

Abstract
BACKGROUND:
Pulmonary fibrosis (PF) is one of the main causes of death in patients with paraquat (PQ) poisoning. This study aimed to evaluate the relationship between mitochondrial fission and oxidative stress in PQ-induced epithelial-mesenchymal transition (EMT) and PF.
METHODS:
C57BL/6 mice and MLE-12 cells were exposed to PQ to construct a PF model in vivo and in vitro. Histological changes in the lungs were examined by hematoxylin and eosin (H&E) staining. Mitochondrial morphology was detected by MitoTracker® Deep Red FM or transmission electron microscopy (TEM). Western blotting and immunofluorescence were used to determine the expression of protein. The migration ability of the cells was detected by the cell scratch test. Mitochondrial DNA (mtDNA) levels were assessed by real-time polymerase chain reaction (PCR). Enzyme-linked immunosorbent assay (ELISA) was applied to detect cytokine levels. Superoxide dismutase (SOD) activity and the levels of glutathione (GSH) and malondialdehyde (MDA) were detected by chemichromatometry.
RESULTS:
PQ exposure caused EMT and PF in vivo and in vitro. PQ destroyed mitochondrial structure and enhanced the expression of dynamin-related protein 1 (Drp1), which were accompanied by oxidative stress. Inhibiting mitochondrial fission using mitochondrial division inhibitor-1 (Mdivi-1), a selective inhibitor of Drp1, attenuated PQ-induced EMT and oxidative damage. Treatment with N-acetyl-L-cysteine (NAC), an antioxidant, reduced Drp1 expression, attenuated mitochondrial structure damage and inhibited PQ-induced EMT and PF. Both Mdivi-1 and NAC treatment markedly suppressed mtDNA release, the expression of Toll-like receptor 9 (TLR9) and phosphorylation (P)-NF-κB p65 as well as cytokines (interleukin 6 [IL-6], interleukin-1β [IL-1β], and tumor necrosis factor-α [TNF-α]) production.
CONCLUSION:
Mutual promotion of mitochondrial fission and oxidative stress contributes to EMT in PQ-induced PF, which is associated with the mtDNA/TLR9/NF-κB pathway.
Keywords: Paraquat, Mitochondrial fission, Oxidative stress, Epithelial-mesenchymal transition, Mitochondrial DNA
INTRODUCTION
Pulmonary fibrosis (PF) is a serious pulmonary interstitial disease.[1] The main pathological characteristics of PF are the proliferation of lung stromal cells and excessive deposition of extracellular matrix, which leads to the destruction of lung structure and function.[2] There are many causes of PF, including pathogenic microorganism infection, drug side effects, and toxicant exposure.[3,4] Some toxic poisons, such as paraquat (PQ), a bipyridine herbicide widely used around the world, accumulate in the lungs after ingestion, leading to lung injury and acute progressive fibrosis.[5,6] Although clinical experts have reached a consensus on the diagnosis and treatment of PQ poisoning, due to the absence of an effective antidote, the mortality rate of PQ poisoning remains as high as 50%–70%.[7-9]
Previous studies have shown that approximately 30% of PF fibroblasts are of epithelial origin, and epithelial-mesenchymal transition (EMT) has been proven to be the core mechanism of PF.[10,11] The existing evidence shows that PQ can induce EMT phenotypes, which may be associated with oxidative injury after poisoning.[6,12] It is generally accepted that PQ causes the production of a significant amount of oxygen free radicals after being reduced to free radicals by a single electron with the aid of nicotinamide adenine dinucleotide phosphate (NADPH).[5-7,13] As a highly dynamic organelle, excessive mitochondrial fission also plays a positive role in the oxidative stress response.[14] Nevertheless, the relationship between mitochondrial fission and oxidative stress in PQ-induced EMT remains unknown.
Pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) are mediators of inflammation in PF.[15] In noninfectious disease-related PF, inflammation is mainly evoked by DAMPs.[16] Mitochondria are not only the energy factories of cells but also the sources of DAMPs. Mitochondria-released DAMPs, particularly mitochondrial DNA (mtDNA), contribute to aseptic inflammation through the Toll-like receptor (TLR)-9 and nuclear factor kappa B (NF-κB) pathways.[16] Some studies have shown the mechanism of mtDNA efflux through the outer membrane of mitochondria.[17,18] In the process of mitochondrial fission, dynamin-related protein 1 (Drp1) forms a spiral oligomer, which wraps and splits the outer membrane of mitochondria.[19] Our previous study found that mitochondrial fission contributes to the release of cytochrome C, which indicates its potential role in the release of other DAMPs.
Using in vivo and in vitro PF models, this study investigated the relationship between mitochondrial fission and oxidative stress in PQ-induced EMT. Additionally, the effect of their crosstalk on the release of mtDNA and its mediating inflammatory response were explored.
METHODS
Animals
Healthy SPF male C57BL/6 mice, 6–8 weeks old, weighing 20–22 g, were purchased from Shanghai Vital River Laboratory Animal Technology Co., Ltd., China. All animal experiments were carried out according to the Guides for the Care and Use of Laboratory Animals, National Academy of Sciences, China. The present animal study was approved by the Laboratory Animal Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (WYYY-AEC-2022-0032).
In vivo experimental procedures
The mice were treated with different concentrations of PQ, and 50 mg/kg PQ was used to induce the PF model as previously described.[6,19]
Experiment 1: The mice were randomly divided into two groups: the control group and the PQ group. At 12 h and days 1, 3 and 7 after exposure to PQ, lung tissue and blood samples were taken for further studies.
Experiment 2: Mitochondrial division inhibitor-1 (Mdivi-1), a specific mitochondrial fission inhibitor, was administered at 6 h after PQ exposure. The mice were randomly divided into four groups: control group, Mdivi-1 group, PQ group, and PQ+Mdivi-1 group.
Experiment 3: The mice were administered N-acetyl-L-cysteine (NAC), a classic antioxidant, at 6 h after PQ exposure. The mice were randomly divided into four groups: control group, NAC group, PQ group, and PQ+NAC group.
In vitro experimental procedures
MLE-12 cells (mouse lung type II epithelial cell line) were purchased from American Type Culture Collection (ATCC). The cells were cultured in DMEM-F12 (11320033, Thermo Fisher Scientific, USA) complete medium containing 10% fetal bovine serum and 1% antibiotics (penicillin and streptomycin). The cells were cultured at 37 °C and 5% CO2. When the density of MLE-12 cells on the plates reached 60%–70%, they were stimulated with PQ. Similar to in vivo studies, Mdivi-1 and NAC were used to inhibit mitochondrial fission and oxidative stress, respectively.
Hematoxylin and eosin (H&E) staining
The slices of left lung tissues were stained according to the instructions of the H&E staining kit (G1120, Beijing Solarbio Science & Technology Company Limited, China) and were viewed under an optical microscope. A semi-quantitative scoring system was introduced to evaluate lung injury, which included alveolar congestion, alveolar hemorrhage, neutrophil infiltration or aggregation in the alveolar cavity or vascular wall, alveolar wall thickening and/or transparent membrane formation. The four variables were summed to represent the lung injury score (total score: 0–16).
Western blotting
Total protein was extracted from MLE-12 cells and lung tissues. The primary antibodies used in this study were against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab8245, Abcam, USA), Drp-1 (8570S, Cell Signaling Technology, USA), E-cadherin (ab76055, Abcam, USA), α-smooth muscle actin (α-SMA) (19245S, Cell Signaling Technology, USA), TLR-9 (ab134368, Abcam, USA), phosphorylated (P)-NF-κB p65 (3033S, Cell Signaling Technology, USA), and NF-κB p65 (8242S, Cell Signaling Technology, USA).
Cell scratch test
MLE-12 cells (5,000 cells/well) were inoculated in 12-well plates and cultured at 37 °C and 5% CO2 to cover the bottom of the plate. Then, a straight line was drawn in the center of each well with a 100 μL sterile micropipette tip, and pictures were taken (0 h). Cells were treated with or without PQ and/or Mdivi-1. Then, 48 h later, the distance remaining was observed with an inverted microscope (Nikon, Japan) after cells of each group migrated from the scratch edge to the center.
Enzyme-linked immunosorbent assay (ELISA)
Mouse lung tissues were homogenized and centrifuge, and the supernatant was collected. The levels of interleukin 6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor-α (TNF-α) in the tissue supernatant were detected by ELISA kits provided by Multi Sciences (EK206, EK201B, EK282HS, Multi Sciences, China).
Superoxide dismutase (SOD) activity and glutathione (GSH) and malondialdehyde (MDA) levels
In brief, mouse lung tissues were homogenized, and the homogenate was centrifuged. The supernatant was used to detect SOD activity and GSH and MDA levels according to the instructions (S0101, S0053, S0131, Beyotime Biotechnology, China).
Mitochondrial morphology
MLE-12 cells (5,000 cells/well) were inoculated in a 12-well plate with glass slides placed inside each well. The cells were cultured at 37 °C and 5% CO2 for 48 h. Then, the cells were stained with MitoTracker® Deep Red FM (200 nmol/L, M22426, Thermo Fisher Scientific, USA) and 6-diamidino-2-phenylindole (DAPI, 4083, Cell Signaling Technology, USA). The slides were viewed under a confocal laser scanning microscope (Nikon, Japan).
Real-time polymerase chain reaction (PCR) for mtDNA
Mouse blood samples were collected, and the plasma was extracted. The mtDNA in plasma and cell culture supernatant was extracted by the blood mtDNA extraction kit (XH007, Beijing Bio Lebo Technology Company Limited, China), and the levels of mtDNA were detected by the mouse mtDNA copy number assay kit (MCN3, Drtroit R&D, USA) based on real-time PCR.
Transmission electron microscopy (TEM)
The lung tissues of mice were fixed with 2.5% glutaraldehyde at 4 °C for 4 h and then fixed with 1% osmium tetroxide at 20 °C for 2 h. Then, the tissue was dehydrated by graded concentrations of ethanol and acetone, embedded in Epon812, sliced using an ultramicrotome, and stained with uranyl acetate and lead nitrate. The sections were observed by TEM (Hitachi, Japan).
Statistical analysis
Statistical analyses were performed using GraphPad 8.0 statistical program (GraphPad software, USA) and SPSS 22.0 statistical software (IBM, USA). The data are represented as the mean±standard deviation (SD). Student’s t-test was used to determine the differences between two groups, and the differences among multiple groups were determined by one-way analysis of variance (ANOVA) followed by least significant difference (LSD) post hoc analysis. The survival rate was analyzed by Kaplan-Meier analysis and compared by the log-rank test. A P-value <0.05 was considered as a statistically significant difference.
RESULTS
PQ caused EMT and PF in vivo and in vitro
The mice were treated with different concentrations of PQ, and 50 mg/kg PQ was used to induce the PF model (supplementary Figure 1A). H&E staining of lung sections showed that the acute lung injury score increased significantly at 12 h but decreased at 7 d after PQ exposure (supplementary Figure 1B). Gradually aggravated diffuse alveolar collapse and thickening suggest the occurrence of PF (supplementary Figure 1B). EMT in PQ-induced PF is evidenced by decreased levels of E-cadherin and increased expression of α-SMA in lung tissues (supplementary Figures 1 C-E). To examine whether PQ could induce EMT in vitro, we incubated MLE-12 cells with PQ for 12, 24 and 48 h. The results showed that PQ suppressed E-cadherin expression and up-regulated α-SMA expression in a time-dependent manner (supplementary Figures 1F-H). The cell scratch test showed decreased migration ability of MLE-12 cells after PQ stimulation, indicating the EMT of the cells (supplementary Figure 1I).
Mitochondrial fission and Drp1 expression increased in PQ-induced PF
At 24 h after PQ exposure, the lung had mitochondria with irregular shapes and disorganized cristae (supplementary Figure 2A). At 7 d after PQ exposure, mitochondrial morphology partially recovered (supplementary Figure 2A). In vitro, PQ-induced mitochondrial fragmentation and the content of mitochondria in MLE-12 cells were also reduced (supplementary Figure 2B). Drp1 is the main mediator of mitochondrial fission. We found that the expression of Drp1 in lung tissue significantly increased at 1 d and 3 d after PQ exposure but slightly decreased at 7 d, which was consistent with the morphological changes in mitochondria (supplementary Figures 2 C and E). In vitro, the peak expression of Drp1 appeared at 48 h after PQ stimulation (supplementary Figures 2 D and F).
Drp1-mediated mitochondrial fission alleviated EMT and PF
To determine the exact relationship between Drp1-mediated mitochondrial fission and EMT in PF, we treated mice with Mdivi-1 at 6 h post PQ exposure. One day later, Drp1 expression and mitochondrial fission were significantly decreased in the lung issues of mice treated with Mdivi-1 compared with those in the control group (Figures 1 A-C). Similarly, in MLE-12 cells exposed to PQ, Mdivi-1 treatment also downregulated the expression of Drp1, reduced mitochondrial fission, and increased mitochondrial content (Figures 1 D-F).
Figure 1.
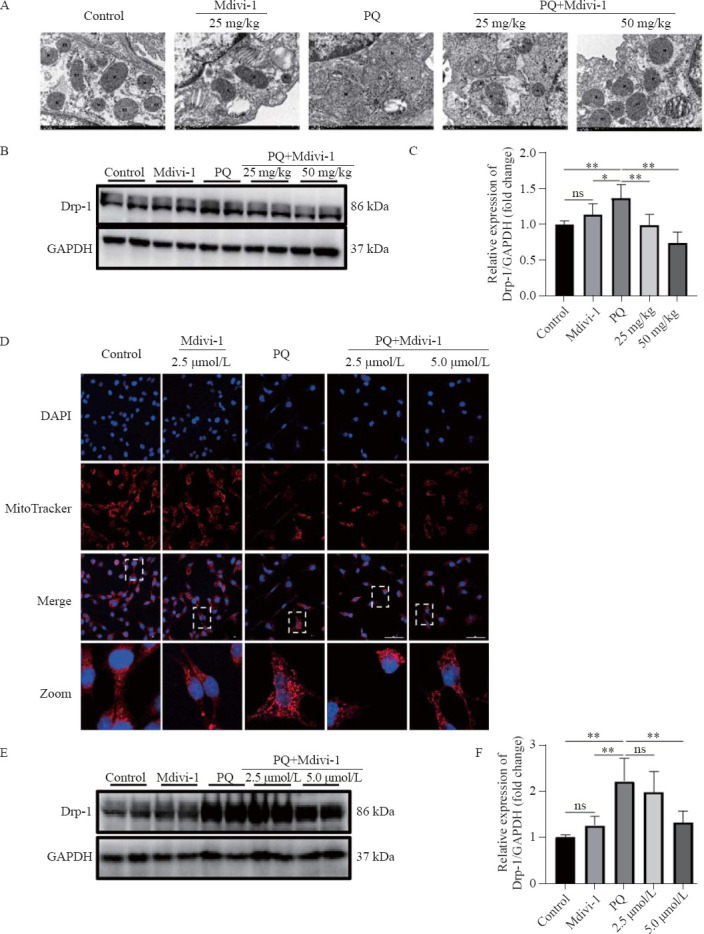
Effects of Midivi-1 treatment on mitochondrial morphology and Drp1 expression in PQ-induced PF. A: morphological changes in mitochondria in lung tissues observed by transmission electron microscopy (original magnification, ×200); B, C: representative immunoblots and quantitative histogram of Drp1 in lung tissues from mice (n=6); D: MitoTracker® Deep Red FM and DAPI used to stain mitochondria and nuclei, and mitochondrial morphology determined using confocal laser microscopy (original magnification, ×200); E, F: representative immunoblots and quantitative histogram of Drp1 in MLE-12 cells. *P<0.05, **P<0.01. All values are denoted as the mean±SD. PQ: paraquat; Drp-1: dynamin-related protein 1; Midivi-1: mitochondrial division inhibitor-1; PF: pulmonary fibrosis; 25 mg/kg: PQ+Mdivi-1 (25 mg/kg); 50 mg/kg: PQ+Mdivi-1 (50 mg/kg); 2.5 μmol/L: PQ+Mdivi-1 (2.5 μmol/L); 5.0 μmol/L: PQ+Mdivi-1 (5.0 μmol/L); GAPDH: glyceraldehyde-3-phosphate dehydrogenase; ns: not significant.
The decrease in diffuse alveolar collapse and thickening indicates the role of Drp1-mediated mitochondrial fission in PF (supplementary Figures 3 A-B), which is confirmed by the decrease in α-SMA levels and increased E-cadherin expression after Mdivi-1 treatment (supplementary Figures 3 C-E). In MLE-12 cells, treatment with Mdivi-1 at 6 h after PQ stimulation significantly enhanced cell migration (supplementary Figure 4A), accompanied by a decrease in α-SMA expression and an increase in E-cadherin expression (supplementary Figures 4 B-D). Taken together, these results supported that Drp1-mediated mitochondrial fission contributed to EMT in PQ-induced PF.
We found that PQ induced increased oxidative stress, as evidenced by elevated MDA production and decreased GSH levels and SOD activity (supplementary Figures 4 E-G). Inhibiting Drp1-mediated mitochondrial fission by Mdivi-1 administration dramatically reduced oxidative stress injury as determined by increases in SOD activity and the levels of GSH and by decreases in MDA production (supplementary Figures 4 E-G).
NAC downregulated Drp1 expression and decreased mitochondrial fission and EMT
NAC administration was associated with a decrease in MDA levels and increases in SOD activity and GSH levels (supplementary Figures 5 A-C). NAC administration attenuated mitochondrial fission, which was determined by the reduction in Drp1 expression and mitochondrial structural damage (supplementary Figures 5 D-F). Compared with the control group, NAC administration decreased EMT, as evidenced by decreased α-SMA expression and increased E-cadherin levels (supplementary Figures 5 G-I). In PF mice, histological analysis showed decreased diffuse alveolar collapse and thickening after treatment with NAC (supplementary Figures 5 J and K).
mtDNA-mediated inflammation decreased after inhibiting Drp1-mediated mitochondrial fission
The over-expression of cytokines and subsequent inflammatory damage are important mechanisms of EMT.[20] In PQ-induced PF, the levels of IL-6, IL-1β and TNF-α in plasma were significantly increased, which was reversed by a Drp1 inhibitor (Figures 2 A-C). The levels of mtDNA, TLR9 expression, and P-NF-κB P65 were significantly increased in PQ-induced PF (Figures 2 E-G). Inhibiting Drp1-mediated mitochondrial fission with a Drp1 inhibitor reduced mtDNA release, as determined by a decrease in mtDNA levels in plasma and decreases in TLR9 and P-NF-κB P65 expression (Figures 2 E-G), but the Drp1 inhibitor did not affect the level of total P65. Together, these results demonstrated that targeting Drp1-mediated mitochondrial fission prevented mtDNA release and its mediated inflammation.
Figure 2.
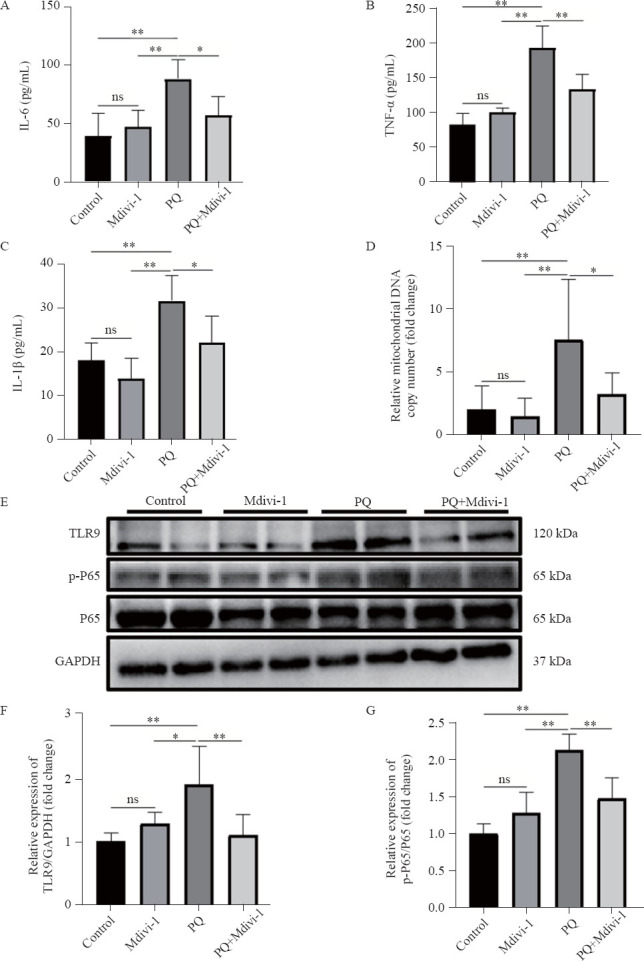
mtDNA-mediated inflammation decreased after inhibiting Drp1-mediated mitochondrial fission. A-C: concentrations of IL-6, TNF-α, and IL-1β from mice measured by ELISA; D: the relative levels of mtDNA in plasma from mice; E-G: representative immunoblots and quantitative histogram of TLR9 and p-P65/P65 in the lung tissues from mice. n=6; *P<0.05, **P<0.01. All values are expressed as the mean±standard deviation. mtDNA: mitochondrial DNA; PQ: paraquat; TLR9: Toll-like receptor 9; P65: NF-κB p65; p-65: (P)-NF-κB p65; GAPDH: glyceraldehyde-3-phosphate dehydrogenase;ns: not significant.
NAC suppressed mtDNA release and activation of the TLR9-NF-κB signaling pathway
We determined whether oxidative stress contributes to mtDNA release. Mice receiving NAC at 6 h after PQ exposure had lower levels of mtDNA in the plasma, indicating that oxidative stress was crucial to the release of mtDNA in PF (supplementary Figure 6 A). NAC treatment also reduced lung inflammation, as evidenced by decreased levels of cytokines, including IL-6, IL-1β and TNF-α (supplementary Figures 6 B-D). Additionally, the levels of TLR9 and P-NF-κB P65 in the lung also decreased after NAC treatment (supplementary Figures 6 E-G). Thus, our results demonstrated that oxidative stress promoted mtDNA-mediated inflammation in PF.
DISCUSSION
Mitochondria are dynamic organelles that undergo fusion and fission to maintain their function and morphology. Currently, an imbalance in mitochondrial fusion and fission has been proven to be involved in the pathogenesis of sepsis.[21] The role of mitochondrial fusion and fission in PF is not completely known. Here, we prove that increased mitochondrial fission significantly induces EMT in the PQ-induced PF mice model. Mechanistically, increased mitochondrial fission contributes to oxidative damage and the activation of the mtDNA/TLR9/NF-κB signaling pathway, which are the two main mechanisms of EMT.[11,22]
Oxidative stress is responsible for PQ-induced lung injury. Previous studies have also illustrated the crucial role of oxidative stress in EMT.[11,22] In diabetic nephropathy, targeted ROS alleviated EMT and renal fibrosis.[23] Chen et al[24] found that fangchinoline decreased ROS production and reversed EMT in non-small cell lung cancer, inhibiting cell invasion and migration. In this study, during PQ-induced EMT, we found that treatment with NAC, a ROS scavenger, not only significantly reduced oxidative damage and EMT but also attenuated mitochondrial fission and Drp1 expression, suggesting that Drp1-mediated mitochondrial fission is involved in oxidative-stress-induced EMT. Mitochondria are one of the sources of ROS in mammalian cells.[13] We found that the inhibition of Drp1-dependent mitochondrial fission by Mdivi-1, indicating increased generation of ROS, requires mitochondrial fission in PQ-induced EMT. Together, the present study showed that mutual promotion of mitochondrial fission and oxidative stress contributes to EMT in PQ-induced PF. Nevertheless, some studies have also shown that a lack of Drp1 leads to mitochondrial elongation and cumulative oxidative damage.[25] Thus, when targeting Drp1 to treat related diseases, the balance of mitochondrial fission and fusion should be fully evaluated.
Blocking TLR9 has also been shown to reduce bleomycin-induced lung injury.[26] To date, oxidative stress and TLR9-mediated inflammation are often used to explain the development of EMT and PF, although one study showed that the ROS inhibitor apocynin could attenuate ox-LDL-mediated mtDNA damage and TLR9 expression.[27] mtDNA shares unmethylated CpG DNA repeats with bacterial DNA and is a ligand for TLR9.[28] We found that treatment with NAC significantly reduced mtDNA release, TLR9 expression and the activation of NF-κB p65. Interestingly, suppression of Drp1-mediated mitochondrial fission also alleviated mtDNA release in vivo and in vitro. Similarly, Zhang et al[29] found that excessive mitochondrial fission was one of the reasons for mtDNA release in LPS-treated Kupffer cells. As mitochondrial fission and Drp1 expression were reduced after NAC treatment in the present study, oxidative stress may contribute to mtDNA release and inflammation through Drp1-mediated excessive mitochondrial fission.
Recently, many studies have reported the mechanism of mtDNA release. In the process of cell apoptosis, Bcl-2-associated X protein (BAX) and Bcl2 antagonist/killer (BAK) oligomerize to form pores in the outer membrane of mitochondria, which mediates the release of mtDNA.[17] In addition, under oxidative stress conditions, mitochondria release short mtDNA fragments through the pores formed by voltage-dependent anion channel (VDAC) oligomers.[18] Increased levels of Drp1 and its interaction with Bax/Bak and VDAC lead to the formation of pores that mediate mtDNA release.[30] Although the mechanisms underlying mtDNA release need to be further ruled out, targeting Bax/Bak or VDAC1 might be a potential treatment strategy for PQ-induced inflammation.
CONCLUSION
Mutual promotion of mitochondrial fission and oxidative stress contributes to EMT in PQ-induced PF, which is associated with the mtDNA/TLR9/NF-κB pathway.
Footnotes
Funding: The study was supported by the Wenzhou Municipal Science and Technology Bureau (Y2020092) and partly by the Key Specialty of Traditional Chinese Medicine of Zhejiang Province in the 13th Five-Year Plan period (Emergency Department).
Ethical approval: The study was approved by the Laboratory Animal Ethics Committee of Wenzhou Medical University (WYYY-AEC-2022-0032).
Conflicts of interest: The authors declare that there are no conflicts of interest regarding the publication of this paper.
Contributors: ZQL conceptualized and drafted the manuscript. JZ, WJL, SQC, and CZ performed the experiments. GJZ and ZQL designed the experiments and supervised and revised the manuscript. All authors made an individual contribution to the final approval of the version published.
All the supplementary files in this paper are available at http://wjem.com.cn.
REFERENCES
- 1.Yi JH, Zhang ZC, Zhang MB, He X, Lin HR, Huang HW, et al. Role of epithelial-to-mesenchymal transition in the pulmonary fibrosis induced by paraquat in rats. World J Emerg Med. 2021;12((3)):214–20. doi: 10.5847/wjem.j.1920-8642.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao M, Wang L, Wang M, Zhou S, Lu Y, Cui H, et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct Target Ther. 2022;7(1):206. doi: 10.1038/s41392-022-01070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruaro B, Matucci Cerinic M, Salton F, Baratella E, Confalonieri M, Hughes M. Editorial:Pulmonary fibrosis:one manifestation, various diseases. Front Pharmacol. 2022;13:1027332. doi: 10.3389/fphar.2022.1027332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun B, Chen YG. Advances in the mechanism of paraquat-induced pulmonary injury. Eur Rev Med Pharmacol Sci. 2016;20(8):1597–602. [PubMed] [Google Scholar]
- 6.Feng MX, Lu YQ. Performance of extracorporeal membrane oxygenation in patients with fatal paraquat poisoning:grasp for straws? World J Emerg Med. 2021;12(3):232–4. doi: 10.5847/wjem.j.1920-8642.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chinese College of Emergency Physicians (CCEP) An expert consensus on diagnosis and treatment of acute paraquat poisoning (2022) Chin J Emerg Med. 2022;31((11)):1435–44. [Google Scholar]
- 8.Hu L, Hong G, Tang Y, Wang X, Wen C, Lin F, et al. Early metabolome profiling and prognostic value in paraquat-poisoned patients:based on ultraperformance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. Chem Res Toxicol. 2017;30(12):2151–8. doi: 10.1021/acs.chemrestox.7b00240. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Hu L, Li H, Hong G, Zhang T, Ma J, et al. An effective machine learning approach for prognosis of paraquat poisoning patients using blood routine indexes. Basic Clin Pharmacol Toxicol. 2017;120(1):86–96. doi: 10.1111/bcpt.12638. [DOI] [PubMed] [Google Scholar]
- 10.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–79. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salton F, Volpe MC, Confalonieri M. Epithelial-mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis. Medicina (Kaunas) 2019;55(4):83. doi: 10.3390/medicina55040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu L, Yu Y, Huang H, Fan H, Hu L, Yin C, et al. Epigenetic regulation of interleukin 6 by histone acetylation in macrophages and its role in paraquat-induced pulmonary fibrosis. Front Immunol. 2017;7:696. doi: 10.3389/fimmu.2016.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao G, Cao K, Xu C, Sun A, Lu W, Zheng Y, et al. Crosstalk between mitochondrial fission and oxidative stress in paraquat-induced apoptosis in mouse alveolar type ii cells. Int J Biol Sci. 2017;13(7):888–900. doi: 10.7150/ijbs.18468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2022;25:1–15. doi: 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S, Li H, Zhang YL, Xin QL, Guan ZQ, Chen X, et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep. 2020;30(13):4370–85e7. doi: 10.1016/j.celrep.2020.02.105. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366(6472):1531–6. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong GL, Tang YH, Li WW, Cao KQ, Tan JP, Hu LF, et al. Vesicle transport related protein Synaptotagmin-1 mediates paraquat transport to antagonize paraquat toxicity. Toxicology. 2022;472:153180. doi: 10.1016/j.tox.2022.153180. [DOI] [PubMed] [Google Scholar]
- 20.Hong GL, Cai QQ, Tan JP, Jiang XZ, Zhao GJ, Wu B, et al. Mifepristone-inducible recombinant adenovirus attenuates paraquat-induced lung injury in rats. Hum Exp Toxicol. 2015;34(1):32–43. doi: 10.1177/0960327114532381. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y, Yao YM, Lu ZQ. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J Mol Med (Berl) 2019;97(4):451–62. doi: 10.1007/s00109-019-01756-2. [DOI] [PubMed] [Google Scholar]
- 22.Phan THG, Paliogiannis P, Nasrallah GK, Giordo R, Eid AH, Fois AG, et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell Mol Life Sci. 2021;78(5):2031–57. doi: 10.1007/s00018-020-03693-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang G, Li S, Zhang C, Chen H, Wang N, Feng Y. Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management. Acta Pharm Sin B. 2021;11(9):2749–67. doi: 10.1016/j.apsb.2020.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen B, Song Y, Zhan Y, Zhou S, Ke J, Ao W, et al. Fangchinoline inhibits non-small cell lung cancer metastasis by reversing epithelial-mesenchymal transition and suppressing the cytosolic ROS-related Akt-mTOR signaling pathway. Cancer Lett. 2022;543:215783. doi: 10.1016/j.canlet.2022.215783. [DOI] [PubMed] [Google Scholar]
- 25.Yamada T, Adachi Y, Fukaya M, Iijima M, Sesaki H. Dynamin-related protein 1 deficiency leads to receptor-interacting protein kinase 3-mediated necroptotic neurodegeneration. Am J Pathol. 2016;186(11):2798–2802. doi: 10.1016/j.ajpath.2016.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alzahrani B, Gaballa MMS, Tantawy AA, Moussa MA, Shoulah SA, Elshafae SM. Blocking Toll-like receptor 9 attenuates bleomycin-induced pulmonary injury. J Pathol Transl Med. 2022;56((2)):81–91. doi: 10.4132/jptm.2021.12.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep. 2013;3:1077. doi: 10.1038/srep01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang C, Wei X, Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016;7(1):11–6. doi: 10.1007/s13238-015-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q, Wei J, Liu Z, Huang X, Sun M, Lai W, et al. STING signaling sensing of DRP1-dependent mtDNA release in kupffer cells contributes to lipopolysaccharide-induced liver injury in mice. Redox Biol. 2022;54:102367. doi: 10.1016/j.redox.2022.102367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenner A, Peña-Blanco A, Salvador-Gallego R, Ugarte-Uribe B, Zollo C, Ganief T, et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 2022;41(8):e108587. doi: 10.15252/embj.2021108587. [DOI] [PMC free article] [PubMed] [Google Scholar]