA) overnight at 70 °C. The resin embedded pellet was then sectioned using a microtome and imaged by TEM.
Transient transfection and luciferase assay
Cells (1.0 × 107) were resuspended in 200 mL of culture media with 10 mg of 3x-NF-κB plasmid and transferred to a 2 mm gap electroporation cuvette (Molecular BioProducts, San Diego, CA). Cells were electroporated using an electro cell manipulator (ECM 630; BTX, San Diego, CA) with the following voltage, capacitance, and resistance: 250 V, 150 mF, and 75 U.38 For each experiment, multiple transfections with the 3x-NF-κB plasmid were pooled in fresh media at 2.0 × 105 cells per mL. Immediately after transfection, the CH12.LX cells were treated with 0.05, 0.5 and 5 mg mL−1 of Au-NPs for 24 or 48 h. For the CH12. IκBα AA cell line, the transfected cells were immediately divided in half and half was treated with 100 mM IPTG for 2 h to activate the IPTG-inducible IκBα AA transgene while the other half was cultured in the absence of IPTG to provide a control that lacked IκBα AA transgene expression. After the 2 h IPTG pre-treatment, the CH12. IκBα AA cells were incubated for 24 h in the absence (negative control) or presence of 5 mg mL−1 Au-NPs. Following the appropriate incubation period, the transfected CH12.LX and CH12. IκBα AA cells were lysed with 1x reporter lysis buffer (Promega, Madison WI) and stored at −80 °C. To measure luciferase activity, the whole cell lysate was thawed and cleared of cellular debris by centrifugation at 14 000 rpm then 20 mL of whole cell lysate was mixed with 100 mL of luciferase assay reagent (Promega) and luciferase activity was measured as relative light units (RLU) with a lumin- ometer (Berthold detection systems). Results are represented relative to the naive control, which was set to 100 percent effect.
ELISA for γ2b and IgA expression
CH12.γ2b-3’IghRR cells were treated with varying concentrations of 10 nm Au-NPs then seeded (2 mL per well) into 48-well plates. To evaluate γ2b protein expression, cells were seeded at a concentration of 3.0 × 104 cells per mL and incubated for 48 h. For IgA protein detection, cells were seeded at a concentration of 2.0 × 105 cells per mL and incubated for 24 h. Following the incubation period, cells were centrifuged at 3000 rpm then lysed with mild lysis buffer (MLB, 1% NP-40, 150 mM NaCl, 10 mM NaPO4, 2 mM EDTA) containing freshly added Complete Mini Protease Inhibitor Cocktail in a 1 : 3 ratio of pellet to MLB. Lysed cells were cleared via centrifugation at 14 000 rpm for 5 min. Supernatants were collected and stored at −80 °C until analysis. To measure γ2b or IgA, cell lysates were thawed on ice and protein concentrations were determined by the Bio-Rad Protein Assay (Bio Rad, Hercules, CA). Samples were diluted to 2 or 3 mg of total protein per 100 mL and analyzed for IgA or γ2b, respectively, by ELISA as previously described33 (all antibodies were purchased from Bethyl Industries, Inc.).
Protein corona analysis from Au-NP surface
Fresh whole cell lysate was isolated from 1.5 × 107 CH12.LX cells using MLB as described above. 10 nm Au-NPs (100 mg mL−1) were added to 1 mL of the cleared lysate and incubated at room temperature with rotation for one hour. Samples were then centrifuged at 14 000 rpm for 10 min. The Au-NP pellet was washed with 1x PBS four times and centrifuged for 7 min at 14 000 rpm between washes. For the final wash, the Au-NP pellet was then transferred to a new 1.5 mL Eppendorf and centrifuged a final time. The supernatant was discarded and 50 mL of 2x loading dye (50 mM Tris–HCl, 10% glycerol, 4% SDS, and 0.4% of 14.2 M stock β-mercaptoethanol) was added to the Au-NP pellet and boiled at 95 °C for 7 min then vortexed for 30 seconds and centrifuged at 14 000 rpm for 10 min. Both the supernatant and Au-NP pellet was collected for further analysis (i.e. UV-Vis, TEM, and Western blot).
Protein isolation
CH12.LX cells (1.0 × 107 cells per treatment) were treated with 5 mg mL−1 of 10 nm Au-NPs over an 8 h time course. Every 15 min, cells were pelleted via centrifugation at 3000 rpm at 4 °C and the supernatant was discarded. Whole cell lysate was isolated using MLB as described above but with the addition of PhosStop Phosphatase Inhibitor Cocktail tablets (Roche Diagonostics, Indianapolis, IN). Nuclear and cytoplasmic fractions were isolated as previously described39 but with the following modifications. The cell pellet was washed twice with 1x PBS and then resuspended in 50 mL ice cold buffer A (10 mM Hepes pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT) and centrifuged at 800 × g for 10 min at 4 °C. The supernatants were collected as cytoplasmic extracts and the pellets were resuspended in 50 mL buffer B (20 mM Hepes pH 7.9, 400 mM NaCl, 1.5 mM MgCl2, 0.1 mM EDTA, 5% glycerol, 0.5 mM PMSF, 0.5 mM DTT) and incubated for 20 min on ice then centrifuged at 10 000 × g for 30 min at 4 °C. The supernatants were collected as nuclear extracts. Protein concentrations were determined using the Bio-Rad Protein Assay. Loading dye was added to 50 mg of protein to a final concentration of 1x and Western blot analysis was performed.
SDS-PAGE and Western blot analysis
Whole cell lysates, cell fractions and the supernatant from the boiled Au-NP pellet were thawed on ice and 50 mg of protein from both the whole cell lysates and cell fractions and 40 mL of eluent from the Au-NPs were electrophoresed through a Mini-PROTEAN TGX 4–20% gradient polyacrylamide gel (Bio-Rad) at 130 volts for 90 min. The proteins were then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA) using an electric current of 100 volts for 75 min. For the evaluation of the Au-NP protein corona, the membrane was immediately washed two times with double distilled water and stained with Sigma Aldrich’s Reversible Protein Detection Kit for Membranes and Polyacrylamide Gels per manufacturer’s directions and imaged using a Fotodyne Foto/Eclipse imager with incandescent background lighting. The membrane was destained and immersed in 3% BSA (bovine serum albumin)–TTBS (tris-buffered saline with 0.05% Tween-20) blocking solution and rocked overnight at 4 °C. The membrane was then incubated at room temperature for 1 h with rabbit anti-IKKα (ab47453, Abcam Cambridge, MA) or anti-IKKβ (ab55404, Abcam), at a 1 : 1000 dilution in 3% BSA–TTBS. The membranes with whole cell lysate and cell fractions, were blocked and then incubated at room temperature for 1 h with either anti-IκBα (sc-371, Santa Cruz Laboratories, Santa Cruz, CA); anti-IκBα phosphorylated S32/36 (ab12135, Abcam); anti-IkBa phosphorylated Y42 (ab24783); anti-RelA (A301-824A, Bethyl Laboratories, Montgomery, TX); anti-p84 (5E10, Genetex, Simpson, PA); or anti b-actin (A5316, Sigma-Aldrich), at a 1 : 1000 (1 : 500 for anti-IκBα phosphorylated S32/36) dilution in 3% BSA–TTBS. After the primary antibody incubation, the blots were washed four times in TTBS at 10 min intervals then incubated with HRP-conjugated secondary antibody (goat anti-rabbit at 1 : 2500 or goat anti-mouse at 1 : 1000) for 1 h. The blots were washed four times in TTBS, then subjected to ECL substrate (Thermoscientific, Waltham, MA) and analyzed on a Fuji LAS-3000 Bioimager (Tokyo, Japan).
Statistical analysis
Luciferase and ELISA data (n ¼ 3 for each treatment group) was normalized to the naive control (set to 100% effect) and the means generated from several experiments were then averaged and represented as the mean ± SEM. As indicated in the figure legends, significant differences at p < 0.05 or p < 0.01 compared to the naive control were determined by a 1-way ANOVA with a Dunnett’s post-hoc test.
3. Results and discussion
Characterization and cellular uptake of 10 nm Au-NPs
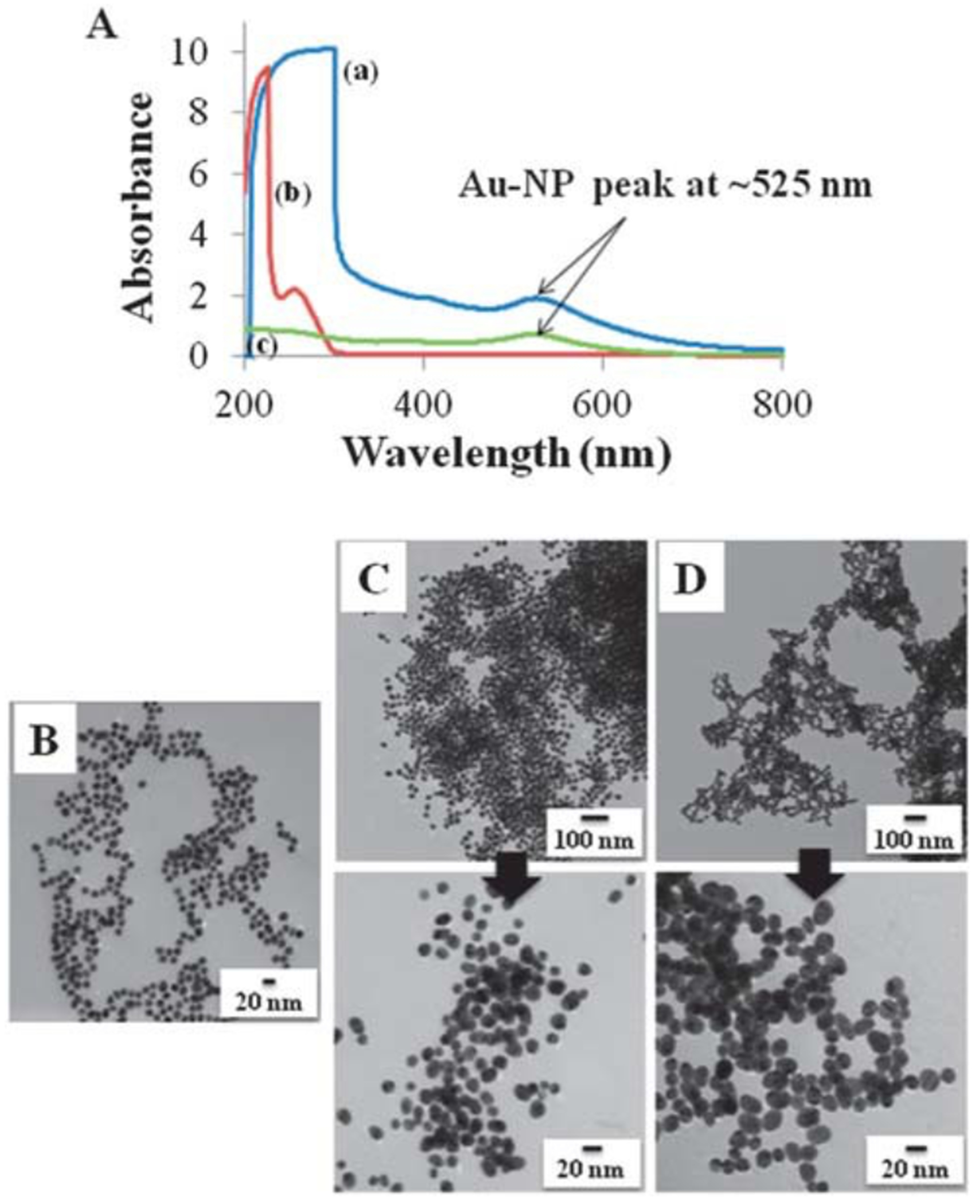
Citrate-stabilized Au-NPs were synthesized and characterized by TEM, DLS and UV-Vis (Fig. 1 and Table 1). The Au-NPs were well dispersed and showed a size distribution of 9 ± 1.5 nm (designated as 10 nm in this manuscript) as determined by TEM (Fig. 1A and Table 1). The hydrodynamic diameter of Au-NPs was 9.7 ± 0.047 nm, as determined by DLS (Table 1) with a polydispersity index (PDI) of 0.352 confirming a uniform size distribution. UV-Vis showed the absorbance spectra of ~525 nm, distinctive to Au-NPs (Fig. 1B).
Fig. 1.
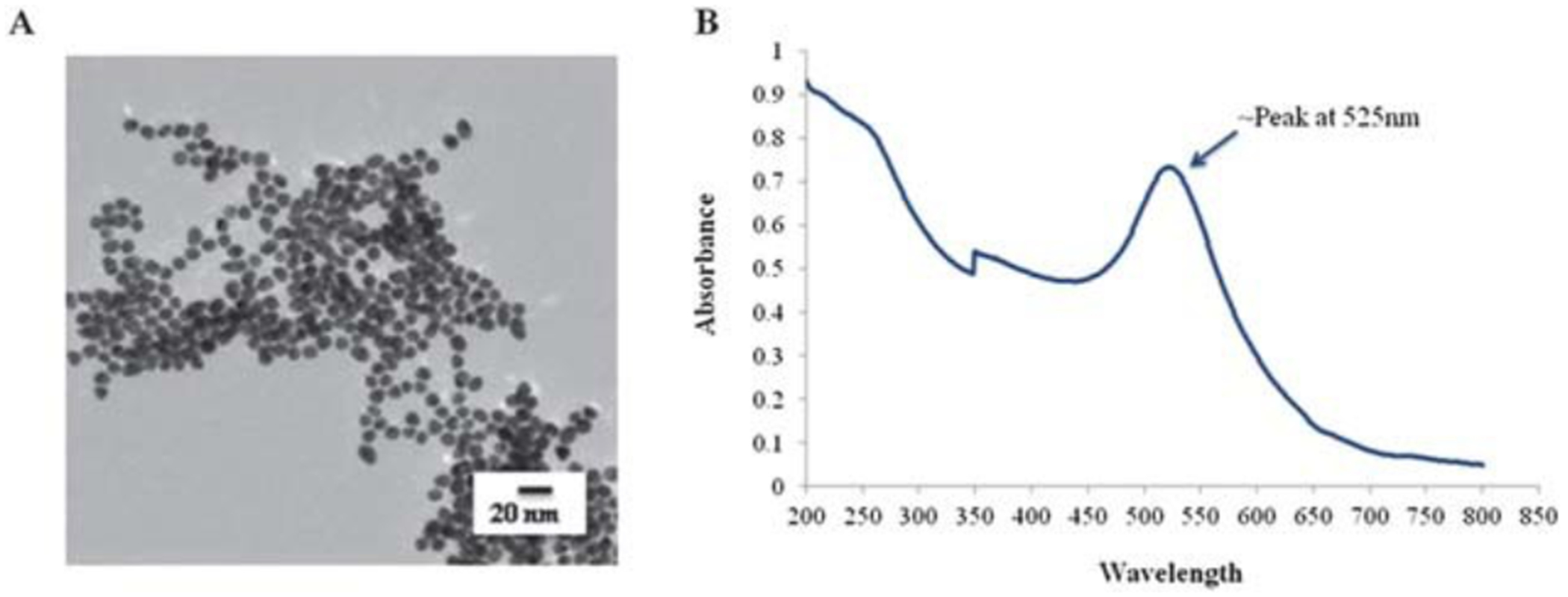
Characterization of gold nanoparticles. Au-NPs were synthesized by the Turkevich method36 but modified to obtain an ~10 nm size. (A) Nanoparticle suspensions were dried on formvar/carbon film-coated Cu grids for TEM analysis of the particle morphology and size. (B) UV-Vis absorption spectra of Au-NPs showing absorption peak at ~525 nm.
Table 1.
Size assessment of citrate-stabilized ~10 nm gold nanoparticlesa
| TEM | DLS | PDI | |
|---|---|---|---|
| Citrate stabilized Au-NPs | D: 9 ± 1.5 nm | DH: 9.7 ± 0.047 nm | 0.352 |
The hydrodynamic diameter (DH) of citrate-stabilized Au-NPs, determined by Dynamic Light Scattering (DLS), confirmed the diameter (D) obtained by TEM. The Polydispersity Index (PDI) was determined to assess uniformity in size distribution of Au-NPs.
Previous studies have demonstrated that Au-NPs can cross the phospholipid bi-layer of the cellular membrane, and localize within intracellular compartments.40–42 To determine the extent of Au-NP uptake into B-lymphocytes, we utilized a B-lymphoma cell line, CH12.LX, and incubated these cells with 50 mg mL−1 of Au-NPs for 1, 3, 4 or 24 h time period. TEM imaging identified cytoplasmic localization of Au-NP by 4 h, which appeared to increase by 24 h (Fig. 2). Aggregates of Au-NPs were observed in membrane-bound cellular organelles, presumably suggesting endoplasmic localization (Fig. 2C). Monodispersed Au-NPs were also seen in the cellular cytoplasm (Fig. 2, thin arrows). Interestingly, there was no obvious impact of Au-NP treatment on cell morphology, viability (data not shown), or proliferation (data not shown) at the tested Au-NP concentrations.
Fig. 2.
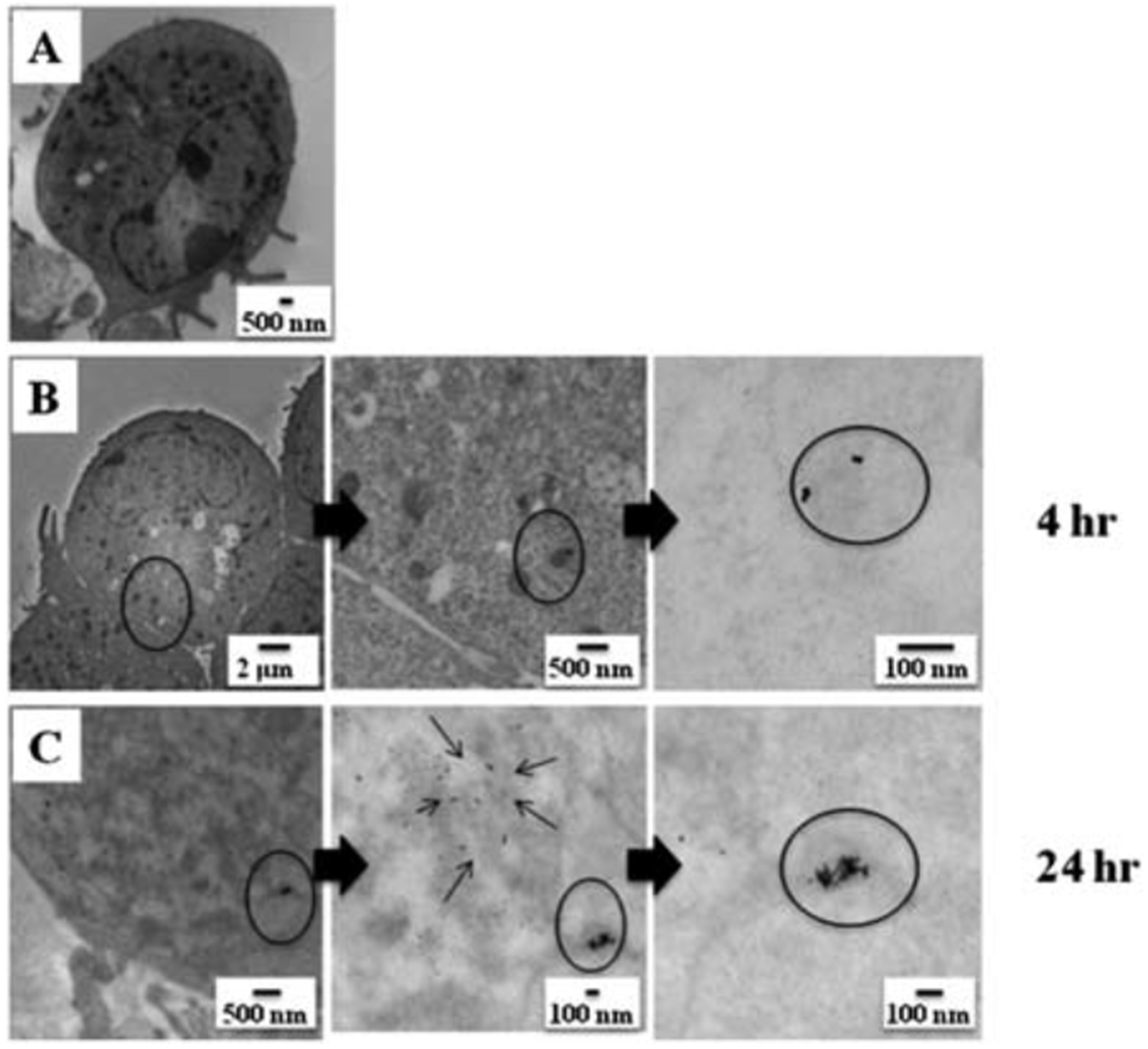
Uptake of gold nanoparticles by CH12.LX cells. CH12.LX cells (5 × 105 cells per mL) were treated with 50 mg mL−1 of 10 nm Au-NPs for varying time intervals (0, 1, 3, 4 or 24 h) then characterized by TEM. Control cell in the absence of Au-NPs (A) and cells incubated with Au-NPs for 4 h (B) or 24 h (C). TEM images are representative of two separate experiments (n ¼ 3 for each treatment and time point).
Au-NPs activate the NF-κB pathway in a B-lymphocyte cell line
A limited number of studies using murine and human primary cells and cell lines have evaluated the effect of either gold compounds or gold nanoparticles on NF-κB activation. These studies demonstrated a decrease in NF-κB activity as evidenced by decreased nuclear translocation and/or protein–DNA binding.20–24,43,44 Using a 3x-NF-κB luciferase reporter plasmid, we evaluated the effect of Au-NPs on NF-κB activity in the CH12.LX B-lymphocyte cell line, which was treated with varying concentrations of 10 nm Au-NPs for 24 or 48 h. Surprisingly, Au-NP significantly induced 3x-NF-κB reporter activity at both time points and this effect was limited to the highest Au-NP concentration (5 mg mL−1) with no effect at lower concentrations (Fig. 3A and B). These results contrast with the above studies identifying a decrease in NF-κB activity. This difference may be attributed to differences in cellular models and/or treatment conditions. For instance a majority of the previous studies utilized murine monocyte/macrophage cellular models but endothelial cells, kidney cells, and a mixed lymphocyte cell population were also utilized.20–24,43,44 Additionally, all of the studies except one with human primary lymphocytes24 evaluated the effects of gold compounds or Au-NPs on NF-κB activity induced by LPS (lipopolysaccharide), TPA (12-O-tetradecanoylphorbol-13-acetate), TNFα (tumor necrosis factor α) or RANKL (receptor activator of nuclear factor-κB ligand) rather than basal activity which may reflect the higher basal activity of NF-κB in lymphocytes as compared to other cell types. Furthermore, only two studies evaluated Au-NPs (both used monocyte/macrophage cellular models) and these particles were either a larger size (i.e. 150 nm vs. 10 nm in our study) or the same size but coated with polyethylene glycol.43,44 Certainly size and surface properties could alter the effects of Au-NPs and NF-κB signaling.
Fig. 3.
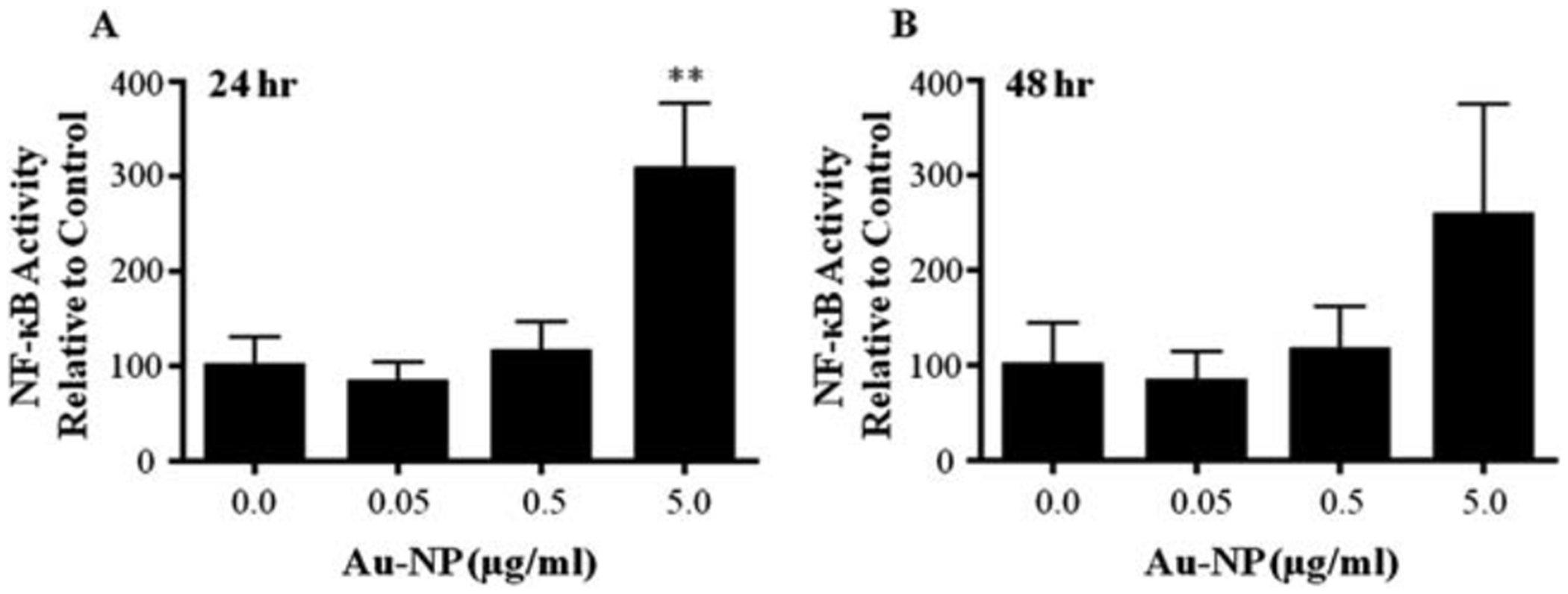
Au-NPs activate the NF-κB signaling pathway. CH12.LX cells were transiently transfected with the 3x-NF-κB luciferase reporter plasmid then cultured at 2.0 × 105 cells per mL with varying concentrations (0.0, 0.05, 0.5 and 5 mg mL−1) of 10 nm Au-NPs. After a 24 h (A) or 48 h (B) incubation, cells were lysed and cellular extracts evaluated for luciferase enzyme activity as measured in relative light units (RLU) and represented on the y-axis as NF-κB activity (mean ± SEM) relative to the naïve control (0.0 mg mlL−1 Au-NP, set to 100%). Mean ± SEM was generated from three separate experiments. ** Statistical significance at p < 0.01 compared to 0.0 mg ml−1 Au-NP control via 1-way ANOVA and a Dunnett’s post-hoc test.
The importance of NF-kB signaling in cellular function underscores the need to clarify the effect of Au-NPs of varying size and surface properties on multiple cell types particularly in light of the limited number of studies currently available and the increasing commercial and medical use of compounds that contain NPs. Additionally, effects of nanomaterials on NF-κB signaling are not limited to gold nanoparticles or to inhibition of NF-κB activity as seen previously in monocyte/macrophage models.43,44 Carboxylated single-walled carbon nanotubes (1–2 nm diameter, 5–30 mm length) have been shown to induce NF-κB nuclear localization and binding to DNA in primary monocytes.45 Therefore, the NF-κB signaling pathway may be a universal target of nanomaterials and the multifaceted biological effects of NF-κB underscore the importance of fully characterizing the effects of nanomaterials on this essential cellular pathway.
Au-NPs increase IgA production and 3’IghRR activity in a B-lymphocyte cell line
The primary function of B-lymphocytes is to produce antibodies (i.e. secreted Ig) with specificity for antigens or non-self molecules. Antibodies opsonize the antigen to facilitate host clearance by other cells of the immune system. Antibodies are polypeptides encoded from the heavy- and light-chain Ig genes and each antibody is composed of two identical heavy chain proteins (IgH) and two identical light chain proteins. One of the transcriptional regulators of IgH expression is the 3’Igh regulatory region (3’IghRR) and there are multiple NF-κB binding sites within the 3’IghRR that have been shown to play a role in 3’IghRR activity.26–28 To determine if treatment with Au-NP leads to a functional effect on Ig transcriptional regulatory elements (i.e. 3’IghRR) and on endogenous antibody production, we utilized a variant of the CH12.LX B-lymphocyte cell line (CH12.γ2b-3’IghRR) that stably expresses an Ig heavy chain (γ2b) under the transcriptional regulation of the 3’IghRR. The CH12.γ2b-3’IghRR cells have been previously characterized and shown to endogenously express IgA antibodies as well as one copy of the γ2b transgene.33
The CH12.γ2b-3’IghRR cells were treated with varying concentrations of 10 nm Au-NPs and evaluated by ELISA for γ2b and IgA protein levels. Similar to the effect on NF-kB, 5 mg mL−1 Au-NP significantly increased 3’IghRR-regulated γ2b transgene expression (Fig. 4A). IgA levels were also significantly increased but at a lower concentration (0.5 mg mL−1) of Au-NP suggesting an enhanced sensitivity of IgA to Au-NPs perhaps through an effect on both heavy and light chain expression or a post-transcriptional modification (Fig. 4B). It is interesting that a classical concentration response curve is not produced for any of the above endpoints (i.e. NF-κB and 3’IghRR activation or IgA expression). The response appears to be threshold-dependent and exhibits an effect at a single concentration. It may be that a more typical concentration response could be attained with a finer concentration range instead of the current log range.
Fig. 4.
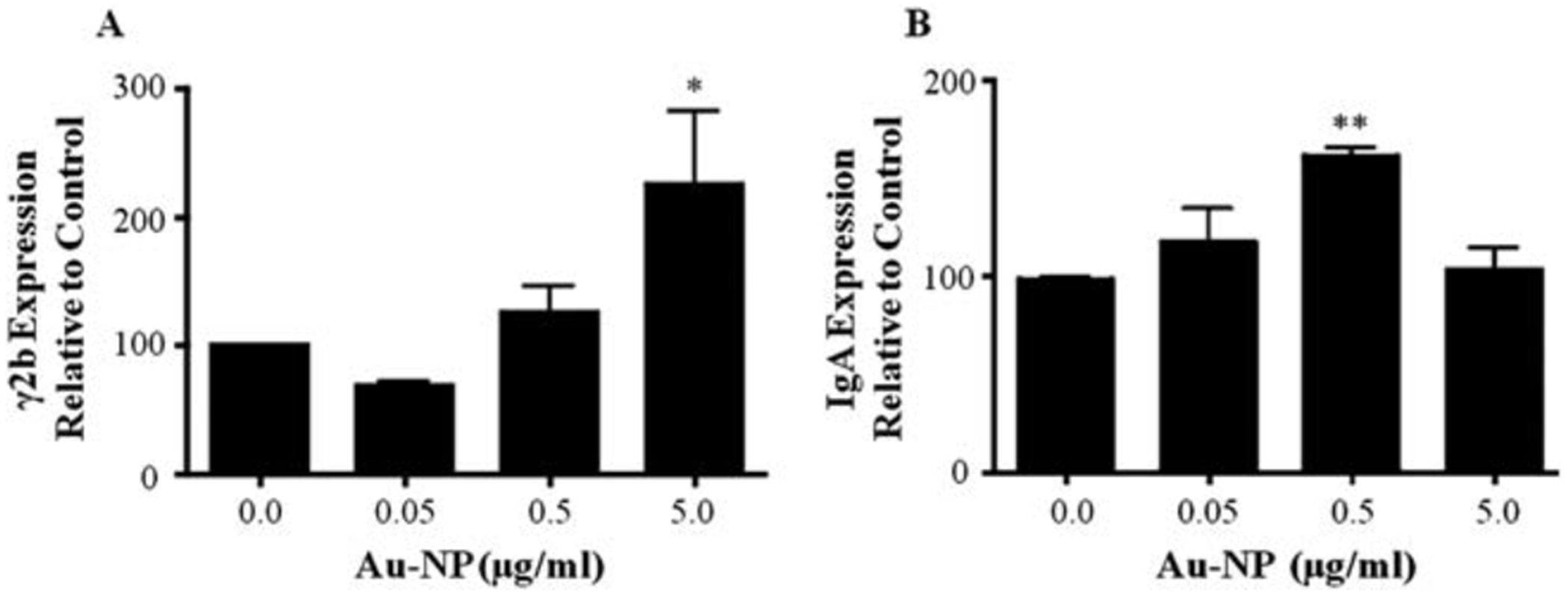
Au-NPs increase 3’IghRR-regulated γ2b transgene expression and IgA secretion. CH12.γ2b-3’IghRR cells at 3.0 × 104 cells per mL were cultured with varying concentrations (0.0, 0.05, 0.5 and 5 mg mL−1) of 10 nm Au-NPs. After a 48 h (γ2b) or 24 h (IgA) incubation, cells were lysed and diluted to 3 or 2 mg whole cell protein per 100 mL and evaluated by ELISA to determine ng of γ2b per 3 mg protein (A) or ng of IgA per 2 mg protein (B). Expression of γ2b or IgA (mean ± SEM) relative to the naïve control (0.0 mg mL−1 Au-NP, set to 100%) is represented on the y-axis. Mean ± SEM was generated from three separate experiments. * and **, statistical significance at p < 0.05 or p < 0.01, respectively, compared to 0.0 mg mL−1 Au-NP control via 1-way ANOVA and a Dunnett’s post-hoc test.
Regardless, we have demonstrated an effect of Au-NPs on NF-κB signaling that translated into an effect on cellular function. Whether the exhibited change in Ig expression would translate into an altered immune response is unknown. However, it is noteworthy that gold compounds which are typically used as immunosuppressive agents have also been shown to induce the expression of auto-antibodies in patients.46 Additionally, animal models treated with gold compounds have developed autoimmune-like disease and an increase in Ig expression.47,48 Since these immuno-pathological effects appear to have a genetic linkage,47 it is reasonable to speculate that a direct effect of Au-NPs on Ig expression as demonstrated here, could induce or exacerbate the autoimmune effects in susceptible individuals.
IKK signal transduction proteins adsorb to Au-NPs
Since the 10 nm Au-NPs are taken up by the CH12.LX B cells (Fig. 2), Au-NPs could interact with membrane-bound and/or intracellular proteins potentially altering signal transduction pathways and normal cellular function. To determine if Au-NPs and cellular proteins physically interact, the dispersion and aggregation pattern of Au-NPs before and after incubation with whole cell protein lysate was assessed using UV-Vis spectroscopy and TEM. Cell lysate from CH12.LX cells, was incubated with Au-NPs for 1 h then either analyzed immediately by UV-Vis (Fig. 5A, curve a) or after proteins that specifically bound the Au-NPs were extracted (Fig. 5A, curve b). The UV-Vis absorption spectra demonstrated a characteristic peak for 10 nm Au-NPs at ~525 nm for the samples containing Au-NPs (compare Fig. 5A, curves a and c to Fig. 1B). Peaks at lower wavelengths likely represent proteins (Fig. 5A, curves a and b) since these peaks are not present when the Au-NPs are analyzed in the absence of cell lysates (Fig. 5A, curve c). Interestingly, proteins extracted from the Au-NP induced more distinct peaks as compared to the whole cell lysate (Fig. 5A, compare curves a and b), which likely reflects selective binding of specific proteins to the Au-NPs. Additionally, no absorption for Au could be seen in the protein sample extracted from the Au-NP pellet after heating under reducing conditions (Fig. 5A, curve b), indicating little or no contamination of the sample with Au-NPs. These results suggest that a protein corona is formed on the surface of the Au-NP which is further supported by TEM analysis of Au-NPs before and after incubation with whole cell protein lysate. After incubation with cell lysate, the Au-NPs are less agglomerated perhaps due to a protein corona which keeps them separated from each other (Fig. 5C). Once the proteins are removed from the surface of the particles by heating under reducing conditions, the Au-NPs agglomerate (Fig. 5D). This agglomeration of Au-NPs can be attributed to the absence of the stabilizing agent (i.e. sodium citrate or protein corona).
Fig. 5.
Protein corona characterization using UV-Vis spectroscopy and TEM imaging. (A) Whole cell lysate from CH12.LX cells was incubated with 10 nm Au-NPs for 1 h then analyzed by UV-Vis either immediately (curve a) or following extraction of proteins that specifically bound the Au-NPs (curve b). Curve c represents the Au-NPs alone control (absence of cell lysate). (B–D) Corresponding TEM imaging for Au-NPs prior to (B) and after (C) incubation with whole cell lysate from CH12.LX cells and following removal of the protein corona (D). Results are representative of three separate experiments.
To further investigate the association of Au-NPs with intracellular proteins, CH12.LX proteins that specifically associated with Au-NPs were eluted by heating under reducing conditions and resolved by SDS-PAGE and Western blot analysis. Reversible membrane staining revealed several protein bands, suggesting a broad spectrum of proteins binding to the Au-NPs (Fig. 6A, lane 3). However, there was less banding as compared to the whole cell lysate control (Fig. 6A, compare lanes 2 and 3) supporting protein specificity for the Au-NPs. Additionally, the absence of detectable bands in the Au-NP only (in lysis buffer) control (Fig. 6A, lane 4) demonstrated that the elucidated bands are not merely an artifact from the Au-NPs or the protein extraction process.
Fig. 6.
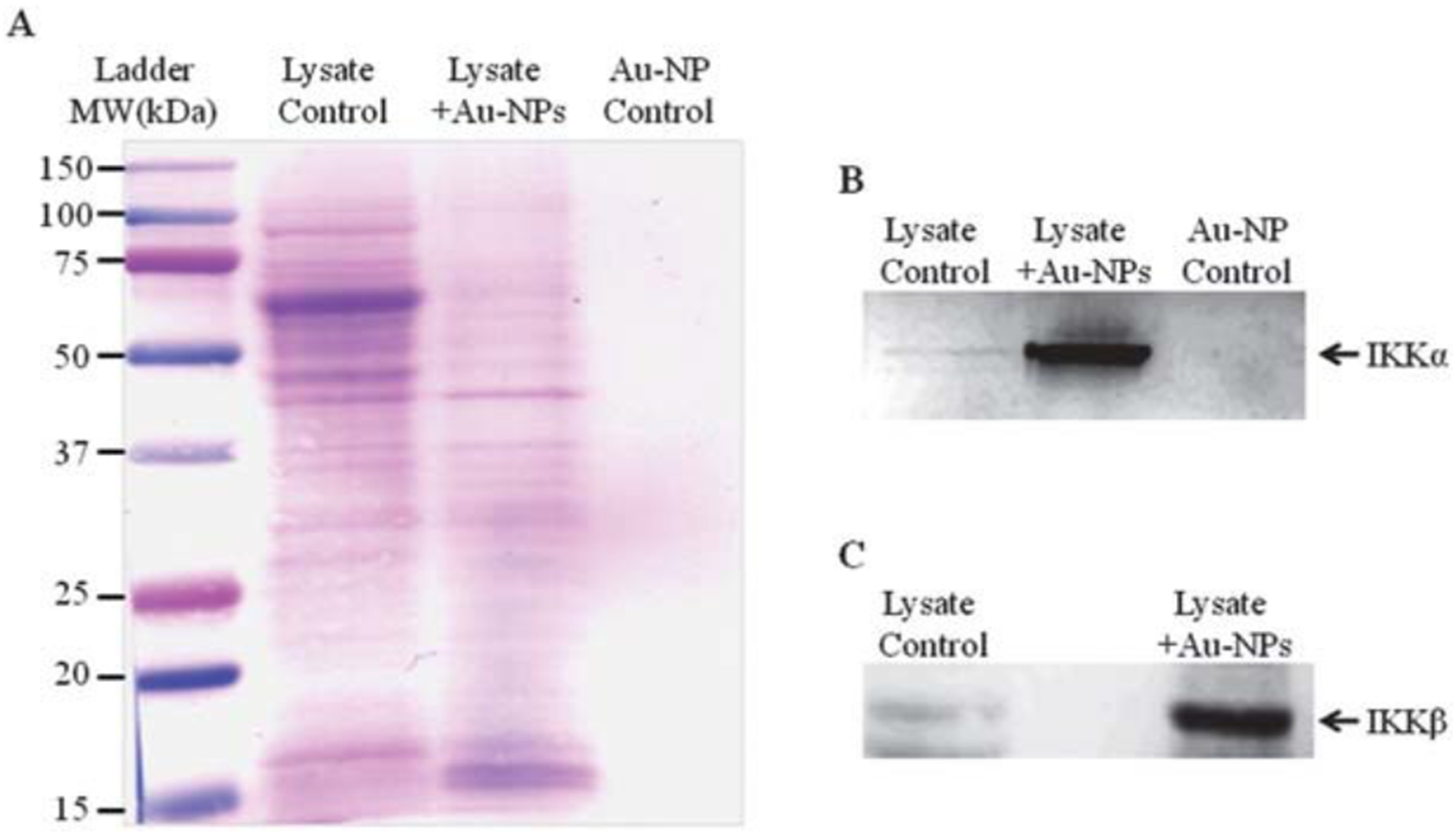
The Au-NP protein corona contains both IKKα and IKKβ. Whole cell lysate from CH12.LX cells was incubated with 10 nm Au-NPs for 1 h. The protein corona was extracted from the Au-NP surface and subjected to Western blot analysis. The membrane was stained with reversible protein staining (A) or probed with antibodies against IKKα (B) or IKKβ (C). The AuNP control represents AuNP in the absence of protein lysate. Results are representative of three separate experiments.
Previous studies have demonstrated inhibition by gold therapeutics (i.e. aurothiomalate) of over-expressed IKK activity in COS cells22 and since an inherent property of gold is to bind to –thiol groups,31 the presence of cysteine residues on IKK makes it a likely protein target for binding to Au-NPs. Therefore, to determine if IKK was one of the many proteins binding to Au-NPs, the Western blot was probed with an antibody specific for IKKα and IKKβ, two catalytic subunits of the IKK complex. Strong IKKα and IKKβ bands were detected in the protein sample eluted from the Au-NPs (Fig. 6B, lane 2 and Fig. 6C, lane 2) that was also detected in the whole cell lysate (Fig. 6B, lane 1 and Fig. 6C, lane 1) but not in the Au-NP in lysis buffer control sample (Fig. 6B, lane 3). These results support a direct interaction between IKK and Au-NPs which may alter NF-κB signaling in the CH12.LX B-lymphocyte cell line and lead to B-lymphocyte dysfunction (i.e. altered Ig expression and antibody formation).
Au-NPs activate the canonical NF-κB signaling pathway
Au-NPs induce NF-κB activation (Fig. 3) and demonstrate the potential to physically interact with IKKα and IKKβ (Fig. 6) perhaps indicating a direct interaction of Au-NPs with the canonical pathway of NF-κB activation. Canonical activation involves IKK-dependent phosphorylation of IκBα at serines 32 and 36, which leads to ubiquitination and degradation of IκBα. Degradation of IκBα releases NF-κB/Rel dimers (i.e. p50/RelA or p50/c-Rel), which translocate to the nucleus, bind DNA, and modulate gene transcription14,49 (see Fig. 10). To determine if Au-NPs activate the canonical pathway, CH12.LX cells were treated with 5 mg mL−1 of 10 nm Au-NPs for up to 8 h. Whole cell lysates were isolated every 15 min then analyzed by Western blot analysis for IκBα and its serine 32 and 36 phosphorylated form (P-IκBα). Initially, we were unable to detect any reactive bands with the P-IκBα antibody (data not shown); however, addition of a phosphatase inhibitor to the lysis buffer resulted in detection of a P-IκBα reactive band at most time points including the untreated control (Fig. 7A and B). Despite this background level of P-IκBα, which is consistent with previous reports with phosphatase inhibitors,50,51 we observed elevated P-IκBα around 45 min with the positive control (1 mg mL−1 LPS) and at later time points (approximately 4.75 to 6 h) with Au-NP treatment (Fig. 7B and A). Correspondingly, IκBα degradation occurred around 6.25 to 7.5 h following Au-NP treatment and rebounded around 6.5 to 7.5 h (Fig. 7A). The window of phosphorylation, degradation and rebound varied slightly between experiments. Unexpectedly there was not a clear separation of the phosphorylated and unphosphorylated forms of IκBα as demonstrated by alignment of the IκBα and P-IκBα blots (data not shown) and by the anti-IκBα antibody, which recognizes both forms of IκBα (Fig. 7A). However, the consistent degradation pattern of IκBα in response to Au-NP exposure supports NF-κB activation through the canonical signaling pathway. Further- more, the delayed kinetics of IκBα degradation by Au-NP as compared to LPS (compare Fig. 7A and C) likely reflects the time for Au-NP to enter the cell (~4 h) as compared to the quicker kinetics of receptor-mediated signaling and amplification through LPS-induced activation of membrane-bound Toll-like receptors (i.e. TLR-4).
Fig. 7.
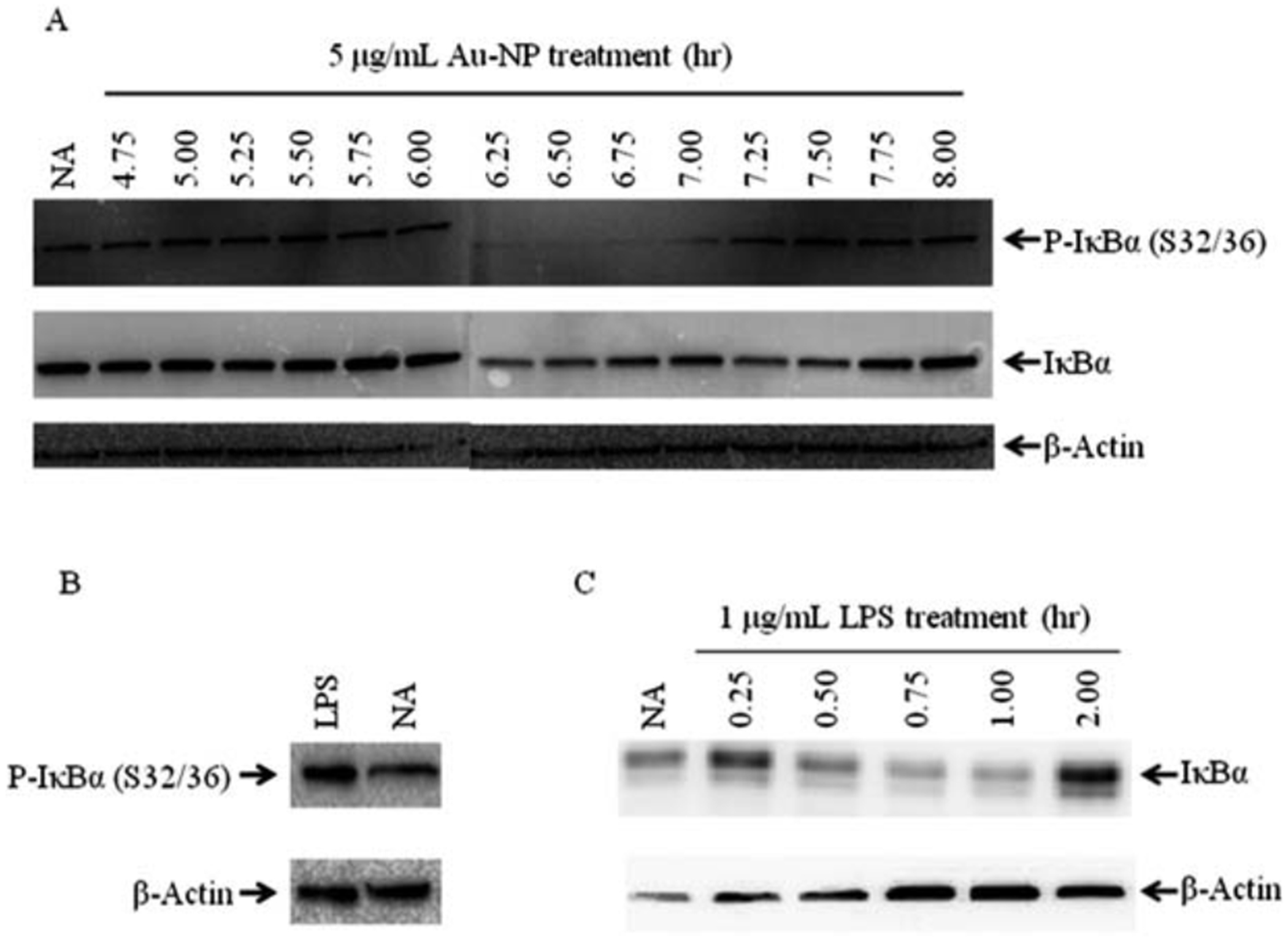
Au-NPs cause phosphorylation and subsequent degradation of IκBα. CH12.LX cells (5.0 × 105 cells per mL) were treated with 5 mg mL−1 of 10 nm Au-NPs (A) or 1 mg ml−1 LPS for 45 min (B) or the indicated time points (C). Cell lysates were prepared and Western blot analysis was performed with antibodies against IκBα, the serine 32 and 36 phosphorylated form of IκBα [P-IκBα (S32/36)], and β-actin. NA denotes the naive untreated control. Results are representative of three separate experiments.
Since IκBα degradation should lead to a rapid increase in nuclear localization of NF-κB, the cytoplasmic and nuclear fractions were isolated from CH12.LX cells treated with 5 mg mL−1 of 10 nm Au-NPs and then probed for RelA (p65). The cell fractions were also probed with b-actin and p84 (a nuclear matrix protein), which served as loading controls for the cytoplasmic and nuclear fractions, respectively. b-Actin has also been shown to be nucleoplasmic52–55 and was detected in the nuclear fraction (Fig. 8); p84 was only detected in the nuclear fraction (data not shown). Overall, there was a general trend for increased nuclear RelA and decreased cytoplasmic RelA around 6.25 h (Fig. 8) that corresponded to the kinetics of IκBα degradation (Fig. 7A). To further confirm these results and therefore the involvement of the canonical pathway in Au-NP-induced activation of NF-κB (see Fig. 10), we evaluated the effect of an IκBαAA super-repressor on activation of the 3x-NF-κB luciferase reporter by Au-NPs. These studies utilized the CH12. IκBαAA B- cell line, which is a variant of the CH12.LX cell line and expresses an IPTG-inducible IκBα super-repressor that is resistant to negative feedback regulation.35 As seen in the CH12.LX cells, Au-NPs activate the 3x-NF-κB luciferase reporter in the CH12.IκBαAA cell line (compare Fig. 3 and 9). However, expression of the IκBαAA super-repressor completely abrogated Au-NP-induced activation of the NF-κB reporter (Fig. 9). Taken together these results support modulation of the canonical NF-κB signaling pathway by Au-NPs leading to induction of NF-κB activity, which correlates with increased activation of the 3’IghRR and Ig expression (see Fig. 10). Previous studies including our own have supported a strong correlation between 3’IghRR activity and Ig expression.27,28,33,56 Moreover, a role of NF-κB in 3’IghRR activation and Ig expression has been supported by various studies including those utilizing the IκBαAA super-repressor.26,27,35,57
Fig. 8.
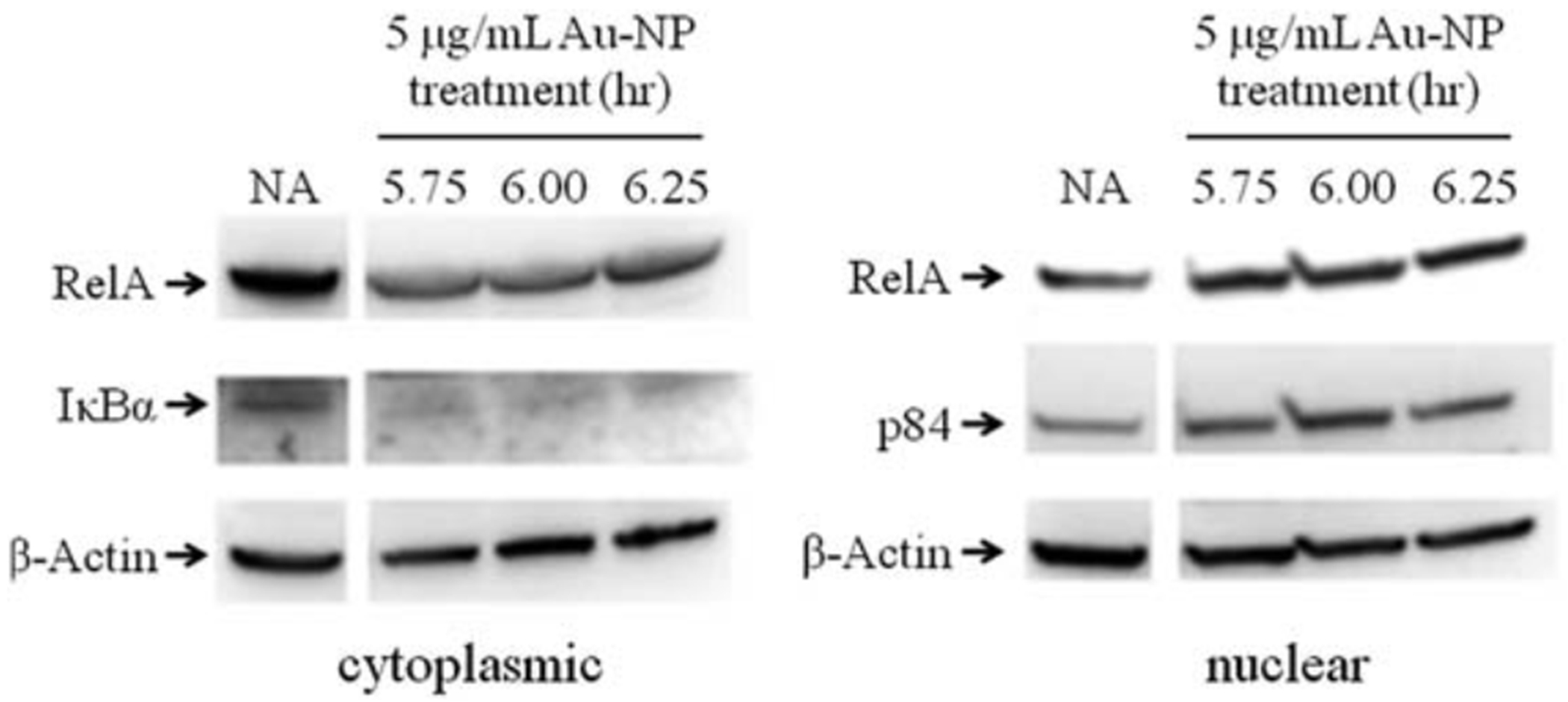
Au-NPs increases nuclear RelA. CH12.LX cells (1 × 107 cells per treatment) were treated with 5 mg mL−1 of 10 nm Au-NPs for the indicated time. Cytoplasmic and nuclear fractions were prepared and Western blot analysis was performed with antibodies against RelA, IκBα, p84 and b-actin. NA denotes the naive untreated control. Results are representative of three separate experiments.
Fig. 9.
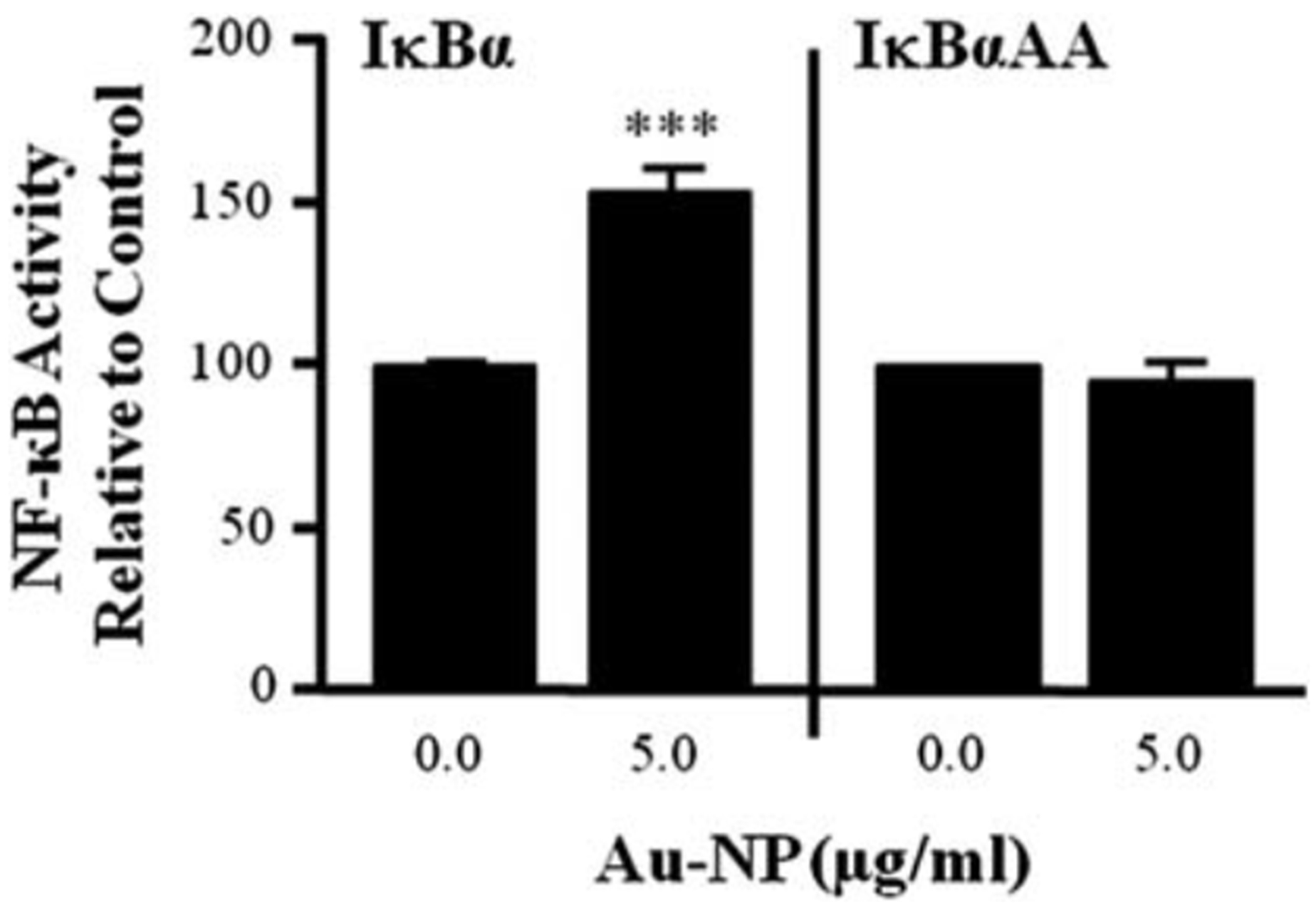
Au-NPs induce NF-κB activation through an IκBα-dependent mechanism. CH12.IkBaAA cells were transiently transfected with the 3x-NFκB luciferase reporter plasmid and multiple transfections were pooled in fresh media. Half of the transfected cells were treated with 100 mM IPTG for 2 h to activate the IPTG- inducible IκBα super-repressor transgene, while the other half was cultured in the absence of IPTG to provide a control that lacked IκBα AA expression. After 2 h, the cells were treated with 5 mg mL−1 Au-NPs or left untreated for a negative control (0.0 mg ml−1 AuNP). After a 24 h incubation, cells were lysed and cellular extracts evaluated for luciferase enzyme activity as measured in relative light units (RLU) and represented on the y-axis as NF-κB activity (mean ± SEM) relative to the naïve control (0.0 mg ml−1 Au-NP, set to 100%). Mean α SEM was generated from three separate experiments. *** Statistical significance at p < 0.001 compared to respective 0.0 mg ml−1 Au-NP control via 2-way ANOVA and a Bonferroni’s post- hoc test.
Although our results support the canonical pathway as a target of Au-NPs, it is unclear if these effects are due to a direct interaction between Au-NPs and IKKs. In support of the physical association between Au-NPs and IKKα and IKKβ, gold compounds have been shown to inactivate the IKK complex, specifically inhibiting both IKKα and IKKβ.22 This would suggest a negative effect on NF-κB activation contrary to the current results; however, inhibition of IKKα activation has been shown to prevent RelA and c-Rel turnover, thereby causing prolonged NF-κB activation.58 Alternatively, Au-NPs have been shown to induce reactive oxygen species (ROS)59,60 and ROS is a well-known modulator of NF-κB transcriptional activity.61 Indeed, our previous results have demonstrated a biphasic effect of H2O2 on 3’IghRR activity and endogenous Ig heavy chain expression (i.e. induction at 30–40 mM concentrations and inhibition at 100–200 mM concentrations).57 Therefore ROS may play an additional role in the effects of Au-NP on NF-κB activation and Ig expression.
4. Conclusions
Modulation of cellular function in the absence of an immediate effect on cell viability suggests molecular interactions between Au-NPs and important signaling pathways. These results highlight the need to study the mode of action of gold nanomaterials before their utilization for in vivo applications. Furthermore, it is likely that nano-bio interactions will be largely dependent on the unique material properties of NPs (i.e. affinity of gold for–thiol groups). Equally important are the particular microenvironments where the NPs are localized. Therefore, emphasis should be placed on extensively characterizing the interactions between NPs and biomolecules, particularly in different cellular models and with different types, sizes, and concentrations of NPs. Characterization of these interactions will likely lead to a clearer understanding of the potential physiological impact of nano-sized materials.
Acknowledgements
This work was partially funded by the Biosciences and Protection Division, Human Effectiveness Directorate, Air Force Research Laboratory under the Oak Ridge Institute for Science and Education (to S.H.), the Student Research Participation Program at the U.S. Air Force Research Laboratory administered by the Oak Ridge Institute for Science and Education (to M. S), and by the National Institute of Environmental Health Sciences grant R01ES014676 (to C.E.W.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding organizations acknowledged above. The authors would like to acknowledge the generous gifts of Dr Geoffrey Haughton (in memoriam, University of North Carolina, Chapel Hill, NC) for the CH12.LX B-cell line, Dr Laurel Eckhardt (Hunter College, The University of New York City, New York, NY) for the γ2b mini-locus model, Dr Ronald Hay (College of Life Sciences, University of Dundee, Dundee, Scotland) for the 3x-NF-κB luciferase reporter plasmid and Dr. Gail Bishop (University of Iowa, Iowa City, Iowa) for the CH12. IκBαAA B-cell line.
Notes and references
- 1.Alkilany AM and Murphy CJ, J. Nanopart. Res, 2010, 12, 2313–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdelhalim MA, Lipids Health Dis., 2011, 10, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdelhalim MA and Jarrar BM, Lipids Health Dis., 2011, 10, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho WS, Cho M, Jeong J, Choi M, Cho HY, Han BS, Kim SH, Kim HO, Lim YT, Chung BH and Jeong J, Toxicol. Appl. Pharmacol, 2009, 236, 16–24. [DOI] [PubMed] [Google Scholar]
- 5.Marquis BJ, Maurer-Jones MA, Braun KL and Haynes CL, Analyst, 2009, 134, 2293–2300. [DOI] [PubMed] [Google Scholar]
- 6.Ng CT, Dheen ST, Yip WC, Ong CN, Bay BH and Lanry Yung LY, Biomaterials, 2011, 32, 7609–7615. [DOI] [PubMed] [Google Scholar]
- 7.Schaeublin NM, Braydich-Stolle LK, Schrand AM, Miller JM, Hutchison J, Schlager JJ and Hussain SM, Nanoscale, 2011, 3, 410–420. [DOI] [PubMed] [Google Scholar]
- 8.Vecchio G, Galeone A, Brunetti V, Maiorano G, Sabella S, Cingolani R and Pompa PP, PLoS One, 2012, 7, e29980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwamoto M, Tokonami S, Shiigi H and Nagaoka T, Res. Chem. Intermed, 2009, 35, 919–930. [Google Scholar]
- 10.Jie G, Liu B, Pan H, Zhu JJ and Chen HY, Anal. Chem, 2007, 79, 5574–5581. [DOI] [PubMed] [Google Scholar]
- 11.Kamei K, Mukai Y, Kojima H, Yoshikawa T, Yoshikawa M, Kiyohara G, Yamamoto TA, Yoshioka Y, Okada N, Seino S and Nakagawa S, Biomaterials, 2009, 30, 1809–1814. [DOI] [PubMed] [Google Scholar]
- 12.Kang T, Hong S, Choi I, Sung JJ, Kim Y, Hahn JS and Yi J, J. Am. Chem. Soc, 2006, 128, 12870–12878. [DOI] [PubMed] [Google Scholar]
- 13.Sivanesan A, Kannan P and Abraham John S, Electrochim. Acta, 2007, 52, 8118–8124. [Google Scholar]
- 14.Perkins ND, Nat. Rev. Mol. Cell Biol, 2007, 8, 49–62. [DOI] [PubMed] [Google Scholar]
- 15.Vallabhapurapu S and Karin M, Annu. Rev. Immunol, 2009, 27, 693–733. [DOI] [PubMed] [Google Scholar]
- 16.Beinke S and Ley SC, Biochem. J, 2004, 382, 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonizzi G and Karin M, Trends Immunol, 2004, 25, 280–288. [DOI] [PubMed] [Google Scholar]
- 18.Hatada EN, Nieters A, Wulczyn FG, Naumann M, Meyer R, Nucifora G, McKeithan TW and Scheidereit C, Proc. Natl. Acad. Sci. U. S. A, 1992, 89, 2489–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayden MS and Ghosh S, Cell, 2008, 132, 344–362. [DOI] [PubMed] [Google Scholar]
- 20.Bratt J, Belcher J, Vercellotti GM and Palmblad J, Clin. Exp. Immunol, 2000, 120, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniel LW, Civoli F, Rogers MA, Smitherman PK, Raju PA and Roederer M, Cancer Res, 1995, 55, 4844–4849. [PubMed] [Google Scholar]
- 22.Jeon KI, Jeong JY and Jue DM, J. Immunol, 2000, 164, 5981–5989. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita M, Ashino S, Oshima Y, Kawamura S, Ohuchi K and Takayanagi M, J. Pharm. Pharmacol, 2003, 55, 245–251. [DOI] [PubMed] [Google Scholar]
- 24.Yang JP, Merin JP, Nakano T, Kato T, Kitade Y and Okamoto T, FEBS Lett, 1995, 361, 89–96. [DOI] [PubMed] [Google Scholar]
- 25.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM and Bravo R, J. Exp. Med, 1998, 187, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaelson JS, Singh M, Snapper CM, Sha WC, Baltimore D and Birshtein BK, J. Immunol, 1996, 156, 2828–2839. [PubMed] [Google Scholar]
- 27.Pinaud E, Marquet M, Fiancette R, Peron S, Vincent-Fabert C, Denizot Y and Cogne M, Adv. Immunol, 2011, 110, 27–70. [DOI] [PubMed] [Google Scholar]
- 28.Vincent-Fabert C, Fiancette R, Pinaud E, Truffinet V, Cogne N, Cogne M and Denizot Y, Blood, 2010, 116, 1895–1898. [DOI] [PubMed] [Google Scholar]
- 29.Nel AE, Madler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V and Thompson M, Nat. Mater, 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]
- 30.Love JC, Estroff LA, Kriebel JK, Nuzzo RG and Whitesides GM, Chem. Rev, 2005, 105, 1103–1169. [DOI] [PubMed] [Google Scholar]
- 31.Nakata S, Kido N, Hayashi M, Hara M, Sasabe H, Sugawara T and Matsuda T, Biophys. Chem, 1996, 62, 63–72. [DOI] [PubMed] [Google Scholar]
- 32.Arnold LW, LoCascio NJ, Lutz PM, Pennell CA, Klapper D and Haughton G, J. Immunol, 1983, 131, 2064–2068. [PubMed] [Google Scholar]
- 33.Henseler RA, Romer EJ and Sulentic CE, Toxicology, 2009, 261, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi X and Eckhardt LA, Int. Immunol, 2001, 13, 1003–1012. [DOI] [PubMed] [Google Scholar]
- 35.Hsing Y and Bishop GA, J. Immunol, 1999, 162, 2804–2811. [PubMed] [Google Scholar]
- 36.Polavarapu L and Xu QH, Nanotechnology, 2009, 20, 185606. [DOI] [PubMed] [Google Scholar]
- 37.Schrand AM, Schlager JJ, Dai L and Hussain SM, Nat. Protoc, 2010, 5, 744–757. [DOI] [PubMed] [Google Scholar]
- 38.Desterro JM, Rodriguez MS and Hay RT, Mol. Cell, 1998, 2, 233–239. [DOI] [PubMed] [Google Scholar]
- 39.Merluzzi S, Moretti M, Altamura S, Zwollo P, Sigvardsson M, Vitale G and Pucillo C, J. Biol. Chem, 2004, 279, 1777–1786. [DOI] [PubMed] [Google Scholar]
- 40.Chithrani BD, Ghazani AA and Chan WC, Nano Lett, 2006, 6, 662–668. [DOI] [PubMed] [Google Scholar]
- 41.Peckys DB and de Jonge N, Nano Lett, 2011, 11, 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya R and Mukherjee P, Adv. Drug Delivery Rev, 2008, 60, 1289–1306. [DOI] [PubMed] [Google Scholar]
- 43.Ma JS, Kim WJ, Kim JJ, Kim TJ, Ye SK, Song MD, Kang H, Kim DW, Moon WK and Lee KH, Nitric Oxide, 2008, 23, 214–219. [DOI] [PubMed] [Google Scholar]
- 44.Sul OJ, Kim JC, Kyung TW, Kim HJ, Kim YY, Kim SH, Kim JS and Choi HS, Biosci., Biotechnol., Biochem, 2010, 74, 2209–2213. [DOI] [PubMed] [Google Scholar]
- 45.Ye S, Zhang H, Wang Y, Jiao F, Lin C and Zhang Q, J. Nanopart. Res, 2011, 13, 4239–4252. [Google Scholar]
- 46.Garner SF, Campbell K, Metcalfe P, Keidan J, Huiskes E, Dong JF, Lopez JA and Ouwehand WH, Blood, 2002, 100, 344–346. [DOI] [PubMed] [Google Scholar]
- 47.Fournie GJ, Saoudi A, Druet P and Pelletier L, Autoimmun. Rev, 2002, 1, 205–212. [DOI] [PubMed] [Google Scholar]
- 48.Havarinasab S, Johansson U, Pollard KM and Hultman P, Clin. Exp. Immunol, 2007, 150, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayden MS and Ghosh S, Genes Dev, 2012, 26, 203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barisic S, Schmidt C, Walczak H and Kulms D, Biochem. Pharmacol, 2010, 80, 439–447. [DOI] [PubMed] [Google Scholar]
- 51.Sun SC, Maggirwar SB and Harhaj E, J. Biol. Chem, 1995, 270, 18347–18351. [DOI] [PubMed] [Google Scholar]
- 52.McDonald D, Carrero G, Andrin C, de Vries G and Hendzel MJ, J. Cell Biol, 2006, 172, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blessing CA, Ugrinova GT and Goodson HV, Trends Cell Biol, 2004, 14, 435–442. [DOI] [PubMed] [Google Scholar]
- 54.Olave IA, Reck-Peterson SL and Crabtree GR, Annu. Rev. Biochem, 2002, 71, 755–781. [DOI] [PubMed] [Google Scholar]
- 55.Xu YZ, Thuraisingam T, Morais DA, Rola-Pleszczynski M and Radzioch D, Mol. Biol. Cell, 2010, 21, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sulentic CEW, Zhang W, Na YJ and Kaminski NE, J. Pharmacol. Exp. Ther, 2004, 309, 71–78. [DOI] [PubMed] [Google Scholar]
- 57.Romer EJ and Sulentic CEW, Free Radical Res, 2011, 45, 796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lawrence T, Bebien M, Liu GY, Nizet V and Karin M, Nature, 2005, 434, 1138–1143. [DOI] [PubMed] [Google Scholar]
- 59.Jia HY, Liu Y, Zhang XJ, Han L, Du LB, Tian Q and Xu YC, J. Am. Chem. Soc, 2009, 131, 40–41. [DOI] [PubMed] [Google Scholar]
- 60.Zhao Y, Gu X, Ma H, He X, Liu M and Ding Y, J. Phys. Chem. C, 2011, 115, 12797–12802. [Google Scholar]
- 61.Morgan MJ and Liu ZG, Cell Res, 2011, 21, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]