

Abstract
Components of the cell division machinery typically function at varying cell cycle stages and intracellular locations. To dissect cellular mechanisms during the rapid division process, small-molecule probes act as complementary approaches to genetic manipulations, with advantages of temporal and in some cases spatial control and applicability to multiple model systems. This review focuses on recent advances in chemical probes and applications to address selected questions in cell division. We discuss uses of both enzyme inhibitors and chemical inducers of dimerization, as well as emerging techniques to promote future investigations. Overall, these concepts may open new research directions for applying chemical probes to advance cell biology.
Editorial Summary
This review summarizes recent progress in small-molecule probes used to address questions in cell division, and appraises some emerging techniques that can be adapted to cell division studies.
A central process for life is to pass the genetic material to offspring through cell division. In eukaryotes, the genetic material is packaged into chromosomes, which replicate and then segregate to daughter cells. For somatic cells or asexual reproduction, daughter cells acquire identical chromosome copies from the parental cell through mitosis. For sexual reproduction, two cycles of meiotic cell division produce haploid gametes from a diploid parent. Multiple molecular machineries are required for successful division in both mitosis and meiosis. Microtubules, consisting of polarized tubulin polymers, organize into a bipolar structure with minus-ends anchored to spindle poles. This dynamic spindle interacts with the chromosomes through kinetochore protein complexes to drive chromosome movement to the spindle equator (congression), ensure that sister chromosomes attach correctly to opposite spindle poles (error correction), and segregate the chromosomes in anaphase. In addition, the spindle assembly checkpoint (SAC) regulates cell cycle progression to promote accurate chromosome segregation by delaying anaphase until all kinetochores are attached to spindle microtubules. Defects in any of these processes can lead to aneuploidy and structural rearrangements of chromosomes, which are highly associated with cancer and developmental diseases1–4. The underlying spatial and temporal regulation of cell division is a long-standing focus of cell, developmental, and reproductive biology studies.
A common cell biology paradigm is to draw inferences by contrasting how cells respond to different perturbations. Genetic perturbations, such as knockout, overexpression, or RNA interference (RNAi), have led to many advances linking molecular functions to phenotypic observations. However, the lack of temporal control (typically ~days) limits studies of dynamic processes in cell division, which is completed within minutes to hours. Moreover, components of the cell division machinery can target different substrates with distinct functions at different intracellular locations, which are difficult to dissect by genetic perturbations. As a complementary approach, small molecule probes provide seconds-to-minutes temporal control and can also be designed for spatial control and reversibility. Using enzymatic inhibitors, for example, acute effects can be observed immediately after adding a small molecule, avoiding potential secondary effects (including lethality in some cases) that can accumulate over time after genetic perturbations. Furthermore, as a single protein can have multiple functions, enzymatic inhibition can help uncover non-enzymatic mechanisms. With the continual development of new small molecule inhibitors, mechanistic studies have taken advantage of these probes to address a variety of questions in cell division.
In recent years, cell biologists have increasingly exploited chemical inducers of dimerization, which bring multiple proteins together (Figure 1a) and provide experimental control over the local concentrations of a specific protein target and its associated partners. Among a variety of dimerizers, established platforms used in cell division studies include rapamycin and the trimethoprim-halo ligand (TMP-Halo, TH) system. Rapamycin dimerizes FKBP and the FKBP-rapamycin-binding (FRB) protein5, and TH dimerizes the Escherichia coli dihydrofoliate reductase (eDHFR) and the Halo enzyme (Figure 1b)6. By fusing these protein tags to a desired anchor and effector pair, dimerization can be accomplished with minute-scale kinetics upon chemical addition. Moreover, these probes have been engineered to be light responsive, allowing recruitment with spatial control. For instance, TH dimerizers have been derived to three platforms: a coumarin-cage for light-induced recruitment (CTH, Figure 1c), an NVOC-insertion for light-induced cleavage (TNH, Figure 1d), and the combination of the two7,8 (CTNH). These photo-inducible methods can respond to light in a few seconds on top of the gained spatial control, and thus provide a powerful toolkit to study cell division events occurring in a short time window at defined intracellular sites.
Figure 1. Chemically induced dimerization recruits effector proteins to anchors.
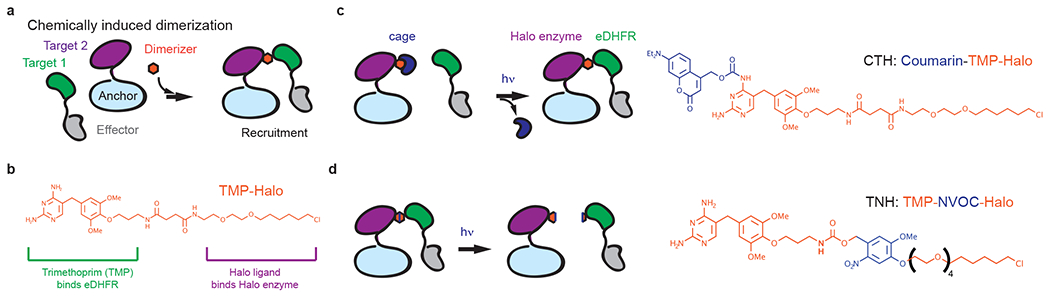
(a) A small-molecule dimerizer binds to a pair of protein targets, each of which is fused to an effector or anchor protein. If the anchor is localized to an intracellular structure, dimerizer addition recruits the effector to that structure. (b) TMP-Halo dimerizer. The bifunctional linker includes a TMP moiety and a Halo ligand to dimerize proteins fused to an Escherichia coli dihydrofoliate reductase (eDHFR) and a Halo enzyme. (c) Schematic of light-induced dimerization using CTH. A coumarin-caged dimerizer initially binds to the Halo-tagged anchor protein. Upon light-uncaging, the exposed TMP moiety recruits eDHFR-tagged effectors to anchors. (d) Schematic of light-induced cleavage using TNH. The photo-cleavable moiety, NVOC, inserted between TMP and Halo, releases the bound effectors from anchors upon light activation.
Photo-caged chemical dimerizers such as CTH or NTH are typically irreversible upon light exposure, or reversible for a single cycle of uncaging followed by photo-cleavage in the case of CTNH. In contrast, the protein conformational changes underlying genetically encoded light-inducible dimerization systems are generally reversible9, which enables the system to be perturbed repetitively but can be a limitation if continuous light exposure is required to maintain dimerization over time. Photo-caged dimerizers remain active after uncaging by a single pulse of light, which can have advantages for long-term perturbations10 or if a subcellular structure such as a randomly oscillating kinetochore is difficult to target repeatedly11. Thus, genetically encoded and small molecule-based approaches have complementary advantages and limitations.
In this review, we summarize recent advances using small molecule probes to dissect cell division mechanisms (Table 1, 2) and envision future techniques and potential strategies for probe design. Instead of a comprehensive review of chemical approaches, we focus on chemical probes and strategies leading to advances that would otherwise be difficult to achieve using only genetic perturbations. Topics covered here focus on prometaphase and metaphase events in both mammalian and yeast mitosis, but the tools and underlying concepts should be transferrable to other cell stages, meiotic studies, and other model systems.
Table 1.
Enzymatic inhibitors and their applications in cell division studies.
| Inhibitors | Function | Applications in cell division |
|---|---|---|
| 5-Iodotubercidin | Haspin inhibitor | Deplete centromeric Aurora B pool56. |
| Apcin | APC/C inhibitor | Prolong or shorten metaphase at low or high SAC activity, respectively70,71. |
| Centrinone | Plk4 inhibitor | Modulate centriole numbers18, generate asymmetric spindles19,20. |
| FCPT BRD9876 |
Eg5 rigor inhibitor | Spindle crosslinking40, block poleward microtubule flux38,39, reduce inter-kinetochore tension11. |
| GSK923925 | CENP-E rigor inhibitor | Generate pole-proximal chromosomes7,61. |
| GW108X | KIF15 inhibitor | Deplete spindle-bound KIF1542. |
| KIF15-IN-1 | KIF15 rigor inhibitor | Reduce inter-kinetochore tension11. |
| MLN8054 MLN8237 |
Aurora A inhibitor | Inhibit Aurora A activity after mitotic entry61,62. |
| proTAME | APC/C inhibitor | Prolong metaphase69. |
| Reversine | MPS1 inhibitor | Shorten metaphase and generate segregation errors65. |
Table 2.
Chemical dimerizers and their applications in cell division studies.
| Dimerizers | Function | Applications in cell division |
|---|---|---|
| AP20187 | FKBP homo-dimerization | Dimerize MPS1 for trans-activation75. |
| Auxin/IAA | Degrade AID-tagged protein by recruiting ubiquitin ligases | Rapidly deplete proteins without known inhibitors82. |
| CTH | Light-inducible dimerization of eDHFR and Halo enzyme | Recruit kinesin motors7 or Aurora B11 to kinetochores. |
| LoKI | Inhibit kinases proximal to the SNAP-tagged protein | Inhibit pole- or kinetochore-localized pools of Aurora A and Plk187. |
| Rapamycin | FKBP-FRB dimerization | Recruit Aurora B to centromeres58 or MAD1 to kinetochores72,73, test MPS1 function at different locations76,89. |
| TNH | Photo-cleavable dimerizer linking eDHFR and Halo enzyme | Initiate mitotic exit by releasing MAD1 from kinetochores7. |
Polo-like kinase 4 and centrosome duplication
Setting up a dynamic spindle requires microtubules to nucleate and polymerize from their organizing centers. One significant pathway is through centrosomes12,13 (Figure 2a). The interior centriole component organizes pericentriolar material to regulate centrosome maturation14 and duplicates under the regulation of Polo-like kinase 4 (PLK4) during interphase15–17. PLK4 activity controls centriole numbers and microtubule density, making it an attractive target to address how centrosomes regulate cell proliferation signaling and spindle dynamics. The PLK4 inhibitor, centrinone, can block centriole duplication, and thus has been used to generate cells lacking centrosomes18 (Figure 2b) or generate asymmetric spindles19,20 (Figure 2c). These studies exploited several properties of centrinone, as discussed below, that are typical of small molecule inhibitors but difficult to achieve by genetic approaches: applicability across different cell lines and species, acute inhibition, and reversibility after inhibitor washout.
Figure 2. Centrinone inhibits PLK4 activity.

(a) Mitotic spindle formation. After nuclear envelope breakdown (dashed lines), centrosomes act as microtubule organizing centers to shape the spindle. Dynamic spindle microtubules capture chromosomes for later congression and segregation. (b) Centrinone prevents centriole duplication by inhibition of PLK4. Because PLK4 has dual activities of auto-activation and phospho-regulated proteolysis, centrinone washout induces a wave of PLK4 activity that generates over-duplicated centrioles in cancer cells. After several cell cycles, the centriole number eventually returns to the initial “set point”. (c) Centrinone generates asymmetric spindles. By blocking centriole duplication in S phase, the cell contains halved centriole numbers in mitosis (1:1 spindle). In the next cycle, the daughter cells contain only one isolated centriole (1:0 spindle), resulting in asymmetric spindles.
Cells containing supernumerary centrosomes are highly associated with chromosome instability and tumorigenesis21,22, leading to a hypothesis that transformed cells gain a fitness advantage by centrosome over-replication. To test whether proliferation of transformed cells depends on supernumerary centrosomes, centrinone was used to deplete centrioles. Multiple human cancer cells are more resistant to centriole loss following centrinone addition, compared to normal cells18, but overexpression of TRIM37 that degrades pericentriolar materials abolishes this resistance23,24. Thus, transformed cells with amplifications of a region containing the TRIM37 gene are “addicted” to supernumerary centrosomes and vulnerable to Plk4 inhibition. To test whether centrosomes can recover after loss, centrinone was washed out to re-activate PLK4, which showed that centrosome numbers return to the initial “set point” in cancer cells (Figure 2b). In contrast, normal cells lacking centrosomes prolong mitosis and arrest in G1 phase by a p53-mediated pathway, preventing new centriole assembly that would occur in S-phase25–27.
As centrosomes increase mitotic spindle dynamics, centrinone can be used to modulate spindle architecture by partially depleting centrosomes. When cells with a single centrosome enter mitosis, the spindle is asymmetric because only one pole includes a centrosome, providing an internal control between the two halves of the spindle. This approach was used to show that centrosomes not only nucleate microtubules and regulate minus end dynamics at spindle poles, but also control plus-end dynamics at kinetochores, leading to a shorter half spindle on the acentrosomal side19. The astral microtubule density is also asymmetric in spindles with a single centrosome, providing a system to probe the role of these microtubules in spindle elongation. Experiments with Potorous tridactylus (rat kangaroo) cells under physical confinement showed that the centrosome-depleted half-spindle elongates more slowly than its counterpart, supporting a model in which compression forces tighten the cortex-microtubule coupling so that cortical force generators exert more force to slide spindle poles apart20. These studies highlight the use of centrinone to control centrosome numbers, without the need for low-throughput laser-ablation approaches that have also been used to remove centrosomes28.
Kinesin motors on spindle microtubules
Bipolar spindles require a group of molecular motors to organize microtubules in a polarized structure, such that the microtubules from opposite poles act as tracks for chromosome congression (Figure 3a). Eg5 (kinesin-5) and KIF15 (kinesin-12) motors work cooperatively to cross-link microtubules and determine pole-to-pole distances. Because of their unique homotetrameric structure, Eg5 motors can both slide interpolar microtubules apart and cross-link parallel microtubule arrays29–31. As an auxiliary mechanism of spindle assembly, KIF15 maintains spindle bipolarity in the absence of Eg532. Additionally, KIF15 acts predominantly on kinetochore microtubules (k-fibers)33,34, suggesting that it may regulate chromosome dynamics. After a bipolar spindle has been established, chromosomes near the poles require the kinetochore motor protein CENP-E (kinesin-7) for congression, by powering movement towards the metaphase plate35. Failures in these various motor proteins arrest cells in mitosis through SAC activation. Inhibitors of these motors can have different mechanisms of action, such as locking motors on microtubules (rigor inhibition) or weakening microtubule interactions, which has been valuable for dissecting key kinesin functions as illustrated in the examples below.
Figure 3. Kinesin motors on spindle microtubules.

(a) Chromosome congression upon establishing spindle bipolarity. Eg5 (kinesin-5) and KIF15 (kinesin-12) motors cross-link and powerstroke on microtubule arrays to establish spindle bipolarity. CENP-E (kinesin-7) motors translocate uncongressed chromosomes from spindle poles to the equator. (b) Inhibition modes of kinesin motors. Rigor inhibition traps kinesin in a strong-binding state on microtubules, whereas inhibitors inducing a weak-binding state or genetic knockdown decrease the number of microtubule-bound motors. (c) K-fibers are mechanically coupled in the spindle. Kinesin motors cross-link interpolar microtubules, k-fibers, and bridging fibers. (d) Centromere relaxation assay. Centromeres are stretched by k-fibers pulling bi-oriented sister kinetochores in opposite directions, and relax immediately following k-fiber ablation a few microns away from one kinetochore. Strong relaxation indicates weak load-bearing on k-fibers. (e) Chromosome congression assay. CENP-E recruitment to kinetochores of pole-proximal chromosomes drives movement towards the metaphase plate, whereas kinesin-1 transports chromosomes away from the pole in all directions.
Spindle microtubules have been long thought to couple mechanically36. K-fiber mechanics, in particular, have been studied because of the established roles of these microtubules in chromosome segregation37. To test how microtubule cross-linking contributes to k-fiber load bearing, the Eg5 rigor inhibitor, FCPT, provides a useful tool (Figure 3b, c). Rigor inhibition increases microtubule cross-linking by trapping Eg5 in a strong-binding rigor state while diminishing the powerstroke (as indicated by diminished poleward microtubule flux38,39), switching Eg5 from an active force generator to a passive cross-linker40. Movements of non-sister kinetochores are more highly correlated upon FCPT treatment, suggesting a coupled network across k-fibers41. To determine the effects of cross-linking on k-fiber load-bearing, the relaxation of centromere stretch upon k-fiber ablation serves as an experimental readout, where strong relaxation amplitude corresponds to weak load-bearing activity (Figure 3d). FCPT treatment decreases the mechanical relaxation amplitude, indicating that induced cross-linking activity increases k-fiber load-bearing39. Thus, mitotic spindles act as a connected network of cross-linked microtubules, suggesting that spindle function depends on integration of mechanics from each microtubule and motor.
Comparisons between different inhibitors, or between inhibitors and RNAi, can dissect which activity mediates a biological process. This approach was used to determine how KIF15 motors organize k-fibers, using the splaying of microtubules within a k-fiber after severing by laser ablation as an assay42. One inhibitor, GW108X, reduces KIF15 microtubule cross-linking activity by weakening microtubule binding, whereas the rigor inhibitor KIF15-IN-1 only diminishes the powerstroke. GW108X promotes k-fiber splaying upon ablation but KIF15-IN-1 does not, suggesting that the remaining microtubule cross-linking upon rigor inhibition is sufficient to hold k-fibers together. Similarly, to dissect how CENP-E contributes to SAC signaling, phenotypes of the CENP-E rigor inhibitor, GSK923925, were compared to genetic knockdown43. If SAC silencing results directly from CENP-E binding microtubules, as suggested previously44,45, then inhibiting CENP-E in a rigor state should be sufficient to resume mitotic progression. The finding that CENP-E rigor inhibition mirrors the phenotype of genetic knockdown suggests that SAC silencing requires the motor activity of CENP-E for chromosome congression, and microtubule binding is not sufficient.
CENP-E drives congression by transporting chromosomes from the spindle pole towards the equator. This activity requires the motor to selectively walk on k-fibers, which face towards the equator, rather than astral microtubules facing the cortex. A post-translational modification of tubulin, detyrosination, differentiates these microtubule populations, with astral microtubules more tyrosinated and k-fibers more detyrosinated46. To test whether CENP-E can read this “tubulin code”, endogenous CENP-E was depleted by siRNA to generate pole-proximal chromosomes, and the CENP-E motor domain was recruited to kinetochores of chromosomes near one pole using the photo-caged dimerizer, CTH (Figure 1c)7. This spatially defined recruitment leaves the opposite pole unaffected as an internal control. CENP-E motor recruitment triggered selective chromosome movement towards the metaphase plate, whereas the kinesin-1 motor domain did not show this selectivity (Figure 3e). This finding illustrates the use of photo-caged dimerizers for spatial control and shows that CENP-E biochemistry optimizes chromosome congression, supporting the tubulin code hypothesis47.
Chromosome error correction
Correct chromosome segregation requires robust error correction mechanisms, which selectively stabilize correct “bi-orientation” with sister kinetochores attached to opposite spindle poles while destabilizing incorrect attachments (Figure 4a). The interactions between kinetochores and spindle microtubules are regulated by a combination of mechanical and biochemical factors37,48–50. Mechanically, tension generated at kinetochores by pulling forces from opposite spindle poles indicates bi-orientation, whereas low tension signals an error to be corrected. Biochemically, Aurora B kinase phosphorylates kinetochore substrates such as NDC80 to destabilize their interactions with microtubules. One mechanism to bridge mechanics and biochemistry is for tension to locally inhibit Aurora B activity at kinetochores to stabilize correct attachments. The observed inner centromere localization of Aurora B suggests a model in which error correction depends on this centromeric pool. Because tension pulls sister kinetochores apart, away from the inner centromere, this model predicts low Aurora B activity at bi-oriented kinetochores, which would selectively stabilize these attachments. To test this model, small molecule probes have enabled complementary strategies in different model systems to either decrease or increase centromeric Aurora B enrichment while preserving its global kinase activity.
Figure 4. Aurora kinases regulate kinetochore-microtubule interactions.

(a) Lack of tension across a sister kinetochore pair provides a signal to identify incorrect attachments, which are destabilized to allow new attachments to form. This process continues until all attachment errors are resolved. (b-c) Effects of recruiting Aurora B to kinetochores to increase local kinase activity. On monopolar spindles (b), kinetochore recruitment primarily triggers k-fiber depolymerization to move the chromosome toward the attached pole. Conversely, recruiting Aurora B to a single kinetochore of a bi-oriented sister pair (c) primarily induces k-fiber release, with the targeted kinetochore moving away from the pole that it was initially attached to. (d) Aurora A localizes to spindle poles and phosphorylates kinetochores near the pole to destabilize microtubule attachments.
First, in mammalian cells, Aurora B enrichment at centromeres requires haspin and BUB1 kinases phosphorylating histone H3 and H2A, respectively51–55. The haspin-kinase inhibitor, 5-Iodotubercidin56, reduces centromeric Aurora B and was used to test whether tension-sensing depends on this pool of kinase. A live-cell assay using fluorescence lifetime imaging to measure Förster resonance energy transfer (FLIM-FRET) showed that binding of the NDC80 complex to microtubules is regulated by tension and by Aurora B57. Furthermore, tension dependence of NDC80 binding to microtubules was lost after treatment with 5-Iodotubercidin, indicating that centromeric Aurora B is a part of the tension-sensing mechanism. Conversely, other experiments showed minimal effects on phosphorylation of Aurora B substrates at kinetochores after reducing centromeric Aurora B, suggesting that a non-centromere pool of Aurora B can phosphorylate kinetochore substrates51,52.
Second, rapamycin-induced dimerization has been used to enrich centromeric Aurora B (IPL1) in budding yeast. By fusing the IPL1-activator SLI15 (INCENP ortholog) to FRB and the centromeric protein MIF2 (CENP-C ortholog) to FKBP, rapamycin addition recruits SLI15 and IPL1 to centromeres58. Centromere recruitment rescues chromosome bi-orientation when other pathways for localizing IPL1 to centromeres are inhibited, indicating that bi-orientation depends on centromeric Aurora B in yeast. In these experiments, chemically induced dimerization enables acute gain-of-function (~10 minutes) after chromosomes are successfully aligned.
In addition to regulating local Aurora B activity at kinetochores, tension could dictate the outcome of Aurora B activation. Aurora B can destabilize kinetochore-microtubule interactions by two mechanisms: inducing either microtubule release from kinetochores or depolymerization while maintaining attachment48,49. To determine whether these mechanisms depend on tension, Aurora B was recruited to kinetochores using the photo-caged chemical dimerizer CTH11 (Figure 1c). Because microtubule release reduces pulling forces exerted by k-fibers, whereas depolymerization generates pulling forces, the two mechanisms predict chromosome movement in opposite directions. Aurora B recruitment to an individual kinetochore out of a sister pair induces microtubule release on bipolar spindles with high tension. In contrast, recruitment induces microtubule depolymerization while maintaining attachment on monopolar spindles, in which tension is lower (Figure 4b–c). Furthermore, decreasing tension using Eg5 or KIF15 rigor inhibitors (BRD9876 or KIF15-IN-1) slows the microtubule release rate. Thus, phosphorylation converts a catch-bond, in which tension stabilizes attachments59, to a more conventional slip-bond in which kinetochores release microtubules under tension. In these experiments, photocaged dimerizers provide control over local kinase activity with second and sub-micron spatiotemporal precision to probe the biological processes underlying error correction.
The tension-dependent response to Aurora B activity suggests different correction pathways for two distinct attachment errors. A merotelic error, in which a single kinetochore attaches to both spindle poles, is under tension due to microtubules pulling in opposite directions. Phosphorylation in this case would facilitate microtubule release, providing an opportunity to make a new attachment. Conversely, for a syntelic kinetochore pair, in which both sisters attach to the same pole with lower tension, phosphorylation induces k-fiber depolymerization to move chromosomes towards spindle poles60. To determine how this poleward chromosome movement leads to error-correction, the CENP-E rigor inhibitor GSK923925 was used to examine kinetochore phosphorylation on both pole-proximal and aligned chromosomes in mitotic cells61 (Figure 4d). The microtubule tether NDC80 was more highly phosphorylated near spindle poles, and this spatial dependence was diminished by treatment with the Aurora A kinase inhibitor, MLN8237. Consistent with these results, kinetochores close to spindle poles are more likely to detach from microtubules in mouse oocytes in meiosis I, and this spatial dependence also decreases with Aurora A inhibition using MLN805462. Together, these findings indicate that Aurora A at spindle poles phosphorylates kinetochores near the pole to promote microtubule detachment and allow the formation of new attachments. For these experiments, Aurora A inhibitors enabled acute inhibition after establishing spindle bipolarity, without perturbing earlier Aurora A functions in spindle assembly.
Metaphase duration and the SAC
To ensure accurate chromosome segregation, SAC signaling delays anaphase onset in response to kinetochores lacking microtubule attachment (Figure 5a). SAC activation, as typically measured by kinetochore localization of checkpoint proteins such as MAD1 and MAD2, activates the mitotic checkpoint complex (MCC) and inhibits the E3 ubiquitin ligase APC/CCDC20 to prevent cyclin B and securin degradation63. Attachments are stabilized by chromosome bi-orientation, which silences SAC signaling and leads to APC/C activation. Failure to maintain SAC activity results in premature anaphase onset and chromosome segregation errors. Small molecule probes have been used to examine mechanisms of SAC activation and silencing, through both direct inhibition of various components and induced dimerization to manipulate their localization
Figure 5. Activating and silencing the SAC.

(a) The SAC is activated by unattached kinetochores to delay anaphase onset until all chromosomes are attached to spindle microtubules (SAC silencing). (b) End-on microtubule attachment at kinetochores silences the SAC. Microtubule-bound DAM1 complex blocks the interaction between MPS1 at the outer kinetochore and SPC105 at the inner-kinetochore. (c) SAC reactivation by rapamycin-induced dimerization. Recruiting exogenous MPS1 close to SPC105 or recruiting a domain of SPC105 close to MPS1 is sufficient to reactivate the SAC.
Because low levels of SAC proteins are sufficient to catalyze downstream signaling, RNAi experiments can reach different conclusions even with undetectable SAC protein expression64. Furthermore, SAC kinases may have diverse functions beyond their kinase activities. Chemical inhibitors provide an alternative approach to probe interactions between SAC components complementary to RNAi experiments. For example, Aurora B inhibition prevents MPS1 kinase localization at kinetochores, whereas MPS1 inhibition using reversine does not change Aurora B localization but prevents MAD1 localization, suggesting that MPS1 acts downstream of Aurora B to regulate SAC signaling65. Furthermore, inhibition of MPS1 kinase activity generates a high frequency of unaligned chromosomes compared to genetic knockdown66, suggesting that inactive MPS1 competes with microtubules for binding to NDC80 at kinetochores and thereby impedes chromosome alignment67,68. In another example, APC/C inhibition using proTAME prevents the APC/C from loading its activator CDC20 and prolongs metaphase for hours, but only ~20 minutes in MAD2-knockdown cells. This finding indicates cooperative APC/C inhibition by proTAME and SAC activity, suggesting a positive feedback loop between SAC silencing and APC/C activation69. Another inhibitor, apcin, blocks APC/CCDC20 binding to its substrates, and further prolongs the metaphase arrest in combination with proTAME70. When cells are arrested with high SAC activity by the MCC inhibiting APC/C, however, apcin can paradoxically induce mitotic slippage by outcompeting the MCC71. These examples illustrate the use of small molecule inhibitors to test interactions between a target and its regulating factors.
Chemically induced dimerization has been used to probe SAC signaling in greater detail. For example, MAD1, MAD2, MPS1, and several other checkpoint proteins localize to unattached kinetochores, raising the question of which are sufficient for APC/C inhibition. MAD1 recruitment to kinetochores in human cells at metaphase, after the checkpoint has been silenced, reactivated the SAC without an increase in kinetochore MPS1, highlighting the importance of MAD1 localization72,73. Furthermore, as several SAC kinases can phosphorylate themselves (autophosphorylation), dimerization strategies can manipulate this reaction. For instance, chemical dimerization, using AP20187 to dimerize two FKBPF36V domains74, enhances MPS1 kinase activity, suggesting that recruitment to kinetochores activates the kinase by increasing local concentration75. Another question is how the SAC senses microtubule attachments, so that it is selectively activated by unattached kinetochores and silenced at each kinetochore by end-on microtubule binding. To determine whether this process depends on the spatial relationship between signaling components, MPS1 and its protein substrate were recruited to varying positions in budding yeast kinetochores76 (Figure 5b–c). Recruiting MPS1 to the inner kinetochore successfully activated the SAC, whereas recruitment to the outer kinetochore (farther from the centromeric nucleosome) did not. These findings indicate that physical proximity between MPS1 and inner kinetochore components activates the SAC on unattached kinetochores. Consistent with this interpretation, the SAC was also activated by recruiting the key MPS1 substrate, a domain of the kinetochore protein SPC105 (mammalian KNL1 ortholog), to the outer-kinetochore. Together, these findings suggest a model in which a physical barrier blocks the interaction between MPS1 and SPC105 upon microtubule attachment, silencing the SAC by preventing SPC105 phosphorylation. A likely candidate for this barrier is the DAM1 complex, which localizes to yeast kinetochores after end-on microtubule binding and is positioned in between MPS1 and SPC105. Overall, these experiments illustrate applications of rapamycin-induced dimerization to multiple proteins in different model systems, and the ability of this approach to recapitulate physiological features by ectopic protein recruitment to probe a complex biological process.
Leading technology and optogenetics
In this section, we appraise several unexplored strategies for cell division studies. We anticipate that chemically induced dimerization and optogenetics can further broaden the applicability of these strategies by providing spatiotemporal control. For example, chemical probes that induce proteasome-assisted proteolysis have been developed to regulate intracellular protein levels by modifying the targeted proteins with multiple ubiquitin proteins on surface-accessible lysine residues. This “ubiquitination” process consists of a series of reactions catalyzed by multiple enzymes, including an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin ligase. To target a protein for degradation, one approach is to recruit such ubiquitination systems proximal to the target. Proteolysis-targeting chimeras (PROTACs), in which a designed bifunctional probe bridges ubiquitin ligases and the target to induce degradation, provide a complementary strategy to small-molecule inhibition77. However, the slow degradation rate of PROTACs (~hours) limits the application for studying more rapid mitotic processes. To resolve this temporal limitation, one solution is to genetically fuse the target protein to a peptide that is recognized by the ubiquitination system as a “degron”. The auxin-inducible degron (AID) system78 provides efficient degradation and high specificity by directly recruiting an E3 ligase (the SCFTIR1 complex) to an AID peptide upon addition of auxin/IAA (Figure 6a). The targeted protein can be degraded within minutes of auxin addition, as shown for a variety of proteins involved in cell division79–81. This promising degradation activity supports assays requiring acute depletion of target proteins, and is especially useful for those lacking known inhibitors. For example, this strategy was applied to target PLK4 before the discovery of centrinone82. Furthermore, because the AID strategy depletes the target protein whereas enzymatic inhibitors do not, phenotypic comparison can uncover non-enzymatic roles of the target protein. However, the AID system lacks spatial control. As an alternative, a photo-sensitive degron system adopts optogenetics to spatially control degron activity, and can degrade target proteins within an hour83,84 (Figure 6b). Using a similar strategy, the AID degron could be engineered to gain spatial control by adding a photocage moiety to auxin derivatives. This chemical approach has the potential advantage of requiring genetic fusion of only a peptide degron rather than a larger light-responsive protein, which can cause steric constraints for large protein complexes.
Figure 6. Prospective strategies for modulating enzymatic activities.

(a) Inducible protein degradation. Auxin/IAA addition recruits AID-tagged target proteins to the AID-recognition component SCFTIR1, which catalyzes poly-ubiquitination to degrade the target. (b) Photo-sensitive degron. Light triggers a conformational change in the LOV2 domain, which exposes the degron to recruit E3 ligases and degrade the target. (c) Recognition-inhibition strategy. A low-affinity inhibitory protein displaces the original high-affinity substrate by increasing its local concentration by recruitment to the target. Possible candidates for inhibitory proteins include user-designed nanobodies or monobodies. (d) Local kinase inhibition (LoKI) strategy. Kinase inhibitors conjugated with the SNAP-tag ligand CLP can be locally concentrated proximal to SNAP-tagged proteins. In this example, by fusing a SNAP tag to the centrosome-localized PACT domain98, inhibitors are concentrated at spindle poles. (e) Droplet-assisted recruitment. A peptide induces droplet formation upon surpassing its critical concentration, thereby increasing the recruitment of effectors. (f) Hook effect of chemically induced dimerization. Scenarios of limiting or excessive dimerizers reduce dimerization efficacy.
Another limitation of degron systems is that degradation efficacy relies on high activity of the degron and low expression level of the target protein. Compared to degrons, more acute inhibition can be achieved by directly blocking substrate accessibility using synthetic proteins or substrate-mimetic molecules (nanobodies or monobodies, Figure 6c). This strategy is especially suitable for highly expressed targets. For instance, designed monobodies rapidly block Aurora A kinase binding to its activator TPX2 and thereby reduce Aurora A activity in vitro85. However, this method offers limited intracellular control, as monobodies block their targets immediately after expression. One potential strategy is to combine a low-affinity monobody with other manipulation tools to tune substrate affinity or local concentrations. For instance, light-responsive optobodies have been engineered from a pair of split nanobody fragments with low target affinity, fused to optogenetic dimerization domains. Light exposure immediately increase inhibition affinity by an order-of-magnitude86 and can provide spatial control. Increasing local concentrations by recruiting low-affinity monobodies can also allow the monobody to outcompete the original substrate. Here, chemically induced dimerizers are candidates for tuning substrate affinity or local concentrations.
Similarly, enzymatic inhibitors can be linked to protein anchors to gain spatial control. One strategy, termed Local Kinase Inhibition (LoKI), is to genetically fuse the protein anchor to a SNAP tag, and conjugate a kinase inhibitor with the SNAP-binding ligand chloropyrimidine (CLP) to locally concentrate inhibitors around SNAP-tagged anchors87 (Figure 6d). In cells expressing an anchor localized to kinetochores or spindle poles, the inhibitor conjugate successfully targeted local Aurora A or Polo-like kinase 1 activity. Kinetochore-localized Aurora A inhibition generated more unphosphorylated kinetochores than centrosome-localized inhibition, supporting the idea of a kinetochore-localized pool of Aurora A88. LoKI provides a powerful approach to dissect localized kinase functions during cell division, with the possibility of incorporating light-sensitive moieties for improved temporal control.
Finally, we propose a strategy to improve the magnitude of recruitment for chemically induced dimerization studies. Tandem repeats of dimerization tags or effector proteins have been used successfully7,11,58,76,87,89,90, but these genetic manipulations have limitations associated with increased protein size. Alternatively, peptides inducing liquid droplet formation could be used to enrich recruitment. These peptides commonly consist of multivalent domains forming low-affinity interactions, which enable binding to multiple partners at critical concentrations91. Manipulating the local concentration of the peptide around its critical concentration can thus efficiently modulate droplet formation using optogenetics92,93 or chemically induced dimerization94. Cargo fused to these peptides can thus recruit additional cargos to enrich recruitment after surpassing the critical concentration (Figure 6e). Moreover, multiple repeats of droplet-inducing peptides are structurally compatible with most proteins, as these peptides are typically short and disordered.
Closing remarks
The studies described in this review demonstrate how small molecule probes have been designed and applied to address biological questions related to cell division. Enzymatic inhibitors provide several advantages over conventional genetic manipulations: applicability across multiple model systems, temporal control, and in many cases reversibility after inhibitor washout. Dimerization-inducing probes provide another level of control over genetically tagged proteins, in some cases with light-responsive modifications for spatial and temporal precision that is especially applicable to rapid and localized processes in cell division. Furthermore, chemical and genetic perturbations can work together to modulate multiple targets at the same time, as these modulations are typically bio-orthogonal.
Although small-molecule probes can provide clear advantages, there are also significant limitations. Designing enzymatic inhibitors with high specificity remains a challenging problem for structural and chemical biologists. One way to minimize potential off-target effects is to limit the dose and timing of inhibitor addition using assays focused on a narrowly defined biological process. Furthermore, two orthogonal approaches targeting the same enzyme or process are advantageous, as their potential artifacts are likely to be different. Chemically induced dimerization strategies avoid some drawbacks of enzymatic inhibitors, as their designed ligands are often bioorthogonal to the chosen model system. However, dimerization can cause steric hindrance that disrupts the functions of the anchor or the recruited effector. Structural orientation, interaction range of the recruited protein, and linker lengths can be considered in the design, although optimization often relies on trial-and-error. Moreover, chemical dimerizers often show a “hook effect” with a three-body system: reduced recruitment efficacy at high concentrations of probes95 (Figure 6f). The excessive bridging probes outnumber the two proteins, so that the two proteins bind to different population of probes and never come together. The hook effect can be avoided using “molecular glue” probes, such as rapamycin96,97, or caged molecules6. Molecular glue probes have a strong binding cooperativity when the two proteins dimerize, whereas their binding affinities to the individual proteins are weak. With caged molecules, a wash-in step can saturate the first receptor, followed by a washout step to remove unbound molecules, thus avoiding the hook effect. Understanding the advantages and limitations of various approaches will help bridge the gap between chemical probe design and experimental needs for biological studies.
Acknowledgments
The authors thank Dr. Damian Dudka for commenting on this article. This article is supported by the National Institutes of Health (GM-122475), National Cancer Institute (U54-CA193417), the Emerson Collective Cancer Research Fund, and the Penn Center for Genome Integrity.
References
- 1.Levine MS & Holland AJ The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 32, 620–638 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sansregret L, Vanhaesebroeck B & Swanton C Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol 15, 139–50 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Leibowitz ML, Zhang C-Z & Pellman D Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu. Rev. Genet 49, 183–211 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Knouse KA, Davoli T, Elledge SJ & Amon A Aneuploidy in Cancer: Seq-ing Answers to Old Questions. Annu. Rev. Cancer Biol 1, 335–54 (2017). [Google Scholar]
- 5.Choi J, Chen J, Schreiber SL & Clardy J Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 273, 239–242 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA & Chenoweth DM Localized light-induced protein dimerization in living cells using a photocaged dimerizer. Nat. Commun 5, 5475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H et al. Optogenetic control of kinetochore function. Nat. Chem. Biol 13, 1096–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports photo-caged and photo-cleavable chemical optogenetic probes and their use for kinetochore studies. For example, recruitment of the motor protein CENP-E to kinetochores near spindle poles demonstrates selective transport towards the spindle equator.
- 8.Aonbangkhen C, Zhang H, Wu DZ, Lampson MA & Chenoweth DM Reversible Control of Protein Localization in Living Cells Using a Photocaged-Photocleavable Chemical Dimerizer. J. Am. Chem. Soc 140, 11926–11930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rost BR, Schneider-Warme F, Schmitz D & Hegemann P Optogenetic Tools for Subcellular Applications in Neuroscience. Neuron 96, 572–603 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Ballister ER, Ayloo S, Chenoweth DM, Lampson MA & Holzbaur ELF Optogenetic control of organelle transport using a photocaged chemical inducer of dimerization. Curr. Biol 25, R407–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G-Y et al. Tension promotes kinetochore-microtubule release by Aurora B kinase. J. Cell Biol (2021). doi: 10.1083/jcb.202007030 [DOI] [PMC free article] [PubMed] [Google Scholar]; Using chemical optogenetics to recruit Aurora B to an individual kinetochore, this report demonstrates that tension dictates contrasting chromosome error-correction pathways: microtubules release at high tension or depolymerize at low tension. Furthermore, decreased inter-kinetochore tension using rigor inhibitors of mitotic kinesin motors slows microtubule release.
- 12.Nigg EA & Stearns T The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol 13, 1154–1160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heald R & Khodjakov A Thirty years of search and capture: The complex simplicity of mitotic spindle assembly. J. Cell Biol 211, 1103–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkham M, Müller-Reichert T, Oegema K, Grill S & Hyman AA SAS-4 is a C. elegans centriolar protein that controls centrosome size. Cell 112, 575–587 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Habedanck R, Stierhof YD, Wilkinson CJ & Nigg EA The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol 7, 1140–1146 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Bettencourt-Dias M et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol 15, 2199–2207 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Aydogan MG et al. An Autonomous Oscillation Times and Executes Centriole Biogenesis. Cell 181, 1566–1581.e27 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong YL et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 348, 1155–1160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports development of the PLK4 inhibitor centrinone and applications in depleting centrosomes. Compared to other approaches, centrinone offers advantages such as acute inhibition, reversibility, and applicability across different model systems.
- 19.Dudka D, Castrogiovanni C, Liaudet N, Vassal H & Meraldi P Spindle-Length-Dependent HURP Localization Allows Centrosomes to Control Kinetochore-Fiber Plus-End Dynamics. Curr. Biol 29, 3563–3578.e6 (2019). [DOI] [PubMed] [Google Scholar]; This study used centrinone to deplete a centrosome and generate an asymmetric mitotic spindle. Comparing the spindle-bound molecules on the two halves of the spindle revealed an increased in the microtubule stabilizer, HURP, on the shorter half-spindle.
- 20.Guild J, Ginzberg MB, Hueschen CL, Mitchison TJ & Dumont S Increased lateral microtubule contact at the cell cortex is sufficient to drive mammalian spindle elongation. Mol. Biol. Cell 28, 1975–1983 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganem NJ, Godinho SA & Pellman D A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silkworth WT, Nardi IK, Scholl LM & Cimini D Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 4, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeow ZY et al. Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature 585, 447–452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meitinger F et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 585, 440–446 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambrus BG et al. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J. Cell Biol 214, 143–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meitinger F et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J. Cell Biol 214, 155–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fong CS et al. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 5, 1–18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magidson V, Lončarek J, Hergert P, Rieder CL & Khodjakov A Laser Microsurgery in the GFP Era: A Cell Biologist’s Perspective. Methods Cell Biol. 82, 239–66 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapitein LC et al. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature 435, 114–8 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Uteng M, Hentrich C, Miura K, Bieling P & Surrey T Poleward transport of Eg5 by dynein-dynactin in Xenopus laevis egg extract spindles. J. Cell Biol 182, 715–726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimamoto Y, Forth S & Kapoor TM Measuring Pushing and Braking Forces Generated by Ensembles of Kinesin-5 Crosslinking Two Microtubules. Dev. Cell 34, 669–681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturgill EG, Norris SR, Guo Y & Ohi R Kinesin-5 inhibitor resistance is driven by kinesin-12. J. Cell Biol 213, jcb.201507036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sturgill EG & Ohi R Kinesin-12 differentially affects spindle assembly depending on its microtubule substrate. Curr. Biol 23, 1280–1290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drechsler H, McHugh T, Singleton MR, Carter NJ & McAinsh AD The Kinesin-12 Kif15 is a processive track-switching tetramer. Elife 3, e01724 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapoor TM et al. Chromosomes can congress to the metaphase plate before biorientation. Science. 311, 388–91 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ingber DE Tensegrity I. Cell structure and hierarchical systems biology. J. Cell Sci 116, 1157–73 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Nicklas RB How cells get the right chromosomes. Science. 275, 632–7 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Needleman DJ et al. Fast Microtubule Dynamics in Meiotic Spindles Measured by Single Molecule Imaging: Evidence That the Spindle Environment Does Not Stabilize Microtubules. Mol. Biol. Cell 21, 323–333 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elting MW, Prakash M, Udy DB & Dumont S Mapping Load-Bearing in the Mammalian Spindle Reveals Local Kinetochore Fiber Anchorage that Provides Mechanical Isolation and Redundancy. Curr. Biol 27, 2112–2122.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groen AC et al. A novel small-molecule inhibitor reveals a possible role of kinesin-5 in anastral spindle-pole assembly. J. Cell Sci 121, 2293–300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vladimirou E et al. Nonautonomous Movement of Chromosomes in Mitosis. Dev. Cell 27, 411–424 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Begley MA et al. K-fiber bundles in the mitotic spindle are mechanically reinforced by Kif15. BioRxiv 10.1101/2020.05.19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wood KW et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl. Acad. Sci 107, 5839–5844 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports the CENP-E rigor inhibitor GSK923925 and its use to prevent chromosome congression and activate the SAC.
- 44.Yao X, Abrieu A, Zheng Y, Sullivan KF & Cleveland DW CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol 2, 484–491 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Tanudji M et al. Gene silencing of CENP-E by small interfering RNA in HeLa cells leads to missegregation of chromosomes after a mitotic delay. Mol. Biol. Cell 15, 3771–81 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barisic M et al. Microtubule detyrosination guides chromosomes during mitosis. Science. 348, 799–803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barisic M & Maiato H The Tubulin Code: A Navigation System for Chromosomes during Mitosis. Trends Cell Biol. 26, 766–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarangapani KK & Asbury CL Catch and release: how do kinetochores hook the right microtubules during mitosis? Trends Genet. 30, 150–159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lampson MA & Grishchuk EL Mechanisms to Avoid and Correct Erroneous Kinetochore-Microtubule Attachments. Biol. 6, 1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broad AJ & Deluca JG The right place at the right time: Aurora B kinase localization to centromeres and kinetochores. Essays Biochem. EBC20190081 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broad AJ, DeLuca KF & DeLuca JG Aurora B kinase is recruited to multiple discrete kinetochore and centromere regions in human cells. J. Cell Biol 219, e201905144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hadders MA et al. Untangling the contribution of Haspin and Bub1 to Aurora B function during mitosis. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F et al. Haspin inhibitors reveal centromeric functions of Aurora B in chromosome segregation. J. Cell Biol 199, 251–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Antoni A, Maffini S, Knapp S, Musacchio A & Santaguida S A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J. Cell Biol 199, 269–284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang C et al. Centromere-localized Aurora B kinase is required for the fidelity of chromosome segregation. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eswaran J et al. Structure and functional characterization of the atypical human kinase haspin. Proc. Natl. Acad. Sci 106, 20198–20203 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoo TY et al. Measuring NDC80 binding reveals the molecular basis of tension-dependent kinetochore-microtubule attachments. Elife 7, 1–34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; A method using fluorescence lifetime microscopy and Förster resonance energy transfer was developed to measure binding between the NDC80 complex and microtubules in live cells. Combining this method with 5-Iodotubercidin to inhibit haspin kinase shows that tension dependence of NDC80 binding requires centromere-localized Aurora B.
- 58.García-Rodríguez LJ, Kasciukovic T, Denninger V & Tanaka TU Aurora B-INCENP Localization at Centromeres/Inner Kinetochores Is Required for Chromosome Bi-orientation in Budding Yeast. Curr. Biol 29, 1536–1544.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; Centromeric enrichment of Aurora B using rapamycin-induced dimerization rescues chromosome segregation fidelity when other pathways for localizing IPL1 to centromeres are inhibited.
- 59.Akiyoshi B et al. Tension directly stabilizes reconstituted kinetochore-microtubule attachments. Nature 468, 576–579 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lampson MA, Renduchitala K, Khodjakov A & Kapoor TM Correcting improper chromosome – spindle attachments during cell division. Nat. Cell Biol 6, 232–237 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Ye AA et al. Aurora A Kinase Contributes to a Pole-Based Error Correction Pathway. Curr. Biol 25, 1842–1851 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chmátal L, Yang K, Schultz RM & Lampson MA Spatial Regulation of Kinetochore Microtubule Attachments by Destabilization at Spindle Poles in Meiosis I. Curr. Biol 25, 1835–1841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lara-Gonzalez P, Westhorpe FG & Taylor SS The spindle assembly checkpoint. Curr. Biol 22, R966–R980 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Musacchio A & Salmon ED The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol 8, 379–393 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Santaguida S, Tighe A, D’Alise AM, Taylor SS & Musacchio A Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol 190, 73–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dou Z et al. Dynamic localization of Mps1 kinase to kinetochores is essential for accurate spindle microtubule attachment. Proc. Natl. Acad. Sci 112, E4546–E4555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ji Z, Gao H & Yu H Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science. 348, 1260–4 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Hiruma Y et al. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science. 348, 1264–1267 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Zeng X et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18, 382–395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sackton KL et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 514, 646–649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richeson KV et al. Paradoxical mitotic exit induced by a small molecule inhibitor of APC/CCdc20. Nat. Chem. Biol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ballister ER, Riegman M & Lampson MA Recruitment of Mad1 to metaphase kinetochores is sufficient to reactivate the mitotic checkpoint. J. Cell Biol 204, 901–908 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuijt TEF, Omerzu M, Saurin AT & Kops GJPL Conditional targeting of MAD1 to kinetochores is sufficient to reactivate the spindle assembly checkpoint in metaphase. Chromosoma 123, 471–480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clackson T et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci 95, 10437–10442 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kang J, Chen Y, Zhao Y & Yu H Autophosphorylation-dependent activation of human Mps1 is required for the spindle checkpoint. Proc. Natl. Acad. Sci 104, 20232–20237 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aravamudhan P, Goldfarb AA & Joglekar AP The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat. Cell Biol 17, 868–879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using rapamycin-induced dimerization, the SAC can be re-activated by recruiting MPS1 kinase close to its substrate, SPC105. This study proposes that a physical barrier between MPS1 and SPC105 silences the SAC when kinetochores achieve end-on microtubule attachment.
- 77.Burslem GM & Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T & Kanemaki M An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 (2009). [DOI] [PubMed] [Google Scholar]
- 79.Holland AJ, Fachinetti D, Han JS & Cleveland DW Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci 109, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Natsume T, Kiyomitsu T, Saga Y & Kanemaki MT Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 15, 210–218 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Daniel K et al. Conditional control of fluorescent protein degradation by an auxin-dependent nanobody. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lambrus BG et al. P53 protects against genome instability following centriole duplication failure. J. Cell Biol 210, 63–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Renicke C, Schuster D, Usherenko S, Essen LO & Taxis C A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol 20, 619–626 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Hermann A, Liewald JF & Gottschalk A A photosensitive degron enables acute light-induced protein degradation in the nervous system. Curr. Biol 25, R749–R750 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Zorba A et al. Allosteric modulation of a human protein kinase with monobodies. Proc. Natl. Acad. Sci. 116, 13937–13942 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu D et al. Optogenetic activation of intracellular antibodies for direct modulation of endogenous proteins. Nat. Methods 16, 1095–1100 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Bucko PJ et al. Subcellular drug targeting illuminates local kinase action. Elife 8, e52220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports a strategy to inhibit kinase activity locally by conjugating kinase inhibitors to the SNAP-binding ligand, CLP, and fusing a SNAP-tag to proteins that localize to specific intracellular structure. The bifunctional probe inhibits kinase activity proximal to the SNAP-tagged proteins.
- 88.DeLuca KF et al. Aurora A kinase phosphorylates Hec1 to regulate metaphase kinetochore-microtubule dynamics. J. Cell Biol 217, 163–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aravamudhan P, Chen R, Roy B, Sim J & Joglekar AP Dual mechanisms regulate the recruitment of spindle assembly checkpoint proteins to the budding yeast kinetochore. Mol. Biol. Cell 27, 3405–3417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen C et al. Ectopic Activation of the Spindle Assembly Checkpoint Signaling Cascade Reveals Its Biochemical Design. Curr. Biol 29, 104–119.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hyman AA, Weber CA & Jülicher F Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol 30, 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Shin Y et al. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bracha D et al. Mapping Local and Global Liquid Phase Behavior in Living Cells Using Photo-Oligomerizable Seeds. Cell 175, 1467–1480.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang H et al. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 31, 2048–2056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Douglass EF, Miller CJ, Sparer G, Shapiro H & Spiegel DA A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc 135, 6092–6099 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schreiber SL Immunophilin-sensitive protein phosphatase action in cell signaling pathways. Cell 70, 365–368 (1992). [DOI] [PubMed] [Google Scholar]
- 97.Banaszynski LA, Liu CW & Wandless TJ Characterization of the FKBP-rapamycin-FRB ternary complex. J. Am. Chem. Soc 127, 4715–4721 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Gillingham AK & Munro S The PACT domain, a conserved centrosomal targeting motif in the coiled-coil proteins AKAP450 and pericentrin. EMBO Rep. 1, 524–529 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]