

Abstract
Previous studies have developed vascularized tumor spheroid models to demonstrate the impact of intravascular flow on tumor progression and treatment. However, these models have not been widely adopted so the vascularization of tumor spheroids in vitro is generally lower than vascularized tumor tissues in vivo. To improve the tumor vascularization level, a new strategy is introduced to form tumor spheroids by adding fibroblasts (FBs) sequentially to a pre‐formed tumor spheroid and demonstrate this method with tumor cell lines from kidney, lung, and ovary cancer. Tumor spheroids made with the new strategy have higher FB densities on the periphery of the tumor spheroid, which tend to enhance vascularization. The vessels close to the tumor spheroid made with this new strategy are more perfusable than the ones made with other methods. Finally, chimeric antigen receptor (CAR) T cells are perfused under continuous flow into vascularized tumor spheroids to demonstrate immunotherapy evaluation using vascularized tumor‐on‐a‐chip model. This new strategy for establishing tumor spheroids leads to increased vascularization in vitro, allowing for the examination of immune, endothelial, stromal, and tumor cell responses under static or flow conditions.
Keywords: chimeric antigen receptor‐T cells, continuous flow, fibroblasts, tumor‐on‐a‐chip, vascularization
A new strategy to form tumor spheroids with fibroblasts in the peripheral region improves the vascularization level of the tumor‐on‐a‐chip model. This vascularized tumor model can be used for molecule and cell perfusion studies of drug transport kinetics, cell trafficking, and chimeric antigen receptor‐T cell response evaluation.
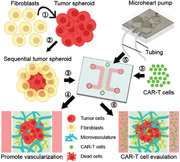
1. Introduction
Tumor vasculature is a key component of the tumor microenvironment. Nutrients, oxygen, therapeutic drugs, and immune cells are transported through tumor vessels to the tumor site. These tumor vessels have unique features that affect the efficiency of chemotherapy and immunotherapy.[ 1 , 2 ] To better understand tumor microenvironments, develop potent anti‐cancer drugs, and establish efficient immunotherapies, there is a need for physiologically relevant in vitro models that can better serve as reliable tumor modeling tools. Microphysiological systems have recently attracted tremendous attention in organ and disease modeling. There have been several reviews summarizing in vitro tumor models, especially tumor‐on‐a‐chip models with vasculature.[ 3 , 4 , 5 , 6 , 7 , 8 ] These vascularized tumor models are used to study drug delivery and screening,[ 9 , 10 ] nanoparticle assessment,[ 11 ] vasculature activation,[ 12 ] and immune cell function (e.g., monocyte,[ 13 ] macrophage,[ 14 ] natural killer cell,[ 15 ] T cell[ 16 ]).
In these vascularized tumor models, tumor cells embedded in a 3D matrix,[ 17 , 18 ] tumor cell monolayers,[ 16 ] microtumors,[ 19 , 20 , 21 ] and tumor spheroids,[ 22 ] or organoids[ 23 ] are used. Among these different tumor model formats, the tumor spheroids have their own unique features. For example, tumor spheroids include 3D cues lacking in tumor cell monolayers, and have a higher cell population complexity that can be formed as tumor cells alone or in combination with other cell types (such as, fibroblasts (FBs),[ 22 ] endothelial cells (ECs),[ 24 ] and pericytes[ 11 ]). Compared to organoids, tumor spheroid models are easier to generate and can be better controlled. Thus, tumor spheroid is a useful format to generate vascularized tumor models in vitro.
Various methods have been used to produce vasculature in the tumor‐on‐a‐chip models including formation of an endothelial monolayer that lines a large‐diameter channel,[ 25 , 26 ] angiogenic sprouting,[ 27 ] or vasculogenesis.[ 9 , 28 ] Although these methods produce vessels that are close to the tumor region, the general tumor vascularization level in most studies is still lower than what is observed in vivo. Thus, there exists a need to improve the vascularization level of tumor‐on‐a‐chip models to mimic better the blood vessel‐rich tumor microenvironment in vivo.
Previous studies have indicated that FBs play a beneficial role in tumoral vascularization in vitro. For instance, co‐seeded FBs in a side‐channel could induce angiogenesis toward the tumor.[ 13 ] Also, tumor spheroids composed of MCF7 or Eca‐109 cells mixed with FBs induced angiogenesis and vessel growth into the tumor spheroids.[ 29 , 30 ] Although it has been shown that FBs are beneficial for tumoral vascularization in vitro, it is not clear whether the location of FBs in the tumor spheroid would further influence tumoral vascularization.
Chimeric antigen receptor (CAR)‐T cell therapy is a powerful tool in cancer treatment, especially for hematological diseases.[ 31 , 32 ] However, many challenges such as modest anti‐tumor activity, limited tumor infiltration, and restricted trafficking still limit the therapeutic efficacy of CAR‐T cells in solid tumors and hematological malignancies.[ 33 , 34 ] Furthermore, CAR‐T cell function could be suppressed by the tumor microenvironment.[ 35 , 36 , 37 ] CAR‐T cells also respond differently in various patients, as cancer patients have unique pathological features including gene expression profile, tumor microenvironment composition, treatment history, etc.[ 32 , 33 ] In order to overcome these challenges, new strategies to engineer, screen, and characterize more powerful CAR‐T cells with improved anti‐tumor activity and decreased toxicity for personalized medicine are necessary. In addition to the traditional systems, for example, cell co‐culture in dish and xenografts in mice, tumor‐on‐a‐chip systems could be a powerful tool that contributes to these aims. For example, tumor spheroids and organoids have been used to evaluate CAR‐T cell infiltration and cytotoxicity[ 38 , 39 , 40 ] and 3D tumor models have explored CAR‐T cell function under hypoxic conditions.[ 41 ] Mouse T cells have been perfused through a multilayer‐based microfluidic device to mimic CAR‐T cell infiltration into the tumor region.[ 16 ] As a pre‐clinical step, evaluating CAR‐T cell responses in a tumor model that recapitulates tumor microenvironments by using human cells, adding various other cell types, and including chemical and physical features could reduce the risk of clinical failure. However, there is currently no vascularized tumor model for CAR‐T cell response evaluation.
In this study, we introduce a new strategy to generate tumor spheroids by combining FBs either initially, mixed with tumor cells (co‐mixed) or after formation of the tumor spheroid (sequential) (Figure 1a). FBs in the co‐mixed spheroid are distributed uniformly or enriched in the center, whereas they tend to be located peripherally in the sequential tumor. By comparing the vascularization level of tumor spheroids made with tumor cells alone, tumor cells co‐mixed with FBs, and the sequential strategy, we found that sequential tumor spheroids have the highest vascularization level. Importantly, more vessels in the sequential tumor regions are perfusable. Finally, we demonstrate the advantages of this method by adding a micropump to circulate CAR‐T cells in the vascularized tumor spheroid and evaluate responses under continuous flow.
Figure 1.
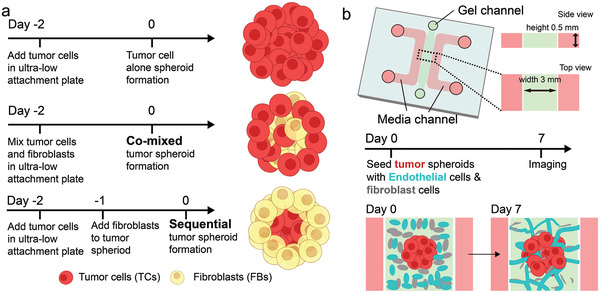
Three methods of generating tumor spheroids for the vascularized tumor microfluidic model. a) Experimental procedures to form a tumor spheroid with tumor alone, a co‐mixed spheroid, and a sequential spheroid. b) Schematic diagram of a microfluidic device to create a vascularized tumor spheroid.
2. Results
2.1. Three Methods to Generate Tumor Spheroids for Vascularized Tumor‐on‐a‐Chip Models
We first found that spheroids containing FB alone are highly vascularized in vitro (Figure S1, Supporting Information). This led us to consider the contribution of FBs to the vascularization of tumor spheroids. To test this concept and further evaluate the contribution of the physical location of the FB within tumor spheroid on vascularization, we generated tumor spheroids of three types: 1) With tumor cells alone, 2) with tumor cells and FBs mixed from the beginning (co‐mixed), 3) by forming the tumor spheroid first and then adding FBs to the pre‐formed tumor spheroid to generate a higher concentration of FBs in the outer layers (sequential) (Figure 1a). These tumor spheroids were then seeded in a microfluidic device immersed in a gel solution containing ECs and FBs (Figure 1b). A microvascular network (MVN) comprised of these vascular cells subsequently formed around the tumor spheroid within 7 days.
2.2. Sequential Tumor Spheroids Show Superior Vascularization
We first tested the abovementioned three methods for tumor spheroid formation in a kidney cancer cell line, SN12C. In vivo, SN12C induced microvessel formation in the severe combined immunodeficient mouse model at the density about 10 blood vessels per high power field image at 400× magnification,[ 42 ] and induced about 30 vessels per image field at 200× magnification in nude mice.[ 43 ] In vitro, SN12C formed spheroids when seeded alone (Figure 2a). When SN12C cells were co‐mixed with FBs, the majority of FBs were located at the center of the spheroid, while SN12C were in the peripheral region (Figure 2a), similar to previous studies using different tumor types.[ 44 ] On the contrary, in the sequential tumor spheroids, FBs were in the peripheral region (Figure 2a). When seeded in the microfluidic devices, both the tumor alone and the co‐mixed tumor spheroid groups resulted in vessels excluded from the tumor region (Figure 2b). Thus, we analyzed the vessel percentage not only in the tumor spheroid region, but also the nearby 50 µm distance region as a measure for tumor spheroid vascularization (Figure 2c). Tumor spheroids formed by the sequential method showed the highest vascularization level in comparison to the other methods (Figure 2b–e). Unlike the tumor alone and co‐mixed groups, vessels were found in the tumor region and nearby 50 µm distance region of the sequential tumor spheroid (Figure 2d,e), although the majority of the vessels were at the periphery of the tumor spheroid. Moreover, tumor spheroid‐associated FBs in the sequential tumor spheroid had the largest area across the three types of tumor spheroids (Figure 2f), indicating a strong microenvironmental remodeling and potentially attracting more ECs to increase vascularization, thus better mimicking a vascularized tumor in vivo. We repeated these experiments using cancer patient‐derived thyroid FBs and found similar results (Figure S2, Supporting Information).
Figure 2.
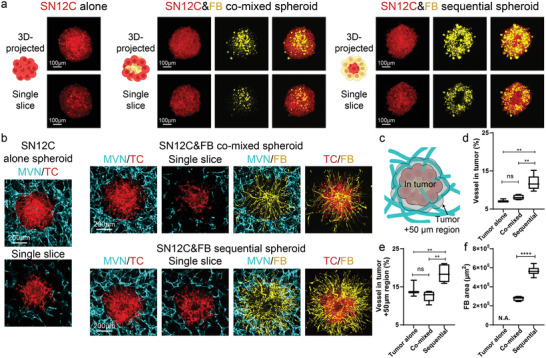
Sequential SN12C tumor spheroid shows the highest vascularization level. a) Schematic, 3D‐projected image, and single slice image of SN12C tumor spheroids formed by tumor alone, co‐mixed, or sequential methods. b) 3D‐projected images of vascularized tumor spheroid formed by tumor alone, co‐mixed, or sequential methods. TC is short for tumor cells. c) Schematic showing the tumor region and nearby 50 µm distance region for vascularization analysis. d) Statistical analysis of vessel percentage in the tumor region or e) tumor +50 µm region. f) Area analysis of FBs from tumor spheroid. Bars represent mean ± S.D. One‐way ANOVA was performed for the statistical comparison in (d) (p < 0.001) and (e) (p < 0.001). Significance was determined using Tukey's multiple comparisons test of mean values between each group. Two‐tailed t tests were performed for the statistical comparisons in figure (f). Data were collected from at least 6 tumor spheroids for each group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We further confirmed our findings using two additional cancer cell lines, lung cancer (H69M) and ovarian cancer (OV90). In vivo, subcutaneously inoculated H69M induced high levels of vessel density (38 to 50 CD31 positive structures per sample slice).[ 45 ] In our system, H69M cells alone were able to form tumor spheroids. In the co‐mixed tumor spheroid, FBs and H69M cells were evenly distributed, while FBs were more concentrated in the peripheral region of sequential tumor spheroids (Figure S3a, Supporting Information). As time progressed, the level of vascularization of sequential tumor spheroids became significantly higher than the other methods (day 7 and 11) (Figure S3b,c, Supporting Information). We also noticed vessel regression in all the groups with the least regression in the sequential tumor group at later time points (Figure S3b, Supporting Information), which might partially explain why the vascularization levels in both tumor alone spheroid and co‐mixed tumor spheroid decreased on days 7 and 11 compared to day 4 (Figure S3d, Supporting Information). FBs in co‐culture in the sequential tumor spheroid migrated more in the microfluidic device compared with the ones in co‐mixed tumor spheroid group (Figure S3e, Supporting Information).
The ovarian cancer cell line, OV90, induced about 7 microvessels per image field at 200x magnification in nude mice.[ 46 ] When seeded in the ultra‐low attachment (ULA) plate, OV90 was able to form tumor spheroids alone (Figure S4a, Supporting Information). In the co‐mixed tumor spheroid, the majority of FBs gathered in the center of the tumor spheroid, with few FBs located at the periphery (Figure S4a, Supporting Information). Interestingly, when FBs were sequentially added to the OV90 spheroids, FBs formed two large clusters at the edges of the tumor spheroid (Figure S4a, Supporting Information). After 7 days seeded in the microfluidic devices, tumor spheroids were surrounded by vessels in all three groups with the highest vessel percentage in the sequential tumor spheroid group (Figure S4b–d, Supporting Information). In the microfluidic devices, unlike SN12C or H69M, most of the FBs were excluded from the OV90 spheroids in the co‐mixed condition (Figure S4b, Supporting Information), implicating a unique interaction between FBs and OV90 tumor cells.
These results suggest that tumor spheroids formed by the sequential strategy are better vascularized, and better mimic the vessel‐rich tumor microenvironment in vivo.
2.3. Perfusion is Better in Vascularized Sequential Tumor Spheroids than Co‐mixed Tumor Spheroids
Next, to evaluate tumor vessel perfusability, we focused on the SN12C tumor spheroids and perfused them by adding fluorescent dextran in the MVNs (Figure 3a). Dextran was detected in the distal region of MVNs in both the co‐mixed spheroid and sequential spheroid, indicating the high perfusability of the MVNs far away from the tumor spheroid (Figure 3b). However, perfusable vessels close to the tumor spheroid were only found in the sequential tumor spheroid group (Figure 3b,c). This indicated that the spatial arrangement of FBs at the periphery of the tumor spheroid has a profound effect on more vessels are perfusability of vessels close to tumor spheroids.
Figure 3.
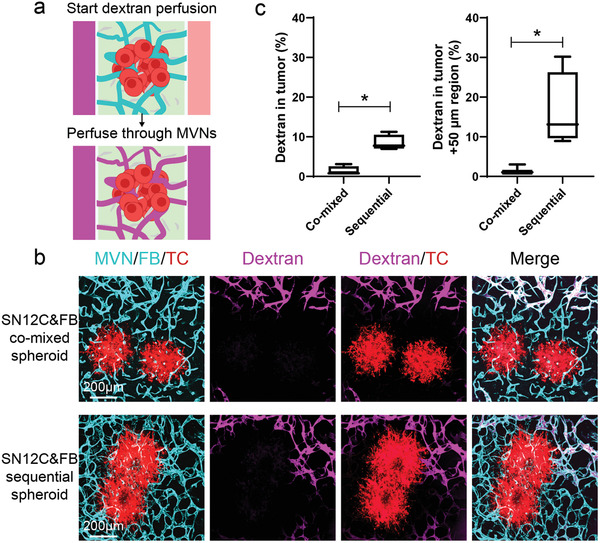
Perfusability of tumoral vessels in co‐mixed and sequential tumor spheroids. a) Schematic showing dextran perfusion in tumor vessels. b) Representative confocal images of vascularized SN12C co‐mixed or sequential tumor spheroids perfused with fluorescein dextran (10 kDa) on day 7. Device was imaged within 5 min after perfusing dextran. c) Statistical analysis of dextran percentage in the tumor region and tumor +50 µm region. Bars represent mean ± S.D. Two‐tailed t tests were performed for the statistical comparisons. Data were collected from at least 6 tumor spheroids for each group. *p < 0.05.
2.4. Sequentially‐Formed Vascularized Tumor Spheroids Recruit more T Cells under Continuous Flow
Lastly, we demonstrated how these vascularized tumor models can be used to evaluate CAR‐T cell recruitment, killing capacity, and inflammatory response. It has been shown that mesothelin CAR‐T cells respond to ovarian cancer cell line Skov3.[ 47 , 48 ] Thus, we tested mesothelin CAR‐T cells in a Skov3 vascularized tumor‐on‐a‐chip model. In previously published nude mouse models, Skov3 induced about 17 microvessels per imaging field (100× magnification)[ 49 ] to 23 microvessels per imaging field (160× magnification).[ 50 ] In our platforms, similar to the other tumor cell lines, Skov3 cells formed tumor spheroids by themselves (Figure 4a), and when co‐seeded, Skov3 cells and FBs were evenly distributed through the spheroid. When seeded with the sequential method, Skov3 cells were more concentrated in the center while FBs were in the peripheral region (Figure 4a). Similar to previous tumor cell lines, Skov3 tumor spheroids made with the sequential method showed the highest vascularization level (Figure 4b,c).
Figure 4.
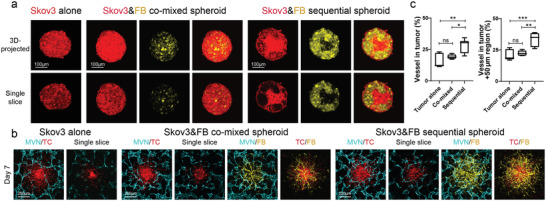
Vascularized Skov3 tumor spheroid model. a) Representative confocal images of Skov3 tumor spheroids formed by the three different methods. b) Representative confocal images of vascularized Skov3 tumor spheroids on day 7. c) Statistical analysis of vessel percentage in the tumor region (left) and tumor +50 µm region (right). Bars represent mean ± S.D. One‐way ANOVA was performed for the statistical comparison. Vessel in tumor, p < 0.01 and tumor +50 µm region, p < 0.001. Significance determined by Tukey's multiple comparisons test of mean values between each group. Data were collected from at least 5 tumor spheroids for each group. *p < 0.05, **p < 0.01, ***p < 0.001.
To mimic the circulation of T cells in vivo, a recirculating pump[ 51 ] was connected to the microfluidic device to introduce continuous flow for 4 days (Figure 5a–c). Control T cells or CAR‐T cells (about 25% CAR positive rate, data not shown) were perfused from the media channel, from which they flowed into the tumoral MVNs, extravasated, and interacted with tumor cells (Figure 5a–c). Both dead cell staining and cytokine secretion were used to assess T cell response. In the sequential tumor spheroid group, CAR‐T cells flowed into the MVNs, adhered to the apical EC surface (Figure 5d, Movie S1, Supporting Information), extravasated (Figure 5e, Movie S2, Supporting Information), and migrated to the tumor region (Figure 5f). After 96 h of perfusion, higher T cell and dead cell densities were found in the CAR‐T cell groups than in the control T cell group in both co‐mixed and sequential tumor spheroids (Figure 6a–c). Also, more T cells were recruited in the sequential tumor spheroids than the co‐mixed tumor spheroids, probably due to the increased vascularization (Figure 6a,b). The higher CAR‐T cell density in the sequential tumor spheroid group led to a higher dead cell density in the tumor region (Figure 6a,c). Media from devices were analyzed for IFNγ using the enzyme‐linked immunoassay (ELISA) and Luminex multiplex ELISA which revealed a stronger response from CAR‐T cells in the sequential tumor spheroid group (Figure 6d,e). All these data demonstrate that this vascularized tumor spheroid model can effectively be used to evaluate CAR‐T cell responses to tumor cells in vitro.
Figure 5.
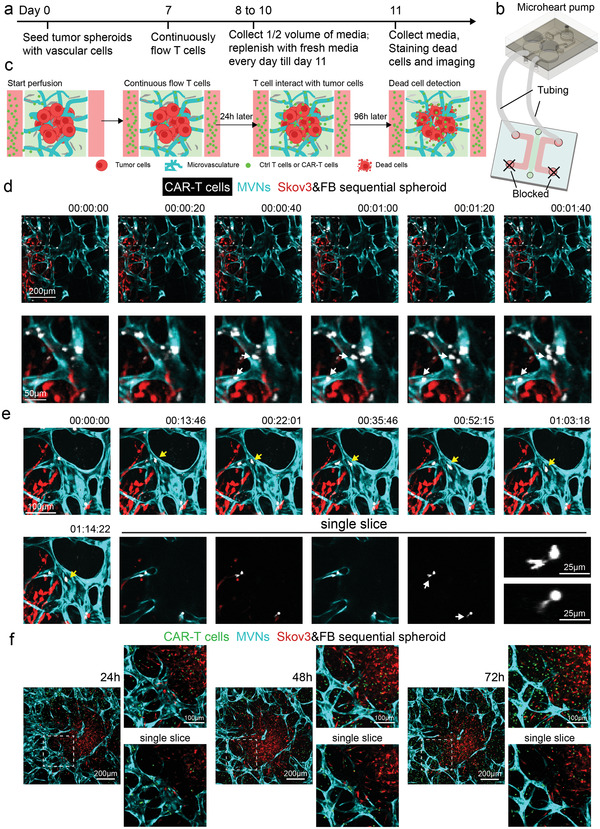
Vascularized tumor spheroid model can be used to study CAR‐T cell transport. a) Experimental procedure for continuous flow of T cells in vascularized tumor spheroid models. Control T cells or CAR‐T cells are perfused on day 7 and recirculated for 4 days. Half‐volume of conditioned media in the devices is collected every 24 h and replenished. On day 11, devices are stained with Nuclear Blue DCS1 to indicate dead cells. b,c) Sketch of continuously flow T cells in the vascularized tumor spheroid model. One reservoir of each media channel is connected to the microheart pump by tubing, while the two other reservoirs are blocked to create a closed loop and continuous flow. d) Time lapse images of CAR‐T cells flowing into the MVNs. A majority of the CAR‐T cells travel through the MVNs quickly with some of the cells adhering to the apical surface of the vessels close to the tumor region. White arrows point to freshly adhered T cells during the time of imaging. e) Time lapse images of CAR‐T cells migration and extravasation. Yellow arrows identify a T cell patrolling in the vessels. Single slice images highlight T cells undergoing extravasation. White arrows identify the portions of the cell that have extravasated. Zoomed‐in images of these two T cells are provided. f) Representative images of CAR‐T cell responses in the vascularized sequential tumor spheroid group at 24, 48, and 72 h time points.
Figure 6.
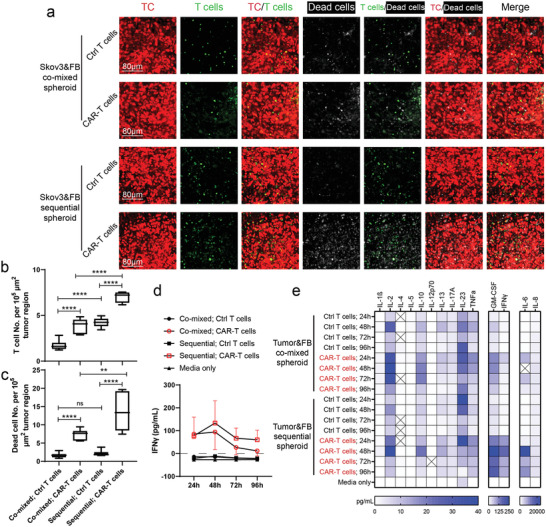
Vascularized tumor spheroid model can be used to assess CAR‐T cell killing efficiency and cytokine secretion. a) Representative 3D projected confocal images of T cells and dead cells in vascularized tumor spheroids formed by co‐mixed or sequential methods. ECs were not labeled. Statistical analysis of b) T cell or c) dead cell number per 105 µm2 tumor region. Bars represent mean ± S.D. Two‐tailed t tests were performed for the statistical comparisons. Data were collected from at least 6 tumor spheroids for each group. **p < 0.01, ****p < 0.0001. d) IFNγ concentration of conditioned media in each group over 96 h. Samples were collected from 3 devices for each group and tested using traditional ELISA methods. e) Luminex multiplex ELISA assay of conditioned media from co‐mixed or sequential tumor spheroid group perfused with control T cells or CAR‐T cells for 24, 48, 72, and 96 h. Pooled media from 3 devices in each group was used for analysis.
3. Discussion
Tumor sites are highly vascularized in vivo, so it is imperative that tumor model systems containing higher fractions of functional vasculature should be used in tumor studies that explore drug delivery to the tumor, immune cell trafficking, and immunotherapy. Building on previous studies demonstrating that FBs positively impact tumoral vascularization in vitro,[ 13 ] we find that adding FBs to a pre‐formed tumor spheroid further improves tumor vascularization in vitro. The developed vasculature is perfusable, and when combined with a pumping system it provides for the continuous circulation of immune cells, which could be used for pre‐clinical CAR‐T cell evaluation. We propose that the increased tumor vasculature in sequentially formed tumor spheroids is a consequence of the location and cell number of FBs. In sequential tumor spheroids, FBs tend to be concentrated at the periphery of the tumor spheroid so they are better able to communicate with ECs in the surrounding extracellular matrix in a microfluidic device. These FBs may provide growth factors, such as vascular endothelial growth factor (VEGF), fibroblast growth factor, and platelet‐derived growth factor, and matrix substrates for ECs to initiate an angiogenic response.[ 52 , 53 , 54 ] In contrast, FBs concentrated at the center of the co‐mixed tumor spheroids have restricted access to the surrounding vasculature causing fewer vessels to be formed in close proximity to the tumor. In addition, more FBs were found in the sequential tumor spheroids than the co‐mixed ones made with SN12C or Skov3 cell lines (Figure S5, Supporting Information), which would further contribute to EC recruitment. The higher FB number in the sequential tumor spheroids also results in the larger area corresponding to FBs in the microfluidic device (Figure 2b).
An interesting phenomenon is that the location of FBs is different in tumor spheroids of different tumor cell types. We used 7 types of tumor cells to form tumor spheroids using our co‐mixed or sequential methods (Figure S6, Supporting Information). When co‐mixed with SN12C, OV90, A375, or MCF7, FBs are located in the center of the spheroid, but when co‐mixed with H69M, Skov3, or HepG2, FBs are more evenly distributed (Figure S6, Supporting Information). When subsequently added to a pre‐formed tumor spheroid made of SN12C, H69M, or Skov3, FBs locate in the peripheral region with a shell‐like structure. When added to OV90 or A375 spheroids, while FBs are still concentrated in the peripheral region, they tend to form clusters. And when FBs were added to HepG2 or MCF7 spheroids, FBs migrate inward from the peripheral region, and within 24 h form embedded FB aggregates that are covered by tumor cells (Figure S6, Supporting Information). These location differences imply distinct relationships between different tumor cells and FBs. Such patterning between two cell types has been extensively studied through both experimental and computational models, indicating the important roles of adhesion molecules and motor proteins.[ 55 , 56 , 57 , 58 ] One explanation for these differences is that different tumor cell lines regulate FB function in distinctive ways, for example, there were fewer FBs in SN12C co‐mixed spheroids than in H69M or OV90 co‐mixed tumor spheroids (Figure S5, Supporting Information), suggesting that the proliferation rate of FBs may be regulated differently depending on tumor types. It is also likely that the type and amount of adhesion molecule expression varies among tumor cell lines, thus affecting the relative FB‐FB versus FB‐ tumor cell affinities. The relative ability of the different tumor cell lines to form spheroids is a clear indication of different TC‐TC adhesive properties. Most of the tumor cell lines tested in this study are epithelial cells in origin, so E‐cadherin expression is critical.[ 59 ] Also, members of the CD44 family are often important and have been extensively studied.[ 60 , 61 , 62 ] Tumor cell‐FB and tumor cell‐matrix adhesions are mediated by a variety of adhesion processes involving members of the integrin family as well as heterotypic E‐cadherin/N‐cadherin bonds, among others.[ 61 ] Another factor is the role that integrin activation plays in regulating cell‐secreted matrix.[ 63 ] FBs secrete matrices to provide adhesion sites for both tumor and FBs when cultured in the ULA plate. The expression profiles of adhesion molecules that match to these matrices may vary in these tumor cell lines. Such adhesion molecule differences have been found to be essential in morphogenesis.[ 64 , 65 ] A future study that systematically investigates the adhesion molecules in tumor spheroids formed by co‐mixed or sequential methods would serve to better elucidate the interaction between tumor cells and stromal cells, and further improve the vascularized tumor‐on‐a‐chip model. To study primary tumor cells using our tumor spheroid formation methods, these different patterns of tumor cell/FB distribution probably will also be observed because of the following reasons. Primary tumor cells are able to convert normal FBs to different phenotypic cancer‐associated FBs through various mechanisms[ 66 ] and primary tumor cells have distinct adhesion molecule expression profile.[ 67 ] Such pattern differences may limit the applicability of our methods, as discussed at the end of discussion.
An important result of this study is that more control T cells and CAR‐T cells are recruited to the tumor region in the sequential tumor group compared to the co‐mixed group. Certainly, the increased vascularization level and improved perfusion vessels close to the tumor region in the sequential tumor group is a contributing factor. T cells are transported through these vasculatures under continuous flow, where they have a higher probability of adhering to and extravasating into the tumor region. Another possible contributing factor is that the immunosuppressive microenvironment has not been established, because only human normal lung FBs were used to form tumor spheroids to test CAR‐T cell efficiency. It is possible that these FBs have not been converted to immunosuppressive, cancer‐associated FBs. Future studies using tissue‐specific stromal cells and patient‐derived stromal cells, such as cancer‐associated FBs, to generate MVNs and tumor spheroids, respectively, would better recapitulate tumor microenvironments in vivo.
Luminex multiplex ELISA results showed that GM‐CSF, IFNγ, IL‐6, and IL‐8 were highly secreted relative to other cytokines. The concentration of IL‐6, IFNγ, and GM‐CSF were higher when CAR‐T cells were perfused into the MVNs embedded with the sequential tumor spheroids than the co‐mixed tumor spheroids. Such concentration differences may be caused by the tumor cell interaction dependent CAR‐T cell activation. As discussed above, the increased vascularization in the sequential tumor group lead to a high accessibility of CAR‐T cells to tumor cells. Such interactions resulted in the enhanced cytokine secretion. Another possible factor contributing to such differences is the vascular cell responses to CAR‐T cell secreted cytokines. For example, GM‐CSF has been found to be produced by FBs and ECs upon immunogenic stimuli.[ 68 ] In the clinical setting, CAR‐T cell cytokine release syndrome (CRS) is critical for balancing tumor treatment efficacy and patient safety. These elevated cytokines are not only secreted by CAR‐T cells, but usually released by other immune cells, such as macrophages, when responding in part to CAR‐T cell activation.[ 69 ] IL‐6, IFNγ, and GM‐CSF have been implicated as elevated cytokines during CRS, and blocking these cytokines has proven to reduce CRS and improve CAR‐T cell function.[ 70 , 71 , 72 , 73 ] Our system has the potential to be used for CRS prediction and cytokine blocking tests by including other immune cell types in the microfluidic device. Both the IFNγ ELISA and Luminex multiplex ELISA results showed decreased cytokine secretion in CAR‐T cell groups at 72 and 96 h, compared to 48 h. This could be due to exhaustion of the CAR‐T cells at the later time points.[ 74 ] Moreover, the culture medium has not been optimized for a long‐term culture to maintain T cell activity. Future studies using this vascularized tumor model to evaluate T cell activation under various culture conditions with direct comparison to in vivo experiments are needed for full validation.
In this work, only lung FBs and patient‐derived thyroid FBs are used to form MVNs. It has been shown that lung FBs, dermal FBs, patient‐derived thyroid FBs, pericytes, bone marrow‐derived mesenchymal stem cells, and astrocytes with brain pericytes can support MVN formation in vitro.[ 75 , 76 , 77 ] In the future, organotypic matched stromal cells can be used to model vascularized tumor from different organs. Also, fibrin gel is used to support MVN formation in this study, consistent with its use in other self‐organized perfusable MVNs formed in microfluidic devices.[ 78 , 79 , 80 , 81 , 82 ] Some studies, however, have used mixed matrices, such as fibrin/collagen and fibrin/matrigel mixture. Kim and colleagues were able to form perfusable MVNs in fibrin gel mixed with a low concentration of collagen I,[ 83 ] but others either failed[ 84 ] or found that pure fibrin gel scaffolds are preferable to collagen gel or collagen/fibrin combinations, because it significantly reduces matrix retractions during MVN maturation.[ 85 ] Other studies used synthesized matrices,[ 86 , 87 ] however the perfusability of these MVNs was not demonstrated. Although vascular cells were seeded initially in fibrin gel in the present work, these cells are able to remodel the matrix by secreting matrices, such as collagen I, collagen IV, laminin, and elastin.[ 88 , 89 , 90 ] Future studies forming MVNs with primary vascular cells from patients or seeding in patient‐derived matrix would improve the pathological relevance of the models.
Although we have shown that the sequential tumor spheroid method benefits vascularization using four different tumor cell lines (SN12C, H69M, OV90, and Skov3), this sequential strategy has limitations. First, this method requires a pre‐formed tumor aggregate. If tumor cells cannot form a spheroid by themselves, as is the case for MD‐MBA‐231 (Figure S7, Supporting Information), this sequential tumor spheroid method has little effect. Second, the interaction between FBs and tumor cells is dynamic, and it is possible that the peripheral FBs in the sequential tumor spheroid may continue to evolve and approach a distribution more similar to the co‐mixed spheroid and we found that in some cases the FB distribution in the sequential and co‐mixed spheroids converge over time (data not shown). Thus, to benefit from the beneficial FB distribution in the sequential method, the spheroids would need to be seeded into microfluidic devices earlier, within about 2 days to avoid FB relocation to the center. However, this short co‐culture time would lead to a less compacted spheroid. Third, in some instances (e.g., HepG2 and MCF7), FBs migrate inward from the peripheral region in as little as 24 h. Once this happens, the benefits of the sequential method wane and the vascularization level may not be increased. All these limitations should be considered when testing a new type of tumor cell, especially for primary tumor samples, for this vascularized tumor‐on‐a‐chip model.
4. Conclusion
In summary, forming tumor spheroids with sequentially added FBs improves vascularization, which mimics better the highly vascularized tumor microenvironment in vivo. This vascularized, sequential tumor spheroid model can be used to enhance both drug delivery and cell trafficking, producing a vascularized in vitro model of drug transport kinetics, cell trafficking, and CAR‐T cell responses.
5. Experimental Section
Cell Culture
Immortalized human umbilical vein endothelial cells (ImHUVECs) and ImHUVECs expressing green fluorescent protein or blue fluorescent protein (BFP, P20‐P30)[ 88 ] were cultured in VascuLife VEGF Endothelial Medium (Lifeline Cell Technology). Lung FBs (Lonza, P7) and lung FBs expressing BFP (P7) were cultured in FibroLife S2 Fibroblast Medium (Lifeline Cell Technology). Cancer patient‐derived FBs were isolated from patient thyroid cancer surgical resections in accordance with a protocol approved by the Institutional Review Board of Dana Farber Cancer Institute (approval number 09‐472 following our previous protocol.[ 75 ] Briefly, specimens were minced, followed by collagenase P, DNAse, and dispase digestion. Patient‐derived FBs were cultured in a gelatin‐coated flask with FibroLife S2 Fibroblast Medium (Lifeline Cell Technology). SN12C expressing red fluorescent protein (RFP), OV90 expressing mCherry, A375 expressing mCherry, and MCF7 expressing mCherry were cultured in DMEM (Thermo Fisher) with 10% FBS. H69M cells expressing mCherry were cultured in RPMI1640 (Thermo Fisher) with 10% FBS and 1% penicillin‐streptomycin (Sigma). Skov3 tumor cells expressing RFP were cultured in McCoy's 5A (modified) medium with 10% FBS (Thermo Fisher). HepG2 expressing mCherry were cultured in EMEM media with 10% FBS. Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor's blood by the monocyte core at MIT. PBMCs were treated with Dynabeads Human T‐Activator CD3/CD28 for T Cell expansion and activation (Thermo Fisher), along with IL‐2 (18.3 ng mL−1), IL‐7 (5 ng mL−1), and IL‐15 (5 ng mL−1) in RPMI1640 with 10% heat inactivated FBS for 6 days. On day 2, expanded cells were infected with lentivirus of mouse single chain anti‐human mesothelin specific CAR overnight. Dynabeads were removed on day 6, and transfected cells were further cultured for another 2 days without cytokines. On day 9, 99% of cells were CD3 positive (BioLegend) and ready for experiments.
Tumor Spheroid Formation
Tumor cells and FBs were detached and then resuspended in tumor cell culture media at 5 × 104 cells mL−1. Cells were cultured in ULA 96‐well plates (Wako Chemicals USA). For tumor cells alone spheroid, 50 µL of tumor cells (2.5 × 103 cells) were loaded in each well of the ULA plate, and an additional 50 µL tumor cell suspension (2.5 × 103 cells) were added (in total 100 µL, 5 × 103 cells), and cultured for 2 days. For co‐mixed tumor spheroid, 50 µL of tumor cells (2.5 × 103 cells) followed by 50 µL of FBs (2.5 × 103 cells) were loaded in each well of the ULA plate, and cultured for 2 days. For the sequential tumor spheroid, 50 µL of tumor cells (2.5 × 103 cells) were first loaded to ULA plate and culture for 24 h, and then 50 µL of FBs (2.5 × 103 cells) were added to the tumor spheroid in each well of ULA plate and cultured for another 24 h. Prior to spheroid imaging, these spheroids were fixed and cleared using RapiClear 1.49 (SUNJin Lab Optical Clearing Innovation). Patient‐derived FBs were stained with CellTracker Green (Thermo Scientific) to detect cell location. For FB cell number analysis, tumor spheroids were fixed, permeabilized with 2% Triton X‐100, and then stained with Nuclear Green DCS1 (Cayman Chemical). Nuclei were counted using Imaris software (Oxford Instruments).
Microfluidic Device Fabrication
Microfluidic devices were assembled following previous publications.[ 9 ] In short, PDMS made of a 10:1 ratio of base to cross‐linker (Ellsworth) was cured in a mold at 60 °C for a minimum of 2 h. Devices were sterilized prior to air‐plasma bonding (Harrick) to clean glass slices, followed by subsequent curing overnight. This microfluidic device contains three parallel channels: A central gel channel flanked by two media channels. Partial walls separate the fluidic channels and serve to confine the liquid gelling solution in the central channel by surface tension before polymerization. The gel channel is 3 mm wide and 0.5 mm tall.
Vascularized Tumor‐on‐a‐Chip Seeding
Tumor spheroids were co‐seeded with ECs and FBs into the device as previously described.[ 88 ] Briefly, tumor spheroids were transferred to microcentrifuge tubes from the ULA plate using a large orifice 200 µL pipette tip. Supernatant was removed carefully leaving tumor spheroids in the bottom of the tube. ECs and FBs were concentrated in VascuLife containing thrombin (4 U mL−1). Cell mixture solution was added into the tube containing tumor spheroids, and then further mixed with fibrinogen (3 mg mL−1 final concentration) at a 1:1 ratio and quickly pipetted into the device through the gel inlet with a final concentration of 7 × 106 mL−1 for ECs and 1 × 106 mL−1 for FBs. The device was placed in a humidified tip box to polymerize at 37 °C for 15 min in a 5% CO2 incubator. After fibrin gel was cured, VascuLife culture medium was added and changed daily in the device. After 7 days, vascularized tumor spheroid devices were ready for further experiments.
Microvascular Network Perfusion, Imaging, and Analysis
To confirm the perfusability of MVNs in tumor devices, the culture medium in one media channel was aspirated, followed by injection of 100 µL of 10 µg mL−1 10 kDa MW fluorescein dextran solution (Invitrogen). The process was then repeated for the other media channel. The device was imaged within 5 min under a confocal microscope. For confocal imaging, an Olympus FLUOVIEW FV1200 confocal laser scanning microscope with a 10× objective was used. Z‐stack images were acquired with a 5 µm step size. All images shown are collapsed Z‐stacks, displayed using range‐adjusted Imaris software, unless otherwise specified. Vessel percentages in the tumor region and tumor nearby 50 µm distance region were measured using ImageJ (NIH, U.S., demonstrated in Figure S8, Supporting Information). FB area was also quantified using ImageJ (NIH, U.S.).
T Cell Perfusion under Continuous Flow in the Vascularized Tumor Device
Control T cells or CAR‐T cells were labeled with CellTracker Green (Thermo Scientific) and then resuspended at a concentration of 1 × 106 mL−1 in culture medium (50% volume of VascuLife and 50% volume of RPMI1640 mixture). T cell suspension was primed into microheart pump,[ 51 ] and then connected to one reservoir of each media channel in the microfluidic device through tubing. The other reservoir of each media channel was blocked. After removing any air bubbles in the system, the micropump was started to maintain continuous flow. Half the volume of conditioned media (200 µL) was collected from devices every 24 h for 4 days and used for IFNγ ELISA (R&D systems) and Luminex ELISA assay (Eve Technologies). Fresh media was added after conditioned media collection. 96 h later, devices were stained with Nuclear Blue DCS1 (AAT Bioquest) to detect dead cells and imaged after washing.
Statistical Analysis
All error bars are shown as mean ± SD. Statistical analysis was conducted by Student's t‐test or one‐way ANOVA with GraphPad Prism. Tukey's multiple comparisons test was used, comparing the mean of each group with every other group as the post hoc test method for one‐way ANOVA analysis. Significance is shown in each figure. Sample numbers and p‐values are provided in the figures or figure legends.
Conflict of Interest
R.D.K. is the co‐founder of and holds a significant financial interest in AIM Biotech, a company that produces microfluidic devices. R.D.K. also receives research support from Amgen, Novartis, Boehringer Ingelheim, GSK, AbbVie, and Roche. D.A.B. is a co‐founder of and holds a significant financial interest in Xsphera Biosciences, and is a consultant for Qiagen/N of One. D.A.B. has also received research funding from Novartis, Gilead Sciences, BMS, and Lilly Oncology.
Supporting information
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Acknowledgements
Z.W. and M.F. contributed equally to this work. Z.W. is supported by a Ludwig Center Fund Post‐Doctoral Research Fellowship from the Koch Institute for Integrative Cancer Research and by National Cancer Institute (U01 CA214381). C.X. is supported by funds from National Natural Science Foundation of China (81903158 and 32170940), Guangzhou Science and Technology Project (201904010042), and Guangdong Natural Science Foundation (2022A1515012658). This work was supported by a National Cancer Institute Physical Sciences‐Oncology Network supplement to award U01 CA214381. The PBMCs were kindly provided by Prof. Bryan Bryson. This work was supported in part by the Koch Institute Support (core) Grant P30‐CA14051 from the National Cancer Institute.
Wan Z., Floryan M. A., Coughlin M. F., Zhang S., Zhong A. X., Shelton S. E., Wang X., Xu C., Barbie D. A., Kamm R. D., New Strategy for Promoting Vascularization in Tumor Spheroids in a Microfluidic Assay. Adv. Healthcare Mater. 2023, 12, 2201784. 10.1002/adhm.202201784
Contributor Information
Chenguang Xu, Email: xuchenguang@smu.edu.cn.
David A. Barbie, Email: david_barbie@dfci.harvard.edu.
Roger D. Kamm, Email: rdkamm@mit.edu.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Liu Z., Wang Y., Huang Y., Kim B. Y. S., Shan H., Wu D., Jiang W., Trends Pharmacol. Sci. 2019, 40, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baghban R., Roshangar L., Jahanban‐Esfahlan R., Seidi K., Ebrahimi‐Kalan A., Jaymand M., Kolahian S., Javaheri T., Zare P., Cell Commun. Signaling 2020, 18, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sontheimer‐Phelps A., Hassell B. A., Ingber D. E., Nat. Rev. Cancer 2019, 19, 65. [DOI] [PubMed] [Google Scholar]
- 4. Piccolo N. D., Shirure V. S., Bi Y., Goedegebuure S. P., Gholami S., Hughes C. C. W., Fields R. C., George S. C., Adv. Drug Delivery Rev. 2021, 175, 113798. [DOI] [PubMed] [Google Scholar]
- 5. Liu X., Fang J., Huang S., Wu X., Xie X., Wang J., Liu F., Zhang M., Peng Z., Hu N., Microsyst. Nanoeng. 2021, 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shang M., Soon R. H., Lim C. T., Khoo B. L., Han J., Lab Chip 2019, 19, 369. [DOI] [PubMed] [Google Scholar]
- 7. Shirure V. S., Hughes C. C. W., George S. C., Annu. Rev. Biomed. Eng. 2021, 23, 141. [DOI] [PubMed] [Google Scholar]
- 8. Wu Y., Zhou Y., Qin X., Liu Y., Biomicrofluidics 2021, 15, 061503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haase K., Offeddu G. S., Gillrie M. R., Kamm R. D., Adv. Funct. Mater. 2020, 30, 2002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paek J., Park S. E., Lu Q., Park K.‐T., Cho M., Oh J. M., Kwon K. W., Yi Y.‐S., Song J. W., Edelstein H. I., Ishibashi J., Yang W., Myerson J. W., Kiseleva R. Y., Aprelev P., Hood E. D., Stambolian D., Seale P., Muzykantov V. R., Huh D., ACS Nano 2019, 13, 7627. [DOI] [PubMed] [Google Scholar]
- 11. Straehla J. P., Hajal C., Safford H. C., Offeddu G. S., Boehnke N., Dacoba T. G., Wyckoff J., Kamm R. D., Hammond P. T., Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2118697119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campisi M., Sundararaman S. K., Shelton S. E., Knelson E. H., Mahadevan N. R., Yoshida R., Tani T., Ivanova E., Cañadas I., Osaki T., Lee S. W. L., Thai T., Han S., Piel B. P., Gilhooley S., Paweletz C. P., Chiono V., Kamm R. D., Kitajima S., Barbie D. A., Front. Immunol. 2020, 11, 2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim D., Hwang K. S., Seo E. U., Seo S., Lee B. C., Choi N., Choi J., Kim H. N., Adv. Healthcare Mater. 2022, 11, 2102581. [Google Scholar]
- 14. Bi Y., Shirure V. S., Liu R., Cunningham C., Ding L., Meacham J. M., Goedegebuure S. P., George S. C., Fields R. C., Integr. Biol. 2020, 12, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ayuso J. M., Rehman S., Virumbrales‐Munoz M., Mcminn P. H., Geiger P., Fitzgerald C., Heaster T., Skala M. C., Beebe D. J., Sci. Adv. 2021, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J., Kim S.‐E., Moon D., Doh J., Lab Chip 2021, 21, 2142. [DOI] [PubMed] [Google Scholar]
- 17. Xiao Y., Kim D., Dura B., Zhang K., Yan R., Li H., Han E., Ip J., Zou P., Liu J., Chen A. T., Vortmeyer A. O., Zhou J., Fan R., Adv. Sci. 2019, 6, 1801531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Michna R., Gadde M., Ozkan A., Dewitt M., Rylander M., Biotechnol. Bioeng. 2018, 115, 2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sobrino A., Phan D. T. T., Datta R., Wang X., Hachey S. J., Romero‐López M., Gratton E., Lee A. P., George S. C., Hughes C. C. W., Sci. Rep. 2016, 6, 31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y., Kankala R. K., Zhang J., Hao L., Zhu K., Wang S., Zhang Y. S., Chen A., Adv. Sci. 2020, 7, 2002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agarwal P., Wang H., Sun M., Xu J., Zhao S., Liu Z., Gooch K. J., Zhao Y., Lu X., He X., ACS Nano 2017, 11, 6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dey M., Ayan B., Yurieva M., Unutmaz D., Ozbolat I. T., Adv. Biol. 2021, 5, e2100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benjamin F. L. L., Lu Rick X., Hu Y., Davenport H. L., Dou W., Wang E. Y., Radulovich N., Tsao M. S., Sun Y., Radisic M., Adv. Funct. Mater. 2020, 30, 2000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park S., Kim T., Kim S., You S., Jung Y., Cancers 2021, 13, 3930.34439103 [Google Scholar]
- 25. Silvani G., Basirun C., Wu H. J., Mehner C., Poole K., Bradbury P., Chou J., Adv. Ther. 2021, 4, 2100106. [Google Scholar]
- 26. Hassell B. A., Goyal G., Lee E., Sontheimer‐Phelps A., Levy O., Chen C. S., Ingber D. E., Cell Rep. 2017, 21, 508. [DOI] [PubMed] [Google Scholar]
- 27. Ko J., Ahn J., Kim S., Lee Y., Lee J., Park D., Jeon N. L., Lab Chip 2019, 19, 2822. [DOI] [PubMed] [Google Scholar]
- 28. Hachey S. J., Movsesyan S., Nguyen Q. H., Burton‐Sojo G., Tankazyan A., Wu J., Hoang T., Zhao D., Wang S., Hatch M. M., Celaya E., Gomez S., Chen G. T., Davis R. T., Nee K., Pervolarakis N., Lawson D. A., Kessenbrock K., Lee A. P., Lowengrub J., Waterman M. L., Hughes C. C. W., Lab Chip 2021, 21, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nashimoto Y., Okada R., Hanada S., Arima Y., Nishiyama K., Miura T., Yokokawa R., Biomaterials 2020, 229, 119547. [DOI] [PubMed] [Google Scholar]
- 30. Hu Z., Cao Y., Galan E. A., Hao L., Zhao H., Tang J., Sang G., Wang H., Xu B., Ma S., ACS Biomater. Sci. Eng. 2022, 8, 1215. [DOI] [PubMed] [Google Scholar]
- 31. June C. H., O'connor R. S., Kawalekar O. U., Ghassemi S., Milone M. C., Science 2018, 359, 1361. [DOI] [PubMed] [Google Scholar]
- 32. June C. H., Sadelain M., N. Engl. J. Med. 2018, 379, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hou A. J., Chen L. C., Chen Y. Y., Nat. Rev. Drug Discovery 2021, 20, 531. [DOI] [PubMed] [Google Scholar]
- 34. Martinez M., Moon E. K., Front. Immunol. 2019, 10, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sterner R. C., Sterner R. M., Blood Cancer J. 2021, 11, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boulch M., Cazaux M., Loe‐Mie Y., Thibaut R., Corre B., Lemaître F., Grandjean C. L., Garcia Z., Bousso P., Sci. Immunol. 2021, 6. [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez‐Garcia A., Palazon A., Noguera‐Ortega E., Powell D. J., Guedan S., Front. Immunol. 2020, 11, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paterson K., Paterson S., Mulholland T., Coffelt S. B., Zagnoni M., IEEE Open J. Eng. Med. Biol. 2022, 3, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jacob F., Salinas R. D., Zhang D. Y., Nguyen P. T. T., Schnoll J. G., Wong S. Z. H., Thokala R., Sheikh S., Saxena D., Prokop S., Liu D.‐A., Qian X., Petrov D., Lucas T., Chen H. I., Dorsey J. F., Christian K. M., Binder Z. A., Nasrallah M., Brem S., O'rourke D. M., Ming G.‐L., Song H., Cell 2020, 180, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michie J., Beavis P. A., Freeman A. J., Vervoort S. J., Ramsbottom K. M., Narasimhan V., Lelliott E. J., Lalaoui N., Ramsay R. G., Johnstone R. W., Silke J., Darcy P. K., Voskoboinik I., Kearney C. J., Oliaro J., Cancer Immunol. Res. 2019, 7, 183. [DOI] [PubMed] [Google Scholar]
- 41. Ando Y., Siegler E. L., Ta H. P., Cinay G. E., Zhou H., Gorrell K. A., Au H., Jarvis B. M., Wang P., Shen K., Adv. Healthcare Mater. 2019, 8, e1900001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pan J., Mestas J., Burdick M. D., Phillips R. J., Thomas G. V., Reckamp K., Belperio J. A., Strieter R. M., Mol. Cancer 2006, 5, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang D., Ding Y., Zhou M., Rini B. I., Petillo D., Qian C.‐N., Kahnoski R., Futreal P. A., Furge K. A., Teh B. T., Cancer Res. 2010, 70, 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amann A., Zwierzina M., Koeck S., Gamerith G., Pechriggl E., Huber J. M., Lorenz E., Kelm J. M., Hilbe W., Zwierzina H., Kern J., Sci. Rep. 2017, 7, 2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cañadas I., Rojo F., Taus Á., Arpí O., Arumí‐Uría M., Pijuan L., Menéndez S., Zazo S., Dómine M., Salido M., Mojal S., De Herreros A. G., Rovira A., Albanell J., Arriola E., Clin. Cancer Res. 2014, 20, 938. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y., Elechalawar C. K., Yang W., Frickenstein A. N., Asfa S., Fung K.‐M., Murphy B. N., Dwivedi S. K., Rao G., Dey A., Mater. Today 2022, 56, 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Z., Jiang D., Yang H., He Z., Liu X., Qin W., Li L., Wang C., Li Y., Li H., Xu H., Jin H., Qian Q., Cell Death Dis. 2019, 10, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morello A., Sadelain M., Adusumilli P. S., Cancer Discov 2016, 6, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shao M., Hollar S., Chambliss D., Schmitt J., Emerson R., Chelladurai B., Perkins S., Ivan M., Matei D., Mol. Cancer Ther. 2012, 11, 1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Agarwal A., Covic L., Sevigny L. M., Kaneider N. C., Lazarides K., Azabdaftari G., Sharifi S., Kuliopulos A., Mol. Cancer Ther. 2008, 7, 2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Offeddu G. S., Serrano J. C., Chen S. W., Shelton S. E., Shin Y., Floryan M., Kamm R. D., J. Biomech. 2021, 119, 110330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Newman A. C., Nakatsu M. N., Chou W., Gershon P. D., Hughes C. C. W., Mol. Biol. Cell 2011, 22, 3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murakami M., Simons M., Curr. Opin. Hematol. 2008, 15, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nowak‐Sliwinska P., Alitalo K., Allen E., Anisimov A., Aplin A. C., Auerbach R., Augustin H. G., Bates D. O., Van Beijnum J. R., Bender R. H. F., Bergers G., Bikfalvi A., Bischoff J., Böck B. C., Brooks P. C., Bussolino F., Cakir B., Carmeliet P., Castranova D., Cimpean A. M., Cleaver O., Coukos G., Davis G. E., De Palma M., Dimberg A., Dings R. P. M., Djonov V., Dudley A. C., Dufton N. P., Fendt S.‐M., et al., Angiogenesis 2018, 21, 425.29766399 [Google Scholar]
- 55. Torisawa Y.‐S., Mosadegh B., Luker G. D., Morell M., O'shea K. S., Takayama S., Integr. Biol. 2009, 1, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krieg M., Arboleda‐Estudillo Y., Puech P.‐H., Käfer J., Graner F., Müller D. J., Heisenberg C.‐P., Nat. Cell Biol. 2008, 10, 429. [DOI] [PubMed] [Google Scholar]
- 57. Foty R. A., Steinberg M. S., Dev. Biol. 2005, 278, 255. [DOI] [PubMed] [Google Scholar]
- 58. Sivakumar N., Warner H. V., Peirce S. M., Lazzara M. J., bioRxiv 2021, 10.1101/2021.08.05.455232. [DOI] [Google Scholar]
- 59. Van Roy F., Berx G., Cell. Mol. Life Sci. 2008, 65, 3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murray P., Frampton G., Nelson P., BMJ 1999, 319, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Labernadie A., Kato T., Brugués A., Serra‐Picamal X., Derzsi S., Arwert E., Weston A., González‐Tarragó V., Elosegui‐Artola A., Albertazzi L., Alcaraz J., Roca‐Cusachs P., Sahai E., Trepat X., Nat. Cell Biol. 2017, 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Janiszewska M., Primi M. C., Izard T., J. Biol. Chem. 2020, 295, 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salmenperä P., Kankuri E., Bizik J., Sirén V., Virtanen I., Takahashi S., Leiss M., Fässler R., Vaheri A., Exp. Cell Res. 2008, 314, 3444. [DOI] [PubMed] [Google Scholar]
- 64. Gumbiner B. M., Nat. Rev. Mol. Cell Biol. 2005, 6, 622. [DOI] [PubMed] [Google Scholar]
- 65. Harris T. J. C., Tepass U., Nat. Rev. Mol. Cell Biol. 2010, 11, 502. [DOI] [PubMed] [Google Scholar]
- 66. Sahai E., Astsaturov I., Cukierman E., Denardo D. G., Egeblad M., Evans R. M., Fearon D., Greten F. R., Hingorani S. R., Hunter T., Hynes R. O., Jain R. K., Janowitz T., Jorgensen C., Kimmelman A. C., Kolonin M. G., Maki R. G., Powers R. S., Puré E., Ramirez D. C., Scherz‐Shouval R., Sherman M. H., Stewart S., Tlsty T. D., Tuveson D. A., Watt F. M., Weaver V., Weeraratna A. T., Werb Z., Nat. Rev. Cancer 2020, 20, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kowalski P. J., Rubin M. A., Kleer C. G., Breast Cancer Res. 2003, 5, R217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kumar A., Khani A. T., Ortiz A. S., Swaminathan S., Front. Immunol. 2022, 13, 901277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frey N., Porter D., Biol. Blood Marrow Transplant. 2019, 25, e123. [DOI] [PubMed] [Google Scholar]
- 70. Sterner R. M., Sakemura R., Cox M. J., Yang N., Khadka R. H., Forsman C. L., Hansen M. J., Jin F., Ayasoufi K., Hefazi M., Schick K. J., Walters D. K., Ahmed O., Chappell D., Sahmoud T., Durrant C., Nevala W. K., Patnaik M. M., Pease L. R., Hedin K. E., Kay N. E., Johnson A. J., Kenderian S. S., Blood 2019, 133, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xue L., Yi Y., Xu Q., Wang L., Yang X., Zhang Y., Hua X., Chai X., Yang J., Chen Y., Tao G., Hu B., Wang X., Cell Discov. 2021, 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kang L., Tang X., Zhang J., Li M., Xu N., Qi W., Tan J., Lou X., Yu Z., Sun J., Wang Z., Dai H., Chen J., Lin G., Wu D., Yu L., Exp. Hematol. Oncol. 2020, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bailey S. R., Vatsa S., Larson R. C., Bouffard A. A., Scarfò I., Kann M. C., Berger T. R., Leick M. B., Wehrli M., Schmidts A., Silva H., Lindell K. A., Demato A., Gallagher K. M. E., Frigault M. J., Maus M. V., Blood Cancer Discovery 2022, 3, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wei J., Liu Y., Wang C., Zhang Y., Tong C., Dai G., Wang W., Rasko J. E. J., Melenhorst J. J., Qian W., Liang A., Han W., Signal Transduction Targeted Ther. 2020, 5, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wan Z., Zhong A. X., Zhang S., Pavlou G., Coughlin M. F., Shelton S. E., Nguyen H. T., Lorch J. H., Barbie D. A., Kamm R. D., Small Methods 2022, 6, e2200143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Margolis E. A., Cleveland D. S., Kong Y. P., Beamish J. A., Wang W. Y., Baker B. M., Putnam A. J., Lab Chip 2021, 21, 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kosyakova N., Kao D. D., Figetakis M., López‐Giráldez F., Spindler S., Graham M., James K. J., Won Shin J., Liu X., Tietjen G. T., Pober J. S., Chang W. G., npj Regener. Med. 2020, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Moya M. L., Hsu Y.‐H., Lee A. P., Hughes C. C. W., George S. C., Tissue Eng., Part C 2013, 19, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang X., Phan D. T. T., Sobrino A., George S. C., Hughes C. C. W., Lee A. P., Lab Chip 2016, 16, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bang S., Lee S.‐R., Ko J., Son K., Tahk D., Ahn J., Im C., Jeon N. L., Sci. Rep. 2017, 7, 8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang S., Wan Z., Pavlou G., Zhong A. X., Xu L., Kamm R. D., Adv. Funct. Mater. 2022, 32, 2206767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mykuliak A., Yrjänäinen A., Mäki A.‐J., Gebraad A., Lampela E., Kääriäinen M., Pakarinen T.‐K., Kallio P., Miettinen S., Vuorenpää H., Front. Bioeng. Biotechnol. 2022, 10, 764237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim S., Lee H., Chung M., Jeon N. L., Lab Chip 2013, 13, 1489. [DOI] [PubMed] [Google Scholar]
- 84. Sugihara K., Yamaguchi Y., Usui S., Nashimoto Y., Hanada S., Kiyokawa E., Uemura A., Yokokawa R., Nishiyama K., Miura T., PLoS One 2020, 15, e0240552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Park Y. K., Tu T.‐Y., Lim S. H., Clement I. J. M., Yang S. Y., Kamm R. D., Cell Mol. Bioeng. 2014, 7, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cuchiara M. P., Gould D. J., Mchale M. K., Dickinson M. E., West J. L., Adv. Funct. Mater. 2012, 22, 4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Brown A., He H., Trumper E., Valdez J., Hammond P., Griffith L. G., Biomaterials 2020, 243, 119921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wan Z., Zhang S., Zhong A. X., Shelton S. E., Campisi M., Sundararaman S. K., Offeddu G. S., Ko E., Ibrahim L., Coughlin M. F., Liu T., Bai J., Barbie D. A., Kamm R. D., Biomaterials 2021, 276, 121032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Davis G. E., Senger D. R., Circ. Res. 2005, 97, 1093. [DOI] [PubMed] [Google Scholar]
- 90. Sephel G. C., Davidson J. M., J. Invest. Dermatol. 1986, 86, 279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Movie 1
Supplemental Movie 2
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.