

Summary paragraph:
The catalytic asymmetric construction of Csp3–Csp3 bonds remains one of the foremost challenges in organic synthesis.1 Metal-catalyzed cross-electrophile couplings (XEC) have emerged as a powerful tool for C–C bond formation.2–5 However, coupling two distinct Csp3-electrophiles with high cross- and stereoselectivity continues as an unmet challenge. Here, we report a highly chemo- and enantioselective Csp3–Csp3 XEC between alkyl halides and nitroalkanes catalyzed by flavin-dependent ‘ene’-reductases. Photoexcitation of the enzyme-templated charge-transfer complex between an alkyl halide and flavin cofactor enables the chemoselective reduction of alkyl halide over the thermodynamically favored nitroalkane partner. The key C–C bond-forming step occurs via the reaction of an alkyl radical with an in situ generated nitronate to form a nitro radical anion that collapses to form nitrite and an alkyl radical. An enzyme-controlled hydrogen atom transfer affords high levels of enantioselectivity. This reactivity is unknown in small molecule catalysis and highlights the potential for enzymes to use new mechanisms to address long-standing synthetic challenges.
Catalytic cross-couplings to forge Csp2–Csp2 bonds have revolutionized organic synthesis, enabling the rapid construction of molecules for the pharmaceutical and agrochemical industries.6,7 As target compounds become more complex — correlating with a higher percentage of sp3-hybridized atoms and stereocenters — there is a need for technologies to forge Csp3–Csp3 bonds stereoselectively.8,9 Cross-electrophile couplings (XEC) involving two distinct Csp3-electrophiles are an attractive alternative to the traditional cross-couplings because they have broad functional group tolerance and avoid the need for sensitive organometallic reagents.10–12 However, these reactions often form homo-coupled products because the metal catalysts struggle to distinguish between the two Csp3-electrophiles. This issue can be diminished using alkyl halides that react at different rates with the metal catalyst.13,14 Moreover, while there has been significant progress toward catalytic asymmetric Csp2–Csp3 XEC,4,5 stereoselective Csp3–Csp3 XEC is underdeveloped (Fig. 1a).15,16 To overcome these limitations, previously unappreciated mechanistic steps and catalytic strategies need to be explored.17–19
Fig. 1. Photoenzymatic asymmetric cross-electrophile coupling reactions.
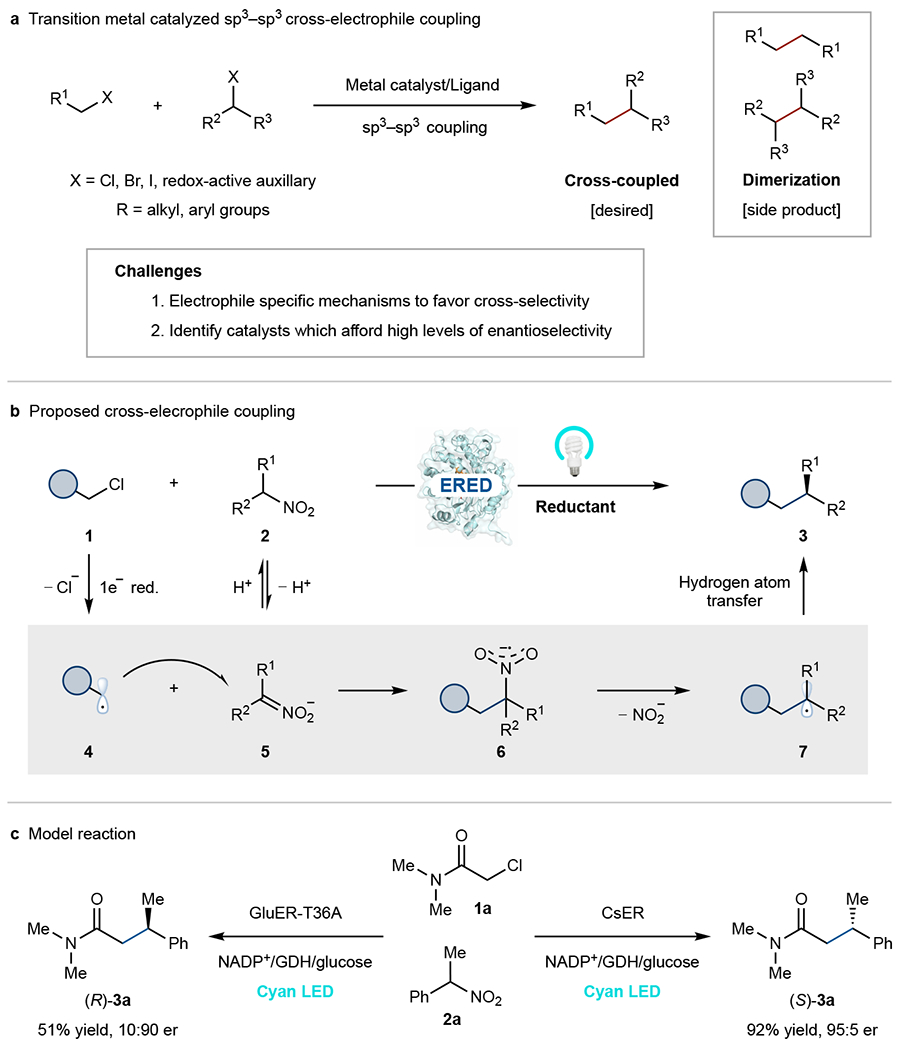
a, Challenges associated with sp3–sp3 XEC. b, Proposed photoenzymatic asymmetric XEC. c, Two stereocomplimentary ‘ene’-reductases catalyze the model reaction at pH 9.0. Enantiomeric ratio (er) refers to as the ratio of (S)- to (R)-enantiomer. ERED, ‘ene’-reductase; GDH, glucose dehydrogenase; NADP+, nicotinamide adenine dinucleotide phosphate.
We questioned whether an enzyme could catalyze an asymmetric Csp3–Csp3 XEC. The high level of selectivity associated with biocatalytic reactions makes them attractive scaffolds for this challenge.20,21 However, as natural enzymes do not catalyze cross-coupling reactions, we needed to develop a novel XEC mechanism that is compatible with existing enzymatic machinery.22,23 Nitroalkanes are unique and ubiquitous reagents in organic synthesis but are not used as electrophiles for cross-couplings.24 We recognized that the reactivity of nitronates could be used for a biocatalytic XEC. Nitronates react with open-shell electrophiles to forge a C–C bond and a nitro radical anion.25,26 If this intermediate were to cleave mesolytically, the resulting radical could be quenched via hydrogen atom transfer (HAT) to afford the cross-coupled product.27 The key to achieving this previously unknown reaction is identifying an enzyme to facilitate C–C bond formation, C–N bond mesolytic cleavage, and HAT (Fig. 1b). We envision a mechanism where reduction of the alkyl halide 1 forms an alkyl radical 4 that can react with an in situ generated nitronate 5 to forge a new C–C bond and a nitro radical anion 6. Enzyme-mediated homolytic cleavage of the C–N bond generates nitrite and an alkyl radical 7 that can be terminated via HAT to afford the cross-coupled product 3 (Fig. 1b). The proposed mechanism is attractive because the orthogonal reactivity of nitroalkanes and alkyl halides avoids undesired dimerization products.
Precise control over the chemoselectivity of the electron transfer events is required for the proposed reaction. Reduction of the nitroalkanes is thermodynamically favored by comparison to all but the most electronically activated alkyl halides (nitroalkanes Ep/2 = ~ −0.9 V vs. saturated calomel electrode, SCE;28 alkyl halides Ep/2=−1.1 V to −2.5 V vs. SCE29).30 To achieve the desired reaction, we require a catalyst that will preferentially reduce alkyl halides instead of the nitroalkanes. We and others recently demonstrated that flavin-dependent ‘ene’-reductases (EREDs) can reduce alkyl halides using protein templated charge-transfer (CT) complexes.31–33 Protein templated complexes provide the opportunity for substrate binding to override the inherent thermodynamic preference in electron transfer events. If the protein only forms the CT complex with the alkyl halide, it would be selectively reduced over the nitroalkane. Finally, EREDs can precisely control the radical terminating HAT step, enabling the formation of products with high levels of enantioselectivity.34,35
We initiated our studies by exploring the photoenzymatic coupling of α-chloroamide 1a (Ep/2 = −1.65 V vs. SCE)31 with 1-nitroethylbenzene 2a (Ep/2 = −0.89 V vs. SCE)28 catalyzed by a panel of EREDs under cyan light irradiation (λmax = 497 nm) (Supplementary Table 1). To our delight, many of the enzymes provided the desired cross-coupled product 3a, with none providing the nitrated product (Supplementary Table 1). The most promising catalyst was the ‘ene’-reductase from Caulobacter segnis (CsER), providing product 3a with moderate yield (28%) but excellent enantioselectivity, with 95:5 enantiomeric ratio (er) of (S)- to (R)-enantiomer of the product. We hypothesized that the modest yield is likely due to inadequate concentration of the nitronate at pH 8.0 (nitroalkane/nitronate = 200:1, pKa of 2a is 10.3).36 Indeed, in moving to the more basic reaction conditions at pH 9.0 (nitroalkane/nitronate = 20:1), the desired product is formed in high yield and excellent enantioselectivity (92% yield, 95:5 er) for the (S)-enantiomer, outperforming other tested EREDs (Fig. 1c and Supplementary Table 1). The ERED variant from Gluconobacter oxydans (GluER-T36A) favors the formation of the (R)-enantiomer of the product (51% yield, 10:90 er), providing a complimentary catalyst for accessing both enantiomers of the product (Fig. 1c). Control experiments confirmed that ERED, cyan light, and NADPH regeneration system (GDH/NADP+/glucose) are crucial for the desired reactivity (Supplementary Table 2). Notably, this reaction can be run on a preparative scale and afford product 3a in 72% isolated yield from a 0.1 mmol-scale reaction with no changes in enantioselectivity. We solved the crystal structure of wild-type CsER, and the docking model of 3a with CsER suggests the (S)-preference of CsER in this reaction (Supplementary Fig. 4). Notably, the coupled product is not formed when the same reaction is attempted using photoredox catalysts.37 Instead, we observed nitroalkane reduction to the oxime using Ir(ppy)3 as a photoredox catalyst, highlighting that this reactivity is unique to biocatalysis.
With the optimized reaction parameters in hand, we sought to explore the scope and limitations of this photoenzymatic transformation (Fig. 2). A variety of α-aryl nitroalkanes are well accepted as XEC partners with α-chloroamide 1a. α-Aryl nitroethanes possessing electron-donating or electron-withdrawing substituents at the meta- and para-position were efficiently converted to the desired enantioenriched β-stereogenic amide products (9-16) in yields of 80–98% with excellent enantioselectivity (>95:5 er). Ortho-substituted α-aryl nitroalkanes were less tolerated in the reaction, only the ortho-fluoro substituted nitroalkane was accepted to provide product 8 in 28% yield and 94:6 er (Fig. 2, and Supplementary Fig. 2). In addition, the larger substrate α-naphthalenyl nitroethane was well tolerated in this reaction, providing the corresponding product 17 with 58% yield and excellent enantioselectivity (99:1 er). This enzyme, however, was limited to relatively small alkyl substituents at the α-position. While the ethyl group was well accepted (19, 64% yield and 98:2 er), larger groups, such as n-propyl, were poorly reactive. Pleasingly, CsER could also accommodate heterocycles, including the electron-rich α-thiophene and the electron-deficient α-pyridine nitroethanes, providing the respective β-heterocycle substituted chiral amide products (22-24) in 40–95% yield and high enantioselectivity (up to 99:1 er). The compatibility of this photoenzyme was further highlighted by the acceptance of α-nitroester as a coupling partner, giving β-stereogenic 1,4-dicarbonyl product 25 in 96% yield and 82:18 er (Fig. 2).
Fig. 2. Scope of the photoenzymatic cross-electrophile couplings.
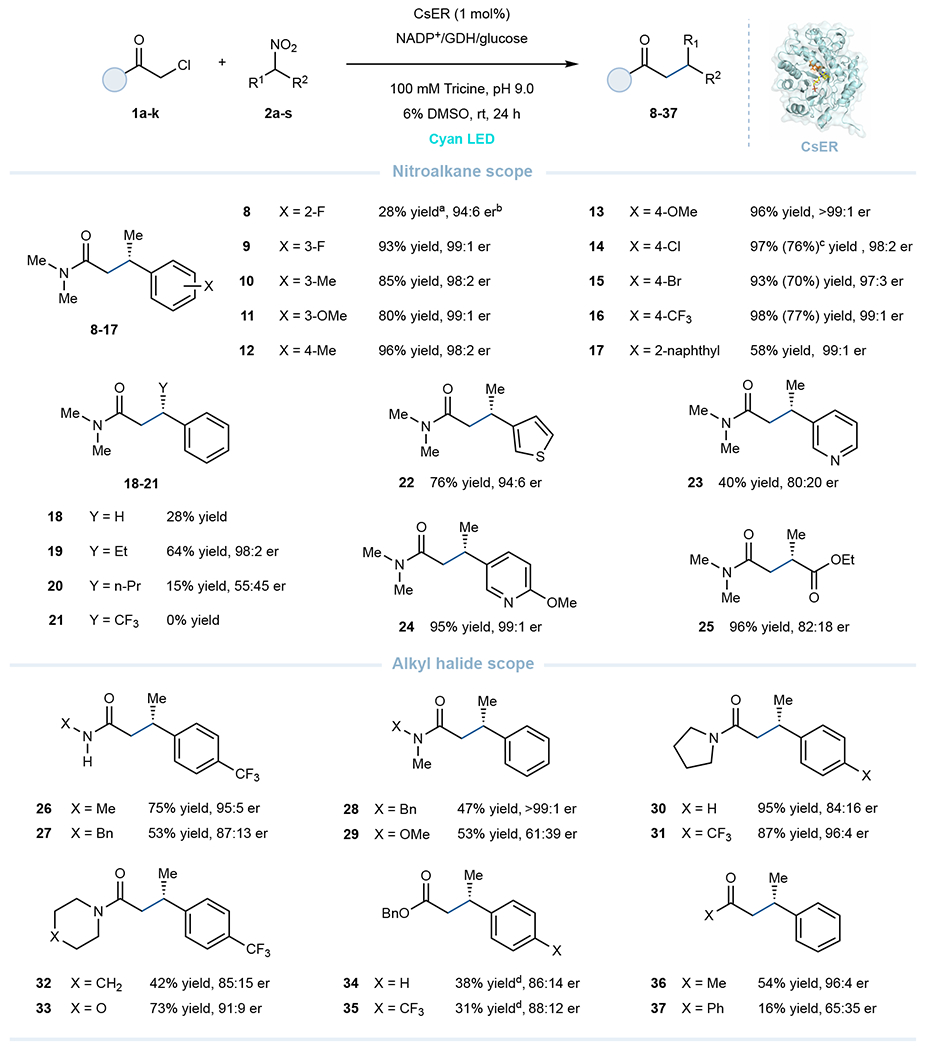
Reaction conditions: α-chloro carbonyl substrate (10 μmol, 2 equiv), nitroalkane (5 μmol, 1 equiv), GDH-105 (0.3 mg), NADP+ (0.05 μmol, 1 mol%), glucose (25 μmol) and purified ‘ene’-reductases (0.05 μmol, 1 mol% based on nitroalkane) in tricine buffer (100 mM, pH 9.0), with 6% DMSO as cosolvent, final total volume is 800 μL. Reaction mixtures were irradiated with cyan LEDs under anaerobic conditions at room temperature for 24 h. a Yields determined via LCMS relative to an internal standard (TBB). b Enantiomeric ratio (er) refers to as the ratio of (S)- to (R)-enantiomer, er determined by HPLC on a chiral stationary phase. c Isolated yields were based on 0.10 mmol-scale reaction. d 3 equiv of α-bromo ester (15 μmol) were used.
As for the alkyl halide scope, secondary amides with methyl or benzyl are well accepted when coupled with α-(p-CF3)phenyl nitroethane (2j) respectively, affording the products (26-27) in 53-75% yields and good enantioselectivities (up to 95:5 er) (Fig. 2). Tertiary amides are also well tolerated by the reaction, with cyclic pyrrolidine, piperidine, morpholine, and linear Weinreb amides providing the corresponding products (28-33) in moderate to good yields and enantioselectivities (42%-95% yields, up to 99:1 er). Pleasingly, different classes of carbonyls as coupling electrophiles are also feasible, as exemplified by the coupling of α-halo ester and α-halo ketones with α-aryl nitroalkanes, giving the respective enantioenriched β-chiral substituted esters and ketones (34-37, Fig. 2). Notably, the β-chiral amide 3a can be reduced to the corresponding γ-chiral amine 41 with good yield and no erosion of stereoselectivity (70% yield, 95:5 er, Fig. 3). Additionally, the enzymatic product 16 can be hydrolyzed to give β-chiral acid 42 and further reduced to γ-chiral alcohol 43 in good yield and excellent stereoretention (99:1 er, Fig. 3).
Fig. 3. Derivatization of the enzymatic products.
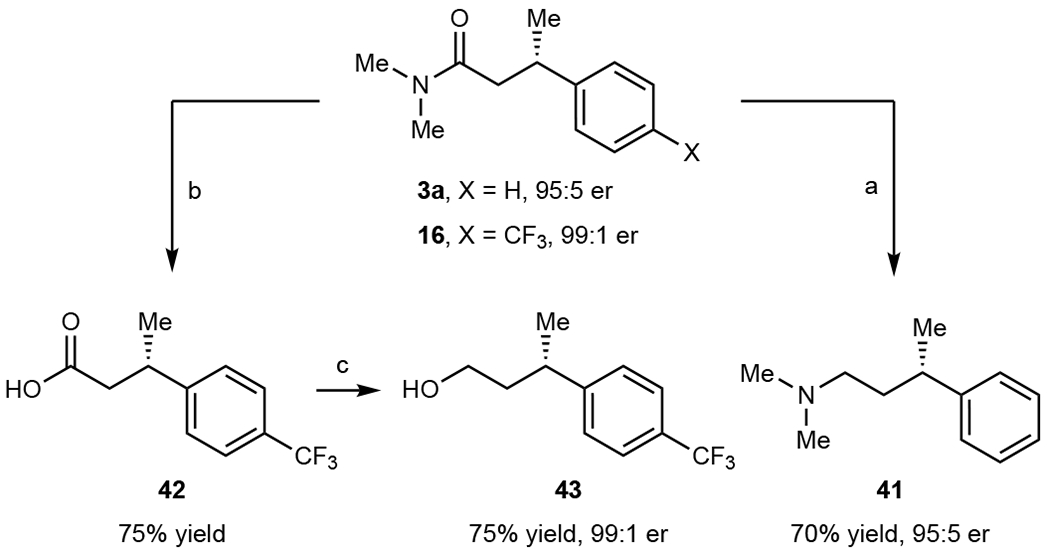
Reaction conditions: a BH3·Me2S (3.0 equiv), THF, 0 to 65 °C, 5 h. b H2SO4 (4 M)/HOAc, 150 °C, 16 h. c BH3·Me2S (3.0 equiv), THF, 0 to 45 °C, 5 h.
Mechanistic studies were conducted to determine the mode of radical initiation. Specifically, we were interested in understanding why the less oxidizing α-chloroamide (1a, Ep/2 = −1.65 V vs. SCE) is reduced preferentially to the nitroalkane (2a, Ep/2 = −0.89 V vs. SCE). We hypothesized that an enzyme templated charge-transfer (CT) complex controlled the electron transfer events.26,27 To probe this possibility, the cofactor FMN in CsER was entirely reduced to FMNhq with sodium dithionite, which revealed negligible absorption around 500 nm (Fig. 4a). Upon the addition of chloroamide 1a, a new broad absorption band (λmax = 480 nm) was observed, suggesting the formation of a CT complex between the FMNhq and 1a. Interestingly, we found nitroalkane 2a can oxidize the ground state FMNhq to generate a flavin feature with an absorption band around 450 – 500 nm. We attribute this feature to a mixture of flavin quinone and flavin semiquinone (Supplementary Fig. 10). Interestingly, when nitroalkane 2a is mixed with CsER and cofactor turnover mix under visible light irradiation, we do not observe reduction to the oxime, hydroxylamine, or hydrodenitrated products. This suggests that initial reduction of the nitroalkane can occur, in contrast to examples with photoredox catalysts,31 but subsequent electron transfers to form oximes and hydroxylamines do not transpire, making this single electron reduction event reversible under the reaction conditions. As the first irreversible step is the reduction of the alkyl halide, reversible nitroalkane reduction does not have a detrimental effect on the reaction.
Fig. 4. Mechanistic experiments.
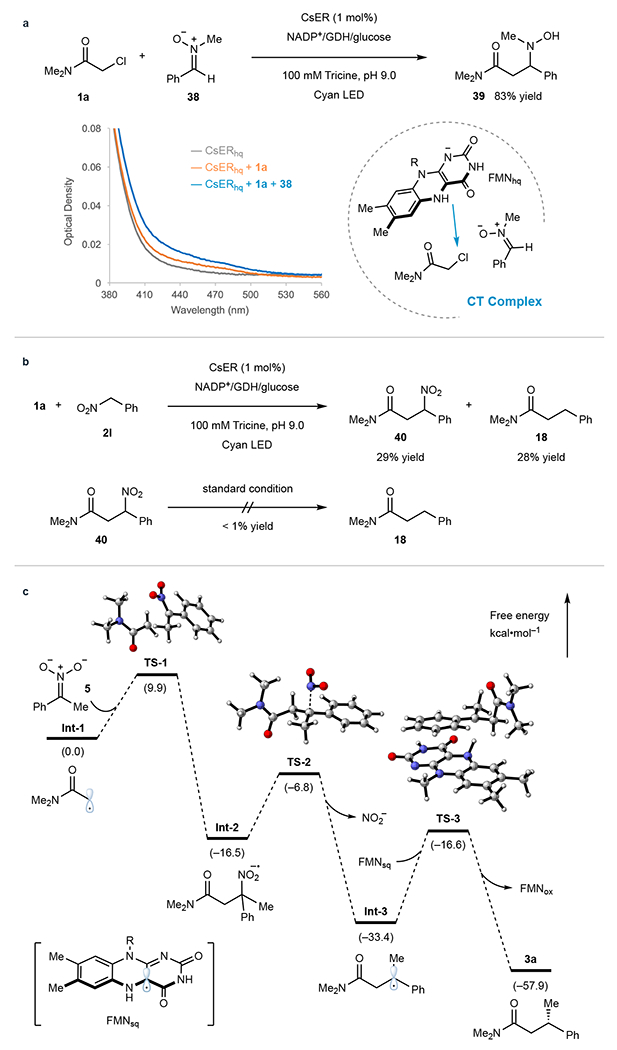
a, UV-Vis spectra of reduced CsER (FMNhq) in the presence of substrates. b, Feed experiment. c, Density functional theory (DFT) calculations of the model reaction.
Another exciting feature of these reactions is the minimal formation of the enzyme-dependent hydrodehalogenated product (Supplementary Table 2), suggesting that alkyl halide reduction occurs when the nitronate is present in the enzyme active site. However, oxidation of FMNhq by the nitroalkane obscures the observation of a higher-order CT complex (Supplementary Fig. 10). To avoid this issue, nitrone 38, a close analog of nitronate 5, was used in the UV-Vis spectra experiments because 38 can also readily react with 1a (Fig. 4a), mimicking the radical initiation step of the model XEC reaction. Intriguingly, a further enhancement of the CT complex spectra was observed when nitrone 38 was added to a sample containing 1a and the reduced CsER, indicating a quaternary CT complex was formed to enable efficient radical formation and prolongation. No CT complex was observed using free FMNhq with 1a and 38, suggesting the CT complex is formed within the enzyme active site (Supplementary Fig. 11).
Next, we were interested in understanding the denitration step of the reaction. While we propose a mechanism where the alkyl radical reacts with the nitronate to form the unstable radical anion, which rapidly undergoes mesolytic cleavage, we recognized the possibility of a two-step mechanism where the coupled nitroalkane is included in a redox-neutral process. The coupled nitroalkane intermediate is a substrate for reductive denitration in this scenario. To probe this possibility, we considered the reaction using nitromethylbenzene (2l), which under standard photoenzymatic conditions forms a 1:1 mixture of cross-coupling product 18 (28% yield) and compound 40 retaining a NO2 group (29% yield) (Fig. 4b). When the nitroalkane product 40 is resubjected to the reaction conditions, no cross-coupling product 18 was observed, indicating that the nitroalkane is not an intermediate in the denitrative coupling reaction. As nitro radical anions are proposed intermediates in non-enzymatic radical reactions but are not reported to undergo mesolytic cleavage,21 we postulate the protein facilitates the mesolytic cleavage event.
The reaction with nitromethylbenzene 2l indicates a competition between denitration and electron transfer to FMNsq under the reaction conditions. To better understand the distinctive feature of this reaction, we conducted density functional theory (DFT) calculations on the model reaction (Fig. 4c) and the one involving nitromethylbenzene 2l (Supplementary Fig. 13). We found the initial addition step of α-amidyl radical Int-1 to nitronate 5 is occurring rapidly with a free energy barrier of only 9.9 kcal mol−1 for the model reaction. The resulting radical anion Int-2 readily undergoes irreversible denitration (a free energy barrier of 9.6 kcal mol−1) to give the radical Int-3, which is terminated by HAT from FMNsq, as supported by deuterium labeling experiments (Supplementary Fig. 8), to provide the final product 3a (Fig. 4c). Next, the same DFT calculations were conducted with nitromethylbenzene 2l (Supplementary Fig. 13). While a similar free energy barrier (10.0 kcal mol−1) was observed for the initial addition step, we found a higher energy barrier (13.4 kcal mol−1) for the denitration step, indicating a slower denitration step when compared to the model reaction with 2a. Thus, as a competitive pathway to the desired cross-coupling, the radical anion Int-2’ can be terminated by oxidation by FMNsq to provide product 40 (Supplementary Fig. 13).
In summary, we have established an unprecedented photoenzymatic enantioselective sp3–sp3 cross-electrophile coupling between the readily available alkyl halides and nitroalkanes. This new enantioconvergent Csp3–Csp3 bond formation reaction is powered by EREDs, highlighting the unparallel capability of biocatalysts in differentiating Csp3 electrophile substrates and controlling stereoselectivity. By using non-traditional coupling partners and mechanisms, our work addresses the long-standing selectivity challenge in transitional-metal catalyzed cross-electrophile couplings by exploiting the promiscuous unnatural reactivity of EREDs, thus expanding the biocatalyst toolbox for asymmetric C–C bond formations.
Supplementary Material
Acknowledgements:
We thank Phil Jeffrey for assistance with X-ray structure determination, and the staff of NSLS-II beamline AMX (17-ID-1) for help with data collection. We thank the Stache group and Musser group for use of their equipment and the Collum group for use of their computational resources. The authors thank Dr. Yucong Zheng for assistance with docking and thank Dr. Shangzheng Sun and Josh Turek-Herman for discussion. The research reported here was supported by the National Institutes of Health National Institute of General Medical Sciences (R01GM127703). This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF though MRI Award CHE-1531632.
Footnotes
Competing interests: The authors declare no competing interests.
Supporting Information: Detailed experimental procedures, characterization data (NMR spectra and HPLC traces).
Data and materials availability:
The data that support the findings in this study are available from the corresponding author upon reasonable request. Crystallographic models and structure factors have been deposited in the Protein Data Bank with accession number 7TNB for CsER.
References
- 1.Choi J & Fu GC Transition metal-catalyzed alkyl-alkyl bond formation: another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Everson DA & Weix DJ Cross-electrophile coupling: principles of reactivity and selectivity. J. Org. Chem 79, 4793–4798 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gu J, Wang X, Xue W & Gong H Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations. Org. Chem. Front 2, 1411–1421 (2015). [Google Scholar]
- 4.Lucas EL & Jarvo ER Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem 1, 0065 (2017). [Google Scholar]
- 5.Poremba KE, Dibrell SE & Reisman SE Nickel-catalyzed enantioselective reductive cross-coupling reactions. ACS Catal. 10, 8237–8246 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biffis A, Centomo P, Del Zotto A & Zecca M Pd metal catalysts for cross-couplings and related reactions in the 21st century: a critical review. Chem. Rev 118, 2249–2295 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Magano J & Dunetz JR Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals. Chem. Rev 111, 2177–2250 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Lovering F, Bikker J & Humblet C Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Lovering F Escape from flatland 2: complexity and promiscuity. Medchemcomm 4, 515–519 (2013). [Google Scholar]
- 10.Qian X, Auffrant A, Felouat A & Gosmini C Cobalt-catalyzed reductive allylation of alkyl halides with allylic acetates or carbonates. Angew. Chem. Int. Ed 50, 10402–10405 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Liu JH et al. Copper-catalyzed reductive cross-coupling of nonactivated alkyl tosylates and mesylates with alkyl and aryl bromides. Chem. – A Eur. J 20, 15334–15338 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Sanford AB et al. Nickel-catalyzed alkyl-alkyl cross-electrophile coupling reaction of 1,3-dimesylates for the synthesis of alkylcyclopropanes. J. Am. Chem. Soc 142, 5017–5023 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu X, Yang T, Wang S, Xu H & Gong H Nickel-catalyzed reductive cross-coupling of unactivated alkyl halides. Org. Lett 13, 2138–2141 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Xu H, Zhao C, Qian Q, Deng W & Gong H Nickel-catalyzed cross-coupling of unactivated alkyl halides using bis(pinacolato)diboron as reductant. Chem. Sci 4, 4022–4029 (2013). [Google Scholar]
- 15.Erickson LW, Lucas EL, Tollefson EJ & Jarvo ER Nickel-catalyzed cross-electrophile coupling of alkyl fluorides: stereospecific synthesis of vinylcyclopropanes. J. Am. Chem. Soc 138, 14006–14011 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Tollefson EJ, Erickson LW & Jarvo ER Stereospecific intramolecular reductive cross-electrophile coupling reactions for cyclopropane synthesis. J. Am. Chem. Soc 137, 9760–9763 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Smith RT et al. Metallaphotoredox-catalyzed cross-electrophile Csp3-Csp3 coupling of aliphatic bromides. J. Am. Chem. Soc 140, 17433–17438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W et al. Electrochemically driven cross-electrophile coupling of alkyl halides. Nature 604, 292–297 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jana SK, Maiti M, Dey P & Maji B Photoredox/nickel dual catalysis enables the synthesis of alkyl cyclopropanes via C(sp3)-C(sp3) cross electrophile coupling of unactivated alkyl electrophiles. Org. Lett 24, 1298–1302 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Bell EL et al. Biocatalysis. Nat. Rev. Methods Primers 1, 46 (2021). [Google Scholar]
- 21.Zhou Q, Chin M, Fu Y, Liu P & Yang Y Stereodivergent atom-transfer radical cyclization by engineered cytochromes. Science 374, 1612–1616 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A et al. An enantioselective artificial Suzukiase based on the biotin–streptavidin technology. Chem. Sci 7, 673–677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierron J et al. Artificial metalloenzymes for asymmetric allylic alkylation on the basis of the biotin–avidin technology. Angew. Chem. Int. Ed 47, 701–705 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Ballini R, Bosica G, Fiorini D, Palmieri A & Petrini M Conjugate additions of nitroalkanes to electron-poor alkenes: recent results. Chem. Rev 105, 933–972 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Gildner PG, Gietter AAS, Cui D & Watson DA Benzylation of nitroalkanes using copper-catalyzed thermal redox catalysis: toward the facile C-alkylation of nitroalkanes. J. Am. Chem. Soc 134, 9942–9945 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gietter AAS, Gildner PG, Cinderella AP & Watson DA General route for preparing β-nitrocarbonyl compounds using copper thermal redox catalysis. Org. Lett 16, 3166–3169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornblum N, Carlson SC & Smith RG Replacement of the nitro group by hydrogen. J. Am. Chem. Soc 100, 289–290 (1978). [Google Scholar]
- 28.Tanner DD et al. On the mechanism of the radical chain transformation of nitroalkanes to alkanes using triaryl- or trialkyltin hydrides. J. Org. Chem 55, 3321–3325 (1990). [Google Scholar]
- 29.Roth HG, Roth HG, Romero NA & Nicewicz DA Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett. 27, 714–723 (2016). [Google Scholar]
- 30.Durchschein K et al. Reductive biotransformation of nitroalkenes via nitroso-intermediates to oxazetes catalyzed by xenobiotic reductase A (XenA). Org. Biomol. Chem 9, 3364–3369 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Biegasiewicz KF et al. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–1169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Page CG et al. Quaternary charge-transfer complex enables photoenzymatic intermolecular hydroalkylation of olefins. J. Am. Chem. Soc 143, 97–102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang X et al. Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 584, 69–74 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Fu H et al. Ground-state electron transfer as an initiation mechanism for biocatalytic C-C bond forming reactions. J. Am. Chem. Soc 143, 9622–9629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandoval BA, Meichan AJ & Hyster TK Enantioselective hydrogen atom transfer: discovery of catalytic promiscuity in flavin-dependent ‘ene’-reductases. J. Am. Chem. Soc 139, 11313–11316 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Fukuyama M et al. The thermodynamic and kinetic acidity properties of nitroalkanes. correlation of the effects of structure on the ionization constants and the rate constants of neutralization of substituted 1-phenyl-1-nitroethanes. J. Am. Chem. Soc 92, 4689–4699 (1970). [Google Scholar]
- 37.Cai S, Zhang S, Zhao Y & Wang DZ New approach to oximes through reduction of nitro compounds enabled by visible light photoredox catalysis. Org. Lett 15, 2660–2663 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings in this study are available from the corresponding author upon reasonable request. Crystallographic models and structure factors have been deposited in the Protein Data Bank with accession number 7TNB for CsER.