

Abstract
The nucleosome acidic patch is a major interaction hub for chromatin, providing a platform for enzymes to dock and orient for nucleosome-targeted activities. To define the molecular basis of acidic patch recognition proteome-wide, we performed an amino acid resolution acidic patch interactome screen. We discovered that the histone H3 lysine 36 (H3K36) demethylase KDM2A, but not its closely related paralog, KDM2B, requires the acidic patch for nucleosome binding. Despite fundamental roles in transcriptional repression in health and disease, the molecular mechanisms governing nucleosome substrate specificity of KDM2A/B, or any related JumonjiC (JmjC) domain lysine demethylases, remain unclear. We used a covalent conjugate between H3K36 and a demethylase inhibitor to solve cryo-EM structures of KDM2A and KDM2B trapped in action on a nucleosome substrate. Our structures show that KDM2-nucleosome binding is paralog-specific and facilitated by dynamic nucleosomal DNA unwrapping and histone charge shielding that mobilize the H3K36 sequence for demethylation.
Keywords: chromatin, nucleosome, demethylase, JmjC, KDM2A, KDM2B, cryo-EM, acidic patch, proteomics
Reversible histone lysine methylation regulates genome-templated processes, including transcription, through local recruitment or exclusion of chromatin binding proteins1. Lysine methylation is reversed by two classes of lysine demethylases (KDMs): flavin-dependent amine oxidases and Fe(II)-α-ketoglutarate (α-KG)-dependent JumonjiC (JmjC) domain demethylases2–5. In contrast to the amine oxidase class that contains only two members, the JmjC domain demethylase class is more expansive, comprising twenty enzymes that are subclassified into KDM2-8 families6,7. JmjC domain lysine demethylases often exist within multi-subunit complexes and can activate or repress transcription based on their specific sites of action within histones1.
The KDM2 subfamily includes two paralogs, KDM2A (JHDM1A/FBXL11) and KDM2B (JHDM1B/FBXL10), that repress gene expression through preferential demethylation of dimethylated H3K36 (H3K36me2)8. KDM2A and KDM2B function in distinct heterochromatin silencing pathways through association with heterochromatin protein 1 (HP1)8–10 or the variant Polycomb repressive complex 1.1 (PRC1.1)11–17, respectively. Due to a central role in regulating gene repression, overexpression of KDM2 family demethylases is linked to diverse hematologic and solid tumors18,19.
Structures of KDM2A in complex with histone peptides demonstrate that H3K36 specificity is in part dictated by local recognition of the H3 sequence20. However, limited structural or mechanistic information is available to describe how KDM2A and KDM2B, or any other JmjC domain proteins, demethylate specific histone lysines within a nucleosomal context. Using an amino acid resolution nucleosome acidic patch interactome screen, we discovered that KDM2A, but not KDM2B, requires the acidic patch for nucleosome binding. We then used a nucleosomally incorporated H3K36-JmjC inhibitor covalent conjugate to stabilize KDM2A- and KDM2B-nucleosome complexes for cryo-EM structure determination. Our structures illustrate paralog-specific acidic patch recognition, dynamic nucleosomal DNA unwrapping, and histone chaperone-like charge shielding that dictate H3K36 specificity.
Results
Proteome-wide footprinting of acidic patch interactions
We previously reported a nucleosome affinity proteomics screen that defined universal principles of nucleosome recognition21. While identifying a role for the acidic patch in the majority of nucleosome interactions, the coarse mutation of the entire patch used in this screen precluded exploration of detailed molecular mechanisms. We have now completed a 2nd-generation nucleosome affinity proteomics screen using a comprehensive library of point mutations, including recently identified oncohistone mutations22, allowing amino acid-resolution footprinting of acidic patch interactions proteome-wide (Extended Data Fig. 1a and Extended Data Files 1–2). Altogether, our library included a wild-type nucleosome, 12 alanine point mutant nucleosomes to scan the acidic patch surface, three oncohistone mutant nucleosomes, and an acidic patch-neutralized (ΔAP) nucleosome with seven combined mutations within the acidic patch (Extended Data Fig. 1a–b). We performed triplicate pulldowns from HEK293T nuclear lysates using reconstituted, biotinylated nucleosomes and analyzed bound proteins using quantitative multiplexed mass spectrometry with tandem mass tags (TMT) across four mixtures (Extended Data Fig. 1a–c). Inclusion of identical wild-type samples in each mixture allowed for normalization and quantitative comparison across the library.
Proteins typically engage the acidic patch with one or more arginines classified as arginine anchors and variant arginines based on their binding locations (Fig. 1a)23. Principle component analysis (PCA) of our screen results showed clear patterns of binding sites within the acidic patch, delineating the arginine anchor cavity (H2AE61, D90, E92) from common variant arginine binding sites (type 1: H2AE56 and H2BQ47, E113; type 2: H2AE64) (Fig. 1b). H2AE61A and D90A mutations closely phenocopy the ΔAP mutation, indicating a dominant role of arginine anchors in acidic patch binding proteome-wide (Fig. 1b–c and Extended Data Fig. 2a). Interestingly, while H2AE92 is often included with E61 and D90 in an arginine anchor binding triad, the E92 side chain participated far more modestly in acidic patch recognition as evidenced by alanine mutations (Fig. 1c). Many proteins that required H2AE61 and H2AD90 for nucleosome binding also relied on H2AE64 and/or H2AE56, H2AQ47, and H2BE113, indicating acidic patch recognition by an arginine anchor in combination with one or more variant arginines. Unexpectedly, H2AN68A mutant nucleosomes exhibited similar PCA patterns to H2AQ47A and H2BE113A mutants despite spatially distinct locations within the acidic patch. Mutations of H2AQ24, D72, N89, and E91, which occupy the outer margins of the acidic patch were well tolerated and therefore demarcate the functional acidic patch zones (Fig. 1c). Our quantitative proteomics data closely matches results from equivalent pulldown experiments analyzed by western blot for select proteins (Extended Data Fig. 1d).
Fig. 1 |. KDM2 paralog-specific acidic patch binding identified by amino acid resolution nucleosome affinity proteomics.
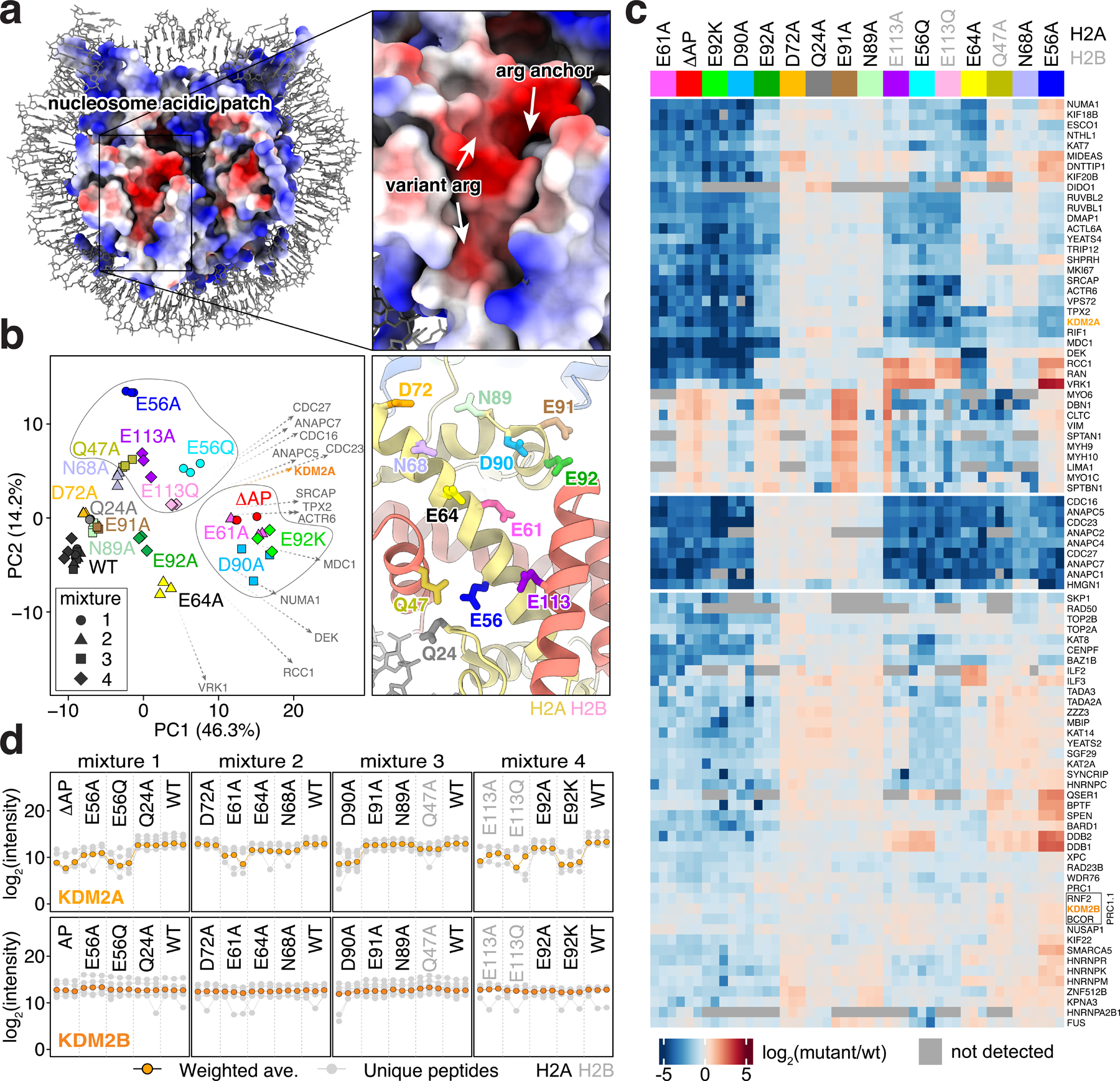
a, Electrostatic surface of nucleosome with arginine anchor cavity and common variant arginine binding positions indicated on zoomed view. b, Principal component analysis of triplicate pulldowns from wild-type (WT) and mutant nucleosomes, left. Arginine anchor (center right of PCA) and variant 1 (top left of PCA) clusters circled. Loading factors for top 14 proteins contributing to PCA patterns indicated by vectors reflected to point in the direction of less binding. ΔAP mutant contains seven mutations across the acidic patch. Positions of mutations shown at right in identical zoomed view to panel a. c, Clustered heatmap showing log2 fold-change for selected proteins binding to each mutant nucleosome relative to wild-type. H2A mutations in black; H2B mutations in grey. Grey squares indicate proteins not quantitated in a sample. d, Profile plots showing intensities of individual unique peptides (grey) and weighted averages (orange) for KDM2A and KDM2B.
Analysis of whole genome sequences of cancer samples led to the recent identification of thousands of mutations in canonical histones22,24. While many of these mutations reside at histone-DNA or histone-histone interfaces where they influence nucleosome stability and remodeling22,24–26, or in histone tails where they disrupt patterns of histone modifications27, several of the most prevalent oncohistone mutations are within the nucleosome acidic patch, likely influencing nucleosome binding. We tested three of the acidic patch oncohistone mutations, H2AE56Q, H2AE92K, and H2BE113Q, together with the corresponding alanine mutations. This allowed simultaneous discovery of loss-of-function and gain-of-function effects. These oncohistone mutations were among the most deleterious mutations we tested, leading to broad loss of protein binding that was typically in excess of that observed for the corresponding alanine mutations (Fig. 1c and Extended Data Fig. 2b–c). Such gain-of-function effects were most pronounced for the H2AE92K charge swap mutation when compared to the E92A side chain truncation. While many proteins lost binding to all three oncohistone mutant nucleosomes, specific proteins were more sensitive to H2AE56Q and H2BE113Q or H2AE92K, suggesting distinct consequences of these cancer-associated mutations in chromatin-based processes.
We examined loading factors to identify proteins that correlated with trends observed by principal component analysis of our screen results, which highlighted a major contribution by KDM2A (Fig. 1b). Despite strong similarities in domain structure and function, KDM2A, but not KDM2B, was sensitive to alanine or oncohistone mutations of H2AE56, E61, D90, E92 and H2BE113 (Fig. 1d). Interestingly, KDM2B clustered with BCOR and RNF2, suggesting that an intact PRC1.1 complex was detected (Fig. 1c). Similar clustering of several other complexes (e.g., the Anaphase Promoting Complex and NuA4 complex) was enabled by the information-rich content of the screen (Extended Data Fig. 2).
Inhibitor stabilizes KDM2-nucleosome complexes for cryo-EM
In order to understand the molecular mechanisms driving KDM2 family paralog-specific acidic patch recognition, we wanted to solve structures of KDM2A and KDM2B bound to the nucleosome. KMD2A and KDM2B are modular proteins that in addition to the JmjC domain include a CxxC domain that binds to non-methylated CpG sequences in internucleosomal linker DNA (Fig. 2a)10,17,28. Despite stable binding of a KDM2B(2–734) JmjC-CxxC-PHD fragment to a nucleosome assembled with 185 bp of DNA, we observed no evidence of H3K36 engagement in single particle cryo-EM 2D classes even in glutaraldehyde crosslinked samples (Extended Data Fig. 3). We envisioned that KDM2B could bind to linker DNA heterogeneously through its CxxC domain10,17,28, but that the JmjC domain might engage the H3 target sequence only transiently, preventing visualization in class averages. Therefore, we wanted to locally stabilize the H3K36-JmjC domain interaction to solve the structure of KDM2B in a functional pose on the nucleosome surface. We tackled this challenge by covalently linking an active site KDM inhibitor directly to the H3K36 site through alkylation of an H3K36C mutant histone. For inhibitor design, we were inspired by published hydroxamate compounds that bind the α-KG cofactor site of KDM2-family demethylases. Much like α-KG, these compounds chelate the Fe(II) metal ion, but also contain long alkyl chains directed toward the methyllysine binding channel29–31. Using KDM2A JmjC-H3(29-41) structures as a guide20, we performed flexible docking of H3K36C-hydroxamate compound conjugates designed with variable flexible alkyl and more rigid aryl linkers, ultimately identifying compounds UNC8016 (1), UNC 8017 (2), and UNC8018 (3) for synthesis (Extended Data Fig. 4a). UNC8016 (1) and UNC8017 (2), but not UNC8018 (3), partially inhibited KDM2B-mediated demethylation of an H3(29-41)K36me2 peptide substrate (Extended Data Fig. 4b). We observed a dose dependent inhibition of KDM2B activity by UNC8016 (1), albeit with an IC50 in the high micromolar range (Extended Data Fig. 4c).
Fig. 2 |. Stabilization of JmjC-nucleosome interaction enables structural analysis of KDM2A/B-nucleosome interaction.
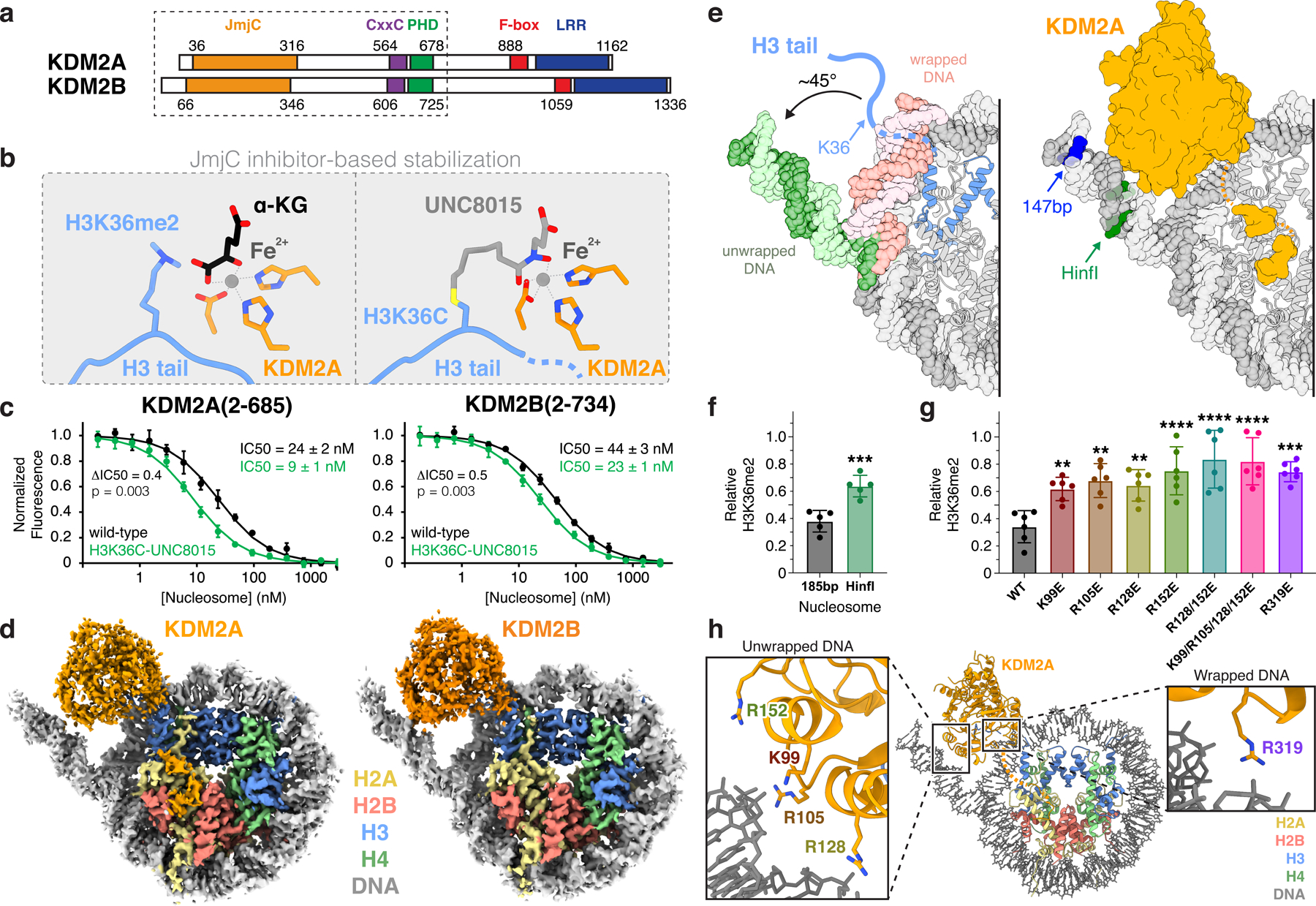
a, Protein domain maps of KDM2A and KDM2B with box depicting region used in structural and biochemical studies. b, Comparison of KDM2A active site in α-ketoglutarate (α-KG)/H3K36me2-bound (left; PDB:4QX7) and H3K36C-UNC8015-bound (right) crystal structures illustrating inhibitor-based stabilization strategy. c, TR-FRET nucleosome binding competition experiment with KDM2A (left) and KDM2B (right) using wild-type or H3K36C-UNC8015 nucleosomes as competitor. Mean ± SD are shown (n=3 assay technical replicates). Ratio of UNC8015/WT IC50 (ΔIC50) values and p-values, calculated using an unpaired two-tailed t-test, indicated d, Cryo-EM reconstructions of KDM2A- (left) and KDM2B- (right) H3K36C-UNC8015 nucleosome complexes. e, DNA unwrapping observed in KDM2A-nucleosome structure in comparison to an unbound nucleosome with fully wrapped DNA (left; PDB:6ESF) and KDM2A-nucleosome structure with terminal DNA positions for 147 bp and HinfI-digested nucleosomes highlighted (right). f, KDM2A demethylation of unmodified wildtype 185 bp and HinfI-digested H3K36me2-nucleosomes, p = 0.009. g, Demethylation of H3K36me2-nucleosomes by KDM2A DNA interface mutants, p-values for comparison to WT are 0.0097, 0.0012, 0.0040, <0.0001, <0.0001, <0.0001, and 0.0001. For panels f and g, relative residual H3K36me2 plotted as individual data points (n=5 assay technical replicates for panel f or n=6 assay technical replicates for panel g) along with mean ± SD, **p ≤0.01, ***p ≤0.001, and ****p ≤0.0001 for statistical differences between wild-type (WT) and mutant samples using an unpaired two-tailed t-test in panel f and an ordinary one-way ANOVA in panel g. h, Basic KDM2A residues positioned to contact unwrapped and wrapped nucleosomal DNA.
We next synthesized UNC8015 (4), an alkyl bromide derivative of UNC8016 (1), and subsequently prepared both H3(29–41)K36C- and full-length H3K36C-UNC8015 conjugates via cysteine alkylation (Extended Data Fig. 4d–e and Supplementary Note). In order to confirm that binding to H3 was maintained upon covalent attachment of UNC8015, we solved a crystal structure of the KDM2A JmjC domain in complex with the H3K36C(29–41)-UNC8015 (5) conjugate (Extended Data Fig. 5 and Supplementary Table 1). The structure shows that UNC8015 occupies the α-KG cofactor site with the hydroxamate positioned to chelate the metal ion (Fig. 2b and Extended Data Fig. 5). The H3 residues 29–39 are clearly visualized in the structure and bind nearly identically to previously reported structures (backbone atom RMSD of 0.34 Å as compared to H3K36me2 structure PDB:4QX7), giving us confidence to explore UNC8015 conjugates in a nucleosome context.
We next incorporated H3K36C-UNC8015 into recombinant nucleosomes. Using time-resolved fluorescence resonance energy transfer (TR-FRET) competitive nucleosome binding assays32, we showed that UNC8015-derivatized nucleosomes displaced KDM2A and KDM2B from unmodified nucleosomes at modestly lower concentrations (i.e., lower IC50s) than equivalent wild-type, unmodified nucleosomes (Fig. 2c). This result indicates a small improvement in overall KDM2-nucleosome binding affinity that was driven by UNC8015 conjugation and is suggestive of local engagement of the UNC8015 by the JmjC domains in a nucleosomal context. Notably, the small change in binding affinity is consistent with the low potency of the inhibitor paired with the high affinity KDM2-nucleosome interaction. We reconstituted and purified complexes of KDM2A(2–685) and KDM2B(2–734) bound to H3K36C-UNC8015 nucleosomes for structural analysis by single particle cryo-EM (Fig. 2a and Extended Data Fig. 3a). Initial 2D classification of particles in these datasets clearly showed the presence of new binding modes not observed in the absence of H3K36C-UNC8015 that localized KDM2A/B near the origin of the H3 tail (Extended Data Fig. 3b). These modes were not observed in the absence of glutaraldehyde crosslinking. Our cryo-EM reconstructions of KDM2A- and KDM2B-nucleosome complexes allowed for unambiguous docking and subsequent model building of the KDM family JmjC domains into the cryo-EM maps (Fig. 2d, Extended Data Fig. 6 and 7, and Supplementary Table 2).
JmjC domain binding requires DNA unwrapping
In the nucleosome core particle, H3K36 is located within the N-terminal tail of H3, immediately after it emerges from the nucleosome disk by threading between the two gyres of DNA (Fig. 2e). Rather than binding to H3K36 in the context of the fully wrapped nucleosome, the KDM2A- and KDM2B-nucleosome structures demonstrated that JmjC engagement of H3K36 is paired with an ~45° unwrapping of the terminal nucleosomal DNA beginning at super-helical location (SHL) -5.5. DNA unwrapping exposes the underlying sequence of the H3 tail adjacent to H3K36. A basic surface on the JmjC domains that is conserved in character between KDM2A and KDM2B is positioned to interact with the unwrapped nucleosomal DNA (Extended Data Fig. 8). In contrast, conservation of this surface is not maintained in the most closely related JmjC-domain KDMs, KDM7A and PHF8 (KDM7B), which target non-H3K36 positions.
To assess the effect of perturbations to KDM2-nucleosome interfaces on H3K36 demethylation, we prepared H3K36me2 nucleosomes using the H3K36-specific methyltransferase, NDS2, which afforded quantitatively dimethylated H3 without evidence of any other methyl-forms (Supplementary Fig. 1a–b). In our hands, both full length KDM2B and KDM2B(2–734) were less efficient nucleosome demethylases than KDM2A(2–685) and had only minimal activity under identical conditions used to test KDM2A, suggesting that KDM2B may require other factors for optimal demethylase activity (Supplementary Fig. 2a). Therefore, we focused our mechanistic investigations primarily on KDM2A.
We first explored the role of DNA unwrapping on KDM2A function by pre-treating H3K36me2 nucleosomes with HinfI, which removes ~25 bp from one end of the nucleosomal DNA, including all unwrapped DNA sequences positioned to interact with the KDM2A JmjC domain on one side of the nucleosome (Fig. 2e and Supplementary Fig. 3). KDM2A was roughly twice as active on undigested nucleosomes as compared to HinfI treated nucleosomes (Fig. 2f and Supplementary Fig. 2b). This result suggests that rather than wrapped DNA being purely repressive toward demethylation by preventing H3 tail mobilization, the unwrapped DNA plays an active role in KDM2A function. Consistent with this conclusion, the individual charge swap mutations of KDM2A K99E, R105E, R128E, or R152E, and combinations thereof, significantly decreased demethylation of nucleosomal H3K36me2 (Fig. 2g–h and Supplementary Fig. 2b). Loss of activity due to these mutations may result from impaired nucleosome binding, as the single R152E mutation and the combined K99E/R105E/R128E/R152 mutation reduced KDM2A-nucleosome binding affinity ~4-fold and ~20-fold, respectively (Extended Data Fig. 8c). In addition to binding unwrapped DNA ends, KDM2A closely approaches wrapped nucleosomal DNA near the nucleosome dyad at SHL1 (Fig. 2h). A single R319E mutation at this interface also significantly decreased KDM2A activity, highlighting critical interactions not only between KDM2A and unwrapped DNA but also a secondary contact point with wrapped nucleosomal DNA (Fig. 2g and Supplementary Fig. 2b).
KDM2A-nucleosome binding is also heavily dependent on nucleosomal DNA length. Using our nucleosome binding affinity competition assay, we show that 147 bp nucleosomes lacking linker DNA disrupt KDM2A-nucleosome binding with a ~10-fold higher IC50 than 185 bp nucleosomes (Extended Data Fig. 8d). However, we do not observe interactions between the JmjC domain and linker DNA in our cryo-EM structures. We hypothesize that linker DNA binding by the KDM2A CxxC domain at least partially accounts for higher affinity binding to the 185 bp nucleosomes that include CpG dinucleotides in the linker segments. We expect such interactions to be heterogeneous and therefore not resolved by single particle analysis. Further truncation of the nucleosomal DNA to 119 bp largely rescues loss of binding, likely in part due to mobilization of the H3 N-terminal tail (Extended Data Fig. 8d).
KDM2 paralog-specific acidic patch recognition
Despite similar DNA binding by the KDM2A and KDM2B JmjC domains and consistent with our proteomics screen results, the two demethylase paralogs diverged in their recognition of the histone disk surface (Fig. 3a). In both full cryo-EM reconstructions and focused refinements of the nucleosome density, the KDM2A-nucleosome complex includes a small well-defined, discontinuous density that bridges from the JmjC domain to the acidic patch (Extended Data Fig. 9a). In contrast, the KDM2B-nucleosome reconstruction shows no evidence of acidic patch binding (Extended Data Fig. 9b). In order to confirm paralog-specific acidic patch-dependent binding of KDM2 demethylases using an orthogonal method, we performed TR-FRET competitive nucleosome binding assays with wild-type and acidic patch-neutralized (ΔAP) mutant nucleosomes. We measured a 3.7x higher IC50 for disrupting the KDM2A-wild-type nucleosome complex with ΔAP nucleosomes relative to wild-type nucleosomes, consistent with a KDM2A-acidic patch interaction (Fig. 3b). A much smaller IC50 change (1.4x) was observed in identical experiments with KDM2B. This establishes a direct mechanism for a stronger requirement of the acidic patch for nucleosome binding by KDM2A than KDM2B. In agreement with this observation, KDM2A failed to demethylate ΔAP H3K36me2 nucleosomes (Fig. 3c and Supplementary Fig. 2c).
Fig. 3 |. KDM2A interacts with nucleosome acidic patch.
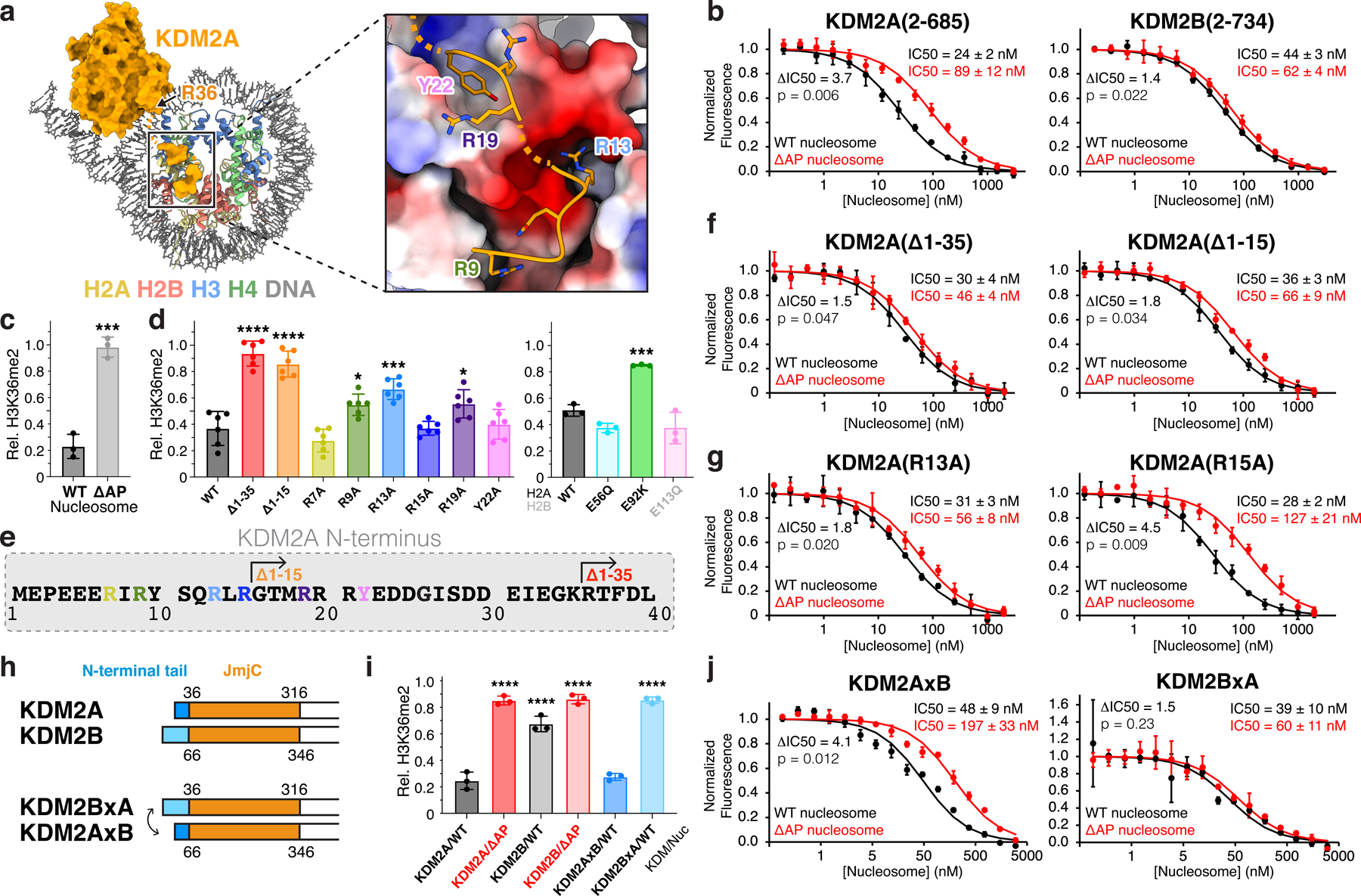
a, N-terminal KDM2A tail residues bind nucleosome acidic patch. b, TR-FRET nucleosome binding competition experiments with KDM2A (left) and KDM2B (right) using wild-type (WT) or acidic patch-neutralized (ΔAP) nucleosomes as competitor. c, KDM2A demethylation of WT and ΔAP H3K36me2-nucleosomes, p = 0.004. d, Demethylation of H3K36me2 nucleosomes by KDM2A N-terminal truncations and mutants (left, p-values for comparison to WT are <0.0001, <0.0001, not significant (ns), 0.468, 0.0002, ns, 0.0361, ns, and <0.0001) and demethylation of oncohistone mutant H3K36me2 nucleosomes by WT KDM2A (right, p-values for comparison to WT are ns, 0.0006, an ns). e, KDM2A N-terminal sequence, with truncations and mutations used in activity and binding assays denoted. f, Nucleosome binding competition experiment with KDM2A truncations using WT or ΔAP nucleosomes as competitor. g, Nucleosome binding competition experiment with KDM2A mutant proteins using WT or ΔAP nucleosomes as competitor. h, Schematic of KDM2 N-terminal tail swap chimeric proteins. i, Demethylation of WT or ΔAP H3K36me2 nucleosomes by WT and chimeric enzymes, KDM2BxA and KDM2AxB, p-values for comparison to KDM2A/WT are <0.0001, <0.0001, <0.0001, ns, <0.0001. j, Nucleosome binding competition experiment with chimeric KDM2 proteins using WT or ΔAP nucleosomes as competitor. For demethylase assays in panels c, d and i, relative residual H3K36me2 (Rel. H3K36me2) plotted as individual data points (n=3 assay technical replicates for panel c and panel d, right, and i, n=6 assay technical replicates for panel d, left) along with mean ± SD, **p ≤0.01, ***p ≤0.001, and ****p ≤0.0001 for statistical differences between WT and mutant samples using an unpaired two-tailed t-test in panel c and an ordinary one-way ANOVA in panels d and i. For binding assays in panels b, f g, and j, mean ± SD are shown (n=3 assay technical replicates) and ratio of ΔAP/WT IC50 (ΔIC50) values and p-values, calculated using an unpaired two-tailed t-test, indicated.
The KDM2A acidic patch-binding density in our reconstructed map shows two adjacent segments of EM density, one packing against the C-terminal end of the H2A α2 and α3 helices, and the other straddling the middle of the H2A α2 helix (Fig. 3a and Extended Data Fig. 9c). These segments of density are not continuous with the JmjC domain density and also not resolved to a degree that allowed for unambiguous a priori sequence assignment. However, the location of the N-terminus of the JmjC domain (last structured residue of JmjC domain is R36) made it likely that the acidic patch-binding density was contributed by a region within the N-terminal 35 residues of KDM2A (Fig. 3a). The N-terminal sequence is poorly conserved between KDM2A and KDM2B, further supporting unique functional roles of this region between the two paralogs (Extended Fig. 9d). We prepared a set of truncations and point mutations of the KDM2A N-terminus and measured the effects on nucleosomal H3K36me2 demethylation and acidic patch-dependent nucleosome binding. Truncation of the N-terminal 35 (Δ1–35) or 15 (Δ1–15) residues of KDM2A nearly abolished H3K36me2 demethylation (Fig. 3d–e and Supplementary Fig. 2d). These two truncations also reduced the ratio of IC50 values when competing with ΔAP versus wild-type nucleosomes as measured by TR-FRET, from 3.7x for the full length KDM2A (Fig. 3b) to 1.5x and 1.8x, respectively, for the truncated versions (Fig. 3f). These IC50 differences more closely resemble the 1.4x difference observed for KDM2B (Fig. 3b). As such, we concluded that the most important acidic patch-binding residues of KDM2A are contained within this N-terminal sequence.
Chromatin factors nearly universally use arginines when binding to the acidic patch23. The KDM2A acidic patch-binding density includes two likely arginine side chains. One arginine inserts into the canonical arginine anchor cavity surrounded by H2AE61, D90, and E92; the other arginine, a type 1 variant arginine, binds in proximity to H2AE56 and H2BE113. As there are seven closely spaced arginines in the KDM2A N-terminal sequence, we systematically tested point mutations to confidently build our atomic model. KDM2A R9A, R13A, and R19A mutations significantly decreased H3K36me2 demethylation, with R13A causing the most severe defect (Fig. 3d and Supplementary Fig. 2d). In contrast, KDM2A R7A and R15A mutants exhibited wild-type demethylase activity. Moreover, we observed a reduction in acidic patch-dependent nucleosome binding for the R13A mutant (1.8x difference in IC50 with ΔAP versus wild-type nucleosomes) (Fig. 3g) that was identical to that observed for the Δ1–15 truncation (Fig. 3f). The KDM2A R15A mutant retained wild-type acidic patch-dependent binding. These results together with our reconstructed map are consistent with only one molecular model that assigns KDM2A R13 as the canonical arginine anchor, which makes charged hydrogen bonds with H2AE61, D90, and E92, and KDM2A R9 as the variant arginine (Fig. 3a and Extended Data Fig. 9c). The intervening residues, S11 and Q12, make additional side-chain hydrogen bonds with H2BE113 and H2AE64, respectively. The adjacent segment of KDM2A density, bridging toward the JmjC domain, is best modeled with the R19-Y22 sequence. Interestingly, Y22 packs into a small hydrophobic cleft between the H2A α2 and α3 helices above H2AA86 but contributes minimally to KDM2A function (Fig. 3d and Extended Data Fig. 9c).
To corroborate this result, we generated chimeric versions of KDM2A and KDM2B with swapped N-terminal tails (Fig 3h). We observed higher H3K36me2 demethylase activity and a greater acidic patch dependence for wild-type KDM2A than wild-type KDM2B (Fig. 3i and Supplementary Fig. 2e). The KDM2A(2–35)-KDM2B(66–734) chimeric protein (KDM2AxB) gained demethylase activity under conditions in which KDM2B was only minimally active (Fig. 3i and Supplementary Fig. 2e). Moreover, grafting the KDM2A N-terminus on KDM2B resulted in acidic patch-dependent nucleosome binding in TR-FRET competition assays, with a 4.1x difference in IC50 values between ΔAP and wild-type nucleosomes (Fig. 3j). In contrast, we observed little demethylase activity for the KDM2B(2–65)-KDM2A(36–685) chimera (KDM2BxA) and only a 1.5x difference in IC50 values between ΔAP and wild-type nucleosomes in competition experiments similar to wild-type KDM2B (Fig. 3i–j and Supplementary Fig. 2e).
We next examined how KDM2A functions on nucleosomes assembled with oncohistone mutants. KDM2A demethylated H2AE56Q and H2BE113Q nucleosomes at wild-type levels (Fig. 3d and Supplementary Fig. 2f). In contrast, H2AE92K mutant nucleosomes were poor substrates for KDM2A. As expected from existing structures of H3K36 methyltrasferase-nucleosome complexes33,34, we observed no defects in enzymatic methylation of oncohistone mutant nucleosomes (Supplementary Fig. 1b), raising the possibility of an imbalance of H3K36 methylation and demethylation in H2AE92K cancers. Furthermore, these results emphasize the likelihood of distinct consequences of different oncohistone mutations on the chromatin landscape.
KDM2A histone charge shielding
Our model contains a gap of KDM2A residues 23–35 between Y22 at the edge of the acidic patch-binding region and R36, the first structured residue of the JmjC domain (Fig. 4a). In this region of the structure, unwrapping of the nucleosomal DNA exposes an underlying basic histone surface. H3K36 methyltransferases Set2 and NSD2/3 unwrap the terminal DNA in an almost identical manner as KDM2A/B and shield the exposed basic histone surfaces through direct interaction with structured regions of the catalytic SET domains33,34 (Extended Data Fig. 10a–c). In contrast, the JmjC domain of KDM2A is positioned too far away from this basic histone surface to make direct contacts. Interestingly, the unresolved KDM2A residues 23–35 include seven acidic aspartic/glutamic acids (Fig. 4a). The N-terminal sequences of KDM2A and KDM2B are poorly conserved, but the overall character and inclusion of seven acidic resides is shared (Extended Data Fig. 10d). We predicted that these acidic residues could make electrostatic interactions to shield the exposed histone surface. Indeed, neutralization of all seven acidic residues in KDM2A significantly reduced KDM2A demethylase activity (Fig. 4b and Supplementary Fig. 2d). We favor a model in which KDM2 family demethylases use a low-complexity acidic sequence in heterogeneous conformations not visible in cryo-EM reconstructions to shield positive charge exposed upon DNA unwrapping and prevent DNA re-wrapping during demethylation.
Fig. 4 |. Dynamic and multivalent KDM2A-nucleosome interactions.
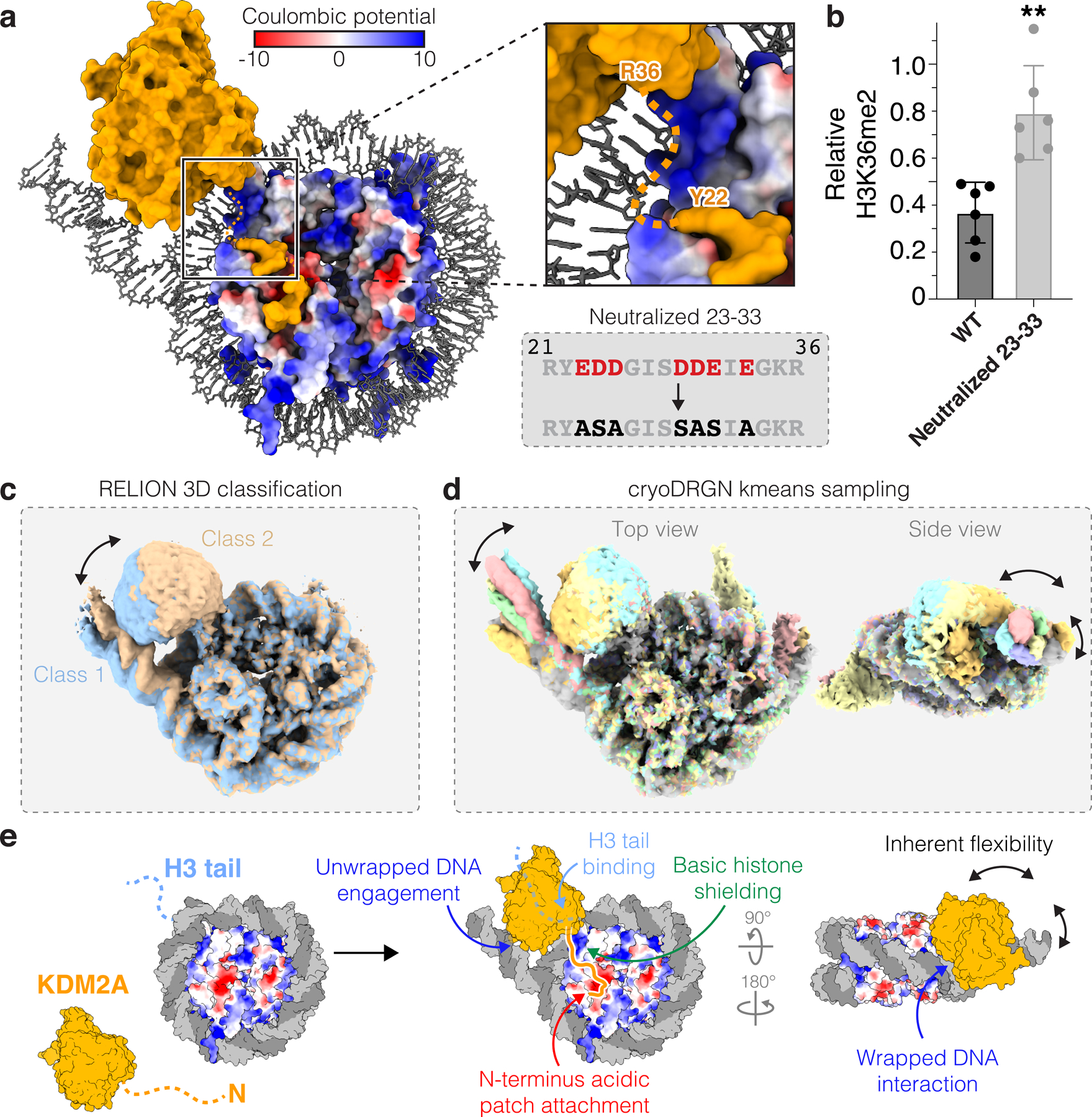
a, Basic histone surface exposed by DNA unwrapping between JmjC domain and acidic patch-binding region of KDM2A. b, Demethylation of H3K36me2-nucleosomes by WT and neutralized 23–33 mutant KDM2A. Relative residual H3K36me2 plotted as individual data points (n=6 assay technical replicates) along with mean ± SD, **p ≤0.01 (0.0014) using an unpaired two-tailed t-test. c-d, 3D classification in RELION (c) and heterogeneity analysis with cryoDRGN kmeans sampling (d) reveals correlated heterogeneity in regions of structure corresponding to JmjC domain and unwrapped DNA. e, Model of KDM2A-nucleosome binding includes dynamic and multivalent interactions with multiple nucleosome surfaces.
Multivalency underlies dynamic nucleosome recognition
Despite making critical interactions with the nucleosome acidic patch, unwrapped and wrapped nucleosomal DNA, exposed basic histones surfaces, and the H3 tail, KDM2A is still rather flexibly bound to the nucleosome. The JmjC domain densities of both KDM2A and KDM2B required multiple rounds of masked classification to become well-defined in reconstructions and still suffered from limited resolution (Extended Data Fig. 6 and 7). Consistently, multiple forms of heterogeneity analysis revealed regional flexibility, with JmjC domain position strongly correlated with motion of the unwrapped DNA, both moving as a unit across multiple dimensions of freedom (Fig. 4c–d and Supplementary Fig. 4). As a result of this heterogeneity, the regions of the cryo-EM maps corresponding to the JmjC domains did not resolve at high enough resolution to observe clear density for the H3 tail within the KMD2A/B active sites. However, based on the last observed density for H3 on the nucleosome surface (H3P43), the positioning of the JmjC domain, as well as our crystal structure of KDM2A bound to a H3K36C-UNC8015 (5) peptide showing flexibility C-terminal to H3H39, we believe that H3K36C-UNC8015 (5) engagement within the JmjC active site is consistent with our model.
Discussion
Our amino acid resolution nucleosome affinity proteomics screen establishes proteome-wide requirements of acidic patch binding and underscores the broad consequences for chromatin binding profiles in cells expressing cancer-associated oncohistone mutations within the acidic patch that complement recent targeted functional and phenotypic studies35. Mutation-associated loss of chromatin binding for protein complexes controlling vital cellular processes, such as cell division, gene expression or DNA damage repair, suggests complex pathogenic mechanisms. Building from our screen, we identified a set of shared and paralog-specific multivalent interactions that define H3K36 specificity of KDM2 family lysine demethylases (Fig. 4e). Although acidic patch binding is essential for KDM2A-mediated H3K36me2 demethylation, the acidic patch interaction contributes only modestly to KDM2A-nucleosome binding. We suspect that acidic patch binding constrains and orients the KDM2A catalytic domain to efficiently target H3K36me2 even though affinity is primarily driven by non-acidic patch interactions. This is similar to our observations with the H3K79 methyltransferase DOT1L, for which disruption of acidic patch binding leads to near complete loss of enzymatic activity but only minor changes to global nucleosome binding36,37. Consistent with this hypothesis, KDM2B is unable to bind the acidic patch and exhibits low activity despite high affinity nucleosome binding. It is likely that KDM2B relies on accessory proteins to position its catalytic domain for efficient demethylation.
To overcome challenges of transient nucleosome binding, we trapped the JmjC domain demethylases in active poses on the nucleosome by site-specific covalent attachment of an α-KG cofactor analog, UNC8015, to the nucleosome. We anticipate that similar substrate-inhibitor conjugates will provide a generalizable approach for solving cryo-EM structures of other complexes that are otherwise intractable due to dynamic and heterogeneous interactions. This approach was successful despite low potency inhibition, demonstrating that even inhibitors with low specificity and/or potency could be broadly repurposed for structural biology.
Methods
Plasmid construction
Point mutant histones, H2A: Q24A, E56A, E56Q, E61A, E64A, N68A, D72A, N89A, D90A, E91A, E92A, E92K, K119C and H2B: Q47A, E113A, E113Q were cloned by site-directed mutagenesis into pST50Tr vectors38. Genes for the acidic patch-neutralized histone H2A(E61A, E64A, N68A, D72A, N89A, D90A, E91A) and histone H3(K36C, C110A) were synthesized by IDT as gBlock gene fragments and cloned into pST50Tr vectors by traditional restriction enzyme-based cloning. cDNA for human KDM2A and KDM2B were obtained from the DNASU Plasmid Repository (HsCD00860177) and VectorBuilder, respectively. KDM2A(2–685) and KDM2B(2–734) were cloned into the pST50Tr vector38 with an N-terminal MBP tag and C-terminal 6xHIS fusion by Gibson assembly and traditional restriction enzyme-based cloning, respectively. KDM2A mutations, truncations and the KDM2A(2–35)-KDM2B(66–734) and KDM2B(2–65)-KDM2A(36–685) chimeric proteins were cloned by either site-directed mutagenesis or Gibson assembly. The cDNA for SKP1 was obtained from the DNASU Plasmid Repository (HsCD00506424). Full length KDM2B was cloned into the pBIG1b biGBac expression vector39 with an N-terminal 3xHALO-3xFLAG-3xMYC-2xSTR tag, along with SKP1, by Gibson assembly. The plasmid containing a gene fragment encoding NSD2(976–1240) with an N-terminal Strep-10xHIS tag in the pST50Tr vector was obtained as a gift from Song Tan. Gene fragments corresponding to mouse KDM2A(36–364) and KDM2A(450–517) were ordered as gBlock gene fragments and cloned into the pST44 polycistronic vector38 with an N-terminal 10xHIS-SUMO tag on KDM2A(36–364) by Gibson assembly. Plasmids containing tandem repeats of 119, 147, or 185 bp Widom 601 DNA were gifts from Song Tan. Biotinylated 185 bp DNA was prepared exactly as previously described except that the NotI restriction recognition sequence (GCGGCCGC) was replaced with the EcoRI sequence (GCGAATTC), allowing digest by EcoRI and ligation of end biotinylated annealed oligonucleotides21.
Nucleosome and protein preparation
Wild-type, acidic patch-neutralized and point mutant histones, H2A, H2B, H3, and H4 were expressed in E. coli BL21(DE3)pLysS cells, purified from insoluble inclusion bodies, and reconstituted into H2A-H2B dimers and H3-H4 tetramers exactly as previously described21. The histone H3(K36C, C110A) was expressed as above, extracted from inclusion bodies in extraction buffer (20 mM Tris-Cl, pH 8.0, 10 mM DTT, 7 M Guanidine-HCl), purified by reverse phase chromatography (39–44% acetonitrile in water containing 0.1% trifluoroacteic acid) using a C18 prep scale column (Vydac), and lyophilized. The histone H2A(K119C)-H2B dimer was biotinylated by resuspending in unfolding buffer (10 mM Tris-Cl, pH 7.5, 7 M Guanidine-Cl, 100 mM TCEP-Cl) at 2 mg/mL and incubating for 2 hours at room temperature with a 1:1 molar ratio of biotin-maleimide (Millipore). An additional molar equivalent of biotin-maleimide was then added and the reaction was allowed to proceed for another 2 hours, after which the reaction was quenched with 20 mM DTT and dialyzed into refolding buffer (10 mM HEPES, pH 7.5, 50 mM NaCl, 10 mM 2-mercaptoethanol) overnight. Refolded dimer was purified by cation exchange chromatography using a Source S resin. The 119, 147 and 185 bp DNA fragments containing Widom 601 positioning sequence40 were purified from E. coli exactly as previously described41,42. For nucleosome affinity pulldowns, the 185 bp DNA fragments were digested with EcoRI and subsequently ligated with a pair of complementary oligonucleotides with one harboring a 5’ biotin modification as previously described21. Nucleosomes were reconstituted and purified as previously described21,43,44. Briefly, either wild-type, acidic patch-neutralized (ΔAP: H2A E61A, E64S, N68A, D72S, N89A, D90A, E91S), point mutant (H2A: Q24A, E56A, E56Q, E61A, E64A, N68A, D72A, N89A, D90A, E91A, E92A, E92K, K119C and H2B: Q47A, E113A, E113Q) or biotinylated H2A-H2B dimers, wild-type H3-H4 or H3K36C-UNC8015-H4 tetramers, and 119, 147, 185 or biotinylated 185 bp DNA fragments were combined and dialyzed from high salt (10 mM Tris-Cl, pH 7.5, 2 M KCl, 1 mM EDTA, 1 mM DTT) to low salt (10 mM Tris-Cl, pH 7.5, 250 mM KCl, 1 mM EDTA, 1 mM DTT) buffers using a gradient dialysis pump, then purified by anion exchange chromatography using a Source Q resin.
KDM2A(2–685), KDM2A truncations and mutants, KDM2B(2–734), mouse KDM2A(36–364;450–517)20, and NSD2(976–1240) were expressed in E. coli BL21(DE3)pLysS cells at 18°C for 16–18 hours and purified over TALON Metal Affinity resin. With the exception of KDM2A(36–364;450–517), all KDM2 proteins were prepared with a permanent C-terminal 6xHis tag. TEV protease was used to cleave N-terminal tags, and cleaved protein was purified by ion exchange chromatography. KDM2A(2–685), KDM2A truncations and mutants, KDM2B(2–734), and NSD2(976–1240) were purified by cation exchange chromatography using Source S resin. KDM2A(2–685), KDM2A truncations and mutants, and KDM2B(2–734) were additionally purified by size exclusion chromatography using a Superdex 200 increase 10/300 column. Mouse KDM2A(36–364;450–517) was purified by anion exchange chromatography using a Source Q resin and subsequently purified by size exclusion chromatography using a Superdex 75 increase 10/300 column. Full length KDM2B was co-expressed with SKP1 in S. frugiperda Sf9 insect cells at 27°C for 48 hours. Nuclear extraction from Sf9 cells was carried out as previously described16, after which KDM2B/SKP1 was purified using Strep-Tactin resin (IBA) using manufacturers protocols. Purity of all proteins and nucleosomes was confirmed by analysis on SDS-PAGE (Extended Data Fig. 1b and Supplementary Fig. 5).
Nucleosome affinity pulldowns and TMT labeling
Human embryonic kidney 293 cells (HEK 293T) nuclear lysates were prepared using the standard salt extraction protocol45 and nucleosome affinity pulldowns were conducted essentially as previously described21. Briefly, 20 μg of biotinylated nucleosomes were immobilized on streptavidin T1 magnetic dynabeads (MyOne, Thermofisher) in BB120 buffer (20 mM HEPES, pH 7.5, 120 mM NaCl, 10% glycerol, 1 mM DTT, 0.1% NP-40) and incubated with 500 μg HEK 293T nuclear lysate with rotation for 2 h at 4°C. Following two brief washes and one 15 min wash in BB120 buffer, beads with nucleosome-bound nuclear proteins were moved to new tubes and after carefully aspirating and discarding any remaining BB120 buffer, nucleosome-nuclear protein complexes were eluted from the beads with 15 μl 2x gel loading buffer (Bio-Rad). Eluted samples were placed in a 100°C heat block for 5 min and then run on 4–20% 12-well Mini-PROTEAN TGX (Bio-Rad) gels (pre-run for 2 min at 250V) for 2 min at 250V. Proteins with molecular weights over ∼20 kDa, which resolved from the strong histones and monomeric streptavidin bands (Extended Data Fig. 1c) were excised from the gel and subjected to in-gel trypsin digestion following manufacturer’s protocols (Thermofisher). The concentration of extracted tryptic peptides was measured using the Pierce Quantitative Fluorometric Peptide Assay (Thermofisher) prior to Tandem Mass Tag (TMT) labeling (Thermofisher, Cat. no. A44522, Lot: WB314414).
Tryptic peptides representing seventeen different conditions in triplicate were labeled with sixteen unique TMT tags to allow deconvolution by mass-spectrometry in four independent runs/mixtures (Extended Data Fig. 1a). Building on experience from our first-generation nucleosome interactome screen, where the acidic patch-neutralized (ΔAP) mutant nucleosome pulled down less total protein than the other mutant nucleosomes21, we anticipated that point mutations in the critical arginine anchor cavity (H2A: E61, D90, E92) may behave in similar fashion. To avoid having the most deleterious mutations in the same mixture, which could cause inconsistencies in protein coverage between the mixtures, we divided the ΔAP and H2A E61, D90, and E92 mutant nucleosomes between the four mixtures. Wild-type tryptic peptides, labeled with the 133N, 133C, 134N TMT tags, were included in each run to allow comparison between the mixtures (Extended Data Fig. 1a). Mixture 1 included: ΔAP (127N, 127C, 128N), H2AQ24A (128C, 129N, 129C), H2AE56A (130N, 130C, 131N) and H2AE56Q (131C, 132N, 132C) mutant nucleosomes. Mixture 2 included: H2AE61A (127N, 127C, 128N), H2AE64A, (128C, 129N, 129C), H2AN68A (130N, 130C, 131N) and H2AD72A (131C, 132N, 132C) mutant nucleosomes. Mixture 3 included H2AN89A (127N, 127C, 128N), H2AD90A, (128C, 129N, 129C), H2AE91A (130N, 130C, 131N) and H2BQ47A (131C, 132N, 132C) mutant nucleosomes. Mixture 4 included: H2AE92A (127N, 127C, 128N), H2AE92K, (128C, 129N, 129C), H2BE113A (130N, 130C, 131N) and H2BE113Q (131C, 132N, 132C) mutant nucleosomes. Additionally, each mixture included a bridge sample consisting of equal amount of tryptic peptides from each of the pulldowns, mixed and labeled with TMT 126 (Extended Data Fig. 1a). This bridge sample was included for normalization across the four mixtures.
Proteomics data acquisition and analysis
TMT-labeled peptides were separated via reverse-phase nano-HPLC using an RSLCnano Ultimate 3000 (Thermo Fisher Scientific). The mobile phase consisted of water + 0.1% formic acid as buffer A and acetonitrile + 0.1% formic acid as buffer B. Peptides were loaded onto a µPAC™ Trapping column (PharmaFluidics) operated at 35°C and flowing at 1 ul/min, and then separated on a 50 cm µPAC™ column (PharmaFluidics) at 300 nL/min using a segmented gradient as follows: 30 sec from 2–5% buffer B, 1.5 min from 5–7% buffer B, 90.5 min from 7–25% buffer B, and 20 min from 25–45% buffer B. Mass spectrometry analysis was performed on an Orbitrap Eclipse (Thermo Fisher Scientific) using a TMT-RTS-MS3 data acquisition method. MS1 scans were obtained at 120,000 resolution in the Orbitrap with a scan range from 350–2000 m/z, 50 ms max injection time, and automated gain control (AGC) target of 1e6. All precursors were filtered for monoisotopic peaks, charge states 2–7, mass between 800 and 8000 Da, dynamic exclusion of 60 seconds with 10 ppm mass tolerance excluding isotopes, and a precursor fit of 50% with a window width of 1.2 m/z. Precursors selected for MS2 were isolated in the quadrupole with an isolation window width of 0.7 m/z. MS2 ions were obtained in the ion trap with 35 ms max injection time and 1e4 AGC and fragmented with collision induced dissociation (CID) collision energy of 35% with 10 ms activation time. Precursors selected for MS3 scans were based on real-time search synchronous precursor selection (RTS-SPS, Orbitrap Eclipse 3.4.3072.18). MS2 spectra were searched against the UniProtKB/Swiss-Prot homo sapiens sequence database with isoforms (downloaded on August 2019) appended with mutant histone and contaminant sequences. For all analyses, the following RTS parameters were used: specific tryptic digestion with up to 1 missed cleavage, carbamidomethyl and TMTpro16plex fixed modifications, and variable methionine oxidation. Real-time FDR filtering was used along with the following scoring thresholds: xcorr ≥ 2, dCN ≥ 0.1, and precursor ppm ≤ 10. MS3 scans were obtained in the Orbitrap at 50,000 resolution with a 100–500 m/z scan range, 86 ms max injection time, higher-energy collision dissociation (HCD) collision energy of 55%, and an MS2 isolation window of 3 m/z.
After data acquisition, raw files were processed for peptide identifications by Comet (2021.01 rev. 0) and Percolator (version 3.05.0) with the same database used for RTS and the following differences in parameters: semi- tryptic digestion with ≤ 2 missed cleavages, and a clear m/z range from 125.5 to 134.5. Identifications with percolator q-values < 0.01 that matched the RTS identifications (same sequence, charge state and MS2 scan) were kept. TMT lot-specific purity correction was performed using non-negative least squares regression and missing values were imputed with spectrum noise levels using an in-house script. MS3 scans were filtered for those with a signal for the bridge channel and summed signal-to-noise ≥ 50. Histones were excluded from downstream analysis due to sequence overlap between WT and mutant histones. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD032791.
Downstream analysis was achieved with an in-house R script and MSstatsTMT (version 2.1.15). Only peptides that were unique to a gene were considered for protein quantification. For redundant MS3 spectra, the ones with highest total TMT intensity were used. Median normalization was performed within each mixture. Specifically, intensity for each condition was multiplied by the median of their respective condition replicates and divided by the median of their respective mixture. Protein summarization was performed by MSstatsTMT (with reference normalization, and without global normalization). All reporter intensities then underwent log2 transformation for statistical analysis. PCA clustering was done based on these normalized data. Proteins were further filtered for heatmap visualization and differential expression analysis. Namely, we required proteins to be quantified in at least three mixtures and having ≥ 2 peptide-spectrum-matches in at least two of those mixtures. Each protein intensity was corrected by subtracting the median of the protein’s intensity in WT channels from the mutant conditions of the same mixture. Fold-changes, ANOVA, t-tests, and figures were based on these normalized data.
Immunoblotting
Nucleosome-bound nuclear protein complexes were obtained from affinity pulldowns as described above, run on 4–20% 12-well Mini-PROTEAN TGX gels (Bio-Rad), transferred to a nitrocellulose membrane (GE Healthcare) in transfer buffer (25 mM Tris-Cl, pH 8.3, 193 mM glycine, 20% MeOH, 0.025% SDS), and immunoblotted using standard procedures. Primary antibodies included rabbit polyclonal histone H3 (Sigma, H0164, 1:5000), rabbit polyclonal HMGN2 (ABclonal, A6156, 1:500), rabbit polyclonal Ran (ABclonal, A0976, 1:1000), mouse monoclonal RCC1 (Sigma, SAB1403666, 1:1000), rabbit polyclonal VRK1 (ABclonal, A7745, 1:500), and rabbit polyclonal TRIP12 (Aviva Systems Biology, OAAB20005, 1:500). Secondary antibodies included IRDye 680RD goat anti-rabbit IgG secondary antibody (LI-COR) and IRDye 800CW goat anti-mouse IgG secondary antibody (LI-COR), both used at 1:10,000 dilution. Blots were imaged on an Odyssey imager (LI-COR).
Preparation of NSD2-methylated H3K36me2 nucleosomes
Wild-type or acidic patch mutant 185 bp nucleosomes were enzymatically methylated by incubating the respective unmodified nucleosomes with a 4:1 molar excess of NSD2(976–1240) for 3 hours at 30°C in methylation buffer (20 mM Tris-Cl, pH 8.0, 50 mM NaCl, 10 μM ZnSO4, 1 mM DTT, 20% glycerol), followed by anion exchange purification using Source Q resin. The methylation state of histone H3 was analyzed by LCMS using an Agilent 6520 Accurate Mass Quadrupole Time-of-Flight mass spectrometer, which indicated uniformly dimethylated histone H3. HinfI-digested nucleosomes were prepared from wild-type NSD2-methylated nucleosomes by incubating a 4:1 molar excess of HinfI with 4 μM nucleosome in CutSmart buffer (NEB) for 1 hour at 37°C, followed by quenching with 15 mM EDTA and anion exchange purification using a Source Q resin.
Alkylation of H3 peptide and full-length protein with UNC8015
UNC8015 (4) was synthesized and characterized as described in the Supplementary Note. The histone H3(A29-Y41, K36C) peptide was ordered from GenScript. The alkylation of the H3(A29–41, K36C) peptide with UNC8015 (4) was carried out as previously described46. Briefly, the H3 peptide was dissolved in alkylation buffer (4 M Guanidine-HCl, 1 M HEPES, pH 7.8, 10 mM D/L-methionine) and 1M DTT was added to bring the reaction to 20 mM DTT. After incubation at 37°C for 1 hour, 5 molar equivalents of UNC8015 (4) were added and the reaction was incubated at 37°C for 2.5 hours. An equivalent volume of 1 M DTT was added to the reaction (now at 40 mM DTT), the temperature was increased to 50°C and the reaction was allowed to proceed for an additional 2.5 hours before quenching by addition of 2-mercaptoethanol to ~0.7 M and incubation at room temperature for 30 minutes. The reaction was transferred to a glass scintillation vial and concentrated under reduced pressure. The resulting crude product was purified via flash chromatography (10–100%, MeOH in water containing 0.1% TFA) to yield the alkylated H3K36C-UNC8015 peptide (5), which was subsequently lyophilized. Alkylation reaction of full-length H3(C110A,K36C) with UNC8015 (4) was carried out as above, but with 50 molar equivalents of UNC8015 (4) and two, 2.5-hour incubations at 50°C. Following the reaction, the alkylated histone was directly dialyzed into 2 mM 2-mercaptoethanol and lyophilized. Purity of the starting H3(C110A, K36C) reagent and H3K36C-UNC8015 product was confirmed by LCMS, which showed ~80% reaction completion at the 5-hour reaction endpoint.
Crystallization and X-ray crystallography
Mouse KDM2A(36–364, 450–517) concentrated to 20 mg/mL was combined with 3 molar equivalents of H3(A29-Y41, K36C) peptide-UNC8015 (5) conjugate, incubated at room temperature for 10 min, and subsequently centrifuged at 17,000 x g for 5 min at room temperature. Crystals were grown by sitting drop vapor diffusion in 100 mM Tris-Cl, pH 8.5 (Hampton), 150 mM lithium sulfate (Hampton), 20% PEG3350 (Millipore Sigma) at room temperature and appeared after approximately 5 days of equilibration. Crystals were harvested into cryoprotectant solution consisting of the base crystallization solution supplemented with 40% ethylene glycol for 15 minutes and flash frozen in liquid nitrogen. Diffraction data were collected at Advanced Photon Source’s SER-CAT 22-ID beamline (1 Å, 100K) equipped with an Eiger 16 M detector. Data were collected with a detector distance of 197 mm, with 180° rotation in 0.3° wedges, an exposure time of 0.25 s, a beam aperture of 50 µm and a transmission of 20%. Reflections were indexed, integrated and scaled using XDS and aimless47,48. Data were truncated at 1.98 Å to reflect the overall resolution limit as defined by I/σI > 2. Analysis of the data with Xtriage indicated a translational non-crystallographic symmetry (tNCS) peak of ~48% of the origin which did not preclude molecular replacement with PhaserMR using chains A and B from PDB 4QX7 and iterative refinement by manual model building in Coot and automatic refinement using Refmac549–51. Fe2+ was modeled into the active site, but not confirmed by crystallographic methods. The structure of mouse KDM2A(36–364, 450–517) bound to the H3K36C-UNC8015 peptide (5) was solved at a resolution of 1.98 Å with Rwork/Rfree = 18.6%/23.7%. The final model possesses 0.94% Ramachandran outliers and 97.50% Ramachandran favored dihedral angles. The final model includes two copies of mouse KDM2A (36–364, 450–517) bound to one Fe2+ ion each in chains A:B, D:E, H3K36C-UNC8015 (A29-H39) in chain C and H3K36C-8015 (T32-K37) in chain F. Due to weaker electron density in chains D, E, and F, we based our conclusions within the text on chains A, B, and C of the complex.
Cryo-EM sample preparation and data collection
To prepare KDM2A/B-nucleosome complexes for cryo-EM structure determination, 4 molar equivalents of KDM2A(2–685) or KDM2B(2–734) were added to H3K36C-UNC8015 nucleosomes in reconstitution buffer (10 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM DTT, 0.1 mM PMSF) in four steps, separated by 5 minute intervals. Complexes were purified by size exclusion chromatography using a Superdex 200 increase 10/300 column, then concentrated to ~10 mg/mL. Each complex was crosslinked by addition of glutaraldehyde (Sigma-Aldrich) to a final concentration of 0.1% and incubation at room temperature for 5 minutes. Crosslinking was quenched by addition of Tris-Cl, pH 7.5 to a final concentration of 20 mM and incubation for 15 minutes at room temperature. Samples were subsequently buffer exchanged into crosslinking buffer (10 mM HEPES, pH 7.5, 50 mM NaCl) using a Zeba spin desalting column (Thermo Fisher) and diluted to 1 mg/mL in freezing buffer (10 mM HEPES, pH 7.5, 30 mM NaCl) immediately before spotting onto grids. Cryo-EM grids were prepared on a Vitrobot Mark IV (Thermo Fisher) set to 4°C, 100% humidity, and -10 to 0 blot force. Quantifoil R1.2/1.3 grids were plasma cleaned with a Tergeo EM plasma cleaner for 60 seconds, after which 3 μL of sample was applied directly to the carbon side of the grid, blotted for 4 seconds with Whatman 595 filter paper, and plunge frozen into a 40/60% ethane/propane mixture. Imaging of grids was carried out on a Thermo Fisher Scientific 200kV TEM Talos Arctica G3 equipped with a Gatan K3 direct electron detector. 60-frame movies were collected in counting mode using SerialEM with a multi-shot 5 x 5 regular pattern, at 45,000x nominal magnification corresponding to a pixel size of 0.91 Å, a defocus range of -0.5 to -3.0 μm, and a total dose of 45–50 e–/Å2. A total of 9,075 movies from one grid and 15,671 movies from four grids were collected for the KDM2A- and KDM2B-nucleosome complexes, respectively.
Cryo-EM data analysis
Movies were imported into optics groupings in RELION-3.1-beta52 based on the beam-image shifts used for data collection. Motion correction and CTF estimation was carried out using the RELION implementation of MotionCor2 and CTFFIND-4.153, respectively, and any micrographs with an estimated resolution over 5 Å were removed from subsequent processing. RELION Laplacian-of-Gaussian and template-based particle picking followed by iterative rounds of 2D and one round of 3D classification resulted in initial reconstructions of the KDM2A- and KDM2B-nucleosome complexes with only a modest globular density corresponding to the JmjC domains. Following per-particle CTF refinement and Bayesian particle polishing in RELION, two rounds of JmjC domain-masked classification were carried out, which resulted in particle subsets with better resolved densities corresponding to the JmjC domains of KDM2A/B. Multibody refinement was carried out by masking the nucleosome and unwrapped DNA-JmjC domains after the first round of masked classification, which showed principal components of motion between the two bodies and resulted in a nucleosome-focused reconstruction with slightly improved resolution for the nucleosome and KDM2A acidic patch-binding residues. Additional heterogeneity analysis was carried out on the KDM2A-nucleosome complex dataset using cryoDRGN54. Briefly, KDM2A-bound particles (~250,000 particles) from an initial round of 3D classification were downsized to an image size of D=256 and the model was trained on a large architecture (1024 x 3) for 50 epochs. Both kmeans and principal component sampling of latent space showed similar heterogeneity in the unwrapped DNA-JmjC domain portions of the complex.
KDM2A/B-nucleosome complex model building
An initial model of the KDM2A-nucleosome complex was constructed by manually docking the structures of the KDM2A(36–364, 450–517)-H3(A29-Y41, K36C)-UNC8015 conjugate (PDB: 7UVA), the nucleosome from our previously published cGAS-nucleosome structure (PDB: 7JO9), and unwrapped nucleosomal DNA from the NSD3-nucleosome structure (PDB: 7CRQ) into the cryo-EM maps in chimera. Changes to the DNA sequence and humanizing the KDM2A sequence relative to the initial models was performed in Coot, and this corrected model was refined through iterative rounds of real space refinement in Phenix55 and manual model building in Coot. Reference model restraints were applied to the region of the model corresponding to the JmjC domain of KDM2A and H3 N-terminal tail due to the limited resolution in this region of the map. Models and map-model correlations were validated in MolProbity56 and Phenix EMRinger. As no crystal structures of the KDM2B JmjC domain exist, a model of the KDM2B-nucleosome complex was constructed by aligning a homology model of the KDM2B JmjC domain (obtained from SWISS-MODEL) to the KDM2A JmjC domain in the KDM2A-nucleosome complex model, which agreed well with the cryo-EM reconstruction of the KDM2B-nucleosome complex.
H3K36me2 peptide and nucleosome demethylation assays
JmjC inhibitors UNC8015–8 were synthesized and characterized as described in the Supplementary Note. Wild-type histone H3(A29–41) and H3(A29-Y41)K36me2 peptides were synthesized by solid-phase peptide synthesis at the UNC High-Throughput Peptide Synthesis and Array Facility. Demethylation assays using the H3(A29-Y41)K36me2 substrate peptide were conducted with the Invitrogen Histone Demethylase Fluorescent Activity Kit (Thermo Fisher). Assays were run at 40 μL in a black 96-well half area plate (Thermo Fisher) in the kit-included Jumonji-type assay buffer with 5 μM KDM2B(2–734), 200 μM H3(A29-Y41) K36me2 peptide substrate or H3(A29-Y41) unmodified control, 1 mM α-KG disodium salt dihydrate (Sigma-Aldrich), 2 mM L-ascorbic acid (Sigma-Aldrich), and 50 μM ammonium iron(II) sulfate hexahydrate (Sigma-Aldrich). Assays were conducted at 37°C for 2 hours, then developed with 10 μL Formaldehyde Reagent for 30 minutes before scanning with an EnVision 2103 plate reader at excitation/emission wavelengths of 260/520 nm. All assays were performed in triplicate distinct samples, and demethylase signal was corrected by subtracting the average background fluorescence intensity signal measured in the control assay with all assay components but using unmethylated H3(A29-Y41) as a substrate.
Nucleosome demethylation assays were performed with 4 µM (chimeric protein and oncohistone assays) or 2 μM demethylase (all other assays) and 4 μM NSD2-methylated nucleosomes at 30°C for 4 hours in demethylation buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM α-KG, 2 mM L-ascorbic acid, 50 μM iron(II) sulfate, 1 mM DTT). 5 μL of reaction diluted 1:1 with SDS-page sample loading buffer was separated on a 4–20% Mini-PROTEAN TGX gel (Bio-Rad) and immunoblotted as described above with the mouse monoclonal histone H3 (1:5000, Cell Signaling Technologies, 3638) and rabbit monoclonal H3K36me2 (1:5000, obtained as a gift from Brian Strahl) primary antibodies. Blots were quantitated using ImageJ and H3K36me2 was normalized to the wild-type H3 signal from the equivalent sample. Immunoblot assays were each replicated 3–6 times with distinct samples, and statistical significance was determined using Prism 9 (GraphPad).
TR-FRET nucleosome binding assays
For nucleosome competition experiments, 100 nM KDM2A(2–685), 5 nM H2AK119-biotinylated 185 bp nucleosomes, 2 nM LANCE Eu-W1024 Streptavidin (Perkin Elmer), 10 nM Ultra ULight α-6xHIS antibody (Perkin Elmer), and 0–3,000 nM of competing nucleosome were mixed in a total volume of 10 μL TR-FRET-150 buffer (10 mM Tris-Cl, pH 7.5, 150 mM NaCl, 5 % glycerol, 0.01% NP-40, 0.01% CHAPS, 5 mM DTT, 100 µg/mL BSA) in white low-volume non-binding 384-well plates (Greiner-BioOne). Plates were mixed at 1000 rpm on a tabletop plate shaker for 1 minute, centrifuged at 1000 x g for 1 minute, then incubated at room temperature for 30 minutes before reading on an EnVision 2103 plate reader using 320 nm excitation and simultaneous dual emission measurements at 615 and 665 nm with a 100 μs delay between excitation and emission measurement. All titration points were performed in triplicate distinct samples and final values were calculated by averaging the ratio of 665/615 nm emission signals. Non-linear curve fitting and IC50 calculation was performed in MATLAB R2020b with the fit non-linear regression model (fitnlm) function by fitting the values to a two-state competition model:
Binding isotherms were conducted as above, but without nucleosome competitor, with 0–750 nM wild-type or mutant KDM2A(2–685), and using a 1:25 molar ratio of α-6xHIS antibody:KDM2A. For binding isotherms, FRET signal (665 nm) was corrected for donor bleed-through by subtracting the product of the donor emission signal (615 nm) and the ratio of FRET signal to donor emission signal in the absence of acceptor. FRET signal was also corrected for direct acceptor excitation by subtracting the signal from equivalent titration wells lacking nucleosomes. Non-linear curve fitting and KD calculation was performed in MATLAB R2020b as above but using a two-state binding model:
Individual data points for TR-FRET datasets and fits to the model are shown in Supplementary Fig. 6.
Code Availability
In-house R script used for proteomics data analysis are available for download at GitHub (https://github.com/GoldfarbLab/NucleosomeProteomeAnalysis).
Extended Data
Extended Data Fig. 1 |. Amino acid resolution nucleosome affinity proteomics screen.
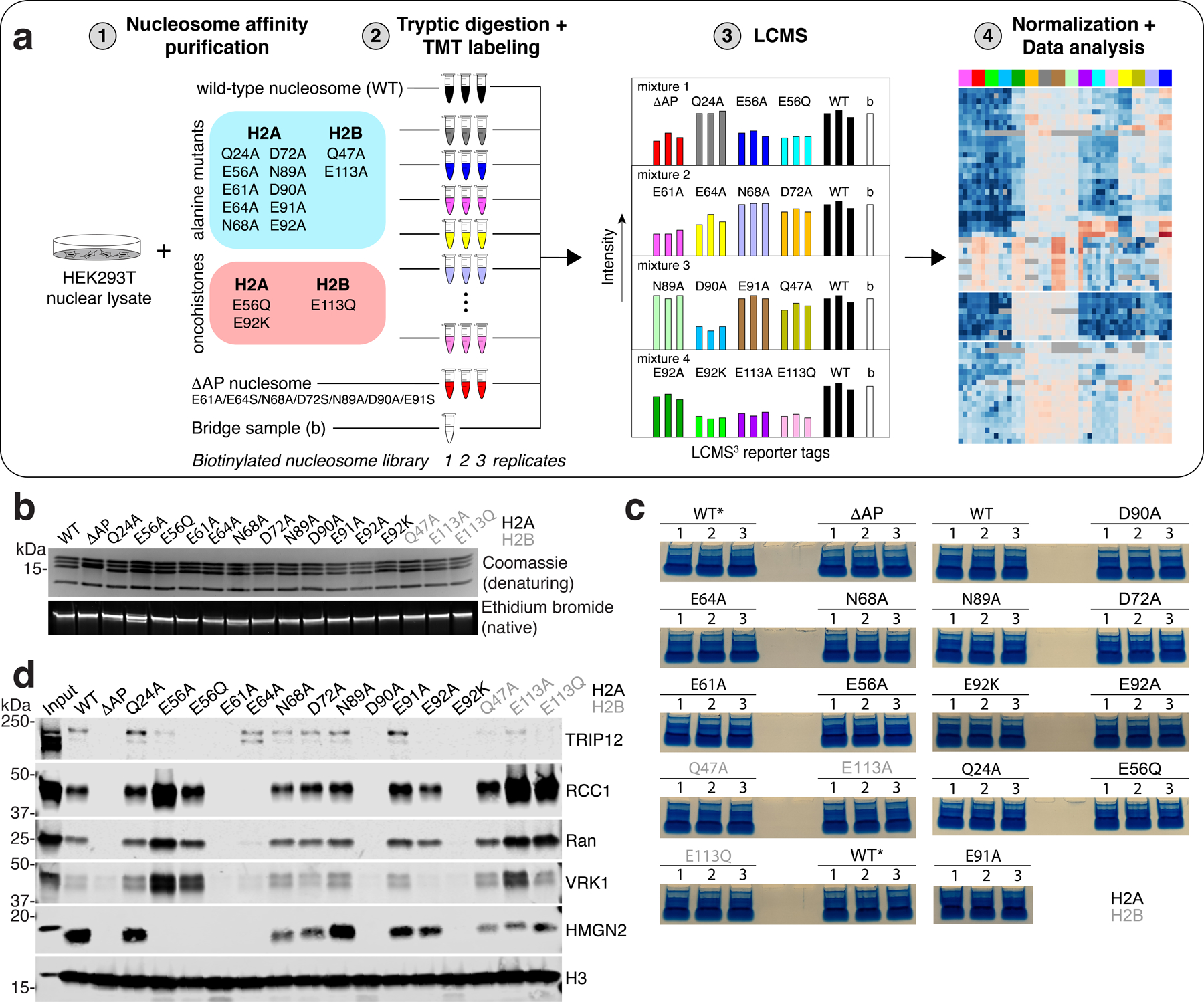
a, a, Schematic of nucleosome affinity proteomics screen. b, Coomassie-stained SDS-polyacrylamide denaturing gel (top) and ethidium bromide-stained native gel bottom of biotinylated nucleosome library. These experiments were repeated independently two times with similar results. c, Triplicate pulldowns run briefly on SDS polyacrylamide gels and stained with Coomassie blue prior to excision and in-gel trypsin digestion. Unused replicates of wild-type (WT) nucleosome pulldowns marked with an asterisk. d, Western blots following pulldowns from HEK293T nuclear lysates using indicated biotinylated nucleosomes. These experiments were performed once.
Extended Data Fig. 2 |. Oncohistone mutations have distinct effects on nucleosome binding.
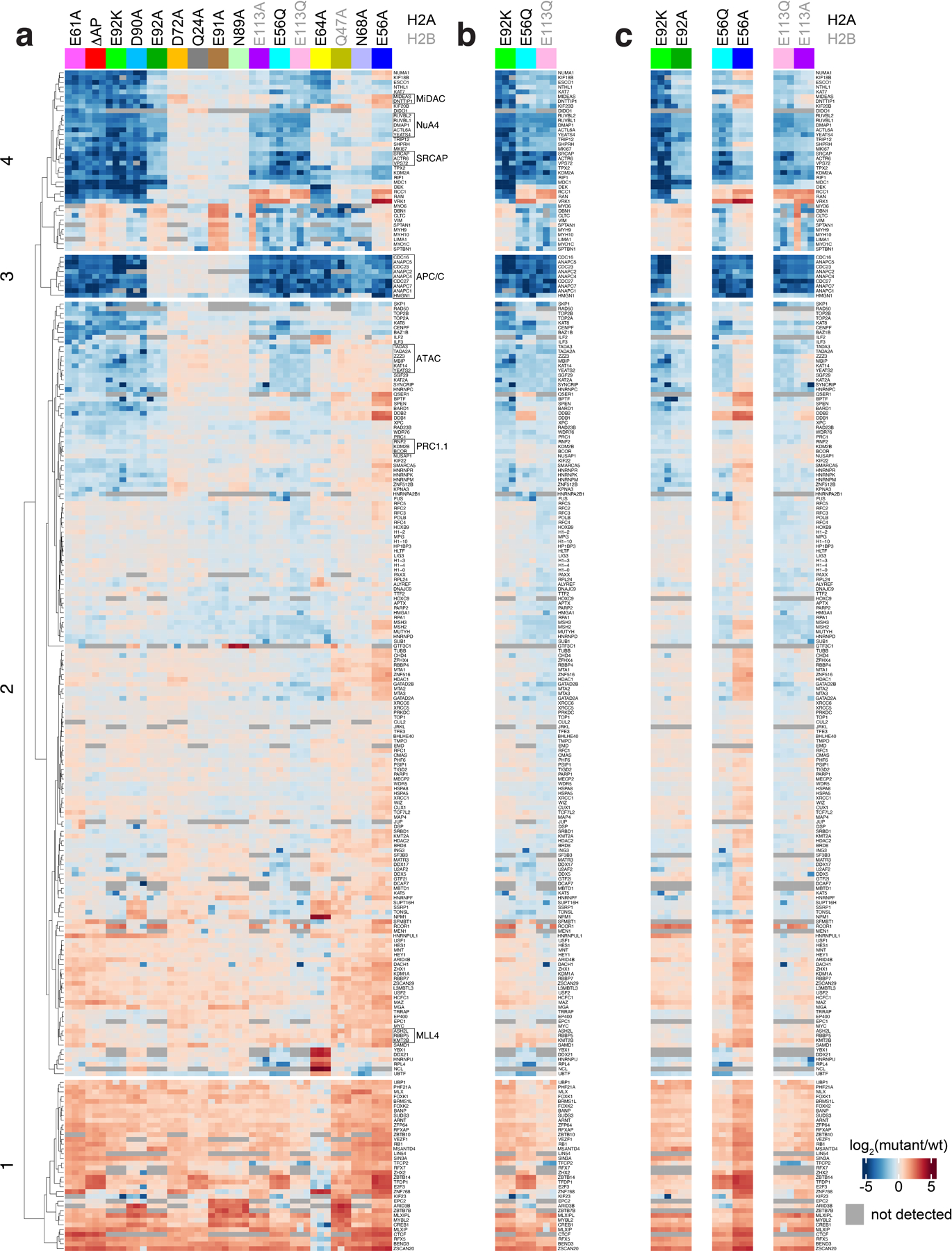
Full heat map showing log2-fold change relative to wild-type nucleosome of quantified proteins listed at right across screen, clustered by protein and nucleosome mutant. b, Heat map of oncohistone mutant nucleosomes, clustered as in panel a. c, Heat map of each oncohistone mutation relative to the corresponding alanine mutation, clustered as in panel a.
Extended Data Fig. 3 |. KDM2A/B-nucleosome complex purification and 2D classification.
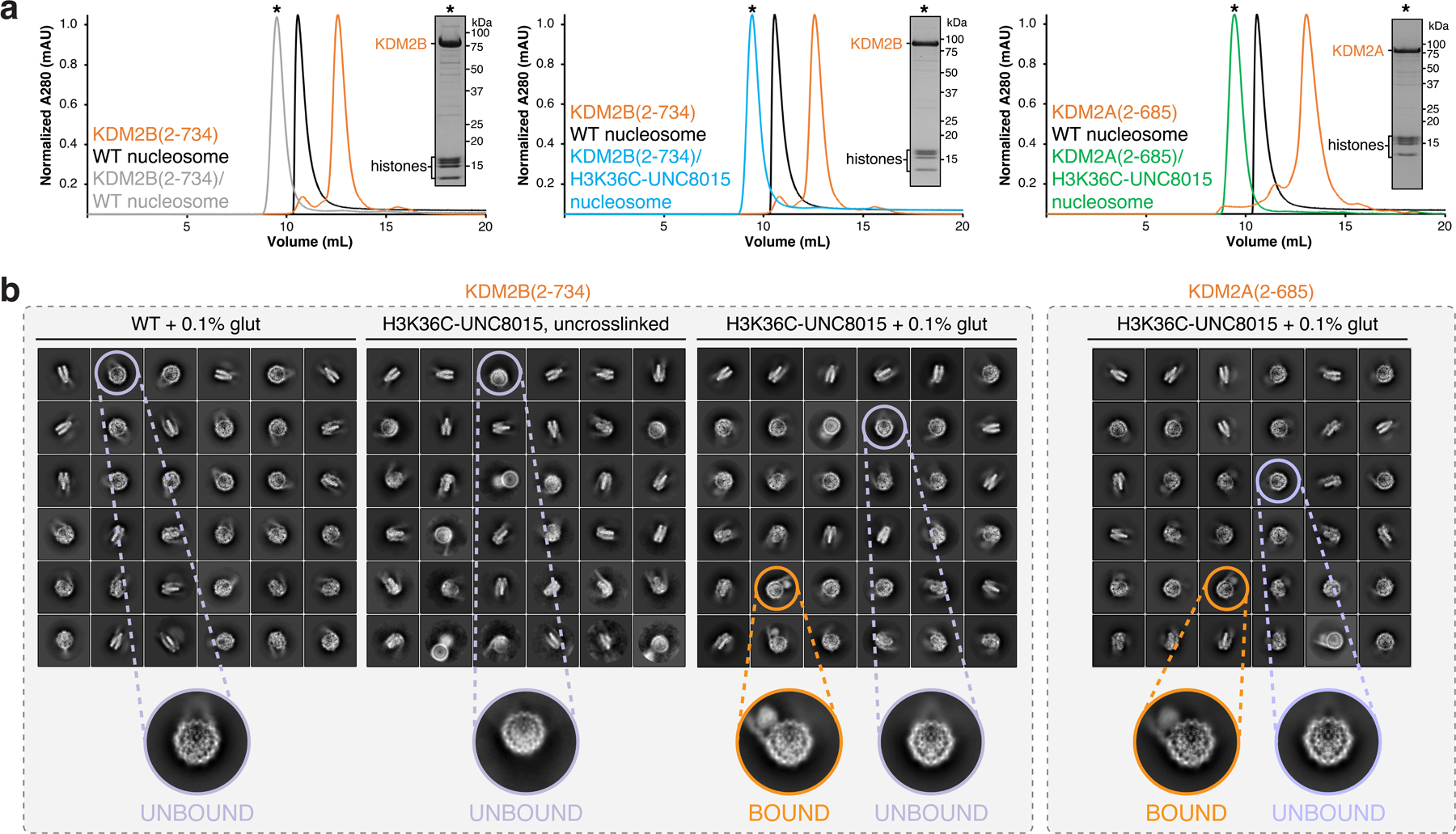
a, Chromatograms displaying normalized 280 nm absorbance from size exclusion purification of KDM2B/WT nucleosome (left), KDM2B/H3K36C-UNC8015 nucleosome (middle), and KDM2A/H3K36C-UNC8015 nucleosome (right) complex reconstitutions. These experiments were performed once. b, Highest populated 2D classes attained from cryo-EM analysis of indicated samples, with selected top views of KDM2A/B-unbound and bound nucleosomes.
Extended Data Fig. 4 |. Design and synthesis of H3K36-JmjC inhibitor covalent conjugate.
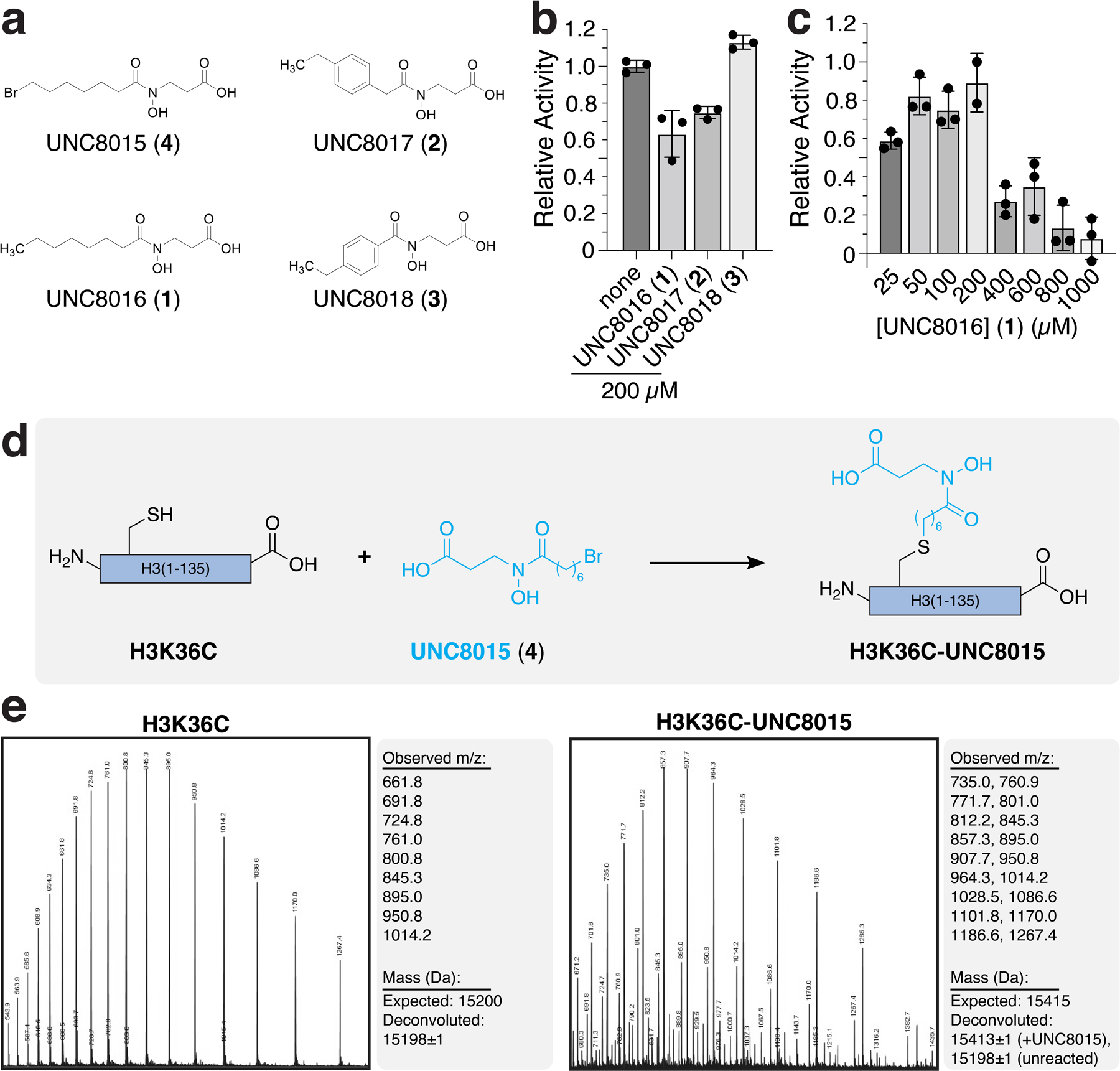
a, Chemical structures of JmjC inhibitors. b, Inhibition assays using H3K36me2 peptide substrates with KDM2B(2–734). c, Dose response inhibition of KDM2B(2–734) by UNC8016 on H3K36me2 peptide substrates. In panels b and c, mean ± SD are shown (n=3 assay technical replicates). d, Synthetic scheme for alkylation reaction of H3K36C mutant with UNC8015. e, LCMS spectra and mass deconvolution (determined using ESIprot) of unreacted H3K36C (left) and reacted H3K36C-UNC8015 (right; 5-hour endpoint).
Extended Data Fig. 5 |. Crystal structure of KDM2A bound to H3K36-UNC8015 peptide covalent conjugate.
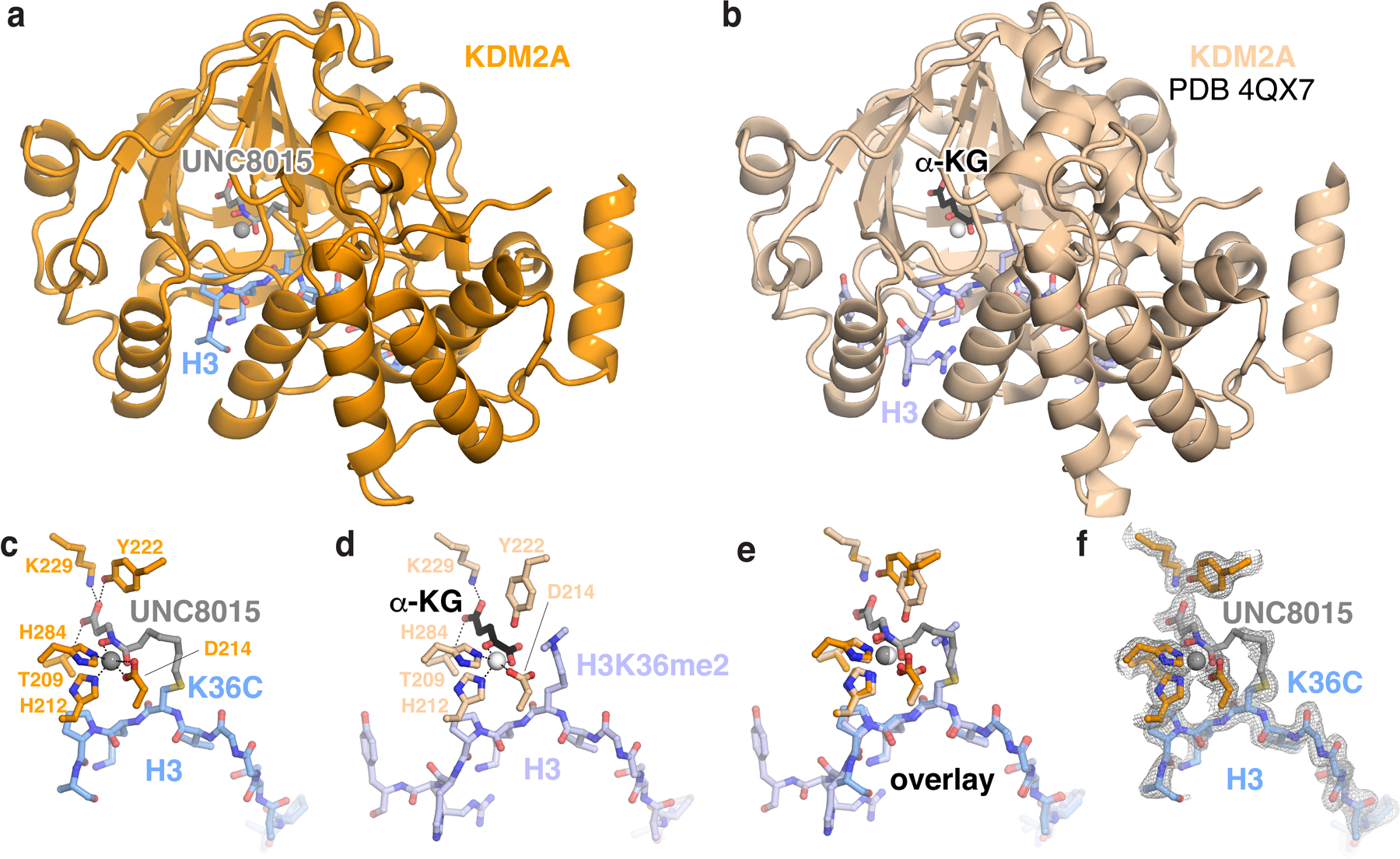
a, Structure of KDM2A catalytic domain in complex with H3K36C-UNC8015. b, Identical view of structure of KDM2A catalytic domain in complex with H3K36me2 peptide and α-ketoglutarate (α-KG). c-e, Zoomed views of c, H3K36C-UNC8015 peptide, d, H3K36me2 peptide and α-KG, and e, overlayed aligned structures. f, 2Fo-Fc electron density map of H3K36C-UNC8015 peptide at 1σ.
Extended Data Fig. 6 |. Cryo-EM data analysis for KDM2A-nucleosome complex.
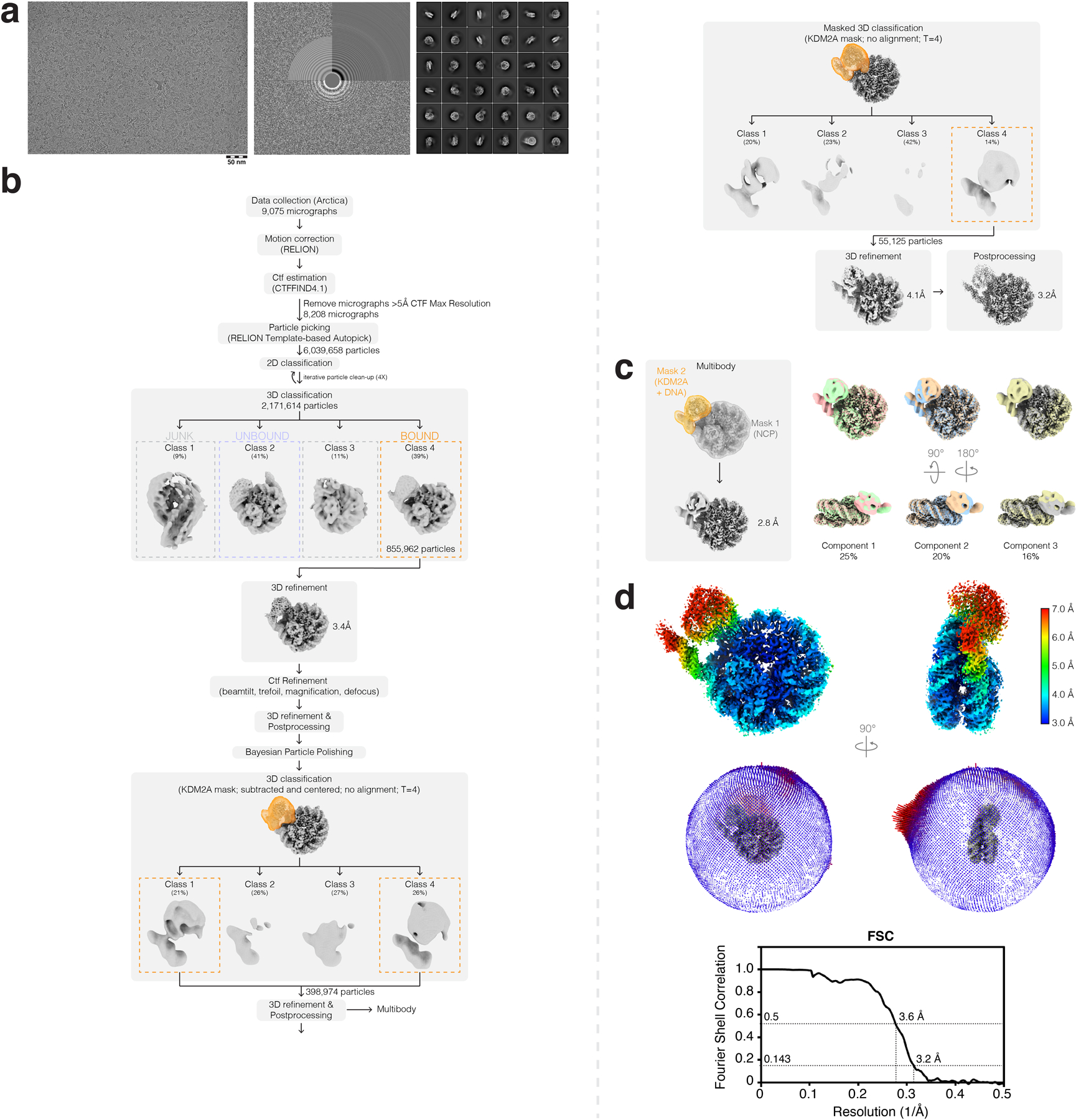
a, Representative micrograph (left), CTF estimation (middle), and 2D classes (right) from KDM2A-nucleosome complex dataset. b, Cryo-EM data analysis workflow carried out in RELION-3.1. c, Multibody refinement analysis results from RELION. d, Final postprocessed map colored by local resolution (top), angular distribution of particles (middle), and FSC curve (bottom) for KDM2A-nucleosome complex.
Extended Data Fig. 7 |. Cryo-EM data analysis for KDM2B-nucleosome complex.
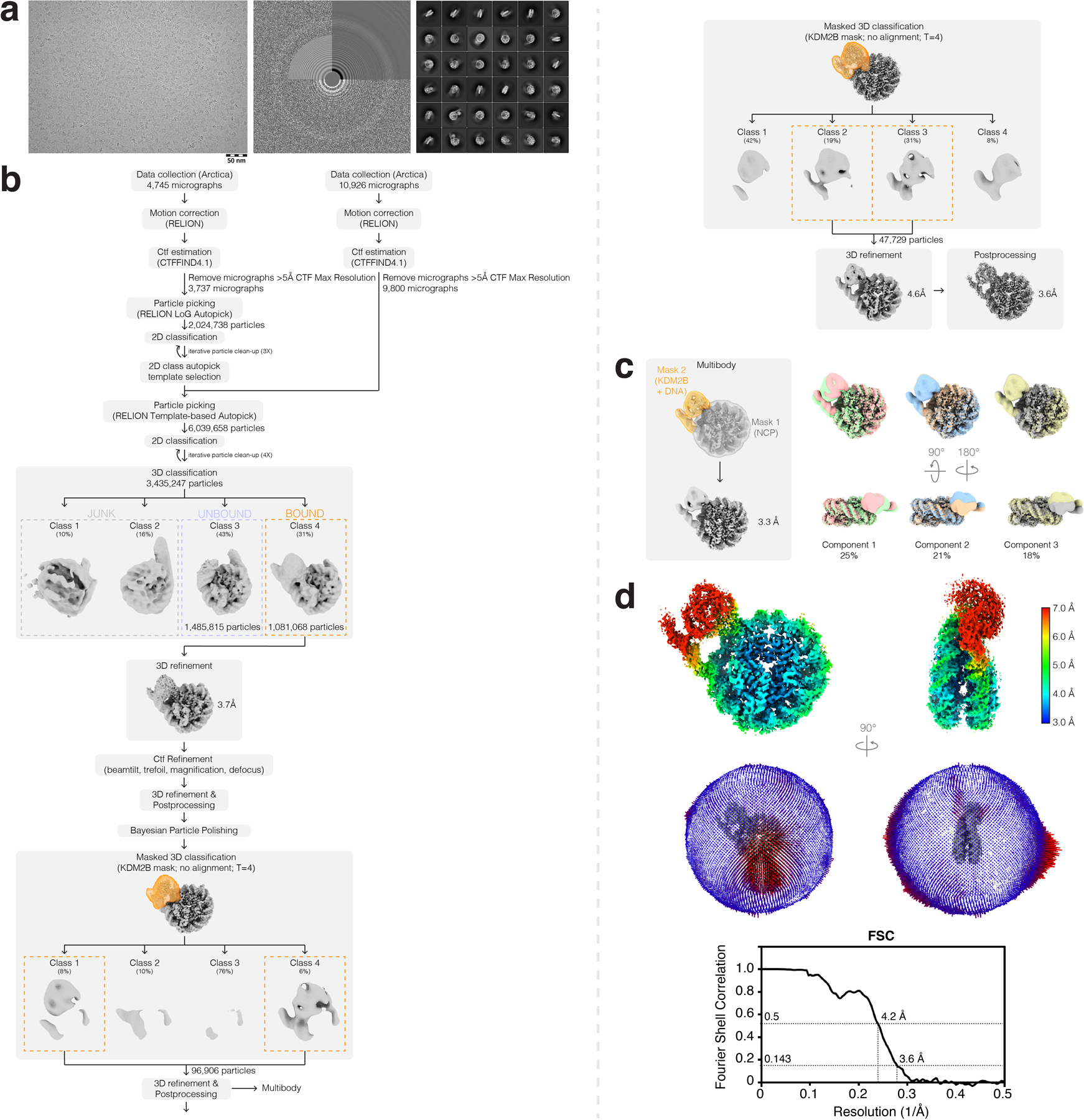
a, Representative micrograph (left), CTF estimation (middle), and 2D classes (right) from KDM2B-nucleosome complex dataset. b, Cryo-EM data analysis workflow carried out in RELION-3.1. c, Multibody refinement analysis results from RELION. d, Final postprocessed map colored by local resolution (top), angular distribution of particles (middle), and FSC curve (bottom) for KDM2B-nucleosome complex.
Extended Data Fig. 8 |. Unwrapped DNA-binding interfaces of KDM2A/B.
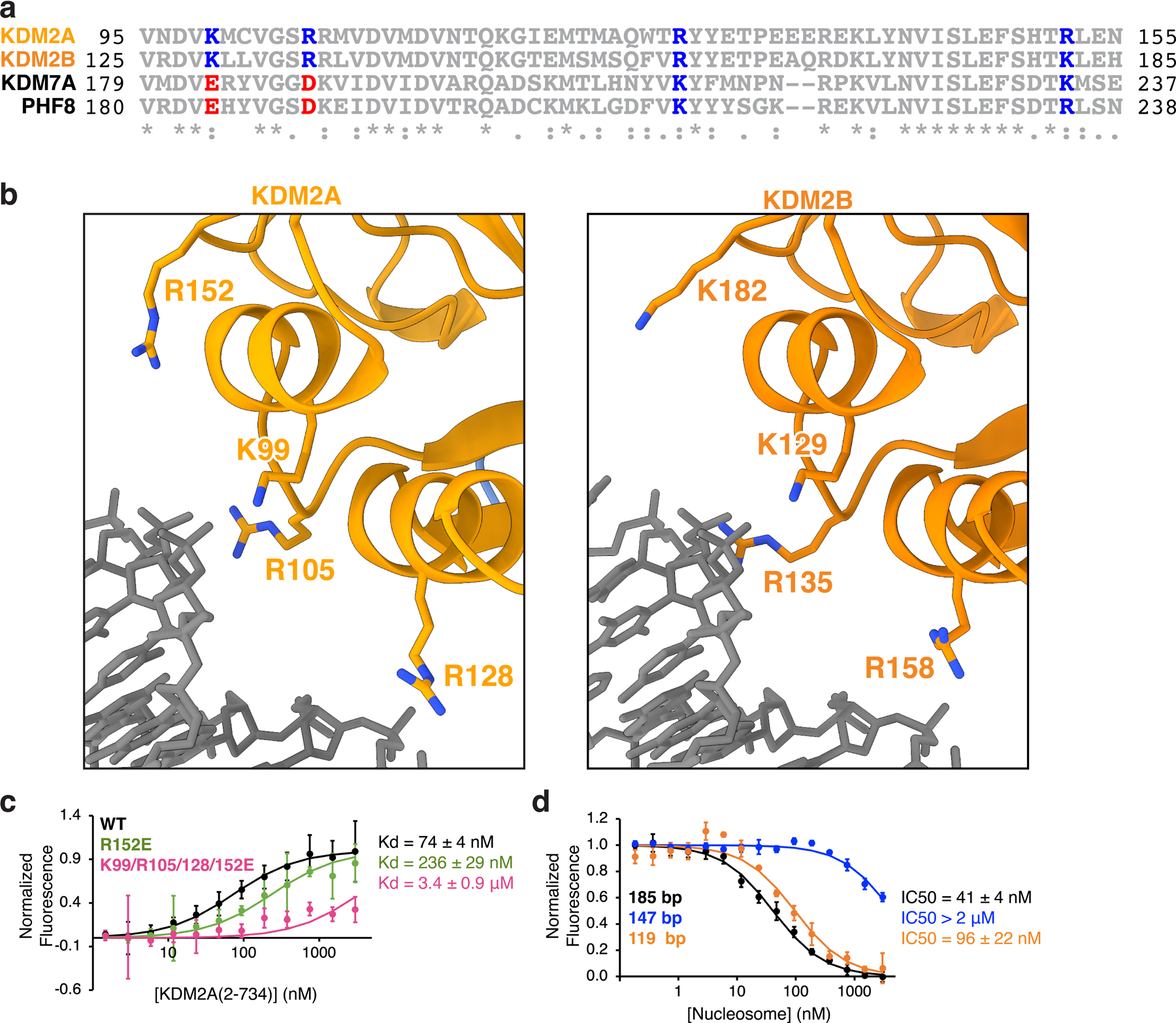
a, Sequence alignment of KDM2A and KDM2B DNA-binding interfaces with closely related KDM7A and PHF8 (KDM7B), with conserved basic amino acids and charge swapped amino acids colored blue and red, respectively. b, Zoomed view of unwrapped DNA-binding interface of KDM2A structure (left) and KDM2B structure (right). c, TR-FRET binding isotherms with KDM2A DNA interface mutants and unmodified wild-type 185 bp nucleosomes. d, TR-FRET nucleosome binding competition experiments using unmodified wildtype 185, 147, or 119 bp nucleosomes. In panels c and d, mean ± SD are shown (n=3 assay technical replicates).
Extended Data Fig. 9 |. Acidic patch-localized density of KDM2A/B.
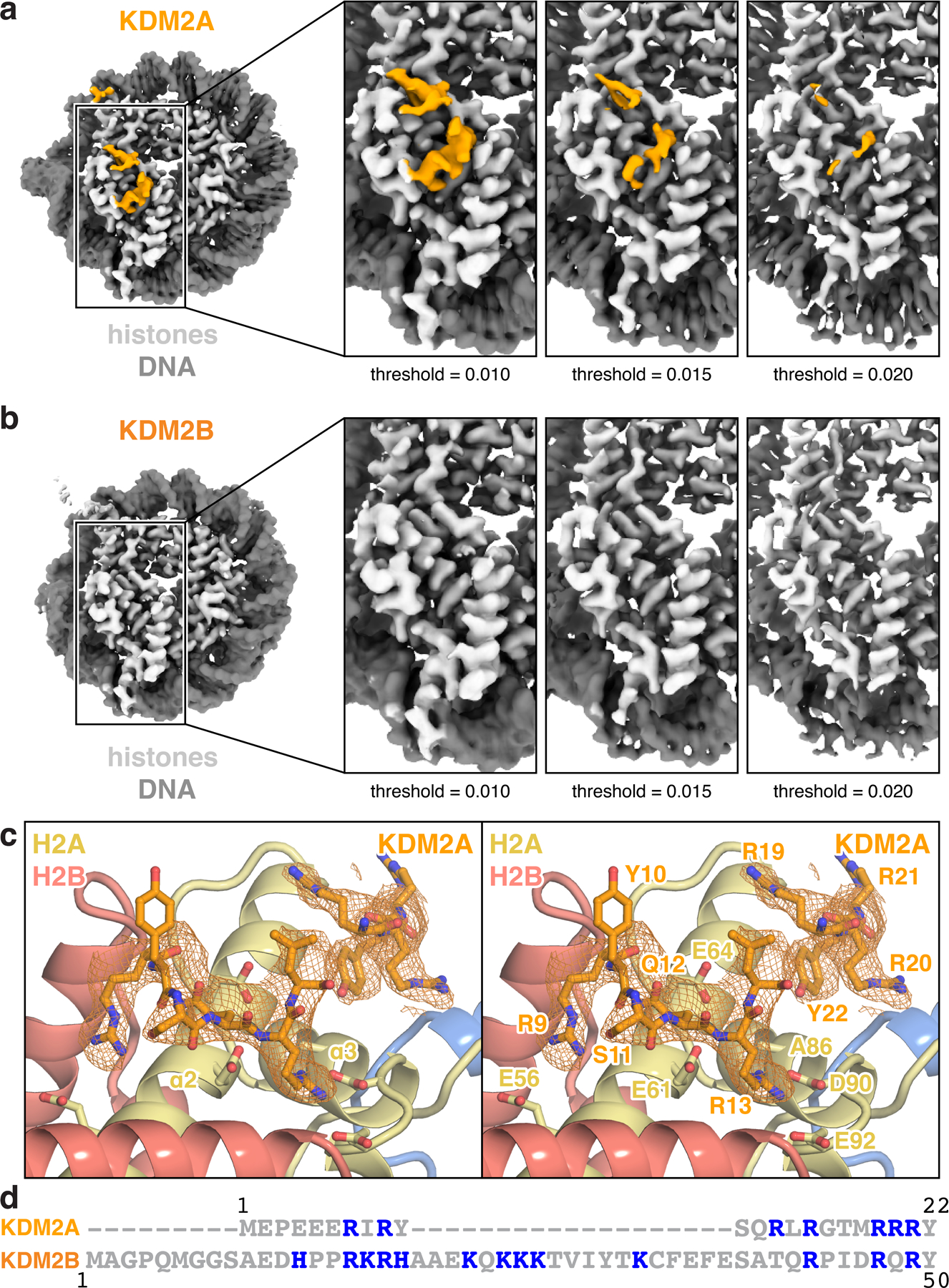
a-b, Acidic patch-localized density in nucleosome-focused cryo-EM maps from multibody analysis of (a) KDM2A- and (b) KDM2B-nucleosome complexes at indicated map thresholds. c, Overlay of acidic patch-localized density from nucleosome-focused map shown in (a) with equivalent region of modeled KDM2A. d, Sequence alignment of KDM2A and KDM2B N-termini, with basic amino acids colored blue.
Extended Data Fig. 10 |. Comparison of H3K36-bound enzyme structures.
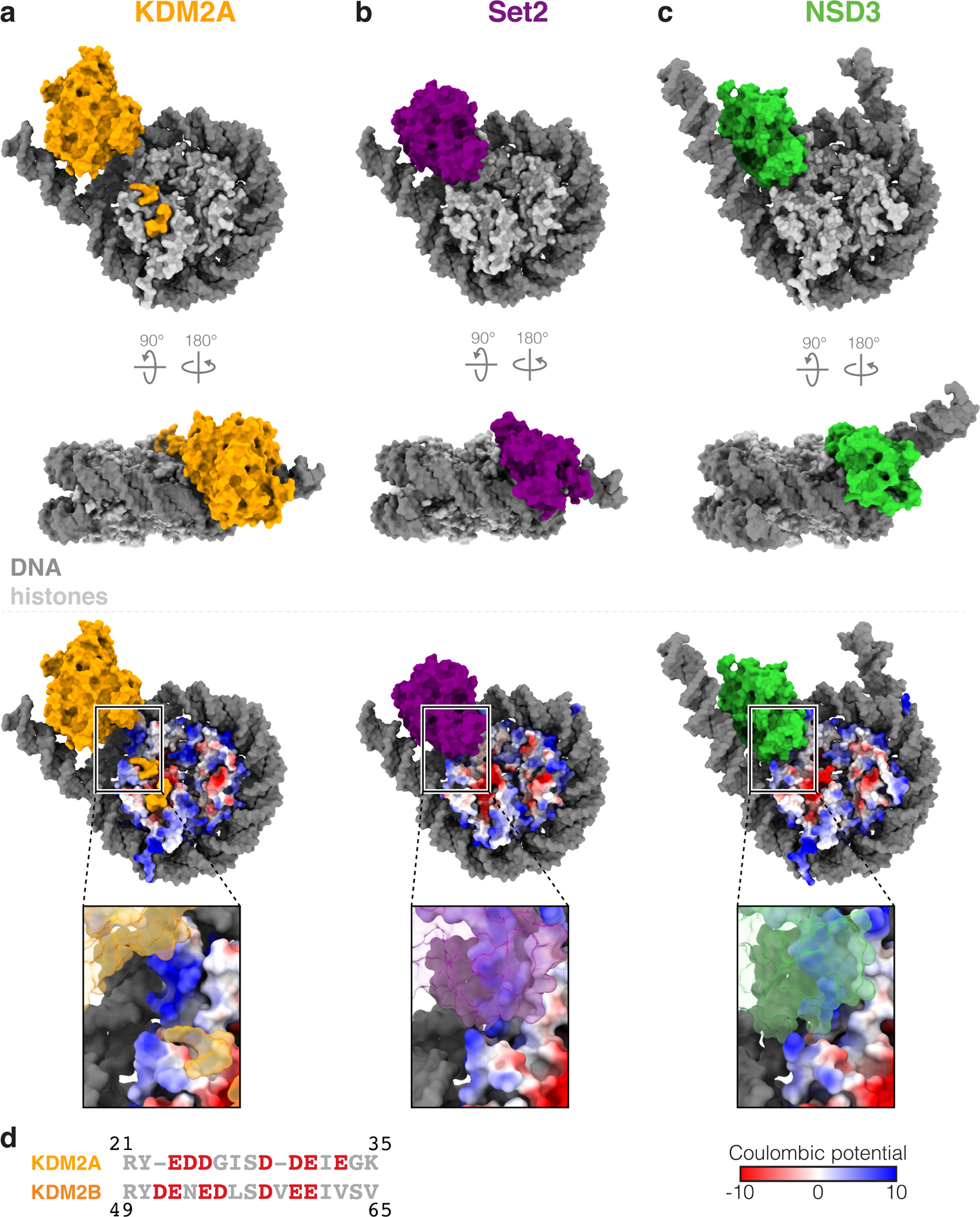
a, Structures of H3K36 lysine demethylase KDM2A, b, H3K36 lysine methyltransferase Set2 (PDB:6NZO) and c, H3K36 lysine methyltransferase NSD3 (PDB:7CRR) bound to the nucleosome. d, Sequence alignment of KDM2A and KDM2B JmjC-adjacent sequences, with acidic amino acids colored red.
Supplementary Material
Acknowledgements
Single particle cryo-EM data were collected with assistance from Jared Peck and Dr. Joshua Strauss at the University of North Carolina at Chapel Hill School of Medicine Cryo-Electron Microscopy Facility, which is partially supported by NIH P30CA016086 and is part of the Molecular Microscopy Consortium of the University of North Carolina at Chapel Hill, Duke University, and the NIEHS. Crystallography data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions, and NIH equipment grants S10_RR25528, S10_RR028976, and S10_OD027000. Plasmids containing 601 nucleosome positioning DNA fragments and NSD2 were gifts from Song Tan. We thank members of the McGinty, Goldfarb, Frye, and James laboratories for critical comments on the manuscript. This work was supported by NIH grants R35GM133498 to R.K.M., F99CA253730 to C.J.S., R35GM139514 to S.V.F., R01CA010305 to L.I.J. and R01GM132299 to D.K., American Cancer Society grant 132609-PF-18-153-01-DMC to A.S., and NSF grant DGE-1650116 to N.A.W..
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD032791. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-26809 (KDM2A:H3K36C-UNC8015 nucleosome complex) and EMD-26810 (KDM2B:H3K36C-UNC8015 nucleosome complex. Atomic coordinates of the KDM2A:H3K36C-UNC8015 nucleosome and KMD2A:H3(29–41)K36C-UNC8015 complexes have been deposited in the Protein Data Bank with accession codes 7UV9 and 7UVA, respectively. Homology model of KDM2B:H3K36C-UNC8015 nucleosome is included as Supplementary File 3. Other structures used in this study are available through the Protein Data Bank with accession codes 4QX7, 6ESF, 6NZO, 7CRR, and 7JO9. All materials in this study are available upon request.
References
- 1.Hyun K, Jeon J, Park K & Kim J Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49, e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsukada Y-I et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Shi Y et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Klose RJ & Zhang Y Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol 8, 307–318 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Shi Y & Whetstine JR Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 25, 1–14 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Black JC, Van Rechem C & Whetstine JR Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Punnia-Moorthy G, Hersey P, Emran AA & Tiffen J Lysine Demethylases: Promising Drug Targets in Melanoma and Other Cancers. Front Genet 12, 680633 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borgel J et al. KDM2A integrates DNA and histone modification signals through a CXXC/PHD module and direct interaction with HP1. Nucleic Acids Res 45, 1114–1129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frescas D et al. KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state cc 7, 3539–3547 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartke T et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Z et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gearhart MD, Corcoran CM, Wamstad JA & Bardwell VJ Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol. Cell. Biol 26, 6880–6889 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagarou A et al. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev 22, 2799–2810 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sánchez C et al. Proteomics analysis of Ring1B/Rnf2 interactors identifies a novel complex with the Fbxl10/Jhdm1B histone demethylase and the Bcl6 interacting corepressor. Molecular & Cellular Proteomics 6, 820–834 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Johansen JV & Helin K Fbxl10/Kdm2b Recruits Polycomb Repressive Complex 1 to CpG Islands and Regulates H2A Ubiquitylation. Mol. Cell 49, 1134–1146 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Farcas AM et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 1, e00205–e00205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackledge NP et al. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell 38, 179–190 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vacík T, Lađinović D & Raška I KDM2A/B lysine demethylases and their alternative isoforms in development and disease. Nucleus 9, 431–441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan M, Yang X, Wang H & Shao Q The critical role of histone lysine demethylase KDM2B in cancer. Am J Transl Res 10, 2222–2233 (2018). [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Z et al. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev 28, 1758–1771 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skrajna A et al. Comprehensive nucleosome interactome screen establishes fundamental principles of nucleosome binding. Nucleic Acids Res 48, 9415–9432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nacev BA et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGinty RK & Tan S Principles of nucleosome recognition by chromatin factors and enzymes. Curr. Opin. Struct. Biol 71, 16–26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett RL et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov 9, 1438–1451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan YCE et al. Cancer-associated histone mutation H2BG53D disrupts DNA-histone octamer interaction and promotes oncogenic phenotypes. Signal Transduct Target Ther 5, 27–4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arimura Y et al. Cancer-associated mutations of histones H2B, H3.1 and H2A.Z.1 affect the structure and stability of the nucleosome. Nucleic Acids Res 46, 10007–10018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinberg DN, Allis CD & Lu C Oncogenic Mechanisms of Histone H3 Mutations. Cold Spring Harb Perspect Med 7, a026443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou JC, Blackledge NP, Farcas AM & Klose RJ Recognition of CpG island chromatin by KDM2A requires direct and specific interaction with linker DNA. Mol. Cell. Biol 32, 479–489 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamada S et al. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J. Med. Chem 53, 5629–5638 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Suzuki T et al. Identification of the KDM2/7 histone lysine demethylase subfamily inhibitor and its antiproliferative activity. J. Med. Chem 56, 7222–7231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo X et al. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J. Am. Chem. Soc 133, 9451–9456 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wesley NA et al. Time Resolved-Fluorescence Resonance Energy Transfer platform for quantitative nucleosome binding and footprinting. Protein Sci 31, e4339 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 590, 498–503 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bilokapic S & Halic M Nucleosome and ubiquitin position Set2 to methylate H3K36. Nat. Commun 10, 3795–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bagert JD et al. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol 17, 403–411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spangler CJ et al. DOT1L activity in leukemia cells requires interaction with ubiquitylated H2B that promotes productive nucleosome binding. CellReports 38, 110369 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson CJ et al. Structural Basis for Recognition of Ubiquitylated Nucleosome by Dot1L Methyltransferase. CellReports 26, 1681–1690.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 38.Tan S, Kern RC & Selleck W The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expression and Purification 40, 385–395 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Weissmann F et al. biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc. Natl. Acad. Sci. U.S.A 113, E2564–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowary PT & Widom J New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Kim S-A, Chatterjee N, Jennings MJ, Bartholomew B & Tan S Extranucleosomal DNA enhances the activity of the LSD1/CoREST histone demethylase complex. Nucleic Acids Res 43, 4868–4880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makde RD, England JR, Yennawar HP & Tan S Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luger K, Rechsteiner TJ & Richmond TJ Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304, 3–19 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM & Richmond TJ Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol 272, 301–311 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Dignam JD, Lebovitz RM & Roeder RG Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11, 1475–1489 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pieters BJGE et al. Installation of Trimethyllysine Analogs on Intact Histones via Cysteine Alkylation. Bioconjugate Chem 30, 952–958 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans PR An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 282–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murshudov GN, Vagin AA & Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 52.Zivanov J, Nakane T & Scheres SHW Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong ED, Bepler T, Berger B & Davis JH CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Afonine PV et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr D Struct Biol 74, 814–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD032791. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-26809 (KDM2A:H3K36C-UNC8015 nucleosome complex) and EMD-26810 (KDM2B:H3K36C-UNC8015 nucleosome complex. Atomic coordinates of the KDM2A:H3K36C-UNC8015 nucleosome and KMD2A:H3(29–41)K36C-UNC8015 complexes have been deposited in the Protein Data Bank with accession codes 7UV9 and 7UVA, respectively. Homology model of KDM2B:H3K36C-UNC8015 nucleosome is included as Supplementary File 3. Other structures used in this study are available through the Protein Data Bank with accession codes 4QX7, 6ESF, 6NZO, 7CRR, and 7JO9. All materials in this study are available upon request.