

Abstract
Complement factor D (FD) is a rate-limiting enzyme of the alternative pathway (AP). Recent studies have suggested that it is synthesized as an inactive precursor and its conversion to enzymatically active FD is catalyzed by mannan-binding lectin-associated serine protease 3 (MASP3). However, whether MASP3 is essential for AP complement activity remains uncertain. It has been shown that Masp1/3 gene knockout did not prevent AP complement overactivation in a factor H knockout mouse, and a human patient lacking MASP3 still retained AP complement activity. In this study, we have assessed AP complement activity in a Masp3 knockout mouse generated by CRISPR/Cas9 editing of the Masp1/3 gene. We confirmed specific Masp3 gene inactivation by showing intact MASP1 protein expression and absence of mature FD in the mutant mice. Using several assays including lipopolysaccharide- and zymosan-induced C3b deposition and rabbit red blood cell (RBC) lysis tests, we detected plasma concentration-dependent AP complement activity in Masp3 gene-inactivated mice. Thus, although not measurable in 5% plasma, significant AP complement activity was detected in 20–50% plasma of Masp3 gene-inactivated mice. Furthermore, whereas FD gene deletion provided more than 90% protection of CD55/Crry-deficient RBCs from AP complement-mediated extravascular hemolysis, Masp3 gene deletion only provided 30% protection in the same study. We also found pro-FD to possess intrinsic catalytic activity, albeit at a much lower level than mature FD. Our data suggest that MASP3 deficiency reduces but does not abrogate AP complement activity and this is explained by intrinsic pro-FD activity which can be physiological relevant in vivo.
Introduction:
The complement system is composed of a group of plasma proteins that can be activated in a cascade of reactions to produce effectors for host defense (1, 2). Many of the complement proteins are dormant inactive proteases that are activated either by autocatalysis through conformational change or by cleavage with upstream proteases (1, 2). Complement activation occurs via three pathways, the classical, lectin and alternative pathways (1, 2). C1r, C1s and C2 are key proteases of the classical pathway that is triggered by immune complexes (1, 2). Mannose-binding lectin (MBL)-associated serine proteases (MASPs) are proteases of the lectin pathway (LP) that is triggered by pattern-recognition molecules such as MBL and ficolins (3). The constitutively active alternative pathway (AP) is driven by two proteases, factor B (FB) and factor D (FD), with FD being upstream of FB and a rating-limiting enzyme (4–6). Many of the proteases of the complement system including C1s, MASPs, FB and FD are targets of anti-complement drugs under development for human diseases (7–9).
Three MASP enzymes in the lectin pathway have been identified, MASP1, MASP2 and MASP3 (3). MASP2 is encoded by the Masp2 gene (10, 11), whereas MASP1 and MASP3 are derived from a single Masp1/3 gene via alternative splicing (12). Until relatively recently, the relationship among the 3 MASP enzymes and their role in LP activation were not clear (13). While studies in mice and human clearly showed that MASP2 is critical for LP complement activity (14, 15), gene knockout of Masp1/3, which eliminated the production of both MASP1 and MASP3, also abrogated LP activity (13). Surprisingly, Masp1/3 gene inactivation additionally inhibited AP complement activity in mice (16). Subsequent experiments based on exon-specific Masp1/3 gene mutation in mice, leading to selective MASP1 or MASP3 enzyme deficiency, established that MASP1, working upstream of MASP2, but not MASP3, is required for LP complement activation. On the other hand, selective Masp3 gene inactivation, like non-specific Masp1/3 gene knockout but unlike selective Masp1 gene inactivation, impaired AP complement activity (17).
It is now understood that the mechanism by which MASP3 regulates AP complement activity is through its role in FD maturation by cleaving off 5 amino acids at the N-terminus of a FD precursor referred to as pro-FD (16). Historically FD has been thought to be constitutively active and is regarded as the only protease of the complement pathways that does not require activation (4). That MASP3 plays a key role in the maturation of FD has renewed our knowledge of FD biosynthesis and function and places MASP3 in the AP complement cascade (16).However, several uncertainties remain, chiefly among them is the degree to which AP complement activity is dependent on MASP3. In both the original report of a Masp1/3 gene knockout mouse and a subsequent paper on a Masp3 single knockout mouse, AP complement activation was described to be abrogated (16, 17). However, a separate study of the same Masp1/3 knockout mouse and a Masp1/3-Factor H double knockout mouse revealed high AP complement activity in both mouse strains, as assessed by a rabbit red blood cell (RBC) lysis test and C3 glomerulopathy phenotyping, respectively (18). Furthermore, it has been reported that a human 3MC (Mingarelli, Malpeuch, Michels, and Carnevale syndromes) patient who was deficient in MASP3 still retained significant AP complement activity (19, 20). In the current study, we have evaluated AP complement activity in a new Masp3-specific knockout mouse generated by CRISPR/Cas9. We found that MASP3 deficiency reduced but did not abrogate AP complement activity and this can be explained by intrinsic pro-FD activity in the absence of MASP3. Our data clarify the uncertainties regarding the role of MASP3 in AP complement activation and have implications for therapeutic target selection in the treatment of AP complement-mediated diseases.
Materials and Methods
Generation of Masp3 knockout mice
MASP3 mRNA and MASP1 mRNA are transcribed from the same Masp1/3 gene via alternative splicing (12). In order to generate a Masp3-specific knockout mouse without disrupting the normal production of MASP1 which plays a critical role in the lectin pathway, we used the CRISPR/Cas9 system to selectively delete exon 12 of the Masp1/3 gene (NCBI reference ID NC_000082.7). Exon 12 encodes the serine protease (SP) domain of MASP3 but is not required for MASP1 mRNA transcript (12). Two CrRNAs targeting the MASP3 SP domain (CrRNA1 CTCCCGAACCACTTGTCGTT and CrRNA3 GCCAGCTATGAGTCTCGGTC) were designed by using the ZHANG lab CRISPR design tool and synthesized at Integrated DNA Technologies (IDTDNA, Iowa, USA), Cas9 (IDTDNA), CrRNAs and tracrRNA (IDTDNA) ribonuclease complex were prepared according to IDTNDA protocols (21). Fertilized oocytes, harvested from C57BL/6J mice, were injected with Cas9 ribonuclease complex and implanted into pseudo-pregnant foster mice at the Transgenic and Chimeric Mouse Facility of the University of Pennsylvania School of Medicine. Masp3 knockout mouse pups were genotyped by PCR using tail DNA and the following primers: Masp3ex12F2, 5’-AAACCTGGTGAAGAGAATCATCGGAG-3’ and Masp3ex12R2, 5’-TGCTTGCTACCACATTCTTCAGGTCC-3’. In this PCR screening, wild-type (WT) Masp3 gene allele is represented by a 756 bp fragment whereas the knockout allele is represented by a 252 bp PCR fragment. The wildtype and mutant mouse Masp1/3 gene sequences are provided in Supplemental Figure 1 and 2.
Recombinant expression and purification of MASP3, MASP1 and pro-fD
Using a commercial cDNA clone in pCDNA3.1 (Genscript #Omu15900CM-1) as a template, an expressing cDNA fragment of mouse MASP3 was amplified by PCR using 5’-TTTTGGCAAAGAATT-CATGAGGTTCCTGTCTTTCTG-3’ (forward) and 5’-CCTGAGGAGTGAATTCTCAATG-GTGATGGTGATGAT-3’ (reverse) as primers. The cDNA was subcloned into the pCAGGS expression vector (22) with a C-terminal 6x His tag. To clone an expression cDNA of mouse MASP1, we first made an S627A mutation in the protease domain (23) of a commercial cDNA plasmid (Transomic, # BC106945) by PCR using 5’-GGCCTCCAGCGTCACCAGCACAG-3’ (forward) and 5’-GTGACGCTGGAGGCCCTATGGTGAC-3’ (reverse) as primers. Using the mutated plasmid as a template, we then amplified the expression cDNA using 5’-TTTTGGCAAAGAATTCGGCAGAAGGAACCAAAGCC-3’ (forward) and 5’-CCTGAGGAGTGAATTCTCAATGGTGATGGTGATGGTGGTTCCTCACCC-3’ (reverse) as primers. The expression cDNA of mouse MASP1 was cloned into the pCAGGS vector with C-terminal His tag. Mouse pro-FD cDNA with an N-terminal His tag was synthesized by IDTDNA and cloned into the pCAGGS vector. Cloning of all protein expression cDNAs into pCAGGS at EcoRI restriction site was carried out using Infusion cloning method (24). For protein expression, Expi-CHO cells were transfected with cDNA plasmids by following manufacture’s transfection and culture protocols (Thermo Fisher Scientific# A29133) in the presence of leupeptin. MASP3 and MASP1 proteins were purified using cOmplete His-Tag resin according to manufacture’s protocols (Roche# 05893682001), followed by a second-round purification using CM-Sepharose chromatography (Sigma#CCF100). Pro-FD was purified by cOmplete His-Tag resin first, followed by immuno-affinity column chromatography using Sepharose beads (Cytiva life sciences, # 17098101) coupled with an in-house generated mouse anti-mouse FD mAb clone 14F11-3.
Generation of recombinant mouse FD
Mouse mature FD cDNA was amplified by nested PCR using mouse FD cDNA plasmid (Sino biologicals cat #MG50539-M) as a template 5’-GGAGCGGCTGTATGTGCAGCAATT-CTGGGTGGCCAGGAGGCC-3’ (forward-1), 5’-CCCGAATTCGCCACCATGCACAG-CTCCGTGTACTTC-3’ (forward-2) and 5’-CCCGAATTCCCCTCAGGATGTCATGT-TACCATTTGTGATGTT-3’ (reverse) as primers. The cDNA was cloned into the pCAGGS vector with a C-terminal His tag and recombinant protein expressed in HEK293 cells by cDNA transfection with Polyethylenimine (PEI). Recombinant FD was purified from culture medium using Ni-NTA His tag resin (Qiagen# 30210).
Generation of anti-mouse MASP3 and anti-mouse FD antibodies
Rabbit anti-mouse MASP3 and anti-mouse FD polyclonal antibodies were generated by Cocalico Biologicals (Stevens,PA, USA) using purified recombinant mouse MASP3 or mouse FD protein as an immunogen (450 μg/rabbit in 5 immunizations). Total IgG from antiserum was purified by the Caprylic acid method (25). From total IgG antibodies, MASP3 or FD reacting IgGs were enriched by affinity column chromatography using Sepharose beads coupled with recombinant mouse MASP3 or FD.
LPS AP assay
Micro titer plates (Nunc Maxisorb# 442404) were coated with lipopolysaccharide (Salmonella typhosa LPS; Sigma) (2μg/well) in PBS 1hr at 37°C. After washing the plate wells with PBST (phosphate buffered saline and 0.05% tween) 3 times, wells were treated with a blocking buffer (1% BSA in PBS) for 1hr at RT. Mouse plasma (50 μg/ml Bivalirudin final), collected using bivalirudin (Hospira Inc., Lake Forest, IL, 50 μg/ml) as an anticoagulant and diluted to the desired percentage with Mg2+-EGTA GVB2+ buffer, was then added to the plate wells (50 μl/well). Plasma samples diluted in the same way but containing EDTA (10 mM) were used as negative controls of AP complement activation. AP complement activation in the plate was allowed to proceed for 1 hr at 37°C. After washing 3 times with PBS-Tween, plate wells were incubated with an HRP-conjugated goat anti-mouse C3 polyclonal antibody (MP biologicals # 0855557) (1:4000 diluted in blocking buffer) for 1hr at room temperature. Plate was washed 3 times with PBS-T and developed with HRP substrate (100 μl 1step Ultra TMB, Thermo Scientific). After 5 min, reaction was stopped with 50 μl of 2N H2SO4 and plate was read at 450 nm in a micro plate reader. In some experiments, the tested mouse plasma was pre-treated with sodium polyanethole sulfonate (SPS, Sigma # P2008, 150μg/ml) before being added to the plate. SPS was used to inhibit CP and AP complement activities.
Chicken RBC lysis assay
Mouse serum was diluted to 10% with GVB2+ buffer, mixed and incubated with Ab-sensitized chicken RBC (catalog no. R401-0050; Rockland Immunochemicals) (2.5×106 cells per reaction, final volume 50 μl) at 37°C for 30 min. Ab-sensitized chicken RBCs were prepared by incubating the cells with a rabbit anti-chicken RBC Ab (150 μg/ml, catalog no. 103-41390; Rockland Immunochemicals) on ice for 1 h. Percentage of lysis was calculated by normalizing the cell-free hemoglobin OD value to that of a completely lysed RBC sample through hypotonic lysis in water. To check the effect of SPS on chicken RBC lysis, serum was pre-mixed with different concentrations of SPS before hemolytic assays.
Rabbit RBC lysis assay
Rabbit RBC lysis assay was performed with mouse Bivalirudin plasma diluted to 20% or 50% in GVB-Mg2+EGTA (10 mM) buffer. Plasma was mixed with 5 μl of rabbit RBC cell suspension (5×108/ml, Rockland, R403-0100) in a final volume of 50 μl and incubated at 37°C for 30 min. In the mouse FD mAb blocking assay, plasma was pre-treated with 500 μg/ml mouse anti-mouse FD mAb (clone 14F11-3, generated inhouse) for 30 min at 4°C before incubating with rabbit RBCs. Reaction was stopped with 100 μl of cold 10 mM EDTA in PBS. Cells were centrifuged at 1500 rpm for 5 min at 4°C. OD of the collected supernatant was measured at 405 nm.
Mannan LP assay
Micro titer plates (Nunc Maxisorb# 442404) were coated with 50μl/well mannan (Sigma, 20μg/ml) in sodium bicarbonate buffer pH 9.0 overnight at 4°C. After washing the plate wells with PBST 3 times, wells were treated with a blocking buffer (1% BSA in PBS) for 1hr at RT. Mouse Bivalirudin plasma, diluted to the desired percentage with GVB2+ buffer containing SPS (150 μg/ml final), was added to the plate wells (50 μl/well). Samples diluted in the same way but containing EDTA (10 mM) were used as negative controls of complement activation. Complement activation in the plate wells was conducted for 1 hr at 37°C. After washing 3 times with PBST, plate wells were incubated with an HRP-conjugated goat anti-mouse C3 polyclonal antibody (1:4000 diluted in blocking buffer) for 1hr at room temperature. The plate was washed 3 times with PBST and developed with HRP substrate (100 μl 1step Ultra TMB, Thermo scientific). After 5 min, the reaction was stopped with 50 μl of 2N H2SO4 and the plate was read at 450 nm in a micro plate reader.
Ova/anti-Ova IC CP assay:
OVA/anti-OVA immune complexes for complement activation were prepared as previously described (26). Briefly, 10% mouse Bivalirudin plasma was diluted with GVB2+ buffer and 50 μl was added to each well of ELISA plates pre-fixed with OVA/anti-OVA immune complexes. The plates were incubated at 37° C for 1 hr followed by detection of plate-bound activated C3 using HRP conjugated goat anti-mouse C3 polyclonal antibody.
Zymosan AP assay:
Zymosan-induced AP complement activation assay was performed as described previously (26). Briefly, 2.5 μl zymosan (2.5mg/ml) was mixed and incubated with 50 μl 20% Bivalirudin plasma diluted in Mg2+-EGTA GVB2+ for 15 minutes at 37°C, and C3 deposition was detected by FITC-conjugated Goat anti-mouse C3 (MP biologicals #0855500) (1:250 dilution) and analyzed by fluorescence-activated cell sorting (FACS).
Extravascular hemolysis (EVH) test
EVH test was performed as previously described (27). Briefly, CD55−/−Crry−/−C3−/− mouse erythrocytes (from 150 μl blood for each recipient mouse) were labeled ex vivo with CFSE (Molecular Probes) in 1 ml PBS containing 5 mM CFSE. The labeling reaction was carried out at RT for 5 min, and then cells were washed several times in PBS. CFSE-labeled CD55−/−Crry−/−C3−/− erythrocytes were transfused intravenously (retro-orbital route) into WT, Masp-3 knockout or FD knockout mice. Blood was collected from the tail vein at 5 min and various subsequent time points as indicated, and the percentage of labeled erythrocytes present was calculated after FACS analysis and normalized to values at 5 min in each mouse.
Western analysis:
For detection of MASP3, mouse EDTA plasma (2μl) was resolved on 4–12% Express plus gradient polyacrylamide gel (Genscript #M41212) under non-reducing conditions and transferred to PVDF membranes. MASP3 was detected by sequential incubation with purified rabbit anti-mouse MASP3 IgG (1μg/ml), HRP-conjugated goat anti-rabbit IgG (1:4000 dilution; Sigma-Aldrich) and the ECL chemiluminescent detection system (Amersham Pharmacia Biotech, Uppsala, Sweden). For detection of mouse FD and pro-FD, 2μl Bivalirudin plasma was first deglycosylated with the PNGase F kit from New England Biolabs (cat# P0704S) and proteins were resolved on a 4–20% Express plus gradient polyacrylamide gel (Genscript #M42012) under reducing conditions. FD was detected by using in-house generated rabbit polyclonal anti-mouse FD IgGs (2μg/ml) as the primary antibody and same procedures as in MASP3 detection.
Functional testing of mouse FD in normal human serum
To test if mouse FD can replace human FD and support AP complement activity in normal human serum (Complement Technology, Inc, Cat #NHS), LPS-induced AP complement activation assay was performed using 10% NHS in which human FD was blocked with a neutralizing mAb (AFD, inhouse generated as a recombinant human IgG4 mAb according to published sequences (28, 29) (10 μg/ml final concentration) and recombinant mouse FD (in-house generated and C-terminal His-tagged [unpublished]) added to various concentrations. C3b deposition was detected by HRP conjugated goat anti-human C3 polyclonal antibody (MP biologicals # 0855237).
Assessment of conversion of recombinant pro-FD to FD in vitro by recombinant MASP3
To test the conversion of recombinant pro-FD to mature FD by recombinant MASP3, 2μg of recombinant pro-FD with an N-terminal 6xHis-tag was incubated with 1μg recombinant MASP3 in 20 μl PBS at 37°C for different lengths of time. The samples were then subjected to deglycosylation treatment using the PNGase kit (New England Biolabs # P0704S) before Western blot detection using an anti-6xHis-tag mAb (R&D#MAB050H) for the presence or absence of the 6xHis-tag in pro-FD and mature FD, respectively.
Factor B (FB) cleavage assay
Different amounts of pro-FD or MASP3-treated pro-FD (mature FD) were incubated with 2μg human FB (Complement Tech #A135) and 2.5 μg human C3b (Complement Tech #A113) in a total volume of 25 μl PBS containing 5 mM Mg+2 at 37°C for 1hr. FB cleavage by pro-FD or mature FD was assessed by running the reaction mixtures on an 4–12% Express plus gradient polyacrylamide gel under reducing conditions and staining the gel with Coomassie blue. The degree of FB cleavage was measured by densitometry of Bb band intensity using the Li-COR Odessy Fc system (Li-COR biosystems).
Results:
MASP3 and MASP1 mRNAs are transcribed by alternative splicing from a single Masp1/3 gene (12). To generate a Masp3-specific knockout (Masp3−/−) mouse without disturbing Masp1 gene expression, we used CRISPR/Cas9 ribonuclease-mediated gene targeting to delete exon 12 of the Masp1/3 gene (Fig 1a). Exon 12 is specific to Masp3 and encodes the serine protease domain of MASP3 (12). Two CrRNAs were designed to excise a 470 bp fragment from exon 12 (Fig 1a). Mice borne out of zygotes injected with the Cas9 ribonuclease complex were genotyped by PCR using tail DNA. Intact Masp1/3 gene allele was indicated by a 756 bp band whereas exon 12-targted allele by a 252 bp band (Fig 1b). The expected deletion of 470 bp from exon 12 was also confirmed by sequencing of the mutant allele (Fig 1a). To help confirm MASP3 deficiency in the mutant mice, we recombinantly expressed mouse MASP3 as a His-tagged protein and used it to generate a polyclonal rabbit anti-mouse MASP3 antibody (Fig 1c, 1d). Since MASP3 and MASP1 share a common heavy chain, the rabbit anti-mouse MASP3 antibody recognized both recombinant MASP3 and recombinant MASP1 which was expressed as His-tagged protein with a S627A mutation in the protease domain (23) (Fig 1c, 1d). Using this antibody, we performed western blot analysis of mouse plasma and confirmed that MASP3 protein was completely absent in Masp3−/− mice whereas MASP1 protein was present at normal levels (Fig 1e). This data demonstrated selective inactivation of Masp3 without affecting Masp1 gene expression in Masp3−/− mice. We also analyzed the effect of MASP3 deficiency on FD maturation. As shown in Fig 1f, Western blot analysis of deglycosylated plasma samples detected FD in Masp3−/− mice to have a slightly higher molecular weight than FD in wildtype mice, consistent with lack of processing of pro-FD to mature FD in the absence of MASP3. These results collectively showed that Masp3 gene was specifically and completely inactivated in the mutant mice, and that no MASP3 protein and mature FD were present in them.
Fig1: Generation and characterization of Masp3 gene knockout mice.
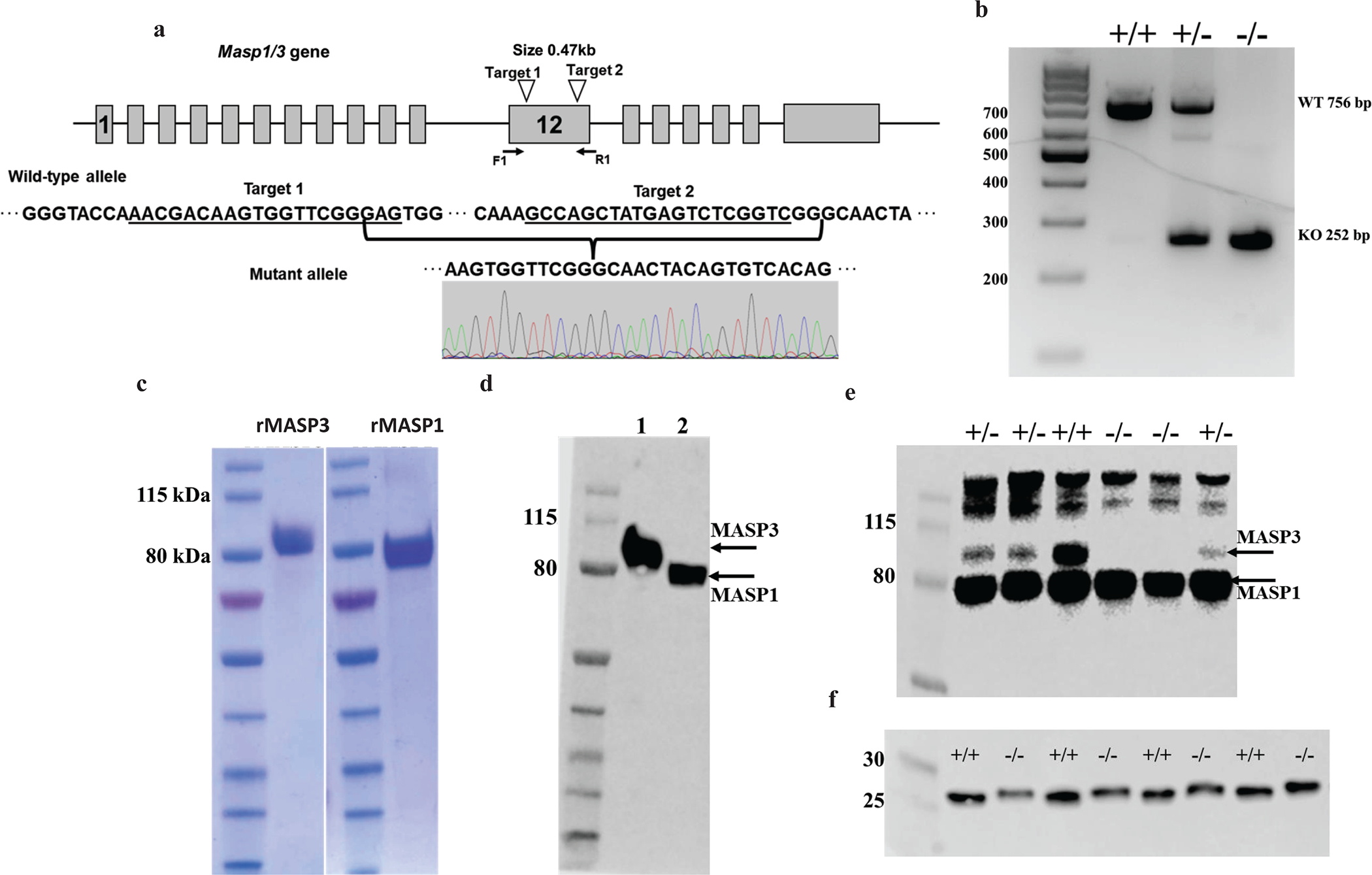
(a) Schematic representation of the mouse Masp1/3 gene locus and Masp3 gene targeting strategy. Grey boxes indicate the location of exons. The locations of CrRNA 1 and CrRNA 2 targeting sites and sequences are indicated by open arrowheads and underlines, respectively. Locations of PCR primers (F1 and R1) used for mouse genotyping are shown by solid arrows. Nucleotide sequence of the two targeting sites and recombinant sequence after partial exon 12 deletion (470 bp) are also shown. (b) Representative result of PCR genotyping of wildtype (+/+), heterozygous (+/−) and homozygous (−/−) Masp3 gene knockout mice. Wildtype (WT) allele is represented by a 756 bp band whereas the mutant allele is represented by a 252 bp band. (c) SDS-PAGE analysis of purified recombinant mouse MASP3 and MASP1(S627A) under non-reducing condition. (d) Western blot analysis showing a rabbit anti-mouse MASP3 antibody recognized both recombinant MASP3 (lane1) and MASP1(S627A) (lane2) as they share the same heavy chain. (e) Western blot analysis of mouse plasma (2 μl/lane) using a rabbit anti-MASP3 polyclonal antibody showing MASP3 protein was absent in homozygous Masp3 knockout mice (−/−) and severely reduced in heterozygous Masp3 knockout mice (+/−) compared with wildtype mice (+/+). In contrast, MASP1 protein levels were similar in all three genotypes of mice (representative of 4 experiments). (f) Western blot analysis of mouse plasma (2 μl/lane) using a rabbit anti-mouse FD polyclonal antibody showing FD protein in Masp3−/− mice to have a slightly higher molecular weight, suggesting it was pro-FD. Note there was no trace of mature FD in Masp3−/− mice. Plasma samples were deglycosylated for 2h before being analyzed on 4–20% gradient polyacrylamide gel under reducing condition (representative of 3 independent experiments). Data in panels b) to d) are representative of at least two independent experiments.
To assess the impact of MASP3 deficiency on complement activity, we performed pathway-specific in vitro complement activation assays using 10% Bivalirudin anti-coagulated plasma from Masp3−/− mice. Because mannan can activate CP and AP complement, to assay mannan-induced LP complement activity specifically, we tested and used a known chemical inhibitor of human CP and AP complement, sodium polyanethole sulfonate (SPS) (30), in wildtype mouse plasma. As shown in Fig2a, we found SPS dose-dependently inhibited mouse CP complement in a chicken RBC lysis assay, achieving complete inhibition at 150 μg/ml. At this concentration, SPS also efficiently blocked LPS-induced mouse AP complement activation (Fig 2b). We next tested mannan-induced LP complement activity in Masp3−/− mice in the presence of SPS. Fig 2c shows that Masp3−/− mice retained normal LP complement activity, suggesting that MASP3 is not required for LP complement activation. We also tested CP complement activity using an OVA/anti-OVA immune complex-based ELISA assay (in GVB buffer and without SPS) and found it to be partially impaired in Masp3−/− mice (Fig 2d), likely due to reduced contribution from AP amplification as we have demonstrated previously with properdin- and FB-deficient mice (26).
Fig 2: Assessment of lectin and classical pathway complement activity in Masp3−/− mouse plasma.

(a) Sodium polyanethole sulfonate (SPS) dose-dependently inhibited mouse classical pathway complement activity in a chicken red blood cell (cRBC) hemolytic assay. Percentage of cRBC lysis was normalized to a cRBC sample completely lysed by hypotonic shock in double-distilled water. (b) SPS at 150 μg/ml also effectively inhibited mouse AP complement activity in an LPS-induced C3b deposition ELISA assay. (c) Assessment of lectin pathway (LP) complement activity in a mannan-based C3b deposition ELISA assay in the presence of 150 μg/ml SPS. The result showed normal LP activity in Masp3−/− mice (Masp3KO). (d) Assessment of classical pathway (CP) complement activity in an OVA/anti-OVA immune complex-based C3b deposition ELISA assay. The result showed reduced CP activity in Masp3−/− mice (Masp3KO), likely reflecting impaired amplification by the alternative pathway. Panels a-d all used 10% plasma from wildtype (WT), Masp3−/− (Masp3KO), C5 knockout (C5KO), C4 knockout (C4KO), factor B knockout (fBKO) and C3 knockout (C3KO) mice. WT plasma with EDTA added, and C5KO, C4KO, fBKO and C3KO mouse plasma were used as negative controls. Results in panels a and b are representative of two independent experiments each, and each bar represents the average of duplicate assays. Results in panels c and d shown are mean values with SD of triplicate assays and representative of at least two independent experiments. * p =0.0104, ** p=0.0074
Assay of LPS-induced AP complement activation in 10% plasma of Masp3−/− mice showed severely impaired activity compared with wildtype mice (Fig 3a), but the degree of reduction varied between different experiments (data not shown). The impairment in AP complement activation could be corrected by the addition of recombinant mouse MASP3 (Fig 3a). To more definitively establish if MASP3 deficiency abrogated AP complement activity, we performed LPS-induced complement activation assay using different concentrations of plasma. As shown in Fig3b–3d, we found partial AP complement activity in 20% and 50% but not 5% Masp3−/− mouse plasma. This was in clear contrast to FD knockout mouse plasma which showed no AP complement activity at any concentrations. These data suggested that AP complement activity in Masp3−/− mice was substantially reduced but not abrogated as in FD knockout mice.
Fig 3: Detection of alternative pathway (AP) complement activity in Masp3−/− mouse plasma.
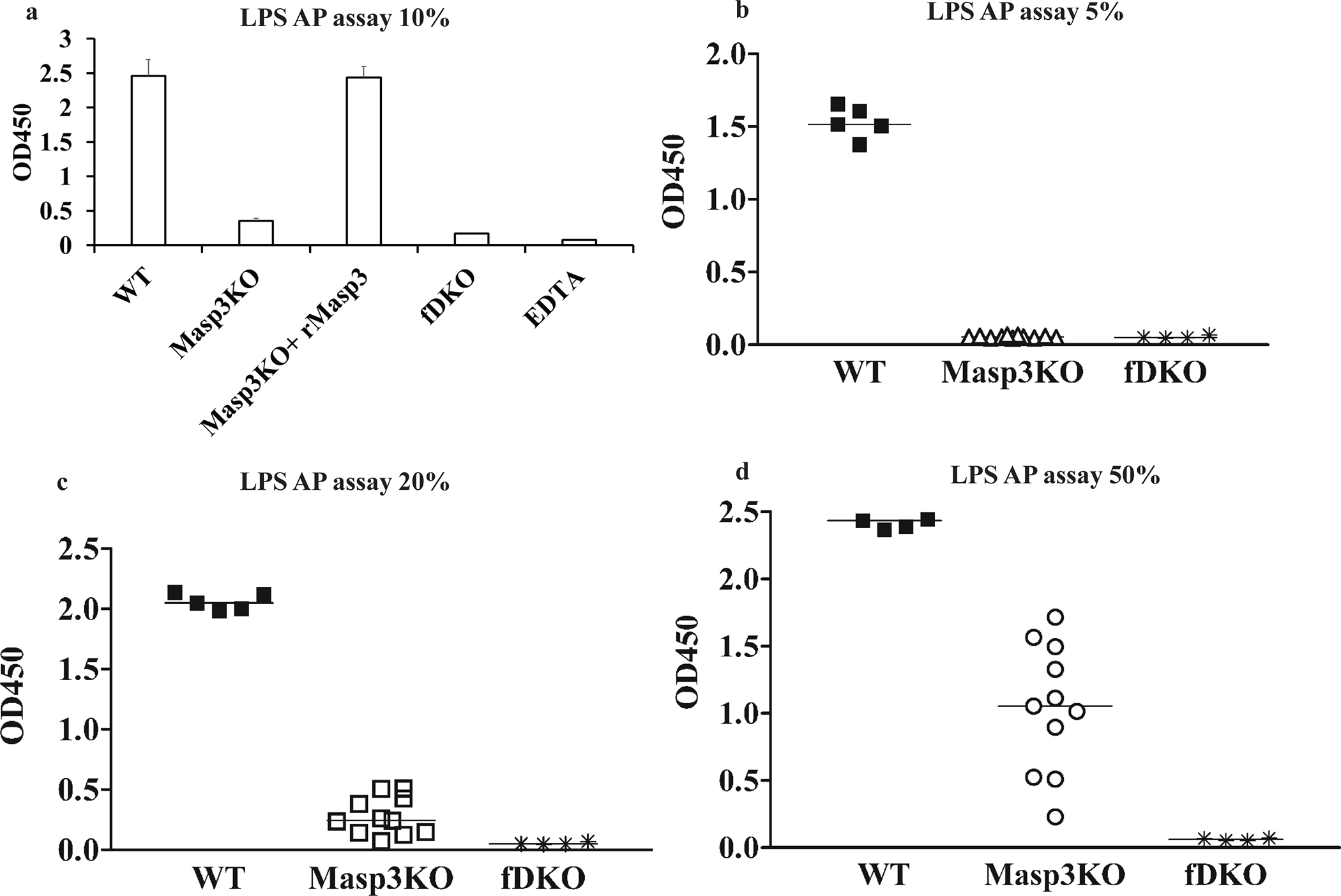
(a) LPS-induced AP complement activity in 10% Masp3−/− mouse plasma was markedly reduced but restored by reconstitution with recombinant mouse MASP3 protein (10 μg/ml final concentration). Note the OD value in Masp3−/− plasma assay did not go down to background levels of FD knockout mouse plasma or wildtype EDTA plasma assays (Bar values are mean with SD of triplicate assays using pooled plasma from 2–3 mice). Data are representative of at least two independent experiments. (b-d) LPS-induced AP complement activity assay using 5%, 20% or 50% wildtype (WT), Masp3−/− (Masp3KO) or FD knockout (fDKO) mouse plasma. The data showed plasma concentration-dependent AP complement activity in Masp3−/− mice, with no activity detectible in 5% plasma but significant activity detected in 50% plasma. In contrast, no AP activity was detected at any plasma concentrations in FD knockout mice. Each symbol represents the average of duplicate assays of plasma from an individual mouse. WT (n=5), Masp3KO (n=11), fDKO (n=4).
To verify the above conclusion, we measured AP complement activity in Masp3−/− mice using several other in vitro and in vivo assays. Fig 4a and 4b show the result of zymosan-induced AP complement activation in 20% plasma, using as readout C3b deposition of zymosan particles measured by FACS. As expected, no complement activation occurred on zymosan with FD knockout mouse plasma. In contrast, significant C3b opsonization was detected on zymosan incubated with Masp3−/− mouse plasma, although it was much less than that detected on zymosan incubated with wildtype mouse plasma. We detected a similar plasma concentration-dependent AP complement activity in Masp3−/− mice in the rabbit RBC hemolytic assay. Although a varied and generally lower than wildtype activity was detected in 20% Masp3−/− mouse plasma, 50% Masp3−/− mouse plasma was almost as active as wildtype mouse plasma in lysing rabbit RBCs (Fig 5a and 5b). Finally, we used an in vivo assay, involving transfusion and extravascular hemolysis of CFSE-labeled CD55/Crry-deficient RBCs (27), to assess AP complement activity in Masp3−/− mice. In this assay, when RBCs harvested from CD55/Crry/C3 triple knockout mice are transfused into complement-sufficient recipient mice, they are susceptible to, and rapidly eliminated by, AP complement-mediated phagocytosis (31). Fig 4c shows that while CD55/Crry/C3 knockout mouse RBCs were largely protected from elimination over a 3-day period after transfusion into FD knockout recipient mice, approximately 70% of such transfused cells were eliminated in Masp3−/− recipient mice, an outcome that was closer to that observed in wildtype recipients than in FD knockout recipients.
Fig 4: Assessment of AP complement activity in Masp3−/− mice using zymosan-based and red blood cell-based C3b opsonization assays.
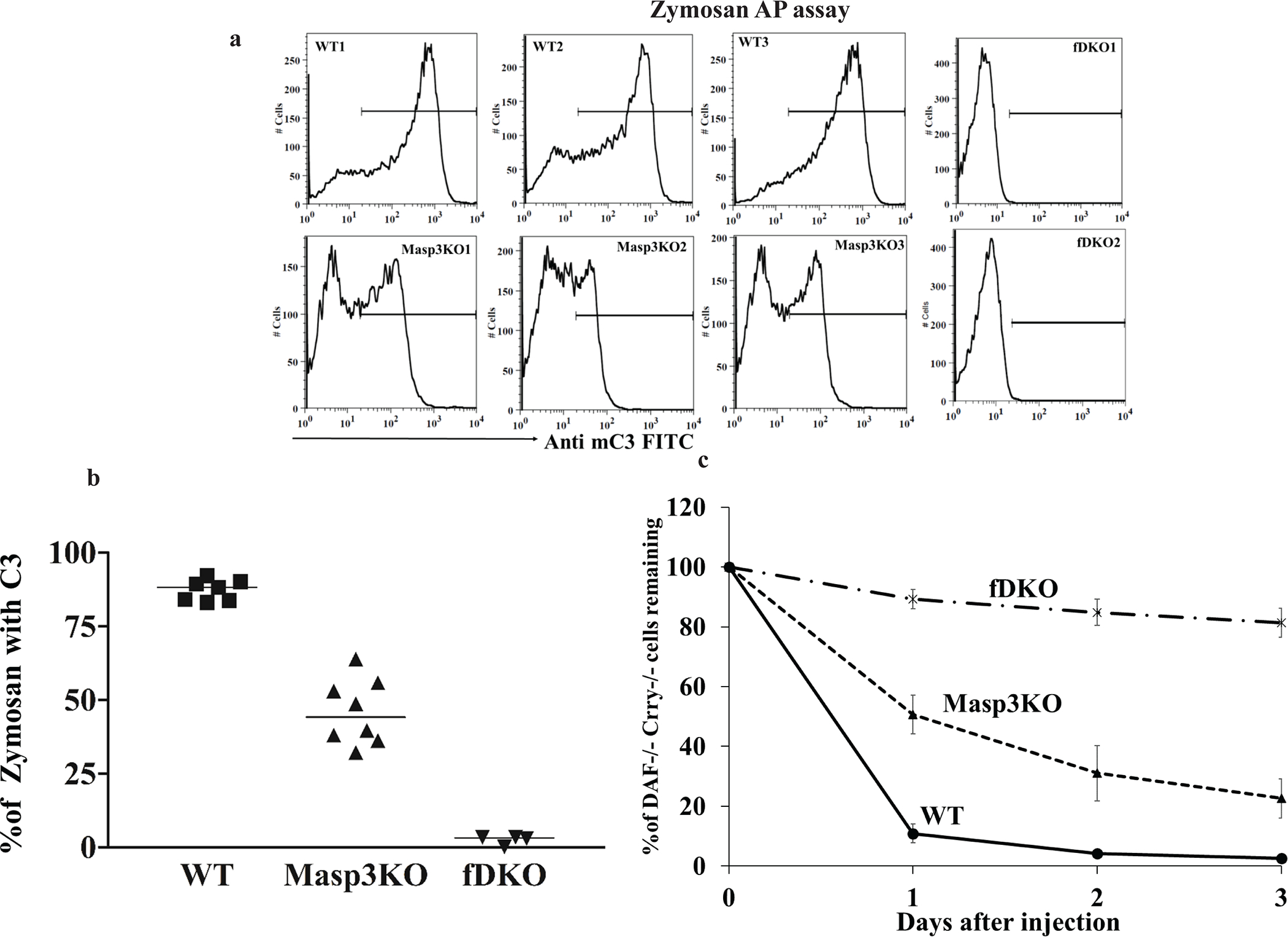
(a) Representative FACS analysis of C3b opsonization of zymosan particles (gated for >98% events on forward and side scatter plots) after in vitro incubation with 20% plasma from wildtype mice (WT, showing 3 out of 7 mice tested), Masp3−/− mice (Masp3KO, showing 3 out of 8 mice tested) or FD knockout mice (fDKO, showing 2 out of 4 mice tested). C3b opsonization by Masp3−/− mouse plasma was markedly reduced compared with that of wildtype mouse plasma but did not reach background levels of FD knockout mice. (b) Quantitation of % zymosan particles with C3b deposition reaching to a specified range as defined by the horizontal lines in panel (a). Each symbol represents data from an individual mouse plasma sample. WT (n=7), Masp3KO (n=8) and fDKO (n=4). (c) Survival of CFSC-labeled CD55−/−/Crry−/−/C3−/− mouse erythrocytes when transfused into wildtype (WT), Masp3−/− (Masp3KO) or FD knockout (fDKO) recipient mice (n=4 recipients for each group). Elimination of CD55−/−/Crry−/−/C3−/− mouse erythrocytes in wildtype recipients was mediated by EVH after AP complement-dependent C3b opsonization of the cells (31). EVH of CD55−/−/Crry−/−/C3−/− mouse erythrocytes was largely prevented in FD knockout mouse recipients but only moderately ameliorated in Masp3−/− mouse recipients. Percentage of transfused cells remaining at various time points was normalized to that at 5 min after transfusion (31).
Fig 5: Assessment of AP complement activity in Masp3−/− mouse plasma using a rabbit red blood cell (RBC) hemolytic assay.
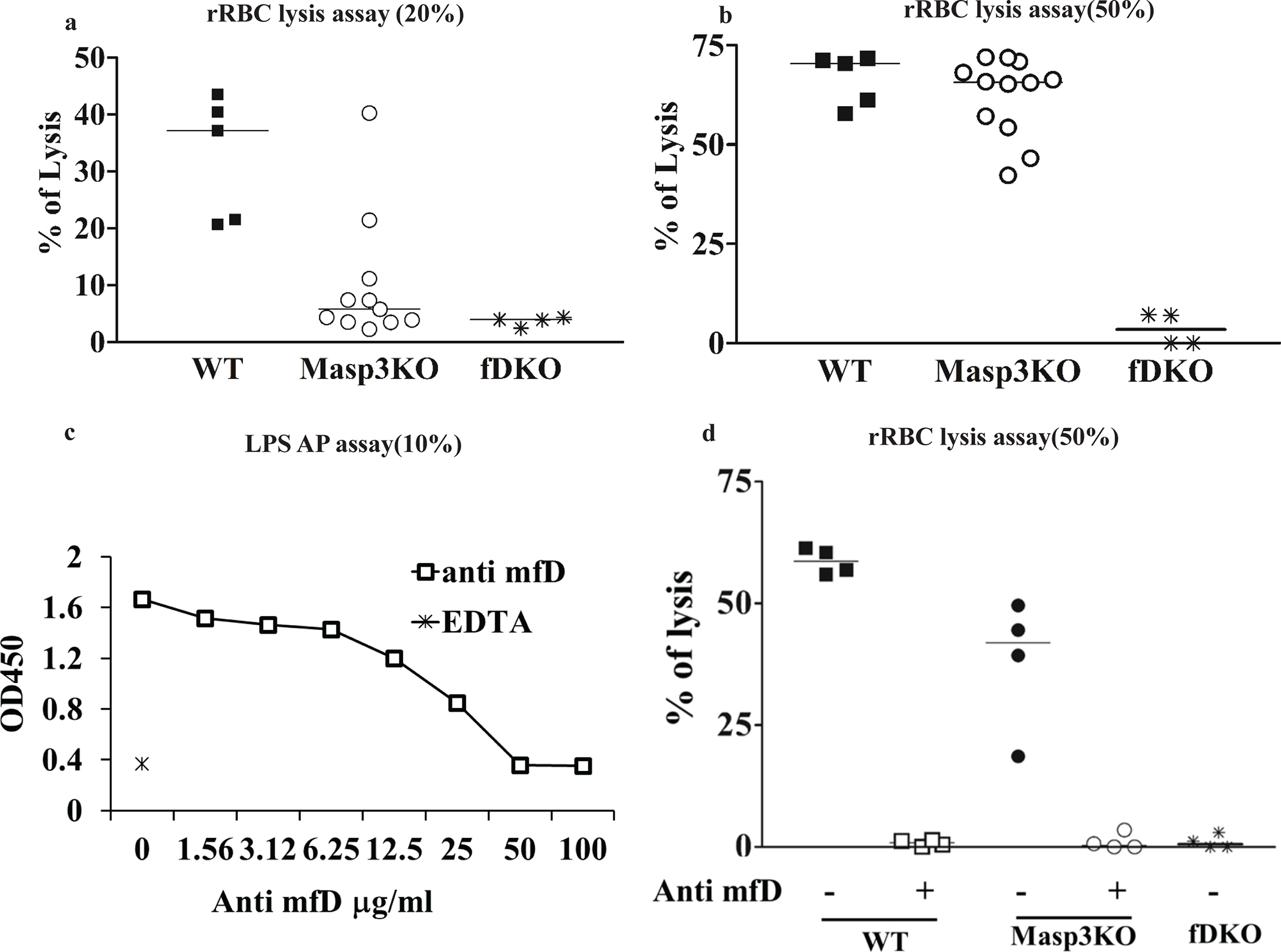
(a, b) Rabbit RBC lysis assays using 20% or 50% wildtype (WT), Masp3−/− (Masp3KO) or FD knockout (fDKO) mouse plasma. Rabbit RBC were easily lysed by wildtype mouse plasma but as expected, not by FD knockout mouse plasma. In contrast, significant lytic activity was detected in 20% plasma of some Masp3−/− mice, and close to wildtype mouse lytic activity was detected in 50% plasma of Masp3−/− mice. Each symbol represents data of plasma from an individual mouse. WT (n=5), Masp3KO (n=11) and fDKO (n=4). (c) Demonstration of dose-dependent inhibition of LPS-induced AP complement activity in 10% wildtype mouse plasma by a function-blocking mouse anti-mouse FD mAb (clone 14F11-3). At 50 or 100 μg/ml, mAb 14F11-3 completely inhibited mouse AP activity. EDTA serum was used as a positive control for complement inhibition. Values are average of duplicate assays using pooled plasma from 3–4 mice. (d) Effect of anti-FD mAb 14F11-3 (500 μg/ml) on rabbit RBC lysis by 50% wildtype (WT, n=4) or Masp3−/− (Masp3KO, n=4) mouse plasma. The data shows that AP complement activity in wildtype and Masp3−/− mouse plasma was completely blocked by mAb 14F11-3, suggesting that the AP activity detected in Masp3−/− mice was dependent on pro-FD and not on other nonspecific protease(s). FD knockout (fDKO) mouse plasma was used as a negative control which produced no hemolysis.
Partial AP complement activity in Masp3−/− mice may arise from a FD bypass mechanism via other protease(s) that can cleave FB or from intrinsic activity of pro-FD. To distinguish these two hypotheses, we used a function-blocking anti-mouse FD mAb (14F11-3) developed inhouse (Fig 5c) to investigate if the partial AP complement activity in Masp3−/− mice could be blocked by FD neutralization. Fig 5d shows that at 500 μg/ml, mAb 14F11-3 completely blocked rabbit RBC lysis by 50% plasma of wildtype or Masp3−/− mice. This result suggested that the partial AP complement activity in Masp3−/− mice was dependent on pro-FD and not on other non-specific proteases. To establish directly that mouse pro-FD has intrinsic FB-cleaving activity to support AP complement activation, we performed an in vitro FB cleavage assay by mixing human FB with human C3b and mouse pro-FD or pro-FD pre-treated with recombinant mouse MASP3. We used human FB and C3b due to their easy availability as commercial reagents, and after confirming that mouse FD can substitute for human FD to support AP complement activation in normal human serum (Fig 6a). We also confirmed by Western blot that mouse pro-FD was effectively converted to mature FD, as indicated by the loss of N-terminal His-tag, after incubating with mouse MASP3 (Fig 6b, 6c). In this FB cleavage assay, we found that both pro-FD and mature FD were able to cleave FB, providing direct evidence that pro-FD has intrinsic enzymatic activity towards C3b-complexed FB. It was clear however that mature FD had higher activity than pro-FD, especially when present at a lower concentration in the reaction (Fig 6d, 6e).
Fig 6: Intrinsic factor B-cleaving activity of pro-FD.
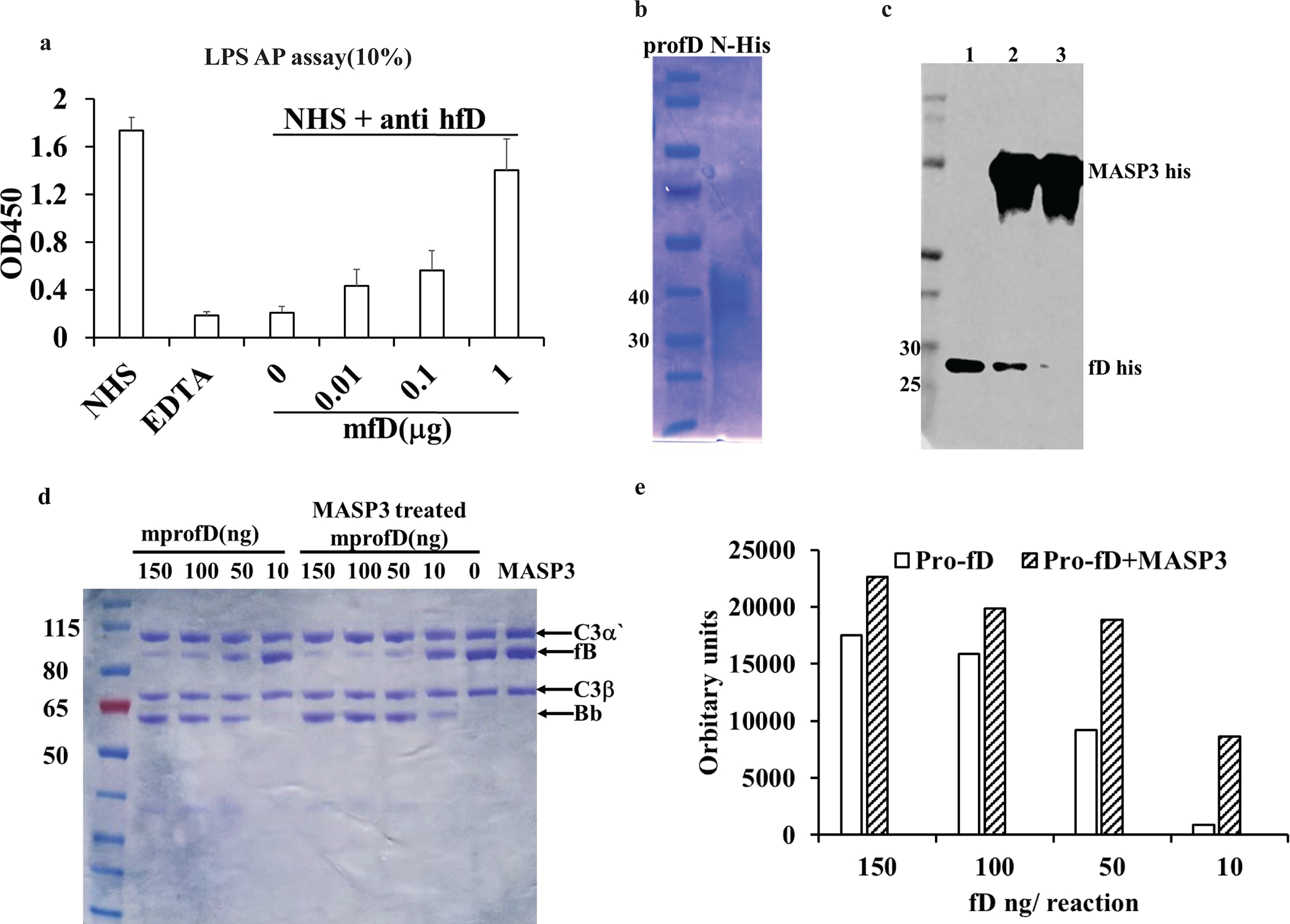
(a) demonstration of function of mouse factor D in human AP complement system, i.e. its activity to cleave human FB, as assessed by LPS-induced C3b deposition ELISA using 10% normal human serum (NHS). Human FD was functionally blocked by a neutralizing mAb (AFD, 5 μg/ml final concentration) and different amounts of mouse FD was added. The data shows that mouse FD dose-dependently restored AP complement activity in human serum. Bar values (mean plus SD) are from triplicate assays using pooled normal human serum (NHS). (b) SDS-PAGE analysis of recombinant N-terminal His-tagged mouse pro-FD sequentially purified from cell culture medium using NiNTA agarose and anti-mouse FD immunoaffinity Sepharose. (c) Kinetic analysis of the conversion of mouse pro-FD to mature FD by recombinant His-tagged mouse MASP3. Western blot was performed to detect N-terminal His tag before and after pro-FD treatment with MASP3. Samples were subjected to deglycosylation treatment before gel analysis under reducing condition. Lane1: mouse pro-FD with N-terminal His tag. Lane2: pro-FD after incubation with His-tagged MASP3 for 1hr. Lane3: pro-FD after incubation with His-tagged Masp3 for 2 hr, showing loss of N-terminal His tag and suggesting complete conversion to mature FD. (d) In vitro assay by SDS-PAGE under reducing condition and Coomassie blue staining showing cleavage of human C3b-complexed FB by mouse pro-FD and mature FD (pro-FD pretreated with MASP3 for 2hr). The data shows a dose-dependent generation of Bb fragment by both pro-FD and mature FD, but mature FD was more efficient than pro-FD when they were present at lower concentrations. Data in b)-d) are representative of at least two independent experiments. (e) Quantification of Bb fragment in panel (d) by densitometry using LiCor Odessy Fc system.
Discussion:
One of the surprising findings in the study of MASP enzymes was a previously unrecognized role of MASP3 in converting pro-FD to mature FD by cleaving 5 amino acids at the N-terminus. This discovery was originally made in a Masp1/3 gene knockout mouse that lacked both MASP1 and MASP3 proteins and had impaired LP and AP complement activity (13, 16). Subsequent studies of Masp1- and Masp3-specific knockout mice established that MASP1 but not MASP3 was required for LP, and conversely, MASP3 but not MASP1 was implicated in AP complement activity (17). In these studies, both Masp1/3 gene knockout and Masp3-specific knockout mice were described to be lacking AP complement activity (16, 17), leading to the conclusion that pro-FD is non-functional and its conversion to catalytically active FD by MASP3 is critical to AP complement. This concept has led to effort to target MASP3 as a therapeutic approach to treat AP complement-mediated human diseases (7).
The role of MASP3 in AP complement activation became somewhat controversial as other contradictory evidence emerged (18–20). First, a human 3MC syndrome patient whose blood lacked both MASP1 and MASP3 proteins was found to have fully functional AP complement activity (19, 20). This raised the question as to whether the finding made in mice was species-specific and cast doubt on a critical role of MASP3 in human AP complement activation. Secondly, an independent study by a different research group of the same Masp1/3 gene knockout mouse showed close to normal AP complement activity in 30% serum using a rabbit RBC hemolytic assay, directly contradicting the result initially reported by Takahashi et al (16). Furthermore, when the Masp1/3 gene knockout mouse was crossed with a factor H (FH) knockout mouse which was known to develop C3 glomerulopathy (C3G), with systemic C3 and FB depletion and glomerular C3 deposition, due to uncontrolled AP complement activation, no impact on the C3G phenotype was observed, contrary to what might be expected if MASP3 were indispensable to AP complement activity (18).
In the present study, we generated and characterized an independent line of Masp3-specific knockout mouse and provided data to reconcile the discrepancies in literature. Using the CRISPR/Cas9 method to edit the Masp1/3 gene which encodes both MASP3 and MASP1 through alternative splicing, we selectively inactivated Masp3 gene expression without affecting plasma MASP1 protein level (Figure 1). We confirmed the conclusion of Hayashi et al that MASP3 is not required for LP complement activity (17). However, unlike what has been described previously (16, 17), we found that the role of MASP3 in AP complement activity was not absolute and that significant activity could be detected in higher and physiologically relevant concentrations of Masp3−/− mouse plasma (>20%) (Figures 3 and 4). In previous studies of Masp1/3−/− and Masp3−/− mice, only low concentrations of mouse plasma (5–10%) were used for AP activity assays (16, 17) and this may explain why no activity was detected. Indeed, we also failed to detect or detected trace levels of AP activity in our line of Masp3−/− mice at lower plasma concentrations (5–10%), and the activity we detected at higher plasma concentrations, while significant, was clearly lower than that detected in wildtype mouse plasma (Figure 3–5). Thus, a major conclusion of the present study is that MASP3 deficiency reduced, but did not abrogate, AP complement activity in mice.
Although partial AP complement activity in Masp3−/− mice could have several plausible explanations, including a FD bypass mechanism for FB cleavage or a MASP3 bypass mechanism for FD maturation, our data showed that the mostly likely mechanism is intrinsic enzymatic activity of pro-FD. The partial AP activity was completely blocked by a neutralizing anti-FD mAb (Figure 5), suggesting that it was not due to a FD bypass mechanism. A MASP3 bypass mechanism in FD maturation was also unlikely as we detected no mature FD in Masp3−/− mice by Western blot (Figure 1). Using an in vitro FB cleavage assay, we provided direct evidence that mouse pro-FD was functionally active in cleaving C3b-complexed FB, albeit with less activity than mature FD generated by MASP3 pre-treatment (Figure 6). The difference in activity between pro-FD and mature FD was especially pronounced at lower concentrations of the two proteins, an observation consistent with in vivo AP complement activity assays where the difference between wildtype and Masp3−/− mice was more striking at low plasma concentrations (Figure 3). Our finding of low intrinsic FB-cleaving activity of pro-FD may not be unique to the mouse complement system. Indeed, it has been found in a recent study of human pro-FD that it too was catalytically active, with an estimated 800 fold lower activity than human mature FD in one assay but potentially having physiological relevance (32). At 50-fold of the physiological FD concentration, the human pro-FD could restore half maximal AP activity of FD-depleted human serum on zymosan (32).
The realization of a role of MASP3 in FD maturation and AP complement activation has stimulated interests in therapeutically targeting human MASP3 for AP complement-mediated diseases (7). Our results presented in this study, if replicated in the human complement system as has been implied by a recent study (32), suggest that such an approach is unlikely to inhibit AP complement completely. While a partial inhibition of AP activity afforded by MASP3 inhibitors may produce clinical benefit, as illustrated by a murine arthritis model in Masp1/3 knockout mice and mice with Masp3 knockdown with siRNA (33, 34), it may not achieve clinical efficacy in other diseases where pro-FD activity is sufficient to cause full or partial disease pathology. The latter scenario finds examples in the murine model of C3 glomerulopathy as reported by Ruseva et al (18) and in the murine EVH model described in this study with relevance to AP complement-driven human renal diseases and paroxysmal nocturnal hemoglobinuria, respectively.
Supplementary Material
Key Points:
Mouse MASP3 converts pro-FD to mature FD, consistent with previous findings.
Unlike what has been reported earlier, mouse pro-FD has catalytic activity.
MASP3 deficiency in mice reduces but does not abrogate AP complement activity.
Acknowledgments
This work is supported in part by National Institutes of Health grants AI-103965, AI-44970 and AI-085596.
References
- 1.Walport MJ 2001. Complement. First of two parts. The New England journal of medicine 344: 1058–1066. [DOI] [PubMed] [Google Scholar]
- 2.Dunkelberger JR, and Song WC. 2010. Complement and its role in innate and adaptive immune responses. Cell Res 20: 34–50. [DOI] [PubMed] [Google Scholar]
- 3.Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, and Skjoedt MO. 2016. A journey through the lectin pathway of complement-MBL and beyond. Immunological reviews 274: 74–97. [DOI] [PubMed] [Google Scholar]
- 4.Volanakis JE, and Narayana SV. 1996. Complement factor D, a novel serine protease. Protein Sci 5: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsson B, and Nilsson Ekdahl K. 2012. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology 217: 1106–1110. [DOI] [PubMed] [Google Scholar]
- 6.Harboe M, and Mollnes TE. 2008. The alternative complement pathway revisited. Journal of cellular and molecular medicine 12: 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobo J, Kocsis A, and Gal P. 2018. Be on Target: Strategies of Targeting Alternative and Lectin Pathway Components in Complement-Mediated Diseases. Frontiers in immunology 9: 1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garred P, Tenner AJ, and Mollnes TE. 2021. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharmacol Rev 73: 792–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mastellos DC, Ricklin D, and Lambris JD. 2019. Clinical promise of next-generation complement therapeutics. Nature reviews. Drug discovery 18: 707–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stover CM, Thiel S, Thelen M, Lynch NJ, Vorup-Jensen T, Jensenius JC, and Schwaeble WJ. 1999. Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J Immunol 162: 3481–3490. [PubMed] [Google Scholar]
- 11.Stover C, Endo Y, Takahashi M, Lynch NJ, Constantinescu C, Vorup-Jensen T, Thiel S, Friedl H, Hankeln T, Hall R, Gregory S, Fujita T, and Schwaeble W. 2001. The human gene for mannan-binding lectin-associated serine protease-2 (MASP-2), the effector component of the lectin route of complement activation, is part of a tightly linked gene cluster on chromosome 1p36.2–3. Genes Immun 2: 119–127. [DOI] [PubMed] [Google Scholar]
- 12.Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, Vorup-Jensen T, and Jensenius JC. 2001. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity 15: 127–135. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi M, Iwaki D, Kanno K, Ishida Y, Xiong J, Matsushita M, Endo Y, Miura S, Ishii N, Sugamura K, and Fujita T. 2008. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J Immunol 180: 6132–6138. [DOI] [PubMed] [Google Scholar]
- 14.Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, Dudler T, Parent B, Lhotta K, Wallis R, Farrar CA, Sacks S, Lee H, Zhang M, Iwaki D, Takahashi M, Fujita T, Tedford CE, and Stover CM. 2011. Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America 108: 7523–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szakacs D, Kocsis A, Szasz R, Gal P, and Pal G. 2019. Novel MASP-2 inhibitors developed via directed evolution of human TFPI1 are potent lectin pathway inhibitors. The Journal of biological chemistry 294: 8227–8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, Homma Y, and Fujita T. 2010. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor D. The Journal of experimental medicine 207: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi M, Machida T, Ishida Y, Ogata Y, Omori T, Takasumi M, Endo Y, Suzuki T, Sekimata M, Homma Y, Ikawa M, Ohira H, Fujita T, and Sekine H. 2019. Cutting Edge: Role of MASP-3 in the Physiological Activation of Factor D of the Alternative Complement Pathway. J Immunol 203: 1411–1416. [DOI] [PubMed] [Google Scholar]
- 18.Ruseva MM, Takahashi M, Fujita T, and Pickering MC. 2014. C3 dysregulation due to factor H deficiency is mannan-binding lectin-associated serine proteases (MASP)-1 and MASP-3 independent in vivo. Clinical and experimental immunology 176: 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, and Thiel S. 2012. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol 189: 3957–3969. [DOI] [PubMed] [Google Scholar]
- 20.Atik T, Koparir A, Bademci G, Foster J 2nd, Altunoglu U, Mutlu GY, Bowdin S, Elcioglu N, Tayfun GA, Atik SS, Ozen M, Ozkinay F, Alanay Y, Kayserili H, Thiel S, and Tekin M. 2015. Novel MASP1 mutations are associated with an expanded phenotype in 3MC1 syndrome. Orphanet J Rare Dis 10: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harms DW, Quadros RM, Seruggia D, Ohtsuka M, Takahashi G, Montoliu L, and Gurumurthy CB. 2014. Mouse Genome Editing Using the CRISPR/Cas System. Curr Protoc Hum Genet 83: 15 17 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miwa T, Sato S, Gullipalli D, Nangaku M, and Song WC. 2013. Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol 190: 3552–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zundel S, Cseh S, Lacroix M, Dahl MR, Matsushita M, Andrieu JP, Schwaeble WJ, Jensenius JC, Fujita T, Arlaud GJ, and Thielens NM. 2004. Characterization of recombinant mannan-binding lectin-associated serine protease (MASP)-3 suggests an activation mechanism different from that of MASP-1 and MASP-2. J Immunol 172: 4342–4350. [DOI] [PubMed] [Google Scholar]
- 24.Zhu B, Cai G, Hall EO, and Freeman GJ. 2007. In-fusion assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques 43: 354–359. [DOI] [PubMed] [Google Scholar]
- 25.Harlow E, and Lane D. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 726 pp. [Google Scholar]
- 26.Kimura Y, Miwa T, Zhou L, and Song WC. 2008. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood 111: 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DD, Miwa T, and Song WC. 2006. Retrovirus-mediated over-expression of decay-accelerating factor rescues Crry-deficient erythrocytes from acute alternative pathway complement attack. J Immunol 177: 5558–5566. [DOI] [PubMed] [Google Scholar]
- 28.Katschke KJ Jr., Wu P, Ganesan R, Kelley RF, Mathieu MA, Hass PE, Murray J, Kirchhofer D, Wiesmann C, and van Lookeren Campagne M. 2012. Inhibiting alternative pathway complement activation by targeting the factor D exosite. The Journal of biological chemistry 287: 12886–12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva JP, Vetterlein O, Jose J, Peters S, and Kirby H. 2015. The S228P mutation prevents in vivo and in vitro IgG4 Fab-arm exchange as demonstrated using a combination of novel quantitative immunoassays and physiological matrix preparation. The Journal of biological chemistry 290: 5462–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palarasah Y, Skjoedt MO, Vitved L, Andersen TE, Skjoedt K, and Koch C. 2010. Sodium polyanethole sulfonate as an inhibitor of activation of complement function in blood culture systems. J Clin Microbiol 48: 908–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molina H, Miwa T, Zhou L, Hilliard B, Mastellos D, Maldonado MA, Lambris JD, and Song WC. 2002. Complement-mediated clearance of erythrocytes: mechanism and delineation of the regulatory roles of Crry and DAF. Decay-accelerating factor. Blood 100: 4544–4549. [DOI] [PubMed] [Google Scholar]
- 32.Dani R, Oroszlán Gábor; Martinusz Róbert; Dobos Bernadett;, and E. Z. Vadas Péter; Gál Péter; Dobó József. 2022. Quantification of the zymogenicity and the substrate-induced activity enhancement of complement factor D. Molecular immunology 150: 195–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banda NK, Takahashi M, Levitt B, Glogowska M, Nicholas J, Takahashi K, Stahl GL, Fujita T, Arend WP, and Holers VM. 2010. Essential role of complement mannose-binding lectin-associated serine proteases-1/3 in the murine collagen antibody-induced model of inflammatory arthritis. J Immunol 185: 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banda NK, Desai D, Scheinman RI, Pihl R, Sekine H, Fujita T, Sharma V, Hansen AG, Garred P, Thiel S, Borodovsky A, and Holers VM. 2018. Targeting of Liver Mannan-Binding Lectin–Associated Serine Protease-3 with RNA Interference Ameliorates Disease in a Mouse Model of Rheumatoid Arthritis. ImmunoHorizons 2: 274–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.