

Abstract
T cells and natural killer (NK) cells have complementary roles in tumor immunity, and dual T cell and NK cell attack thus offers opportunities to deepen the impact of immunotherapy. Recent work has also shown that NK cells play an important role in recruiting dendritic cells to tumors and thus enhance induction of CD8 T cell responses, while IL-2 secreted by T cells activates NK cells. Targeting of immune evasion mechanisms from the activating NKG2D receptor and its MICA and MICB ligands on tumor cells offers opportunities for therapeutic intervention. Interestingly, T cells and NK cells share several important inhibitory and activating receptors that can be targeted to enhance T cell– and NK cell–mediated immunity. These inhibitory receptor-ligand systems include CD161-CLEC2D, TIGITCD155, and NKG2A/CD94-HLA-E. We also discuss emerging therapeutic strategies based on inhibitory and activating cytokines that profoundly impact the function of both lymphocyte populations within tumors.
Keywords: T cells, natural killer cells, cancer immunotherapy
1. TUMOR HETEROGENEITY AS A CHALLENGE FOR CANCER IMMUNOTHERAPY
Cancer cell heterogeneity is one of the major challenges for every approach to cancer therapy, including immunotherapy. Heterogeneity can arise by acquisition of new mutations resulting in the emergence of therapy-resistant tumor subclones (1, 2). A second dimension of heterogeneity is less appreciated, the ability of cancer cells to adapt to therapeutic pressure by shifting their cellular state, such as a shift to a quiescent state in response to drugs that kill rapidly proliferating cells (3, 4). Thus, researchers must consider which cancer cell mutations and adaptations give rise to resistance to proactively design immunotherapies that prevent selection of resistant tumor cells.
Tumor-specific CD8 T cells detect and kill cancer cells by T cell receptor (TCR) recognition of tumor cell–derived peptides presented by MHC class I (MHC-I) proteins (5, 6). IFN-γ secreted by activated T cells sensitizes tumor cells to T cell attack by inducing upregulation of many genes in the MHC-I antigen presentation pathway, thus increasing the cell surface density of these TCR ligands (7). CD8 T cells can recognize target cells with a high degree of sensitivity: Even recognition of one or a few peptide-MHC ligands is sufficient for induction of T cell activation (8–10). However, this powerful recognition system is entirely dependent on continued expression of MHC-I proteins by tumor cells. Tumor cells that have lost MHC-I expression by mutational or epigenetic mechanisms thus become invisible to the TCR on CD8 T cells (11, 12).
The major hypothesis we investigate here is that a coordinated response by T cells and natural killer (NK) cells is advantageous because these lymphocyte populations use distinct recognition strategies to identify cancer cells. Consequently, NK cells can kill cancer cells that have escaped recognition by CD8 T cells. In fact, loss of MHC-I expression sensitizes tumor cells to NK cell cytotoxicity because several MHC-I proteins serve as ligands for inhibitory receptors on NK cells (13, 14). We discuss the distinct roles of T cells and NK cells in tumor immunity, the pathways that inhibit T cell and NK cell function, and therapeutic approaches that can engage both cytotoxic lymphocyte populations. NK cells also play an important role in recruiting dendritic cells (DCs) to tumors and therefore contribute to induction of T cell responses (15, 16). We embed this discussion of T cell and NK cell biology in the context of their rich cellular interactions with other immune cell populations required for protective antitumor immunity.
2. COMPLEMENTARY CONTRIBUTIONS OF T CELLS AND NK CELLS TO TUMOR IMMUNITY
The importance of T cell–mediated immunity is well documented, both in humans and many experimental animal models. Major therapeutics approved by the US Federal Drug Administration (FDA) enhance T cell–mediated immunity. These include PD-1 and CTLA-4 antibodies that block the function of major inhibitory receptors on T cells (17–19), as well as genetically engineered T cells that express tumor-specific chimeric antigen receptors (CARs) or TCRs (20, 21). The central importance of cytotoxic T cells in tumor immunity is explained by sensitive TCR recognition of tumor peptides presented by MHC-I proteins, which enables efficient induction of tumor cell apoptosis by released perforin and granzymes (5). While cytotoxic T cells certainly play a critical role in tumor immunity, it is also important to consider the major contribution of CD4 T cells. The requirement for CD4 T cells has been elegantly demonstrated in a nonimmunogenic sarcoma model: Introduction of an MHC-I-presented neoantigen for CD8 T cells into these sarcoma cells is not sufficient to confer sensitivity to PD-1 plus CTLA-4 blockade (22). Response to combination checkpoint blockade is only observed when sarcoma cells express both MHC-I and MHC-II neoantigens. CD4 T cells are required not only for initial priming of the cytotoxic state of CD8 T cells but also in the tumor microenvironment (TME), where they reprogram inhibitory myeloid cell populations to a more immunostimulatory state (22).
NK cells recognize tumor cells through a set of germ line–encoded activating receptors that bind to ligands upregulated on stressed cells, as discussed in detail in Section 4 (23, 24). The majority of circulating human NK cells are in a poised cytotoxic effector state (CD16+CD56dim cells), while a smaller population (CD56highCD16−) is in a less differentiated state (25). NK cells can thus respond during an early stage of the antitumor immune response, while T cell responses develop with a delay due to the requirement for substantial clonal expansion from small numbers of antigen-reactive naive precursors (26). NK cells play a critical role in protecting against metastatic dissemination, which has been documented with a variety of experimental approaches (26, 27). For example, NK cell depletion substantially increases the number of spontaneous metastases in immunocompetent mouse models following orthotopic implantation of aggressive syngeneic tumor cells. An important role of NK cells against metastatic tumor cells has also been documented in genetically engineered mouse strains that lack expression of major NK cell activating receptors, including the NKp46 receptor (28). The important role of NK cells against metastatic disease is explained by their abundance in blood and bone marrow as well as in perivascular spaces of organs that are frequently seeded by metastatic tumor cells, such as the lung (29). In patients with different types of solid tumors, large numbers of circulating tumor cells have been identified, but only a very small fraction of these circulating tumor cells successfully establish macrometastases (30, 31).
The high frequency of NK cells in the blood and bone marrow also explains their important role in the therapy of hematological malignancies. Antibody-mediated cellular cytotoxicity (ADCC) by NK cells is an important effector mechanism for FDA-approved antibodies, such as rituximab, a CD20 monoclonal antibody (mAb) approved for the treatment of B cell malignancies, including relapsed non-Hodgkin lymphoma (32). ADCC is primarily mediated by NK cells through the activating CD16A receptor (encoded by FCGR3A) that recognizes tumor cell–bound IgG antibodies; CD16A is also expressed by macrophages (33). Genetic evidence from clinical trials directly implicates the CD16A receptor in the therapeutic activity of such antitumor mAbs. The CD16A receptor has a functionally important polymorphic residue (valine or phenylalanine at position 158) that affects the cytotoxic activity of NK cells against antibody-decorated tumor cells (34). NK cells homozygous for valine (V/V) versus phenylalanine (F/F) at position 158 have a higher affinity for human IgG1 and higher cytotoxic activity against antibody-decorated tumor cells (35). This polymorphism is clinically relevant: For example, treatment of follicular lymphoma with rituximab is associated with a higher response rate in patients with V/V compared to F/F at position 158 of CD16A (36, 37).
3. AN NK CELL—DENDRITIC CELL AXIS IN T CELL–MEDIATED TUMOR IMMUNITY
NK cells not only serve as cytotoxic effector cells but also recruit DCs into solid tumors, thereby shaping T cell–mediated tumor immunity (15, 16) (Figure 1). Conventional DCs are currently divided into two major subsets, cDC1 and cDC2 (38). The cDC1 subset is essential for protective antitumor immunity by presenting cell-associated antigens from apoptotic tumor cells to CD8 and CD4 T cells (39, 40). cDC1 can be identified by the selective expression of the C-type lectin receptor CLEC9A and the XCR1 receptor that binds the XCL1 and XCL2 chemokines (41, 42). cDC1 are particularly efficient in taking up apoptotic tumor cells and transporting these antigens to tumor-draining lymph nodes, where they can transfer antigens to other DC populations (39, 43). In addition to this trafficking role, cDC1 also play a key role within tumors by recruiting and activating tumor-specific CD8 T cells (44). Many studies have shown that protective antitumor immunity, including the efficacy of immune checkpoint blockade (ICB), is abolished in Batf3 knockout mice that lack cDC1 (40, 45–47).
Figure 1.
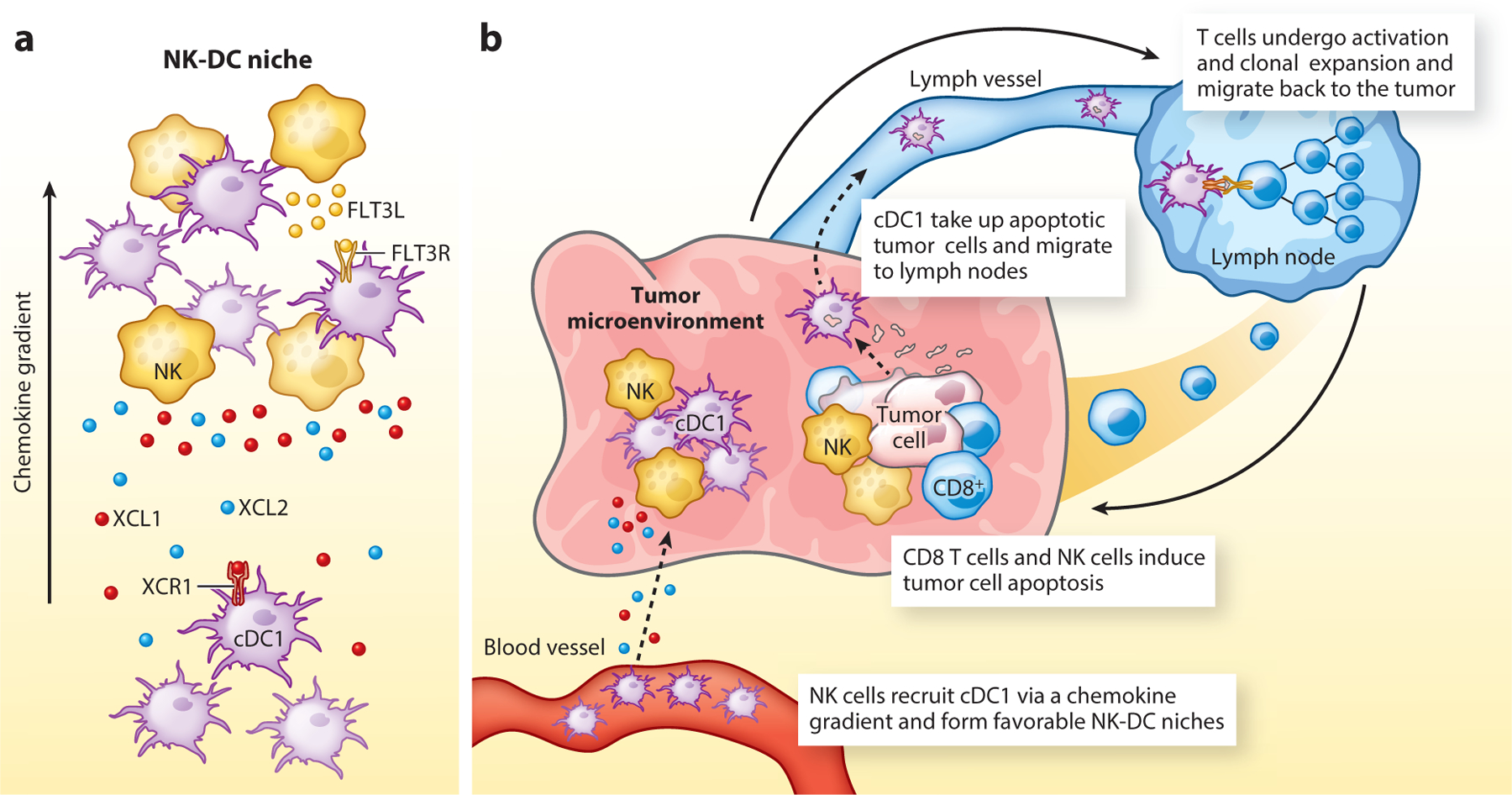
NK cells recruit dendritic cells into tumors. (a) NK cells form cellular niches with cDC1 in tumors. Cross-presenting cDC1 express the XCR1 chemokine receptor, enabling recruitment by NK cells that secrete XCL1 and XCL2. NK cells also secrete FLT3L, which induces dendritic cell differentiation. (b) cDC1 recruited by NK cells are important for priming of CD8 and CD4 T cells to cell-associated antigens. cDC1 take up apoptotic fragments from dying tumor cells and transport these antigens to tumor-draining lymph nodes, where they activate CD8 and CD4 T cells. NK cells thereby contribute to the induction of T cell–mediated immunity against tumors. Abbreviations: cDC1, type 1 conventional DC; DC, dendritic cell.
Two major studies have demonstrated that NK cells serve a critical role in recruiting cDC1 to tumors. The first study showed that NK cells secrete chemokines that mediate cDC1 recruitment into tumors, in particular XCL1, XCL2, and CCL5 (15). This is an important finding because cDC1 selectively express the XCR1 chemokine receptor for XCL1/2 (Figure 1a). Prostaglandin E2 (PGE2) strongly inhibits CCL5 and XCL1 secretion by NK cells and thereby inhibits cDC1 recruitment (48, 49). The second study focused on FLT3 ligand (FLT3LG gene), a critical cytokine that regulates DC differentiation and survival (16). In human melanomas, cDC1 infiltration correlates with FLT3LG mRNA levels, which in turn positively correlate with longer patient survival. In an FLT3 ligand reporter mouse, expression of this important cytokine is detected within NK cells. Interestingly, live cell imaging demonstrates close interactions between NK cells and cDC1 within specialized niches within tumors, and depletion of NK cells (but not T cells) greatly diminishes cDC1 recruitment to tumors (Figure 1). In human melanoma and other cancer types, cDC1 and NK cell infiltration are strongly correlated (16, 50). These studies demonstrate an important connection between NK cell and T cell responses in tumors: NK cells recruit and support the survival of DCs, which in turn are critical for T cell–mediated tumor immunity.
In solid tumors, T cells tend to be significantly more abundant than NK cells. Single-cell analyses have identified NK cells in common human cancer types, including melanoma and lung cancer (51, 52). These single-cell data have identified multiple cellular states, including a highly cytotoxic state shared with circulating NK cells and a state characterized by XCL1 and XCL2 expression (51). The latter population may be involved in recruiting cDC1 to tumors. It will be important to develop therapeutic strategies that enhance NK cell infiltration into solid tumors to enhance recruitment of DCs as well as direct targeting of tumor cells by NK cells.
4. MOLECULAR LOGIC OF TUMOR CELL RECOGNITION BY T CELLS VERSUS NK CELLS
T cells and NK cells differ fundamentally in the molecular mechanisms of tumor cell recognition. T cells identify tumor cells by TCR-mediated recognition of MHC-bound peptide antigens. All key functions of T cells are under the control of the TCR, including cytotoxicity, cytokine production, and proliferation (53). Tumors can thus escape immunity by CD8 T cells through downregulation or loss of MHC-I expression (12) (Figure 2a). In contrast, NK cells utilize an array of germ line–encoded activating and inhibitory receptors to identify stressed and transformed cells. No single receptor is essential for tumor cell recognition; rather, NK cell–mediated cytotoxicity is controlled by integration of signals from multiple surface receptors (24) (Figure 2b). The fact that none of the individual activating receptors is absolutely required for tumor cell recognition provides greater flexibility for recognition of stressed and transformed cells compared to T cells. T cells and NK cells thus exert distinct selection pressures on tumor cells, providing the rationale for dual targeting of tumors by T cells and NK cells.
Figure 2.
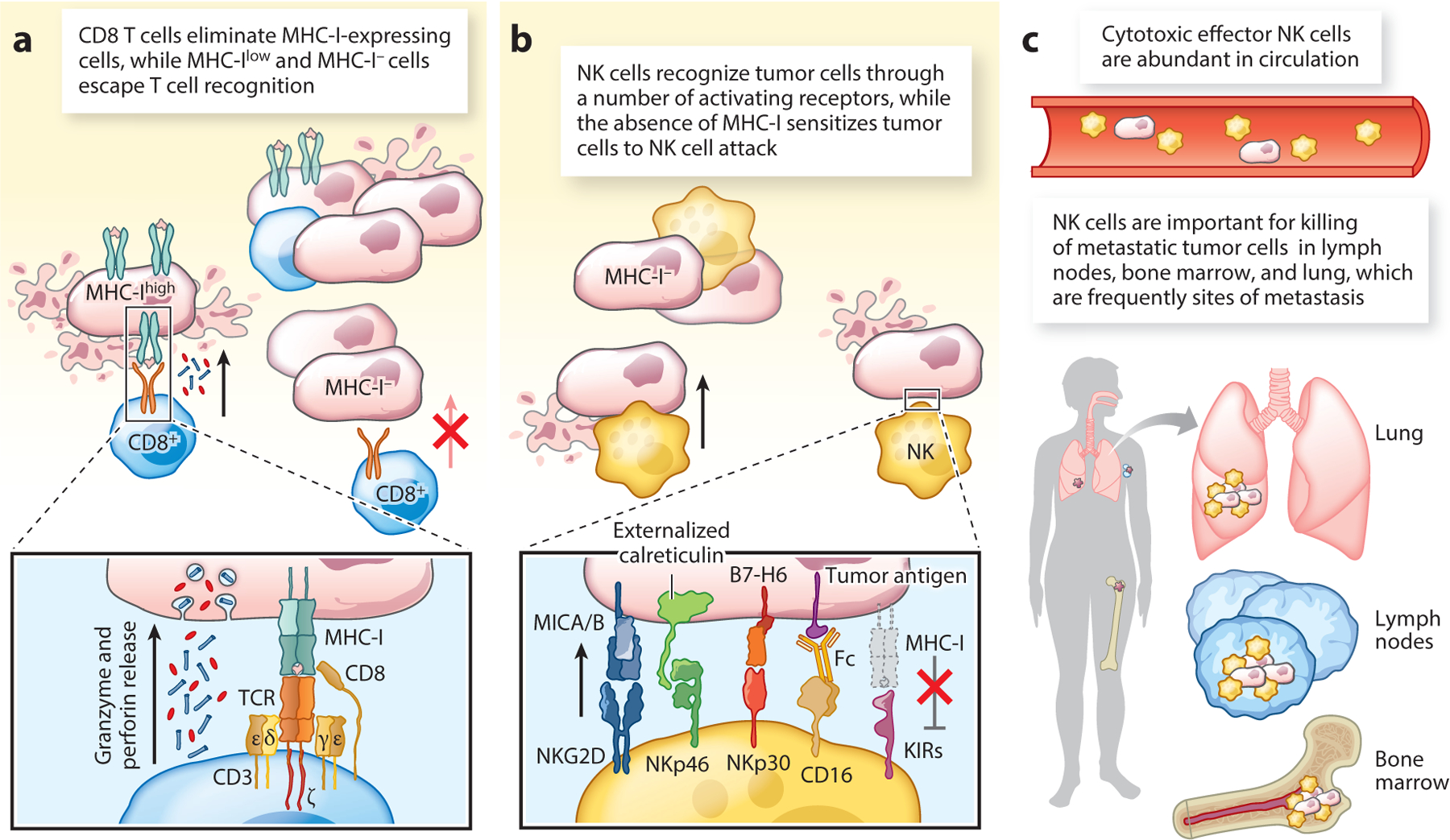
Complementary roles of CD8 T cells and NK cells in tumor immunity. (a) MHC-I proteins present tumor-derived peptides to CD8 T cells, resulting in T cell activation and release of perforin and granzymes that induce tumor cell apoptosis. CD8 T cells exert a powerful selection pressure that can result in outgrowth of MHC-I-deficient cells that are no longer recognized by CD8 T cells. (b) NK cells recognize tumor cells through a set of germ line–encoded activating receptors, including NKG2D, NKp46, NKp30, and CD16. The activity of NK cells against tumor cells is attenuated by inhibitory receptors against MHC-I proteins, including inhibitory KIRs in humans. Consequently, MHC-I-deficient tumor cells are more readily killed by NK cells. (c) Role of NK cells against metastatic tumor cells. NK cells are abundant in the blood, bone marrow and perivascular spaces of the lung, a frequent site of metastasis. The majority of circulating NK cells are in a poised cytotoxic effector state, enabling rapid killing of migrating tumor cells. Abbreviations: KIR, killer cell immunoglobulin-like receptor; TCR, T cell receptor.
TCR activation is of course modulated by a complex set of surface receptors, and these costimulatory and coinhibitory receptors regulate early T cell signaling initiated by the TCR. The CD28 costimulatory receptor plays a central role in priming of tumor-specific T cells and binds to the CD80 and CD86 ligands expressed on the surface of activated DCs (54). The coinhibitory receptors CTLA-4 and PD-1 have become major drug targets. The CTLA-4 blocking mAb ipilimumab confers a significant survival benefit in patients with metastatic melanoma. The durability of this survival benefit is striking, with patients alive at ten years despite metastatic disease at the initiation of treatment (55, 56). Antibodies targeting PD-1 or its ligand PD-L1 have become leading oncology drugs and have now been approved by the FDA for more than 20 cancer indications (57–59).
Illustrations frequently depict these costimulatory and coinhibitory receptors on the surface of a T cell interacting with a target cell, suggesting a high level of redundancy. However, each of these receptors plays a unique role in T cell biology dictated by the temporal and spatial patterns of receptor and ligand expression. An excellent example is the comparison of the CTLA-4 and PD-1 receptors. The CTLA-4 inhibitory receptor binds to the same ligands (CD80 and CD86) on antigen-presenting cells (APCs) as CD28, but with a higher affinity (60–62). It is transported to the cell surface following initial TCR triggering and attenuates early T cell activation by APCs. CTLA-4 inhibition thus enhances T cell priming in lymph nodes and T cell activation by DCs within tumors (63, 64). In contrast, expression of the PD-1 receptor is highly upregulated following repetitive TCR activation within tumors and sites of chronic infection (65–67). PD-L1 and PD-L2 are expressed by APCs, yet PD-L1 is also broadly expressed by nonhematopoietic cells (19, 68). Importantly, PD-L1 expression is highly upregulated on cancer cells by IFN-γ released by activated T cells, resulting in a negative feedback mechanism termed adaptive resistance (69).
LAG-3 is a third inhibitory receptor on T cells that has proven relevant in human cancer. It is a CD4 homolog that binds to MHC-II proteins with a higher affinity compared to CD4 (70). In murine model systems, LAG-3 antibodies enhance the efficacy of PD-1 blockade (71). In a recent phase 2–3 clinical trial, a combination of LAG-3 and a PD-1 mAb was shown to confer a significant survival benefit compared to PD-1 monotherapy, and this combination therapy has recently been approved by the FDA for the treatment of unresectable or metastatic melanoma (72).
NK cells can recognize tumors cells through multiple activating receptors that bind to ligands upregulated by stressed cells, including the NKG2D, NKp46, NKp30, and NKp44 receptors (Figure 2b). The NKG2D receptor (discussed in more detail in Section 5) binds ligands induced in tumor cells by DNA damage and cGAS-STING signaling (73–75). The NKp46 receptor is expressed by virtually all NK cells and recognizes the P-domain of calreticulin mislocalized to the cell surface during endoplasmic reticulum stress (76). The human NKp30 activating receptor recognizes the B7-H6 protein expressed by tumor cell lines from a variety of cancer types (77, 78). The NKp44 receptor recognizes PDGF-DD, a growth factor released by cancer cells that promotes angiogenesis. PDGF-DD engagement of NKp44 triggers secretion of IFN-γ and TNF-α, which arrest tumor cell growth (79).
Another important feature of NK cell recognition is that MHC-I proteins serve as ligands for inhibitory receptors, including inhibitory KIRs (killer cell immunoglobulin-like receptors) in humans, Ly49 receptors in mice, and the NKG2A/CD94 receptor in both species (80–82). This means that loss of MHC-I expression sensitizes tumor cells to killing by NK cells, providing an important rationale for pursuing immunotherapy strategies that engage both T cells and NK cells (Figure 2b,c).
5. RECEPTORS SHARED BY T CELLS AND NK CELLS THAT REPRESENT THERAPEUTIC OPPORTUNITIES
T cell and NK cell biology is frequently presented as the dichotomy of adaptive and innate recognition. However, there is also substantial overlap in the receptor–ligand systems of NK cells and CD8 T cells. Activating and inhibitory receptors expressed by both CD8 T cells and NK cells thus offer opportunities to engage both cytotoxic lymphocyte populations in antitumor immunity.
5.1. Inhibitory CD161 Receptor and Its Ligand, CLEC2D
The CD161 receptor (encoded by the KLRB1 gene) belongs to the family of C-type lectin receptors and forms a homodimer. It was initially identified as an inhibitory receptor on NK cells that blocks killing of tumor cells that express the CLEC2D ligand (83, 84). More recently, CD161 has been identified as an important inhibitory receptor for tumor-infiltrating T cells (85, 86). An early report had suggested that CD161 cross-linking enhanced cytokine production by MAIT (mucosal-associated invariant T) cells, but these experiments were performed with beads coated with CD3, CD28, and CD161 antibodies, rather than with target cells that expressed the natural CLEC2D ligand (87). CLEC2D, a C-type lectin receptor, is expressed by tumor cells and infiltrating myeloid cells in several human cancer types, including solid tumors and hematological malignancies (85, 88) (Figure 3a). The CLEC2D ligand of CD161 is expressed at a high level by germinal center B cells (89). Consequently, B cell lymphomas that originate from germinal center B cells can express high levels of CLEC2D, including follicular lymphoma, Burkitt lymphoma, and a subtype of diffuse large B cell lymphoma with a germinal center B cell–like gene expression signature (88). CLEC2D is also expressed by DCs and B cells following Toll-like receptor (TLR) activation (90). Targeting of this inhibitory receptor can thus enhance the antitumor activity of T cells and NK cells.
Figure 3.
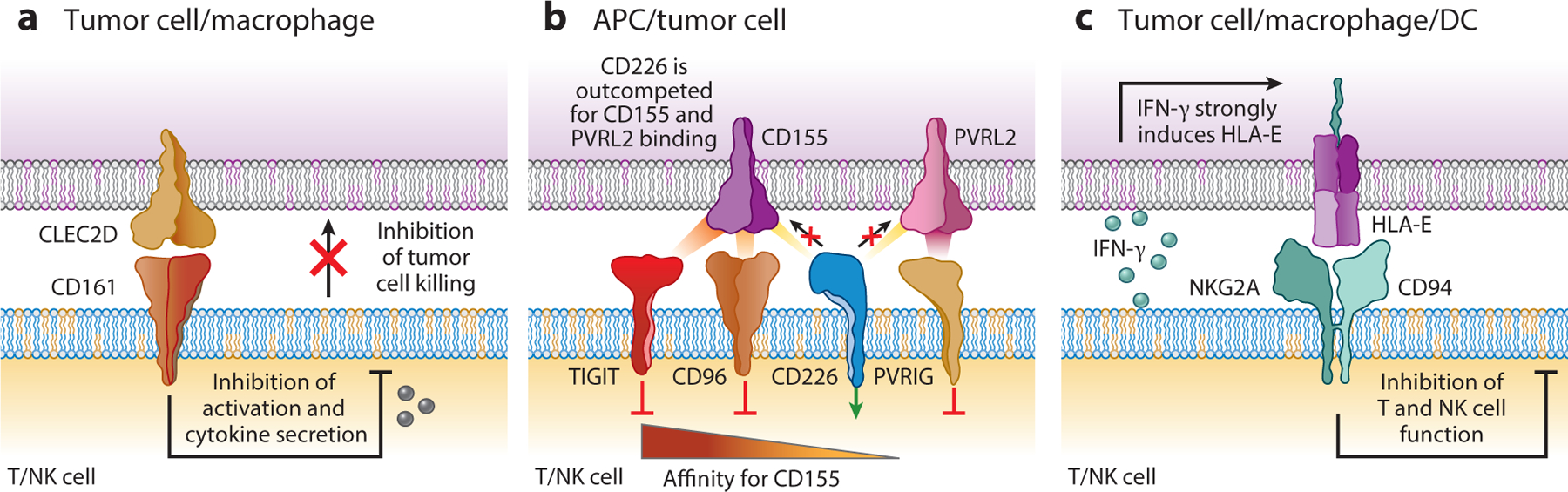
Inhibitory receptors shared by T cells and NK cells. (a) The CD161 inhibitory receptor is expressed by NK cells, and its expression is highly upregulated by tumor-infiltrating CD8 and CD4 T cells. It binds to the CLEC2D ligand, which can be expressed by tumor cells and immune cells in the tumor microenvironment. CD161 signaling inhibits the cytotoxic activity of NK cells and CD8 T cells, and it also inhibits cytokine production by both lymphocyte populations. (b) The CD226 activating receptor is regulated by three inhibitory receptors in NK cells and T cells: TIGIT, CD96, and PVRIG. CD226 binds to CD155, a protein that is highly expressed in a number of human cancer types. TIGIT and CD96 bind to CD155 with a higher affinity compared to CD226, thus outcompeting the activating receptor for ligand binding. The primary ligand for the PVRIG inhibitory receptor is PVRL2. (c) The inhibitory NKG2A/CD94 receptor is expressed by NK cells and a subset of CD8 T cells. It binds HLA-E, whose expression is induced by IFN-γ secreted by activated T cells and NK cells. Abbreviation: APC, antigen-presenting cell.
Recent single-cell RNA-seq studies have demonstrated high-level expression of KLRB1 by tumor-infiltrating T cells in multiple cancer types, including glioblastoma, hepatocellular cancer, colorectal cancer, and melanoma (85, 86). In glioblastoma, the KLRB1 gene is expressed at a higher level by clonally expanded versus nonexpanded CD8 T cells, indicating that its expression is upregulated by CD8 T cells that proliferate in response to antigen recognition (85). Furthermore, expression of cytotoxicity genes by CD8 T cells correlates with expression of several NK cell genes, including KLRB1. These data indicate that tumor-infiltrating CD8 T cells upregulate expression of several (but not all) NK cell receptors in response to cues from the microenvironment. Flow cytometry analysis demonstrates that the CD161 protein is expressed by both CD8 and CD4 T cells. Coculture experiments with primary human T cells equipped with a tumor-specific TCR (NY-ESO-1 TCR) show that editing with a KLRB1 compared to a control guide RNA enhances T cell–mediated killing of glioblastoma cells as well as cytokine secretion. T cell function is also enhanced when this pathway is inhibited with a CD161 blocking mAb. Importantly, KLRB1-edited compared to control-edited T cells confer a significant survival benefit in two humanized mouse models of glioblastoma (85).
A pan-cancer program of KLRB1+ CD8 T cells includes several other NK cell genes and the two genes that encode the IL-18 receptor (IL18R1, IL18RAP). KLRB1 is most highly expressed by tumor-infiltrating CD8 and CD4 T cells with an effector-memory signature, rather than highly exhausted T cells (85). Consistent with these findings, circulating CD161+ memory T cells have higher cytotoxic activity and cytokine production than their CD161− counterparts (91). RNA-seq data show that circulating CD161+ T cells, including MAIT cells, CD8 T cells, CD4 T cells, and γδ T cells, share a core transcriptional program with innate-like features, including upregulated expression of the genes encoding the IL-12 and IL-18 receptors (87). Targeting this inhibitory receptor may thus enhance the function of CD8 and CD4 T cells, including tissue-resident T cells. Finally, a recent study of CAR T cell exhaustion demonstrated upregulation of several NK cell receptors, including CD161, in an exhausted state induced by repetitive in vitro challenge with tumor cells (92). Targeting the CD161 receptor may therefore also be relevant for CAR T cell therapies.
5.2. Inhibitory NKG2A/CD94 Receptor and Its Ligand HLA-E
HLA-E is a nonclassical MHC-Ib molecule that binds to the inhibitory NKG2A/CD94 receptor expressed by NK cells and a subset of CD8 T cells (93) (Figure 3c). Human HLA-E and its murine Qa-1 homolog bind peptides derived from MHC-I signal sequences and thus function as part of the “missing self” recognition mode of NK cells because transcriptional downregulation of MHCI genes sensitizes cells to NK cell cytotoxicity due to reduced surface levels of HLA-E (94–96). In many solid tumors, HLA-E is overexpressed by cancer cells, and expression of this protein is also detected on tumor-infiltrating macrophages and DCs (93). The NKG2A/CD94 receptor is constitutively expressed by the majority of circulating and tumor-infiltrating NK cells. While only a small fraction of blood CD8 T cells express NKG2A/CD94, its expression is upregulated by a subset of tumor-infiltrating CD8 T cells that coexpress PD-1 and other inhibitory receptors (97).
The relevance of this pathway has been studied in several murine tumor models. Qa-1 is strongly upregulated on the surface of tumor cells by IFN-γ secreted by activated T cells. In a murine model of head and neck squamous cell carcinoma (HNSCC), an NKG2A-blocking mAb enhances the efficacy of a peptide-based vaccine (98). A humanized NKG2A-blocking antibody (monalizumab) is in clinical development. This mAb enhances NK cell–mediated ADCC against HNSCC cells in vitro in the presence of the EGFR-blocking mAb cetuximab. This mAb showed limited efficacy as a monotherapy in HNSCC and gynecological malignancies, but in an interim analysis of monalizumab combined with cetuximab in HNSCC induced partial responses based on RECIST (response evaluation criteria in solid tumors) in 31% of patients (99). Also, a recent phase 2 clinical trial showed that the combination of monalizumab and durvalumab (a PD-1 mAb) resulted in a higher response rate and prolonged progression-free survival compared to monotherapy with durvalumab (100). These findings will need to be further investigated in phase 3 clinical trials.
5.3. CD226 Receptor and Inhibitory Counterparts TIGIT, CD96, and PVRIG
The CD226 receptor is an important costimulatory receptor expressed by both NK cells and CD8 T cells (101, 102). Its relevance in antitumor immunity has been demonstrated by increased tumor growth in CD226-deficient mice (103). The major ligand for this activating receptor, CD155, is broadly expressed by human cancer cells; CD226 also binds to a second ligand, PVRL2 (CD112). Interestingly, the activity of the activating CD226 receptor is antagonized by three inhibitory receptors, TIGIT, CD96, and PVRIG. The TIGIT and CD96 inhibitory receptors have a higher affinity than CD226 for the shared CD155 ligand and can outcompete it for ligand binding (104) (Figure 3b).
Among these three inhibitory receptors, TIGIT has been most extensively studied. TIGIT is expressed by NK cells, CD8 T cells, and regulatory T cells (Tregs). TIGIT is highly expressed by human tumor-infiltrating CD8 T cells in different solid tumor types and lymphomas (105). TIGIT is also an important inhibitory receptor in NK cells and contributes to NK cell dysfunction in tumors (106). Inhibition of TIGIT and PD-L1 enhances tumor immunity in multiple murine models and reinvigorates the antitumor function of NK cells (107). A phase 2 clinical trial of a TIGIT-blocking mAb (tiragolumab) randomized patients with previously untreated non-small-cell lung cancer (NSCLC) to receive either TIGIT plus PD-L1 or placebo plus PD-L1 mAbs. Combination therapy resulted in an improved overall response rate (31.3% for combination therapy versus 16.2% for monotherapy) (108). This approach is currently being evaluated in a phase 3 clinical trial in patients with NSCLC. Also, in NSCLC patients treated with a PD-L1 mAb, bulk RNA-seq analysis showed that higher mRNA levels of CD226, but not CD28, were associated with longer overall survival. Both PD-1 and TIGIT inhibit CD226, but through distinct mechanisms: PD-1 dephosphorylates Tyr-322 in the cytoplasmic domain of CD226, while TIGIT outcompetes CD226 for CD155 binding (109).
The CD96 and PVRIG receptors have been studied less extensively. In murine models, inactivation of the CD96 receptor in either CD8 T cells or NK cells enhances antitumor immunity, but its role in human cells is less clear (110, 111). PVRIG is also expressed by T cells and NK cells, and its major ligand is PVRL2 (CD112). Targeting of this receptor enhances the function of human T cells and NK cells in vitro, and a PVRIG-blocking mAb is currently being evaluated in a phase 1 clinical trial (104, 112).
5.4. NKG2D Receptor and Its Stress-Induced Ligands
The NKG2D receptor is expressed by human NK cells, CD8 T cells, and innate T cells [NK T (NKT) cells and γδ T cells] (113). Upon ligation, NKG2D signals through the adaptor protein DAP10 to induce perforin-dependent cytolysis by NK cells, γδ T cells, and NKT cells; it also provides a costimulatory signal for CD8 T cells (114) (Figure 4a). The NKG2D receptor recognizes a set of ligands upregulated by stressed and transformed cells. In humans, these ligands include the MICA/MICB (MICA/B) and ULBP1–6 proteins; in mice these ligands include Rae1α–ε, H60a–c, and Mult1 (115). This receptor-ligand system is important for tumor immunity because these ligands are upregulated by DNA damage and cGAS-STING signaling but are rarely expressed by healthy cells (73, 75). In essence, upregulation of NKG2D ligands flags stressed cells for elimination by cytotoxic lymphocytes. NKG2D ligand expression is frequently detected in multiple solid and hematologic malignancies, including prostate, ovarian, and breast cancers as well as melanoma and multiple myeloma (73, 116, 117). The importance of this pathway is also underscored by enhanced tumor susceptibility of NKG2D-deficient mice (118).
Figure 4.
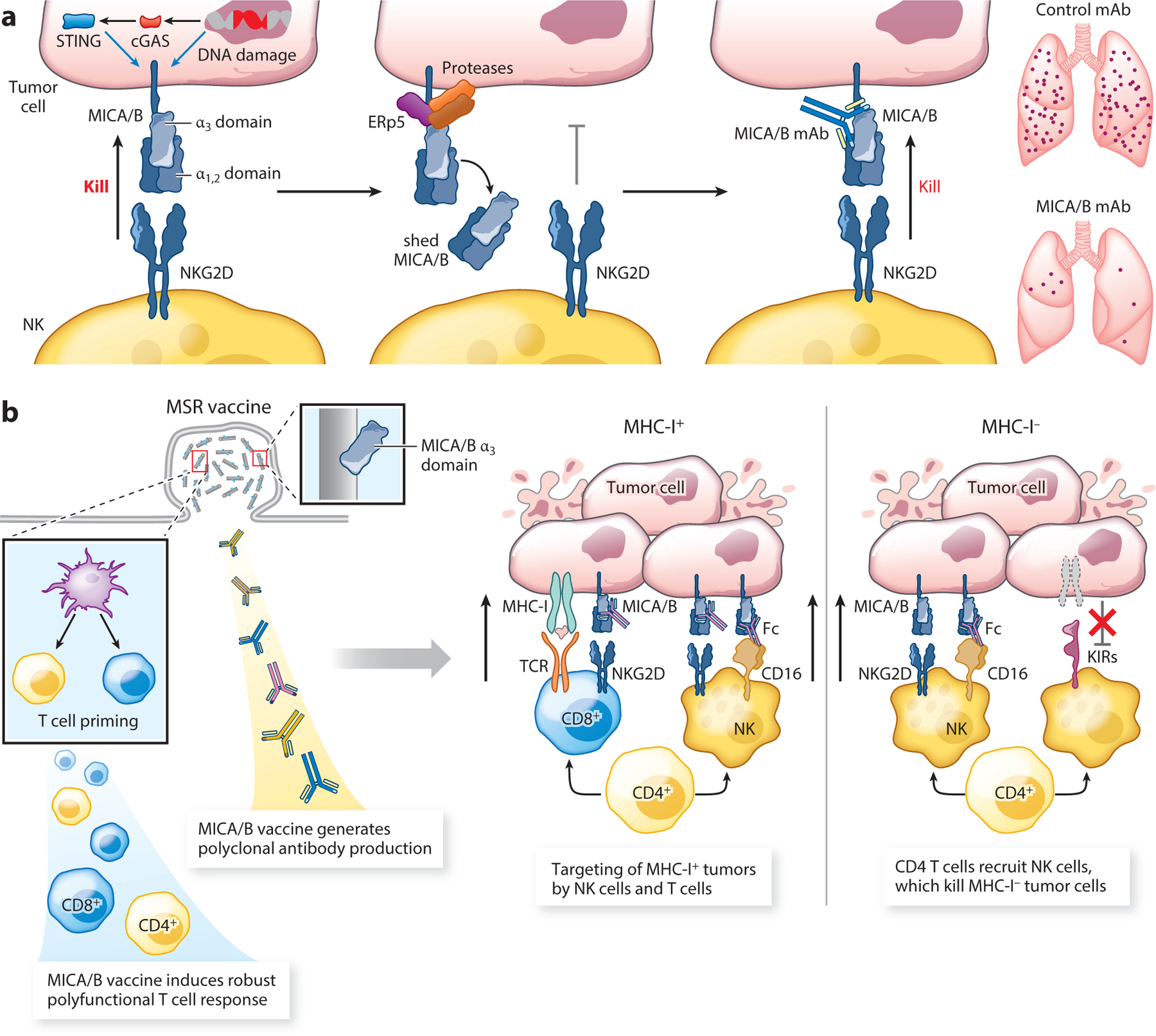
Therapeutic targeting of the NKG2D-MICA/B pathway. (a) The activating NKG2D receptor is expressed by human NK cells, CD8 T cells, NKT cells, and γδ T cells. It binds to the stress-induced ligands MICA and MICB (MICA/B) on tumor cells, but tumors evade an NKG2D-mediated immune attack by proteolytic shedding of MICA/B through the coordinated action of the ERp5 disulfide isomerase and the proteases ADAM10 and ADAM17. A monoclonal antibody against the MICA/B α3 domain inhibits proteolytic shedding and induces an NK cell–mediated immune attack against metastases. (b) A vaccine targeting the MICA/B α3 domain induces a T cell response against this stress protein as well as polyclonal antibodies that inhibit MICA/B shedding. This vaccine retains activity against MHC-I-deficient tumor cells based on the coordinated action of CD4 T cells and NK cells. CD4 T cells recruit NK cells into tumors, while NK cells kill tumor cells following activation through the NKG2D and CD16 receptors. Abbreviations: DC, dendritic cell; KIR, killer cell immunoglobulin-like receptor; mAb, monoclonal antibody; TCR, T cell receptor.
Proteolytic shedding of MICA/B results in immune escape by substantially diminishing the density of these stimulatory NKG2D ligands on the surface of tumor cells (119, 120). The shed proteins have also been reported to induce internalization of the NKG2D receptor on cytotoxic lymphocytes (121). The disulfide isomerase ERp5 is involved in initiation of shedding: It disrupts a structural disulfide bond in the MICA/B α3 domain, rendering it accessible to proteolysis by ADAM-10/17 and MMP14 (119) (Figure 4a). Shed MICA is strongly associated with disease progression in many human cancers, but it is not detected in the serum of healthy subjects (122).
Proteolytic shedding of MICA/B can be inhibited with a mAb that binds to the α3 domain. Treatment of human cancer cell lines with such a MICA/B mAb substantially increases the surface density of these NKG2D ligands and induces their killing by human NK cells (120). Murine tumor cells expressing human MICA/B have been used as an animal model because human MICA/B bind to the murine NKG2D receptor. In mouse models of metastasis, treatment with such a MICA/B mAb substantially reduces the number of metastases by an NK cell–dependent mechanism (Figure 4a). Use of an Fc region that binds with high affinity to activating Fc receptors, including the CD16 receptor on NK cells, further enhances the therapeutic activity of this MICA/B mAb by inducing NK cell activation through both NKG2D and CD16 receptors (120). This mAb is also active against metastases resistant to cytotoxic T cells through loss of MHC-I expression (123).
6. A CANCER VACCINE THAT INDUCES DUAL T CELL AND NK CELL ATTACK AGAINST RESISTANT TUMORS
Most current cancer vaccines focus on peptide epitopes, necessitating personalization due to the vast diversity of MHC alleles between individuals (124). Also, cytotoxic T cells exert substantial selection pressure, resulting in the emergence of tumor clones that have downregulated or lost MHC-I expression. All critical aspects of T cell function, including cytotoxicity, cytokine production, and proliferation are under the control of the TCR, and MHC-I-deficient tumor cells thus become invisible to the TCR on CD8 T cells (12). A vaccine that elicits a dual attack by T cells and NK cells could prevent the emergence of MHC-I-deficient tumor clones because loss of MHCI expression renders tumor cells more susceptible to killing by NK cells. Human tumors evade recognition by the activating NKG2D receptor through proteolytic shedding of the activating MICA and MICB ligands on tumor cells, as discussed above. A cancer vaccine that targets the MICA/B α3 domain involved in proteolytic shedding induces high-titer antibodies that inhibit shedding and increases the density of the stimulatory MICA/B proteins on the surface of tumor cells. This vaccine induces a striking influx of diverse T cell and NK cell populations into murine tumors (125). This vaccine shows activity in a clinically important setting: Immunization following surgical removal of highly metastatic primary tumors substantially reduces the later outgrowth of metastases. Interestingly, this vaccine also remains effective against tumors resistant to CD8 T cells due to inactivating mutations in genes in the MHC-I presentation pathway (B2m) or IFN-γ receptor signaling (Ifngr1) (Figure 4b). Immunity against MHC-I-deficient tumors requires both NK cells and CD4 T cells: CD4 T cells enable recruitment of NK cells that serve as key effector cells following activation of the NKG2D receptor (MICA/B ligands) and CD16A receptors (tumor cell surface–bound MICA/A Abs). Vaccine-induced antibodies serve important roles in this antitumor immune response: Tumor-bound MICA/B antibodies enhance cross-presentation of tumor antigens by DCs to CD8 T cells and increase NK cell–mediated killing of tumor cells through the activating CD16 Fc receptor on NK cells. These findings also highlight an important general point: Therapeutic approaches that focus on NK cells require a CD4 T cell response to enable efficient recruitment of NK cells into tumors (125).
Vaccines can thus be designed to induce both T cell and NK cell responses against tumors. Immunization against tumor surface proteins can elicit T cell and antibody responses, and tumor-bound antibodies can enhance tumor cell killing by NK cells through the activating CD16A receptor. It is important to ensure that CD4 T cell epitopes are present in such vaccines because they enhance differentiation of CD8 T cells into a cytotoxic effector state, reprogram immunosuppressive myeloid cells in the TME, and induce recruitment of NK cells.
7. TARGETING OF INHIBITORY CYTOKINES AND SMALL-MOLECULE MEDIATORS TO ENHANCE T CELL AND NK CELL FUNCTION
7.1. TGF-β
TGF-β is a major immunosuppressive cytokine that potently inhibits both T cell and NK cell function within tumors. TGF-β strongly inhibits T cell proliferation and effector functions (126, 127). In NK cells, TGF-β signaling inhibits key metabolic programs regulated by mTOR (mammalian target of rapamycin), resulting in inhibition of proliferation and cytotoxicity (128). TGF-β also inhibits the surface expression of multiple activating receptors by tumor-infiltrating NK cells, including the NKG2D receptor (129, 130).
However, it has been challenging to target the TGF-β pathway in tumor immunity due to its pleiotropic biology. Two novel approaches have yielded promising data that are based on the unusual mechanisms regulating TGF-β function. Most cytokines are regulated by the rate of secretion, but TGF-β is deposited in abundant quantities as an inactive precursor on the extracellular matrix. Active TGF-β is released from this latent complex by integrin αv receptors through a force-dependent mechanism. Integrins αvβ6 and αvβ8 have the highest affinity for the latent complex and are most potent in releasing active TGF-β (131, 132). Integrin β6 is selectively expressed by epithelial cells, but its expression is low in healthy epithelia. However, integrin αvβ6 is highly expressed by epithelial cancer cells, and its expression correlates with tumor invasiveness and reduced survival (133). Antibody-mediated targeting of the integrin αvβ6 receptor thus offers an approach for more selective inhibition of TGF-β activation in epithelial cancers. An integrin αvβ6/8 mAb substantially enhances T cell–mediated killing of human and murine triple-negative breast cancer cells in vitro and sensitizes two aggressive murine models of triple-negative breast cancer to PD-1 inhibition by inducing a substantial influx of CD8 T cells (134, 135). The second approach is based on the finding that TGFB1 is the most prevalent isoform expressed at the mRNA level in many types of human tumors (136). This mAb blocks TGF-β1 activation from the latent complex and induces a substantial survival benefit in combination with a PD-1 mAb in murine tumor models refractory to ICB. Importantly, this TGF-β1 mAb does not induce damage to heart valves, a major side effect of small-molecule inhibitors that block signaling induced by TGF-β1, TGF-β2, and TGF-β3.
7.2. Prostaglandin E2
PGE2 plays an important role in immunosuppression and is also associated with enhanced cancer cell survival and invasiveness. Cyclooxygenase (COX)-1 and 2, the enzymes critical for PGE2 synthesis, are frequently overexpressed in a number of human cancers, including colorectal cancer (137). Inhibition of PGE2 synthesis in murine melanoma cell lines through inactivation of Ptgs1 and Ptgs2 genes results in a profound shift in the TME away from tumor-promoting cytokines (IL-1β and IL-6) to cytokines/chemokines involved in protective antitumor immunity (IL-12, IFN-γ, and CXCL10) (48). Importantly, CD103+ cDC1 are significantly increased in the TME of these Ptgs1/2−/− tumors and express major activation markers (CD40, CD86). As discussed in Section 3, NK cells play a central role in the recruitment of cDC1 by secreting XCL1 and CCL5, and a substantial increase in NK cell infiltration is observed early in Ptgs1/2−/− tumors (15). Consequently, Ptgs1/2−/− tumors are spontaneously rejected in fully immunocompetent but not immunodeficient mice. COX inhibition with aspirin reduces tumor growth in combination with PD-1 blockade in models in which either drug alone is ineffective, although the effect of aspirin is not as pronounced as inactivation of the Ptgs1 and Ptgs2 genes in tumor cells, potentially due to partial enzyme inhibition (48, 138).
In human melanoma, high PTGS2 (COX-2) mRNA levels are associated with higher mRNA levels of tumor-promoting cytokines (IL1b, IL6, IL8), while low PTGS2 mRNA levels are associated with increased levels of genes involved in antitumor immunity, including many interferon-inducible genes (48). In addition, a substantial number of observational studies have shown that long-term use of aspirin is associated with a lower incidence of colon cancer (139). This concept has also been tested in a randomized trial in patients with Lynch syndrome, which is associated with an increased risk of colon cancer and other cancers due to germ line mutations in DNA mismatch repair genes. Patients received aspirin or placebo for two years, and after ten years of follow-up a significantly reduced hazard ratio of 0.65 was observed for colon cancer in the aspirin versus the placebo group (140). The potential use of aspirin for cancer prevention requires further study, including large randomized clinical trials.
8. ENGINEERING STIMULATORY CYTOKINES FOR T CELL– AND NK CELL–MEDIATED TUMOR IMMUNITY
Several major cytokines act on both T cells and NK cells, including IL-2, IL-12, IL-15, and IL-18, providing opportunities to enhance both T cell and NK cell function. A number of approaches are being taken to engineer variants of IL-2 that stimulate effector T cells and NK cells but not FoxP3+ Tregs. The IL-2 receptor is composed of IL-2Rβ (CD122) and the γ common chain (γc), which form the intermediate-affinity IL-2 receptor expressed by effector T cells and NK cells (141). IL-2Rα (CD25) associates with IL-2Rβ and γc to form the trimeric, high-affinity IL-2 receptor complex. CD25 is expressed at the highest level by Foxp3+ CD4 Tregs, which preferentially expand when IL-2 is limiting. Yeast display and targeted evolution have been used to design a variant of IL-2 (super2) that activates the intermediate-affinity IL-2 receptor on effector T cells and NK cells but lacks CD25 binding (142, 143). NKTR-214 is a pegylated form of IL-2 that slowly releases the active cytokine with limited binding to the IL-2Rα subunit compared to unmodified IL-2. This design increases tumor exposure and substantially expands the number of tumor-infiltrating CD8 T cells and NK cells in murine tumor models (144). In a phase 1 clinical trial, monotherapy with NKTR-214 induced disease stabilization in 14 of 26 patients (53.8%) as well as substantial proliferation of circulating CD4 T cells, CD8 T cells, and NK cells. Bulk RNA-seq analysis of pre- and on-treatment tumor biopsies showed increased mRNA levels of genes expressed by T cells and NK cells, including genes for cytotoxicity and other effector programs. Also, flow cytometry and immunohistochemistry analysis demonstrated increased accumulation of CD8 T cells and NK cells in on-treatment compared to pretreatment tumor biopsies (145).
Substantial efforts are also dedicated to developing IL-15 as a therapeutic agent because it activates effector T cells and NK cells but not FoxP3+ Tregs. IL-15 has a unique mechanism of action: It is trans-presented by DCs and other cell types bound to the IL-15Rα chain and activates the IL-2Rβγ complex (shared by IL-2 and IL-15) on effector T cells and NK cells. IL-15 is important for survival of memory T cells and induces NK cell proliferation and activation (146). An IL-15 super-agonist has been developed in which IL-15 is fused to the sushi domain of the IL-15Rα chain and the Fc region of IgG1, which enhances its activity and extends its half-life (147). In a phase 1 clinical trial in patients with relapse of a hematological malignancy following bone marrow transplantation, the IL-15 super-agonist ALT-803 induced therapeutic responses in 19% of treated patients. Consistent with its mechanism of action, ALT-803 induced striking proliferation of NK cells and CD8 T cells in the blood combined with enhanced expression of activating receptors (NKG2D, NKp30) and granzyme B by circulating NK cells (148). Many other creative approaches are being taken to harness the important biology of IL-2 and IL-15 and are the subjects of excellent recent reviews (149, 150).
IL-18 is a member of the IL-1 cytokine family and is activated by caspase-1 downstream of NLRP3 inflammasome activation. It activates T cells and NK cells through MYD88 and downstream NF-κB signaling. IL-18Rα is widely expressed by NK cells and markedly upregulated by tumor-infiltrating CD4 and CD8 T cells (151). However, the clinical application of IL-18 has been hampered by negative regulation of IL-18 through a decoy receptor called IL-18–binding protein (IL-18BP) that efficiently neutralizes IL-18. A decoy-resistant variant of IL-18 (DR-18) has been developed by protein engineering (152). DR-18 protein exhibits significant therapeutic activity in multiple murine tumor models through the action of CD8 T cells, NK cells, and IFN-γ. Mechanistically, treatment with DR-18 shifts intratumoral T cells into an effector state and simultaneously reduces markers of T cell exhaustion. DR-18 also exhibits significant therapeutic activity against MHC-I-deficient tumors by inducing an activated effector state of tumor-infiltrating NK cells. This engineered form of IL-18 is therefore of significant interest for enhancing T cell– and NK cell–mediated tumor immunity (152).
9. COMBINATION THERAPIES THAT ENGAGE T CELLS AND NK CELLS
A systematic analysis of combination therapies that effectively treat large tumors in murine model systems has identified a combination of four drugs abbreviated as AIPV, which is composed of an antitumor antibody (A), half-life-extended IL-2 (I), anti-PD-1 (P), and a peptide vaccine (V) (153). This regimen can be further streamlined into a single dose of AIP (antitumor antibody, IL-2, and PD-1 mAb) followed by ICB with PD-1 plus CTLA-4 mAbs. Single-dose AIP therapy induces a rapid early response detectable on day 1 with a prominent role of NK cells and a contribution by macrophages, including upregulated expression of proinflammatory chemokines and cytokines (26). Upregulated expression of factors required for recruitment of DCs and effector T cells, including FLT3L, XCL1, CXCL9, and CXCL10, is greatly reduced when NK cells are depleted prior to AIP therapy. Early immune activation also results in greater uptake of tumor antigens by cDC1 and increases the activation state of these DCs. Tumor-bound antibody (A) and extended-half-life IL-2 (I) thus contribute to early NK cell activation, which enables recruitment of DCs and CD8 T cells. This work illustrates how early activation of NK cells and macrophages induces innate immune activation within tumors and thereby sensitizes aggressive murine tumors to ICB (26). More generally, new insights into pathways shared by T cells and NK cells can now be used to rationally design combination therapies that engage both T cell and NK cell populations.
Innate immune stimulators also provide opportunities for activation of CD8 T cells and NK cells. STING agonists induce powerful antitumor responses mediated by CD8 T cells and NK cells in murine models (154). A STING agonist shows strong synergy with an IL-2 superkine in challenging MHC-I-deficient and MHC-I-positive murine tumors by mobilizing T cells and NK cells (155).
10. SUMMARY AND FUTURE DIRECTIONS
Recent discoveries have shown that the biology of T cells in tumors is highly interconnected with that of NK cells. NK cells play an important role in T cell–mediated tumor immunity by recruiting DCs and providing a niche that supports their survival. Even though NK cells and T cells belong to the innate and adaptive branches of the immune system, they also share important activating and inhibitory receptor-ligand systems, providing opportunities for immunotherapies that engage both cytotoxic lymphocyte populations: CD8 T cells and NK cells.
Future studies will investigate the environmental niches formed by DCs, NK cells, and T cells in murine and human tumors and define the molecular mechanisms that inhibit NK cell and T cell recruitment and their function within tumors. Emerging spatial technologies may provide critical insights into dynamic interactions between DCs, NK cells, and T cells in the TME, including in human malignancies that are responsive or refractory to immunotherapies (156, 157). A deeper molecular understanding of such defining features of human tumors may identify novel immunotherapy targets and guide the development of combination therapies that effectively engage DCs, NK cells, and T cells.
ACKNOWLEDGMENTS
We would like to thank Dr. Lucas Ferrari de Andrade for reading the manuscript and providing helpful suggestions. K.W.W. is a member of the Parker Institute for Cancer Immunotherapy (PICI) and the Ludwig Center at Harvard Medical School. This work was supported by NIH grants R01 CA238039, R01 CA251599, R01 CA234018, P01 CA163222 and P01 CA236749, a sponsored research agreement with Novartis, a grant from the Ben and Catherine Ivy Foundation as well as research support by James and Tania McCann (to K.W.W.).
Footnotes
DISCLOSURE STATEMENT
K.W.W. serves on the scientific advisory board of T-Scan Therapeutics, SQZ Biotech, Bisou Bioscience Company, DEM Biopharma, and Nextechinvest and receives sponsored research funding from Novartis. He is a cofounder of Immunitas, a biotech company.
LITERATURE CITED
- 1.Turajlic S, Sottoriva A, Graham T, Swanton C. 2019. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet 20:404–16 [DOI] [PubMed] [Google Scholar]
- 2.Vitale I, Shema E, Loi S, Galluzzi L. 2021. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med 27:212–24 [DOI] [PubMed] [Google Scholar]
- 3.Yuan S, Norgard RJ, Stanger BZ. 2019. Cellular plasticity in cancer. Cancer Discov 9:837–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vegliante R, Pastushenko I, Blanpain C. 2022. Deciphering functional tumor states at single-cell resolution. EMBO J 41:e109221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiery J, Lieberman J. 2014. Perforin: a key pore-forming protein for immune control of viruses and cancer. Subcell. Biochem 80:197–220 [DOI] [PubMed] [Google Scholar]
- 6.Garcia KC, Teyton L, Wilson IA. 1999. Structural basis of T cell recognition. Annu. Rev. Immunol 17:369–97 [DOI] [PubMed] [Google Scholar]
- 7.Alspach E, Lussier DM, Schreiber RD. 2019. Interferon gamma and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb. Perspect. Biol 11:a028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, et al. 2013. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 39:846–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, et al. 2010. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 463:963–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sykulev Y, Cohen RJ, Eisen HN. 1995. The law of mass action governs antigen-stimulated cytolytic activity of CD8+ cytotoxic T lymphocytes. PNAS 92:11990–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. 2017. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168:707–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, et al. 2016. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375:819–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ljunggren HG, Karre K. 1990. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 11:237–44 [DOI] [PubMed] [Google Scholar]
- 14.Hilton HG, Parham P. 2017. Missing or altered self: human NK cell receptors that recognize HLA-C. Immunogenetics 69:567–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, et al. 2018. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172:1022–37.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, et al. 2018. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med 24:1178–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma P, Allison JP. 2015. The future of immune checkpoint therapy. Science 348:56–61 [DOI] [PubMed] [Google Scholar]
- 18.Leach DR, Krummel MF, Allison JP. 1996. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–36 [DOI] [PubMed] [Google Scholar]
- 19.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. 2016. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol 34:539–73 [DOI] [PubMed] [Google Scholar]
- 20.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. 2018. CAR T cell immunotherapy for human cancer. Science 359:1361–65 [DOI] [PubMed] [Google Scholar]
- 21.Sadelain M 2015. CAR therapy: the CD19 paradigm. J. Clin. Investig 125:3392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, et al. 2019. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574:696–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morvan MG, Lanier LL. 2016. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 16:7–19 [DOI] [PubMed] [Google Scholar]
- 24.Wolf NK, Kissiov DU, Raulet DH. 2022. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol In press [DOI] [PubMed] [Google Scholar]
- 25.Cooper MA, Fehniger TA, Caligiuri MA. 2001. The biology of human natural killer-cell subsets. Trends Immunol 22:633–40 [DOI] [PubMed] [Google Scholar]
- 26.Wang C, Cui A, Bukenya M, Aung A, Pradhan D, et al. 2021. Reprogramming NK cells and macrophages via combined antibody and cytokine therapy primes tumors for elimination by checkpoint blockade. Cell Rep 37:110021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guillerey C, Huntington ND, Smyth MJ. 2016. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol 17:1025–36 [DOI] [PubMed] [Google Scholar]
- 28.Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, et al. 2012. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J. Immunol 188:2509–15 [DOI] [PubMed] [Google Scholar]
- 29.Dogra P, Rancan C, Ma W, Toth M, Senda T, et al. 2020. Tissue determinants of human NK cell development, function, and residence. Cell 180:749–63.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paoletti C, Hayes DF. 2016. Circulating tumor cells. Adv. Exp. Med. Biol 882:235–58 [DOI] [PubMed] [Google Scholar]
- 31.Massague J, Obenauf AC. 2016. Metastatic colonization by circulating tumour cells. Nature 529:298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salles G, Barrett M, Foa R, Maurer J, O’Brien S, et al. 2017. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv. Ther 34:2232–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, et al. 2017. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol 95:347–55 [DOI] [PubMed] [Google Scholar]
- 34.Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. 1997. FcγRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell FcγRIIIa, independently of the FcγRIIIa-48L/R/H phenotype. Blood 90:1109–14 [PubMed] [Google Scholar]
- 35.Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, et al. 2004. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res 64:4664–69 [DOI] [PubMed] [Google Scholar]
- 36.Weng WK, Levy R. 2003. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol 21:3940–47 [DOI] [PubMed] [Google Scholar]
- 37.Treon SP, Hansen M, Branagan AR, Verselis S, Emmanouilides C, et al. 2005. Polymorphisms in FcγRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenstrom’s macroglobulinemia. J. Clin. Oncol 23:474–81 [DOI] [PubMed] [Google Scholar]
- 38.Balan S, Saxena M, Bhardwaj N. 2019. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol 348:1– 68 [DOI] [PubMed] [Google Scholar]
- 39.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, et al. 2016. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30:324–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, et al. 2020. cDC1 prime and are licensed by CD4+ T cells to induce anti-tumour immunity. Nature 584:624–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, et al. 2009. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458:899–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, et al. 2009. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31:823–33 [DOI] [PubMed] [Google Scholar]
- 43.Ruhland MK, Roberts EW, Cai E, Mujal AM, Marchuk K, et al. 2020. Visualizing synaptic transfer of tumor antigens among dendritic cells. Cancer Cell 37:786–99.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spranger S, Dai D, Horton B, Gajewski TF. 2017. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31:711–23.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, et al. 2016. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov 6:71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, et al. 2008. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, et al. 2010. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J. Exp. Med 207:823–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, et al. 2015. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162:1257–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, et al. 2020. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity 53:1215–29.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. 2020. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol 20:7–24 [DOI] [PubMed] [Google Scholar]
- 51.de Andrade LF, Lu Y, Luoma A, Ito Y, Pan D, et al. 2019. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight 4:e133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, et al. 2017. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169:750–65.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wucherpfennig KW, Gagnon E, Call MJ, Huseby ES, Call ME. 2010. Structural biology of the T-cell receptor: insights into receptor assembly, ligand recognition, and initiation of signaling. Cold Spring Harb. Perspect. Biol 2:a005140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharpe AH. 2009. Mechanisms of costimulation. Immunol. Rev 229:5–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, et al. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 363:711–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, et al. 2013. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. N. Y. Acad. Sci 1291:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pauken KE, Torchia JA, Chaudhri A, Sharpe AH, Freeman GJ. 2021. Emerging concepts in PD-1 checkpoint biology. Semin. Immunol 52:101480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ribas A, Wolchok JD. 2018. Cancer immunotherapy using checkpoint blockade. Science 359:1350–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Topalian SL, Drake CG, Pardoll DM. 2015. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27:450–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Greenwald RJ, Latchman YE, Sharpe AH. 2002. Negative co-receptors on lymphocytes. Curr. Opin. Immunol 14:391–96 [DOI] [PubMed] [Google Scholar]
- 61.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3:541–47 [DOI] [PubMed] [Google Scholar]
- 62.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. 2016. CD28 costimulation: from mechanism to therapy. Immunity 44:973–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krummel MF, Allison JP. 1995. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med 182:459–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chambers CA, Kuhns MS, Egen JG, Allison JP. 2001. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol 19:565– 94 [DOI] [PubMed] [Google Scholar]
- 65.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, et al. 2018. Role of PD-1 during effector CD8 T cell differentiation. PNAS 115:4749–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–87 [DOI] [PubMed] [Google Scholar]
- 67.McLane LM, Abdel-Hakeem MS, Wherry EJ. 2019. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol 37:457–95 [DOI] [PubMed] [Google Scholar]
- 68.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, et al. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192:1027–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taube JM, Anders RA, Young GD, Xu H, Sharma R, et al. 2012. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med 4:127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. 2019. Lymphocyte-activation gene 3 (LAG3): the next immune checkpoint receptor. Semin. Immunol 42:101305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, et al. 2012. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72:917–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, et al. 2022. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med 386:24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gasser S, Orsulic S, Brown EJ, Raulet DH. 2005. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436:1186–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. 2013. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol 31:413–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Bert N, Lam AR, Ho SS, Shen YJ, Liu MM, Gasser S. 2014. STING-dependent cytosolic DNA sensor pathways regulate NKG2D ligand expression. Oncoimmunology 3:e29259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Santara SS, Crespo Â, Lee D-J, Hu JJ, Zhang Y, et al. 2021. The NK receptor NKp46 recognizes ectocalreticulin on ER-stressed cells. bioRxiv 2021.10.31.466654, Nov. 3
- 77.Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, et al. 2011. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med 17:700–7 [DOI] [PubMed] [Google Scholar]
- 78.Kaifu T, Escaliere B, Gastinel LN, Vivier E, Baratin M. 2011. B7-H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cell Mol. Life Sci 68:3531–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, et al. 2018. Natural killer cells control tumor growth by sensing a growth factor. Cell 172:534–48.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. 2019. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol 16:430–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dohring C, Colonna M. 1996. Human natural killer cell inhibitory receptors bind to HLA class I molecules. Eur. J. Immunol 26:365–69 [DOI] [PubMed] [Google Scholar]
- 82.Yokoyama WM, Kim S. 2006. Licensing of natural killer cells by self-major histocompatibility complex class I. Immunol. Rev 214:143–54 [DOI] [PubMed] [Google Scholar]
- 83.Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, Lanier LL. 2005. Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J. Immunol 175:7796–99 [DOI] [PubMed] [Google Scholar]
- 84.Aldemir H, Prod’homme V, Dumaurier MJ, Retiere C, Poupon G, et al. 2005. Cutting edge: Lectin-like transcript 1 is a ligand for the CD161 receptor. J. Immunol 175:7791–95 [DOI] [PubMed] [Google Scholar]
- 85.Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, et al. 2021. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184:1281–98.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun Y, Wu L, Zhong Y, Zhou K, Hou Y, et al. 2021. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 184:404–21.e16 [DOI] [PubMed] [Google Scholar]
- 87.Fergusson JR, Smith KE, Fleming VM, Rajoriya N, Newell EW, et al. 2014. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep 9:1075–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Germain C, Guillaudeux T, Galsgaard ED, Hervouet C, Tekaya N, et al. 2015. Lectin-like transcript 1 is a marker of germinal center-derived B-cell non-Hodgkin’s lymphomas dampening natural killer cell functions. Oncoimmunology 4:e1026503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Llibre A, Lopez-Macias C, Marafioti T, Mehta H, Partridge A, et al. 2016. LLT1 and CD161 Expression in human germinal centers promotes B cell activation and CXCR4 downregulation. J. Immunol 196:2085–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Germain C, Meier A, Jensen T, Knapnougel P, Poupon G, et al. 2011. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-gamma contributes to modulate immune responses. J. Biol. Chem 286:37964–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fergusson JR, Huhn MH, Swadling L, Walker LJ, Kurioka A, et al. 2016. CD161intCD8+ T cells: a novel population of highly functional, memory CD8+ T cells enriched within the gut. Mucosal Immunol 9:401–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, et al. 2021. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 184:6081–100.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Borst L, van der Burg SH, van Hall T. 2020. The NKG2A-HLA-E axis as a novel checkpoint in the tumor microenvironment. Clin. Cancer Res 26:5549–56 [DOI] [PubMed] [Google Scholar]
- 94.Lee N, Llano M, Carretero M, Ishitani A, Navarro F, et al. 1998. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. PNAS 95:5199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, et al. 1998. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 391:795–99 [DOI] [PubMed] [Google Scholar]
- 96.Jensen PE, Sullivan BA, Reed-Loisel LM, Weber DA. 2004. Qa-1, a nonclassical class I histocompatibility molecule with roles in innate and adaptive immunity. Immunol. Res 29:81–92 [DOI] [PubMed] [Google Scholar]
- 97.Abd Hamid M, Wang RZ, Yao X, Fan P, Li X, et al. 2019. Enriched HLA-E and CD94/NKG2A interaction limits antitumor CD8+ tumor-infiltrating T lymphocyte responses. Cancer Immunol. Res 7:1293–306 [DOI] [PubMed] [Google Scholar]
- 98.van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, et al. 2018. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell 175:1744–55.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, et al. 2018. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175:1731–43.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Herbst RS, Majem M, Barlesi F, Carcereny E, Chu Q, et al. 2022. COAST: an open-label, phase II, multidrug platform study of durvalumab alone or in combination with oleclumab or monalizumab in patients with unresectable, stage III non-small-cell lung cancer. J. Clin. Oncol In press [DOI] [PubMed] [Google Scholar]
- 101.O’Donnell JS, Madore J, Li XY, Smyth MJ. 2020. Tumor intrinsic and extrinsic immune functions of CD155. Semin. Cancer Biol 65:189–96 [DOI] [PubMed] [Google Scholar]
- 102.de Andrade LF, Smyth MJ, Martinet L. 2014. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol 92:237–44 [DOI] [PubMed] [Google Scholar]
- 103.Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, et al. 2008. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med 205:2959–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alteber Z, Kotturi MF, Whelan S, Ganguly S, Weyl E, et al. 2021. Therapeutic targeting of checkpoint receptors within the DNAM1 axis. Cancer Discov 11:1040–51 [DOI] [PubMed] [Google Scholar]
- 105.Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, et al. 2015. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig 125:2046–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, et al. 2018. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol 19:723–32 [DOI] [PubMed] [Google Scholar]
- 107.Anderson AC, Joller N, Kuchroo VK. 2016. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44:989–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, et al. 2022. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol 23:781–92 [DOI] [PubMed] [Google Scholar]
- 109.Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, et al. 2022. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity 55:512–26.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mittal D, Lepletier A, Madore J, Aguilera AR, Stannard K, et al. 2019. CD96 is an immune checkpoint that regulates CD8+ T-cell antitumor function. Cancer Immunol. Res 7:559–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, et al. 2014. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol 15:431–38 [DOI] [PubMed] [Google Scholar]
- 112.Whelan S, Ophir E, Kotturi MF, Levy O, Ganguly S, et al. 2019. PVRIG and PVRL2 are induced in cancer and inhibit CD8+ T-cell function. Cancer Immunol. Res 7:257–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lanier LL. 2015. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res 3:575–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu J, Song Y, Bakker AB, Bauer S, Spies T, et al. 1999. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285:730–32 [DOI] [PubMed] [Google Scholar]
- 115.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, et al. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727–29 [DOI] [PubMed] [Google Scholar]
- 116.Spear P, Wu M-R, Sentman M-L, Sentman CL. 2013. NKG2D ligands as therapeutic targets. Cancer Immun 13:8. [PMC free article] [PubMed] [Google Scholar]
- 117.Raulet DH, Marcus A, Coscoy L. 2017. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol. Rev 280:93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, et al. 2008. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28:571–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, et al. 2007. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 447:482–6 [DOI] [PubMed] [Google Scholar]
- 120.Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, et al. 2018. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 359:1537–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Groh V, Wu J, Yee C, Spies T. 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419:734–38 [DOI] [PubMed] [Google Scholar]
- 122.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296:1323–26 [DOI] [PubMed] [Google Scholar]
- 123.Ferrari de Andrade L, Kumar S, Luoma AM, Ito Y, Alves da Silva PH, et al. 2020. Inhibition of MICA and MICB shedding elicits NK-cell-mediated immunity against tumors resistant to cytotoxic T cells. Cancer Immunol. Res 8:769–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shemesh CS, Hsu JC, Hosseini I, Shen BQ, Rotte A, et al. 2021. Personalized cancer vaccines: clinical landscape, challenges, and opportunities. Mol. Ther 29:555–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Badrinath S, Dellacherie MO, Li A, Zheng S, Zhang X, et al. 2022. A vaccine targeting resistant tumors by dual T cell plus NK cell attack. Nature 606(7916):992–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Batlle E, Massague J. 2019. Transforming growth factor-beta signaling in immunity and cancer. Immunity 50:924–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.MO Li, Flavell RA. 2008. TGF-beta: a master of all T cell trades. Cell 134:392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, et al. 2016. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 9:ra19. [DOI] [PubMed] [Google Scholar]
- 129.Lazarova M, Steinle A. 2019. Impairment of NKG2D-mediated tumor immunity by TGF-beta. Front. Immunol 10:2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, et al. 2003. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. PNAS 100:4120–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, et al. 2017. Force interacts with macromolecular structure in activation of TGF-beta. Nature 542:55–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, et al. 1999. The integrin αvβ6 binds and activates latent TGF β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96:319–28 [DOI] [PubMed] [Google Scholar]
- 133.Niu J, Li Z. 2017. The roles of integrin αvβ6 in cancer. Cancer Lett 403:128–37 [DOI] [PubMed] [Google Scholar]
- 134.Eberlein C, Kendrew J, McDaid K, Alfred A, Kang JS, et al. 2013. A human monoclonal antibody 264RAD targeting αvβ6 integrin reduces tumour growth and metastasis, and modulates key biomarkers in vivo. Oncogene 32:4406–16 [DOI] [PubMed] [Google Scholar]
- 135.Bagati A, Kumar S, Jiang P, Pyrdol J, Zou AE, et al. 2021. Integrin αvβ6-TGFβ-SOX4 pathway drives immune evasion in triple-negative breast cancer. Cancer Cell 39:54–67.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, et al. 2020. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med 12:eaay8456. [DOI] [PubMed] [Google Scholar]
- 137.Kalinski P 2012. Regulation of immune responses by prostaglandin E2. J. Immunol 188:21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, et al. 2015. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med 21:327–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Algra AM, Rothwell PM. 2012. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 13:518–27 [DOI] [PubMed] [Google Scholar]
- 140.Burn J, Sheth H, Elliott F, Reed L, Macrae F, et al. 2020. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet 395:1855–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leonard WJ, Lin JX, O’Shea JJ. 2019. The γc family of cytokines: basic biology to therapeutic ramifications. Immunity 50:832–50 [DOI] [PubMed] [Google Scholar]
- 142.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, et al. 2012. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 484:529–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, et al. 2019. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565:186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, et al. 2016. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res 22:680–90 [DOI] [PubMed] [Google Scholar]
- 145.Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, et al. 2019. A first-in-human study and biomarker analysis of NKTR-214, a novel IL2Rβγ-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov 9:711–21 [DOI] [PubMed] [Google Scholar]
- 146.Guo Y, Luan L, Patil NK, Sherwood ER. 2017. Immunobiology of the IL-15/IL-15Rα complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev 38:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, et al. 2006. Converting IL-15 to a superagonist by binding to soluble IL-15Rα. PNAS 103:9166–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, et al. 2018. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 131:2515–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Spangler JB, Moraga I, Mendoza JL, Garcia KC. 2015. Insights into cytokine-receptor interactions from cytokine engineering. Annu. Rev. Immunol 33:139–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Conlon KC, Miljkovic MD, Waldmann TA. 2019. Cytokines in the treatment of cancer. J. Interferon Cytokine Res 39(1):6–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kaplanski G 2018. Interleukin-18: biological properties and role in disease pathogenesis. Immunol. Rev 281:138–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, et al. 2020. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583:609–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, et al. 2016. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med 22:1402–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nicolai CJ, Wolf N, Chang IC, Kirn G, Marcus A, et al. 2020. NK cells mediate clearance of CD8+ T cell-resistant tumors in response to STING agonists. Sci. Immunol 5:eaaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wolf NK, Blaj C, Picton LK, Snyder G, Zhang L, et al. 2022. Synergy of a STING agonist and an IL-2 superkine in cancer immunotherapy against MHC I-deficient and MHC I+ tumors. PNAS 119:e2200568119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schurch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, et al. 2020. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182:1341–59.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, et al. 2018. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174:968–81.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]