

Abstract
Genome mining of biosynthetic pathways with no identifiable core enzymes can lead to discovery of the so-called unknown (biosynthetic route)-unknown (molecular structure) natural products. In this work, we focused on a conserved fungal biosynthetic pathway (ank) that lacks a canonical core enzyme, and used heterologous expression to identify the associate natural product to be a highly modified cyclo-arginine-tyrosine dipeptide. Biochemical characterization of the ank pathway led to identification of a new arginine cyclodipeptide synthase (RCDPS), which was previously annotated as a hypothetical protein (HP) and has no sequence homology to nonribosomal peptide synthetase (NPRS) or bacterial cyclodipeptide synthase (CDPS). RCDPS homologs are widely encoded in fungal genomes and we showed other members of this family can synthesize diverse cyclo-arginine-Xaa dipeptides. Characterization of a cyclo-Arg-Trp (cRW) RCDPS showed the enzyme is aminoacyl-tRNA dependent, and represents the first report of such CDPS-like enzyme from fungi. Further characterization of the biosynthetic pathway anchored by the cRW synthase led to discovery of new compounds of which the structures would not have been predicted without knowledge of RCDPS function. Our work demonstrates the importance of examining unknown-unknown biosynthetic pathways towards discovering new scaffold-forming enzymes and expanding the chemical space of natural products.
Introduction
Genome mining has brought a renaissance to natural product discovery.1,2 Using homology-based functional predictions, bioinformatics algorithms can readily extract potential biosynthetic gene clusters (BGCs) from sequenced microbial genomes.3,4 It is estimated that over 1 million BGCs can be predicted from the current genome database, and can be mined for natural products using synthetic biology approaches.5 Anchored by canonical core enzymes such as polyketide synthases (PKSs), nonribosomal peptide synthetases (NRPSs), terpene synthases, and prenyltransferases, predicted BGCs that have no associated metabolites have been referred to as unknown cluster/known metabolite category (unknown-knowns).6,7 Despite the increased sophistication of microbial genome mining expeditions in the past few years,8 it remains highly challenging to identify and categorize potential BGCs that have no core enzymes or no proteins with sequence similarity to known core enzymes. Collectively referred to as the unknown cluster/unknown metabolite category (unknown-unknowns),6 these BGCs represent the true biosynthetic dark matter that are underexplored with respect to new structures and biological activities. Therefore, efforts aimed at identification and characterization of unknown-unknown BGCs and potentially new scaffold-forming enzymes are much needed.
In the search for new BGCs that have unknown metabolite category, one needs to deemphasize the presence of open reading frames (ORFs) in BGCs that are predicted to be known scaffold-building enzymes, and instead, search for clusters of ORFs that do not produce recognizable primary or secondary metabolites.9,10 For example, a BGC that contains a cluster of tailoring enzymes and no canonical core enzyme is a reasonable starting point, as evidenced by recent characterization of bacterial BGCs of altemicidin,10 fluopsin,11 and guanitoxin.12 These tailoring enzymes can include redox enzymes such as oxidoreductases,13 transferring enzymes such as methyl-, acetyl-, or glycosyl-transferases;14 and other enzymes that are found frequently in BGCs, such as PLP-dependent15 or ATP-grasp enzymes.16,17 In addition, the presence of ORFs that are designated as hypothetical proteins (HPs)18 or proteins predicted to have domains of unknown function (DUFs)19 is predictive of new enzymatic function. Bioinformatics identification of potential unknown-unknown clusters, followed by comparison of homologous BGCs in the genome database, can further highlight conserved ORFs and define cluster boundaries. In this work, we demonstrate such workflow to decode one family of unknown-unknown BGCs in filamentous fungi. We identified a new eukaryotic, tRNA-dependent dipeptide synthase that produces Arg-Xaa diketopiperazine (DKP)20 compounds very rarely found in this family of natural products. This previously designated HP has no sequence homology to the known bacterial cyclodipeptide synthase (CDPS),21,22 and is widely conserved in fungi. Using this new core-forming enzyme as a genome mining beacon, new arginine-containing natural products were discovered.
Results and Discussion
Genome mining of the unknown-unknown ank cluster
To identify candidates for the aforementioned unknown-unknown BGCs, we started by searching for clusters that contain a PLP-dependent enzyme with sequence homology to cystathionine-γ-synthase. Previous work in both fungi and bacteria showed that homologs of these enzymes can catalyze PLP-dependent γ-replacement reactions using O-acyl-l-homoserine as substrate23, 24 to form C-N, C-C, and C-S bonds.15,25,26 We reasoned that BGCs without a core enzyme could utilize such enzymes to build molecular scaffolds. Using our published algorithm that searches for BGCs that include and/or exclude specific enzymes,27 we located one such unknown-unknown BGC from several different fungal species, including the ank cluster from Aspergillus thermomutatus (Fig. 1a and S1, Table S1). The encoded PLP-dependent enzyme AnkD shows 50% identity to the PLP-dependent enzymes CndF23 and FlvA28 (Table S1), which catalyze C-C bond formation. In addition to ankD, other conserved genes include those encoding a cytochrome P450 (ankB), a FAD-dependent monooxygenase (ankC), a NRPS-independent siderophore (NIS) synthetase (ankE), an O-methyltransferase (OMT) (ankF), and a homolog of the ATP-dependent glutathione synthetase (ankG). The ankA gene encodes a 518-aa hypothetical protein, which has no conserved domain or predicted function, and is also conserved among all clusters shown in Fig. 1a. Without a canonical core enzyme, no prediction of metabolite structure from this cluster can be made.
Fig 1. Reconstitution of the ank pathway from A. thermomutatus leads to new natural product.
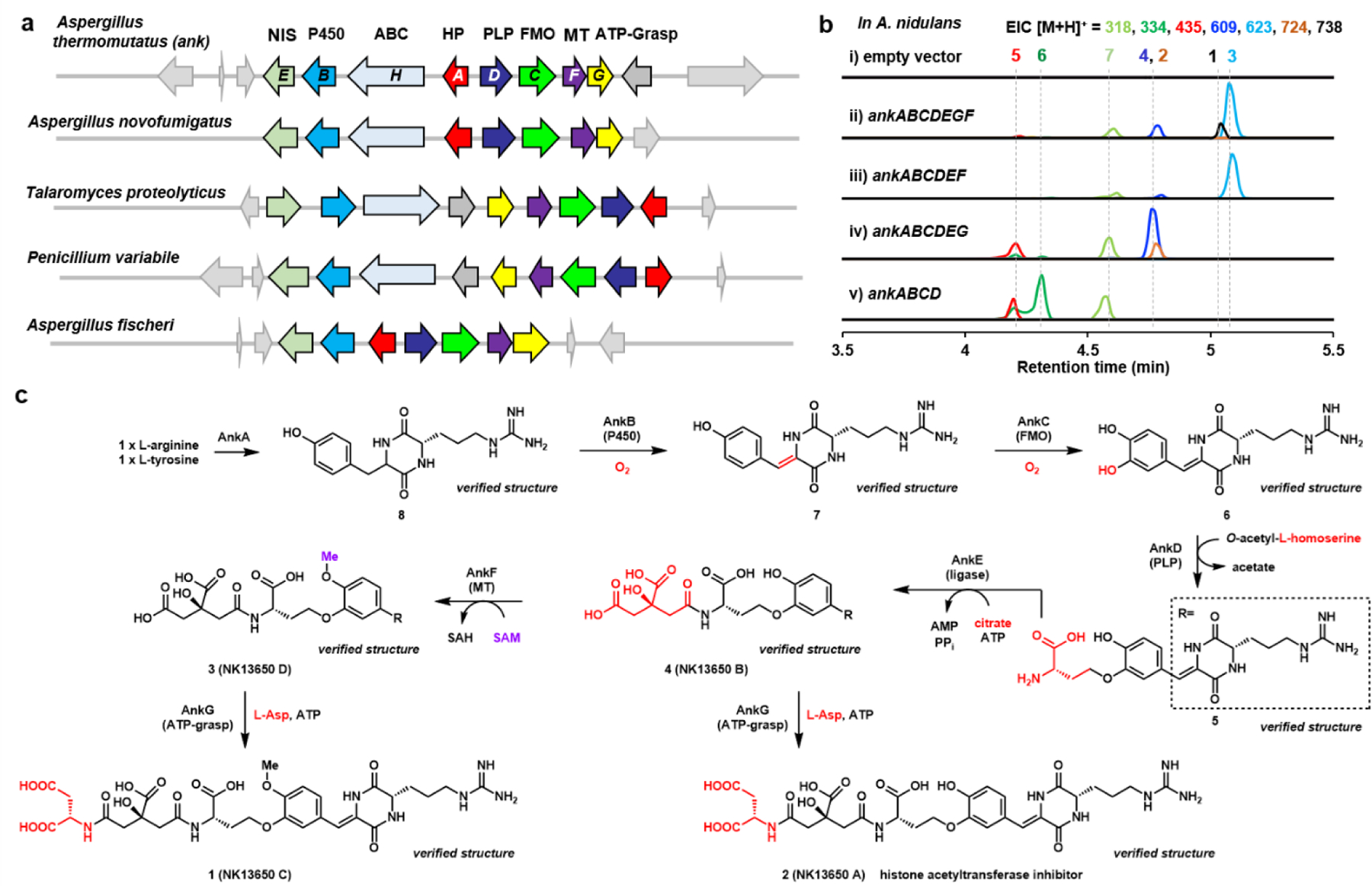
a) The ank BGC encodes the PLP-dependent enzyme AnkD flanked by genes encoding potential tailoring enzymes. This gene cluster is conserved in a number of fungi as shown. Abbreviations: HP: hypothetical protein; P450: cytochrome P450 monooxygenase; FMO: flavin-dependent monooxygnease; PLP: pyridoxal-5’-phosphate dependent enzyme; NIS: NRPS-independent siderophore synthetase. No core enzyme is present, including PKS, NRPS or terpene cyclase. b) QTOF analysis of metabolites produced by different gene combinations in A. nidulans heterologous host are shown in ii) to v). The selected ions are shown and the colors of the traces match to the indicated mass and compounds. A. nidulans transformed with the empty vectors is the control in i). c) Biosynthetic pathway to 1 and 2. The compounds indicated with “verified structure” have been fully characterized by NMR (see Supporting Information). 2 was previously reported to be a p300-selective histone acetyltransferase inhibitor.30
The genes ankA-G were expressed in the heterologous host Aspergillus nidulans A1145 ΔEMΔST for metabolite analysis.29 This resulted in the emergence of several new metabolites, including 1 (molecular weight, MWT: 737), 3 (MWT: 622), 4 (MWT: 608) and 7 (MWT: 317) (Fig. 1b, ii). These compounds were purified and the structures were solved with NMR analysis. 1 with the highest MWT was elucidated to be a linear compound connecting cyclo-arginine-dehydrotyrosine dipeptide, homoserine, citrate and aspartate (Fig. S25-S29, Table S7). A compound database search showed that 1 (named here NK13650 C) is the O-methylated version of the p300-histone acetyltransferase (HAT) inhibitor 2 (NK13650 A).30 3 (named here NK13650 D) was solved to be the precursor of 1 prior to attachment of the l-Asp residue (Fig. S33-S37, Table S9), while 4 corresponded to the O-desmethylated version of 3 (NK13650 B, Fig. S43-S45, Table S11).30 We also detected and isolated an ornithine version of 3 from the extract (3b), which we propose to derive from 3 via the activities of the host arginase31 (Fig. S10, S38-S42, Table S10). Lastly from this strain, NMR analysis of 7 confirmed the compound to be cyclo-Arg-dehydro-Tyr (Fig. S94) with the Z olefin configuration.32
The structures of these compounds suggest 1 is biosynthesized in a step-wise fashion starting with the cyclo-Arg-Tyr diketopiperazine (cRY) 8. To assign functions to the enzymes encoded in the gene cluster, individual genes were removed stepwise from the fully reconstituted pathway, followed by metabolite detection and structural analysis. We first assigned the O-methyltransferase ankF to be responsible for methylation of the C17 phenol group. Coexpression of the entire biosynthetic cassette except ankF led to the formation of 4, as well as the expected emergence of 2 (NK13650 A, MWT: 723) (Fig. S30-S32, Table S8),30 confirming the role of AnkF (Fig. 1b, iv). The NMR data and optical rotation measurement of 2 matched those in the previous publication,30 and led to the complete assignment of stereochemistries of the different building blocks in 2, as well as those in 1. In parallel, comparing the structures of 1 and 3 suggests that amidation of 3 with l-Asp is likely the last step. This could be catalyzed by the predicted ATP-grasp enzyme AnkG, which was initially predicted as a hypothetical protein but displayed the closest secondary structure homology33 to glutathione synthetase, which produces ADP and Pi during catalysis.34 Removal of ankG from the pathway led to disappearance of 1 and predominant accumulation of 3 (Fig 1b, iii). The accumulation of both 2 and 4 in the strain without AnkF suggests AnkG can also use 4 as a substrate.
The next enzyme involved in the biosynthesis of 4 is likely the predicted NIS homolog AnkE. NIS homologs are predominantly found in bacteria and are involved in the biosynthesis of citrate-containing siderophores, such as staphyloferrin A and aerobactin.35,36 Typically, one carboxylate of citrate is adenylated followed by amide bond formation with amine nucleophiles.37 We propose that AnkE has an analogous function to ligate citrate to the precursor 5. Indeed, expression of the remaining genes (ankABCD) without ankE led to the disappearance of 4 and emergence of 5 (MWT: 434) and 6 (MWT: 333) (Fig. 1b, v). Characterization of 5 by NMR confirmed the proposed structure, in which the side chain of l-homoserine is attached to the DKP via an aryl ether linkage (Fig. S46-S50, Table S12). We also isolated and characterized 6, which is the precursor to 5 without the l-homoserine attachment (Fig. S51-S55, Table S13). The conversion of 6 to 5 requires expression of the PLP-dependent enzyme AnkD, as expression of ankABC only resulted in formation of 6 (Fig. S11, iii). Recombinant AnkD purified from E. coli (Fig. S9) was confirmed to convert 6 to 5 in an O-acetyl-l-homoserine dependent fashion (Fig. S12). This is the first confirmed example of C-O bond formation through γ-substitution by a PLP-dependent enzyme, and the mechanism is shown in Fig. S5.
The remaining unassigned enzymes AnkA (HP), AnkB (P450) and AnkC (FMO) are involved in the early steps of the pathway that form 6. Expression of AnkA alone in A. nidulans led to the emergence of a polar compound 8 (Fig. S13, ii), which was structurally verified to be cRY dipeptide (Fig. S95).38 Expression of AnkA in Saccharomyces cerevisiae JHY65139 also led to the accumulation of 8 (Fig. S14). Expression of AnkA and AnkB afforded the previously characterized dehydro-cyclodipeptide 7 (Fig. S11, iv), which indicates the P450 enzyme is responsible for desaturation, likely through hydroxylation of the benzylic position in 8 followed by dehydration.40,41 Finally, AnkC is assigned as the hydroxylase that installs the m-OH in 6, through a canonical flavin-dependent aromatic hydroxylation mechanism.42
Collectively, reconstitution of the biosynthesis of 1 from the ank BGC shows that a bioactive natural product with considerable structural complexity can be generated without a bona fide core enzyme. Primary cellular metabolites, including l-Arg, l-Tyr, l-Asp, l-homoserine, and citrate were combined in sequential order to furnish the final product 1. This finding supports additional efforts to mine such unknown-unknown BGCs for new natural products.
AnkA and Homolog are Cyclo-Arginine-Xaa Dipeptide Synthases
The most unexpected finding in the biosynthesis of 1 is that AnkA is responsible for formation of the cyclodipeptide 8. Given that the remaining pathway builds on 8, AnkA can be considered as the scaffold generating enzyme. Cyclodipeptides are generally formed via two routes: i) an NRPS-dependent pathway present in both bacteria43 and fungi;44,45 and ii) a tRNA-dependent pathway catalyzed by cyclodipeptide synthase (CDPS) only found in bacteria.21,22 AnkA is a 518-residue protein that has no predicted function, no characterized sequence homolog, and no structural homolog detected through Phyre2.33
We searched for AnkA homologs that may catalyze formation of other cyclodipeptides. A tBLASTn search using ankA as query against the JGI ascomycete genome database, NCBI nucleotide collection, as well as in-house fungal genome sequences identified more than 100 hits for homologs with greater than 20% identity to ankA (Table S6). Selected sequences were used to generate a phylogenetic tree using MAFFT software (Fig. 2).46 Analysis of the genes surrounding the homologs showed that while some are standalone with no predicted tailoring enzymes nearby, others are flanked by diverse tailoring enzymes as seen in the ank cluster (Fig. S3). Six of these homologs were chosen for functional analysis, including AnoA from Aspergillus nomius; AvaA from A. versicolor; PthA from Penicillium thymicola; AteA from A. terreus; AmaA from Apiospora montagnei; and EshA from Eupenicillium shearii. A sequence alignment of these to AnkA can be found in Fig. S4, which showed limited sequence homology.
Fig. 2. AnkA and homologs from fungi are cyclo-arginine-Xaa-dipeptide synthases (CRDPS).
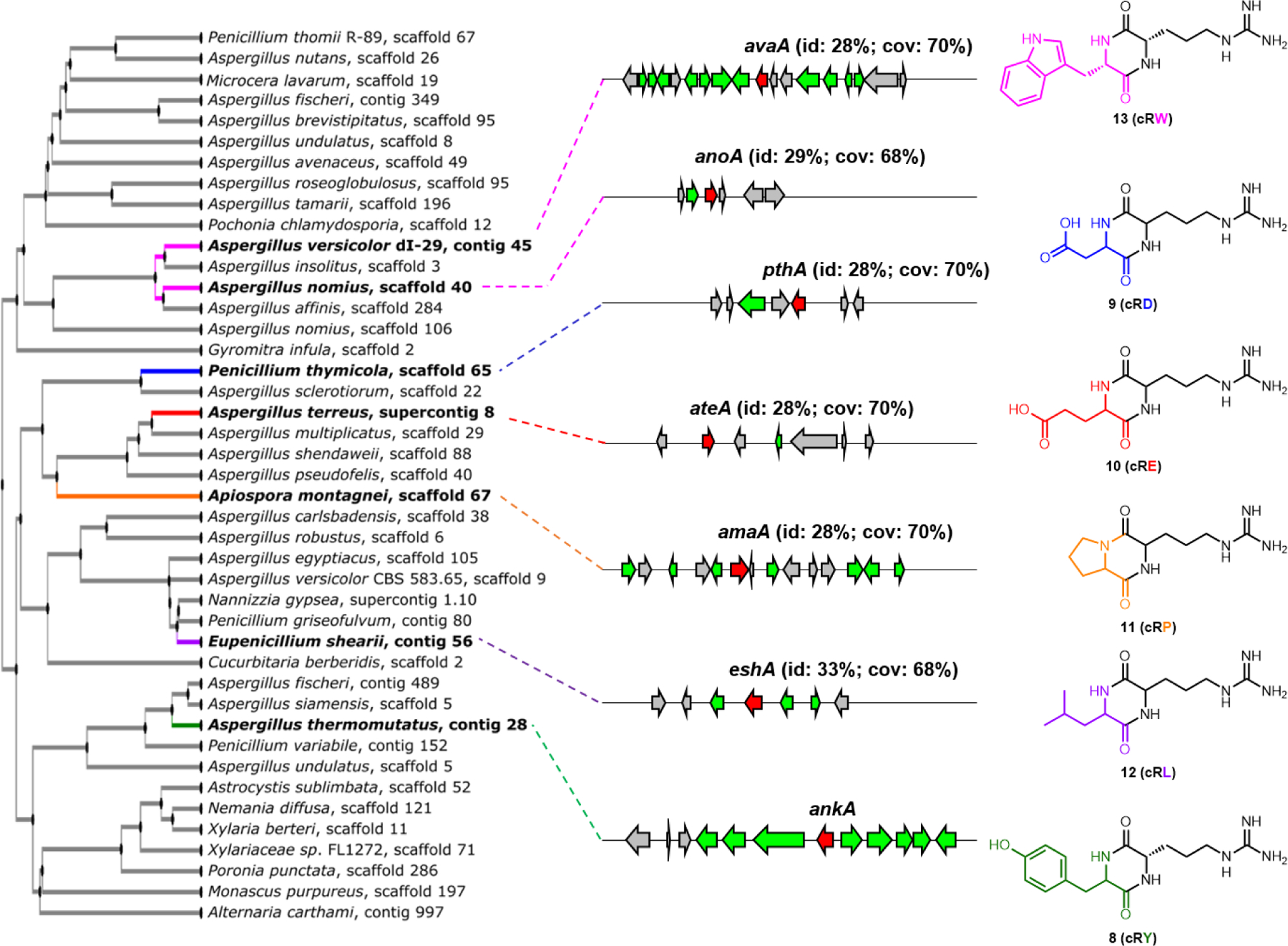
MAFFT phylogenetic tree of select AnkA homologs from sequenced fungal genomes. Six additional gene clusters encoding the analogs are shown on the right. Genes encoding AnkA homologs (AvaA, AnoA, PthA, AteA, AmaA and EshA) are shown in red, while genes encoding putative tailoring enzymes are shown in green. The % identity and % coverage of these homologs to AnkA are shown in parenthesis. Structures of arginine-containing cyclodipeptides synthesized by expression of the CRDPS are shown on the right.
Each of the AnkA homologs was expressed in A. nidulans followed by metabolite analysis (Fig. S13). We observed a variety of arginine containing DKPs, including: cyclo-Arg-Asp (cRD, 9) from PthA (Fig. S56-S60, Table S14); cyclo-Arg-Glu (cRE, 10) from AteA (Fig. S61-S65, Table S15); cyclo-Arg-Pro (cRP, 11)47 from AmaA (Fig. S96); cyclo-Arg-Leu (cRL, 12)48 from EshA (Fig. S97); and cyclo-Arg-Trp (cRW, 13)49 from both AvaA and AnoA (Fig. S66-S67, Table S16). Notably, 11 is the only example of an arginine containing DKP produced by a bacterial CDPS from Lysobacter antibioticus.50 Halogenated and ring-modified variants of cRP have been previously isolated from marine sponges,51,52 and the sequence of AmaA could be used to mine the metagenome for the BGCs of these derivatives. The stereochemistry of 13 was confirmed by Marfey’s analysis and NMR comparison to synthetic cyclo-l-Arg-l-Trp49 (Fig. S20, Table S16). Therefore, it is evident that all active AnkA homologs utilize arginine as one of the amino acid building blocks, while the second amino acid can vary. With the exception of cRP,50 arginine-containing DKPs have been conspicuously absent from microbial metabolomes. Our discovery of the AnkA family of enzymes, which we rename here to be arginine-containing cyclodipeptide synthases (RCDPSs), shows Nature can indeed synthesize a variety of such guanidine-containing DKPs. Based on the product structures and phylogenetic relationship shown in Fig. 2, each subclade has different substrate specificity for the second amino acid (D, E, P, W, L, Y). Exploring the biosynthetic products of RCDPSs from other clades should therefore give rise to additional arginine-containing DKPs.
RCDPSs are tRNA-dependent cyclodipeptide synthases
To study the mechanism of RCDPs, we attempted to express the homologs from Fig. 2 from either yeast or E. coli as soluble proteins. While several of these enzymes functioned in yeast to produce the expected cyclodipeptide (Fig. S14-S15), the cRW 13-producing AvaA functioned in both E. coli and yeast robustly (Fig. S16 and S18). As a result, subsequent biochemical studies focused on AvaA as a representative member.
We first assayed the activity of AvaA in lysate from the yeast expression strain. Trp-(indole)-d5 was added to the lysate to distinguish newly synthesized cRW-d5 from those produced during yeast culturing. The 13-d5 product (MWT: 348) was observed from the lysate (Fig. 3a), in contrast to the negative control in which no Trp-d5 was added. Given that AvaA bears no similarity to NRPS, we started off by determining if the formation of 13 is RNA-dependent. Treating the lysate with RNase A prior to substrate addition completely abolished 13-d5 formation. In lysates treated with RNase A followed by addition of the RNase A inhibitor RNasin, formation of 13-d5 was only observed after addition of total yeast RNA, indicating the reaction is RNA-dependent (Fig. 3a). In assays with lysate that was first desalted to remove small molecules and salts, addition of ATP and MgCl2, which are necessary for aminoacyl-tRNA synthetase function,53,54 was required for product formation (Fig. S17). These observations are consistent with AvaA being a RNA-dependent enzyme, functionally analogous to bacterial CDPSs.55,56
Fig. 3. Biochemical characterization of AvaA.
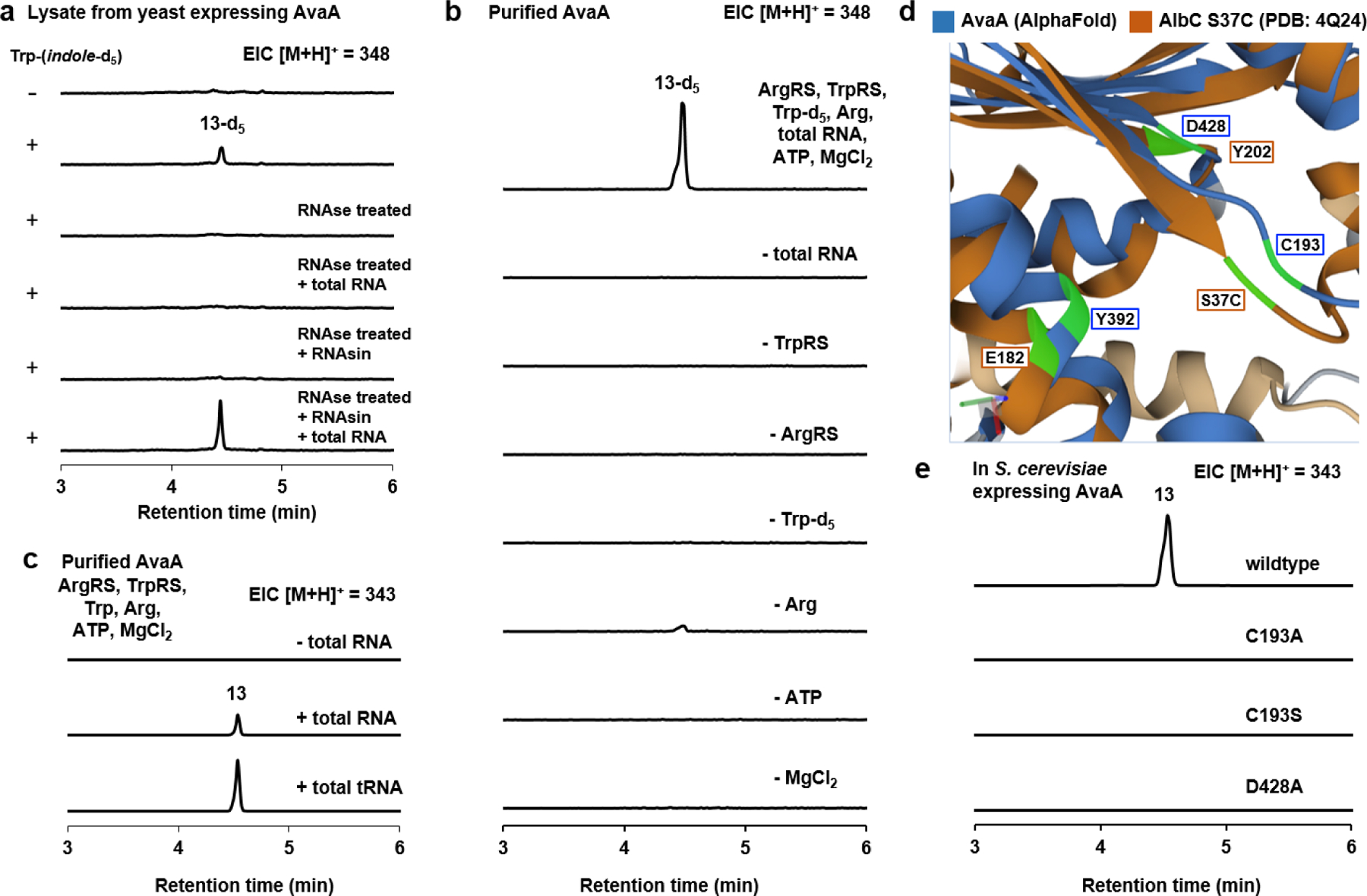
a) QTOF analysis of yeast lysate containing AvaA assayed in the presence of l-Trp-(indole)-d5. In samples treated with RNase, amino acid substrates, RNasin and total RNA were added after RNase treatment as indicated. b) In vitro assays with purified AvaA, ArgRS, TrpRS, deacylated yeast total RNA, amino acids and cofactors. Removal of any component led to loss of cyclodipeptide synthesis. The residual amount in the – Arg sample is attributed to incomplete deacylation of the total RNA sample. c) In vitro assays with purified AvaA, ArgRS, TrpRS, purified yeast tRNA, unlabeled substrates, and cofactors. d) Overlay of AlbC S37C mutant (brown) and AvaA model (blue) generated by AlphaFold. AlbC S37C structure (4Q24) was used because a suicide substrate analogue was co-crystallized in the enzyme active site. The dark blue and dark brown portions correspond to regions with similar structure, whereas the gray (AvaA) and beige (AlbC) correspond to regions with low similarity. The active site residues that are compared are highlighted in green. Predicted distances between the compared residues using RCSB Pairwise Structure Alignment (http://https://www.rcsb.org/alignment) are: AvaA C193/AlbC S37C: 2.06 Å; AvaA D428/AlbC Y202: 2.34 Å; AvaA Y392/AlbC E182: 2.58 Å. e) QTOF analysis of the extracts from yeast expression of wild type AvaA and C193A, C193S, and D428A mutants.
We then performed in vitro reactions with recombinant AvaA expressed and purified from E. coli (Fig. S9). The yeast arginine-tRNA synthetase (ArgRS, YDR341C)57 and tryptophan-tRNA synthetase (TrpRS, YOL097C)58 were also expressed and purified from E. coli (Fig. S9). Production of 13-d5 was observed in the complete reconstitution that contained AvaA, ArgRS, TrpRS, l-Trp-d5, l-Arg, deacylated total yeast RNA, ATP, and MgCl2 (Fig. 3b). Omitting ArgRS or TrpRS, as well as MgCl2 or ATP, completely abolished cyclodipeptide formation, consistent with the requirement of aminoacylated tRNA for AvaA function. Omission of l-Trp-d5 abolished product formation, whereas exclusion of l-Arg resulted in trace amounts of 13-d5 being formed. This background activity may be due to our inability to completely deacylate l-Arg-tRNA in the total RNA pool. To confirm tRNA is required, we isolated tRNA from yeast total RNA using PAGE electrophoresis (Fig. S22); followed by confirmation of isolated tRNA through RT-PCR (Fig. S24). Gratifyingly, replacement of total RNA with purified tRNA in the complete reconstitution assay above supported formation of 13 (Fig. 3c). In contrast, addition of other purified RNA fractions from PAGE, including the 5S RNA and degraded RNA front, did not support 13 synthesis (Fig. S23). Collectively, these biochemical assays confirmed AvaA and by inference, other members of the RCDPS family are tRNA-dependent cyclodipeptide synthases with the mechanism shown in Fig. S6.
CDPSs, including the well-characterized AlbC (cLF synthase, PDB: 4Q24),56 share a conserved catalytic triad of Ser, Glu and Tyr residues, of which Ser forms the covalent aminoacyl adduct followed by intramolecular DKP cyclization .56 Alignment of the six functionally verified RCDPS in Fig. 2 led to identification of only few conserved residues, including E432 and Y515, of which E432 is part of a D428D429XXE432 motif (Fig. S4). To investigate the functions of these residues in RCDPS, we expressed single-residue mutants of AvaA in yeast and evaluated cRW production. Both E432A and Y515A mutations led to complete loss of cyclodipeptide formation (Fig. S18, v-x). Yeast expressing Y515F mutation also significantly decreased the formation of 13 by 89% (Fig. S18, xi-xiii.). To gain more insight into RCDPS catalysis, using the AlphaFold59 structural prediction tool, parts of the predicted RCDPS (518 aa) structure showed a striking resemblance (RMSD 3 Å) to that of the much smaller AlbC (239 aa), with 32% coverage of AvaA (Fig. 3d and S8). The core of RCDPS adopts a Rossmann fold, which is observed for bacterial CDPSs.60 Structural comparison revealed that a conserved cysteine residue in RCDPS (C193 in AvaA) aligns with the active site serine (S37) in AlbC, and conserved D428 and D429 that are part of the DDXXE motif are predicted to be at the same location as the general base Y202 in AlbC (Fig. 3d, S8d). In addition, Y392 in AvaA is predicted to be at the same position in the active site as E182 in AlbC, which is the general base that deprotonate the positively charged amino group of the acyl acceptor for the first amide bond formation (Fig. 3d, S8e). Introducing C193A, C193S or C193T mutations completely abolished AvaA activity (Fig. 3e; S19a). In addition, D428A, D429A, Y392A and Y392F mutations also abolished AvaA activity (Fig. 3e; S19). Overall, while the full structure of RCDPS will be needed to assign the roles to these residues, C193, DDXXE, Y392 and Y515 are important for the catalytic activity of RCDPSs, and conservation of these residues should be a criterion when mining for additional RCDPS homologs. A proposed role of these residues is shown in Fig. S8.
Genome mining of ava cluster led to new natural products
To determine if the cRW cyclodipeptide 13 can be modified into more elaborate natural products, we performed coexpression of nearby genes from the A. versicolor cluster (Fig. 4a). Immediately flanking avaA is avaB that encodes an FMO with 28% identity to dimethylaniline monooxygenase; avaC that encodes a kynurenine formamidase (KFA), and avaD that encodes a protein with 38% identity to Gcn5-related N-acetyltransferases (GNAT)61 (Fig. 4a). Other genes nearby encode additional tailoring enzymes, including five P450s, a beta-lactamase, and a dioxygenase, etc. (Table S2). Most of these genes are conserved in homologous clusters from three other fungal species, suggesting their involvement in the pathway (Fig. S2). However, given the size of the entire cluster (31 kB) poses challenges for heterologous reconstitution, we chose to first examine the immediate genes flanking avaA as shown in Fig. 4a.
Fig. 4. Heterologous expression of ava tailoring enzymes from A. versicolor dI-29.

a) The ava BGC containing the RCDPS avaA flanked by tailoring genes. b) Structures of cyclo-Arg-Trp derivatives isolated from A. nidulans heterologous upon expression of immediate flanking genes.
Coexpression of avaA-D in A. nidulans resulted in a number of compounds 14–18 (Fig. S21), which were isolated and structurally characterized (Fig. 4b). Compound 14 is a pyrroloindole DKP with a fused 6/5/5/6 ring system (Fig. S68-S72, Table S17). 15 was determined to be a derivative of 14 upon hydrolysis of the guanidine group (Fig. S73-S77, Table S18), which could be catalyzed by A. nidulans arginase.31 We propose 14 is derived from AvaB catalyzed epoxidation of the indole ring in 13 to 19, followed by epoxide opening to generate the hydroxyiminium 20 that can be captured intramolecularly as a shunt pathway by the amide nitrogen (Fig. S7).62,63 On the other hand, the structures of 16 and 17 revealed cleavage of the indole ring, followed by oxidative rearrangements. 16 is the cyclo-Arg-kynurenine dipeptide (Fig. S78-S82, Table S19), while 17 contains a 10 e- conjugated benzene-isoxazole bicyclic anthranil ring structure (Fig. S83-S87, Table S20). Anthranils have emerged as privileged synthons to construct a diverse array of C-N bonds.64 To the best of our knowledge, this is the first report of an anthranil functional group found in natural products. We propose AvaB is a multifunctional flavoenzyme that is responsible for generating the cyclo-Arg-formylkynurenine DKP 21, which can be deformylated by AvaC to give 16 (Fig. S7). An additional N-oxidation of 16 by AvaB followed by cyclization and dehydration gives 17. Lastly, 18 was solved by NMR analysis to be a derivative of 17 following N-acetylation of the guanidine group (Fig. S88-S93, Table S21). Stepwise reconstitution confirmed N-acetylation is catalyzed by the Gcn5 homolog AvaD (Fig. S21). Although a handful of natural products containing acetylated arginine have been isolated previously,65,66 this is the first identification of an arginine N-acetyltransferase. Additional genes in the ava cluster were introduced into the heterologous host. However, no modification of the products could be detected. The roles of these enzymes in the ava pathway are under investigation.
Conclusions
The first discovery of aminoacyl-tRNA-dependent natural product biosynthesis was made in 2008.67 Since then, an increasing number of such enzymes have been identified from bacteria and studied.60 Leveraging aminoacyl-tRNA in natural product biosynthesis blurs the line between primary and secondary metabolism, exemplifying Nature’s expansive toolkit to promote molecular diversity. Our discovery of a new family of enzymes termed RCDPS fills in the blank of biosynthetic machineries constructing Arg-containing cyclodipeptides, across both prokaryotic and eukaryotic kingdoms. The RCDPS constitutes a new class of core genes in natural product biosynthesis, and together with the additional tailoring enzymes found here, can broaden the scope of future genome mining endeavors for novel natural products. Our work demonstrates the power of bioinformatics-guided genome mining in unlocking the vast biosynthetic potential of unknown-unknown natural product BGCs in microbial genomes.
METHODS
1. Strains and culture conditions
Aspergillus thermomutatus, Aspergillus versicolor dI-29, Apiospora montagnei, Aspergillus terreus, Penicillium thymicola, Aspergillus nomius, and Penicillium subrubescens were grown on PDA (potato dextrose agar, BD) at 28 °C for 3 days for cell proliferation or in liquid PDB medium (PDA medium without agar) for isolation of genomic DNA. Aspergillus nidulans A1145 ΔSTΔEM68 was grown at 28 °C on CD medium (1 L: 10 g of glucose, 50 mL of 20X nitrate salts, 1 mL of trace elements, pH 6.5, and 20 g/L of agar for solid cultivation) for heterologous expression of the gene cluster, compound production, and RNA extraction. For preparation of 20X nitrate salts, 120 g of NaNO3, 10.4 g of KCl, 10.4 g of MgSO4•7H2O, 30.4 g of KH2PO4 were dissolved in 1 L of double distilled water. For preparation of the trace element solution, 2.20 g of ZnSO4•7H2O, 1.10 g of H3BO3, 0.50 g of MnCl2•4H2O, 0.16 g of FeSO4•7H2O, 0.16 g of CoCl2•5H2O, 0.16 g of CuSO4•5H2O, and 0.11 g of (NH4)6Mo7O24•4H2O were dissolved in 100 mL of double-distilled water, and the pH was adjusted to 6.5.2 All Escherichia coli strains were cultured in LB media at 37 °C. Yeast strains were cultured in YPD media (yeast extract 1%, peptone 2%, glucose 2%) at 28 °C.
2. General DNA manipulation techniques
E. coli TOP10 was used for cloning, following standard recombinant DNA techniques. DNA restriction enzymes were used as recommended by the manufacturer (New England Biolabs, NEB). PCR reactions were performed using Q5 High-Fidelity DNA Polymerase (NEB), Phusion High-Fidelity DNA Polymerase (NEB), and PFX High-Fidelity DNA Polymerase (Invitrogen). The gene-specific primers are listed in Table S5. PCR products were confirmed by DNA sequencing. E. coli BL21(DE3) (Novagen) was used for protein expression. The Saccharomyces cerevisiae strain JHY65139 was used as the yeast host for in vivo homologous recombination to construct the A. nidulans expression plasmids.
For isolation of RNA from A. nidulans transformants containing the ank gene cluster, the strains were grown on CD agar for 3 days at 28 °C. The RNA extraction was performed using RiboPure™ Yeast RNA Isolation Kit (Ambion) following the manufacturer’s instructions. Residual genomic DNA in the extracts was digested by DNase I (2 U/μL) (Invitrogen) at 37 °C for 4 hours. SuperScript III First-Strand Synthesis System (Invitrogen) was used for cDNA synthesis with oligo-dT primers following directions from the user manual.
3. Heterologous expression of the ank and ava gene clusters as well as AnkA homologs from A. montagnei, A. terreus, P. thymicola, A. nomius, and E. shearii in A. nidulans
To construct plasmids for heterologous expression in A. nidulans A1145, the plasmids pYTU, pYTP, and pYTR69 with auxotrophic markers for uracil (pyrG), pyridoxine (pyroA), and riboflavin (riboB), respectively, were used as backbones to insert genes. Genes from the ank and ava gene clusters as well as AnkA homologs from A. montagnei, A. terreus, P. thymicola, A. nomius, and E. shearii and their native terminators were amplified by PCR with overhang primers using the genomic DNA (gDNA) from the native hosts as the template. Constitutive gpdA promoters from A. niger (gpdAp), Penicillium oxalicum (POgpdAp), and Penicillium expansum (PEgpdAp) as well as coxAp from A. niger were amplified by PCR. pYTP and pYTR were digested with PacI/NotI and pYTU was digested with PshAI/NotI. The overlapping DNA fragments and their corresponding digested vectors were co-transformed into S. cerevisiae JHY65139 to assemble the expression plasmids in vivo by yeast homologous recombination. The plasmids were extracted from yeast using Zymoprep™ Yeast Plasmid Miniprep I (Zymo Inc. USA), and transformed into E. coli TOP10 by electroporation to isolate single plasmids. After extraction from E. coli, the plasmids were sequenced to confirm correct assembly.
To prepare protoplasts, A. nidulans A1145 ΔSTΔEM68 was initially grown on CD agar plates supplemented with 10 mM of uridine, 5 mM of uracil, 0.5 μg/ml of pyridoxine HCl and 2.5 μg/ml of riboflavin at 30 °C for 5 days. Fresh spores of A. nidulans A1145 were inoculated into 50 mL of liquid CD media in a 250-mL flask and germinated at 30 °C, 250 rpm for 16 h. Mycelia were harvested by centrifugation at 3,500 rpm for 10 min and washed with 10 mL of osmotic buffer (1.2 M of MgSO4, 10 mM of sodium phosphate, pH 5.8). The mycelia were transferred into 10 mL of osmotic buffer containing 20 mg of lysing enzymes from Trichoderma and 15 mg of Yatalase in a 1250-mL flask. The cells were digested for 6–8 hours at 30 °C, 80 rpm and were then poured through a sterile cell strainer (Fisher, Cat No. 22363547) into a sterile 50 mL Falcon tube. The filtrate containing the digested cells was centrifuged at 4300 x g for 20 min at 4 oC. The supernatant was decanted and the pellet was washed with 10 mL of STC buffer (1.2 M of sorbitol, 10 mM of CaCl2, 10 mM of Tris-HCI, pH 7.5). The protoplasts were then resuspended in 1 mL of STC buffer.
For each transformation, plasmids were added to 100 μl of the A. nidulans A1145 protoplast suspension prepared above, and the mixture was incubated for 60 min on ice. Then 600 μl of PEG solution (60% PEG, 50 mM of calcium chloride, and 50 mM of Tris-HCl, pH 7.5) was added to the protoplast mixture, followed by additional incubation at room temperature for 20 min. The mixture was dropped on CD sorbitol agar plates (CD solid medium with 1.2 M of sorbitol and the appropriate supplements: 10 mM of uridine, 5 mM of uracil, 0.5 μg/mL of pyridoxine HCl, and/or 2.5 μg/mL of riboflavin according to the markers in the transformed plasmids) and incubated at 37 °C for 2–3 days.
4. Chemical analysis and compound isolation.
For small scale metabolite analysis in A. nidulans, transformants were grown on CD agar for 2–6 days at 28 °C and then extracted with methanol. A. nidulans strains expressing AvaABD, AvaA-D, and AvaACD were grown on CD agar for 2 days at 28 °C and then extracted with acetone. For small scale analysis of production of compounds 8, 11 and 13 in yeast, S. cerevisiae JHY651 strains expressing AnkA, AmaA, and AvaA (wildtype and mutants) were inoculated in 1 mL of dropout media for 24 hours. 100 μL of starter culture was used to inoculate 3 mL of YPD. The cells were grown at 28 °C, 250 rpm for 48 hours and the cell pellets were extracted with acetone for 8 and 11 and methanol for 13, respectively. The organic phases were dried and dissolved in 50% water 50% methanol for analysis. LC-MS analyses were performed on a Shimadzu 2020 EV LC-MS with a reverse-phase column (Phenomenex Kinetex, C18, 1.7 μm, 100 Å, 2.1 × 100 mm) using positive-and negative-mode electrospray ionization with a linear gradient of 5–95% acetonitrile-H2O (containing 0.1% formic acid) in 15 min followed by 95% acetonitrile for 3 min with a flow rate of 0.3 ml/min. QTOF analysis was performed on an Agilent Quadrupole Time of Flight LC/MS (6545 LC/Q-TOF) with a reverse-phase column (Agilent Poroshell, 120 EC-C18, 2.7 μm, 3.0 × 50 mm) using positive-mode electrospray ionization with a linear gradient of 1–95% acetonitrile-H2O (containing 0.1% formic acid) in 9 min followed by 95% acetonitrile for 3 min with a flow rate of 0.6 mL/min.
For isolation of 3 and 3b, an A. nidulans transformant expressing AnkA-F was grown on 4 L of solid CD media for 5 days at 28 °C and then extracted with methanol. For isolation of 4, an A. nidulans transformant expressing AnkA-E was grown on 4 L of solid CD media for 5 days at 28 °C and then extracted with methanol. For isolation of 2, an A. nidulans transformant expressing AnkABCDEG was grown on 4 L of solid CD media for 3 days at 28 °C and then extracted with methanol. For isolation of 1, an A. nidulans transformant expressing AnkA-G was grown on 4 L of solid CD media for 3 days at 28 °C and then extracted with methanol. The extracts were concentrated and the methanol was removed by a rotary evaporator. The pH of the aqueous extracts was lowered to 4, and the extracts were mixed with Diaion® HP-20 resin for 1 hour. The resin mixtures were then poured through a column, and the flowthrough was discarded. The resin was washed with water and 25% methanol with 0.1% formic acid. The compounds were eluted from the resin with 50% methanol. The eluent was concentrated and extracted with chloroform to remove impurities. The resulting aqueous extracts were used for purification by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and methanol (B) with 0.1% formic acid using a gradient of 0–20 min 25% B; 25–32 min 100% B; 32–39 min 25% B. For 3b, fractions containing the target compound were combined and further purified by HPLC with a semi-preparative reverse-phase column (Kinetex 5 μm C18 100 Å, 250 × 10.0 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–83 min 5–10% B; 83–90 min 100% B; 90–97 min 5% B. For 2 and 4, fractions containing the target compounds were combined and further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-MS-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–60 min 17% B; 60–67 min 100% B; 67–74 min 17% B. For 1, fractions containing the target compound were combined and further purified by HPLC with a semi-preparative reverse-phase column (Kinetex 5 μm C18 100 Å, 250 × 10.0 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–85 min 5–10% B; 85–92 min 100% B; 92–99 min 5% B.
For isolation of 7, 8, and 12, A. nidulans transformants expressing AnkA and AnkAB, respectively, were grown on 4 L of solid CD media for 4 days at 28 °C and then extracted with acetone. For isolation of 11, amaA was cloned into the yeast expression vector XW554 through yeast homologous recombination. In this plasmid, expression of amaA was under the control of the ADH2 promoter. S. cerevisiae JHY65139 expressing AmaA was inoculated in 80 mL of dropout media for 24 hours. The starter culture was used to inoculate 4 L of YPD, and the cells were shaken at 28 °C, 250 rpm for 48 hours. The cell pellet was harvested by centrifugation and extracted with acetone. The crude extracts were absorbed with 3 g of Celite, which was purified with the CombiFlash system (Teledyne) using a 100 g HP C18 column (RediSepRf) and reverse phase gradient elution with water (A) and methanol (B) using a gradient of 0–5 min 5% B; 5–125 min 5–100% B; 125–135 min 100% B. For compound 12, acetonitrile was used instead of methanol during CombiFlash purification. For compound 7, fractions containing the target compound were subject to purification by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–17 min 20% B; 17–24 min 100% B; 24–31 min 20% B. A second HPLC step was performed with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 0% B; 5–65 min 0–20% B; 65–72 min 100% B; 72–79 min 0% B. For 12, fractions containing the target compound from the CombiFlash were combined and purified by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–45 min 5–10% B; 45–54 min 100% B; 54–63 min 5% B. For 11, fractions containing the target compound from the CombiFlash were combined and purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–25 min 5–95% B; 25–32 min 95% B; 32–39 min 5% B.
For isolation of 9 and 10, A. nidulans transformants expressing PthA and AteA, respectively, were grown on 4 L of solid CD media for 4 days at 28 °C and then extracted with methanol. The crude extracts were absorbed with 3 g of Celite, which was purified with the CombiFlash system (Teledyne) using a 100 g HP C18 column (RediSepRf) and reverse phase gradient elution with water (A) and methanol (B) using a gradient of 0–40 min 5% B; 40–55 min 100% B. For 9, fractions containing the target compound were combined and purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 0% B; 5–20 min 0–1% B; 20–27 min 100% B; 27–34 min 0% B. The compound was purified with two more rounds of HPLC with the same column and solvent system using a gradient of 0–20 min 0% B; 20–27 min 100% B; 27–34 min 0% B. For 10, fractions containing the target compound from the CombiFlash were combined and purified by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–8 min 5% B; 8–15 min 100% B; 15–22 min 5% B. The compound was further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 0% B; 5–20 min 0–4% B; 20–27 min 100% B; 27–34 min 0% B.
For isolation of 5 and 6, an A. nidulans transformant expressing AnkABCDF was grown on 4 L of solid CD media for 3 days at 28 °C and then extracted with methanol. The extracts were concentrated and the methanol was removed by a rotary evaporator. The pH of the aqueous extracts was lowered to 4, and the extracts were mixed with Diaion® HP-20 resin for 1 hour. The resin mixture was then poured through a column, and the flowthrough was discarded. The resin was washed with water and 10% methanol with 0.1% formic acid. The compounds were eluted from the resin with 30% methanol. The eluent was concentrated and extracted with chloroform to remove impurities. The resulting aqueous extract was used for purification by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–40 min 5–13.8% B; 40–47 min 100% B; 47–54 min 5% B. Fractions containing 6 were further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 2% B; 5–105 min 2–12% B; 105–112 min 100% B; 112–117 min 2% B. Fractions containing 5 were further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 2% B; 5–50 min 2–13.7% B; 50–57 min 100% B; 57–64 min 2% B.
For isolation of 13, an A. nidulans transformant expressing AvaA was grown on 4 L of solid CD media for 4 days at 28 °C and then extracted with methanol. For isolation of 14 and 15, an A. nidulans transformant expressing AvaABC was grown on 4 L of solid CD media for 3 and 6 days, respectively, at 28 °C and then extracted with methanol. For isolation of 16 and 17, an A. nidulans transformant expressing AvaA-D was grown on 4 L of solid CD media for 3 days at 28 °C and then extracted with methanol. The extracts were concentrated by a rotary evaporator. The aqueous extracts were mixed with Diaion® HP-20 resin for 1 hour. The resin mixtures were then poured through a column, and the flowthrough was discarded. The resin was washed with water and 25% and 50% methanol. The compounds were eluted from the resin with 75% methanol. The eluent was concentrated and extracted with chloroform to remove impurities. The resulting aqueous extracts were used for purification by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and methanol (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–45 min 5–19% B; 45–52 min 100% B; 52–59 min 5% B. For 13 and 14, fractions containing the target compounds were further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 6% B; 5–65 min 6–13% B; 65–72 min 100% B; 72–79 min 6% B. For 15, fractions containing the target compound were purified by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–25 min 5% B; 25–32 min 100% B; 32–39 min 5% B. For 16, fractions containing the target compound were further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 2% B; 5–65 min 2–20% B; 65–72 min 100% B; 72–79 min 2% B. For 17, fractions containing the target compound were further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 2% B; 5–65 min 2–14% B; 65–72 min 100% B; 72–79 min 2% B.
The stereochemistry of 13 was determined by Marfey’s analysis and proton NMR spectroscopy. The proton NMR spectra of 13 corresponded to that of l-l or d-d cyclo-Arg-Trp.49 For Marfey’s analysis, 0.2 mg of 13 was hydrolyzed with 500 μL of 6 M HCl with the addition of 1% beta-mercaptoethanol70 to prevent degradation of tryptophan for 1 hour. Standards of l-Trp and d-Trp and hydrolyzed 13 were derivatized with Marfey’s reagent.71 The samples were analyzed on the QTOF (Fig. S20), indicating 13 contains l-Trp. Together, we concluded 13 is composed of l-Arg and l-Trp.
For isolation of 18, an A. nidulans transformant expressing AvaA-D was grown on 8 L of solid CD media for 2 days at 28 °C and then extracted with acetone. The extracts were concentrated by a rotary evaporator. The aqueous extract was mixed with Diaion® HP-20 resin for 1 hour. The resin mixture was then poured through a column, and the flowthrough was discarded. The resin was washed with water and the target compound was eluted with 25% acetonitrile. The eluent was concentrated and extracted with chloroform to remove impurities. The resulting aqueous extract was used for purification by HPLC with a semi-preparative reverse-phase column (Cosmosil 5C18-AR-II, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 5% B; 5–45 min 5–15% B; 45–52 min 100% B; 52–59 min 5% B. Fractions containing the target compound were further purified by HPLC with a semi-preparative reverse-phase column (Cosmosil PBr, 10ID x 250 mm) with water (A) and acetonitrile (B) with 0.1% formic acid using a gradient of 0–5 min 1% B; 5–125 min 1–15% B; 125–132 min 100% B; 132–139 min 1% B. To determine the position of the acetyl group in 18, the ROESY correlation between H13 to H19 indicated the guanidine group is connected to C13. The HMBC correlation from H17 to C16 and chemical shift of C16 (172.6 ppm) showed the presence of an acetyl group. ROESY correlations between H14 and H18 and between H17 and H18 indicated the acetyl group is connected to the guanidine group.
NMR spectra were obtained with a Bruker AV500 spectrometer with a 5-mm dual cryoprobe at the UCLA Molecular Instrumentation Center (1H 500 MHz, 13C 125 MHz). For compounds 1, 3, 3b, 4, 5, 6, 12, 16, and 17, NMR spectra were measured in D2O with the addition of 0.5% TFA-d to increase compound solubility. For compounds 2 and 4, NMR spectra were measured in DMSO-d6 with 0.5% TFA-d. The NMR spectra of compounds 7 and 18 were measured in DMSO-d6. The NMR spectra of compounds 8 and 13 were measured in CD3OD. The NMR spectra of compounds 9, 10, 14, and 15 were measured in D2O. Optical rotations were measured on a Rudolph Research Analytical Autopol III Automatic Polarimeter.
5. Preparation of yeast lysates containing AvaA and in vitro lysate assays
AvaA was cloned into the yeast expression vector XW5572 through yeast homologous recombination. In this plasmid, expression of avaA was under the control of the ADH2 promoter. Overexpression of AvaA was performed as follows:73 the yeast strain JHY65139 harboring the expression plasmid was grown overnight in 3 × 1 mL cultures of uracil dropout medium at 28 °C. The starter culture was used to inoculate 24 × 3 mL cultures of YPD. The cells were grown at 28 °C, 250 rpm for 24 hours. Cells were harvested by centrifugation and resuspended in lysis buffer (50 mM of Tris-HCl, 150 mM NaCl, pH 8.0, 10% glycerol). Cells were disrupted by beads (zirconia beads from RiboPure™-Yeast Kit) for 2 min with 30 s shaking intervals on ice. The lysate was centrifuged at 4 °C, 15,000 g for 10 minutes to pellet cell debris.
Lysate assays were performed in 50 mM Tris-HCl, 150 mM NaCl, pH 8.0 with a final volume of 50 μL. For the assays in Fig. S17, the lysates were first desalted using a spin desalting column (Zeba, 40K MWCO). The assays contained 0–2 mM ATP, 0–10 mM MgCl2, 5 mM l-Arg, and 5 mM Trp-d5. For the assays in Figure 3a, lysates with the addition of 6 mM DTT with and without 0.3 μg/mL RNase A (Qiagen) were incubated at room temperature for 20 min. RNasin (Promega) was added to the RNase treated samples at a final concentration of 20 U/mL, followed by incubation at room temperature for 5 min. The assays contained the treated lysates, 0.5 mM ATP, 10 mM MgCl2, 5 mM l-Arg, and 5 mM Trp-d5, with and without 400 ng/μL of total RNA purified from yeast.74 The reactions were incubated at room temperature for 18 hours, and an equal volume of methanol was added to quench the reactions. Samples were centrifuged and the supernatant was analyzed by QTOF as described previously.
6. Expression and purification of AnkD, AvaA, ArgRS, and TrpRS from E. coli BL21(DE3) and in vitro assays
The ankD gene was amplified with overhang primers from the cDNA of A. nidulans expressing AnkA-F. The avaA gene was amplified with overhang primers from the genomic DNA of Aspergillus versicolor dI-29. The argRS (YDR341C) and trpRS (WRS1) genes were amplified with overhang primers from the genomic DNA of S. cerevisiae JHY651. The expression vector pET28a was digested with NdeI/XhoI (NEB) for N-His tag expression and NcoI/XhoI (NEB) for C-His tag expression. The plasmids p3028 (N-His6-ankD), p3029 (N-His6-avaA), p3030 (trpRS-C-His6), and p3031 (argRS-C-His6) were constructed through Hifi assembly (NEB). The sequences of the assembled plasmids were confirmed by DNA sequencing.
Overexpression and subsequent protein purification of AnkD, AvaA, ArgRS, and TrpRS were performed as follows:75 BL21(DE3) harboring the expression plasmid was grown overnight in 16 × 5 mL of LB medium with 50 μg/mL of kanamycin at 37 °C. The starter cultures were used to inoculate 4 × 1 L of fresh LB medium and shaken at 37 °C until the optical density at 600 nm (OD600) reached 0.8. Then expression of the gene was induced with 0.1 mM of isopropylthio-β-d-galactoside (IPTG) at 16 °C. After 20 hours, cells were harvested by centrifugation, resuspended in lysis buffer (50 mM of Na2HPO4, 150 mM of NaCl, 10% glycerol, pH 8.0), and lysed on ice by sonication. The lysate was centrifuged at 14,000 g for 15 min at 4 °C to remove cellular debris. Purification of the recombinant His6-tagged proteins using affinity chromatography with Ni-NTA agarose resin (Qiagen) was carried out according to the manufacturer’s instructions. Purified proteins were concentrated and exchanged into storage buffer (50 mM Na2HPO4, 150 mM NaCl, 10% glycerol, pH 8.0) by using Centriprep filters (Amicon). Purified proteins were analyzed by SDS-PAGE. Bradford Protein Assay (Bio-Rad) was used to measure protein concentration. Aliquots of purified enzymes were flash frozen and stored at −80 °C. For the assays in Figure 3b, ArgRS and AvaA were further purified by FPLC with a size exclusion column (HiLoad™ 16/600 Superdex™ 75 pg) with a flow rate of 1 mL/min for 120 and 240 min, respectively, using phosphate buffer (50 mM Na2HPO4, 150 mM NaCl, 10% glycerol, pH 8.0) as the solvent.
Enzyme assays of AnkD were performed in 50 mM Tris-HCl (pH 8.0) buffer with a final volume of 50 μL. The assays contained ~100 μM of compound 6, 100 μM PLP, 1 mM O-acetyl homoserine, and 4 μM of purified AnkD. The reactions were incubated at room temperature for 30 min, and two volumes of methanol were added to quench the reactions. Samples were centrifuged and the supernatant was analyzed by QTOF as described previously.
For the assays in Figure 3b, total yeast RNA was deacylated by incubating 160 μg of total RNA in 50 μL of 1 M Tris (pH 8.8) with RNasin (20 U/μL) at 37 °C for 3 hours.76 Deacylated RNA was recovered by ethanol precipitation, and the pellet was washed with 75% ethanol before resuspension in 50 mM Na2HPO4, 150 mM NaCl buffer (pH 8.0). Enzyme assays with AvaA were performed in 50 mM Na2HPO4, 150 mM NaCl buffer with a final volume of 20 μL. The assays contained 3 mM MgCl2, 2.5 mM ATP, 0.8 mM l-Arg, 0.8 mM Trp-d5, 400 ng/μL deacylated total yeast RNA, 0.3 μM TrpRS, 0.4 μM ArgRS, and 0.1 μM AvaA. The reactions were incubated at room temperature for 18 hours, and an equal volume of methanol was added to quench the reactions. Samples were centrifuged and the supernatant was analyzed by QTOF as described previously.
For the assays in Figure 3c, yeast tRNA was purified from yeast total RNA by neutral RNA polyacrylamide gel electrophoresis.77 The tRNA band was cut out of the gel and crushed followed by overnight incubation in 3 volumes of 0.3 M NaCl at 4 °C with constant agitation to elute the tRNA. The samples were centrifuged, and the supernatant was collected. The tRNA was recovered by ethanol precipitation, and the pellet was resuspended in 50 mM Na2HPO4, 150 mM NaCl buffer (pH 8.0). To confirm the identity of the tRNA band, RT-PCR was performed using Superscript III (Invitrogen) and specific primers that anneal to a Trp tRNA containing an intron. The Trp tRNA cDNA product was amplified by two rounds of overhang PCR, and the spliced sequence was confirmed by Sanger sequencing. Enzyme assays with AvaA were performed in 50 mM Na2HPO4, 150 mM NaCl buffer with a final volume of 35 μL. The assays contained 3 mM MgCl2, 2.5 mM ATP, 0.8 mM l-Arg, 0.8 mM l-Trp, 50 ng/μL total yeast RNA or 16 ng/μL purified yeast tRNA, and 3 μM each of purified TrpRS, ArgRS, and AvaA. The reactions were incubated at room temperature for 18 hours, and an equal volume of methanol was added to quench the reactions. Samples were centrifuged and the supernatant was analyzed by QTOF as described previously.
Supplementary Material
Acknowledgement
This work was supported by the NIH grant R35GM118056 to Y.T. D.A.Y. is supported by the UCLA Dissertation Year Fellowship. K. N. is supported by an overseas postdoctoral fellowship from the Uehara Memorial Foundation in Japan.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Harvey AL, Edrada-Ebel R & Quinn RJ The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov 14, 111–129 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Kenshole E, Herisse M, Michael M & Pidot SJ Natural product discovery through microbial genome mining. Curr. Opin. Chem. Biol 60, 47–54 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Blin K et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47, W81–W87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khaldi N et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol 47, 736–741 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kautsar SA, Blin K, Shaw S, Weber T & Medema MH BiG-FAM: the biosynthetic gene cluster families database. Nucleic Acids Res 49, D490–D497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.M. Gilchrist CL, Li H & Chooi Y-H Panning for gold in mould: can we increase the odds for fungal genome mining? Org. Biomol. Chem 16, 1620–1626 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Biermann F & Helfrich EJN Hidden Treasures: Microbial Natural Product Biosynthesis off the Beaten Path. mSystems 6, e00846–21 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Ziemert N, Alanjary M & Weber T The evolution of genome mining in microbes – a review. Nat. Prod. Rep 33, 988–1005 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Hannigan GD et al. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res 47, e110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barra L et al. β-NAD as a building block in natural product biosynthesis. Nature 600, 754–758 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Patteson JB et al. Biosynthesis of fluopsin C, a copper-containing antibiotic from Pseudomonas aeruginosa. Science 374, 1005–1009 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lima ST et al. Biosynthesis of Guanitoxin Enables Global Environmental Detection in Freshwater Cyanobacteria. J. Am. Chem. Soc (2022) doi: 10.1021/jacs.2c01424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang MC, Zou Y, Watanabe K, Walsh CT & Tang Y Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev 117, 5226–5333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rix U, Fischer C, Remsing LL & Rohr J Modification of post-PKS tailoring steps through combinatorial biosynthesis. Nat. Prod. Rep 19, 542–580 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Du Y-L & Ryan KS Pyridoxal phosphate-dependent reactions in the biosynthesis of natural products. Nat. Prod. Rep 36, 430–457 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Zhao G et al. Structural Basis for a Dual Function ATP Grasp Ligase That Installs Single and Bicyclic ω-Ester Macrocycles in a New Multicore RiPP Natural Product. J. Am. Chem. Soc 143, 8056–8068 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fawaz MV, Topper ME & Firestine SM The ATP-grasp enzymes. Bioorganic Chem 39, 185–191 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bösch NM et al. Landornamides: Antiviral Ornithine-Containing Ribosomal Peptides Discovered through Genome Mining. Angew. Chem 132, 11861–11866 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Pan G et al. Discovery of the leinamycin family of natural products by mining actinobacterial genomes. Proc. Natl. Acad. Sci 114, E11131–E11140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacques IB et al. Analysis of 51 cyclodipeptide synthases reveals the basis for substrate specificity. Nat. Chem. Biol 11, 721–727 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Canu N, Moutiez M, Belin P & Gondry M Cyclodipeptide synthases: a promising biotechnological tool for the synthesis of diverse 2,5-diketopiperazines. Nat. Prod. Rep 37, 312–321 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Borgman P, D. Lopez R & L. Lane A The expanding spectrum of diketopiperazine natural product biosynthetic pathways containing cyclodipeptide synthases. Org. Biomol. Chem 17, 2305–2314 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Liu CT & Tang Y Discovery and Biocatalytic Application of a PLP-Dependent Amino Acid γ-Substitution Enzyme That Catalyzes C–C Bond Formation. J. Am. Chem. Soc 142, 10506–10515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brzovic P, Holbrook EL, Greene RC & Dunn MF Reaction mechanism of Escherichia coli cystathionine .gamma.-synthase: direct evidence for a pyridoxamine derivative of vinylgloxylate as a key intermediate in pyridoxal phosphate dependent .gamma.-elimination and .gamma.-replacement reactions. Biochemistry 29, 442–451 (1990). [DOI] [PubMed] [Google Scholar]
- 25.Faulkner JR et al. On the Sequence of Bond Formation in Loline Alkaloid Biosynthesis. ChemBioChem 7, 1078–1088 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Hai Y, Chen M, Huang A & Tang Y Biosynthesis of Mycotoxin Fusaric Acid and Application of a PLP-Dependent Enzyme for Chemoenzymatic Synthesis of Substituted l-Pipecolic Acids. J. Am. Chem. Soc 142, 19668–19677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu N et al. Targeted Genome Mining Reveals the Biosynthetic Gene Clusters of Natural Product CYP51 Inhibitors. J. Am. Chem. Soc 143, 6043–6047 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yee DA et al. Genome Mining of Alkaloidal Terpenoids from a Hybrid Terpene and Nonribosomal Peptide Biosynthetic Pathway. J. Am. Chem. Soc 142, 710–714 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu N et al. Identification and Heterologous Production of a Benzoyl-Primed Tricarboxylic Acid Polyketide Intermediate from the Zaragozic Acid A Biosynthetic Pathway. Org. Lett 19, 3560–3563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tohyama S et al. Discovery and Characterization of NK13650s, Naturally Occurring p300-Selective Histone Acetyltransferase Inhibitors. J. Org. Chem 77, 9044–9052 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Bartnik E & Weglenski P Regulation of arginine catabolism in Aspergillus nidulans. Nature 250, 590–592 (1974). [DOI] [PubMed] [Google Scholar]
- 32.Tsukamoto S, Kato H, Hirota H & Fusetani N Pipecolate derivatives, anthosamines A and B, inducers of larval metamorphosis in ascidians, from a marine sponge Anthosigmella aff. raromicrosclera. Tetrahedron 51, 6687–6694 (1995). [Google Scholar]
- 33.Kelley LA, Mezulis S, Yates CM, Wass MN & Sternberg MJE The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meister A 23. Glutamine Synthetase of Mammals. in The Enzymes (ed. Boyer PD) vol. 10 699–754 (Academic Press, 1974). [Google Scholar]
- 35.Cotton JL, Tao J & Balibar CJ Identification and Characterization of the Staphylococcus aureus Gene Cluster Coding for Staphyloferrin A. Biochemistry 48, 1025–1035 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Challis GL A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. Chembiochem Eur. J. Chem. Biol 6, 601–611 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Mydy LS, Bailey DC, Patel KD, Rice MR & Gulick AM The Siderophore Synthetase IucA of the Aerobactin Biosynthetic Pathway Uses an Ordered Mechanism. Biochemistry 59, 2143–2153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasaki Y, Akutsu Y, Suzuki K, Sakurada S & Kisara K Structure and analgesic activity relationship of cyclo-tyrosyl-arginyl and its three stereoisomers. Chem. Pharm. Bull. (Tokyo) 29, 3403–3406 (1981). [DOI] [PubMed] [Google Scholar]
- 39.Harvey CJB et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao HJ et al. Insights into the Desaturation of Cyclopeptin and its C3 Epimer Catalyzed by a non-Heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chem. Int. Ed 57, 1831–1835 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Bollinger JM Jr. et al. CHAPTER 3. Mechanisms of 2-Oxoglutarate-Dependent Oxygenases: The Hydroxylation Paradigm and Beyond. in Metallobiology (eds. Schofield C & Hausinger R) 95–122 (Royal Society of Chemistry, 2015). doi: 10.1039/9781782621959-00095. [DOI] [Google Scholar]
- 42.Walsh CT & Wencewicz TA Flavoenzymes: Versatile Catalysts in Biosynthetic Pathways. Nat. Prod. Rep 30, 10.1039/c2np20069d (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Healy FG, Krasnoff SB, Wach M, Gibson DM & Loria R Involvement of a Cytochrome P450 Monooxygenase in Thaxtomin A Biosynthesis by Streptomyces acidiscabies. J. Bacteriol 184, 2019–2029 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maiya S, Grundmann A, Li S-M & Turner G The Fumitremorgin Gene Cluster of Aspergillus fumigatus: Identification of a Gene Encoding Brevianamide F Synthetase. ChemBioChem 7, 1062–1069 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Lazos O et al. Biosynthesis of the Putative Siderophore Erythrochelin Requires Unprecedented Crosstalk between Separate Nonribosomal Peptide Gene Clusters. Chem. Biol 17, 160–173 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Katoh K, Rozewicki J & Yamada KD MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform 20, 1160–1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Izumida H, Imamura N & Sano H A Novel Chitinase Inhibitor from a Marine Bacterium, Pseudomonas sp. J. Antibiot. (Tokyo) 49, 76–80 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Furukawa T et al. Cyclic dipeptides exhibit potency for scavenging radicals. Bioorg. Med. Chem 20, 2002–2009 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Li X et al. Determination of Absolute Configuration and Conformation of a Cyclic Dipeptide by NMR and Chiral Spectroscopic Methods. J. Phys. Chem. A 117, 1721–1736 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Gondry M et al. A Comprehensive Overview of the Cyclodipeptide Synthase Family Enriched with the Characterization of 32 New Enzymes. Front. Microbiol 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vergne C et al. Verpacamides A−D, a Sequence of C11N5 Diketopiperazines Relating Cyclo(Pro-Pro) to Cyclo(Pro-Arg), from the Marine Sponge Axinella vaceleti: Possible Biogenetic Precursors of Pyrrole-2-aminoimidazole Alkaloids. Org. Lett 8, 2421–2424 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Tilvi S et al. Agelastatin E, Agelastatin F, and Benzosceptrin C from the Marine Sponge Agelas dendromorpha. J. Nat. Prod 73, 720–723 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Cusack S Aminoacyl-tRNA synthetases. Curr. Opin. Struct. Biol 7, 881–889 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Ibba M & Söll D Aminoacyl-tRNA Synthesis. Annu. Rev. Biochem 69, 617–650 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Gondry M et al. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond–forming enzymes. Nat. Chem. Biol 5, 414–420 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Moutiez M et al. Unravelling the mechanism of non-ribosomal peptide synthesis by cyclodipeptide synthases. Nat. Commun 5, 5141 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Sissler M, Eriani G, Martin F, Giegé R & Florentz C Mirror Image Alternative Interaction Patterns of the Same tRNA with Either Class I Arginyl-tRNA Synthetase or Class II Aspartyl-tRNA Synthetase. Nucleic Acids Res 25, 4899–4906 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.John TR, Ghosh M & Johnson JD Identification and Expression of the Saccharomyces cerevisiae Cytoplasmic Tryptophanyl-tRNA Synthetase Gene. Yeast 13, 37–41 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moutiez M, Belin P & Gondry M Aminoacyl-tRNA-Utilizing Enzymes in Natural Product Biosynthesis. Chem. Rev 117, 5578–5618 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Dyda F, Klein DC & Hickman AB GCN5-Related N-Acetyltransferases: A Structural Overview. Annu. Rev. Biophys. Biomol. Struct 29, 81–103 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ye Y et al. Fungal-derived brevianamide assembly by a stereoselective semipinacolase. Nat. Catal 3, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li S et al. Biochemical Characterization of NotB as an FAD-Dependent Oxidase in the Biosynthesis of Notoamide Indole Alkaloids. J. Am. Chem. Soc 134, 788–791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zou L-H et al. Copper-Catalyzed Ring-Opening/Reconstruction of Anthranils with Oxo-Compounds: Synthesis of Quinoline Derivatives. J. Org. Chem 84, 12301–12313 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Arai N et al. Argadin, a New Chitinase Inhibitor, Produced by Clonostachys sp.FO-7314. Chem. Pharm. Bull. (Tokyo) 48, 1442–1446 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Houard J et al. Cabanillasin, a new antifungal metabolite, produced by entomopathogenic Xenorhabdus cabanillasii JM26. J. Antibiot. (Tokyo) 66, 617–620 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Garg RP, Qian XL, Alemany LB, Moran S & Parry RJ Investigations of valanimycin biosynthesis: Elucidation of the role of seryl-tRNA. Proc. Natl. Acad. Sci 105, 6543–6547 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sato M et al. Involvement of Lipocalin-like CghA in Decalin-Forming Stereoselective Intramolecular [4+2] Cycloaddition. ChemBioChem 16, 2294–2298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan Y et al. Resistance-gene-directed discovery of a natural-product herbicide with a new mode of action. Nature 559, 415–418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ng LT, Pascaud A & Pascaud M Hydrochloric acid hydrolysis of proteins and determination of tryptophan by reversed-phase high-performance liquid chromatography. Anal. Biochem 167, 47–52 (1987). [DOI] [PubMed] [Google Scholar]
- 71.Ekanayake DI et al. Broomeanamides: Cyclic Octapeptides from an Isolate of the Fungicolous Ascomycete Sphaerostilbella broomeana from India. J. Nat. Prod 84, 2028–2034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cacho RA & Tang Y Reconstitution of Fungal Nonribosomal Peptide Synthetases in Yeast and In Vitro. in Nonribosomal Peptide and Polyketide Biosynthesis: Methods and Protocols (ed. Evans BS) 103–119 (Springer, 2016). doi: 10.1007/978-1-4939-3375-4_7. [DOI] [PubMed] [Google Scholar]
- 73.Hang L et al. Reversible Product Release and Recapture by a Fungal Polyketide Synthase Using a Carnitine Acyltransferase Domain. Angew. Chem 129, 9684–9688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collart MA & Oliviero S Preparation of Yeast RNA. Curr. Protoc. Mol. Biol 23, 13.12.1–13.12.5 (1993). [DOI] [PubMed] [Google Scholar]
- 75.Ohashi M et al. SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature 549, 502–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janssen BD, Diner EJ & Hayes CS Analysis of aminoacyl- and peptidyl-tRNAs by gel electrophoresis. Methods Mol. Biol. Clifton NJ 905, 291–309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petrov A, Tsa A & Puglisi JD Chapter Sixteen - Analysis of RNA by Analytical Polyacrylamide Gel Electrophoresis. in Methods in Enzymology (ed. Lorsch J) vol. 530 301–313 (Academic Press, 2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.