

Abstract
Summary
PyHMMER provides Python integration of the popular profile Hidden Markov Model software HMMER via Cython bindings. This allows the annotation of protein sequences with profile HMMs and building new ones directly with Python. PyHMMER increases flexibility of use, allowing creating queries directly from Python code, launching searches, and obtaining results without I/O, or accessing previously unavailable statistics like uncorrected P-values. A new parallelization model greatly improves performance when running multithreaded searches, while producing the exact same results as HMMER.
Availability and implementation
PyHMMER supports all modern Python versions (Python 3.6+) and similar platforms as HMMER (x86 or PowerPC UNIX systems). Pre-compiled packages are released via PyPI (https://pypi.org/project/pyhmmer/) and Bioconda (https://anaconda.org/bioconda/pyhmmer). The PyHMMER source code is available under the terms of the open-source MIT licence and hosted on GitHub (https://github.com/althonos/pyhmmer); its documentation is available on ReadTheDocs (https://pyhmmer.readthedocs.io).
1 Introduction
Protein similarity search and annotation is a key part of biological sequence analysis, allowing for the discovery of protein function from sequence data. Improving on Position-Specific Scoring Matrices that were used historically to search for motifs, Profile Hidden Markov Models (pHMMs) were introduced in HMMER (Eddy 2011) to model protein sequence families. Since then, HMMER has become a de facto standard for protein domain annotation, with several protein domain databases such as Pfam (Mistry et al. 2021) or TIGRFAM (Haft et al. 2013) being distributed as pHMMs.
HMMER provides different utilities with a command line interface (CLI), and only offers an application programming interface (API) for the C language (Kernighan and Ritchie 1978). This makes it rather cumbersome to use with modern languages such as Python, which are now more popular among scientists (Perkel 2015). Here we describe PyHMMER, a library providing Python bindings to the C API of HMMER using Cython (Behnel et al. 2011), allowing seamless integration of HMMER in larger Python programs, or in Jupyter notebooks (Kluyver et al. 2016). Such literate programming efforts help to address the reproducibility crisis, allowing for exact versioning and reproducible scripts for pHMM generation, among other recommended practices (Rule et al. 2019).
2 Implementation
The original HMMER codebase is organized into several components: a general purpose library for biological sequence manipulation named easel, a core library specific to HMMER named libhmmer, and dedicated CLI tools for building HMMs and performing sequence searches (hmmbuild, hmmsearch, phmmer, etc.).
The Cython language, a superset of Python that can be compiled into C or C++ extension modules, allows defining foreign function interfaces to the library components of HMMER. Using this particular feature, we developed the pyhmmer.easel and pyhmmer.plan7 extension modules to wrap the most relevant types from the easel and libhmmer libraries, respectively. In both submodules these types are exposed as Python classes, allowing the user to create and manipulate a Sequence or an HMM object directly (Listing 1).
Listing 1.
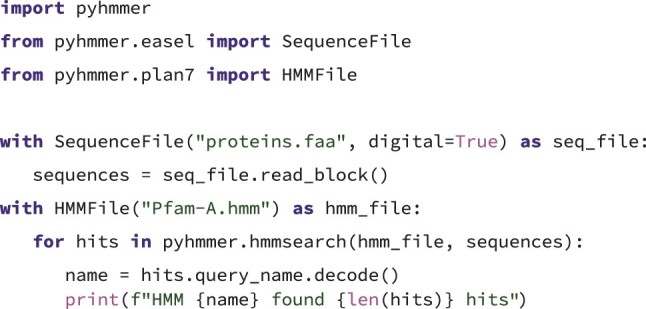
A Python code snippet demonstrating how to run hmmsearch with the PyHMMER API. The target sequences are pre-fetched from the proteins.faa file before running the search loop while the query HMMs are loaded iteratively from Pfam-A.hmm. More code examples can be found at https://pyhmmer.readthedocs.io/en/latest/examples/.
Classes in PyHMMER implement the relevant methods from the Python data model: containers like TopHits or Domains support the usual Python syntax for iterating or indexing, while file readers support the context manager protocol to be used inside a with statement and the iteration protocol to read the file content with a simple for loop. Subclassing is supported to allow end-users to implement additional functionalities if needed. Numeric collections, such as VectorF which wraps a one-dimensional array of floating point numbers, all implement the buffer protocol. They can be used seamlessly with the entire NumPy ecosystem (Harris et al. 2020), using the numpy.asarray function to wrap them into an ndarray without copying the underlying data. Type hints are provided for all public classes and functions, allowing a static analyzer such as MyPy (https://mypy-lang.org) to detect type errors ahead of runtime. These type annotations also make PyHMMER more pleasant to use inside an Integrated Development Environment (IDE), where the function signatures can be suggested and corrected automatically.
For sensible types, the corresponding Python class also exposes internal attributes through Python properties that can be used for introspection. This includes some attributes that were not originally available in the HMMER results, such as the uncorrected p-value for each alignment (HMMER would only report the Bonferroni-corrected P-values in the output tables). HMM objects can be modified manually, for instance to set bitscore cutoffs using an externally computed threshold.
3 Results
The functionality of several of the CLI tools from HMMER was rewritten as pure Python functions using the API from the pyhmmer.easel and pyhmmer.plan7 modules.
Computational efficiency of the new parallelization code was benchmarked against the original implementation for the hmmsearch task (Fig. 1b–d). It shows much better performance for smaller target databases such as the proteome of a single microbial species, which can be entirely loaded into memory before querying. For extreme cases where none of the HMM or sequence databases fit into memory, a fallback using file readers is implemented to support larger searches at the cost of I/O overhead.
Figure 1.
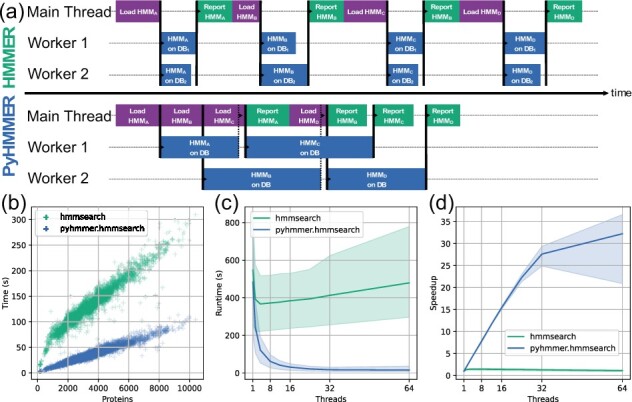
Comparison of the parallelization strategy for running hmmsearch. (a) A summary of the job dispatching between the HMMER and the PyHMMER implementations of hmmsearch. HMMER breaks the target database into chunks and synchronizes the queries; PyHMMER runs multiple queries in parallel threads while using the main thread for I/O. Blocking and non-blocking inter-thread communications are shown in plain and dashed lines, respectively. (b) Runtime comparison of hmmsearch annotation with six threads. Proteins were obtained from representative microbial genomes contained in the proGenomes2 database (Mende et al. 2020), and annotated with Pfam version 33.1 (Mistry et al. 2021) grouped by genome. Each command was run once on an Intel i7-10710U processor with six physical cores. (c) Average runtime and (d) speedup of hmmsearch implemented in PyHMMER compared to HMMER. Proteins of 10 different species were downloaded from proGenomes and annotated with Pfam version 35.0. Shown are the averages of three runs on an Intel Xeon E5-2683 processor with 16 physical cores. More benchmarks can be found at https://pyhmmer.readthedocs.io/en/latest/benchmarks.html.
The parallel section in particular was reimplemented with plain Python threads and a different model for balancing load across worker threads (Fig. 1a), the default number of which is chosen as the number of physical cores to avoid resource contention. The hmmpgmdb client was adapted to use Python sockets for network communication, although message encoding and decoding uses the HMMER code for consistency.
The new threading strategy reduces latency caused by the filesystem as it introduces pre-fetching of query HMMs from the HMM file while the worker threads run the one-to-many comparison pipeline. Modeling the speedup with Amdahl’s law (Amdahl 1967) suggests that the original hmmsearch task is not taking full advantage of multi-core machines, with only around 35% of the code being truly parallel (Fig. 1d). In comparison, PyHMMER has ∼96% of the code in parallel sections. In practice, using PyHMMER to annotate a large protein set on a six-core machine reduced the runtime by 72%, totaling only 27 h where HMMER took 97 h (Fig. 1b). Similarly, hmmscan tasks benefit from the PyHMMER parallelization strategy, which makes it possible to annotate 1 million proteins with 32 threads within ∼2.5 h compared to an estimate of >7 days the original HMMER implementation would take.
At the time of writing, PyHMMER has already been integrated into several projects covering various areas of bioinformatics, including long-read transcriptome sequencing (Lienhard et al. 2021); average amino-acid identity estimation (Konstantinidis et al. 2022) or clustering of biosynthetic gene clusters (Kautsar et al. 2021).
Acknowledgements
The authors thank Sean R. Eddy and his team for the development of HMMER, which features outstanding documentation and quality standards; Rob Finn and Nicolo Lazzaro for their invitation to work on the jackhmmer and hmmpgmdb re-implementations; Antonio P. Camargo and Felix Langer for their suggestions during the early stage of development; Troy Sincomb for fixing errors in the documentation; Valentyn Bezshapkin and Zachary Kurtz for their contributions; and all individuals who reported issues on the GitHub tracker.
Contributor Information
Martin Larralde, Structural and Computational Biology Unit, EMBL, Meyerhofstraße 1, Heidelberg 69117, Germany.
Georg Zeller, Structural and Computational Biology Unit, EMBL, Meyerhofstraße 1, Heidelberg 69117, Germany.
Conflict of interest
None declared.
Funding
This work was supported by the European Molecular Biology Laboratory; the SFB 1371 of the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) [395357507] and the Federal Ministry of Education and Research (BMBF) [031L0181A].
Data availability
The data underlying this article are available on GitHub at https://github.com/althonos/pyhmmer. The datasets for benchmarking were derived from sources free for academic and non-commercial use: proGenomes 3 (https://progenomes.embl.de), Pfam 33.1 (https://www.ebi.ac.uk/interpro/).
References
- Amdahl GM. Validity of the single processor approach to achieving large scale computing capabilities. In: Proceedings of the April 18–20, 1967, Spring Joint Computer Conference, AFIPS ’67 (Spring). New York, NY: Association for Computing Machinery, 1967, 483–5.
- Behnel S, Bradshaw R, Citro C. et al. Cython: the best of both worlds. Comput Sci Eng 2011;13:31–9. 10.1109/MCSE.2010.118. [DOI] [Google Scholar]
- Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol 2011;7:e1002195. 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, Richter RA. et al. TIGRFAMs and genome properties in 2013. Nucleic Acids Res 2013;41:D387–95. 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ. et al. Array programming with NumPy. Nature 2020;585:357–62. 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautsar SA, van der Hooft JJJ, de Ridder D. et al. BiG-SLiCE: a highly scalable tool maps the diversity of 1.2 million biosynthetic gene clusters. GigaScience 2021;10:giaa154. 10.1093/gigascience/giaa154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernighan BW, Ritchie DM.. The C Programming Language. Englewood Cliffs, NJ: Prentice-Hall, 1978. [Google Scholar]
- Kluyver T, Ragan-Kelley B, Perez F. et al. Jupyter Notebooks—A Publishing Format for Reproducible Computational Workflows. In: Loizides F, Schmidt B (eds.), Positioning and Power in Academic Publishing: Players, Agents and Agendas. Amsterdam, The Netherlands: IOS Press, 2016, 87–90. [Google Scholar]
- Konstantinidis K, Ruiz-Perez C, Gerhardt K. et al. FastAAI: efficient estimation of genome average amino acid identity and phylum-level relationships using tetramers of universal protein. Research Square 2022. 10.21203/rs.3.rs-1459378/v1. [DOI]
- Lienhard M, Van den Beucken T, Claiment F. et al. Long-read transcriptome sequencing analysis with IsoTools Genetics. bioRxiv 2021. 10.1101/2021.07.13.452091. [DOI] [PMC free article] [PubMed]
- Mende DR, Letunic I, Maistrenko OM. et al. proGenomes2: an improved database for accurate and consistent habitat, taxonomic and functional annotations of prokaryotic genomes. Nucleic Acids Res 2020;48:D621–5. 10.1093/nar/gkz1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J, Chuguransky S, Williams L. et al. Pfam: the protein families database in 2021. Nucleic Acids Res 2021;49:D412–9. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkel JM. Programming: pick up Python. Nature 2015;518:125–6. 10.1038/518125a. [DOI] [PubMed] [Google Scholar]
- Rule A, Birmingham A, Zuniga C. et al. Ten simple rules for writing and sharing computational analyses in Jupyter Notebooks. PLoS Comput Biol 2019;15:e1007007. 10.1371/journal.pcbi.1007007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data underlying this article are available on GitHub at https://github.com/althonos/pyhmmer. The datasets for benchmarking were derived from sources free for academic and non-commercial use: proGenomes 3 (https://progenomes.embl.de), Pfam 33.1 (https://www.ebi.ac.uk/interpro/).