

Abstract
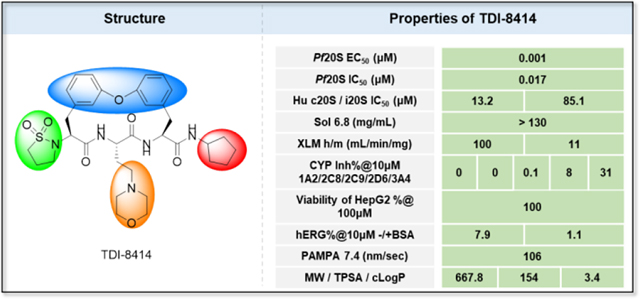
With increasing reports of resistance to artemisinins and artemisinin-combination therapies, targeting the Plasmodium proteasome is a promising strategy for antimalarial development. We recently reported a highly selective Plasmodium falciparum proteasome inhibitor with anti-malarial activity in the humanized mouse model. To balance the permeability of the series macrocycles with other drug-like properties, we conducted further structure-activity relationship studies on the biphenyl ether tethered macrocyclic scaffold. Extensive SAR studies around the P1, P3, and P5 groups and peptide backbone identified compound TDI-8414. TDI-8414 showed nanomolar antiparasitic activity, no toxicity to HepG2 cells, high selectivity against the Plasmodium proteasome over the human constitutive proteasome and immunoproteasome, improved solubility and PAMPA permeability, and enhanced metabolic stability in microsomes and plasma of both humans and mice.
Keywords: Malaria, antimalaria, Plasmodium proteasome, species-selective parasite proteasome inhibitors, pharmacokinetics
Graphical Abstract
Introduction
Malaria is one of the most prevalent infectious diseases in the world. Africa carries the heaviest malaria burden, accounting for ~95% of malaria cases and deaths, primarily in young children.1 Among the Plasmodium species causing human malaria, P. falciparum (Pf)is the most virulent, and infections are commonly life-threating. During symptomatic disease, parasites replicate rapidly and can reach > 1012 parasites in an infected individual,2 setting the stage for selection of resistance to antimalarials. In fact, resistance to most antimalarials in clinical use is reported globally, and resistance has even been detected to some drugs at the clinical trials stage. It is alarming that resistance to artemisinins and artemisinin-combination therapies is widespread in the Greater Mekong Region of southeast Asia and now emerging independently in sub-Saharan Africa.3–5 The loss of effectiveness of artemisinins would be disastrous for global malaria control. It is an urgent priority to develop antimalarials that target novel parasite proteins and/or demonstrate synergy with artemisinins so as to prolong their clinical effectiveness, overcome existing resistance and minimize the emergence of resistance.
Proteasomes of pathogenic microbes are novel targets for discovery and development of antimicrobials,6 starting with the development of species selective proteasome inhibitors of Mycobacterium tuberculosis (Mtb)7, followed by those of Plasmodium8, Trypanosoma, and Leishmania.9 Genetic studies validated the essentiality of the Plasmodium proteasome10 and its pharmacological inhibition by small molecule inhibitors, six represents shown in Figure 1a8, 11–16. In addition to action against the erythrocytic stage, which is responsible for human disease, several studies also established that proteasome inhibitors are active against the liver stage, gametocytes, and gametes (Figure 1b).13, 16, 17 Compared to wild type strains, artemisinin resistant parasites (with K13 mutations) were slightly more sensitive to proteasome inhibitors, and proteasome inhibitor resistant mutants were slightly more sensitive to artemisinins.16, 18 Synergy between proteasome inhibitors and artemisinins has been demonstrated by multiple groups 12, 16, 19–21.
Figure 1.

a) Representative Pf20S inhibitors. b) Macrocyclic peptides selectively inhibit Pf20S over human c20S and i20S. Proteasome inhibitors are active against the Plasmodium parasites at multiple stages of their life cycle and are transmission blocking.
We recently developed a proteasome inhibitor TDI8304 that is highly selective for the P. falciparum proteasome over both the human constitutive proteasome (c20S) and immunoproteasome (i20S) (Figure 1a), with metabolic stability and in vivo efficacy in a humanized mouse model of P. falciparum infection.11 Starting from this reported cyclic peptide 1 (CP1),14 the monophenyl linked macrocycle TDI8304 was developed as a lead compound with a good balance of potency,11 selectivity, solubility, plasma stability, and microsome stability, but it suffered from modest PAMPA permeability. We hypothesized that macrocyclic peptides with a biphenyl tether, which has a high lipophilicity, might balance the PAMPA permeability with other pharmacokinetic properties (Figure 2).22 In this paper, we present our second structure-activity relationship study of biphenyl ether tethered Pf20S selective macrocycles in an attempt to improve the permeability and other pharmacokinetic properties of this class of antimalarials.
Figure 2.

Optimization strategy for biphenyl ether tethered macrocycles as antimalarial proteasome inhibitors. Note: TDI-8414 is in Table 3.
RESULTS AND DISCUSSION
Macrocycle 1 showed remarkable anti-parasitic activity and PAMPA permeability; however, it was rapidly metabolized by mouse microsomes (Table 1).11, 14 We started by replacing the P3 homophenylalanine (homo-Phe) of 1 with hydrogen (2), methoxy methyl (3) or trifluoropropyl (4) groups to reduce lipophilicity (cLogP = 2.2 to 3.1), all of which resulted in much improved liver microsomal stability, but also complete loss of potency. Replacing the P3 homo-Phe of 1 with propyl (5) or isobutyl (6) groups significantly decreased potency against parasites. Replacing the P3 homo-Phe of 7 with a phenyl group (8) also resulted in marked potency loss, yet replacement with a piperidine (9) maintained the anti-parasitic activity and dramatically improved the solubility with decreased cLogP. This modification paved the way for further optimization of 9.
Table 1.
Optimization of P3
|
EC50 (μM) |
β5 IC50 (μM) | Permeability | Metabolic stabilityc m/hLM Clint | Kinet. Solu. | Cytotox. | cLogP | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | P5 | P3 | 3D7 | Pf20S | c-20S | i-20S | PAMPA | Ratio /MDR1 | ||||
| 1* | A |
|
0.003 | 0.014 | 4.0 | >100 | 151 | >0.77/<4 | 270/11.2 | <1.4 | 93 | 4.7 |
| 2 | A | H | >2.8 | >100 | 3.7 | 2.7 | <18 | 4.1/0 | −4/−17.4 | 4.1 | 104 | 2.2 |
| 3 | A |
|
>2.8 | 14.6 | >100 | >100 | 44 | >8.7/<1 | −11.7/−12.7 | 14 | 98 | 2.4 |
| 4 | A |
|
>2.8 | >100 | >100 | >100 | 73 | 14/0 | −11.7/−2 | <1.2 | 98 | 3.1 |
| 5 | A |
|
0.59 | 0.9 | >100 | >100 | 81 | >2.5/<2 | 37.7/37.7 | <1.9 | 91 | 3.8 |
| 6 | A |
|
0.62 | 3.8 | >100 | >100 | 95 | >39/<1 | 25.2/−4 | <1.4 | 94 | 4.2 |
| 7** | B |
|
0.0002 | 0.003 | 0.38 | 33.9 | 214 | 31/5 | >768/644 | 3.8 | 77 | 5.3 |
| 8 | B |
|
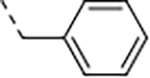
1.7 | 5.9 | 2.9 | 7.2 | 237 | 47/2 | >768/522 | 22 | 105 | 4.8 |
| 9 | B |
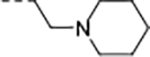
|
0.010 | 0.095 | 1.5 | 9.6 | 30 | 26/1 | 73.9/15.5 | >140 | 97 | 4.0 |
| 10 | B |
|
0.001 | 0.014 | 0.14 | 1.4 | 264 | 46/3 | >768/687 | >140 | 110 | 3.4 |
| 11 | B |
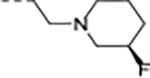
|
0.003 | 0.013 | 17 | 54 | 171 | 100/1 | >768/238 | >130 | - | 3.7 |
: compound reported in REF 22
: compound reported in REF 22; PAMPA unit: nm/sec at pH 7.4; MDR unit: ratio (B→A/A→B); A→B, nm/sec; Metabolic stability m/hLM Clint: mouse/human liver microsomes and unit- μL/min/mg; Kinet. Solu.: Kinetic solubility at pH 6.8 (μg/mL); Cytotox.: cytotoxicity, % viability of HepG2 cells at 30 μM. “-”: not tested.
Compound 9 was rapidly cleared within 30 min post cassette-dosing at 0.3 mg/kg, i.v. (Table S1).23 Compound 9 showed high stability after incubation with human plasma and microsomes, but fast clearance in mouse plasma and microsomes. 9 showed good potency, solubility, and passive permeability, and was selected as the preferred compound for further structural modifications to improve metabolic and plasma stability. The piperidine was considered to be a potential metabolic liability. We introduced a metabolically stable diF at the 4-position (10) and a fluorine atom at the 3-position (11) of the piperidine of 9 to lower the electron density as well as block potential oxidative metabolism at this site. These two macrocycles showed improved anti-parasitic activity over 9; however, both suffered significantly reduced microsome stability.
To understand the rapid microsomal clearance of 9, we investigated the metabolites generated in the presence of mouse liver microsomes. After incubating 9 with mouse liver microsomes for 60 min in the presence of NADPH and uridine diphosphoglucuronic acid, LC-MS/MS indicated that 9 was extensively metabolized, mostly via hydroxylation (Figure 3a, b). M659a is proposed to be formed via hydroxylation of the biphenyl linker, while M659b and M659c could be formed through oxidative metabolism of the P5 lactam by hepatic cytochrome P450 in a NADPH dependent manner. The P5 lactam appears to be the major site for oxidative metabolism. No glucuronide conjugates of 9 or its primary hydroxylated metabolites were detected, suggesting that 9 is not the substrate for UDP-glucuronosyltransferases (UGTs).
Figure 3.

Metabolite Profile of 9 in mouse liver microsome (MLM) and mouse plasma. a) LC/MS profile of 9 after incubation with MLM or MLM + NADPH + UDPGA; b) Chemical structures of metabolites of 9 in mouse liver microsome; c) Chemical structures of metabolites of 9 in mouse plasma.
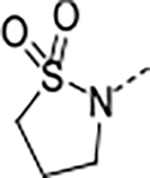
Based on the metabolite profiling of 9, the next optimization process was focused on improving mouse microsomal stability through structural modification of the P5 group while maintaining potency and favorable biochemical properties. Blocking the metabolic soft spot and reducing the electron density of the P5 group via increased polarity are two common strategies to prevent oxidative metabolism and improve microsomal stability. We first replaced the adjacent methylene of the lactam group in macrocycle 9 with an oxygen atom in 12 to block the potential metabolic site (Table 2). This modification was tolerated for potency but failed to improve the metabolic stability, likely due to the oxazolidinone group of 12 that has higher electron density and is more prone to oxidative metabolism than the lactam group. We next directed our efforts to introducing polar substituents and decreasing electron density of the P5 group. Replacing the lactam group of 13 with a polar methyl sulfonamide group afforded macrocycle 14, which showed an improved metabolic stability across species (m/hLM, 19/6 μL/min/mg), but a marked loss of antiparasitic activity (IC50 >2.7 μM). Cyclizing the methyl sulfonamide group of 14 provided compound 15 with a sultam as P5, which recovered anti-parasitic activity and maintained metabolic stability across species, but still suffered from poor permeability (Table 2). As shown in Figure 4, the improved mouse microsomal stability of 15 translated into low turnover in mouse hepatocytes (Clint = 2.8 μL/min/106 cells).
Table 2.
Optimization of P5
|
EC50 (μM) |
β5 IC50 (μM) | Permeability | Metabolic stability m/hLM Clint | Kinet. Solu. | Cyto- tox. | cLogP | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | P1 | P2 | 3D7 | Pf20S | c-20S | i-20S | PAMPA | Ratio /MDR1 | ||||
| 9 | A |
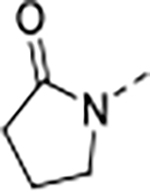
|
0.010 | 0.095 | 1.5 | 9.6 | 30 | 26/1 | 73.9/15.5 | >140 | 97 | 4.0 |
| 12 | A |
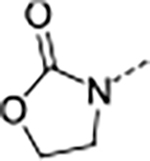
|
0.013 | 0.034 | 23.0 | >100 | 10 | >8/<1 | 112/16 | 65 | 4.1 | |
| 13 | B |
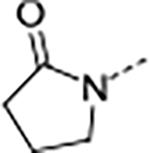
|
0.073 | 0.21 | 33.5 | 88.0 | <1 | >1/<1 | 203/54 | >150 | 3.2 | |
| 14 | B |
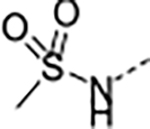
|
>2.8 | 2.0 | >100 | >100 | <1 | N.D. /<1 | 19/6 | >140 | 2.7 | |
| 15 | B |
|
0.085 | 0.46 | >100 | 59.8 | <2 | >0.5/<2 | 8/34 | >140 | 3.1 | |
PAMPA unit: nm/sec at pH7.4; MDR unit: ratio (B→A/A→B); A→B, nm/sec; Metabolic stability m/hLM Clint: mouse / human liver microsomes and unit- μL/min/mg; Kinet. Solu.: Kinetic solubility at pH6.8 (μg/mL); Cytotox.: cytotoxicity, % viability of HepG2 cells at 30 μM. “-”: not tested.
Figure 4.

In vitro metabolic stability of macrocycle 15 in mouse hepatocytes. Diazepam was used as a positive control compound.
After incubation with human and mouse plasma for 120 min, 9 exhibited remarkable stability in human plasma, but was rapidly degraded in mouse plasma (Figure 5a, b). Mouse plasma metabolite characterization of macrocycle 9 was performed using LC/MS. A macrocyclic carboxylic acid was identified as a major metabolite, suggesting that hydrolysis of the P1 amide is a major clearance mechanism (Figure 3c). Although N-methylation of a susceptible amide can often induce resistance to hydrolysis by proteases, our docking model of compound 9 suggested that the P1 amide forms two critical hydrogen bonds with Gly47 and Ser21 residues of the β5 subunit (Figure 6). To improve the plasma stability via altering the electronic character of the P1 amide bond, the electron-withdrawing trifluoromethyl group of 9 was replaced with an electron-donating cyclopropyl group in 16 and 22 (Table 3), which maintained the high antiparasitic activity but failed to improve mouse plasma stability.
Figure 5.

a) Mouse plasma stability of selected macrocycles; b) Human plasma stability of selected macrocycles. Positive control compounds were enalapril (mouse plasma) and procaine (human plasma).
Figure 6.

Docking pose of 9 into Pf20S (PDB: 5FMG). 9 is shown in orange. The β5 subunit is in green. The β6 subunit is in cyan. Hydrogen bonds are indicated by dashed yellow lines.
Table 3.
Optimization of P1
|
EC50 (μM) |
β5 IC50 (μM) | Permeability | Metabolic stability m/hLM Clint | Kinet. Solu. |
Cytotox. | cLogP | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | P1 | 3D7 | Pf20S | c-20S | i-20S | PAMPA | Ratio /MDR1 | ||||
| 9 |
|
0.010 | 0.095 | 1.5 | 9.6 | 30 | 26/1 | 73.9/15.5 | >140 | 97 | 4.0 |
| 16 |
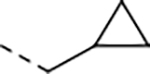
|
0.031 | 0.083 | 30.0 | 33.3 | 19 | >18/<1 | 110/46 | >130 | 102 | 4.2 |
| 17 |
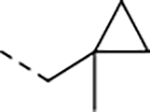
|
0.008 | 0.059 | 11.8 | 22.3 | 97 | >30/<1 | 86/43 | >130 | - | 4.7 |
| 18 |
|
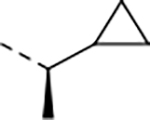
0.57 | 4.3 | >100 | >100 | 137 | >27/<1 | 230/58 | >140 | 103 | 4.5 |
| 19 |
|
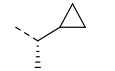
0.078 | 0.8 | 11.6 | 30.9 | 81 | >20/<1 | 104/51 | >120 | - | 4.5 |
| 20 |
|
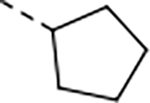
0.030 | 0.077 | 28.6 | 17.9 | 49 | 66/1 | 195/75 | >140 | - | 4.7 |
| 21 |
|
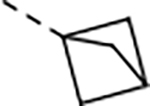
0.61 | 4.5 | 31.3 | 32.4 | 46 | >55/<1 | 109/38 | 110 | - | 3.9 |
| 22 |
|
0.003 | 0.008 | 21.9 | >100 | 101 | >22/<1 | 89/41 | >130 | - | 3.0 |
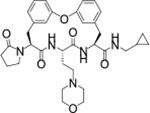
| TDI-8414 |
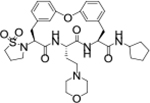
|
0.001 | 0.017 | 13.2 | 85.1 | 106 | >39/<1 | 11/100 | >130 | 105 | 3.4 |
PAMPA unit: nm/sec at pH7.4; MDR unit: ratio (B→A/B→B); A→B, nm/sec; Metabolic stability m/hLM Clint: mouse / human liver microsomes and unit- μL/min/mg; Kinet. Solu.: Kinetic solubility at pH6.8 (μg/mL); Cytotox.: cytotoxicity, % viability of HepG2 cells at 30 μM. “-”: not tested.
We next explored introducing an extra methyl group at the alpha position (18 and 19) or beta position (17) of the amino group of 16 to improve the plasma stability by sterically hindering the approach of proteases in plasma to the susceptible amide bond. All three compounds with the methyl substitution showed improved mouse plasma stability and better PAMPA permeability than 16. Compounds 18 and 19 with alpha-substituted methyl groups are slightly more stable than 17 with a beta-substituted methyl group suggesting that steric hindrance around the amide bond is a stabilizing factor for mouse plasma stability, which confirmed P1 amide as a metabolic soft spot. Compound 19, with an (S)-methyl substitution, was 7.4-fold more potent against the parasite than the R analog, 18. Macrocycle 20, with cyclopentyl as the P1 group, showed high mouse plasma stability and PAMPA permeability as alpha methyl substituted compounds while maintaining high antiparasitic activity, however, 20 showed fast mouse liver microsomal clearance (195 μL/min/mg). Replacing the P1 group with a more bulky 1-bicyclo[1.1.1]pentyl group (21) was detrimental for antiparasitic activity. Replacing the combination of the high mouse microsomal stability of 15 and the high mouse plasma stability and PAMPA permeability of 20 might balance the drug-like properties. Consistent with our hypothesis, macrocycle TDI-8414 achieved a balance of high potency, selectivity, solubility, PAMPA permeability, metabolic stability and plasma stability. Additionally, TDI-8414 showed synergistic effect with dihydroartemisinin (DHA) in a ring-stage survival assay (RSA)16(Figure 7a).
Figure 7.

Synergy and ex vivo data. a) In vitro synergy of TDI-8414 and DHA against P. falciparum 3D7. Date was average of two independent experiments and each in duplicates. FIC = fractional inhibitory concentration. b-d) Ex vivo data of compounds 9 (b), 23 (c) and 25 (d) against Plasmodium clinical isolates from patients in Tororo, Uganda. Each circle was an IC50 obtained from a clinical isolate. Data were shown with geometric means with 95% confidence intervals.
MDR results suggested that this class of compounds suffered poor permeability and a high efflux ratio. The N-H bond of peptides and peptidomimetics were reported as elements that were recognized by efflux transporter.24 We therefore used N-methylation to reduce the number of hydrogen bond donors to 2 (Table 4). We chose 7 as the starting point, and N-methylated the P2-amide (23) and P3-amide (24). Although the N-methylated products showed improved MDR properties, they suffered a 1000-fold loss of potency to 0.21 μM for 23 and a complete loss for 24. Additionally, there was no improvement in metabolic stability over 7. The low efflux ratio and high A to B permeability of compound 23 and 24 provided a clue that reducing the number of hydrogen bond donor to two via N-H methylation would improve the MDR properties of the macrocyclic peptides, especially the N-H methylation of P2 amide that resulted in good MDR properties and moderate antiparasitic activity. Compound 25 was developed as a species-selective inhibitor for the Mtb proteasome (Mtb20S) over human proteasomes and showed cross antimicrobial activity, but still suffered from fast clearance (Table 4).
Table 4.
N-methylated macrocyclic peptides
| ID | EC50 (μM) |
β5 IC50 (μM) | Permeability | Metabolic stability m/hLM Clint | Kinet. Solu. | Cytotox. | cLogP | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3D7 | Pf20S | c-20S | i-20S | PAMPA | Ratio /MDR1 | ||||||
| 23 |
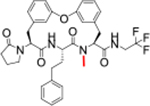
|
0.21 | 3.2 | >100 | >100 | 186 | 5.5/37 | >768/471 | 4.2 | 106 | 6.0 |
| 24 |
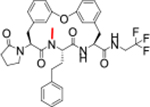
|
>2.8 | >100 | >100 | >100 | 283 | 6.3/23 | >768/>768 | 4 | 100 | 6.0 |
| 25 |
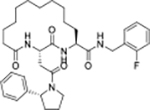
|
0.25 | 3.0 | >100 | >100 | 339 | 11/16 | >768/>768 | <0.85 | 100 | 6.3 |
PAMPA unit: nm/sec at pH7.4; MDR unit: ratio (B→A/A→B); A→B, nm/sec; Metabolic stability Clint unit: μL/min/mg; Kinet. Solu.: Kinetic solubility at pH6.8 (μg/mL); Cytotox.: cytotoxicity, % viability of HepG2 cells at 30 μM. “-”: not tested.
The inhibitory activity of compounds 2-25 and TDI-8414 against the other 4 active subunits of hu-c20S and hu-i20S were determined. All the compounds showed insignificant inhibition against human β1c, β2c, β1i, and β2i (Table S2).
Compounds 9, 23 and 25 were selected for further testing for ex vivo antiparasitic activity against freshly isolated P. falciparum isolates from 38, 35 and 28 malaria patients, respectively in Uganda (Figure 7b–d). The EC50 values of 9 ranged from 6.3–30 nM, with a geometric mean of 15.2 nM, in agreement with the results for P. falciparum laboratory strains (Table 2). Compounds 23 and 25 showed EC50 ranges of 283–1203 nM and 264–860 nM, with geometric means of 732 nM and 505 nM, respectively, representing a 3-fold and 2-fold decrease in potency, respectively, over the results for laboratory strains.
Chemistry
As shown in Scheme 1, a series of biphenyl ether tethered macrocycles with various P3 groups were synthesized.22 The synthesis of advanced intermediates is shown in Scheme S1–3. The fragment 26 and 27a-b underwent the Chan-Lam coupling reaction, affording biphenyl ether 28a-b, which were then subjected to Boc-deprotection and coupling reactions with amino acid 29a-h, yielding dipeptides 30a-h. Subsequent acid-mediated Boc-deprotection and Pd/C mediated benzyl deprotection of compounds 30a-h followed by intramolecular HATU mediated amide coupling reactions yield macrocycles 2, 3, 4, 5, 6, 8, 9, and 10.
Scheme 1.

Synthesis of compound 2, 3, 4, 5, 6, 8, 9, and 10 with various P3 group.
Reagents and conditions: (a) Cu(OAc)2, Et3N, 4A molecular sieve, O2, DCM; (b) (1) HCl/dioxane, (2) HATU, DIPEA, amino acid 29a-h, DMF; (c) (1) HCl/dioxane, (2) Pd/C, H2, THF; (d) HATU, DIPEA, DMF.
As shown in Scheme 2–3, several biphenyl ether tethered macrocycles with various P1 groups were synthesized. The Cu(OAc)2 mediated Chan-Lam coupling reaction between phenylboronic acid 32 and phenols 27b, 33 afforded compounds 34a-b. TFA-mediated Boc-deprotection of compounds 34a-b followed by amide coupling reactions of resulting amines and amino acids 35a-b yielded dipeptides 36a-c, which were then subjected to sequential removal of benzyl and Cbz groups, and intramolecular amidation affording macrocycles 37a-c. Macrocycles 17, 18, 19, and 21 with various P1 groups were prepared via a sequence of hydrolysis of tert-butyl ester 37a and amide coupling reactions with amines. Removal of the tert-butyl group in macrocycle 37b and a subsequent coupling reaction with amines provided macrocycles 38a-d, which were subjected to ceric ammonium nitrate (CAN) mediated hydrolysis of cyclic acetal11 and reductive amination with derivatives of piperidine, yielding macrocycles 11, 13, 16, 20, and 22.
Scheme 2.

Synthesis of compound 17, 18, 19, and 21 with various P1 group.
Reagents and conditions: (a) Cu(OAc)2, Et3N, 4A molecular sieve, O2, DCM; (b) (1) TFA, DCM, 0°C, (2) EDCI, HOBt, DIPEA, DMF, 35a-b; (c) (1) Pd/C, H2, THF, (2) EDCI, HOBt, DIPEA, DMF; (d) (1) TFA, DCM, (2) EDCI, HOBt, Pyridine, R1NH2.
Scheme 3.

Synthesis of compounds 11, 13, 16, 20, and 22 with various P1 and P3 group.
Reagents and conditions: (a) (1) TFA, DCM, (2) EDCI, HOBt, pyridine, R1NH2; (b) (1) ceric ammonium nitrate (CAN), MeCN/H2O, (2) amines, AcOH, Pd/C, H2, MeOH.
As shown in Scheme 4, several biphenyl ether tethered macrocycles with various P5 groups were synthesized. starting from intermediate 37c. Hydrazinolysis of phthalimide 37c provided amine 39, which underwent amidation with 2-chloroethyl chloroformate and subsequent alkylation yielded oxazolidone 40a. Compound 40b was prepared from amine 39 via sulfonamidation with MsCl. Sulfonamidation of 39 with 3-chloropropanesulfonyl chloride and subsequent base-mediated cyclization afforded sultam 40c.25 Compounds 40a-c underwent TFA-mediated removal of the tert-butyl groups and followed by an amide coupling reaction, affording compounds 41a-c, which were subjected to CAN mediated hydrolysis of cyclic acetal and reductive amination with piperidine, yielding macrocycles 12, 14, and 15.
Scheme 4.

Synthesis of compounds 12, 14, and 15 with various P5 group.
Reagents and conditions: (a) N2H4·H2O, THF; (b) (1) 2-chloroethyl chloroformate, Et3N, THF, (2) Cs2CO3, NaI, DMF; (c) MsCl, DCM, DIPEA; (d) (1) 3-chloropropanesulfonyl chloride, Et3N, DCM, (2) K2CO3, NaI, DMF; (e) (1) TFA, DCM, (2) R1NH2, EDCI, HOBt, DMF, DIPEA; (f) (1) ceric ammonium nitrate (CAN), MeCN, water, (2) piperidine, H2, Pd/C, AcOH, MeOH.
The synthesis of macrocycle TDI-8414 using the Chan-Lam coupling reaction as a macrocyclization strategy is described in Scheme 5. Dipeptide 44 was synthesized via an amide coupling reaction of amine 42 and acid 43. Both the Boc and pinacol groups of 44 were removed under Lewis acid ZnBr2, affording compound 45, which was coupled with acid 46 to give tripeptide 47. Reductive debenzylation and intramolecular Chan-Lam coupling of tripeptide 47 afforded macrocycle 48. The tert-butyl group of 48 was removed and the P5 sultam group meanwhile was hydrolyzed with TFA, affording compound 49, which underwent a subsequent amide coupling reaction and POCl3 mediated intramolecular sulfonamidation, yielding macrocycle TDI-8414. The synthesis of macrocycles 23-25 is shown in Scheme S4–6.
Scheme 5.

Synthesis of compound TDI8–414 using Chan-Lam coupling reaction as macrocyclization strategy.
Reagents and conditions: (a) HATU, DIPEA, THF/DMF, 93% yield; (b) ZnBr2, DCM, 53% yield; (c) compound 46, T3P, DIPEA, DMF, 22% yield; (d) (1) Pd/C, H2, MeOH, (2) Cu(OAc)2, Et3N, 4A molecular sieve, O2, DCM, 14% yield over two steps; (e) TFA, DCM, (f) (1) T3P, DIPEA, DMF, cyclopentylamine, (2) POCl3, 5.4% yield over 3 steps.
Conclusion
In summary, a series of macrocycles containing a biphenyl tether were designed, synthesized, and evaluated as highly potent Pf20S-selective inhibitors. Extensive SAR studies around the P1, P3, and P5 groups of the macrocycles were conducted to balance potency with other PK properties (Figure 8). Both saturated and unsaturated P3 substituents were allowed. Chain length of the P3 group was important for antiparasitic activity. Replacing the phenyl group with heterocycle groups improved solubility and metabolic stability. The oxidative hydroxylation of the P5 lactam was demonstrated as the major metabolic pathway by mouse microsomes, and replacing the P5 lactam group with a polar sultam substituent improved mouse microsomal stability via decreasing the electron density of the P5 group. The improved mouse microsomal stability of 15 resulted in low liver hepatocyte intrinsic clearance. Wide ranges of primary and secondary amino P1 groups were tolerated. The metabolic profile suggested that the P1 amide was a soft spot in mouse plasma. Increasing the steric bulk of the P1 group prevented the hydrolysis of the P1 amide in mouse plasma and improved mouse plasma stability. Within this series, macrocycle TDI-8414 demonstrated potent antiparasitic activity, high selectivity over human proteasomes, high PAMPA permeability, high solubility, high plasma stability, high mouse microsomal stability, and low CYP inhibition. Both macrocyclic peptide TDI-8304 and TDI-8414 have high efflux ratios, which impose a challenge for improving oral bioavailability. Reducing the number of hydrogen bond donors via N-H methylation as shown in 23 significantly improves the efflux properties providing a clue that shielding a hydrogen bond donor via noncovalent approaches may improve the oral exposure and maintain the potency and selectivity at the same time. Further optimization is under way.
Figure 8.

Overall SAR summary of macrocyclic Pf20S inhibitors.
Experimental Section
All purchased reagents and starting materials were used as received unless otherwise noted. All non-aqueous reactions were performed under argon in oven- or flamed-dried glassware. 1H- and 19F- NMR spectra were obtained on a Bruker 400/500 MHz system. Chemical shifts δ values are expressed in parts per million, with the solvent resonance as an internal standard (chloroform-d, 1H: 7.26 ppm; Methanol-d4, 1H: 3.31 ppm; DMSO-d6, 1H: 2.50 ppm). NMR data are reported in an order: chemical shift, multiplicity (s: singlet; d: doublet; t: triplet; q: quartet; m: multiplet; br: broad), coupling constant, and integration. Purities of all final compounds were determined on a Waters UPLC/MS and all were > 95%.
Benzyl (S)-2-acetamido-3-(3-(3-((S)-2-((tert-butoxycarbonyl)amino)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoate (28a).
To a solution of compound 26 (477.6 mg, 1.2 mmol, 1.2 eq) in dichloromethane (6.0 mL) was added 4A molecular sieve (1.5 g), copper acetate (277.9 mg, 1.5 mmol, 1.5 eq), triethylamine (1.0 g, 10.2 mmol, 1.4 mL, 10.0 eq) and compound 27a (320.0 mg, 1.0 mmol, 1.0 eq). The mixture was stirred at 25°C for 16 hours and then another batch of compound 26 (39.8 mg, 102.0 μmol, 0.1 eq) was added. The mixture was stirred at 25°C for 2 hours. LCMS showed desired compound was detected. The mixture was filtered and then filter liquor was concentrated to give crude product. The mixture was purified by reverse phase flash chromatograph (TFA) to afford compound 28a (360.0 mg, 522.9 μmol, 51.3% yield, 95.5% purity) as a yellow brown solid. LCMS: RT = 0.97 min, m/z = 680.1 [M+Na]+. 1H NMR (CDCl3, 400 MHz): δ = 7.39 – 7.33 (m, 4H), 7.25 – 7.21 (m, 4H), 6.92 (br.s, 1H), 6.79 – 6.76 (m, 3H), 6.78 – 6.26 (m, 3H), 6.26 (d, J = 8.0 Hz, 1H), 5.15 (d, J = 8.4 Hz, 1H), 5.08 (s, 2H), 5.02–5.01 (m, 1H), 4.52–4.51 (m, 1H), 3.83 – 3.81 (m, 1H), 3.07 – 3.04 (m, 4H), 1.97 (s, 3H), 1.35 (s, 9H).
Benzyl (S)-2-acetamido-3-(3-(3-((S)-2-(2-(((benzyloxy)carbonyl)amino)acetamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoate (30a).
To a solution of compound 28a (800.0 mg, 1.2 mmol, 1.0 eq) in dioxane (5.0 mL) was added hydrochloric acid/dioxane (4 M, 5.0 mL). The mixture was stirred at 25°C for 4 hours. LCMS showed starting material was consumed completely and desired MS was detected. The mixture was concentrated to give compound 28a-amine (770.0 mg, crude, HCl salt) as a yellow brown solid. LCMS: RT = 0.82 min, m/z = 558.2 [M+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 9.27 (t, J = 6.0 Hz, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.33 (br.s, 3H), 7.35 – 7.28 (m, 7H), 7.03 – 6.92 (m, 4H), 6.87 – 6.82 (m, 2H), 5.08 (dd, J1 = 12.8 Hz, J2 =18.4 Hz, 2H), 4.54 – 4.48 (m, 1H), 4.10 – 4.09 (m, 1H), 4.03 – 3.89 (m, 2H), 3.11 – 2.96 (m, 3H), 2.92 – 2.86 (m, 1H), 1.78 (s, 3H). To a solution of Z-glycine (95.4 mg, 456.0 μmol, 0.9 eq) in dimethyl formamide (DMF) (4.0 mL) was added HATU (288.0 mg, 757.6 μmol, 1.5 eq) and diisopropylethylamine (DIPEA) (391.6 mg, 3.0 mmol, 529.2 μL, 6.0 eq). The mixture was stirred at 25°C for 0.25 hourr and then 28a-amine (300.0 mg, 505.0 μmol, 1.0 eq, HCl) was added. The mixture was stirred at 25°C for 0.25 hour. LCMS showed starting material was consumed completely and desired MS was detected. The mixture was poured into water (10 mL) and then extracted by ethyl acetate (3 × 20 mL). The combined organic phase was washed by saturate sodium carbonate (3 × 20 mL), brine (20 mL) and dried over sodium sulfate. After filtration and concentration, the crude product was purified by reverse phase flash (TFA condition) to afford compound 30a (160.0 mg, 213.7 μmol, 42.3% yield) as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ = 8.71 (t, J = 6.4 Hz, 1H), 8.37 (d, J = 7.6 Hz, 1H), 8.18 (d, J = 7.6 Hz, 1H), 7.37 – 7.26 (m, 13H), 7.00 – 6.96 (m, 2H), 6.92 (d, J = 11.6 Hz, 2H), 6.84 (d, J = 8.0 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H), 5.10 (dd, J 1= 12.8 Hz, J 2= 19.2 Hz, 2H), 5.01 (s, 2H), 4.53 – 4.51 (m 2H), 3.89 – 3.62 (m, 2H), 3.60 – 3.53 (m, 2H), 3.05 – 2.86 (m, 3H), 2.79 – 2.74 (m, 1H), 1.78 (s, 3H).
(S)-2-acetamido-3-(3-(3-((S)-2-(2-aminoacetamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoic acid (31a).
To a solution of compound 30a (160.0 mg, 213.7 μmol, 1.0 eq) in tetrahydrofuran (THF) (5.0 mL) was added Pd(OH)2/C (30.1 mg). The mixture was degassed and purged with hydrogen for 3 times, then the mixture was stirred at 25°C for 20 hours under hydrogen atmosphere. LCMS showed starting material and intermediate was consumed completely and desired MS was detected. The mixture was filtered and then filter cake was washed by dichloromethane (10 mL) and methanol (5 mL). The filtrate liquid was concentrated to afford compound 31a (140.0 mg, crude) as a white solid. LCMS: RT = 0.61 min, purity:27.9%, m/z = 525.1 [M+H]+.
(5S,11S)-11-acetamido-7,10-dioxo-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (2).
To a solution of compound 31a (140.0 mg, 266.9 μmol, 1.0 eq) in DMF (6.0 mL) was added DIPEA (69 mg, 533.9 μmol, 93.2 μL, 2.0 eq) and HATU (152.2 mg, 400.4 μmol, 1.5 eq) at 0°C under nitrogen. The mixture was stirred at 0°C for 1 hour. LCMS showed starting material was consumed completely. The mixture was poured into water (10 mL) and then extracted by ethyl acetate (3 × 20 mL). The combined organic phase was washed by brine (20 mL) and dried over sodium sulfate. After filtration and concentration, the crude product was recrystallization by methanol (2 × 2 mL) to afford 2 (10.1 mg, 19.6 μmol, 7.4% yield, 98.5% purity) as a white solid. LCMS: RT = 2.67 min, m/z = 507.1 [M+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.84 (t, J = 6.4 Hz, 1H), 8.13 (d, J = 8.8 Hz, 1H), 7.99 – 7.98 (m, 1H), 7.64 (d, J = 6.4 Hz, 1H), 7.29 – 7.25 (m, 2H), 7.11 (d, J = 7.6 Hz, 1H), 6.99 – 6.98 (m, 1H), 6.87 (d, J = 8.0 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.76 (s, 1H), 3.65 (s, 1H), 4.70 – 4.68 (m, 1H), 4.65 – 4.61 (m, 1H), 3.97 – 3.89 (m, 3H), 3.45 – 3.42 (m, 1H), 3.07 (d, J = 11.2 Hz, 1H), 2.96 (d, J = 12.4 Hz, 1H), 2.81 (dd, J1 = 6.4 Hz, J2 = 12.8 Hz, 1H), 2.69 – 2.63 (m, 1H), 1.89 (s, 3H).
Benzyl (S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3-methoxypropanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoate (30b).
To a solution of Boc-O-methyl-L-serine (150.0 mg, 228.1 μmol, 1.0 eq) in DMF (3.0 mL) was added DIPEA (88.4 mg, 684.2 μmol, 3.0 eq) and HATU (130.1 mg, 342.1 μmol, 1.5 eq) in turn at 0°C. Then compound 28a-amine (150.0 mg, 228.1 μmol, 1.0 eq) was added into the mixture and the reaction was stirred at 0°C for 4 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was added water (10 mL), and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL), dried with anhydrous sodium sulfate. After filtration and concentration, 300.0 mg of crude compound 30b was obtained as yellow oil. LCMS: RT = 0.99 min, purity: 54.7%, m/z = 759.3 [MS+H]+.
(S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-amino-3-methoxypropanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoic acid (31b).
To a solution of compound 30b (300.0 mg, 395.4 μmol, 1.0 eq) in dioxane (3 mL) was added HCl/dioxane (4 M, 5.0 mL, 50.6 eq). The reaction mixture was stirred at 26°C for 2.5 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. The mixture was concentrated in vacuum to afford compound 30b-amine (140.0 mg, crude, HCl salt) as yellow oil. LCMS: RT = 0.72 min, purity: 82.1%, m/z = 659.2 [MS+H]+. To a solution of 30b-amine (140.0 mg, 212.6 μmol, 1.0 eq) in THF (3 mL) was added into Palladium hydroxide (14.9 mg, 0.5 eq). Then the mixture was degassed under vacuum and purged hydrogen for 3 times and the reaction was stirred at 26°C for 3 hours. LCMS showed starting material was consumed completely and desired compound MS was detected, the reaction mixture was filtered, and the filtrate was concentrated in vacuum to afford compound 31b (110.0 mg, crude) as yellow oil. LCMS: RT = 0.72 min, purity: 82.1%, m/z = 569.2 [MS+H]+.
(5S,8S,11S)-11-acetamido-8-(methoxymethyl)-7,10-dioxo-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (3).
To a solution of compound 31b (110.0 mg, crude) in DMF (3.0 mL) was added DIPEA (75.0 mg, 580.4 μmol, 3.0 eq) and HATU (73.6 mg, 193.5 μmol, 1.0 eq) at 0°C. The reaction was stirred at 0°C for 2.5 hours, then HATU (36.8 mg, 96.7 μmol, 0.5 eq) was added into the mixture and the reaction was stirred at 0°C for 4 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was added water (10 mL) and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 20 mL) and dried with anhydrous sodium sulfate. After filtration and concentration, the residue was triturated with methanol (1 mL) to afford 3 (31.0 mg, 27.7% yield) as an off-white solid. LCMS: RT = 2.92 min, purity: 95%, m/z = 551.2 [MS+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.54 – 8.50 (m, 2H), 8.07 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 7.2 Hz, 1H), 7.33 – 7.24 (m, 2H), 7.14 (d, J = 8.0 Hz, 1H), 6.97 (dd, J1 = 1.6 Hz, J2 = 6.4 Hz, 1H), 6.85 (d, J = 7.2 Hz, 2H), 6.80 (d, J = 7.2 Hz, 1H), 6.09 (s, 1H), 4.67 – 4.61 (m, 2H), 4.56 – 4.51 (m, 1H), 4.03 – 3.95 (m, 2H), 3.20 (s, 3H), 3.12 (d, J = 12.0 Hz, 1H), 2.85 – 2.80 (m, 1H), 2.74 – 2.66 (m, 2H), 2.52 (d, J = 1.6 Hz, 2H), 1.88 (s, 3H).
Benzyl (S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-5,5,5-trifluoropentanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoate (30c).
To a solution of Boc-L-trifluoronorvaline in DMF (5.0 mL) was added HATU (211.2 mg, 555.5 μmol, 1.5 eq) and DIPEA (239.3 mg, 1.9 mmol, 323.4 μL, 5.0 eq) at 0°C. The mixture was stirred at 20°C for 30 minutes and then compound 28a-amine (220.0 mg, 370.4 μmol, 1.0 eq, HCl) was added into the mixture and stirred at 0°C for 1.5 hours. LCMS showed the starting material was consumed completely and desired product Ms was detected. The mixture was poured into water (20 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic phase was washed with brine (3 × 30 mL) and dried over sodium sulfate. After filtration and concentration, the crude product was purified with silica gel column (petroleum ether: ethyl acetate=10: 1~2: 1) to provide compound 30c (250.0 mg, 296.8 μmol, 80.1% yield, 96.3% purity) as a yellow solid. LCMS: RT = 0.98 min, m/z = 811.1 [MS+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.34 – 7.32 (m, 3H), 7.27 – 7.23 (m, 5H), 6.93 (td, J1 = 2.0 Hz, J2 = 9.6 Hz, 2H), 6.87 – 6.78 (m, 3H), 6.70 (m, 2H), 6.20 (br. s, 1H), 5.10 (s, 2H), 5.03 – 5.01 (m, 2H), 4.84 – 4.81 (m, 1H), 3.99 – 3.97 (m, 1H), 3.87 – 3.82 (m, 2H), 3.18 (dd, J1 = 5.6 Hz, J2 = 13.6 Hz, 1H), 3.07 (dd, J1 = 5.2 Hz, J2 = 14.4 Hz, 1H), 3.00 – 3.29 (m, 2H), 2.05 – 2.02 (m, 2H), 1.93 (s, 3H), 1.94 – 1.85 (m, 1H), 1.76 – 1.68 (m, 1H), 1.42 (s, 9H).
(S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-amino-5,5,5-trifluoropentanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoic acid (31c).
To a solution of compound 30c (250.0 mg, 308.4 μmol, 1.0 eq) in dioxane (10 mL) was added hydrochloric acid/dioxane (4 M, 10.0 mL, 129.7 eq). The mixture was stirred at 20°C for 1 hour. LCMS showed the starting material was consumed completely. The mixture was concentrated under vacuum to provide compound 30c-amine (220.0 mg, crude) as a white solid. LCMS: RT = 1.45 min, m/z = 711.3 [MS+H]+. To a solution of compound 30c-amine (230.0 mg, 323.6 μmol, 1.0 eq) in THF (10.0 mL) was added Pd(OH)2/C (50.0 mg). The mixture was degassed under vacuum and purged hydrogen for 3 times. The mixture was stirred at 20°C for 2 hours under hydrogen balloon. LCMS showed the starting material was consumed completely. The mixture was filtrated out. The filtrate liquid was concentrated under vacuum to provide compound 31c (180.0 mg, 290.1 μmol, 89.6% yield) as a white solid. 1H NMR (MeOD, 400 MHz): δ = 7.35 – 7.26 (m, 2H), 7.03 – 6.87 (m, 6H), 4.74 – 4.70 (m, 1H), 4.65 – 4.62 (m, 1H), 3.95 – 3.83 (m, 3H), 3.69 – 3.69 (m, 1H), 3.58 – 3.56 (m, 1H), 3.20 – 3.09 (m, 2H), 2.95 – 2.86 (m, 2H), 2.31 – 2.21 (m, 1H), 2.11 – 2.06 (m, 1H), 1.90 (s, 3H).
(5S,8S,11S)-11-acetamido-7,10-dioxo-N-(2,2,2-trifluoroethyl)-8-(3,3,3-trifluoropropyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (4).
To a solution of compound 31c (120.0 mg, 193.4 μmol, 1.0 eq) in DMF (2.0 mL) was added DIPEA (75.0 mg, 580.1 μmol, 101.3 μL, 3.0 eq) and HATU (110.3 mg, 290.1 μmol, 1.5 eq) in turn at 0 °C. The reaction was stirred at 0°C for 1 hour. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was quenched by water (10 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL), dried with anhydrous sodium sulfate. After filtration and concentration, the residue was triturated with methanol (1 mL) to provide 4 (10.0 mg, 8.5% yield) as a white solid. LCMS: RT = 3.26 min, purity: 98.6%, m/z = 603.2 [MS+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.85 (t, J = 6.4 Hz, 1H), 8.50 (d, J = 8.8 Hz, 1H), 8.05 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 6.8 Hz, 1H), 7.34 – 7.26 (m, 2H), 7.12 (d, J = 7.6 Hz, 1H), 6.98 (dd, J1 = 1.6 Hz, J2 = 8.0 Hz, 1H), 6.88 – 6.86 (m, 2H), 6.75 (s, 1H), 6.16 (s, 1H), 4.66 – 4.56 (m, 2H), 4.45 – 4.39 (m, 1H), 3.99 – 3.94 (m, 2H), 3.05 (d, J = 12.4 Hz, 1H), 2.83 – 2.71 (m, 3H), 2.17 – 2.09 (m, 2H), 1.87 (s, 3H), 1.71 – 1.54 (m, 2H).
Benzyl (S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoate (30d).
To a solution of Boc-L-norvaline (73.2 mg, 336.7 μmol, 1.0 eq) in DMF (3.0 mL) was added N,N-DIPEA (130.5 mg, 1.0 mmol, 176.4 μL, 3.0 eq), EDCI (96.8 mg, 505.0 μmol, 1.5 eq) and HOBT (68.2 mg, 505.0 μmol, 1.5 eq) at 0°C, then compound 28a-amine (200.0 mg, 336.7 μmol, 1.0 eq, HCl) was added into the mixture and the reaction was stirred at 26 °C for 6 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. To the reaction mixture was added water (5 mL). The mixture was acidified by HCl (1N) until pH= 4 and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL), dried over anhydrous sodium sulfate. After filtration and concentration, compound 30d (240.0 mg, crude) was obtained as yellow oil. LCMS: RT = 1.05 min, purity: 86.5%, m/z = 757.3 [MS+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.84 (dd, J1 = 8.4 Hz, J2 = 28.0 Hz, 1H), 7.47 – 7.43 (m, 1H), 7.34 – 7.30 (m, 3H), 7.25 – 7.20 (m, 4H), 6.91 – 6.87 (m, 3H), 6.79 (d, J = 7.6 Hz, 1H), 6.75 (br.s, 1H), 6.70 (br.s, 1H), 6.60 (d, J = 8.0 Hz, 1H), 6.27 (br.s, 1H), 5.09 (s, 2H), 4.98 (dd, J1 = 7.2 Hz, J2 = 23.2 Hz, 2H), 4.81 – 4.75 (m, 1H), 3.90 – 3.81 (m, 2H), 3.18 – 3.01 (m, 4H), 2.05 (d, J = 2.0 Hz, 3H), 1.66 – 1.61 (m, 2H), 1.39 (s, 9H), 1.26 (d, J = 1.2 Hz, 2H), 0.97 (t, J = 7.2 Hz, 3H).
(S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-aminopentanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoic acid (31d).
To a solution of 30d (240.0 mg, 317.1 μmol, 1.0 eq) in dioxane (3.0 mL) was added HCl/dioxane (4 M, 4.0 mL, 50.5 eq), the reaction was stirred at 26°C for 1 hour. LCMS showed starting material was consumed completely and desired compound MS was detected. The mixture was concentrated in vacuum to give the compound 30d-amine (270.0 mg, 270.9 μmol, 85.4% yield, 69.5% purity, HCl) as yellow oil. LCMS: RT = 0.78 min, m/z = 657.2 [MS+H]+. To a solution of 30d-amine (270.0 mg, 411.2 μmol, 1.0 eq) in THF (3.0 mL) was added Pd(OH)2/C (28.9 mg, 205.6 μmol, 0.5 eq) under hydrogen balloon (15 psi). Then the mixture was degassed under vacuum and purged hydrogen for 3 times and the reaction was stirred at 26°C for 1.5 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was filtered and the filtrate was concentrated in vacuum to give the compound 31d (110.0 mg, crude) as yellow oil. LCMS: RT = 0.77 min, purity: 82.5%, m/z = 567.2 [MS+H]+.
(5S,8S,11S)-11-acetamido-7,10-dioxo-8-propyl-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (5).
To a solution of 31d (110.0 mg, 194.2 μmol, 1.0 eq) in DMF (2.0 mL) was added DIPEA (75.3 mg, 582.5 μmol, 101.7 μL, 3.0 eq) and HATU (110.7 mg, 291.2 μmol, 1.5 eq) at 0°C, the reaction was stirred at 0°C for 2.5 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was added water (10 mL) and extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 20 mL), dried with anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was triturated with methanol (1 mL), filtered and the cake was collectedto give 5 (21.0 mg, 18.7% yield) as an off-white solid. LCMS: RT = 1.95 min, purity: 95.0%, m/z = 549.2 [MS+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.81 (t, J = 6.0 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.55 (d, J = 7.2 Hz, 1H), 7.33 – 7.21 (m, 2H), 7.14 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 9.2 Hz, 1H), 6.86 – 6.75 (m, 3H), 6.10 (s, 1H), 4.67 – 4.58 (m, 2H), 4.37 – 4.31 (m, 1H), 3.97 – 3.90 (m, 2H), 3.00 (d, J = 13.2 Hz, 1H), 2.86 – 2.67 (m, 2H), 1.88 (s, 2H), 1.45 – 1.41 (m, 1H), 1.36 – 1.30 (m, 1H), 1.21 – 1.14 (m, 3H), 0.80 (t, J = 7.2 Hz, 4H).
Benzyl (S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-4-methylpentanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoate (30e).
To a solution of Z-L-leucine (45.2 mg, 170.3 μmol, 1.1 eq) in DMF (3.0 mL) was added DIPEA (50.0 mg, 387.1 μmol, 67.6 μL, 2.5 eq), HOBt (27.2 mg, 201.3 μmol, 1.3 eq) and EDCI (38.6 mg, 201.3 μmol, 1.3 eq) at 0°C under nitrogen and then compound 28a-amine (100.0 mg, 154.8 μmol, 1.0 eq) was added. The mixture was stirred at 25°C for 16 hours. LCMS showed starting material was consumed completely and desired MS was detected. TLC (dichloromethane: methanol = 10:1) indicated starting material was consumed completely and one new spot formed. The mixture was poured into water (10 mL) and then extracted by ethyl acetate (3 × 10 mL). The combined organic phase was washed by brine (10 mL) and dried over sodium sulfate. After filtration and concentration, the crude product was purified by reverse phase flash (TFA condition) to give compound 30e (70.0 mg, 78.6 μmol, 50.8% yield, 90.4% purity) as a yellow solid. LCMS: RT = 0.96 min, m/z = 805.2 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.34 – 7.32 (m, 9H), 7.24 – 7.19 (m, 4H), 6.88– 6.86 (m, 3H), 6.77 – 6.74 (m, 2H), 6.67 – 6.62 (m, 2H), 6.24 (d, J = 6.4 Hz, 1H), 5.32 (d, J = 6.0 Hz, 1H), 5.10 (m, 3H), 4.99 (d, J = 11.6 Hz, 2H), 4.79 – 4.76 (m, 1H), 4.04 – 4.02 (m, 1H), 3.79 – 3.77 (m, 2H), 3.16– 3.11 (m, 2H), 3.02 – 2.97 (m, 2H), 1.91 (s, 3H), 1.48– 1.34 (m, 3H), 0.85 – 0.71 (m, 6H).
(S)-2-acetamido-3-(3-(3-((S)-2-((S)-2-amino-4-methylpentanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)propanoic acid (31e).
To a solution of compound 30e (300.0 mg, 372.7 μmol, 1.0 eq) in THF (4.0 mL) was added Pd/C (90.0 mg, 10% purity). The mixture was degassed and purged with hydrogen for 3 times, and then the mixture was stirred at 25°C for 23 hours under hydrogen balloon. LCMS showed starting material was consumed completely and desired MS was detected. To the mixture was added dichloromethane (10 mL) and methanol (5 mL). The mixture was filtered and then the filter liquor was concentrated to give crude product. The crude product was triturated by acetonitrile (5 mL) to give compound 31e (100.0 mg, 172.2 μmol, 46.2% yield, 100% purity) as an off-white solid. LCMS: RT = 0.69 min, m/z = 581.3 [M+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 9.78 (br.s, 1H), 8.91 (t, J = 5.6 Hz, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.17 (d, J = 6.4 Hz, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.99 – 6.96 (m, 1H), 6.90 – 6.86 (m, 4H), 6.80 (d, J = 7.6 Hz, 1H), 4.73 – 4.66 (m, 1H), 4.18 – 4.15 (m, 1H), 4.04 – 3.82 (m, 4H), 3.70 (t, J = 7.2 Hz, 1H), 3.02 – 3.01 (m, 2H), 2.93 – 2.87 (m, 1H), 1.82 (s, 3H), 1.51 – 1.45 (m, 1H), 1.33 (t, J = 7.2 Hz, 2H), 0.81 (t, J = 7.6 Hz, 6H).
(5S,8S,11S)-11-acetamido-8-isobutyl-7,10-dioxo-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (6).
To a solution of compound 31e (97.0 mg, 167.1 μmol, 1.0 eq) in DMF (7.0 mL) was added DIPEA (43.2 mg, 334.1 μmol, 58.4 μL, 2.0 eq) and HATU (95.3 mg, 250.6 μmol, 1.5 eq) at 0°C under nitrogen. The mixture was stirred at 0°C for 5 hours. LCMS showed starting material was consumed completely. The mixture was poured into water (10 mL) and then extracted by ethyl acetate (3 × 10 mL). The combined organic phase was washed with brine (20 mL), dried over sodium sulfate. After filtration and concentration, the crude product was recrystallized by acetonitrile (4 mL) to afford 6 (20.1 mg, 35.6 μmol, 21.3% yield, 99.7% purity) as a white solid. LCMS: RT = 3.04 min, m/z = 563.2 [M+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.78 (t, J = 6.0 Hz, 1H), 8.41 (d, J = 9.2 Hz, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 7.2 Hz, 1H), 7.32 – 7.26 (m, 2H), 7.14 (d, J = 7.6 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 8.0 Hz, 1H), 6.83 – 6.81 (m, 2H), 6.15 (s, 1H), 4.65 – 4.63 (m, 1H), 4.57 – 4.54 (m, 1H), 4.41 – 4.39 (m, 1H), 3.99 – 3.92 (m, 2H), 2.99 (d, J = 12.4 Hz, 1H), 2.87 – 2.80 (m, 2H), 2.70 – 2.67 (m, 1H), 1.87 (s, 3H), 1.46 – 1.42 (m, 1H), 1.32 – 1.25 (m, 2H), 0.83 (t, J = 6.8 Hz, 6H).
benzyl (S)-3-(3-(3-((S)-2-((tert-butoxycarbonyl)amino)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (28b).
To a solution of compound 27b (310.0 mg, 785.5 μmol, 1.0 eq) in dichloromethane (5.0 mL) was added compound 1 (674.3 mg, 1.7 mmol, 2.2 eq), copper acetate (214.0 mg, 1.2 mmol, 1.5 eq), triethylamine (794.9 mg, 7.9 mmol, 1.1 mL, 10.0 eq) and 4A molecular sieve (400.0 mg). The mixture was stirred at 25°C in the air for 3 hours. LCMS showed starting material was consumed completely and desired MS was detected. The mixture was filtered and then the filter was concentrated to give crude product. The crude product was purified by silica gel column (petroleum ether: ethyl acetate=3:1 to 3:1) to afford compound 28b (510.0 mg, 677.8 μmol, 86.3% yield, 90.9% purity) as a yellow solid. LCMS: RT = 0.96 min, m/z = 684.2 [M+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.59 – 8.56 (m, 1H), 7.38 – 7.32 (m, 4H), 7.29 – 7.24 (m, 2H), 7.05 – 6.98 (m, 5H), 6.89 (s, 1H), 6.85 – 6.83 (m, 1H), 6.78 – 6.75 (m, 1H), 5.18 (dd, J1 = 12.8 Hz, J2 = 16.0 Hz, 2H), 4.97 (dd, J1 = 5.2 Hz, J2 = 10.8 Hz, 1H), 3.39 – 3.35 (m, 2H), 3.24 – 3.15 (m, 2H), 3.06 (dd, J1 = 11.2, J2 = 14.4 Hz, 1H), 2.93 – 2.89 (m, 2H), 2.80 – 2.71 (m, 2H), 2.20 – 2.02 (m, 2H), 1.85 – 1.69 (m, 2H), 1.31 (d, J = 6.8 Hz, 9H).
Benzyl (S)-3-(3-(3-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3-phenylpropanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (30f).
To a solution of compound 28b (320.0 mg, 468.0 μmol, 1.0 eq) in dioxane (5.0 mL) was added hydrochloric acid/dioxane (4 M, 10.0 mL). The mixture was stirred at 25°C for 1.5 hours. TLC (petroleum ether: ethyl acetate=1:1) indicated starting material was consumed completely and one new spot formed. The mixture was concentrated to afford compound 28b-amine (305.0 mg, 404.8 μmol, 86.5% yield, 82.3% purity, HCl salt) as a yellow solid. LCMS: RT = 0.78 min, purity:82.3%, m/z 584.3[M+H]+. 1H NMR: (DMSO-d6, 400 MHz) δ = 9.22 – 9.19 (m, 1H), 7.40 – 7.26 (m, 8H), 7.02 (d, J = 6.4 Hz, 3H), 6.93 – 6.82 (m, 4H), 5.18 (dd, J1 = 12.8 Hz, J2 = 16.0 Hz, 2H), 4.97 (dd, J1 = 5.2 Hz, J2 = 10.8 Hz, 1H), 4.47 (br.s, 1H), 4.11 – 4.07 (m, 1H), 3.98 – 3.96 (m, 1H), 3.24 – 2.94 (m, 6H), 2.19 – 2.09 (m, 1H), 2.05 – 2.00 (m, 1H), 1.88 – 1.73 (m, 2H). To a solution of Boc-L-phenylalanine (51.8 mg, 195.1 μmol, 1.1 eq) in DMF (2.0 mL) was added DIPEA (68.8 mg, 532.2 μmol, 93.0 μL, 3.0 eq), HATU (101.2 mg, 266.1 μmol, 1.5 eq) at 0°C, then compound 28b-amine (110.0 mg, 177.4 μmol, 1.0 eq, HCl) was added into the mixture and the reaction was stirred at 26°C for 1.5 hours. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was quenched by water (3 mL), acidified by HCl (1N, 4 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL) and dried with anhydrous sodium sulfate. After filtration and concentration, the residue was purified by silica gel column chromatography (Petroleum ether: Ethyl acetate=20:1 to 3:1) to provide compound 30f (140.0 mg, 128.3 μmol, 72.3% yield, 76.2% purity) as yellow oil. LCMS: RT = 1.03 min, m/z = 831.1[M+H]+.
(S)-3-(3-(3-((S)-2-((S)-2-amino-3-phenylpropanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoic acid (31f).
To a solution of compound 30f (140.0 mg, 168.5 μmol, 1.0 eq) in dioxane (2.0 mL) was added HCl/dioxane (4M, 5.0 mL, 118.7 eq). The reaction was stirred at 26°C for 2 hours. LCMS showed starting material was consumed completely and desired compound MS was detected, the residue was concentrated in vacuum to give compound 30f-amine (128.0 mg, crude, HCl) as yellow oil. LCMS: RT = 0.86 min, purity: 82.2%, m/z 731.2 [MS+H]+. To a solution of compound 30f-amine (60.0 mg, 78.2 μmol, 1.0 eq, HCl salt) in THF (5.0 mL) was added Pd(OH)2 (15.0 mg, 10% purity) under hydrogen (15 psi, balloon). The mixture was degassed under vacuum and purged hydrogen for 3 times and the suspension was stirred at 26°C for 1 hour. LCMS showed starting material was consumed completely and desired compound MS was detected. The reaction mixture was filtered and the filtrate was concentrated in vacuum to give crude compound 31f (50.0 mg, 77.9% yield) as yellow oil. LCMS: RT = 0.73 min, purity: 78.0%, m/z = 641.2 [MS+H]+.
(5S,8S,11S)-8-benzyl-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (8).
To a solution of compound 31f (50.0 mg, 73.8 μmol, 1.0 eq, HCl salt) in DMF (3.0 mL) was added DIPEA (30.3 mg, 234.1 μmol, 40.9 μL, 3.2 eq) and HATU (44.5 mg, 117.1 μmol, 1.6 eq) in turn. The reaction was stirred at 0°C for 0.5 hour. LCMS showed starting material was consumed completely and desired MS was detected. The reaction mixture was quenched by water (10 mL), acidified by HCl (1 N, 4 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL), dried with anhydrous sodium sulfate. After filtration and concentration, the residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150 × 25 × 10 μm; mobile phase: [water(0.1%TFA)-ACN];B%: 42%–69%,12min) to give 8 (6.0 mg, 11.9% yield) as a white solid. LCMS: RT = 3.01 min, m/z = 623.2 [MS+H]+. 1H NMR (DMSO-d6, 400 MHz): δ = 8.66 −8.63 (m, 1H), 8.36 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 9.2 Hz, 1H), 7.31 (t, J = 8.0 Hz, 2H), 7.21 – 7.14 (m, 5H), 6.98 – 6.92 (m, 4H), 6.53 (s, 1H), 6.44 (s, 1H), 4.79 – 4.75 (m, 1H), 4.64 – 4.58 (m, 1H), 4.44 (dd, J1 = 2.0 Hz, J2 = 11.6 Hz, 1H), 4.02 – 3.89 (m, 2H), 3.04 – 3.02 (m, 2H), 2.94 – 2.86 (m, 1H), 2.79 – 2.75 (m, 1H), 2.63 – 2.57 (m, 2H), 2.41 (d, J = 10.4 Hz, 1H), 2.28 – 2.25 (m, 1H), 2.17 – 2.06 (m, 2H), 1.72 – 1.69 (m, 1H), 1.59 – 1.56 (m, 1H).
benzyl (S)-3-(3-(3-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-4-(piperidin-1-yl)butanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (30g).
To a solution of compound 29g (95.0 mg, 331.7 μmol, 1.0 eq) in DMF (3.0 mL) was added HOBT (58.3 mg, 431.3 μmol, 1.3 eq), EDCI(82.7 mg, 431.3 μmol, 1.3 eq) and DIPEA (107.2 mg, 829.4 μmol, 144.5 μL, 2.5 eq) at 0°C under nitrogen and then compound 28b-amine (300.0 mg, 483.8 μmol, 1.5 eq, HCl salt) was added. The mixture was stirred at 20°C for 16 hours. LCMS showed starting material was consumed completely and desired MS was detected. The mixture was poured into water (20 mL) and then added 1 N HCl (4 mL). The mixture was extracted by ethyl acetate (3 × 20 mL). The combined organic phase was washed by saturate sodium carbonate (3 × 20 mL), brine (20 mL), and dried over sodium sulfate. After filtration and concentration, the crude product was purified by prep-HPLC (column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water(0.1%TFA)-ACN]; B%: 35%−65%,13min) to afford compound 30g (120.0 mg, 124.8 μmol, 37.6% yield, 88.6% purity) as yellow oil. LCMS: RT = 0.833 min, m/z = 852.4 [M+H]+. 1H NMR (MeOD, 400 MHz): δ = 7.35 – 7.26 (m, 7H), 7.00 – 6.84 (m, 6H), 5.17 (d, J = 2.8 Hz, 2H), 5.08 – 5.06 (m, 1H), 4.75 – 4.73 (m, 1H), 4.15 – 4.05 (m, 1H), 3.89 – 3.87 (m, 2H), 3.48 – 3.45 (m, 3H), 3.36 – 3.34 (m, 1H), 3.09 – 2.85 (m, 7H), 2.26 – 2.16 (m, 2H), 2.01 – 1.69 (m ,10H), 1.43 (m, 10H).
(S)-3-(3-(3-((S)-2-((S)-2-amino-4-(piperidin-1-yl)butanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoic acid (31g).
A solution of compound 30g in THF (4.0 mL) was added Pd(OH)2 (40.0 mg). The suspension was degassed under vacuum and purged hydrogen for 3 times. The resulting mixture was stirred at 20°C for 1 hour under hrdrogen balloon. LCMS showed starting material was consumed completely and desired MS was detected. The mixture was filtered and the filtrated liquid was concentrated to afford compound 30g-acid (95.0 mg, crude) as a white solid. LCMS: RT = 0.77 min, purity: 96.3%, m/z = 762.2 [M+H]+. 1H NMR (MeOD, 400 MHz): δ = 7.30 – 7.24 (m, 2H), 7.99 – 6.85 (m, 6H), 4.87 – 4.76 (m ,2H), 4.07 – 3.90 (m, 3H), 3.44 – 3.31 (m, 3H), 3.13 – 2.91 (m, 8H), 2.28 – 2.21 (m, 2H), 2.03 – 1.83 (m, 8H), 1.69 – 1.55 (m, 2H), 1.42 – 1.29 (m, 10H). Compound 30g-acid (130.0 mg, 170.6 μmol, 1.0 eq) in dioxane (5.0 mL) was added HCl/dioxane (4 M, 10.0 mL). The mixture was stirred at 20°C for 40 minutes. LCMS showed starting material was consumed completely. The mixture was concentrated under vacuum to afford compound 31g (115.0 mg, crude, HCl) as a light yellow solid.
(5S,8S,11S)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (9).
To a solution of compound 31g (110.0 mg, 157.6 μmol, 1.0 eq, HCl) in DMF (10.0 mL) was added DIPEA (50.9 mg, 393.9 μmol, 68.6 μL, 2.5 eq), HOBt (29.8 mg, 220.6 μmol, 1.4 eq) and EDCI (42.3 mg, 220.6 μmol, 1.4 eq) at 0°C under nitrogen and the result mixture was stirred at 20°C for 16 hours. LCMS showed starting material was consumed completely and desired MS was detected. The mixture was poured into water (10 mL) and then extracted by ethyl acetate (3 × 20 mL). The combined organic phase was washed by brine (20 mL) and dried over sodium sulfate. After filtration and concentration, the crude product was purified by prep-HPLC (column: Phenomenex Gemini 10 μm C18 150 × 25mm;mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN]; B%: 35%−65%,12min) to afford 9 (25.8 mg, 38.1 μmol, 24.2% yield, 95.1% purity) as a white solid. LCMS: RT = 2.62 min, m/z = 644.3[M+H]+. 1H NMR (MeOD, 400 MHz): δ = 7.34 – 7.26 (m, 2H), 7.09 (d, J = 8.4 Hz, 1H), 6.98 – 6.91 (m, 3H), 6.65 (s, 1H), 6.50 (s, 1H), 4.77 – 4.68 (m, 2H), 4.34 (t, J = 7.2 Hz, 1H), 4.04 – 3.82 (m, 2H), 3.64 – 3.52 (m, 2H), 3.29 – 3.22 (m, 1H), 3.14 – 3.09 (m, 1H), 3.01 (dd, J1 = 9.2 Hz, J2 = 15.2 Hz, 1H), 2.77 (dd, J1 = 3.2 Hz, J2 =12.4 Hz, 1H), 2.48 – 2.31 (m, 6H), 2.29 (t, J = 7.2 Hz, 2H), 2.09 – 2.01 (m, 2H), 1.85 – 1.68 (m, 2H), 1.57 – 1.55 (m, 4H), 1.49 – 1.38 (m, 2H).
Benzyl (S)-3-(3-(3-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-4-(4,4-difluoropiperidin-1-yl)butanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (30h).
To a solution of compound 29h (155.7 mg, 483.1 μmol, 1.2 eq) in DMF (5.0 mL) was added HOBt (81.6 mg, 603.8 μmol, 1.5 eq), EDCI (115.8 mg, 603.8 μmol, 1.5 eq) and DIPEA (260.1 mg, 2.0 mmol, 350.6 μL, 5.0 eq) at 0°C. Then compound 28b-amine (250.0 mg, 402.6 μmol, 1.0 eq, HCl) was added into the mixture and the mixture was stirred for 16 hours at 20°C. LCMS showed the starting material was consumed completely and desired product was detected. The mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic phase was washed with brine (20 mL) and dried over sodium sulfate. After filtration and concentration, the crude product was purified with prep-HPLC(column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water(0.1%TFA)-ACN];B%: 35%−65%,13min) to provide compound 30h (180.0 mg, 202.7 μmol, 50.4% yield) as colorless oil. 1H NMR (CDCl3, 400 MHz): δ = 7.95 (br.s, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.35 – 7.33 (m, 4H), 7.27 – 7.24 (m ,4H), 6.99 – 6.91 (m, 5H), 6.85 (d, J = 7.6 Hz, 1H), 5.53 (br.s, 1H), 5.31 – 5.29 (m, 1H), 5.16 (dd, J1 = 12.0 Hz, J2 = 17.6 Hz, 2H), 5.00 – 4.93 (m, 1H), 3.86 – 3.72 (m, 2H), 3.46 – 3.31 (m, 4H), 3.11 (dd, J1 = 4.0 Hz, J1 = 9.6 Hz, 1H), 2.95 – 2.64 (m, 5H), 2.33 – 2.20 (m, 4H), 2.00 – 1.75 (m, 8H), 1.45 (s, 9H).
(S)-3-(3-(3-((S)-2-((S)-2-amino-4-(4,4-difluoropiperidin-1-yl)butanamido)-3-oxo-3-((2,2,2-trifluoroethyl)amino)propyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoic acid (31h).
To a solution of compound 30h (150.0 mg, 168.9 μmol, 1.0 eq) in methanol (2.0 mL) was added Palladium hydroxide (37.5 mg, 26.7 μmol, 10% purity). The mixture was degassed under vacuum and purged hydrogen for 3 times. The mixture was stirred at 20°C under hydrogen balloon for 2 hours. LCMS showed the starting material was consumed completely. The mixture was filtrated and the filter liquid was concentrated under vacuum to provide compound 30h-acid (130.0 mg, 163.0 μmol, 96.5% yield) as colorless oil. LCMS: RT = 0.75 min, purity: 89.6%, m/z = 798.4[M+H]+. To a solution of compound 30h-acid (120.0 mg, 150.4 μmol, 1.0 eq) in dioxane (5.0 mL) was added HCl/dioxane (4 M, 5.0 mL, 133.0 eq). The mixture was stirred at 20°C for 0.5 hour. LCMS showed the starting material was consumed completely and desired product was detected. The mixture was filtrated and the filtrate liquid was concentrated under vacuum to provide compound 31h (120.0 mg, crude, HCl) was obtained as a white solid. LCMS: RT = 0.68 min, purity: 89.1%, m/z = 698.3[M+H]+.
(5S,8S,11S)-8-(2-(4,4-difluoropiperidin-1-yl)ethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (10).
To a solution of compound 31h (100.0 mg, 136.6 μmol, 1.0 eq, HCl salt) in DMF (3.0 mL) was added DIPEA (44.1 mg, 341.5 μmol, 59.5 μL, 2.5 eq), HOBt (24.0 mg, 177.6 μmol, 1.3 eq) and EDCI (34.0 mg, 177.6 μmol, 1.3 eq) at 0°C under nitrogen and then the mixture was stirred at 25°C for 16 hours. After 16 hours, LCMS showed starting material was consumed completely and desired MS was detected. The mixture was poured into water (10 mL) and then extracted by ethyl acetate (3 × 20 mL). The combined organic phase was dried over sodium sulfate. After filtration and concentration, the crude product was purified by prep-HPLC(column: Phenomenex Gemini 5 μm C18 250 × 21.2 mm; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN];B%: 35%−65%,12min) to afford 10 (13.5 mg, 19.9 μmol, 14.5% yield, 100.0% purity) as a white solid. LCMS: RT = 2.65 min, m/z = 680.2[M+H]+. 1H NMR (MeOD, 400 MHz): δ = 7.33 (t, J = 7.6 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 6.98 – 6.89 (m, 3H), 6.66 (s, 1H), 6.47 (s, 1H), 4.76 (dd, J1 = 4.0 Hz, J2 = 8.8 Hz, 1H), 4.71 (dd, J1 = 3.2 Hz, J2 = 12.0 Hz, 1H), 4.38 (t, J = 7.2 Hz, 1H), 4.04 – 3.82 (m, 2H), 3.64 – 3.52 (m, 2H), 3.23 (d, J = 12.8 Hz, 1H), 3.13 (dd, J1 = 3.2 Hz, J2 =14.8 Hz, 1H), 2.98 (dd, J1 = 9.2 Hz, J2 = 15.2 Hz, 1H), 2.77 (dd, J1 = 3.2 Hz, J2 = 12.4 Hz, 1H), 2.52 – 2.45 (m, 4H), 2.43 – 2.35.
benzyl (S)-3-(3-(3-((S)-3-(tert-butoxy)-2-((tert-butoxycarbonyl)amino)-3-oxopropyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (34a).
To a solution of compound 32 (8.1 g, 22.1 mmol, 1.5 eq), compound 27b (5.0 g, 14.7 mmol, 1.0 eq), 4A molecular sieve (5.0 g) and triethylamine (7.5 g, 73.7 mmol, 10.3 mL, 5.0 eq) in dichloromethane (100 mL) was added copper acetate (4.0 g, 22.1 mmol, 1.5 eq). The mixture was stirred at 25°C for 18 hours under oxygen (15 psi). LCMS showed 20% of material 27b remianed. The mixture was filtered through a celite pad; the solid was washed with ethyl acetate (4 × 30 mL). The combined filtrates were concentrated in vacuum. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 10/1 ~ 4/1) to give compound 34a (5.7 g, 8.6 mmol, 58.5% yield, 100.0% purity) as light-yellow gum. LCMS: RT = 1.05 min, m/z = 681.1 [M+Na]+. 1H NMR (CDCl3, 400 MHz): δ = 7.38 – 7.31 (m, 5H), 7.25 – 7.21 (m, 2H), 6.94 – 6.91 (m, 2H), 6.85 – 6.83 (m, 4H), 5.19 – 5.09 (m, 4H), 4.44 – 4.41 (m, 1H), 3.39 – 3.31 (m, 3H), 3.04 – 2.98 (m, 3H), 2.32 – 2.23 (m, 2H), 1.96 – 1.77 (m, 2H), 1.41 (s, 9H), 1.40 (s, 9H).
Benzyl (S)-3-(3-(3-((S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-4-(piperidin-1-yl)butanamido)-3-(tert-butoxy)-3-oxopropyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (36a).
To a solution of compound 34a (5.7 g, 8.6 mmol, 1.0 eq) in dichloromethane (120.0 mL) was added trifluoroacetic acid (37.0 g, 324.2 mmol, 24.0 mL, 37.6 eq) at 0°C. The mixture was stirred at 0°C for 4 hours. TLC (petroleum ether : ethyl acetate = 2:1) showed most of starting material was consumed. The mixture was poured into saturated sodium bicarbonate solution (300.0 mL, pH = 7). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, concentrated in vacuum to give compound 34a-amine (4.2 g, 6.8 mmol, 79.4 % yield, 91.4% purity) as yellow gum, which was used for the next step without further purification. LCMS: RT = 0.89 min, m/z 559.1 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.35 – 7.27 (m, 7H), 6.99 – 6.97 (m, 2H), 6.86 – 6.82 (m, 4H), 5.20 – 5.16 (m, 2H), 4.94 (dd, J = 10.8, 5.6 Hz, 1H), 3.58 – 3.56 (m, 1H), 3.46 – 3.40 (m, 1H), 3.33 – 3.29 (m, 2H), 3.05 (dd, J = 14.4, 10.8 Hz, 1H), 2.91 (d, J = 6.8 Hz, 1H), 2.25 – 2.22 (m, 2H), 1.90 – 1.84 (m, 2H), 1.37 (s, 9H). To a solution of compound 35a (231.0 mg, 531.8 μmol, 1.2 eq, trifluoroacetic acid salt), DIPEA (401.0 mg, 3.1 mmol, 540.3 μL, 7.0 eq) in DMF (3.0 mL) was added HOBt (78.0 mg, 576.1 μmol, 1.3 eq) at 0°C, the mixture was stirred at 0°C for 10 minutes. EDCI (340.0 mg, 1.8 mmol, 4.0 eq) was added and then compound 34a-amine (300.0 mg, 443.1 μmol, 1.0 eq) in DMF (1.0 mL) was drop-wise added at 0°C. The reaction mixture was stirred at 0°C for 20 minutes and then stirred at 25°C for another 16 hours under nitrogen atmosphere. LCMS showed the starting material was consumed completely and desired product mass was detected. The mixture was quenched with water (10 mL) and then combined with batch EW1319–1700. The mixture was extracted with ethyl acetate (3 × 15 mL), washed with brine (3 xx 15 mL), dried over anhydrous sodium sulfate, concentrated in vacuum. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 2/1 ~ 0/1) to give compound 36a (450.0 mg, purity 87.5%) as yellow gum. LCMS: RT = 0.95 min, m/z = 861.4 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.35 – 7.23 (m, 12H), 6.98 – 6.96 (m, 2H), 6.88 – 6.77 (m, 4H), 5.17 – 4.99 (m, 5H), 4.58 – 4.57 (m, 1H), 4.21 – 4.20 (m, 1H), 3.44 – 3.42 (m, 1H), 3.31 – 3.30 (m, 1H), 3.27 – 3.24 (m, 1H), 3.08 – 3.97 (m, 9H), 2.28 – 2.17 (m, 3H), 1.96 – 1.84 (m, 2H), 1.76 – 1.75 (m, 4H), 1.59 – 1.48 (m, 3H), 1.41 – 1.39 (m, 9H).
Tert-butyl (5S,8S,11S)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (37a).
To a solution of compound 36a (300.0 mg, 304.7 μmol, 1.0 eq) in isopropyl alcohol (6.0 mL) was added Pd/C (50.0 mg, 10% purity) and Pd(OH)2/C (50.0 mg, 10% purity) under nitrogen atmosphere. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen (15 psi) at 25°C for 8 hours. LCMS showed the starting material was consumed and 51% of the intermediate imine remained. The mixture was filtered. The solid was washed with isopropyl alcohol (3 × 2 mL). Pd/C (50 mg, 10% purity) and Pd(OH)2/C (50 mg, 10% purity) was added into the combined filtrate under nitrogen atmosphere. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen (15 psi) at 25°C for 14 hours. LCMS showed the starting material was consumed and 11.65% of the intermediate imine remained, the desired compound was detected. The residue was purified by reverse flash (trifluoroacetic acid condition). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was lyophilized to give crude compound 36a-amino acid (280.0 mg, crude, Na salt) as a white solid, which was used into the next step without further purification. LCMS: RT = 0.77 min, m/z 637.5 [M+H]+, purity: 98.5%. To a solution of compound 36a-amino acid (280.0 mg, 315.6 μmol, 1.0 eq) in DMF (280 mL) was added DIPEA (286.0 mg, 2.2 mmol, 384.8 μL, 7.0 eq) and HOBt (64.0 mg, 473.4 μmol, 1.5 eq) at 0°C. The mixture was stirred at 0°C for 10 minute. EDCI (303.0 mg, 1.6 mmol, 5.0 eq) was added. The reaction mixture was stirred at 0°C for 20 minutes and then stirred at 25°C for another 16 hours. LCMS showed a part of starting material remained. DIPEA (82.0 mg, 631.3 μmol, 110.0 μL, 2.0 eq) and EDCI (121.0 mg, 631.3 μmol, 2.0 eq) was added at 0°C. The mixture was stirred at 25°C for another 24 hours. LCMS showed the starting material was consumed. The mixture was poured into ice water (200 mL) and then extracted with ethyl acetate (3 × 120 mL). The combined organic layers were washed with brine (4 × 100 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by reverse flash column (trifluoroacetic acid condition) and then re-purified by prep-HPLC (column: Boston pH-lex 10 μm C18 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 26%−56%, 10min) to give compound 37a (40.0 mg, 63.8 μmol, 20.2% yield, 98.7% purity) as a light yellow solid. Meanwhile 20 mg of the diastereoisomer was obtained. LCMS: RT = 0.88 min, m/z 619.3 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.34 (t, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 6.95 – 6.90 (m, 3H), 6.69 – 6.68 (m, 1H), 6.35 (s, 1H), 4.71 (dd, J = 12.0, 4.0 Hz, 1H), 4.62 (dd, J = 8.4, 4.0 Hz, 1H), 4.39 (t, J = 7.2 Hz, 1H), 3.63 – 3.61 (m, 2H), 3.27 – 3.21 (m, 2H), 2.99 (dd, J = 15.6, 8.8 Hz, 1H), 2.81 (dd, J = 12.4, 3.6 Hz, 1H), 2.60 – 2.32 (m, 8H), 2.08 – 2.01 (m, 2H), 1.93 – 1.79 (m, 4H), 1.66 – 1.62 (m, 4H), 1.50 (m, 9H).
(5S,8S,11S)-N-((1-methylcyclopropyl)methyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (17).
To a solution of compound 37a (35.0 mg, 56.6 μmol, 1.0 eq) in dichloromethane (1.0 mL) was added trifluoroacetic acid (0.4 mL) at 0°C. The mixture was stirred at 25°C for 3 hours. TLC showed the starting material was consumed completely. The mixture was concentrated in vacuum to give crude compound 37a-acid (40.0 mg, crude, trifluoroacetic acid) as light-yellow gum, which was used for the next step without further purification. To a solution of 37a-acid (35.0 mg, 51.7 μmol, 1.0 eq, TFA), (1-methylcyclopropyl)methanamine (19.0 mg, 155.2 μmol, 3.0 eq, HCl) and DIPEA (40.1 mg, 310.3 μmol, 54.1 μL, 6.0 eq) in THF (0.3 mL) was added HATU (39.0 mg, 103.5 μmol, 2.0 eq) at 0°C. The mixture was stirred at 20°C for 16 hours under nitrogen atmosphere. LCMS showed the starting material was consumed; the desired compound was detected. The mixture was quenched with water (10 mL) and then adjusted to pH=7 with HCl (1M). The mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by prep-HPLC (column: Phenomenex Gemini 10 μm C18 150 × 25 mm; mobile phase: [water (10mM NH4HCO3)-ACN]; B%: 50%−80%, 8 min) to give 17 (16.0 mg, 25.2 μmol, 48.8% yield, 99.3% purity) as a light yellow solid. LCMS: RT = 2.68 min, m/z = 630.3 [M+H]+. 1H NMR (MeOD, 400 MHz): δ = 7.33 – 7.26 (m, 2H), 7.08 (d, J = 7.6 Hz, 1H), 6.96 – 6.93 (m, 3H), 6.64 (s, 1H), 6.53 (s, 1H), 4.75 – 4.69 (m, 2H), 4.34 (t, J = 7.2 Hz, 1H), 3.59 – 3.54 (m, 2H), 3.26 (t, J = 12.4 Hz, 1H), 3.13 – 3.08 (m, 3H), 3.05 – 2.97 (m, 1H), 2.75 (dd, J = 12.8, 3.2 Hz, 1H), 2.41 – 2.27 (m, 6H), 2.23 (t, J = 7.6 Hz, 2H), 2.07 – 2.01 (m, 2H), 1.81 – 1.71 (m, 2H), 1.57 – 1.53 (m, 4H), 1.44 – 1.43 (m, 2H), 1.07 (s, 3H), 0.46 – 0.44 (m, 2H), 0.32 – 0.30 (m, 2H).
(5S,8S,11S)-N-((R)-1-cyclopropylethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (18).
To a solution of 37a-acid (30.0 mg, 44.3 μmol, 1.0 eq, TFA) in pyridine (0.6 mL) was added HOBt (6.0 mg, 44.3 μmol, 1 eq) at 0°C, the mixture was stirred at 0°C for 15 minutes. (R)-1-Cyclopropylethylamine (11.0 mg, 133.0 μmol, 3.0 eq) and EDCI (26.0 mg, 133.0 μmol, 3.0 eq) were added into the mixture at 0°C and then the mixture was stirred at 25°C for another 2 hours. LCMS showed the starting material was remained. Another EDCI (26.0 mg, 133.0 μmol, 3.0 eq) and (R)-1-cyclopropylethylamine (12.0 mg, 133.0 μmol, 3.0 eq) were added into the mixture at 0°C. The mixture was stirred at 25°C for another 18 hours. LCMS showed the most starting material remained. Another (R)-1-cyclopropylethylamine (38.0 mg, 443.3 μmol, 10.0 eq) and EDCI (51.0 mg, 266.0 μmol, 6.0 eq) was added into the mixture at 0°C. The reaction mixture was stirred at 25°C for another 18 hours. Pyridine (0.6 mL) and EDCI (51.0 mg, 266.0 μmol, 6.0 eq) were added into the mixture. The reaction mixture was stirred at 25°C for another 20 hours. LCMS showed the starting material remained. HATU (67.0 mg, 177.3 μmol, 4.0 eq) was added into the mixture. The reaction mixture was stirred at 20°C for another 24 hours. LCMS showed the most of starting material was consumed, the desired compound was detected. The mixture was poured into water (20 mL) and then extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine (3 × 20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by prep-HPLC (column: Boston pH-lex 10 μm C18 150 × 25 mm; mobile phase: [water(0.1%TFA)-ACN];B%: 35%−59%,8min) and then further purified by prep-HPLC (column: Waters Xbridge 5 μm C18 150 × 25 mm; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN];B%: 50%−68%,10min) to give 18 (8.7 mg, 13.7 μmol, 30.9% yield, 99.3% purity) as a white solid. LCMS: RT = 2.86 min, m/z = 630.4 [M+H]+. 1H NMR (MeOD, 400 MHz): δ = 7.33 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.96 – 6.91 (m, 3H), 6.66 (s, 1H), 6.50 (s, 1H), 4.72 – 4.65 (m, 2H), 4.34 (t, J = 7.2 Hz, 1H), 3.60 – 3.56 (m, 2H), 3.35 – 3.33 (m, 1H), 3.27 – 3.24 (m, 1H), 3.09 – 2.95 (m, 2H), 2.76 (dd, J = 12.4, 2.8 Hz, 1H), 2.42 – 2.25 (m, 8H), 2.07 – 2.01 (m, 2H), 1.81 – 1.72 (m, 2H), 1.56 – 1.55 (m, 4H), 1.45 – 1.44 (m, 2H), 1.23 (d, J = 6.8 Hz, 3H), 0.88 – 0.85 (m, 1H), 0.53 – 0.38 (m, 2H), 0.28 – 0.20 (m, 2H).
(5S,8S,11S)-N-((S)-1-cyclopropylethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (19).
To a solution of 37a-acid (30.0 mg, 44.3 μmol, 1.0 eq, TFA), (S)-1-cyclopropylethylamine (11.0 mg, 133.0 μmol, 3.0 eq) and DIPEA (17.0 mg, 133.0 μmol, 23.2 μL, 3.0 eq) in THF (0.4 mL) was added HATU (33.7 mg, 88.7 μmol, 2.0 eq) at 0°C. The mixture was stirred at 20°C for 16 hours under nitrogen atmosphere. LCMS showed the starting material was consumed; the desired mass was detected. The mixture was quenched with water (10 mL). The mixture was adjusted to pH=7 with HCl (1M) and then extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by prep-HPLC (column: Phenomenex Gemini 10 μm C18 150 × 25 mm; mobile phase: [water (10mM NH4HCO3)-ACN]; B%: 50%−80%, 8min) to give 19 (6.9 mg, purity: 99.3%) as a light-yellow solid. LCMS: RT = 2.68 min, m/z = 630.3 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.33 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.96 – 6.95 (m, 3H), 6.66 (s, 1H), 6.47 (s, 1H), 4.71 – 4.64 (m, 2H), 4.35 (t, J = 7.2 Hz, 1H), 3.64 – 3.53 (m, 2H), 3.31 – 3.27 (m, 2H), 3.08 – 3.00 (m, 2H), 2.75 (dd, J = 12.8, 3.2 Hz, 1H), 2.48 – 2.34 (m, 6H), 2.25 (t, J = 7.6 Hz, 1H), 2.09 – 1.99 (m, 2H), 1.82 – 1.68 (m, 2H), 1.57 – 1.54 (m, 4H), 1.44 – 1.43 (m, 2H), 1.19 (d, J = 6.8 Hz, 3H), 0.93 – 0.87 (m, 1H), 0.55 – 0.45 (m, 2H), 0.36 – 0.30 (m, 1H), 0.25 – 0.19 (m, 1H).
(5S,8S,11S)-N-(bicyclo[1.1.1]pentan-1-yl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (21).
To a solution of compound 37a-acid (40.0 mg, 59.1 μmol, 1.0 eq, trifluoroacetic acid salt) in pyridine (0.5 mL) was added propellamine (21.0 mg, 177.3 μmol, 3.0 eq, HCl salt). The mixture was cooled to 0°C and then added HOBt (8.0 mg, 59.1 μmol, 1.0 eq). The mixture was stirred at 0°C for 10 minutes. EDCI (34.0 mg, 177.3 μmol, 3.0 eq) was added at 0°C. The reaction mixture was stirred at 0°C for 20 minutes and then stirred at 25°C for anothers 1.5 hours under nitrogen atmosphere. LCMS showed the starting material was consumed and desired product mass was detected. The mixture was quenched with water (5 mL). The mixture was concentrated in vacuum; the residue was diluted with water (20 mL) and then extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by prep-HPLC (column: Boston pH-lex 10μm C18 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 23%−53%, 10min) and then re-purified by prep-HPLC (column: Phenomenex Synergi C18 10μm 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 23%−53%, 12min). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was concentrated in vacuum to removed acetonitrile, extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was lyophilized to give 21 (15.0 mg, purity 98.4%) as a white solid. LCMS: RT = 2.42 min, m/z 628.3 [M+H]+ , purity: 98.4%. 1H NMR (Methanol-d4, 400 MHz): δ = 7.34 (t, J = 8.0 Hz, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 6.95 – 6.89 (m, 3H), 6.66 (s, 1H), 6.42 (s, 1H), 4.69 (dd, J = 12.4, 3.6 Hz, 1H), 4.58 – 4.55 (m, 1H), 4.37 (t, J = 6.8 Hz, 1H), 3.62 – 3.59 (m, 2H), 3.33 – 3.31 (m, 1H), 3.24 (t, J = 12.8 Hz, 1H), 3.06 (dd, J = 15.6, 8.0 Hz, 1H), 2.94 (dd, J = 15.2, 8.5 Hz, 1H), 2.82 (dd, J = 12.4, 3.6 Hz, 1H), 2.65 – 2.60 (m, 5H), 2.44 – 2.35 (m, 3H), 2.10 – 2.06 (m, 8H), 1.90 – 1.76 (m, 2H), 1.67 – 1.64 (m, 4H), 1.52 – 1.51 (m, 2H).
Benzyl (S)-3-(3-(3-((S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-3-(1,3-dioxan-2-yl)propanamido)-3-(tert-butoxy)-3-oxopropyl)phenoxy)phenyl)-2-(2-oxopyrrolidin-1-yl)propanoate (36b).
To a solution of compound 35b (2.6 g, 8.3 mmol, 1.2 eq) and DIPEA (3.5 g, 27.4 mmol, 4.8 mL, 4.0 eq) in DMF (45.0 mL) was added HOBt (1.2 g, 8.9 mmol, 1.3 eq) at 0°C. The mixture was stirred at 0°C for 10 minutes. EDCI (2.6 g, 13.7 mmol, 2.0 eq) was added and then compound 34a-amine (4.2 g, 6.8 mmol, 1.0 eq) in DMF (15.0 mL) was added. The reaction mixture was stirred at 0°C for 20 minutes and then stirred at 25°C for another 1.5 hours under nitrogen atmosphere. LCMS showed most of the starting material was consumed and desired compound was detected. The mixture was quenched with ice water (100 mL) and then extracted with ethyl acetate (3 × 60 mL). The combined organic layers were washed with brine (3 × 50 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 6:1 ~ 1:1) to give compound 36b (4.1 g, 4.7 mmol, 68.0% yield, 96.9% purity) as colorless gum. LCMS: RT = 1.01 min, m/z 850.4 [M+H]+. 1H NMR (CDCl3, 400 MHz) δ = 7.36 – 7.30 (m, 10H), 7.24 – 7.19 (m, 2H), 7.00 (d, J = 5.6 Hz, 1H), 6.91(d, J = 6.8 Hz, 2H), 6.83 – 6.81 (m, 4H), 5.94 (d, J = 6.4 Hz, 1H), 5.15 – 5.09 (m, 5H), 4.71 – 4.68 (m, 2H), 4.36 – 4.34 (m, 1H), 4.03 – 4.00 (m, 2H), 3.72 – 3.63 (m, 2H), 3.35 – 3.30 (m, 3H), 3.06 (d, J = 6.0 Hz, 2H), 3.30 – 2.94 (m, 1H), 2.31 – 2.21 (m, 2H), 2.05 – 1.72 (m, 6H), 1.38 (s, 9H).
Tert-butyl (5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (37b).
To a solution of compound 36b (4.1 g, 4.8 mmol, 1.0 eq) in THF (60.0 mL) was added Pd/C (0.4 g, 10% purity) and Pd(OH)2/C (0.4 g, 10% purity) under nitrogen atmosphere. The mixture was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen atmosphere (15 psi) at 25°C for 14 hours. LCMS showed the starting material was consumed and desired product mass was detected. The mixture was filtered, the solid was washed with THF (4 × 10 mL) and ethyl acetate (3 × 10 mL). The combined filtrate was concentrated in vacuum to give compound 36b-amino acid (3.1 g, crude) as a white solid, which was used into the next step without further purification. LCMS: RT = 0.81 min, m/z 626.3 [M+H]+ , purity: 95.1%. 1H NMR (Methanol-d4, 400 MHz): δ = 7.28 – 7.26 (m, 2H), 7.01– 6.98 (m, 2H), 6.86 – 6.84 (m, 4H), 4.84 – 4.82 (m, 1H), 4.75 – 4.71 (m, 1H), 4.60 (dd, J = 9.2, 5.2 Hz, 1H), 4.10 – 4.06 (m, 2H), 4.00 – 3.98 (m, 1H), 3.83 – 3.72 (m, 2H), 3.59 – 3.56 (m, 1H), 3.41 – 3.33 (m, 2H), 3.19 – 3.14 (m, 1H), 2.99 – 2.86 (m, 2H), 2.32 – 2.01 (m, 7H), 1.87 – 1.86 (m, 1H), 1.45 (s, 9H). To a solution of compound 36b-amino acid (1.0 g, 1.6 mmol, 1.0 eq) and DIPEA (1.0 g, 8.0 mmol, 1.4 mL, 5.0 eq) in DMF (80.0 mL) was added HOBt (324.0 mg, 2.4 mmol, 1.5 eq) and EDCI (613.0 mg, 3.2 mmol, 2.0 eq) at 0°C. The mixture was stirred at 25°C for 15 hours. LCMS showed the starting material remained. Another batch of DIPEA (516.0 mg, 4.0 mmol, 695.9 μL, 2.5 eq) and EDCI (613.0 mg, 3.2 mmol, 2.0 eq) were added at 0°C. The reaction mixture was stirred at 25°C for another 15 hours. LCMS showed the starting material was consumed. The mixture was quenched with ice water (100 mL) and then extracted with ethyl acetate (3 × 100 mL). The combined organic layers were washed with brine (3 × 50 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 1/1~0/1) to give 37b (410.0 mg, 564.7 μmol, 35.3% yield, 83.7% purity) as a white solid. LCMS: RT = 0.88 min, m/z = 608.1 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 8.11 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 6.95 – 6.92 (m, 3H), 6.66 – 6.65 (m, 1H), 6.45 (m, 1H), 4.70 (dd, J = 12.0, 3.2 Hz, 1H), 4.66 – 4.61 (m, 1H), 4.52 – 4.48 (m, 2H), 4.00 – 3.97 (m, 2H), 3.75 – 3.53 (m, 5H), 3.26 (t, J = 12.4 Hz, 1H), 3.18 (dd, J = 15.2, 4.0 Hz, 1H), 3.00 (dd, J = 15.2, 8.8 Hz, 1H), 2.73 (dd, J = 12.4, 2.8 Hz, 1H), 2.43 – 2.41 (m, 2H), 2.01 – 1.84 (m, 8H), 1.50 (s, 9H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (38a).
To a solution of 37b (410.0 mg, 564.7 μmol, 1.0 eq) in dichloromethane (5.0 mL) was added trifluoroacetic acid (2.4 g, 21.0 mmol, 1.6 mL, 37.2 eq). The mixture was stirred at 25°C for 5.5 hours. TLC (petroleum ether: ethyl acetate = 0:1) showed most of the starting material was consumed. The mixture was poured into water (50 mL) and then adjusted pH = 4~5 with saturated sodium bicarbonate aqueous. The mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum to give compound 37b-acid (440.0 mg, crude) as a white solid, which was used into the next step without further purification. To a solution of compound 37b-acid (120.0 mg, 217.6 μmol, 1.0 eq) in pyridine (1.5 mL) was added HOBt (29.0 mg, 217.6 μmol, 1.0 eq). The mixture was stirred at 0°C for 10 minutes. 2,2,2-Trifluoroethylamine (43.0 mg, 435.1 μmol, 34.2 μL, 2.0 eq) and EDCI (104.0 mg, 543.9 μmol, 2.5 eq) were added at 0°C. The reaction mixture was stirred at 0°C for 20 minutes and then stirred at 25°C for 6 hours. LCMS showed the starting material was consumed completely and desired product mass was detected. The mixture was quenched with water (10 mL) and then combined with another batch. The mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with 1N HCl solution (30 mL), saturated sodium bicarbonate solution (30 mL) and brine (30 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate/Ethanol = 16/3/1 ~ 12/3/1) to give compound 38a (130.0 mg, purity 93.5%) as a light-yellow solid. LCMS: RT = 0.86 min, m/z 633.2 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.35 – 7.26 (m, 2H), 7.07 (d, J = 7.6 Hz, 1H), 6.98 – 6.94 (m, 3H), 6.64 – 6.63 (s, 1H), 6.56 (s, 1H), 4.76 (dd, J = 9.6, 4.0 Hz, 1H), 4.76 (dd, J = 12.0, 3.2 Hz, 1H), 4.49 (t, J = 5.2 Hz, 1H), 4.44 (t, J = 7.2 Hz, 1H), 4.00 – 3.95 (m, 4H), 3.77 – 3.50 (m, 4H), 3.26 (t, J = 12.4 Hz, 1H), 3.13 (dd, J = 15.2, 3.6 Hz, 1H), 3.00 (dd, J = 15.2, 9.6 Hz, 1H), 2.74 (dd, J = 12.8, 2.8 Hz, 1H), 2.42 – 2.38 (m, 2H), 2.06 – 2.02 (m, 2H), 1.85 – 1.77 (m, 3H), 1.32 – 1.29 (m, 1H).
(5S,8S,11S)-8-(2-((R)-3-fluoropiperidin-1-yl)ethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (11).
To a solution of compound 38a (110.0 mg, 173.9 μmol, 1.0 eq) in acetonitrile (1.1 mL) was added CAN (238.0 mg, 434.7 μmol, 216.7 μL, 2.5 eq) in water (1.1 mL). The mixture was stirred at 70°C for 3 hours. TLC (dichloromethane: methanol =10:1) showed the starting material was consumed and a new spot was detected. The mixture was poured into water (10 mL) and then extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with saturated sodium sulfite solution (2 × 30 mL) and brine (30 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum to give crude product compound 38a-aldehyde (85.0 mg, 146.4 μmol, 84.2% yield, 98.98% purity) as a light-yellow solid, which was used into the next step without further purification. To a solution of compound 38a-aldehyde (85.0 mg, 147.9 μmol, 1.0 eq) and triethylamine (75.0 mg, 739.7 μmol, 103.0 μL, 5.0 eq) in methanol (0.5 mL) was added (R)-3-fluoropiperidine (62.0 mg, 443.8 μmol, 3.0 eq, HCl), acetic acid (18.0 mg, 295.9 μmol, 16.9 μL, 2.0 eq) and Pd/C (50.0 mg, 10% purity) under nitrogen atmosphere. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen atmosphere (15 psi) at 25°C for 16 hours. LCMS showed the starting material and intermedaite were consumed completely and desired product mass was detected. The mixture was filtered. The solid was washed with methanol (4 × 5 mL). The combined filtrate was concentrated in vacuum. The residue was diluted with ethyl acetate (50 mL) and then washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by prep-HPLC (column: Boston pH-lex 10μm C18 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 21%−51%, 10 minutes). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was concentrated in vacuum to removed acetonitrile, extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was lyophilized to give 11 (42.1 mg, purity 96.7%) as a white solid. LCMS: RT = 2.30 min, m/z = 662.2 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.33 (t, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 6.96 – 6.91 (m, 3H), 6.64 (s, 1H), 6.53 (s, 1H), 4.77 (dd, J = 10.0, 4.0 Hz, 1H), 4.70 (dd, J = 12.0, 3.2 Hz, 1H), 4.66 – 4.63 (m, 0.5H), 4.53 – 4.52 (m, 0.5H), 4.37 (t, J = 7.2 Hz, 1H), 4.01 – 3.85 (m, 2H), 3.64 – 3.53 (m, 2H), 3.25 (t, J = 12.4 Hz, 1H), 3.12 (dd, J = 15.2, 3.6 Hz, 1H), 2.99 (dd, J = 15.2, 9.6 Hz, 1H), 2.77 (dd, J = 12.8, 3.2 Hz, 1H), 2.66 – 2.63 (m, 1H), 2.41 – 2.32 (m, 7H), 2.09 – 2.04 (m, 2H), 1.78 – 1.70 (m, 4H), 1.54 – 1.52 (m, 2H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-N-((1-methyl-1H-pyrazol-4-yl)methyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (38b).
To a solution of compound 37b-acid (200.0 mg, 362.6 μmol, 1.0 eq) and (1-methyl-1H-pyrazol-4-yl)methanamine (48.0 mg, 435.1 μmol, 1.2 eq) in pyridine (0.2 mL) was added HOBt (49.0 mg, 362.6 μmol, 1.0 eq) at 0°C. The mixture was stirred at 0°C for 10 minutes. EDCI (174.0 mg, 906.5 μmol, 2.5 eq) was added. The reaction mixture was stirred at 25°C for 16 hours under nitrogen atmosphere. LCMS showed the starting material was consumed and desired product mass was detected. The mixture was diluted with water (20 mL). The mixture was adjusted pH = 6 with 1N HCl solution and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate/Ethanol = 1/1/0 ~ 8/9/3) to give compound 38b (100.0 mg, 155.1 μmol, 42.8% yield, 100.0% purity) as a light-yellow solid. LCMS: RT = 0.764 min, m/z = 645.1 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.51 (s, 1H), 7.41 (s, 1H), 7.32 (t, J = 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.07 (d, J = 7.6 Hz, 1H), 6.97 – 6.95 (m, 2H), 6.87 (d, J = 8.8 Hz, 1H), 6.63 (s, 1H), 6.54 (s, 1H), 4.69 – 4.61 (m, 2H), 4.48 (t, J = 5.2 Hz, 1H), 4.42 (t, J = 7.2 Hz, 1H), 4.24 (s, 2H), 3.95 – 3.93 (m, 2H), 3.84 (s, 3H), 3.72 – 3.50 (m, 4H), 3.24 (t, J = 12.4 Hz, 1H), 3.11 (dd, J = 15.2, 3.6 Hz, 1H), 2.94 (dd, J = 15.2, 9.6 Hz, 1H), 2.74 (dd, J = 12.4, 2.8 Hz, 1H), 2.42 – 2.37 (m, 2H), 2.05 – 2.01 (m, 2H), 1.86 – 1.84 (m, 3H), 1.30 – 1.26 (m, 1H).
(5S,8S,11S)-N-((1-methyl-1H-pyrazol-4-yl)methyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (13).
To a solution of compound 38b (90.0 mg, 139.6 μmol, 1.0 eq) in acetonitrile (0.9 mL) was added CAN (191.0 mg, 349.0 μmol, 173.9 μL, 2.5 eq) in water (0.9 mL). The mixture was stirred at 70°C for 3 hours. TLC (dichloromethane: methanol = 10:1) showed the starting material was consumed completely and desired product mass was detected on LCMS. The mixture was diluted with water (10 mL) and then extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with saturated sodium sulfite solution (20 mL), brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum to give the crude compound 38b-aldehyde (60.0 mg, 102.3 μmol, 73.3% yield) as a light-yellow solid, which was used into the next step without further purification. To a solution of compound 38b-aldehyde (60.0 mg, 102.3 μmol, 1.0 eq) in methanol (5.0 mL) was added piperidine (44.0 mg, 511.4 μmol, 50.5 μL, 5.0 eq) and acetic acid (12.0 mg, 204.6 μmol, 11.7 μL, 2.0 eq). Pd/C (20.0 mg, 10% purity) was added under nitrogen atmosphere. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen (15 psi) at 25°C for 14 hours. LCMS showed the intermediate remained. Pd/C (50.0 mg, 10% purity) was added. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen (15 psi) at 25°C for another 3 hours. LCMS showed the intermediate was consumedand desired product mass was detected. The mixture was filtered through celit pad, the solid was washed with methanol (4 × 4 mL). The combined filtrates were concentrated in vacuum. The residue was diluted with ethyl acetate (40 mL) and then washed with brine (10 mL), dried over anhydrous sodium sulfate, concentrated in vacuum. The residue was purified by prep-HPLC (column: UniSil 10 μm C18 120 × 30 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 21%−51%, 10 mins). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was concentrated in vacuum to removed acetonitrile, extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtated and concentrated in vacuum. The residue was lyophilized to give 13 (12.6 mg) as a light-yellow solid. LCMS: RT = 2.15 min, m/z 656.3 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.52 (s, 1H), 7.41 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 6.98 – 6.93 (m, 2H), 6.84 (d, J = 7.6 Hz, 1H), 6.66 (s, 1H), 6.47 (s, 1H), 4.73 – 4.65 (m, 2H), 4.35 (t, J = 7.2 Hz, 1H), 4.24 (s, 2H), 3.85 (s, 3H), 3.63 – 3.55 (m, 2H), 3.24 (t, J = 12.4 Hz, 1H), 3.08 (dd, J = 15.2, 3.6 Hz, 1H), 2.97 (dd, J = 15.2, 9.2 Hz, 1H), 2.79 (dd, J = 12.8, 3.2 Hz, 1H), 2.50 – 2.36 (m, 8H), 2.07 – 2.04 (m, 2H), 1.62 – 1.58 (m, 4H), 1.49 – 1.48 (m, 2H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-N-(cyclopropylmethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (38c).
To a solution of compound 37b-acid (240.0 mg, 435.1 μmol, 1.0 eq) in pyridine (2.5 mL) was added HOBt (59.0 mg, 435.1 μmol, 1.0 eq) at 0°C. The mixture was stirred at 0°C for 10 minutes. Cyclopropylmethanamine (62.0 mg, 870.2 μmol, 2.0 eq) and EDCI (209.0 mg, 1.1 mmol, 2.5 eq) were added at 0°C. The reaction mixture was stirred at 0°C for 20 minutes and then stirred at 25°C for 16 hours under nitrogen atmosphere. LCMS showed most of the starting material remained. Another batch of EDCI (209.0 mg, 1.1 mmol, 2.5 eq) was added, the reaction mixture was stirred at 25°C for another 18 hours. LCMS showed the starting material was consumed and desired product mass was detected. The reaction mixture was quenched with ice water (20 mL), and then adjusted pH=6~7 with 1 N HCl solution. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with 1 N HCl solution (20 mL), saturated sodium bicarbonate solution (20 mL) and brine (20 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 1/1 ~ 0/1) to give compound 38c (179.0 mg, 282.9 μmol, 65.0% yield, 95.6% purity) as a light yellow solid. LCMS: RT = 0.88 min, m/z = 605.3 [M+H]+ , purity: 95.6%. 1H NMR (Methanol-d4, 400 MHz): δ = 8.11 (t, J = 5.2 Hz, 1H), 7.98 (dd, J = 21.2, 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 6.98 – 6.94 (m, 3H), 6.65 (s, 1H), 6.55 (s, 1H), 4.72 – 4.69 (m, 2H), 4.50 (t, J = 4.8 Hz, 1H), 4.45 – 4.41 (m, 1H), 3.99 – 3.96 (m, 2H), 3.76 – 3.48 (m, 4H), 3.26 (t, J = 12.4 Hz, 1H), 3.11 – 3.04 (m, 4H), 2.75 (dd, J = 12.8, 2.8 Hz, 1H), 2.42 – 2.38 (m, 2H), 2.06 – 1.86 (m, 5H), 1.33 – 1.29 (m, 1H), 1.01 – 0.96 (m, 1H), 0.52 – 0.50 (m, 2H), 0.23 – 0.22 (m, 2H).
(5S,8S,11S)-N-(cyclopropylmethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (16).
To a solution of compound 38c (194.0 mg, 320.8 μmol, 1.0 eq) in acetonitrile (2.0 mL) was added CAN (440.0 mg, 802.1 μmol, 399.7 μL, 2.5 eq) in water (2.0 mL) at 25°C. The mixture was stirred at 70°C for 2.5 hours. LCMS and TLC (petroleum ether: ethyl acetate = 0:1) showed most of the starting material was consumed and desired product was detected. The mixture was quenched with saturated sodium bicarbonate solution (20 mL) and then extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, concentrated in vacuu to give compound 38c-aldehyde (140.0 mg, crude) as a light yellow solid, which was used into the next step without further purification. To a solution of compound 38c-aldehyde (140.0 mg, 256.1 μmol, 1.0 eq) and acetic acid (30.8 mg, 512.3 μmol, 29.3 μL, 2.0 eq) in methanol (5.0 mL) was added piperidine (109.0 mg, 1.3 mmol, 126.5 μL, 5.0 eq) and Pd/C (0.02 g, 10% purity) under nitrogen atmosphere. The mixture was degassed and purged with hydrogen several times. The reaction mixture was stirred at 25°C for 14 hours under hydrogen (15 psi). LCMS showed the starting material was consumed but the intermediate remained. The mixture was filtered and Pd/C (0.02 g, 10% purity) was added into the filtrate under nitrogen atmosphere. The mixture was degassed and purged with hydrogen several times. The reaction mixture was stirred at 25°C for another 2 hours under hydrogen (15 psi). The mixture was filtered through cilte pad and then the solid was washed with methanol (4 × 8 mL). The filtrate was concentrated in vacuum. The residue was purified by prep-HPLC (column: Boston pH-lex 10 μm C18 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 25%−55%, 10min). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was concentrated in vacuum to removed acetonitrile, extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtated and concentrated in vacuum. The residue was lyophilization to give 16 (22.5 mg, 35.1 μmol, 13.7% yield, 96.0% purity) as a white solid. LCMS: RT = 2.28 min, m/z = 616.3 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.34 (t, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H), 6.97 – 6.94 (m, 3H), 6.67 (t, J = 2.0 Hz, 1H), 6.48 (s, 1H), 4.72 – 4.67 (m, 2H), 4.38 (t, J = 7.2 Hz, 1H), 3.62 – 3.60 (m, 2H), 3.25 (t, J = 12.4 Hz, 1H), 3.13 – 3.04 (m, 4H), 2.81 (dd, J = 12.4, 2.8 Hz, 1H), 2.59 – 2.55 (m, 3H), 2.47 – 2.43 (m, 3H), 2.09 – 2.07 (m, 2H), 1.88 – 1.78 (m, 2H), 1.65 – 1.62 (m, 4H), 1.51 – 1.50 (m, 2H), 1.32 – 1.29 (m, 1H), 1.01 – 0.91 (m, 2H), 0.52 – 0.50 (m, 2H), 0.23 – 0.22 (m, 2H).
(5S,8S,11S)-N-(cyclopropylmethyl)-8-(2-morpholinoethyl)-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (22).
To a solution of compound 38c-aldehyde (90.0 mg, 164.7 μmol, 1.0 eq) in methanol (5.0 mL) was added morpholine (72.0 mg, 823.3 μmol, 72. 5 μL, 5.0 eq) and acetic acid (20.0 mg, 329.3 μmol, 18.8 μL, 2.0 eq). Pd/C (20.0 mg, 10% purity) was added under nitrogen atmosphere. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred at 25°C for 16 hours under hydrogen (15 psi). LCMS showed the intermediate remained, Pd/C (50 mg, 10% purity) was added. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred at 25°C for 26 hours under hydrogen (15 psi). LCMS showed the most of intermediate was consumed and desired product mass was detected. The mixture was then filtered throught celite pad. The soild was washed with methanol (3 × 5 mL). The combined filtrate was concentrated in vacuum. The residue was diluted with ethyl acetate (30 mL) and then washed with brine (2 × 10 mL). The organic layers were dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by prep-HPLC (column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 20%−50%, 13 mins). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was concentrated in vacuum to removed acetonitrile, extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, concentrated in vacuum. The residue was lyophilized to give 22 (14.3 mg purity 98.9%) as a white solid. LCMS: RT = 2.25 min, m/z 618.3 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.34 (t, J = 8.0 Hz, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 6.98 – 6.91 (m, 3H), 6.67 (s, 1H), 6.47 (s, 1H), 4.72 – 4.68 (m, 2H), 4.39 (t, J = 7.2 Hz, 1H), 3.67 – 3.64 (m, 6H), 3.27 – 3.24 (m, 1H), 3.09 – 3.02 (m, 4H), 2.77 (dd, J = 12.8, 3.2 Hz, 1H), 2.44 – 2.30 (m, 8H), 2.05 – 2.03 (m, 2H), 1.79 – 1.72 (m, 2H), 1.00 – 0.96 (m, 1H), 0.53 – 0.49 (m, 2H), 0.24 – 0.20 (m, 2H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-N-cyclopentyl-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (38d).
To a solution of compound 37b-acid (200.0 mg, 362.6 μmol, 1.0 eq) and cyclopentanamine (62.0 mg, 725.2 μmol, 71.6 μL, 2.0 eq) in pyridine (2.5 mL) was added HOBt (49.0 mg, 362.6 μmol, 1.0 eq) at 0°C. The mixture was stirred at 0°C for 10 minutes. EDCI (174.0 mg, 906.5 μmol, 2.5 eq) was added. The reaction mixture was stirred at 25°C for 19 hours. LCMS showed the starting material was consumed and desired product mass was detected. The mixture was poured into water (15 mL) and then adjusted to pH = 6. The mixture was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with 1N HCl (10 mL), saturated sodium bicarbonate solution (20 mL) and brine (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, concentrated in vacuum. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 1/1~ 0/1) to give compound 38d (130.0 mg, 210.1 μmol, 58.0% yield, 100.0% purity) as a white solid. LCMS: RT = 0.83 min, m/z = 619.2 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.95 (d, J = 7.2 Hz, 1H),7.33 (t, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.07 (d, J = 7.6 Hz, 1H), 6.98 – 6.93 (m, 3H), 6.65 (s, 1H), 6.56 (s, 1H), 4.69 – 4.63 (m, 2H), 4.49 (t, J = 5.2 Hz, 1H), 4.42 (t, J = 7.2 Hz, 1H), 4.10 – 4.08 (m, 1H), 3.99 – 3.96 (m, 2H), 3.77 – 3.51 (m, 4H), 3.26 (t, J = 12.4 Hz, 1H), 3.10 – 2.96 (m, 2H), 2.76 (dd, J = 12.8, 2.8 Hz, 1H), 2.41 – 2.39 (m, 1H), 2.10 – 2.05 (m, 3H), 1.95 – 1.73 (m, 7H), 1.62 – 1.59 (m, 2H), 1.50 – 1.48 (m, 1H), 1.33 – 1.24 (m, 1H).
(5S,8S,11S)-N-cyclopentyl-7,10-dioxo-11-(2-oxopyrrolidin-1-yl)-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (20).
To a solution of compound 38d (130.0 mg, 210.1 μmol, 1.0 eq) in acetonitrile (1.5 mL) was added CAN (288.0 mg, 525.3 μmol, 261.8 μL, 2.5 eq) in water (1.5 mL). The mixture was stirred at 60~70°C for 3 hours. TLC (dichloromethane: methanol = 10:1) showed the starting material was consumed completely. The mixture was poured into water (10 mL) and then extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with saturated sodium sulfite solution (2 × 20 mL), brine (20 mL), dried over anhydrous sodium sulfate, filtered, concentrated in vacuum to give crude compound 38d-aldehyde (80.0 mg, 142.7 μmol, 67.9% yield, 100.0% purity) as a light yellow solid, which was used into the next step without further purification. To a solution of compound 38d-aldehyde (80.0 mg, 142.7 μmol, 1.0 eq) in methanol (2.0 mL) was added piperidine (61.0 mg, 713.5 μmol, 70.5 μL, 5.0 eq) and acetic acid (17.0 mg, 285.4 μmol, 16.3 μL, 2.0 eq). Pd/C (20.0 mg, 10% purity) was added under nitrogen atmosphere. The suspension was degassed under vacuum and purged with hydrogen several times. The mixture was stirred under hydrogen (15 psi) at 30°C for 16 hours. LCMS showed the starting material was consumed completely. The mixture was filtered through celit pad, the solid was washed with methanol (4 × 4 mL). The combined filtrate was concentrated in vacuum. The residue was diluted with ethyl acetate (40 mL) and then washed with brine (10 mL), dried over anhydrous sodium sulfate, concentrated in vacuum. The residue was purified by prep-HPLC (column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water (0.1%trifluoroacetic acid)-acetonitrile]; B%: 28%−58%, 6min). The fraction was adjusted to pH = 7 with saturated sodium bicarbonate solution. The mixture was concentrated in vacuum to removed acetonitrile, extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, concentrated in vacuum. The residue was lyophilized to give 20 (30.0 mg, 47.4 μmol, 33.2% yield, 99.6% purity) as a white solid. LCMS: RT = 2.38 min, m/z 630.3 [M+H]+. 1H NMR (Methanol-d4, 400 MHz): δ = 7.33 (t, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.97 – 6.90 (m, 3H), 6.66 (s, 1H), 6.49 (s, 1H), 4.72 – 4.65 (m, 2H),4.34 (t, J = 7.2 Hz, 1H), 4.10 – 4.07 (m, 1H), 3.60 – 3.58 (m, 2H), 3.26 (t, J = 12.4 Hz, 1H), 3.09 – 2.94 (m, 2H), 2.77 (dd, J = 12.8, 3.0 Hz, 1H), 2.41 – 2.29 (m, 8H), 2.07 – 1.89 (m, 4H), 1.74 – 1.44 (m, 14H).
Benzyl (S)-3-(3-(3-((S)-3-(tert-butoxy)-2-((tert-butoxycarbonyl)amino)-3-oxopropyl)phenoxy)phenyl)-2-(1,3-dioxoisoindolin-2-yl)propanoate (34b).
To a mixture of compound 33 (2.0 g, 4.3 mmol, 1.0 eq), compound 32 (2.4 g, 6.5 mmol, 1.5 eq), copper acetate (1.2 g, 6.5 mmol, 1.5 eq) and triethylamine (2.2 g, 21. 7 mmol, 3.0 mL, 5.0 eq) in dichloromethane (25.0 mL) was added 4A molecular sieves (5.0 g). The mixture was stirred at 20°C for 16 hours under oxygen atmosphere (15 psi). The reaction mixture was concentrated in vacuo. The residue was purified by column chromatography (petroleum ether: ethyl acetate = 20:1 ~ 3:1) to afford compound 34b (2.3 g, 3.2 mmol, 72.8% yield, 98.9% purity) as yellow gum. LCMS: RT = 1.10 min, m/z = 743.3 [M+Na]+. 1H NMR (CDCl3, 400 MHz): δ = 7.82 – 7.78 (m, 2H), 7.74 – 7.70 (m, 2H), 7.35 – 7.28 (m, 5H), 7.13 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 6.4 Hz, 2H), 6.84 – 6.83 (m, 1H), 6.78 – 6.75 (m, 2H), 6.67 (dd, J = 1.6 Hz, 8.0 Hz, 1H), 5.26 – 5.17 (m, 3H), 5.07 (d, J = 8.0 Hz, 1H), 4.45 – 4.41 (m, 1H), 3.61 – 3.48 (m, 2H), 3.06 – 2.91 (m, 2H), 1.39 – 1.38 (m, 18H).
benzyl (S)-3-(3-(3-((S)-2-((S)-2-(((benzyloxy)carbonyl)amino)-3-(1,3-dioxan-2-yl)propanamido)-3-(tert-butoxy)-3-oxopropyl)phenoxy)phenyl)-2-(1,3-dioxoisoindolin-2-yl)propanoate (36c).
To a solution of compound 34b (2.3 g, 3.2 mmol, 1.0 eq) in dichloromethane (40 mL) was added trifluoroacetic acid (12.3 g, 108.1 mmol, 8.0 mL, 33.9 eq) drop wise at 0°C. The mixture was stirred for 4 hours at 0°C. TLC (petroleum ether: ethyl acetate = 3:1) and LCMS showed the starting material was consumed. The reaction mixture was slowly added to saturated sodium bicarbonate (200 mL), extracted with ethyl acetate (3 × 200 mL). The combined organic phase was washed with brine (100 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 34b-amine (2.0 g, crude) as yellow gum. LCMS: RT = 0.84 min, m/z = 621.3 [M+H]+. 1H NMR (CD3OD, 400 MHz): δ = 7.82 (s, 4H), 7.30 (s, 5H), 7.22 – 7.13 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 6.90 – 6.78 (m, 4H), 6.68 (dd, J = 2.0 Hz, 8.4 Hz, 1H), 5.24 – 5.20 (m, 3H), 4.08 – 4.04 (m, 1H), 3.58 – 3.40 (m, 2H), 3.09 (d, J = 7.2 Hz, 2H), 1.44 – 1.39 (m, 9H). To a solution of compound 35b (1.1 g, 3.2 mmol, 1.0 eq) and DIPEA (1.3 g, 9.7 mmol, 1.7 mL, 3.0 eq) in DMF (13.0 mL) was added HBTU (1.6 g, 4.2 mmol, 1.3 eq) wise-portion at 0°C. The mixture was stirred for 10 minutes at 0°C. A solution of compound 34b-amine (2.0 g, 3.2 mmol, 1.0 eq) in DMF (8.0 mL) was added drop wise at 0°C. The mixture was stirred for 20 minutes at 20°C. LCMS and TLC (petroleum ether: ethyl acetate = 2:1) showed desired product was detected. The reaction mixture was poured into water (100 mL), extracted with ethyl acetate (3 × 100 mL). The combined organic phase was washed with brine (3 × 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by column (petroleum ether: ethyl acetate = 10:1 ~ 1:1), followed by reverse phase column (0.1% trifluoroacetic acid in water/acetonitrile) to afford compound 36c (1.9 g, 2.1 mmol, 64.7% yield, 100.0% purity) as yellow gum. LCMS: RT = 1.13 min, m/z = 912.4 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.81 – 7.77 (m, 2H), 7.72 – 7.69 (m, 2H), 7.34 – 7.26 (m, 10H), 7.13 – 7.04 (m, 3H), 6.87 (t, J = 5.2 Hz, 2H), 6.77 – 6.76 (m, 2H), 6.63 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 6.02 – 6.00 (m, 1H), 5.25 – 5.17 (m, 3H), 5.13 – 5.09 (m, 2H), 4.73 – 4.66 (m, 1H), 4.65 (br. s, 1H), 4.38 (br. s, 1H), 4.01 – 3.97 (m, 2H), 3.69 – 3.56 (m, 4H), 3.06 – 3.05 (m, 2H), 2.04 – 1.96 (m, 2H), 1.38 – 1.37 (m, 9H), 1.27(s, 2H).
tert-butyl (5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-11-(1,3-dioxoisoindolin-2-yl)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (37c).
A solution of compound 36c (1.9 g, 2.1 mmol, 1.0 eq) in THF (30.0 mL) was purged with nitrogen 10 minutes, Pd(OH)2/C (0.2 g, 10% purity on carbon) and Pd/C (0.2 g, 10% purity on carbon) was added in one-portion. The mixture was degassed with hydrogen three times, then stirred for 3 hours at 20°C under hydrogen atmosphere (15 psi). LCMS showed about 20% of intermediate mass was observed. The mixture was stirred for 2 hours at 20°C under hydrogen (15 psi). LCMS showed one main peak with desired mass was detected. The reaction mixture was filtered and washed with methanol (2 × 30 mL) to afford compound 36c-amino acid (1.2 g, 1.7 mmol, 84.0% yield) as a white solid. LCMS: RT = 0.85 min, m/z = 688.2 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.80 (s, 4H), 7.17 – 7.11 (m, 2H), 6.97 – 6.92 (m, 1H), 6.87 – 6.85 (m, 2H), 6.78 – 6.75 (m, 2H), 6.64 – 6.61 (m, 1H), 5.12 (dd, J = 4.8 Hz, 11.6 Hz, 1H), 4.83 – 4.82 (m, 1H), 4.67 (dd, J = 5.6 Hz, 8.8 Hz, 1H), 4.11 – 4.05 (m, 3H), 3.85 – 3.76 (m, 2H), 3.54 – 3.41 (m, 2H), 3.17 – 3.12 (m, 1H), 3.00 – 2.94 (m, 1H), 2.22 – 2.17 (m, 1H), 2.09 – 2.03 (m, 1H), 1.46 – 1.36 (m, 11H). To a solution of compound 36c-amino acid (500.0 mg, 727.0 μmol, 1.0 eq) and DIPEA (371.0 mg, 2.9 mmol, 500.0 μL, 4.0 eq) in DMF (50.0 mL) was added a mixture of EDCI (500 .0mg, 2.6 mmol, 3.6 eq) and HOBt (58.9 mg, 436.2 μmol, 0.6 eq) at 0°C. The mixture was stirred for 12 hours at 20°C. LCMS showed desired mass was detected. The reaction mixture was poured into ice-water (50 mL) and HCl (1N, 10 mL), a lot of white precipitate was formed and collected by filter. The residue was dried under in vacuo and purified by column chromatography (SiO2, petroleum ether : ethyl acetate = 10:1 ~ 1:1) to afford compound 37c (0.1 g, 179.2 μmol, 24.7% yield, 100.0% purity) as a white solid. Notes: This reaction worked well at low concentration (1.5 mol/L) and the Pht group is unstable at base condition. If the reaction is used for the next step directly after work-up, the yield can be improved. LCMS: RT = 1.05 min, m/z = 614.3 [M-t-Bu+H]+; 670.4 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.87 – 7.84 (m, 2H), 7.76 – 7.73 (m, 2H), 7.41 (t, J = 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 7.08 – 7.05 (m, 3H), 7.01 – 7.00 (m, 1H), 6.96 (d, J = 7.6 Hz, 1H), 6.80 – 6.79 (m, 1H), 6.53 (d, J = 7.6 Hz, 1H), 5.14 (dd, J = 2.0 Hz, 11.6 Hz, 1H), 4.73 – 4.68 (m, 2H), 4.57 (dd, J = 4.0 Hz, 6.4 Hz, 1H), 3.96 – 3.83 (m, 3H), 3.64 – 3.49 (m, 2H), 3.25 – 3.20 (m, 2H), 3.05 (dd, J = 6.0 Hz, 14.0 Hz, 1H), 2.29 – 2.23 (m, 1H), 1.87 – 1.81 (m, 1H), 1.44 – 1.42 (m, 9H), 1.29 – 1.22 (m, 2H).
Tert-butyl (5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-11-amino-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (39).
To a solution of compound 37c (180.0 mg, 268.8 μmol, 1.0 eq) in THF (2.0 mL) was added hydrazine hydrate (26.9 mg, 537.5 μmol, 26.1 μL, 2.0 eq). The mixture was stirred for 1.5 hours at 25°C, then heated to 60°C and stirred for 12 hours. LCMS showed desired mass was detected. The reaction mixture was concentrated in vacuo. The residue was triturated with ethyl acetate (5 mL). The organic phase was concentrated in vacuo and then purified by reverse column (0.1% of trifluoroacetic acid in water/acetonitrile). The fraction was basified to pH=8 with saturated aqueous sodium bicarbonate, extracted with dichloromethane (3 × 50 mL). The combined organic phase was washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford 39 (110.0 mg, 203.9 μmol, 75.8% yield, 100.0% purity) as a white solid. LCMS: RT = 0.95 min, m/z = 540.3 [M+Na]+. 1H NMR (CDCl3, 400 MHz): δ = 8.33 (d, J = 8.4 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.09 – 7.05 (m, 2H), 6.97 (d, J = 7.6 Hz, 1H), 6.87 – 6.85 (m, 1H), 6.80 – 6.79 (m, 1H), 6.31 (t, J = 2.0 Hz, 1H), 4.69 – 4.65 (m, 1H), 4.61 (t, J = 5.2 Hz, 1H), 4.55 (t, J = 6.8 Hz, 1H), 4.15 – 4.11 (m, 1H), 4.04 – 4.00 (m, 2H), 3.82 (td, J = 2.4 Hz, 12.0 Hz, 1H), 3.72 (td, J = 2.4 Hz, 12.0 Hz, 1H), 3.26 (d, J = 3.2 Hz, 1H), 3.16 – 3.11(m, 1H), 3.00 (dd, J = 7.6 Hz, 14.4 Hz, 1H), 2.85 (dd, J = 10.4 Hz, 14.0 Hz, 1H), 1.97 – 1.87 (m, 2H), 1.53 (s, 9H), 1.36 – 1.26 (m, 2H).
Tert-butyl (5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-7,10-dioxo-11-(2-oxooxazolidin-3-yl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (40a).
To a solution of 39 (280.0 mg, 518.9 μmol, 1.0 eq) and triethylamine (157.5 mg, 1.6 mmol, 216.7 μL, 3.0 eq) in THF (3.0 mL) was added 2-chloroethyl carbonochloridate (112.0 mg, 783.4 μmol, 80.6 μL, 1.5 eq) at 0°C. The mixture was stirred for 30 minutes at 0°C. LCMS showed most of the starting material was consumed and one main peak with desired mass was observed. The reaction mixture was poured into water (10 mL), extracted with ethyl acetate (2 × 10 mL). The combined organic phase was washed with brine (2 × 10 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by reverse column (0.1% trifluoroacetic acid in water/acetonitrile) to afford compound 39-chloride (230.0 mg, 356.0 μmol, 68.6% yield) as a white solid. LCMS: RT = 1.01 min, m/z 646.4 [M+H]+. 1H NMR (CD3OD, 400 MHz): δ = 7.30 – 7.26 (m, 2H), 6.99 – 6.97 (m, 4H), 6.89 – 6.86 (m, 1H), 6.63 (s, 1H), 6.38 (s, 1H), 4.62 – 4.61 (m, 1H), 4.55 – 4.49 (m, 1H), 4.37 – 4.30 (m, 2H), 4.00 – 3.96 (m, 2H), 3.80 – 3.67 (m, 4H), 3.35 – 3.34 (m, 2H), 3.25 – 3.21 (m, 1H), 2.99 – 2.85 (m, 3H), 1.87 – 1.80 (m, 2H), 1.52 – 1.49 (m, 9H), 1.32 – 1.27 (m, 2H). To a solution of compound 39-chloride (120.0 mg, 185.7 μmol, 1.0 eq) and sodium iodide (41.8 mg, 278.6 μmol, 1.5 eq) in DMF (0.5 mL) was added cesium carbonate (151.3 mg, 464.3 μmol, 2.5 eq) at 0°C. The mixture was stirred for 2 hours at 25°C. LCMS showed one main peak with desired mass was observed. The reaction mixture was poured into water (20 mL), extracted with ethyl acetate (3 × 20 mL). The combined organic phase was washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by column (petroleum ether: ethyl acetate = 10:1 ~ 1:2) to afford compound 40a (70.0 mg, 105.0 μmol, 56.5% yield, 91.4% purity) as a white solid. LCMS: RT = 0.86 min, m/z = 554.3 [M-t-Bu+H]+; 632.3[M+Na]+. 1H NMR (CDCl3, 400 MHz): δ = 7.38 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.03 (dd, J = 2.0 Hz, 8.4 Hz, 1H), 6.95 (dd, J = 1.6 Hz, 8.4 Hz, 1H), 6.88 (s, 1H), 6.76 (d, J = 7.2 Hz, 1H), 6.62 (d, J = 6.0 Hz, 1H), 6.44 (d, J = 7.6 Hz, 1H), 6.23 (s, 1H), 4.61 – 4.57 (m, 2H), 4.50 – 4.45 (m, 2H), 4.38 (t, J = 8.0 Hz, 2H), 4.03 – 3.97 (m, 3H), 3.75 – 3.61 (m, 3H), 3.29 (t, J = 12.4 Hz, 1H), 3.18 (dd, J = 5.2 Hz, 14.0 Hz, 1H), 3.04 (dd, J = 4.0 Hz, 14.0 Hz, 1H), 2.91 – 2.88 (m, 1H), 2.09 – 1.98 (m, 2H), 1.49 (s, 9H), 1.28 – 1.25 (m, 2H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-7,10-dioxo-11-(2-oxooxazolidin-3-yl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (41a).
To a solution of compound 40a (55.0 mg, 90.2 μmol, 1.0 eq) in dichloromethane (2.0 mL) was added trifluoroacetic acid (847.0 mg, 7.4 mmol, 550.0 μL, 82.3 eq) drop wise at 0°C. The mixture was stirred for 2 hours at 25°C. LCMS showed most of the starting material was consumed and the desired mass was observed. The reaction mixture was poured into water (20 mL), adjusted to pH=5 with saturated sodium bicarbonate, extracted with ethyl acetate (2 × 15 mL). The combined organic phase was washed with brine (3 × 20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 40a-acid (50.0 mg, crude) as a white solid. LCMS: RT = 0.82 min, m/z = 554.2 [M+H]+, purity: 50.4%. To a solution of compound 40a-acid (50.0 mg, 90.3 μmol, 1 eq), DIPEA (35.0 mg, 271.0 μmol, 47.2 μL, 3.0 eq) and 2,2,2-trifluoroethanamine (9.0 mg, 90.3 μmol, 7.1 μL, 1.0 eq) in DMF (1.0 mL) was added HOBt (7.3 mg, 54.2 μmol, 0.6 eq) and EDCI (26.0 mg, 135.5 μmol, 1.5 eq) at 0°C. The mixture was stirred for 12 hours at 25°C. LCMS showed the desired mass was observed. The reaction mixture was poured into water (10 mL), extracted with ethyl acetate (3 × 10 mL). The combined organic phase was washed with brine (2 × 20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 41a (50.0 mg, crude) as yellow gum and used directly without further purification. LCMS: RT = 0.78 min, m/z = 635.1 [M+H]+, purity: 83.9%.
(5S,8S,11S)-7,10-dioxo-11-(2-oxooxazolidin-3-yl)-8-(2-(piperidin-1-yl)ethyl)-N-(2,2,2-trifluoroethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (12).
To a solution of compound 41a (50.0 mg, 78.8 μmol, 1.0 eq) in acetonitrile (0.8 mL) was added CAN (108.0 mg, 197.0 μmol, 98.2 μL, 2.5 eq) in water (0.8 mL). The mixture was stirred for 2 hours at 70°C. LCMS showed desired mass was detected. The reaction mixture was poured into water (10 mL), extracted with ethyl acetate (2 × 20 mL). The combined organic phase was washed with saturated sodium sulfite (20 mL), brine (20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 41a-aldehyde (50 mg, crude) as a white solid and used directly without further purification. LCMS: RT = 0.88 min, m/z = 599.3 [M+Na]+. To a solution of compound 41a-aldehyde (50.0 mg, 86.7 μmol, 1.0 eq) and piperidine (14.8 mg, 173.5 μmol, 17.1 μL, 2.0 eq) in methanol (1.0 mL) was added acetic acid (1.0 mg, 17.4 μmol, 1.0 μL, 0.2 eq). The mixture was purged with nitrogen atmosphere 10 minutes, then Pd/C (20.0 mg, 10% purity) was added in one portion. The mixture was degassed with hydrogen three times and stirred for 12 hours at 25°C under hydrogen atmosphere (15 psi). LCMS showed the desired mass was observed. The reaction mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by prep-TLC (petroleum ether: ethyl acetate: ethanol = 1:6:2), followed by prep-HPLC (column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water (0.04%NH3H2O+10mM NH4HCO3)-ACN]; B%: 40%−67%, 10min) to afford 12 (3.5 mg, 5.2 μmol, 6.0% yield, 96.2% purity) as a white solid. LCMS: RT = 1.70 min, m/z = 646.3 [M+H]+. 1H NMR (CDCl3, 400 MHz) δ = 8.06 (s, 1H), 7.46 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.30 – 7.25 (m, 1H), 7.11 (d, J = 8.0 Hz, 1H), 7.09 – 7.05 (m, 2H), 6.98 – 6.97 (m, 1H), 6.54 (s, 1H), 6.47 (s, 1H), 4.89 (br. s, 1H), 4.45 (dd, J = 1.6 Hz, 8.0 Hz, 1H), 4.38 (t, J = 8.0 Hz, 2H), 4.09 – 4.05 (m, 3H), 3.73 – 3.71 (m, 2H), 3.35 – 3.25 (m, 1H), 3.10 – 3.06 (m, 2H), 2.95 – 2.80 (m, 1H), 2.38 – 2.32 (m, 5H), 1.95 – 1.80 (m, 1H), 1.48 – 1.42 (m, 9H).
Tert-butyl (5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-11-(methylsulfonamido)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (40b).
To a solution of 39 (230.0 mg, 426.2 μmol, 1.0 eq) in dichloromethane (3.0 mL) was added DIPEA (148.4 mg, 1.2 mmol, 0.2 mL, 2.7 eq), and then methylsufonyl chloride (0.6 g, 5.1 mmol, 391.9 μL, 11.9 eq) was added at 0°C drop wise. The mixture was stirred at 25°C for 1 hour. TLC (petroleum ether: ethyl acetate = 1:1) showed the starting material was consumed and desired mass was observed on LCMS. The reaction mixture was poured into water (20 mL), extracted with ethyl acetate (3 × 30 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by prep-TLC (SiO2, petroleum ether: ethyl acetate = 1:1) to afford compound 40b (120.0 mg, 194.3 μmol, 45.6% yield, 100.0% purity) as a white solid. LCMS: RT = 0.93 min, m/z 618.3 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.38 (t, J = 8.0 Hz, 1H), 7.30 – 7.29 (m, 1H), 7.16 (d, J = 8.0 Hz, 1H), 7.03 – 6.96 (m, 2H), 6.97 (d, J = 7.6 Hz, 1H), 6.58 (s, 1H), 6.53 – 6.50 (m, 2H), 5.55 (br .s, 1H), 4.66 – 4.64 (m, 2H), 4.54 – 4.52 (m, 1H), 4.20 – 4.15 (m, 1H), 4.00 (dd, J = 4.4 Hz, 11.2 Hz, 2H), 3.74 – 3.63 (m, 2H), 3.28 (dd, J = 4.0 Hz, 10.0 Hz, 1H), 3.16 – 3.10 (m, 1H), 2.97 – 2.87 (m, 5H), 2.11 – 2.05 (m, 1H), 1.95 – 1.87 (m, 1H), 1.51 – 1.44 (m, 9H), 1.33 – 1.25 (m, 2H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-N-((1-methyl-1H-pyrazol-4-yl)methyl)-11-(methylsulfonamido)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (41b).
To a solution of compound 40b (140.0 mg, 226.6 μmol, 1.0 eq) in dichloromethane (1.5 mL) was added trifluoroacetic acid (770.0 mg, 6.8 mmol, 0.5 mL, 29.8 eq) drop wise at 0°C. The mixture was stirred for 2 hours at 20°C. LCMS showed one main peak with desired mass was detected. The reaction mixture was poured into water (10 mL), adjusted to pH = 5 with saturated sodium bicarbonate, extracted with ethyl acetate (3 × 20 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 40b-acid (120.0 mg, crude) as a white solid. LCMS: RT = 0.80 min, m/z 562.2 [M+Na]+. To a solution of compound 40b-acid (120.0 mg, 213.7 μmol, 1.0 eq) and (1-methylpyrazol-4-yl) methanamine (47.5 mg, 427.4 μmol, 2.0 eq) in DMF (3.0 mL) was added a mixture of EDCI (90.0 mg, 469.5 μmol, 2.2 eq) and HOBt (20.2 mg, 149.6 μmol, 0.7 eq) at 0°C. The mixture was stirred for 5 hours at 25°C. LCMS showed one main peak with desired mass was detected. The reaction mixture was concentrated in vacuo. The residue was dissolved in ethyl acetate (20 mL), washed with HCl (0.01N, 2 × 10 mL), brine (3 × 20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was triturated with ethyl acetate: petroleum ether = 1:100 (50 mL) to afford compound 41b (75.0 mg, 103.1 μmol, 48.3% yield, 90.0% purity) as a white solid. LCMS: RT = 0.79 min, m/z = 655.3 [M+H]+, purity: 62.9%. 1H NMR (CDCl3, 400 MHz): δ = 7.88 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.57 – 7.53 (m, 1H), 7.51 – 7.43 (m, 1H), 7.34 – 7.26 (m, 2H), 7.08 – 7.05 (m, 1H), 7.00 – 6.97 (m, 2H), 6.99 – 6.89 (m, 1H), 6.67 – 6.57 (m, 1H), 6.42 – 6.35 (m, 1H), 4.97 – 4.94 (m, 2H), 4.77 – 4.70 (m, 4H), 4.67 – 4.51 (m, 2H), 4.27 – 4.23 (m, 1H), 3.99 – 3.64 (m, 4H), 3.27 – 3.13 (m, 2H), 3.05 – 2.90 (m, 5H), 1.99 – 1.81 (m, 2H), 1.33 – 1.37 (m, 2H).
(5S,8S,11S)-N-((1-methyl-1H-pyrazol-4-yl)methyl)-11-(methylsulfonamido)-7,10-dioxo-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (14).
To a solution of compound 41b (75.0 mg, 114.6 μmol, 1.0 eq) in acetonitrile (1.0 mL) was added a solution of CAN (157.0 mg, 286.4 μmol, 142.7 μL, 2.5 eq) in water (1.0 mL). The mixture was stirred for 2 hours at 70°C. LCMS showed the starting material was consumed. The reaction mixture was poured into ethyl acetate (30 mL), washed with saturated sodium sulfite (2 × 20 mL), brine (2 × 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford compound 41b-aldehyde (70.0 mg, crude) as a white solid. A solution of compound 41b-aldehyde (70.0 mg, 117.3 μmol, 1.0 eq), piperidine (1.4 mg, 16.8 μmol, 1.7 μL, 1.0 eq) and acetic acid (7.1 mg, 117.3 μmol, 6.7 μL, 1.0 eq) in methanol (2.0 mL) was degassed with nitrogen three times and then Pd/C (20.0 mg, 10% purity on carbon) was added in one portion. The mixture was degassed with hydrogen and stirred for 12 hours at 25°C under hydrogen atmosphere (15 psi). LCMS showed one main peak with desired mass was detected. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (Column: Boston pH-lex 10um C18 150 × 25 mm, mobile phase: [water (0.1%TFA)-ACN]; B%: 19%−49 %, 8min). The fraction was basified to pH = 8 with saturated aqueous sodium bicarbonate and extracted with dichloromethane (3 × 30 mL). The combined organic phase was washed with brine (2 × 20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by prep-HPLC (column: Gemini 5 μm C18 150 × 25 mm; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN]; B%: 25%−55%, 12min) to afford compound 14 (2.3 mg, 3.2 μmol, 2.8% yield, 95.1% purity) as a white solid. LCMS: RT = 3.41 min, m/z = 666.3 [M+H]+. 1H NMR (CD3OD, 400 MHz): δ 7.53 (s, 1H), 7.42 (s, 1H), 7.32 – 7.25 (m, 2H), 7.06 (d, J = 7.2 Hz, 1H), 6.96 (d, J = 7.2 Hz, 2H), 6.88 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 6.65 – 6.64 (m, 1H), 6.36 (s, 1H), 4.64 (dd, J = 3.2 Hz, 10.0 Hz, 1H), 4.38 (t, J = 7.2 Hz, 1H), 4.27 – 4.24 (m, 3H), 3.85 (s, 3H), 3.13 – 3.08 (m, 1H), 2.96 – 2.86 (m, 6H), 2.38 – 2.25 (m, 6H), 1.81 – 1.74 (m, 2H), 1.57 – 1.55 (m, 4H), 1.46 – 1.42 (m, 2H).
Tert-butyl (5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-11-(1,1-dioxidoisothiazolidin-2-yl)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (40c).
To a solution of 39 (250.0 mg, 463.3 μmol, 1.0 eq) and triethylamine (140.6 mg, 1.4 mmol, 193.5 μL, 3.0 eq) in dichloromethane (1.0 mL) was added 3-chloropropane-1-sulfonyl chloride (123.0 mg, 694.9 μmol, 84.3 μL, 1.5 eq) at 0°C. The mixture was stirred for 30 minutes at 0°C. LCMS showed most of the starting material was consumed and one main peak with desired mass was observed. The reaction mixture was poured into water (20 mL), extracted with ethyl acetate (2 × 20 mL). The combined organic phase was washed with brine (2 × 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford compound 39-sulfonamide (250.0 mg, crude) as a white solid. LCMS: RT = 1.00 min, m/z = 680.4 [M+H]+, purity: 58.3%. 1H NMR (CDCl3, 400 MHz): δ 7.34 – 7.25 (m, 2H), 7.10 – 7.06 (m, 2H), 6.99 – 6.95 (m, 3H), 6.87 – 6.85 (m, 1H), 6.62 – 6.56 (m, 1H), 6.42 – 6.41 (m, 1H), 4.62 – 4.57 (m, 3H), 4.52 – 4.49 (m, 1H), 4.12 – 4.07 (m, 2H), 3.77 – 3.66 (m, 4H), 3.27 – 3.18 (m, 3H), 2.98 – 2.93 (m, 2H), 2.88 – 2.85 (m, 1H), 2.27 – 2.22 (m, 2H), 1.95 – 1.94 (m, 1H), 1.86 – 1.80 (m, 1H), 1.53 – 1.51 (m, 9H). To a solution of compound 39-sulfonamide (260.0 mg, 382.2 μmol, 1.0 eq) and sodium iodide (85.9 mg, 573.4 μmol, 1.5 eq) in DMF (3.0 mL) was added potassium carbonate (105.7 mg, 764.5 μmol, 2.0 eq) at 25°C. The mixture was stirred for 12 hours at 25°C. LCMS showed one main peak with desired mass was observed. The reaction mixture was poured into water (10 mL), extracted with ethyl acetate (3 × 20 mL). The combined organic phase was washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by column chromatography (SiO2, petroleum ether: ethyl acetate = 5:1 ~ 1:2) to afford compound 40c (160.0 mg, 235.5 μmol, 61.6% yield, 94.8% purity) as a white solid. LCMS: RT = 0.96 min, m/z = 644.3 [M+H]+. 1H NMR (CD3OD, 400 MHz): δ = 7.32 (t, J = 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.96 – 6.89 (m, 3H), 6.59 (t, J = 1.6 Hz, 1H), 6.43 (s, 1H), 4.64 – 4.59 (m, 2H), 4.46 (t, J = 8.0 Hz, 1H), 4.25 (dd, J = 3.2 Hz, 13.0 Hz, 1H), 3.98 (dd, J = 4.0 Hz, 11.6 Hz, 2H), 3.78 – 3.71 (m, 2H), 3.51 (t, J = 6.8 Hz, 2H), 3.24 – 3.21 (m, 2H), 3.19 – 3.16 (m, 2H), 3.04 – 2.99 (m, 1H), 2.89 (dd, J = 3.2 Hz, 12.8 Hz, 1H), 2.37 – 2.32 (m, 2H), 1.85 – 1.80 (m, 2H), 1.49 (s, 9H), 1.32 – 1.29 (m, 2H), 0.94 – 0.85 (m, 2H).
(5S,8S,11S)-8-((1,3-dioxan-2-yl)methyl)-11-(1,1-dioxidoisothiazolidin-2-yl)-N-((1-methyl-1H-pyrazol-4-yl)methyl)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (41c).
To a solution of compound 40c (160.0 mg, 248.6 μmol, 1.0 eq) in dichloromethane (4.0 mL) was added trifluoroacetic acid (1.5 g, 13.5 mmol, 1.0 mL, 54.3 eq) at 0°C. The mixture was stirred for 2 hours at 25°C. LCMS showed desired mass was observed and part of the starting material remained, then the mixture was stirred for 1 hour at 25°C. The reaction mixture was poured into water (20 mL), extracted with ethyl acetate (3 × 30 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford compound 40c-acid (150.0 mg, crude) as a white solid, which was used for the next step directly without further purification. LCMS: RT = 0.74 min, m/z = 588.3 [M+H]+, purity: 35.4%. To a solution of compound 40c-acid (150.0 mg, 255.3 μmol, 1.0 eq), (1-methylpyrazol-4-yl) methanamine (75.4 mg, 510.5 μmol, 2.0 eq, HCl salt) and DIPEA (99.0 mg, 765.8 μmol, 133.4 μL, 3.0 eq) in DMF (2.0 mL) was added EDCI (73.4 mg, 382.9 μmol, 1.5 eq) and HOBt (20.7 mg, 153.2 μmol, 0.6 eq) at 0°C. The mixture was stirred for 12 hours at 25°C. LCMS showed desired mass was detected. The reaction mixture was poured into water (20 mL), extracted with ethyl acetate (2 × 20 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 41c (100.0 mg, crude) as a white solid, which was used for the next step directly without further purification. LCMS: RT = 0.83 min, m/z = 681.3 [M+H]+, purity: 45.0%.
(5S,8S,11S)-11-(1,1-dioxidoisothiazolidin-2-yl)-N-((1-methyl-1H-pyrazol-4-yl)methyl)-7,10-dioxo-8-(2-(piperidin-1-yl)ethyl)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (15).
To a solution of compound 41c (100.0 mg, 146.9 μmol, 1.0 eq) in acetonitrile (1.0 mL) was added a solution of CAN (201.3 mg, 367.2 μmol, 183.0 μL, 2.5 eq) in water (1.0 mL). The mixture was stirred for 2 hours at 70°C. LCMS showed desired mass was detected. The reaction mixture was poured into water (10 mL), extracted with ethyl acetate (2 × 20 mL). The combined organic phase was washed with saturated sodium sulfite (20 mL), brine (20 mL), dried over anhydrous sodium sulfate and concentrated in vacuo to afford compound 41c-aldehyde (100.0 mg, crude) as a white solid, which was used for the next step directly without further purification. LCMS: RT = 0.80 min, m/z = 623.3 [M+H]+, purity: 47.7%. To a solution of compound 41c-aldehyde (100.0 mg, 160.6 μmol, 1.0 eq) and piperidine (27.4 mg, 321.2 μmol, 31.7 μL, 2.0 eq) in methanol (1.5 mL) was added acetic acid (4.8 mg, 80.3 μmol, 4.6 μL, 0.5 eq). The mixture was degassed with nitrogen 10 minutes, then Pd/C (0.1 g, 10% purity) was added in one-portion. The mixture was degassed with hydrogen three times and stirred for 17 hours at 25°C under hydrogen atmosphere (15 psi). LCMS showed one main peak with desired mass was observed. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water (0.1%TFA)-ACN]; B%: 16%−46%, 13min). The fraction was basified to pH = 8 with saturated sodium bicarbonate aqueous, extracted with dichloromethane (3 × 30 mL). The combined organic phase was washed with brine (2 × 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford compound 15 (2.3 mg, 3.0 μmol, 1.9% yield) as a white solid. LCMS: RT = 1.71 min, m/z 692.3 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 8.55 (t, J = 5.0 Hz, 1H), 8.39 (d, J = 8.4 Hz, 0.6H), 8.30 (d, J = 8.4 Hz, 0.3H), 7.53 (s, 1H), 7.41 (s, 1H), 7.36 (t, J = 8.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 7.16 (d, J = 7.2 Hz, 1H), 6.98 – 6.93 (m, 2H), 6.80 (d, J = 7.6 Hz, 1H), 6.65 (s, 1H), 6.36 (s, 1H), 4.61 – 4.58 (m, 1H), 4.46 – 4.43 (m, 1H), 4.36 (dd, J = 3.6 Hz, 12.0 Hz, 1H), 4.25 – 4.24 (m, 2H), 3.85 (s, 3H), 3.71 – 3.67 (m, 1H), 3.63 – 3.59 (m, 1H), 3.52 – 3.44 (m, 2H), 3.25 – 3.21 (m, 2H), 3.19 – 3.15 (m, 2H), 3.12 – 3.06 (m, 2H), 2.99 – 2.94 (m, 2H), 2.86 – 2.82 (m, 2H), 2.42 – 2.31 (m, 2H), 2.11 – 2.03 (m, 1H), 1.94 – 1.87 (m, 3H), 1.85 – 1.79 (m, 1H), 1.75 – 1.68 (m, 2H), 1.50 – 1.46 (m, 1H).
Tert-butyl (S)-2-((S)-2-((tert-butoxycarbonyl)amino)-4-morpholinobutanamido)-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)propanoate (44).
To a solution of compound 43 (1.1 g, 3.8 mmol, 1.0 eq) in THF (10.0 mL) was added HATU (1.7 g, 4.6 mmol, 1.2 eq) and DIPEA (740.0 mg, 5.7 mmol, 996.7 μL, 1.5 eq) at 0°C and the solution was stirred at 0°C for 30 minutes. Compound 42 (1.4 g, 4.0 mmol, 1.0 eq) was added to the solution at 0°C and the solution was stirred at 0°C for 2 hours. LCMS showed the starting material was consumed completely and desired mass was observed. The reaction was quenched with saturated ammonium chloride (20 mL) and the solution was extracted with ethyl acetate (3 × 60 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by reverse phase flash (TFA) to give compound 44 (2.2 g, 3.6 mmol, 93.4% yield) as a light yellow gum. 1H NMR (CD3OD, 400 MHz): δ = 7.63 – 7.49 (m, 2H), 7.40 – 7.29 (m, 2H), 4.62 – 4.55 (m, 1H), 4.22 – 3.99 (m, 3H), 3.83 – 3.64 (m, 2H), 3.52 – 3.40 (m, 2H), 3.24 – 2.97 (m, 6H), 2.19 – 1.86 (m, 2H), 1.46 – 1.34 (m, 26H).
(3-((S)-2-((S)-2-amino-4-morpholinobutanamido)-3-(tert-butoxy)-3-oxopropyl)phenyl)boronic acid (45).
To a solution of compound 44 (2.2 g, 3.6 mmol, 1.0 eq) in dichloromethane (10.0 mL) and dioxane (1.0 mL) was added zinc bromide (4.0 g, 17.8 mmol, 5.0 eq) at 15°C and the solution was stirred at 15°C for 8 hours. LCMS showed trace starting material still remained and desired mass was observed. The reaction was quenched with DIPEA (3.0 mL), and the mixture was concentrated to remove the solvent. The residue was purified by reverse phase flash (TFA) to give compound 45 (1.1 g, 1.9 mmol, 53.1% yield, 94.4% purity, TFA) as an off-white solid. LCMS: RT = 0.67 min, m/z = 436.2 [M+H]+, purity: 94.4%. 1H NMR (CD3OD, 400 MHz): δ = 7.66 – 7.53 (m, 3H), 7.39 – 7.31 (m, 3H), 4.63 (dd, J = 8.4 Hz, 6.4 Hz, 1H), 4.04 (dd, J = 7.2 Hz, 6.0 Hz, 1H), 3.96 – 3.85 (m, 4H), 3.46 – 3.40 (m, 2H), 3.27 – 3.26 (m, 1H), 3.16 – 3.05 (m, 4H), 2.78 – 2.74 (m, 1H), 2.45 – 2.27 (m, 2H), 2.19 – 1.86 (m, 2H), 1.41 (s, 9H).
(3-((S)-2-((S)-2-((S)-3-(3-(benzyloxy)phenyl)-2-(1,1-dioxidoisothiazolidin-2-yl)propanamido)-4-morpholinobutanamido)-3-(tert-butoxy)-3-oxopropyl)phenyl)boronic acid (47).
To a solution of compound 46 (752.0 mg, 2.0 mmol, 1.0 eq) in DMF (10.0 mL) was added T3P (2.6 g, 4.0 mmol, 2.4 mL, 50% purity in ethyl acetate, 2.0 eq) and DIPEA (1.3 g, 10.0 mmol, 1.7 mL, 5.0 eq) at 0°C and the solution was stirred at 0°C for 30 minutes. Then compound 45 (1.1 g, 2.0 mmol, 1.0 eq, TFA) was added to the solution at 0°C and the solution was stirred at 0°C for additional 1.5 hours. LCMS showed the starting material was consumed and desired MS was detected. The reaction mixture was filtered to give a crude product. The crude product was purified by reverse phase flash (TFA) to give compound 47 (0.4 g, 441.5 μmol, 22.1% yield) as a white solid. 1H NMR (CD3OD, 400 MHz): δ 7.46 – 7.25 (m, 10H), 6.95 – 6.80 (m, 3H), 5.10 (s, 2H), 4.59 – 4.35 (m, 3H), 4.10 – 3.90 (m, 2H), 3.74 – 3.42 (m, 5H), 3.25 – 2.85 (m, 11H), 2.85 – 2.48 (m, 2H), 2.31 – 2.21 (m, 3H), 2.05 – 1.99 (m, 2H), 1.41 (s, 9H).
tert-butyl (5S,8S,11S)-11-(1,1-dioxidoisothiazolidin-2-yl)-8-(2-morpholinoethyl)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylate (48).
To a solution of compound 47 (350.0 mg, 441.5 μmol, 1.0 eq) in methanol (3.0 mL) was added Pd/C (40.0 mg, 10% purity) and Pd(OH)2/C (40.0 mg, 10% purity) at 15°C under nitrogen atmosphere and the solution was stirred at 15°C for 12 hours under hydrogen atmosphere (15 psi). LCMS showed the starting material was consumed and desired mass was observed. The reaction was filtered by celite, and the filter cake was washed with methanol (10 mL), then the solution was concentrated under reduced pressuer to give compound 47-phenol (300.0 mg, crude) as a light yellow gum. 1H NMR (CDCl3, 400 MHz): δ = 7.28 – 7.01 (m, 4H), 6.78 – 6.65 (m, 4H), 4.58 – 4.50 (m, 1H), 4.38 – 4.30 (m, 2H), 3.76 – 3.43 (m, 7H), 3.20 – 2.93 (m, 11H), 2.81 – 2.21 (m, 5H), 2.12 – 1.94 (m, 1H), 1.74 – 1.62 (m, 1H), 1.40 (s, 9H). A mixture of compound 47-phenol (200.0 mg, 284.7 μmol, 1.0 eq), copper acetate (103.0 mg, 569.3 μmol, 2.0 eq), 4A molecular sieve (500.0 mg) and triethylamine (144.0 mg, 1.4 mmol, 198.1 μL, 5.0 eq) in dichloromethane (15.0 mL) was stirred at 20°C under air balloon (15 psi) for 13 hours. LCMS showed the starting material was consumed completely and desired mass was observed. The mixture was filtered by celite, and the filter cake was washed with dichloromethane (10 mL), the solution was concentrated under reduced pressure to give a residue. The residue was purified by prep-HPLC (column: Phenomenex Synergi 10 μm C18 150 × 25 mm; mobile phase: [water (0.1%TFA)-ACN]; B%: 22%−52%, 10 minutes) to give compound 48 (30.0 mg, 38.9 μmol, 13.7% yield, 100.0% purity, TFA) as an off-white solid. LCMS: RT = 0.82 min, m/z = 657.3 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.38 (t, J = 8.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 7.02 (dd, J = 8.4 Hz, J = 1.6 Hz, 1H), 6.96 (dd, J = 8.0 Hz, J = 1.6 Hz, 1H), 6.80 – 6.74 (m, 3H), 6.44 (d, J = 7.2 Hz, 1H), 6.08 (br. s, 1H), 4.58 – 4.57 (m, 1H), 4.47 – 4.42 (m, 1H), 4.08 (dd, J = 12.0 Hz, 3.2 Hz, 1H), 3.96 – 3.92 (m, 4H), 3.89 – 3.83 (m, 1H), 3.59 – 3.54 (m, 1H), 3.26 – 3.16 (m, 6H), 3.06 – 2.93 (m, 2H), 2.88 – 2.80 (m, 2H), 2.45 – 2.38 (m, 2H), 1.50 (s, 9H).
(5S,8S,11S)-8-(2-morpholinoethyl)-7,10-dioxo-11-((3-sulfopropyl)amino)-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxylic acid (49).
To a solution of compound 48 (15.0 mg, 22.8 μmol, 1.0 eq) in dichloromethane (0.4 mL) was added trifluoroacetic acid (154.0 mg, 1.4 mmol, 100.0 μL, 59.1 eq) at 0°C. The mixture was stirred for 12 hours at 20°C. LCMS showed desired mass was observed. The reaction mixture was concentrated in vacuo to afford compound 49 (20.0 mg, crude, TFA salt) as yellow gum, which was used for the nest step without further purification.
(5S,8S,11S)-N-cyclopentyl-11-(1,1-dioxidoisothiazolidin-2-yl)-8-(2-morpholinoethyl)-7,10-dioxo-2-oxa-6,9-diaza-1,3(1,3)-dibenzenacyclododecaphane-5-carboxamide (TDI-8414).
To a solution of compound 49 (20.0 mg, 32.3 μmol, 1.0 eq), DIPEA (17.0 mg, 129.3 μmol, 22.5 μL, 4.0 eq) and cyclopentanamine (6.0 mg, 64.7 μmol, 6.4 μL, 2.0 eq) in dichloromethane (0.5 mL) was added T3P (41.0 mg, 64.7 μmol, 38.5 μL, 50% purity in ethyl acetate, 2.0 eq) drop wise at 0°C. The mixture was stirred for 1 hour at 0°C. LCMS showed one main peak with desired mass was observed. The reaction mixture was concentrated in vacuo to afford compound 49- sulfonic acid (25.0 mg, crude) as yellow gum, which was used for the next step without further purification. A solution of compound 49-sulfonic acid (22.0 mg, 32.1 μmol, 1.0 eq, two batches) in phosphorus oxychloride (3.8 g, 24.6 mmol, 2.3 mL, 765.8 eq) was stirred at 0°C for 2 hours. LCMS showed the starting material was consumed and desired mass was observed. The reaction mixture was added to ice-water (20 mL), adjusted to pH~8 with 1N sodium hydroxide, extracted with ethyl acetate (4 × 30 mL). The combined organic phase was washed with brine (2 × 30 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue (two batches) was purified by prep-HPLC (column: Luna 5μM C18 150 × 25 mm; mobile phase: [water (0.225%FA)-ACN]; B%: 23%−43%,7.8min & column: Phenomenex Gemini 10 μm C18 150 × 25mm; mobile phase: [water (10mM NH4HCO3)-ACN]; B%: 40%−70%, 10min) to afford TDI-8414 (4.9 mg, 7.3 μmol, 5.4% yield, 100.0% purity) as a white solid. LCMS: RT = 1.67 min, purity: 100.0%, m/z: = 668.3 [M+H]+. 1H NMR (CDCl3, 400 MHz): δ = 7.41 (t, J = 8.0 Hz, 1H), 7.37 – 7.34 (m, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.13 (d, J = 7.2 Hz, 1H), 7.07 – 7.01 (m, 3H), 6.91 (d, J =7.2 Hz, 1H), 6.58 (s, 1H), 6.38 (s, 1H), 6.19 – 6.16 (m, 1H), 4.73 – 4.69 (m, 1H), 4.19 – 4.06 (m, 3H), 3.87 – 3.82 (m, 1H), 3.69 – 3.95 (m, 2H), 3.59 – 3.48 (m, 3H), 3.22 – 2.98 (m, 6H), 2.49 – 2.31 (m, 6H), 2.99 – 1.89 (m, 3H), 1.72 – 1.72 (m, 4H), 1.58 – 1.42 (m, 4H), 1.32 – 1.22 (m, 1H).
IC50 determination.
IC50 values of all compounds against Pf20S β5, human c-20S β5c and i-20S β5i were determined in 96-well plates as reported.22
Antimalarial activity in the erythrocytic stage.
Parasite growth inhibition assays were performed as reported.22
Ex vivo EC50 values against P. falciparum field isolates in Uganda.
The activity of selected compounds was tested against P. falciparum isolates using a 72-h growth inhibition assay with parasite DNA readout by Sybr Green detection as described.26 These isolates were collected from patients living in the Tororo and Busia Districts, Uganda, who were newly diagnosed with P. falciparum malaria before antimalarial treatment was administered. These studies were approved by the Uganda National Council of Science and Technology, the Makerere University Research and Ethics Committee, and the University of California, San Francisco Committee on Human Research.
Parallel artificial membrane permeability assay (PAMPA).
All PAMPA assays were performed as reported.22 Propranolol was used as positive control compound. Methyclothiazide was used as a negative control compound.
LLC-PK1-MDR1 assay.
Human MDR1 expressing LLC-PK1 cells (hMDR1/LLC-PK1) were used to investigate if compounds are MDR1 substrates. All the MDR1 assays were performed as reported.22 Lucifer Yellow was co-incubated with test compound as a membrane integrity marker. Digoxin was used as positive control compound.
Kinetic solubility.
The kinetic solubility of the compounds was determined as reported.22 Diazepam was included as the positive control in each experiment.
Live microsomal stability.
The metabolic stability of the compounds by human or mouse liver microsomes was determined as reported.22 Flutamide was included as the positive control compound in each experiment.
Plasma stability.
To 100 μL of human/mouse plasma was added 1μL of 10 mM test compound. The mixtures were incubated at 37 °C for 0, 60, 120 min. The reactions were stopped at each time point by adding 200 μL ice-cold methanol containing 50 μM internal standard. For the T0 min samples, 200 μL ice-cold methanol containing internal standard was added to human plasma prior to addition of test compound. Precipitated proteins were pelleted by centrifuging at 13,500 rpm for 20 min at 4 °C. The supernatants were collected and analyzed on an Acquity™ UPLC / MS system, coupled with a PDA detector. Column chromatography was carried out on a C18 Column, 130Å, 1.7 μm, 2.1 mm X 100 mm. The percentage of compounds remaining at each time points relative to starting concentration were calculated using integrated UV peak areas normalized to the internal control. Procaine was included as the positive control compound in each human plasma stability assay experiment. Enalapril was included as the positive control in each mouse plasma stability assay experiment.
Hepatocytes stability.
50 μL diluted compound was added to 50 μL hepatocyte cells (2×106 cells/mL in HT medium) was dispensed into 96-well plated. After incubating at 37 °C in 5% CO2, 100 μL cold acetonitrile containing internal standard was added to the mixture to stop the reaction. For the T0, 100 μl cold acetonitrile containing internal standard was added to hepatocyte cell prior to addition of diluted compound. After centrifugation at 3000 rpm for 10 min at 4 °C, the supernatant was collected and analyzed by LC/MS-MS. Diazepam was included as the positive control in each experiment. The intrinsic clearance (in μL/min/106 cells) was calculated by dividing incubation volume (μL) by number of cells in incubation (×106 cells) and multiplying by elimination rate constant (min−1). The elimination rate constant is the negative gradient, which was calculated from the remaining ratio up to 2 h.
Supplementary Material
ACKNOWLEDGMENT
We are indebted to Drs. Leigh Baxt and Stacia Kargman at TDI for their suggestions. The authors acknowledge Daniel Mota, Liselle Guiang, Ryan Scales and Judith Okoro for assistance with ex vivo testing of compounds in Uganda. We thank Mikayla Herring at Weill Cornell Medicine for conducting synergy assay of TDI-8414 with dihydroartemisinin. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
Funding Sources
The study was supported by NIH grants R01AI143714 (G.L.), R21AI123794 (G.L. and L.A.K.), R01AI139179 (P.J.R, P.K.T, R.A.C.), T37MD003407 (S.C.); The Brockman Foundation (L.A.K.); Weill Cornell Medicine Matching Fund (G.L.); Department of Medicine, Weill Cornell Medicine Seed fund (L.A.K.); Milstein Program in Chemical Biology and Translational Medicine. We gratefully acknowledge in-kind support of the Tri-Institutional Therapeutics Discovery Institute (TDI), a 501(c)(3) organization. TDI receives financial support from TDI’s owners (Weill Cornell Medicine, Memorial Sloan Kettering Cancer Center and The Rockefeller University and), Takeda Pharmaceutical Company, and generous contributions from Mr. Lewis Sanders, Mr. Howard Milstein and other philanthropic sources.
ABBREVIATIONS
- PAMPA
parallel artificial membrane permeability assay
- MDR
MDR1-MDCK permeability
- ER
efflux ratio
- m/hLM
mouse/human liver microsome stability
- cLogP
calculated partition coefficient
- TPSA
topological polar surface area
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge at http://pubs.acs.org.
Synthetic procedures for key intermediates, NMR and HPLC spectra of final compounds, and experimental procedures for the biological assays (PDF)
Molecular formula strings (CSV)
Any additional relevant notes should be placed here.
REFERENCES
- 1.Organization, W. H. World malaria report 2021. 2021.
- 2.Phillips MA; Burrows JN; Manyando C; van Huijsduijnen RH; Van Voorhis WC; Wells TNC Malaria. Nat Rev Dis Primers 2017, 3, 17050. [DOI] [PubMed] [Google Scholar]
- 3.Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han KT; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ; Tracking Resistance to Artemisinin, C. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014, 371, 411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeung S; Socheat D; Moorthy VS; Mills AJ Artemisinin resistance on the Thai-Cambodian border. Lancet 2009, 374, 1418–9. [DOI] [PubMed] [Google Scholar]
- 5.Balikagala B; Fukuda N; Ikeda M; Katuro OT; Tachibana SI; Yamauchi M; Opio W; Emoto S; Anywar DA; Kimura E; Palacpac NMQ; Odongo-Aginya EI; Ogwang M; Horii T; Mita T Evidence of Artemisinin-Resistant Malaria in Africa. N Engl J Med 2021, 385, 1163–1171. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H; Lin G Microbial proteasomes as drug targets. PLoS Pathog 2021, 17, e1010058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin G; Li D; de Carvalho LP; Deng H; Tao H; Vogt G; Wu K; Schneider J; Chidawanyika T; Warren JD; Li H; Nathan C Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 461, 621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PC; Bogyo M Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khare S; Nagle AS; Biggart A; Lai YH; Liang F; Davis LC; Barnes SW; Mathison CJ; Myburgh E; Gao MY; Gillespie JR; Liu X; Tan JL; Stinson M; Rivera IC; Ballard J; Yeh V; Groessl T; Federe G; Koh HX; Venable JD; Bursulaya B; Shapiro M; Mishra PK; Spraggon G; Brock A; Mottram JC; Buckner FS; Rao SP; Wen BG; Walker JR; Tuntland T; Molteni V; Glynne RJ; Supek F Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang M; Wang C; Otto TD; Oberstaller J; Liao X; Adapa SR; Udenze K; Bronner IF; Casandra D; Mayho M; Brown J; Li S; Swanson J; Rayner JC; Jiang RHY; Adams JH Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhan W; Zhang H; Ginn J; Leung A; Liu YJ; Michino M; Toita A; Okamoto R; Wong TT; Imaeda T; Hara R; Yukawa T; Chelebieva S; Tumwebaze PK; Lafuente-Monasterio MJ; Martinez-Martinez MS; Vendome J; Beuming T; Sato K; Aso K; Rosenthal PJ; Cooper RA; Meinke PT; Nathan CF; Kirkman LA; Lin G Development of a Highly Selective Plasmodium falciparum Proteasome Inhibitor with Antimalaria Activity in Humanized Mice. Angew Chem Int Ed Engl 2021, 60, 9279–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie SC; Metcalfe RD; Mizutani H; Puhalovich T; Hanssen E; Morton CJ; Du Y; Dogovski C; Huang SC; Ciavarri J; Hales P; Griffin RJ; Cohen LH; Chuang BC; Wittlin S; Deni I; Yeo T; Ward KE; Barry DC; Liu B; Gillett DL; Crespo-Fernandez BF; Ottilie S; Mittal N; Churchyard A; Ferguson D; Aguiar ACC; Guido RVC; Baum J; Hanson KK; Winzeler EA; Gamo FJ; Fidock DA; Baud D; Parker MW; Brand S; Dick LR; Griffin MDW; Gould AE; Tilley L Design of proteasome inhibitors with oral efficacy in vivo against Plasmodium falciparum and selectivity over the human proteasome. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H; van der Linden WA; Verdoes M; Florea BI; McAllister FE; Govindaswamy K; Elias JE; Bhanot P; Overkleeft HS; Bogyo M Assessing subunit dependency of the Plasmodium proteasome using small molecule inhibitors and active site probes. ACS Chem Biol 2014, 9, 1869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H; Tsu C; Blackburn C; Li G; Hales P; Dick L; Bogyo M Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome. J Am Chem Soc 2014, 136, 13562–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H; Ponder EL; Verdoes M; Asbjornsdottir KH; Deu E; Edgington LE; Lee JT; Kirk CJ; Demo SD; Williamson KC; Bogyo M Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem Biol 2012, 19, 1535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirkman LA; Zhan W; Visone J; Dziedziech A; Singh PK; Fan H; Tong X; Bruzual I; Hara R; Kawasaki M; Imaeda T; Okamoto R; Sato K; Michino M; Alvaro EF; Guiang LF; Sanz L; Mota DJ; Govindasamy K; Wang R; Ling Y; Tumwebaze PK; Sukenick G; Shi L; Vendome J; Bhanot P; Rosenthal PJ; Aso K; Foley MA; Cooper RA; Kafsack B; Doggett JS; Nathan CF; Lin G Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proc Natl Acad Sci U S A 2018, 115, E6863–E6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aminake MN; Arndt HD; Pradel G The proteasome of malaria parasites: A multi-stage drug target for chemotherapeutic intervention? Int J Parasitol Drugs Drug Resist 2012, 2, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenthal MR; Ng CL A Proteasome Mutation Sensitizes P. falciparum Cam3.II K13(C580Y) Parasites to DHA and OZ439. ACS Infect Dis 2021, 7, 1923–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dogovski C; Xie SC; Burgio G; Bridgford J; Mok S; McCaw JM; Chotivanich K; Kenny S; Gnadig N; Straimer J; Bozdech Z; Fidock DA; Simpson JA; Dondorp AM; Foote S; Klonis N; Tilley L Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 2015, 13, e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoo E; Stokes BH; de Jong H; Vanaerschot M; Kumar T; Lawrence N; Njoroge M; Garcia A; Van der Westhuyzen R; Momper JD; Ng CL; Fidock DA; Bogyo M Defining the Determinants of Specificity of Plasmodium Proteasome Inhibitors. J Am Chem Soc 2018, 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simwela NV; Hughes KR; Rennie MT; Barrett MP; Waters AP Mammalian Deubiquitinating Enzyme Inhibitors Display in Vitro and in Vivo Activity against Malaria Parasites and Potentiate Artemisinin Action. ACS Infect Dis 2021, 7, 333–346. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H; Ginn J; Zhan W; Liu YJ; Leung A; Toita A; Okamoto R; Wong TT; Imaeda T; Hara R; Yukawa T; Michino M; Vendome J; Beuming T; Sato K; Aso K; Meinke PT; Nathan CF; Kirkman LA; Lin G Design, Synthesis, and Optimization of Macrocyclic Peptides as Species-Selective Antimalaria Proteasome Inhibitors. J Med Chem 2022, 65, 9350–9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe T; Schulz D; Morisseau C; Hammock BD High-throughput pharmacokinetic method: cassette dosing in mice associated with minuscule serial bleedings and LC/MS/MS analysis. Anal Chim Acta 2006, 559, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hitchcock SA Structural modifications that alter the P-glycoprotein efflux properties of compounds. J Med Chem 2012, 55, 4877–95. [DOI] [PubMed] [Google Scholar]
- 25.Hirashima S; Oka T; Ikegashira K; Noji S; Yamanaka H; Hara Y; Goto H; Mizojiri R; Niwa Y; Noguchi T; Ando I; Ikeda S; Hashimoto H Further studies on hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors toward improved replicon cell activities: benzimidazole and structurally related compounds bearing the 2-morpholinophenyl moiety. Bioorg Med Chem Lett 2007, 17, 3181–6. [DOI] [PubMed] [Google Scholar]
- 26.Tumwebaze PK; Katairo T; Okitwi M; Byaruhanga O; Orena S; Asua V; Duvalsaint M; Legac J; Chelebieva S; Ceja FG; Rasmussen SA; Conrad MD; Nsobya SL; Aydemir O; Bailey JA; Bayles BR; Rosenthal PJ; Cooper RA Drug susceptibility of Plasmodium falciparum in eastern Uganda: a longitudinal phenotypic and genotypic study. Lancet Microbe 2021, 2, e441–e449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.