

Abstract
Heparan sulfate (HS) proteoglycans are extended (-GlcAβ1,4GlcNAcα1,4-)n co-polymers containing decorations of sulfation and epimerization that are linked to cell surface and extracellular matrix proteins. In mammals, HS repeat units are extended by an obligate heterocomplex of two exostosin family members, EXT1 and EXT2, where each protein monomer contains distinct GT47 (GT-B fold) and GT64 (GT-A fold) glycosyltransferase domains. In this study, we generated human EXT1–EXT2 (EXT1–2) as a functional heterocomplex and determined its structure in the presence of bound donor and acceptor substrates. Structural data and enzyme activity of catalytic site mutants demonstrate that only two of the four glycosyltransferase domains are major contributors to co-polymer syntheses: the EXT1 GT-B fold β1,4GlcA transferase domain and the EXT2 GT-A fold α1,4GlcNAc transferase domain. The two catalytic sites are over 90 Å apart, indicating that HS is synthesized by a dissociative process that involves a single catalytic site on each monomer.
Proteoglycans harboring heparan sulfate (HS) chains are ubiquitously expressed on cell surfaces and in extracellular matrices1,2. These glycosaminoglycan (GAG) chains interact with numerous proteins, growth factors, morphogens and extracellular matrix proteins and play crucial roles in tissue homeostasis and signal transduction, where they drive processes such as cell survival, division, migration, differentiation, pathogen binding and cancer development2–6. HS proteoglycan biosynthesis is a complex process involving core protein synthesis, initiation of a GlcAβ1,3Galβ1,3Galβ1,4Xylβ-Ser linker tetrasaccharide7,8, (-GlcAβ1,4GlcNAcα1,4-)n polymer backbone extension (Fig. 1a), polymer modification by GlcA epimerization and addition of N-sulfate and O-sulfate groups2,8,9. Polymer backbone extension involves the alternating transfer of GlcAβ1,4- and GlcNAcα1,4- residues from their respective UDP-sugar donors by the HS co-polymerase, comprised of a complex of EXT1 and EXT2 proteins10,11. Prior data indicated that EXT1 alone exhibited weak activities for both GlcA and GlcNAc transfer10,12, whereas EXT2 exhibited no detectable activity and was relegated to the role of a chaperone for EXT1 (refs. 10, 13,14).
Fig. 1 |. Overall architecture of the EXT1–2 complex.
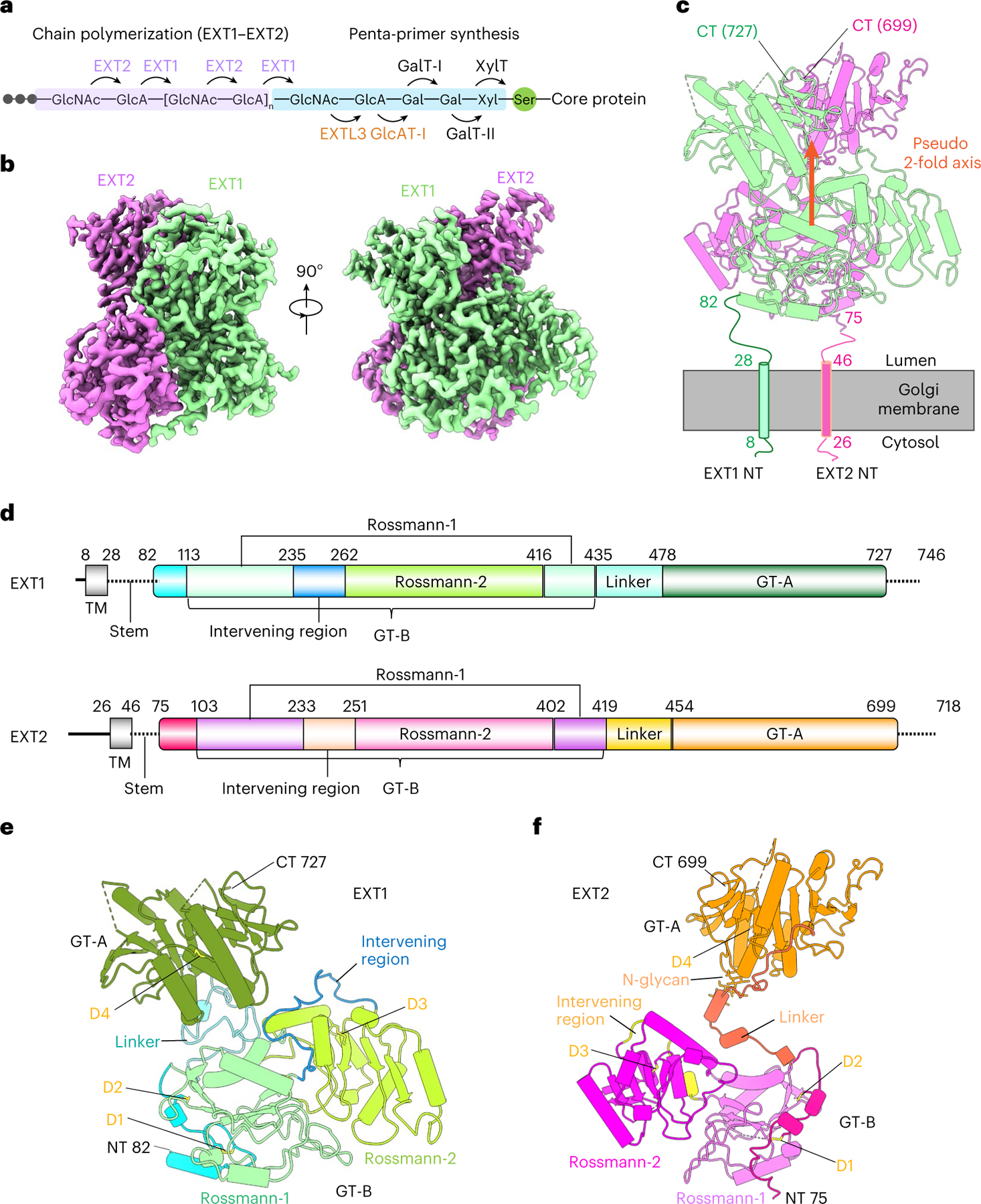
a, Sketch of HS GAG biosynthesis. The GAG is initiated by synthesis of a penta-saccharide primer linked to a serine residue in the core protein. The GAG is elongated by the alternating addition of GlcA and GlcNAc catalyzed by EXT1–2. b, Two orthogonal views of cryo-EM 3D map of EXT1–2. c, The atomic model of EXT1–2 in the cartoon is shown in its likely orientation with respect to the Golgi membrane. The truncated N-terminal transmembrane α-helices are shown as cylinders. The orange arrow indicates a pseudo two-fold symmetry axis relating EXT1 GT-B and EXT2 GT-B. d, Domain organization of EXT1 and EXT2. The intervening loop connects the Rossmann-1 and Rossmann-2 subdomains of the GT-B domain. The linker loop connects the N-terminal GT-B and the C-terminal GT-A domains. e,f, Cartoon view of the atomic models of EXT1 and EXT2 shown separately. The view is the same as in the right panel of b. Domains shown in color are the same as in d; N-glycan on EXT2 Asn637 is in sticks. D1–4 refer to the four disulfide bonds in each domain. TM, transmembrane domain.
EXT1 and EXT2 are homologous enzyme isoforms (35% identity), and each contains an N-terminal transmembrane domain followed by two separate domains: a CAZy GT47 glycosyltransferase domain and a GT64 domain15. Thus, each isoform could contribute both GlcA and GlcNAc transferase activities16, although no studies have yet determined which sites in the EXT1–2 complex are indeed functional. Here we report five cryogenic electron microscopy (cryo-EM) structures of the human EXT1–2 co-complex in an apo form or bound to donor substrate and acceptor analogs. EXT1 and EXT2 form an asymmetric 1:1 heterodimeric complex that is fully functional in HS polymer extension, and the substrate-bound structures indicate that only two of the four potential active sites bind donors and acceptors during HS polymerization. The N-terminal EXT1 GT47 GT-B domain catalyzes β1,4GlcA transfer, whereas the C-terminal EXT2 GT64 GT-A domain catalyzes α1,4GlcNAc transfer. The structure also provides a model for how HS polymer extension is achieved by shuttling between the two remote catalytic sites that are 9 nm apart. Our work illuminates the structure and function of an essential human enzyme that catalyzes HS chain elongation.
Results
Expression and purification of EXT1–2
We generated soluble secreted catalytic domains encoding EXT1 and EXT2 devoid of their cytoplasmic and transmembrane sequences by expression in two different expression vectors encoding either short or longer N-terminal fusion sequences (pGEn1 or pGEn2 vectors17). EXT1 or EXT2, when expressed individually, were minimally secreted into the mammalian cell culture media in either vector (Extended Data Fig. 1a). However, co-expression of EXT1 and EXT2 in either vector combination led to effective secretion of a heterocomplex into the media. Co-expression in vectors encoding short fusion tags led to secretion of ~90-kDa proteins by SDS-PAGE, whereas co-expression in vectors encoding the larger fusion tags resulted in ~115-kDa protein bands, consistent with the respective sizes of the EXT1 and EXT2 fusion proteins. Co-expression of EXT1 and EXT2, each in different fusion vectors, led to secretion of two discrete proteins of ~90 kDa and ~115 kDa in equal abundance (Extended Data Fig. 1a), suggesting that co-expression is required for efficient folding and secretion. Multi-angle light scattering coupled with size exclusion chromatography (SEC-MALS) analysis confirmed that the purified enzyme was a heterodimer of EXT1 and EXT2 (Extended Data Fig. 1b). The fusion tags were removed by TEV protease treatment before cryo-EM studies to reduce structural heterogeneity (Extended Data Fig. 1c).
Cryo-EM structure for EXT1–2 co-complex
Cryo-EM structural analysis of the EXT1–2 apo-enzyme (3.1-Å average resolution) revealed an N-terminal GT-B (GT47) domain followed by a C-terminal GT-A (GT64) domain for each protein (Fig. 1b, Supplementary Table 1 and Supplementary Fig. 1). Most of the main chain and side chains for the two proteins could be traced, except for 64 N-terminal stem residues and 17 C-terminal residues of EXT1 and 35 N-terminal stem residues and 19 C-terminal residues of EXT2 (Fig. 1b–f and Supplementary Fig. 2). EXT1 and EXT2 formed a 1:1 heterodimer with a pseudo two-fold symmetry connecting the two N-terminal GT-B domains and a separate pseudo two-fold symmetry axis connecting the C-terminal GT-A domains (Fig. 1c and Extended Data Fig. 2). As predicted by the 35% sequence identity for EXT1 and EXT2, the two GT-B domains and two GT-A domains superimpose well (root mean square deviation (RMSD) of 0.93 Å and 0.76 Å, respectively; Extended Data Fig. 2). The two GT-B domains assemble back-to-back with their putative active site pockets facing away from each other, whereas the two GT-A domains interact side-by-side, resembling the mouse EXTL2 homodimer18 with adjacent active site pockets facing away from the GT-B domains.
In contrast, the respective linker regions between the GT-B and GT-A domains of EXT1 and EXT2 significantly diverge in structure. As a result, the pseudo two-fold axis linking the two GT-B domains does not coincide with the two-fold axis linking the two GT-A domains (Fig. 1c–f). Although EXT1 has a longer linker (43 residues) than EXT2 (35 residues), the GT-A domain of EXT1 is closer to the GT-B domain, such that EXT1 is more compact than EXT2 (Fig. 1e,f). This leads to a tightly packed globular structure in which EXT1 and EXT2 asymmetrically twist around each other. The interface between EXT1 and EXT2 is extensive, with a total area of 3,400 Å2, and includes an intermolecular anti-parallel β sheet at the C-termini of the GT-A domains (Extended Data Fig. 2d).
Based on the locations of the first resolved residues in EXT1 (Pro82) and EXT2 (Ser75), the EXT1–2 complex would be anchored in the Golgi membrane via their N-terminal transmembrane and stem sequences, with their GT-B domains tethered proximal to the membrane (Fig. 1c). The disordered stem regions (Phe28-Ser81 in EXT1 and Trp46-Asp74 in EXT2) likely afford flexibility for the enzyme to access core protein substrates of different sizes and shapes.
A total of eight conserved disulfide bonds stabilize the structure, with four in EXT1 and four in EXT2 (Fig. 1e,f). We also observed an N-glycan on Asn637 at the predicted N-glycosylation sequon in EXT2 (Fig. 1f).
Only EXT1 GT-B and EXT2 GT-A domains bind donor substrates
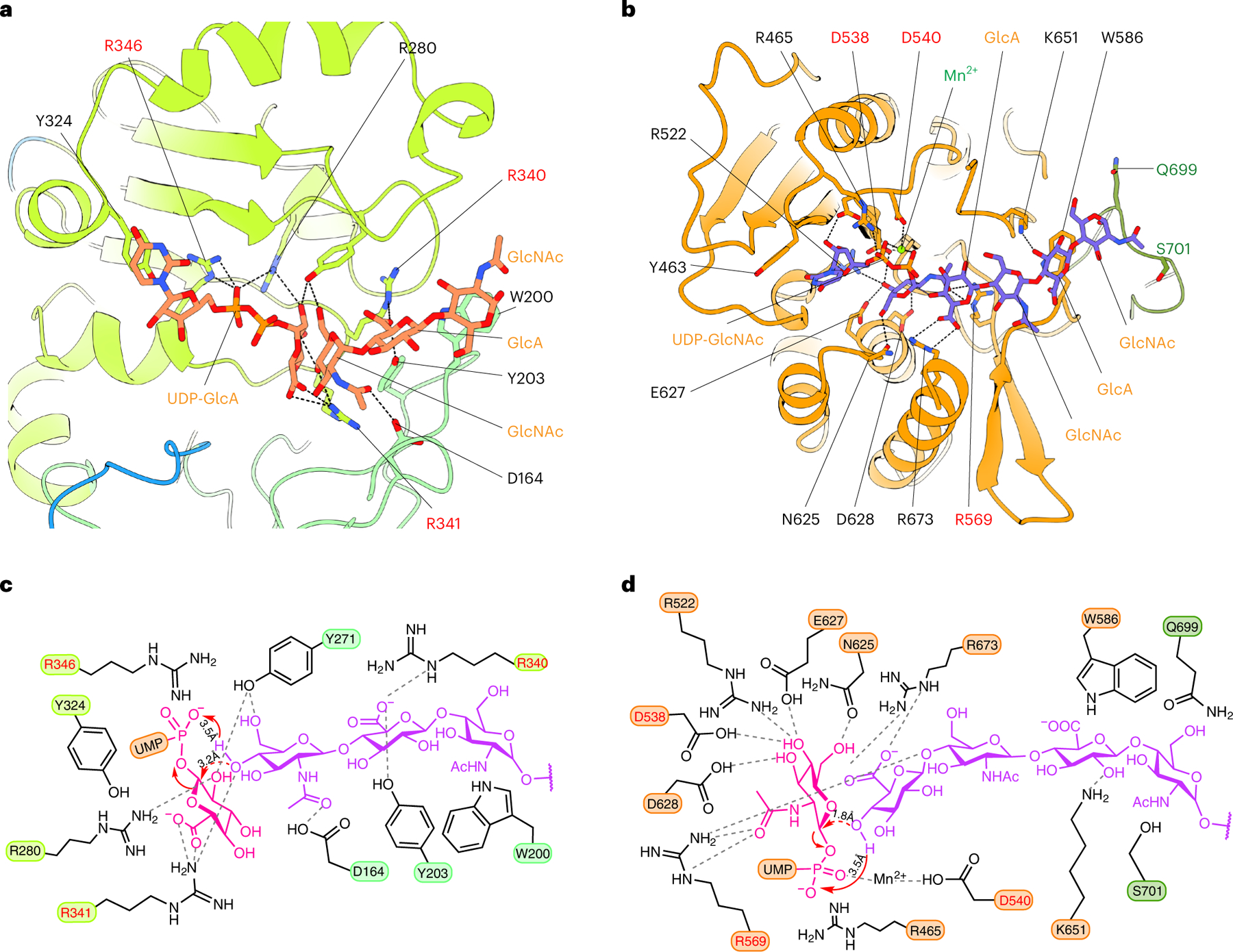
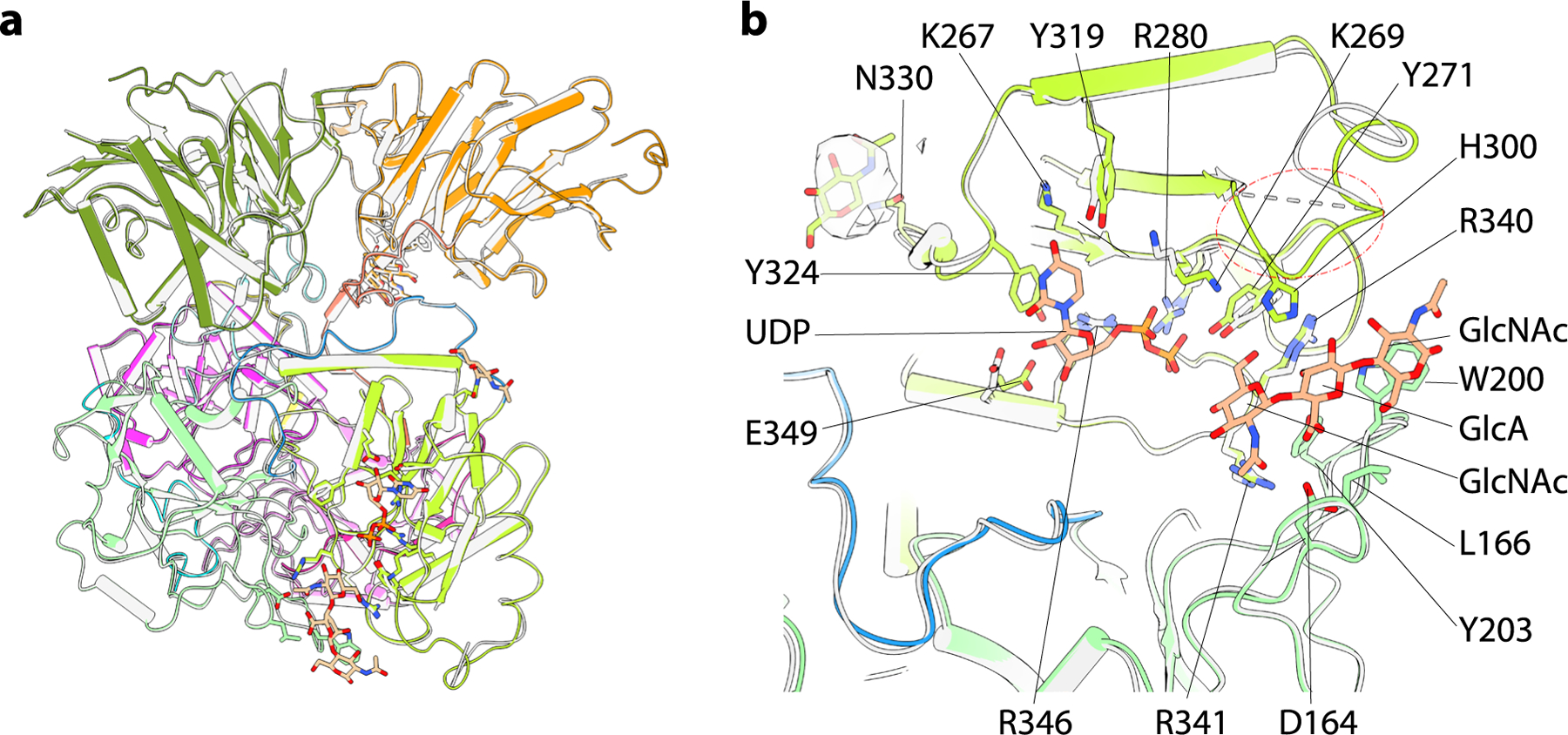
To understand substrate interactions for EXT1 and EXT2, we also determined structures of the EXT1–2 complex in the presence of UDP-GlcA or UDP-GlcNAc (3.0-Å and 3.3-Å average resolutions, respectively; Supplementary Figs. 3 and 4 and Supplementary Table 1). Because the enzyme heterocomplex was active, slow hydrolysis of the donor substrate to UDP was expected. In the presence of 10 mM UDP-GlcA, we observed a UDP density only in the EXT1 GT-B domain, whereas all three other potential donor binding sites (EXT1 GT-A, EXT2 GT-B and EXT2 GT-A) were empty (Fig. 2a,b and Supplementary Fig. 5a). The UDP was bound to the Rossmann-2 subdomain of the dual-domain GT-B fold, with the diphosphate coordinated by Lys269, Tyr271 and a salt bridge with Arg280 (Fig. 2b). The Tyr324 side chain π–π stacks with the uracil, whereas Arg280, Lys267 and Tyr319 also H-bond with the uracil.
Fig. 2 |. Structures of EXT1–2 in the presence of donor substrates.
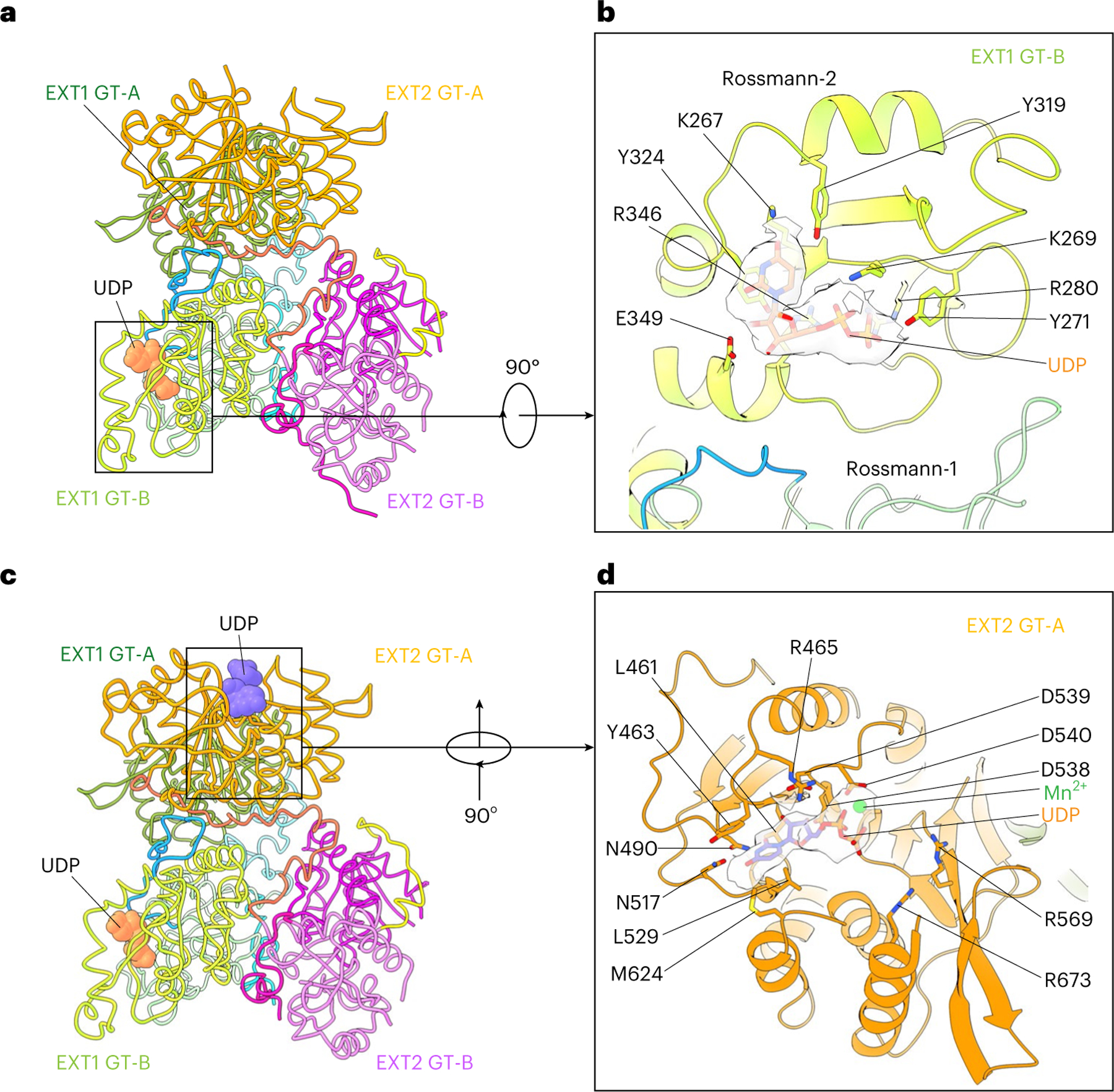
a, Structure of EXT1–2 in the presence of the donor UDP-GlcA. The enzyme is active and has hydrolyzed the donor into UDP and free sugar GlcA. The freed GlcA has likely diffused into solution. Only EXT1 GT-B catalytic pocket is occupied by UDP that is shown in orange spheres, whereas the potential pockets of EXT1 GT-A, EXT2 GT-B and EXT2 GT-A are unoccupied. b, Close-up view of the EXT1 GT-B catalytic pocket superimposed with the UDP density shown as transparent light gray surface. c, Structure of EXT1–2 in the presence of the donor UDP-GlcNAc and MnCl2. Both EXT2 GT-A and EXT1 GT-B pockets are occupied by UDP (orange and purple spheres). EXT1 GT-A and EXT2 GT-B are unoccupied. d, Close-up view of EXT2 GT-A pocket with the UDP and Mn2+ density shown in transparent light gray surface. Key residues lining the catalytic pocket are shown in sticks and labeled in b and d.
In the presence of 10 mM UDP-GlcNAc and 10 mM MnCl2, EXT1 GT-A and EXT2 GT-B domains were empty; the EXT2 GT-A domain was occupied by UDP:Mn2+; and the EXT1 GT-B domain was occupied by UDP, although the UDP density in the EXT1 GT-B domain was weak (Fig. 2c,d and Supplementary Fig. 5b–d). This latter UDP density likely resulted from hydrolysis of the UDP-GlcNAc in the EXT2 GT-A site and diffusion into EXT1 GT-B domain. The uridine in the EXT2 GT-A domain formed π–π stacking interactions with Tyr463 and an H-bond with Asn517, whereas Asp539 H-bonds with the ribose, and Arg465 forms a salt bridge with the α-phosphate (Fig. 2d). The catalytic Mn2+ was coordinated by the UDP α-phosphate and Asp540. Notably, the observed UDP binding modes in EXT1–2 are consistent with typical donor substrate binding for GT-A and GT-B families of glycosyltransferases19,20.
4-mer binds only to the EXT1 GT-B domain
For structural studies on acceptor complexes, we first confirmed that the enzyme was active toward a panel of pNP-tagged heparosan acceptor primers containing either non-reducing terminal GlcNAc (4-mer and 6-mer) or GlcA (5-mer and 7-mer) residues (Supplementary Table 2). The 4-mer and 7-mer were chosen for the structural studies because their non-reducing terminal residues (GlcNAc for the 4-mer, GlcA for the 7-mer) would be anticipated to target selective interactions with the GlcA and GlcNAc transferase active sites, respectively. Substrate binding (UDP, MnCl2 and 4-mer) was performed, and the resulting structure (3.0-Å resolution; Supplementary Fig. 6 and Supplementary Table 1) was similar to the apo enzyme (Fig. 3a–d and Extended Data Fig. 3a,b), except for additional densities of UDP:Mn2+ in the EXT2 GT-A domain donor site, UDP and acceptor ligand in the EXT1 GT-B domain active site and an ordering of a three-residue loop (H300-K302) adjacent to the EXT1 GT-B active site that was disordered in the apo structure (Extended Data Fig. 3b). Notably, no bound acceptor ligand was observed in the EXT1 GT-A domain or in either domain of EXT2 (Fig. 3b–d), and UDP was bound only to the EXT1 GT-B and EXT2 GT-A domains.
Fig. 3 |. Structures of EXT1–2 in the presence of acceptor substrates and UDP.
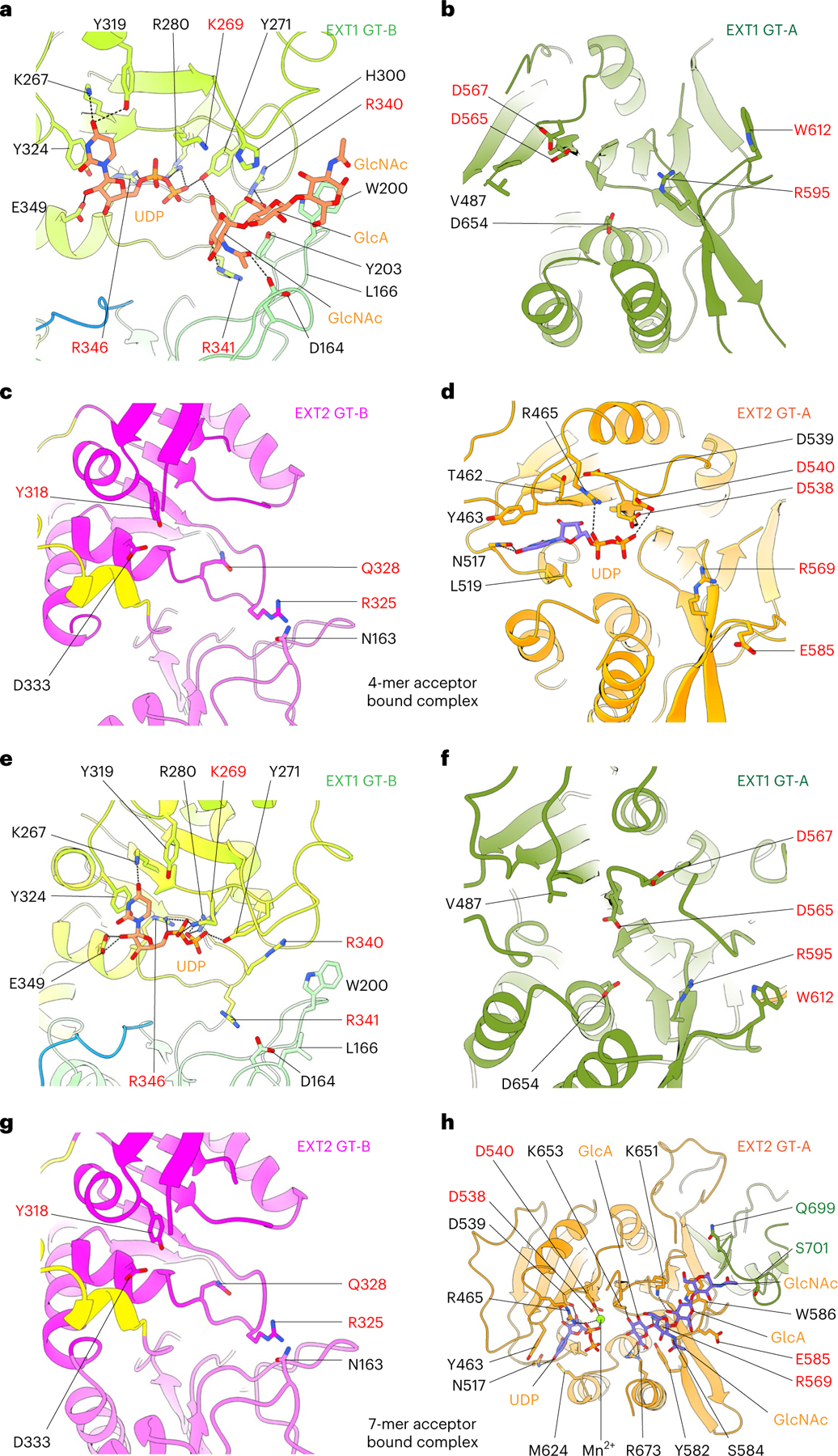
a–d, Close-up view of EXT1 GT-B (a) and GT-A (b) and EXT2 GT-B (c) and GT-A (d) in the 4-mer acceptor-bound EXT1–2 complex structure. The 4-mer binds only to the EXT1 GT-B domain, with three of its four sugars stabilized. e–h, Close-up view of EXT1 GT-B (e) and GT-A (f) and EXT2 GT-B (g) and GT-A (f) in the 7-mer acceptor-bound EXT1–2 complex structure. The 7-mer binds only to the EXT2 GT-A domain with four of the seven sugars stabilized. EXT1 and EXT2 are colored as in Fig. 1e,f. UDP, the 4-mer and 7-mer acceptors and key residues lining the potential active pockets are in sticks; H-bonds and salt bridges are shown as black dashed line.
In the EXT1 GT-B catalytic cleft, densities for three non-reducing terminal GlcNAc-GlcA-GlcNAc residues from the 4-mer acceptor were well resolved (Fig. 3a and Supplementary Fig. 2), but the GlcA-pNP residues at the reducing terminus were not modeled because the density was very weak. The three resolved sugars interact with both Rossmann-1 and Rossmann-2. Specifically, Asp164, Tyr271 and Arg341 form H-bonds with the terminal GlcNAc, and Arg340 and Tyr203 H-bond with the second GlcA, whereas Trp200 forms a π–π stacking interaction with the third sugar unit (GlcNAc) (Fig. 3a). The close proximity between the acceptor GlcNAc O4 hydroxyl nucleophile and the β-phosphate of the bound UDP within the EXT1 GT-B active site confirms our prediction that the 4-mer is positioned appropriately for GlcA transfer and indicates that only the terminal 3 sugars stably interact with the EXT1 GT-B domain. Residues in the EXT1 GT-B and the EXT2 GT-A catalytic clefts that interact with the UDP donor analog were identical to those in the respective EXT1 GT-B:UDP and EXT2 GT-A:UDP:Mn2+ complexes (Figs. 2b,d and 3a,d).
7-mer binds only to the EXT2 GT-A domain
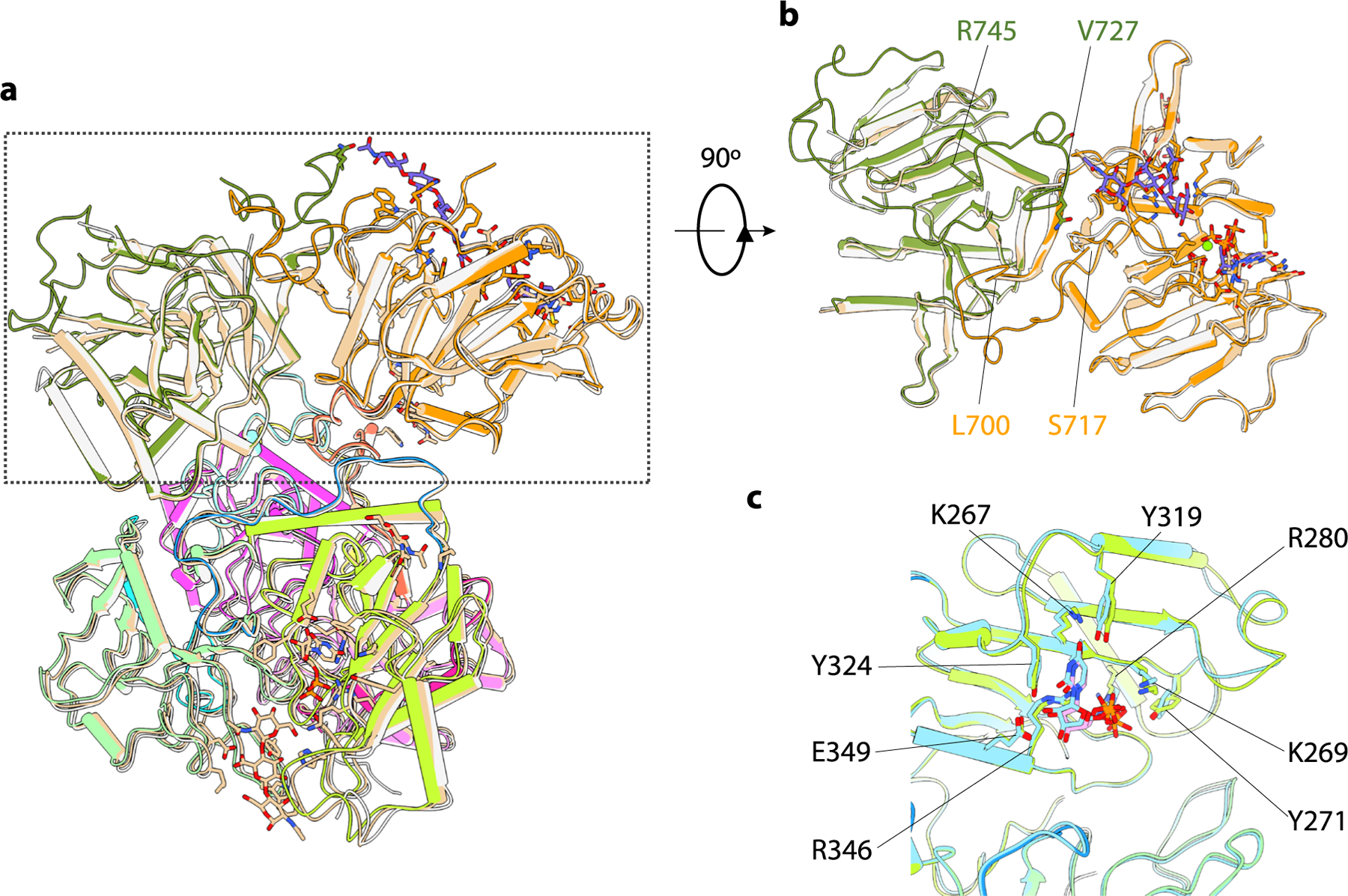
Substrate binding and cryo-EM were also performed with the 7-mer acceptor in the presence of UDP and MnCl2 (2.8-Å resolution; Supplementary Fig. 7 and Supplementary Table 1). The 7-mer complex structure superimposes well with both UDP-GlcA-bound EXT1–2 and 4-mer complex structures, indicating that no major conformational changes occur upon substrate binding (Extended Data Fig. 4a,b). No ligand complexes were observed for the EXT1 GT-A and EXT2 GT-B domains (Fig. 3f,g), and no acceptor was bound to the EXT1 GT-B domain. However, we did observe a UDP bound to the EXT1 GT-B domain (Fig. 3e), with interactions like those found in the UDP complexes (Fig. 2b). In the EXT2 GT-A domain, there was a bound UDP, Mn2+ ion and the non-reducing terminal tetrasaccharide (GlcA-GlcNAc-GlcA-GlcNAc) from the 7-mer acceptor (Fig. 3h and Supplementary Fig. 2). The positioning of the 7-mer non-reducing terminal GlcA residue adjacent to the β-phosphate of the bound UDP also confirms that this acceptor is positioned appropriately for GlcNAc transfer and validates the EXT2 GT-A domain as responsible for GlcNAc transferase activity. The remaining three sugar units of the 7-mer acceptor (GlcA-GlcNAc-GlcA-pNP) had very weak densities and were not modeled. Interestingly, the 7-mer binding stabilized the C-terminal loops of both EXT1 (Val727-Arg745) and EXT2 (Leu700-Ser717) (Fig. 3h and Extended Data Fig. 4).
In the EXT2 GT-A:UDP:Mn2+:7-mer complex, interactions with the UDP are identical to the EXT2 GT-A:UDP:Mn2+ complex, whereas the terminal GlcA in the acceptor site is H-bonded with Arg569, Lys653 and Arg673. Lys651 H-bonds with the third sugar unit (GlcA), and Trp586 forms a π–π stacking interaction with the fourth sugar unit (GlcNAc) (Fig. 3h). Surprisingly, the fourth sugar unit (GlcNAc) also forms van der Waals interactions with Gln732 and Ser734 of the neighboring EXT1.
Mutagenesis confirms the assigned catalytic sites
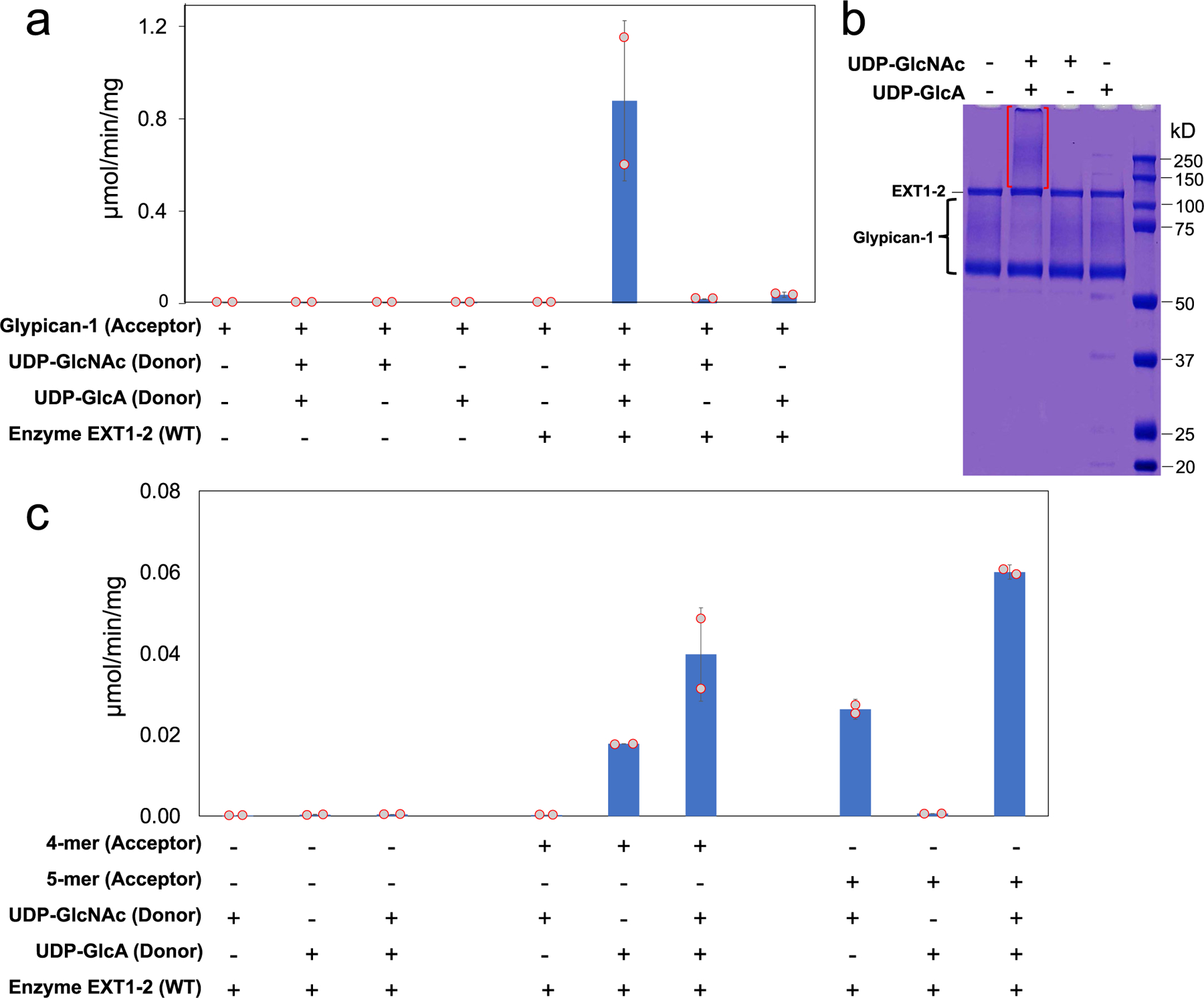
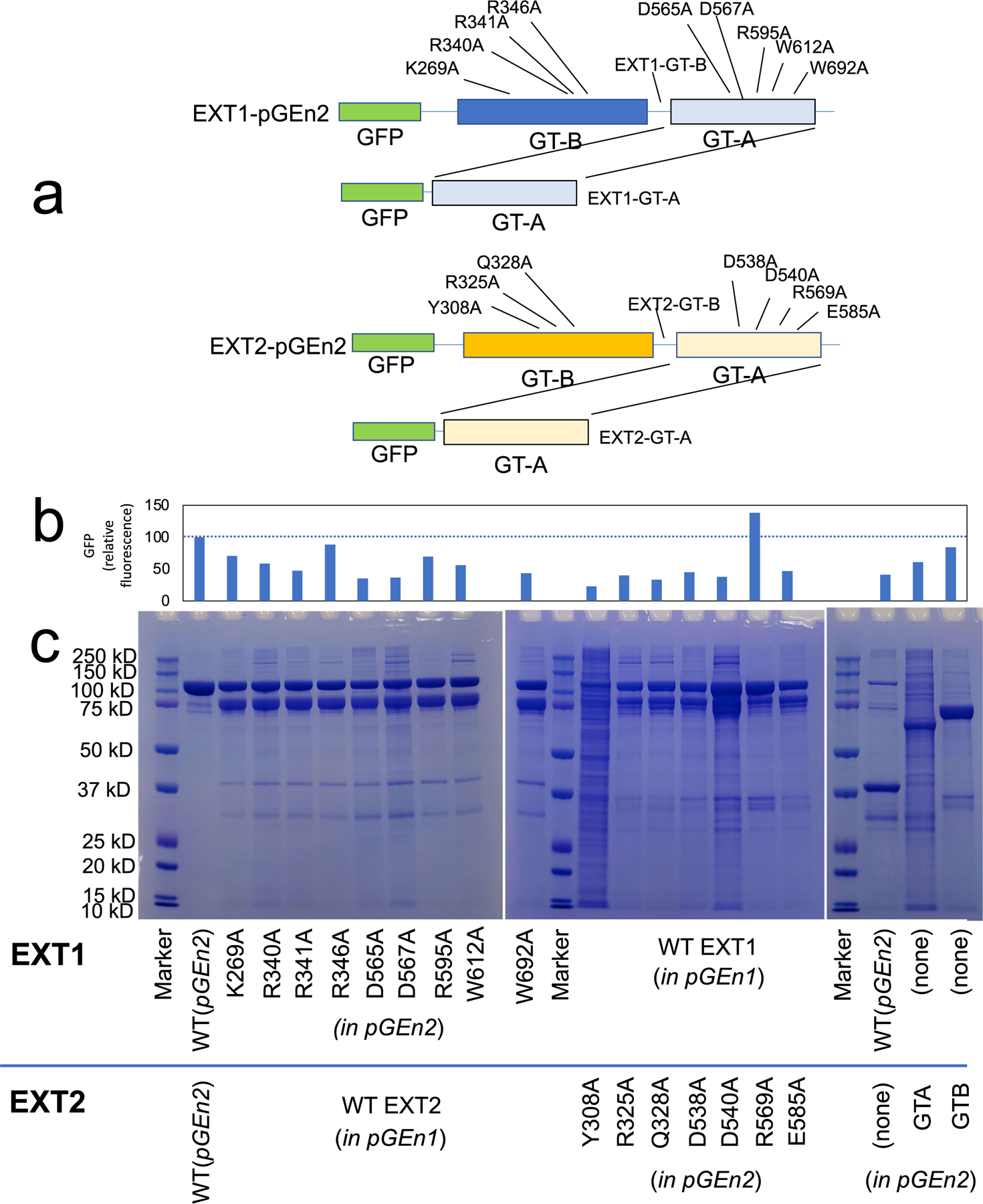
To confirm HS co-polymerase activity for EXT1–2, we employed glypican-1 as an exogenous proteoglycan acceptor and a UDP-Glo assay for detection. Enzymatic extension of the glypican-1 HS chain using either of the two single UDP-GlcNAc or UDP-GlcA donors resulted in a low but significant signal above background from the single sugar transfer events (Extended Data Fig. 5a). However, when both sugar donors were concurrently added, activity was enhanced by ~40-fold, suggesting strong co-polymerase activity toward the acceptor. SDS-PAGE of the co-polymerase product detected conversion to a higher molecular weight species, reflecting polymer extension (Extended Data Fig. 5b). The UDP-Glo co-polymerase assay was then used to test a panel of active site alanine mutations in EXT1–2 (Fig. 4a). Alanine mutants in the EXT1 GT-B or EXT2 GT-A domain active sites either eliminated or nearly eliminated co-polymerase activity, whereas residues peripheral to the active sites (K269A and E585A) had some residual activity. In contrast, mutations in the EXT1 GT-A or EXT2 GT-B domains resulted in wild-type activity levels (R325A), minor reductions (D565A, D567A, R595A, W612A, W612A and Y308A) or slightly enhanced (Q328A) activities (Fig. 4a). These data suggest that the EXT1 GT-B and EXT2 GT-A domains are essential for co-polymerase function, whereas the EXT1 GT-A and EXT2 GT-B domains play minimal catalytic roles in HS synthesis.
Fig. 4 |. Enzyme activity of wild-type and mutant forms of EXT1–2.
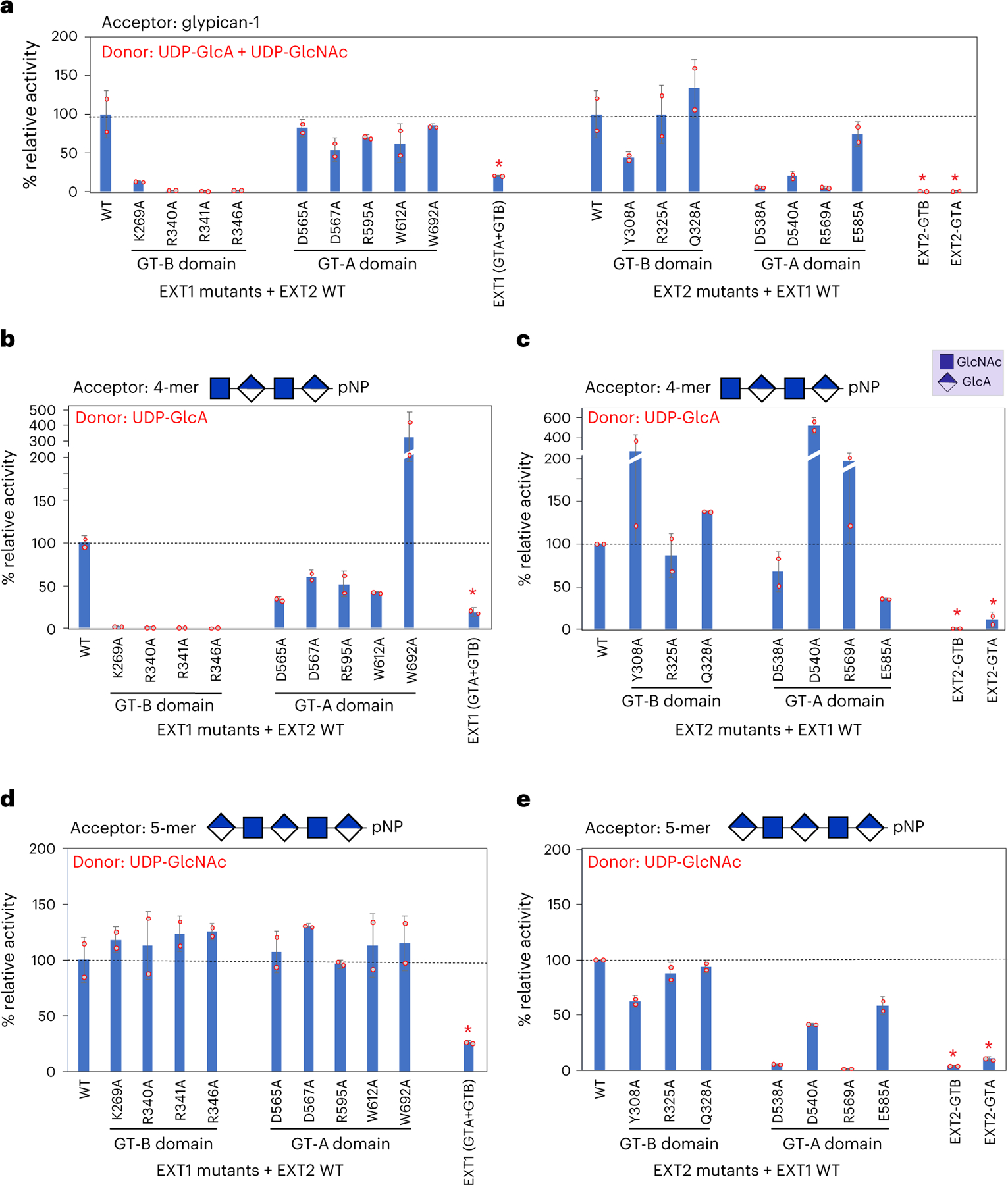
Wild-type, mutant or truncated forms of EXT1 or EXT2 were co-expressed or expressed as single proteins as indicated, and the recombinant products were purified and assayed for enzyme activity using recombinant glypican-1 (a) or HS oligosaccharides (b–e) as acceptors. a, HS co-polymerase assays were performed using glypican-1 as acceptor substrate and UDP-GlcA and UDP-GlcNAc as donors. Enzyme assays were also performed with 4-mer (b,c) or 5-mer (d,e) heparosan primer acceptors for the EXT1 mutants (b,d) or EXT2 mutants (c,e) using either UDP-GlcA (b,c) or UDP-GlcNAc (d,e) as donors. Mutation of the EXT1 GT-B active site led to complete loss of GlcA transferase activity (b), whereas mutations in EXT2 GT-A active site generally led to a loss of GlcNAc transferase activity (e), similarly to the loss on co-polymerase activity (a) for the same mutations. EXT1 expressed alone (EXT1 (GTA + GTB)) led to low but detectable co-polymerase, GlcA and GlcNAc transferase activities, whereas the individual EXT2 GT-A or GT-B domains expressed alone had minimal levels of enzyme activity. In each of the latter cases, the resulting purified proteins exhibited extensive proteolysis (Extended Data Fig. 6) that compromised the ability to quantitate the intact proteins (red asterisks), and the resulting enzyme activities are considered qualitative. Plots show the mean values (bar) ± s.d. (error bars) for n = 2 technical replicates (red circles). WT, wild-type.
Efforts to express individual domains or single subunits of the EXTs led to limited success (Extended Data Fig. 6a–c). Expression of EXT2 alone, single GT-A or GT-B domains from EXT1 or co-expression of GT-A or GT-B domains from EXT1 and EXT2 led to no protein secretion. Very low levels of secretion and co-polymerase activity were obtained for EXT1 alone, and the purified product demonstrated extensive proteolysis that limited protein quantitation (Extended Data Fig. 6c). Thus, enzyme activity for EXT1 alone should be considered qualitative (Fig. 4a). Reduced expression, partial proteolysis and trace levels of enzyme activity were also observed for the purified EXT2 GT-A and GT-B domains.
UDP-Glo assays were also performed using pNP-tagged 4-mer or 5-mer oligosaccharide acceptors using either single-nucleotide sugars as donors or both donors in combination. Similarly to the assays with glypican-1 as acceptor, no GlcA transferase activity was detected for active site mutants in the EXT1 GT-B domain (Fig. 4b), and minimal GlcNAc transferase activity was detected for the D538A and R569A active site mutants in the EXT2 GT-A domain (Fig. 4e) using their respective expected 4-mer and 5-mer acceptors. These patterns match the co-polymerase activities for the respective mutants with glypican-1 as acceptor (Fig. 4a). GlcNAc transferase activities for GT-A or GT-B domains mutants in EXT1 were essentially equivalent to wild-type EXT1–2. GlcA and GlcNAc transferase activities for mutants in the other domains varied from minor reductions in activity to significant stimulation (W692A, Y308A, D540A and R569A) (Fig. 4b–e), suggesting that individual mutants may influence long-range allosteric effects for single sugar transfer assays. However, only the Q328A mutant resulted in a minor increase in co-polymerase activity (Fig. 4a).
We also confirmed that the UDP-Glo assays reflected HS glycan extension by analyzing the enzymatic products by matrix-assisted laser desorption/ionization–mass spectrometry (MALDI–MS) (Extended Data Figs. 7 and 8). Extension of the 4-mer acceptor (containing a non-reducing terminal GlcNAc residue) using a UDP-GlcA donor led to a mass increase consistent with a GlcA addition (Extended Data Fig. 7e). Equivalent reactions employing UDP-GlcNAc as donor led to no extension (Extended Data Fig. 7d). Similarly, extension of the 5-mer acceptor (containing a non-reducing terminal GlcA residue) detected only single sugar addition when UDP-GlcNAc was used as donor (Extended Data Fig. 7g). Reactions containing either acceptor glycan and both donors led to a time-dependent polymer extension consistent with an alternating addition of GlcA and GlcNAc residues to the non-reducing terminus (Extended Data Fig. 8). The predominant products reflected a disaccharide addition containing a non-reducing terminal GlcNAc residue, a result suggesting that GlcA transferase activity was limiting in the reaction. The broad Poisson distribution of mass peaks for the extended products across the reaction time course also suggests a distributive model for polymer extension21,22.
Underlying mechanism for inactive EXT1 GT-A and EXT-2 GT-B
To understand why EXT2 GT-B is inactive, we compared its putative substrate binding pocket structure, surface charge distribution and sequence with the active EXT1 GT-B domain (Extended Data Fig. 9a–d). Although the two domains are superimposable with an RMSD of 0.93 Å, several important differences exist around their respective active site pockets. The pocket of EXT1 GT-B is larger than that of EXT2 GT-B, making substrate interactions more feasible. In addition, the UDP binding pocket is positively charged in EXT1 GT-B, whereas the corresponding EXT2 GT-B pocket is nearly neutral (Extended Data Fig. 9a,b) because key residues are non-conservatively substituted. For example, EXT1 residues Lys269, Arg340 and Arg346 are replaced by Val259, Ala324 and Val330 in EXT2 (Extended Data Fig. 9c,d), respectively. These structural and physiochemical differences likely lead to inactivation of the EXT2 GlcA transferase domain.
Although active site mutations in the EXT1 GT-A domain did not impact glypican-1 co-polymerase activity or GlcNAc transfer to a 5-mer acceptor, EXT1 when expressed alone had low but detectable co-polymerase activity (Fig. 4a,b,d). We compared the structure and sequence of the EXT1 GT-A domain with the EXT2:UDP:Mn2+:7-mer complex and the mouse EXTL2 structure18 (Extended Data Fig. 9e,f). The three domains are highly similar with an RMSD of 0.76 Å between EXT1 and EXT2 GT-A domains and 0.72 Å between EXT1 GT-A and mEXTL2. The EXT1 GT-A domain also harbors key catalytic residues conserved in the EXT2 and mEXTL2 GT-A domains, including the DxD motif (D565-E-D567), Arg595 and Arg701. However, we also noted that the first loop (Val487-Pro495) of EXT1 GT-A is longer (nine versus five residues for EXT2 GT-A and mEXT2L) and moves out and away from the UDP binding pocket compared to EXT2 GT-A and mEXTL2. In EXT2, Tyr463 (mEXTL2 Tyr74) forms π–π stacking with the UDP and provides significant binding energy for the nucleotide23, but this residue is replaced by Val487 in the EXT1 GT-A domain. In addition, EXT2 Arg465 (mEXTL2 Arg76) forms a salt bridge with the UDP phosphate group, and this residue is replaced by Pro489 in EXT1 GT-A. Furthermore, the acceptor substrate binding region, including the C-terminal loop (Q732-L736), is divergent in sequence compared to the EXT2 and EXT2L GT-A domains. For example, Lys651 in the active EXT2 GT-A domain is replaced by Gln677 in the weakly active EXT1 GT-A domain. These active site changes could account for the diminished GlcNAc transferase activity for the EXT1 GT-A domain.
Mapping human disease mutations
The human genetic disease hereditary multiple exostoses (HME) generally results from a heterozygous loss of function in either EXT1 or EXT2 activity and is characterized by benign bony outgrowths in juxta-epiphyseal regions of long bones, termed exostoses or osteochondromas, and, in some cases, chondrosarcomas24 or seizures, scoliosis and macrocephaly/microcephaly syndrome (SSMS)25. Over 800 mutations in the EXT1 gene and over 400 mutations in the EXT2 gene have been reported26, but only a subset of these mutations lead to pathology27. Of these, 22% are missense mutations in the EXT1 gene, and 15% are missense mutations in the EXT2 gene27–29. Mapping of 58 EXT1 and 33 EXT2 disease-related missense mutations from the Human Gene Mutation Database30 onto the EXT1–2 structure revealed that mutations were clustered mainly in the EXT1 GT-B active site pocket and within the core of the EXT2 GT-B domain (Extended Data Fig. 10). Six disease-related mutations in EXT1 have been characterized (D164H, R280G/S and R340C/H/L)24,31–34. Each is in the GT-B domain catalytic pocket and leads to enzyme inactivation31,32,34. One characterized EXT2 HME mutation (D227N34,35) is at the interface between the EXT1 and EXT2 GT-B domains and may impact heterodimer stability. By contrast, there was a surprising under-representation of HME missense mutations within the GT-A domains of either EXT1 or EXT2. Our testing of putative active site mutations in the two EXT1–2 GT-A domains led to protein heterocomplexes that were expressed and secreted at similar levels to wild-type EXT1–2 (Extended Data Fig. 6), suggesting that these mutations do not lead to defects in enzyme folding or stability. Thus, it is unclear why there is such a paucity of HME mutations in the EXT1–2 GT-A domains compared to those found in the respective EXT1–2 GT-B domains.
Mechanisms of the EXT1–2 GlcA and GlcNAc transferase sites
The EXT1 GT-B GlcA transferase domain would be expected to employ an established inverting SN2 catalytic mechanism19,20 to produce a GlcA-β1,4GlcNAc product from the α-linked UDP-GlcA donor. An intact UDP-GlcA donor was computationally docked with Rosetta into the 4-mer bound EXT1 GT-B domain (Fig. 5a,c), and interactions with the UDP leaving group were analogous to the EXT1:UDP complex. Additional interactions include H-bonds between the GlcA O2 and Arg280, bridging H-bonds between Tyr271 and both the donor GlcA O2 and GlcNAc O6 of the acceptor and bridging H-bonds between Arg341 and both the GlcA carboxyl group and GlcNAc O4 of the acceptor. These combined interactions tether the donor and acceptor to orient the donor anomeric carbon within a catalytic distance (3.2 Å) of the GlcNAc O4 acceptor nucleophile (Fig. 5a,c), consistent with an SN2-type inverting catalytic mechanism. However, there is no obvious side chain residue in proximity to the acceptor O4 nucleophile to act as a catalytic base. Prior studies on the inverting GT-B glycosyltransferases, POFUT1 (refs. 36–38) and AtFUT1 (ref. 39), also identified no apparent ionizable side chains in proximity to the acceptor hydroxyl group, and an SN1-like mechanism was proposed. A similar SN1-like mechanism may also occur for the GT-B domain of EXT1.
Fig. 5 |. Catalytic reaction mechanisms of EXT1 GT-B and EXT2-GTA.
a, Modeled Michaelis complex of EXT1 GT-B in the presence of donor UDP-GlcA and the three-sugar acceptor (*GlcNAc-GlcA-GlcNAc). b, Modeled Michaelis complex of EXT2 GT-A in the presence of the donor UDP-GlcNAc and the four-sugar acceptor (*GlcA-GlcNAc-GlcA-GlcNAc). In a and b, the acceptors are from our experimental structures. The binding modes and poses of the donors UDP-GlcA (a) and UDP-GlcNAc (b) were computed by Rosetta and are consistent with UDP binding in our structures and the UDP-sugar binding models in previously determined glycosyltransferase structures. EXT1 GT-B and EXT2 GT-A are colored as in Fig. 1. Substrates are in sticks. Key residues involved in donor and substrate binding are shown in sticks and labeled. c, Sketch for the potential SN1-type inverting reaction mechanism for EXT1 GT-B. d, Sketch for the established SNi-type retaining reaction mechanism of EXT2 GT-A. The residues marked in red are confirmed in our study to have significant impact on EXT1–2 activity.
In contrast, the EXT2 GT-A domain is predicted to employ a retaining, dissociative SNi-type catalytic mechanism analogous to EXTL2 (refs. 18,40) and other GT-A-retaining glycosyltransferases20,41,42. Modeling of UDP-GlcNAc in the 7-mer bound EXT2 GT-A domain indicated that the core catalytic residues that interact with the donor sugar are positioned similarly to equivalent catalytic residues in EXTL2, including Arg673, Arg465 Asp628, Arg522, Asp538, Glu627 and Asn625 (Fig. 5b,d), consistent with a SNi-type reaction mechanism proposed for EXTL2 (refs. 18,40).
Discussion
Deciphering the enzymatic and structural basis for HS synthesis has been a challenge because of the complicated enzymology7,8, numerous enzyme isoforms and potential requirements for assembly into higher-order oligomeric enzyme complexes. Five ‘exostosin’ proteins are thought to play roles in HS synthesis (EXT1, EXT2 and EXTL1–3), but the precise contributions of the EXTL1–3 isoforms remain controversial8,43. Four EXT proteins contain two distinct domains (N-terminal GT47 and C-terminal GT64 domains)15, whereas EXTL2 contains only a C-terminal GT64 domain44. The EXTL2 structure was previously solved as donor and acceptor complexes, and the enzyme forms a classical GT-A fold symmetric homodimer18. While this manuscript was being reviewed, a cryo-EM structure of EXTL3 was also published45. It is also a symmetric homodimer comprised of an N-terminal GT-B and a C-terminal GT-A fold, but the active site of the GT-B fold is altered to block effective substrate binding. Both EXTL2 and EXTL3 exhibit α1,4GlcNAc transferase activity from their respective GT-A domains and add the first GlcNAc residue to the proteoglycan linker tetrasaccharide (GlcNAcT-I activity) and potentially polymer extension (GlcNAcT-II activity), although their exact roles in each activity in vivo remain uncertain8,9,43–46.
By comparison, our expression studies on EXT1 and EXT2 demonstrate that they form an obligate heterodimer that exhibits HS backbone (heparosan) co-polymerase activity on both a mature proteoglycan acceptor (glypican-1) and short heparosan primers. The asymmetry of the EXT1–2 heterodimer arises from the differences in linker regions between their respective GT-B and GT-A domains and combines with differences in complementary interface residues to explain the necessity for heterocomplex formation. The structure also suggests that single EXT1 or EXT2 protein chains or homodimeric assemblies are unlikely to be stable. This is consistent with our inability to express EXT2 alone, and expression of EXT1 alone generates only trace levels of mostly degraded material. However, low levels of co-polymerase activity were detected for the EXT1 expression product, indicating a possibility for HS production from EXT1 alone.
Our structural studies also demonstrated substrate binding only to the EXT1 GT-B and EXT2 GT-A domains but not the EXT1 GT-A or EXT2 GT-B domains. Enzyme assays on active site mutants in the EXT1 GT-B and EXT2 GT-A domains led to loss of both co-polymerase activity and single sugar transfer activity, although enzyme inactivation with the EXT2 GT-A active site mutants was not complete. Mutations in the EXT1 GT-A and EXT2 GT-B domains had variable impacts on co-polymerase and single sugar transfer activities, possibly through long-range allosteric effects. These data confirm that the EXT1 GT-B domain exclusively provides β1,4GlcA transferase activity, whereas the EXT2 GT-A domain contributes most, if not all, of the α1,4GlcNAc transferase activity in the EXT1–2 complex.
Assembly of multi-domain enzyme complexes may have evolved to accomplish the challenging task of efficiently assembling carbohydrate polymers comprised of two alternating sugars. Other proteoglycan-like polymerases have been identified containing homodimers of two GT-A fold domains (bacterial chondroitin polymerase47 and human LARGE1 (refs. 48,49)) or the chlorella virus hyaluronan synthase that employs a single bifunctional GT-A domain for polymer synthesis50. The EXT proteins represent the first glycosyltransferases that harbor both GT-A and GT-B domains for polymer synthesis. The existing structures of the EXT family members suggests a complex evolution. Fusion of GT-A and GT-B domain coding regions likely led to a progenitor EXT enzyme that could enhance the efficiency of HS synthesis by tethering two separate active sites into a single polypeptide chain. Subsequent gene duplications led to four EXT isoforms with two domains each, whereas one additional isoform (EXTL2) likely lost a GT-B domain by truncation. Like chondroitin synthase and LARGE1, formation of EXT homodimers could likely then help stabilize the extended multi-domain proteins. Unlike EXTL2, EXTL3 retained its GT-B domain but evolved to lose GlcA transferase activity while retaining a functional GT-A fold GlcNAc transferase domain. Surprisingly, EXT1 and EXT2 evolved to form an obligate heterodimer that led to apparent loss of function for two of the four domains and catalytic division of labor for β1,4GlcA and α1,4GlcNAc transferase sites between the two subunits.
The cryo-EM studies also noted that the two functional catalytic sites in EXT1–2 are ~90 Å apart. The lack of additional observed HS fragment binding sites elsewhere in the complex, the wide span of the active sites and the apparent distributive model for polymer extension suggest that iterative polymer synthesis likely involves a complete dissociation from one enzyme active site before switching to the other complementary active site during polymer extension (Fig. 6a,b). Additional efficiency for HS extension may also result from anchoring the HS core protein substrates to the EXT1–2 complex during GAG chain polymerization (Fig. 6b), because previous data indicated that the glypican-1 core protein enhances HS polymerization compared to free glycan acceptors14.
Fig. 6 |. Proposed model for GAG polymerization.
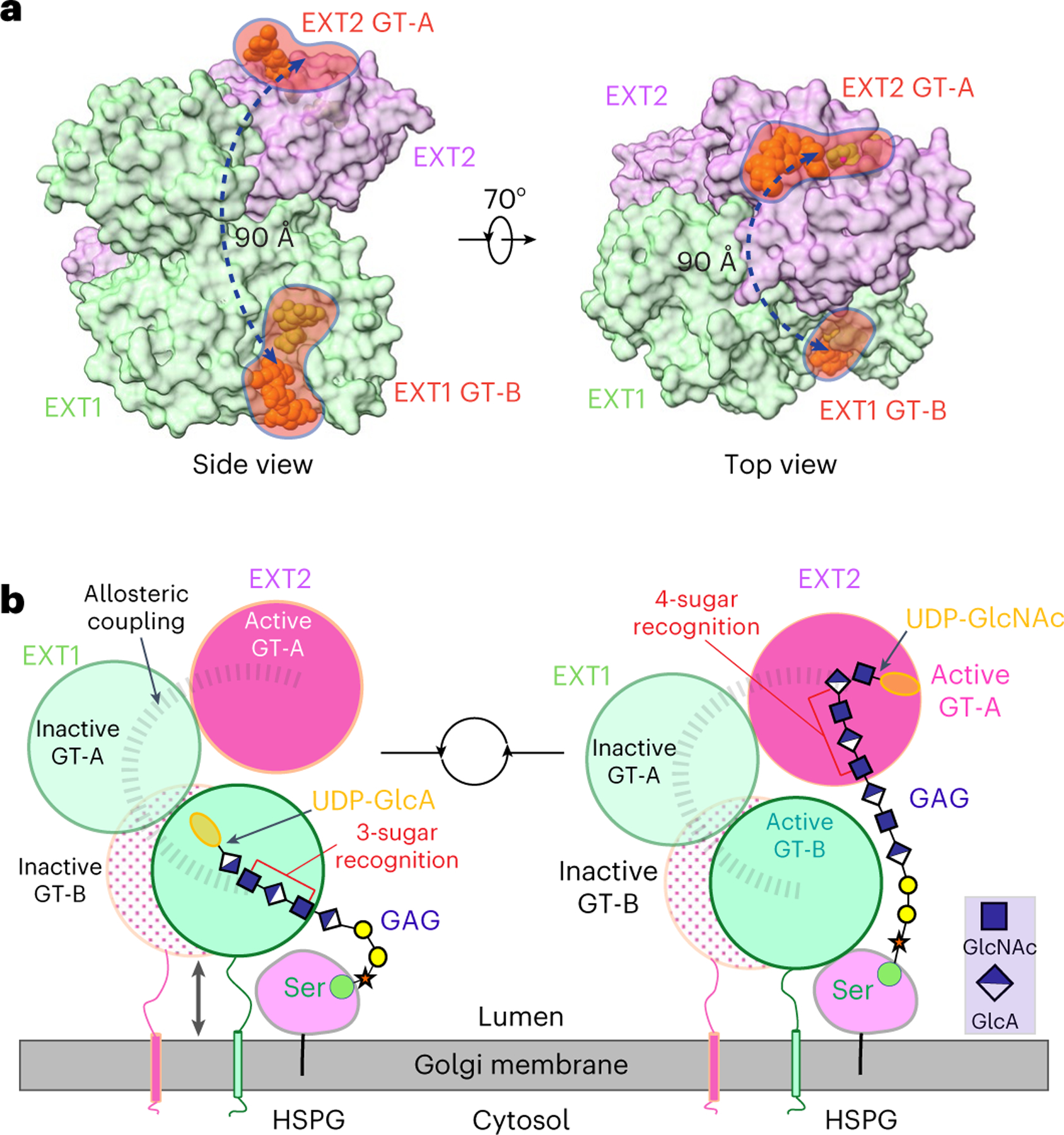
a, Two orthogonal surface views of EXT1–2. The UDP and acceptor substrates in the active pockets of EXT1 GT-B and EXT2 GT-A are shown as spheres. EXT1 and EXT2 are colored as in Fig. 1d. Left panel is in the same view as in Fig. 1c. b, EXT1 and EXT2 form a heterodimer that docks on the Golgi membrane. HSPG, usually a membrane-attached protein, moves close to EXT1–2 complex. The stem region of EXT1 and EXT2 may adopt extended and condensed conformation to accommodate the alternating addition of GlcA and GlcNAc for HS GAG chain elongation.
The broad separation of the two EXT1–2 active sites was unexpected considering previously observed efficiency for HS synthesis in vivo2,43. The ‘GAGosome’ model2 predicts that substrate channeling could contribute to efficient HS biosynthesis based on the observation of higher-order enzyme complexes that appear to be tightly coupled1. Although widely separated active sites for the EXT1–2 structure do not appear to be consistent with such a model, additional higher-order assemblies of enzymatic or structural components may bridge the EXT1–2 complex to upstream linker synthesis or downstream N-deacetylation, sulfation and epimerization to allow more efficient substrate handoff during HS maturation. The present structural and enzymatic studies on the EXT1–2 complex will provide a foundation for future studies on such larger complexes that further test the GAGosome hypotheses for HS biosynthesis.
Methods
Expression and purification of human EXT1–2 co-complex
Expression constructs for human EXT1 (residues 28–746, UniProt Q16394) and EXT2 (residues 46–718, UniProt Q93063) were generated encoding the truncated catalytic domains in the pGEn1 expression vector (encoding an NH2-terminal His8 and StrepII tag, followed by TEV protease recognition site) and the pGEn2 vector (encoding His8, AviTag, GFP and TEV protease recognition site) essentially as described in our previous studies17.
Single or pairwise transient transfections of constructs encoding full-length, truncated or mutant forms of EXT1 and EXT2 were performed in HEK293-F cells (FreeStyle 293-F cells, Thermo Fisher Scientific) as previously described17. For co-expression, plasmid DNAs were added in an equal ratio. Enzyme preparations were adjusted to contain 20 mM HEPES, 20 mM imidazole, 300 mM NaCl, pH 7.5, and subjected to Ni-NTA Superflow (Qiagen) chromatography as described in our previous studies17.
Protein expression for the cryo-EM studies employed co-transfections of EXT1-pGEn1 and EXT2-pGEn2, purification of the complex by Ni2+-NTA chromatography, cleavage of NH2-terminal fusion sequences with TEV protease, further purification by Ni2+-NTA chromatography, buffer exchange into 20 mM HEPES, 150 mM NaCl, pH 7.0, concentration to ~5 mg ml−1 and flash-freezing.
Size exclusion–multi-angle light scattering
The EXT1–2 preparation (1 mg ml−1) was injected on an analytical scale Superdex 75 gel filtration column in a buffer containing 20 mM HEPES (pH 7.4) and 150 mM NaCl. In-line light scattering was measured using a MiniDAWN TREOS detector (Wyatt Technology) and differential refractive index using a Optilab rEx detector (Wyatt Technology). Data analysis was performed using the ASTRA software package 6.0 (Wyatt Technology).
Cryo-EM grids preparation and data collection
First, Quantifoil R2/1 300 mesh gold grids were glow-discharged for 30 seconds. Then, droplets of 3 µl of sample at 0.3 mg ml−1 were applied to the freshly treated grids. The grids were blotted with Whatman 595 filter paper with the blot force set to 2 and blot time set to 3 seconds. Finally, the EM grids were flash-frozen into liquid ethane using an FEI Vitrobot Mark IV with the chamber temperature set to 6 °C and humidity set to 95%. For EXT1–2 complexed with the respective UDP-sugar donor, 10 mM UDP-GlcA or 10 mM UDP-GlcNAc and 10 mM MnCl2 were mixed with protein sample, and then the mixture was applied to grids immediately. For the 4-mer and 7-mer complexes, purified EXT1–2 was mixed with different solutions on ice for 1 hour (that is, 50 × 4-mer, 10 mM UDP, 10 mM MnCl2 or 50 × 7-mer, 10 mM UDP, 10 mM MnCl2). Cryo-EM datasets were collected automatically with SerialEM software in a 300-kV Titan Krios at a nominal magnification of ×105,000. Micrographs were recorded in the Gatan K3 direct detector with a pixel size equivalent to 0.414 Å per pixel at the specimen level. For EXT1–2, the defocus values were set in the range from −1.0 µm to −2.2 µm. Forty one-frame movies were recorded with a dose rate of 44.5 electrons per Å2 per second with an exposure time of 1.5 seconds. For EXT1–2 complexed with UDP-sugar, the defocus values were varied from −1.0 µm to −2.1 µm. Seventy five-frame movies were acquired at a total dose of 62 electrons per Å2 with an exposure time of 1.5 seconds. For the 4-mer bound complex, the defocus values were set in the range from −1.0 µm to −2.2 µm. Fifty-frame movies were recorded with a dose rate of 40 electrons per Å2 per second with an exposure time of 1.5 seconds. For the 7-mer bound complex, two datasets were collected, and the defocus values were set in the range from −0.9 µm to −1.8 µm. Finally, 40/60-frame movies were recorded with a dose rate of 45.3/48 electrons per Å2 per second with an exposure time of 1.5 seconds.
Cryo-EM image processing
The program MotionCorr2 (ref. 51) was used for motion correction, and CTFFind-4.1.10 (ref. 52) was used for the contrast transfer function estimation and correction. All the remaining steps were carried out using Relion-3.1 (ref. 53). For EXT1–2, the 4-mer and 7-mer complexes, approximately 1,000 particles, were manually picked and subjected to two-dimensional (2D) classification to generate templates for subsequent automatic particle picking. For EXT1–2 complexed with UDP-sugar, particles were automatically picked using LoG. The resolution of the three-dimensional (3D) map was estimated by gold standard Fourier shell correlation at the standard threshold of 0.143.
For EXT1–2, a total of 2,980 movies were recorded, and 2,608 images were kept after manual inspection. A total of 3,403,358 particles were picked automatically. After 2D classification, 1,317,525 particles in those classes with clear features were retained and subjected to 3D classification. Based on the quality of the four 3D classes, 429,785 particles were selected for further 3D reconstruction, refinement, CTF refinement and Bayesian polishing. To improve the resolution, these particles were subjected to 3D classification without alignment, after which 120,216 particles in two 3D classes were selected for 3D refinement and post-processing, resulting in a 3.1-Å average resolution EM map.
For EXT1–2 with UDP-GlcA, 5,742 movies were acquired, and 5,572 images were kept after CTF correction and manual inspection. After 2D classification, 2,093,792 particles out of 4,871,613 auto-picking particles were retained and subjected to 3D classification. In total, 824,437 particles in two 3D classes with best feature were kept for 3D reconstruction and refinement. To improve the map quality, one more round of 3D classification without particle alignment was carried out. Finally, a 3.0-Å average resolution 3D map was obtained using 335,313 particles through 3D refinement, followed by Bayesian polishing, CTF refinement and post-processing.
For EXT1–2 with UDP-GlcNAc, 5,460 movies were recorded, and 5,360 images were kept after manual inspection. After 2D classification, 1,619,668 particles out of 4,783,612 auto-picking particles were retained and subjected for 3D classification. In total, 668,776 particles belonging to the best 3D class were retained for 3D reconstruction and refinement. To improve the resolution, another round of 3D classification without alignment was performed, and 161,338 particles in one best class were retained. Further 3D refinement, followed by Bayesian polishing, CTF refinement and post-processing, resulted in a 3.3-Å average resolution 3D map.
For the 4-mer acceptor-bound EXT1–2 complex structure, 3,080 movies were recorded, and 2,991 images were kept after manual inspection. In total, 1,938,290 particles were picked automatically. After 2D classification, 1,253,323 particles belonging to the 2D classes with clear features were retained. The resulting particle images were subjected to two rounds of 3D classification, with 3D reference volume low-pass filtered at either 6 Å or 60 Å, leading to two sets of four 3D classes. In total, 31,440 particles from the best class of each 3D class set were combined, and, after removal of duplicated particles, 451,367 particles were retained for final 3D reconstruction, refinement, CTF refinement, Bayesian polishing and post-processing, resulting in a 3.0-Å average resolution 3D map.
For the 7-mer acceptor-bound EXT1–2 structure, 5,108 movies were recorded, and 4,592 images were kept after manual inspection. In total, 5,509,005 particles were picked automatically. After 2D classification, 2,171,949 particles belonging to the 2D classes with clear features were retained and subjected to 3D classification. Based on the quality of the four 3D classes, 443,302 particles were selected for further 3D reconstruction, refinement, CTF refinement and Bayesian polishing. To improve the resolution, these particles were subjected to two rounds of 3D classification without alignment, after which 83,144 particles belonging to the best 3D class were kept. We next recorded a second dataset of 7,609 movies. In total, 6,341 images were kept after manual inspection, leading to 5,668,467 particles after automatic particle picking using the same previously obtained templates. After 2D classification, 1,808,615 particles in those classes with clear features were retained and subjected to 3D classification. Based on the quality of the four 3D classes, 451,400 particles were selected for further 3D classification without alignment, after which 238,172 particles were kept. We combined the best particles from the two datasets, leading to 321,316 particles for final 3D refinement, CTF refinement, Bayesian polishing and post-processing, resulting in a 2.8-Å average resolution 3D map.
Model building, refinement and validation
The atomic model EXT1–2 was built de novo using Coot54. Bulky residues were used as landmarks, and the secondary structure prediction by PSIPRED server55 was used as a reference during model building. For the four substrate-bound complexes, the coordinates of apo EXT1–2 were used as initial model and docked into the EM maps in Chimera56. Then, UDP and sugar units of the 4-mer acceptor or UDP, Mn2+ and sugar units of the 7-mer acceptor were manually built into the 3D maps of EXT1–2 complexed with UDP, the 4-mer complex and the 7-mer complex using Coot54, respectively. The completed model was refined in real space using PHENIX57 and manually adjusted in Coot54. Finally, the model was validated using MolProbity58. To avoid overfitting, cross-validation through three Fourier shell correlation curves (model versus final map, model versus half1 map and model versus half2 map) was performed. ESPript59 and UCSF ChimeraX56 were used to prepare figures. The ligand structures were drawn in ChemDraw 20.1.
Molecular docking
Sugar donors UDP-GlcA and UDP-GlcNAc were docked into the binding pocket of EXT1 GT-B domain of the 4-mer complex and the EXT2 GT-A domain of the 7-mer complex, respectively. Molecular docking was performed using RosettaLigand. Next, 100 conformers of sugar donors were generated using Frog2 online server60. Then, the ligand parameter file was prepared using Rosetta script ‘molfile_to_params.py’. For protein preparation, the UDP and acceptor analogs were deleted from their pockets. To generate a starting model for RosettaLigand docking, one conformer of sugar donors was docked into its binding pocket based on the UDP orientation in the 4-mer complex and the 7-mer complex in Chimera. Finally, 100 docked poses were generated, and the best one was selected by visual inspection in Chimera.
Generation of mutants for EXT1 and EXT2 using site-directed mutagenesis
Expression constructs for mutant forms of EXT1 and EXT2 were generated in the pGEn2 vector using the Q5 Mutatgenesis Kit (New England Biolabs). The primers for each mutation were designed using NEBaseChanger program, and plasmids were isolated for expression in HEK293-F cells (Thermo Fisher Scientific). Each mutant was generated in the corresponding pGEn2 vector construct and was co-expressed with the wild-type EXT partner monomer in pGEn1 as a secreted soluble protein and was further purified using Ni-NTA chromatography as described above. Glycerol (10%) was added to the purified proteins, and they were stored at −80 °C before further kinetics studies and enzyme assay.
Mutations in the GT-B domain of EXT1 (K269A, R340A, R341A and R346A) and the GT-A domain of EXT1 (D565A, D567A, R595A, W612A and W692A) were generated. EXT1 with only the GT-B domain was generated by mutagenesis to add a termination codon in the EXT1 coding region. The resulting expression construct encoded residues 28–475 of EXT1. The GT-A domain of EXT1 was generated by polymerase chain reaction (PCR) amplification and transferred into the pGEn2 vector to encode residues 476–746 of EXT1. Similarly, mutations in the GT-B domain of EXT2 (Y308A, R325A and Q328A) and the GT-A domain of EXT2 (D538A, D540A, R569A and E585A) were generated. EXT2 with only the GT-B domain was generated by mutagenesis to add a termination codon in the EXT2 coding region. The resulting expression construct encoded residues 46–440 of EXT2. The GT-A domain was generated by PCR amplification and transferred into the pGEn2 vector to encode residues 441–718 of EXT2.
Expression and purification of glypican-1 as the acceptor for co-polymerase activity
The expression construct encoding the GAG core protein human glypican-1 (residues 24–529, UniProt P35052) was generated by gene synthesis and transferred into the pGEn2 vector, similarly to our previous studies17. Glypican-1 was expressed in HEK293-F cells using plasmid DNA and polyethyleneimine as transfection reagent, and the secreted expression product was purified using Ni-NTA chromatography as described above. Fusion tags were removed using TEV protease and digestion (ratio of 1:10) at 4 °C overnight, followed by Ni-NTA chromatography to remove the tags. The glypican-1 preparation was buffer exchanged into PBS and stored in aliquots at −80 °C for further use.
Enzyme assays for EXT1–2 co-complex and mutants for EXT1 and EXT2
Enzyme activity for the EXT1–2 co-complex and their mutants was determined using the UDP-Glo Glycosyltransferase Assay (Promega), which quantified UDP formed as a by-product of the GlcNAc and GlcA transferase reactions. The assays were performed according to the manufacturer’s instructions. Enzyme assays for co-polymerase activity were carried out in reactions that consisted of a universal buffer (100 mM each of MES, MOPS, TRIS, pH 7.0), UDP-GlcNAc or UDP-GlcA (200 µM) as donors, 10 mM MnCl2 and Glypican-1 (9 µM) as an acceptor and wild-type or mutant EXT1–2 enzyme preparations. Enzyme reactions were initiated with the addition of enzyme and carried out for 1 hour at 37 °C, followed by the UDP-Glo assay reagent additions as described previously39. Assays were performed in duplicate.
Enzyme assays to monitor GlcNAc transferase and GlcA transferase activities for wild-type and mutant EXT1–2 were carried out using synthetic oligosaccharide acceptors (4-mer: GlcNAc-GlcA-GlcNAc-GlcA-pNP; 5-mer: GlcA-GlcNAc-GlcA-GlcNAc-GlcA-pNP; 6-mer: GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-pNP; and 7-mer: GlcA-GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-pNP) obtained from Glycan Therapeutics. Assays employed either UDP-GlcA or UDP-GlcNAc as donors, respectively. Enzyme assays using synthetic oligosaccharide acceptors consisted of universal buffer (250 mM each of MES, MOPS, TRIS, pH 7.0), UDP-GlcA (200 µM) as donor for the 4-mer (0.5 mM) acceptor or UDP-GlcNAc (200 µM) as donor for the 5-mer (0.5 mM) acceptor and 10 mM MnCl2. Enzyme reactions were initiated with the addition of enzyme and carried out for 1 hour at 37 °C, followed by UDP-Glo assay reagent additions as described previously39.
Acceptor kinetics were carried out using wild-type EXT1–2 and individual 4-mer, 5-mer, 6-mer and 7-mer synthetic oligosaccharide acceptors (0–1 mM) with either UDP-GlcA (200 µM) as donor for the 4-mer and 6-mer acceptors or UDP-GlcNAc (200 µM) as donor (plus 10 mM MnCl2) for the 5-mer and 7-mer acceptors in reactions for 1 hour at 37 °C, followed by UDP-Glo assay reagent additions as described below. To determine co-polymerase activity using 4-mer and 5-mer acceptors, reactions consisted of a universal buffer (250 mM each of MES, MOPS, TRIS, pH 7.0), UDP-GlcNAc and UDP-GlcA (1 mM) as donors, 10 mM MnCl2, 4-mer or 5-mer (50 µM) as an acceptor and wild-type EXT1–2. Enzyme reactions were initiated with the addition of enzyme and carried out for 1 hour at 37 °C, followed by the UDP-Glo assay as described above.
Detection of co-polymerase action by EXT1–2 on glypican-1 using gel migration assay
To detect co-polymerase activity of wild-type EXT1–2 toward glypican-1 as acceptor, we also employed SDS-PAGE to detect the extension product. Reactions were comprised of universal buffer (100 mM each of MES, MOPS, TRIS, pH 7.0), UDP-GlcNAc and/or UDP-GlcA (1 mM) as donors, 10 mM MnCl2 and glypican-1 (9 µM) as an acceptor and wild-type EXT1–2 as enzyme. Enzyme reactions were initiated with the addition of enzyme and carried out for 6 hours at 37 °C, followed by resolving the reaction products by SDS-PAGE and staining with Coomassie R-250 to detect the change in mobility for the HS chains on glypican-1.
Analysis of enzymatic products using MALDI–MS
The product analysis for single sugar addition on the 4-mer and 5-mer primers and co-polymerase reactions to extend each of the primers by wild-type EXT1–2 was determined by MALDI-time of flight (TOF) MS. To monitor single sugar transfer, enzyme assays were carried out using reaction mixtures comprised of 4-mer (300 µM) or 5-mer (150 µM) with 1 mM of UDP-GlcA or UDP-GlcNAc, respectively, in reactions containing 25 mM HEPES buffer (pH 7.0) and 5 mM MnCl2 at 37 °C. Enzyme reactions were initiated by the addition of wild-type EXT1–2. The product formation was detected after 30 minutes using MALDI analysis. Nafion 117 solution (Sigma-Aldrich) was applied to a Bruker MSP 96 ground steel target and air-dried. Reaction samples were mixed 1:1 with a 20 mg ml−1 of 2,5-dihydroxybenzoic acid matrix solution in 50% methanol. Negative-ion MALDI-TOF MS spectra were acquired using an LT Bruker LT Microflex spectrometer. For detection of co-polymerase activity by wild-type EXT1–2, reactions with 4-mer or 5-mer were supplied with both the donors (UDP-GlcA and UDP-GlcNAc) in reaction conditions as above, and time points were taken to analyze the respective products by MALDI analysis.
Extended Data
Extended Data Fig. 1 |. Expression and purification of the EXT1–2 complex for structural studies.
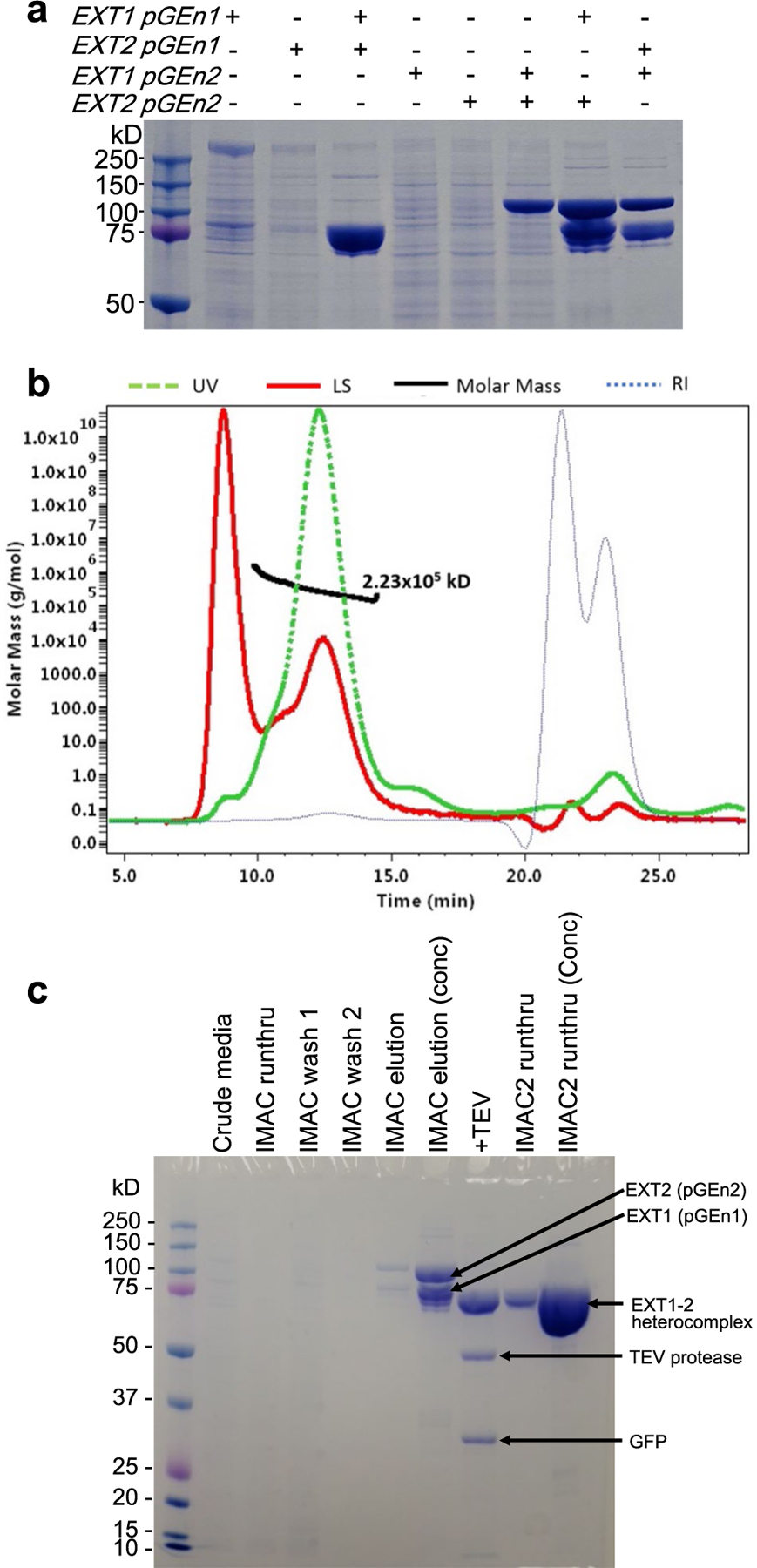
a, Single and co-expression of EXT1 and EXT2 as small N-terminal fusion proteins (pGEn1 vector) and larger N-terminal fusion proteins (pGEn2 vector). SDS-PAGE of the expressed and purified construct combinations is shown indicating only co-expression of EXT1 and EXT2 in either fusion vector format leads to appreciable secretion of the respective fusion proteins. b, The purified EXT1–2 complex prior to TEV cleavage was characterized by size exclusion-multiangle light scattering (SEC-MALS). A280 is shown by the green line, refractive index in blue, light scattering in red and calculated molar mass in black. The molecular mass derived from SEC-MALS analysis (~223 kDa) is in close agreement with the predicted size of the heterodimeric EXT1–2 complex with the respective fusion tags (EXT1: 88 kDa + EXT2: 109 kDa + 4 N-glycans). b, The EXT1-pGEn1 and EXT2-pGEn2 expression constructs were co-transfected into HEK293F cells and the secreted heterocomplex in the media (Crude media) was purified by Ni2+-NTA purification (IMAC run-through, Wash 1, Wash 2) to yield a highly-enriched enzyme preparation (IMAC1 elution). The enzyme was concentrated (IMAC1 elution conc) and cleaved with TEV protease to remove the fusion tag sequences (+TEV). Ni2+-NTA chromatography separated the unbound EXT1–2 complex from the bound tag sequences and TEV protease (IMAC2 runthru), as the latter were all His-tagged. Panels a and b are each representative of data from two biological replicate experiments. Original uncropped images are provided in the Source Data.
Extended Data Fig. 2 |. Structure of EXT1–2.
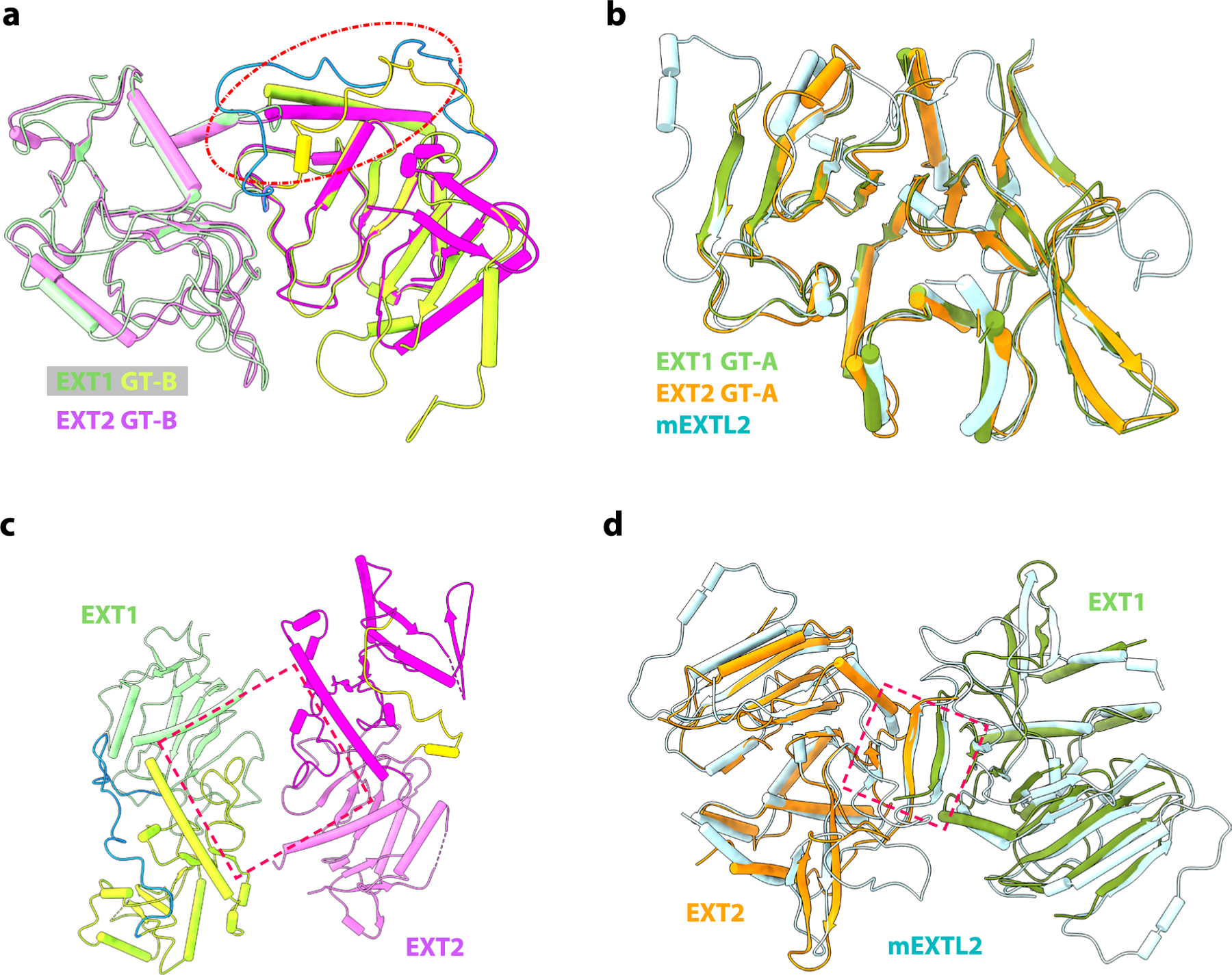
a, Superimposition of GT-B fold of EXT1 and EXT2. The intervening loop in red dash cycle. b, Superimposition of GT-A fold from EXT1, EXT2, and mEXTL2 (PDB ID: 1ON6). c, Top view of GT-B fold in EXT1–2, suggesting a pseudo-twofold symmetry. d, Superimposition of GT-A fold from EXT1–2 and mEXTL2 (PDB ID: 1ON6) in top view, suggesting a pseudo-twofold symmetry for GT-A fold in EXT1–2. EXT1–2 is colored the same as Fig. 1, and mEXTL2 in cyan.
Extended Data Fig. 3 |. Comparison of 4-mer complex with EXT1–2.
a, Superimposition of EXT1–2 and 4-mer complex. b, Superimposition of EXT1 GT-B fold from EXT1–2 and 4-mer complex. 4-mer complex colored the same as in Fig. 1, and EXT1–2 in light gray. Residues involved in active pocket are shown as sticks. The disordered loop H300-C302 in EXT1–2 marked in red dashed oval. The density of NAG at Asn330 is shown as 50% transparency.
Extended Data Fig. 4 |. Comparison of 7-mer complex with EXT1–2, 4-mer complex, and EXT1–2 complexed with UDP-GlcA.
a, Superimposition of EXT1–2, 4-mer and 7-mer complex. b, Orthogonal view highlighting the GT-A fold comparison. 7-mer complex colored the same as in Fig. 1, EXT1–2 in light gray, and 4-mer complex in wheat. c, Superimposition of EXT1 GT-B fold from EXT1–2 complexed with UDP-GlcA and 7-mer complex. 7-mer complex are colored the same as in Fig. 1, and EXT1–2 complexed with UDP-GlcA in cyan. Residues involved in active pocket are shown as sticks.
Extended Data Fig. 5 |. Development of an HS co-polymerase assay for EXT1–2.
A co-polymerase assay was developed using the HS proteoglycan, glypican-1 (a and b), or heparosan primers (4-mer-pNP or 5-mer-pNP) (c) as acceptors and UDP-GlcA and UDP-GlcNAc (200 µM each) as donors. Individual assay components were tested in the reaction mixture and HS polymerization was detected using the UDP-Glo assay format (a and c). Recombinant human glypican-1 was expressed in HEK293 cells and purified for use as an acceptor substrate for HS extension of the proteoglycan. a, Low, but detectable activity was revealed when single UDP sugars were added to the reaction mix reflecting single sugar additions to the HS chains. When both UDP-GlcA and UDP-GlcNAc were added to the reaction, the enzyme activity increased ~40 fold indicating an iterative use of the sugar donors during the HS extension reaction. b, Reaction products were resolved by SDS-PAGE and Coomassie staining. Extended HS polymer product on glypican-1 appearing as a high molecular weight smear (red brackets) was detected only when enzyme and both sugar nucleotide donors were present in the co-polymerase reaction. The data are representative of n = 2 biologically independent samples. Original uncropped images are provided in the Source Data. c, UDP-Glo reactions using 4-mer-pNP and 5-mer-pNP as acceptors. Extension of the 4-mer primer with single sugar nucleotide donors only occurred when UDP-GlcA, but not UDP-GlcNAc, was used as donor as expected for an acceptor containing a non-reducing terminal GlcNAc residue. In contrast, extension of the 5-mer primer with single sugar nucleotide donors only occurred with UDP-GlcNAc, but not UDP-GlcA, was used as donor consistent with the presence of a non-reducing terminal GlcA residue on the acceptor. Addition of both donors in extension reactions with either the 4-mer or 5-mer as acceptor led to enhanced activity indicating an iterative use of the sugar donors during the HS extension reaction. Plots show the mean values (bar) ± s.d. (error bars) for n = 2 technical replicates (red circles).
Extended Data Fig. 6 |. Expression and purification of EXT1 and EXT2 mutants.
a. Diagrammatic representation of the positions of the EXT1 and EXT2 active site mutations that were generated. The respective GT-B and GT-A domains are indicated relative to the N-terminal GFP encoded by the pGEn2 vector. Residues for mutagenesis were chosen based on proximity to the respective GT-A or GT-B domain active sites. Truncation to form expression constructs encoding the respective single domains are also indicated. Termination codons were introduced in the linker regions between the GT-A and GT-B domains of EXT1 and EXT2 to generate the respective single GT-B domains (EXT1-GT-B and EXT2 GT-B). Each of the GT-A domains were isolated by PCR and transferred into the pGEn2 vector (EXT1 GT-A and EXT2 GT-A) to generate the respective single GT-A domains. Each of the mutants in the pGEn2 vector were co-expressed along with wild type versions of the corresponding partner EXT (in pGEn1 vector) to generate a secreted heterocomplex (for example EXT1 K269A mutant co-expressed with wild type EXT2) that was subsequently purified by Ni2+-NTA chromatography and resolved by SDS PAGE c, Initial expression level of the proteins in the culture media was monitored by GFP fluorescence b, since the mutant EXT form harbored a GFP fusion as a result of expression in the pGEn2 vector. The upper band on the SDS-PAGE gel (~115 kDa) corresponds to the mutant EXT form expressed in the pGEn2 vector, while the lower band (~90 kDa) corresponds to the wild-type EXT form (pGEn1 vector) that was co-expressed. Individual EXT1 and EXT2 isoforms and individual domains were also expressed singly or in combination. Of these latter expression tests, only EXT1 alone or the individual EXT2 GT-A or GT-B domains resulted in any appreciable secreted products. Each co-transfection experiment was performed once and two different SDS PAGE gels of the samples were generated. Data presented are representative of the respective experiments. Original uncropped images are provided in the Source Data.
Extended Data Fig. 7 |. Enzymatic extension of 4-mer and 5-mer acceptor primers with single sugar residues.

To complement the UDP-Glo assays performed in Fig. 4 and Extended Data Fig. 5, we analyzed the reaction products for glycan extension by MALDI-MS. Individual spectra were obtained for the indicated enzyme, sugar nucleotide donors (a and b), and the 4-mer-pNP (c) and 5-mer-pNP (e) acceptors. Electrospray mass spectra of each of the synthetic acceptors indicated a predicted parent mass of 897.75 and 1073.87 for the 4-mer-pNP and 5-mer-pNP acceptors, respectively (c and f). However, MALDI-MS of the compounds produced a broad pair of mass peaks where the smaller, higher mass peak matched the predicted mass (singly charged masses of 898 and 1074 for the 4-mer-pNP and 5-mer-pNP acceptor, respectively, insets for panels c and f at the top). For each compound, the more abundant second species was 15 mass units smaller than the predicted mass, consistent with an in-source rearrangement resulting in neutral mass loss during MALDI analysis. The cause of this 15 mass unit neutral loss is not clear at the present time, but it did not impact the ability of the 4-mer and 5-mer to act as acceptors for extension by EXT1–2. Reactions containing the 4-mer acceptor, which harbored a reducing terminal GlcNAc residue, could be extended by the mass of a GlcA unit in the presence of the UDP-GlcA donor (e), while reactions containing the UDP-GlcNAc donor led to no extension (d). By comparison, reactions containing the 5-mer acceptor (harboring a reducing terminal GlcA residue) could be extended by the mass of a GlcNAc residue in the presence of a UDP-GlcNAc donor (g), but reactions containing UDP-GlcA as donor led to no extension (h). Data presented are representative of n > 3 independent reactions.
Extended Data Fig. 8 |. Time course of co-polymer extension of 4-mer and 5-mer primers.

While single sugar residue extensions were observed on 4-mer and 5-mer primers in the presence of UDP-GlcA and UDP-GlcNAc, respectively, a time course of co-polymer extension was examined by the addition of both donors to reactions containing the 4-mer or 5-mer primers. Blank reactions containing boiled enzyme (red asterisk) and both sugar nucleotides (a), 4-mer-pNP (b) or 5-mer-pNP (f) were examined for comparison to reactions containing 4-mer-pNP, enzyme and both donors (c-e) or 5-mer-pNP, enzyme and both donors (g-i) in a time course extension. Both reactions showed a similar set of extension products with the 4-mer being extended by the addition of a GlcA residue to form a 5-mer product followed by a GlcNAc addition and subsequent extension of a disaccharide unit terminating in a GlcNAc sugar residue indicating that the GlcA transferase activity was rate limiting (c-e). A similar extension of the 5-mer-pNP was observed with the extension to an 18-mer over the course of the 1 h reaction (g-i). Data presented are representative of n > 3 independent reactions.
Extended Data Fig. 9 |. Comparison of the catalytic pockets of GT-A domains and GT-B domains explain why some of these domains are inactive.
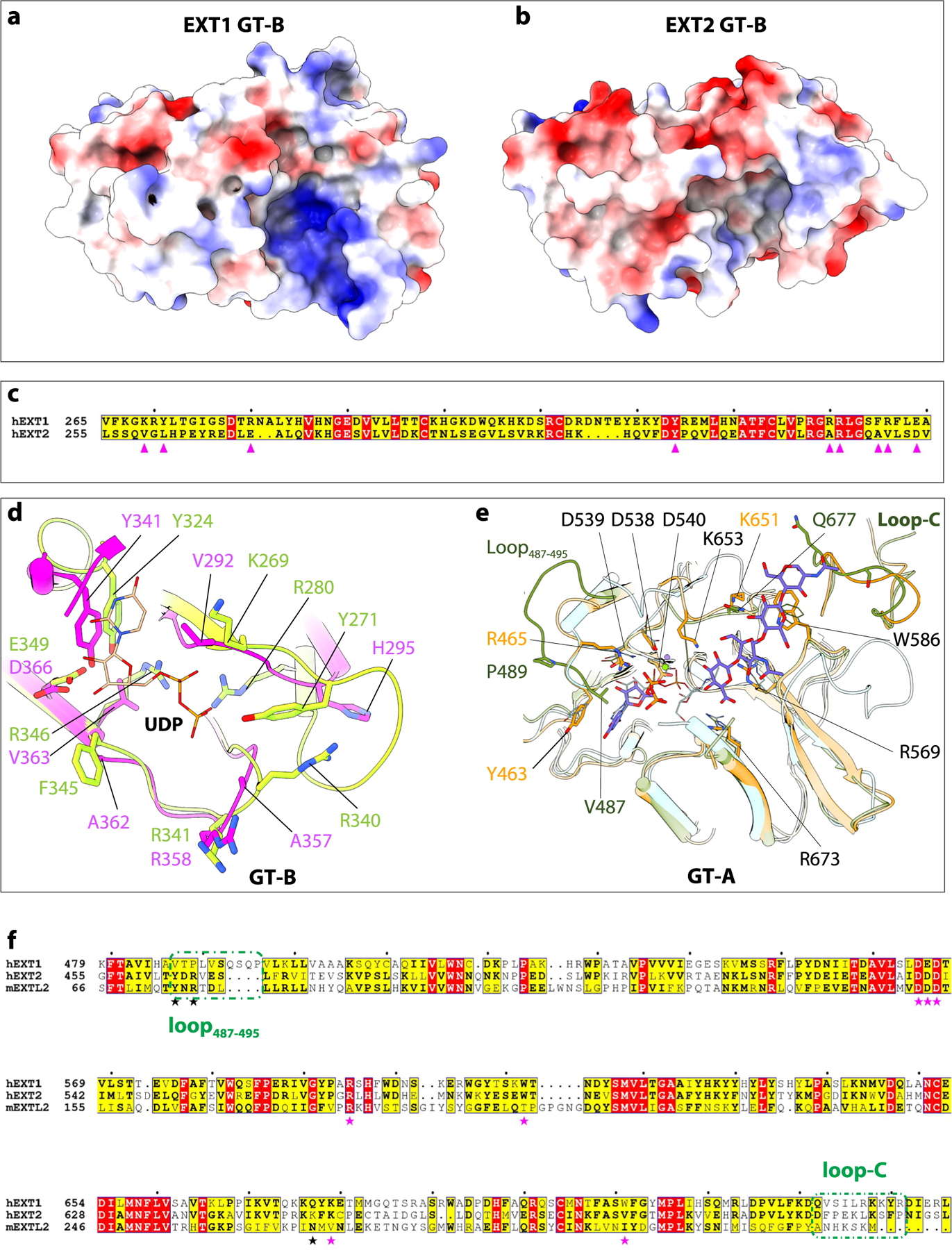
a, b, Electrostatic potential of GT-B cleft in EXT1 and EXT2. Positive charged surface colored in blue, and negative charged surface in red. Superimposition of GT-B fold of EXT1 and EXT2. c, d, Sequence alignment and close-up view of UDP binding pocket of EXT1 and EXT2 GT-B. The key residues are marked and shown in sticks. e, Superimposition of active pocket in hEXT1 GT-A, hEXT2 GT-A, and mEXTL2 (PDB ID: 1ON6). The residues involved in substrate interaction shown in stick, and residues in hEXT2 are labeled, except for V487, P489, and K684 those not conserved in hEXT1. hEXT1 and hEXT2 colored the same as in Fig. 1, mEXTL2 colored in cyan. f, Sequence alignment of hEXT1, hEXT2, and mEXTL2. The residues involved in substrate interaction marked in star, the same one in hEXT1 and hEXT2 in magenta, while different one in black. The first (loop487–495) and last (loop-C) loop are marked in olive dashed frame.
Extended Data Fig. 10 |. Mapping of disease related missense mutations onto the structure of EXT1 and EXT2.
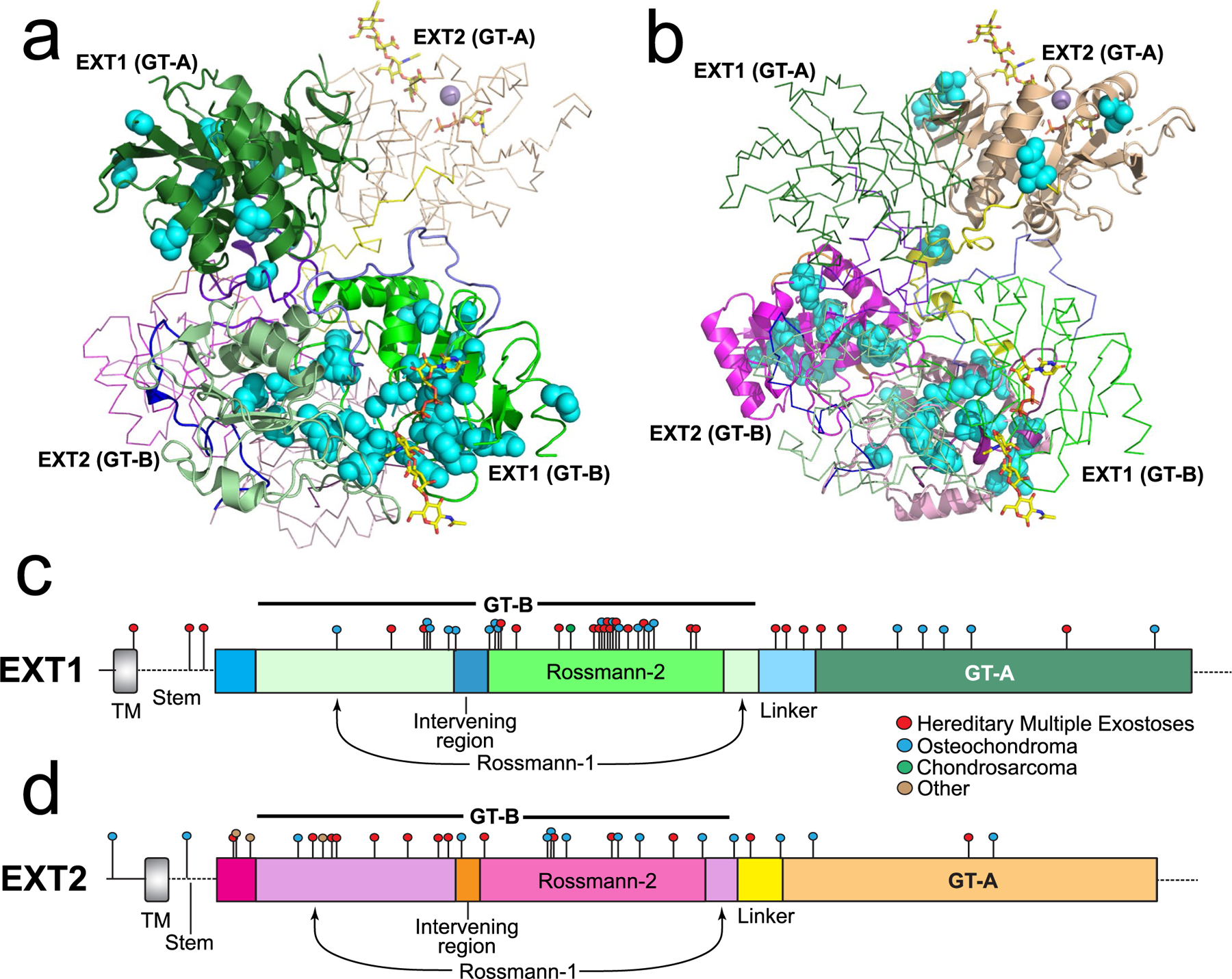
Mutations in the EXT1 and EXT2 coding regions that lead to disease were identified in the Human Gene Mutation Database30 and are shown on the linear representations of the respective protein sequences as colored circles (c and d) and as cyan spheres on the structural representation of EXT1 (a) and EXT2 (b). EXT1–2 are colored the same as Fig. 1. Disease characteristics shown in the legend are based on the annotations for the respective mutations in the Human Gene Mutation Database.
Supplementary Material
Acknowledgements
Cryo-EM images were collected in the David Van Andel Advanced Cryo-Electron Microscopy Suite at the Van Andel Institute. We thank G. Zhao and X. Meng for facilitating data collection, R. Weiss for helpful discussions on HS biology and S. Archer-Hartman for LC–MS analysis of oligosaccharide substrates (supported by National Institutes of Health (NIH) grant R24GM137782 to P. Azadi). This work was supported by NIH grants R01GM130915 and P41GM103390 (to K.W.M.) and R01CA231466 (to Huilin Li) and the Van Andel Institute (to Huilin Li). R.A.A was supported by funding from the US Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, under award DE-SC0015662. Materials generated in this study are freely available upon sending a request to the corresponding authors.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-022-01220-2.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41589-022-01220-2.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41589-022-01220-2.
Peer review information Nature Chemical Biology thanks Kamil Godula and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM 3D maps of human exostosin-1 and exostosin-2 heterodimer in the apo form (3.1 Å), bound to a 4-mer acceptor substrate (3.0 Å), bound to a 7-mer acceptor substrate (2.8 Å), complexed with UDP-GlcNAc (3.3 Å) or complexed with UDP-GlcA (3.0 Å), have been deposited in the Electron Microscopy Data Bank under accession codes EMD-25035, EMD-25036, EMD-25037, EMD-26701 and EMD-26702, respectively. The associated atomic models have been deposited in the Protein Data Bank (PDB) under accession codes 7SCH, 7SCJ, 7SCK, 7UQX and 7UQY, respectively. The crystal structure of the mouse EXTL2 (PDB accession code 1ON6) was used for comparison with human EXT1–2 structure. These data are available from the authors upon reasonable request. Source data are provided with this paper.
References
- 1.Carlsson P & Kjellen L in: Heparin—A Century of Progress (eds Lever R, Mulloy B & Page CP) 23–40 (Springer, 2012). [Google Scholar]
- 2.Esko JD & Selleck SB Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem 71, 435–471 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Sarrazin S, Lamanna WC & Esko JD Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol 3, a004952 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop JR, Schuksz M & Esko JD Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Kim SH, Turnbull J & Guimond S Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol 209, 139–151 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Busse-Wicher M, Wicher KB & Kusche-Gullberg M The exostosin family: proteins with many functions. Matrix Biol 35, 25–33 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Sugahara K & Kitagawa H Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol 10, 518–527 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Sugahara K & Kitagawa H Heparin and heparan sulfate biosynthesis. IUBMB Life 54, 163–175 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Annaval T et al. Heparan sulfate proteoglycans biosynthesis and post synthesis mechanisms combine few enzymes and few core proteins to generate extensive structural and functional diversity. Molecules 25, 4215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCormick C, Duncan G, Goutsos KT & Tufaro F The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl Acad. Sci. USA 97, 668–673 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitagawa H & Nadanaka S in: Handbook of Glycosyltransferases and Related Genes (eds Taniguchi N et al. ) 905–923 (Springer Tokyo, 2014). [Google Scholar]
- 12.Busse M & Kusche-Gullberg M In vitro polymerization of heparan sulfate backbone by the EXT proteins. J. Biol. Chem 278, 41333–41337 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Wei G et al. Location of the glucuronosyltransferase domain in the heparan sulfate copolymerase EXT1 by analysis of Chinese hamster ovary cell mutants. J. Biol. Chem 275, 27733–27740 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Kim BT, Kitagawa H, Tanaka J, Tamura J & Sugahara K In vitro heparan sulfate polymerization: crucial roles of core protein moieties of primer substrates in addition to the EXT1–EXT2 interaction. J. Biol. Chem 278, 41618–41623 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Drula E et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 50, D571–D577 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senay C et al. The EXT1/EXT2 tumor suppressors: catalytic activities and role in heparan sulfate biosynthesis. EMBO Rep 1, 282–286 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moremen KW et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol 14, 156–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedersen LC et al. Crystal structure of an α1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. J. Biol. Chem 278, 14420–14428 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Lairson LL, Henrissat B, Davies GJ & Withers SG Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem 77, 521–555 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Moremen KW & Haltiwanger RS Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol 15, 853–864 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keys TG et al. Engineering the product profile of a polysialyltransferase. Nat. Chem. Biol 10, 437–442 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Urbanowicz BR, Pena MJ, Moniz HA, Moremen KW & York WS Two Arabidopsis proteins synthesize acetylated xylan in vitro. Plant J 80, 197–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang A, Singh S, Phillips GN Jr. & Thorson JS Glycosyltransferase structural biology and its role in the design of catalysts for glycosylation. Curr. Opin. Biotechnol 22, 800–808 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hecht JT et al. Hereditary multiple exostoses (EXT): mutational studies of familial EXT1 cases and EXT-associated malignancies. Am. J. Hum. Genet 60, 80–86 (1997). [PMC free article] [PubMed] [Google Scholar]
- 25.Farhan SM et al. Old gene, new phenotype: mutations in heparan sulfate synthesis enzyme, EXT2 leads to seizure and developmental disorder, no exostoses. J. Med. Genet 52, 666–675 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Fokkema I et al. The LOVD3 platform: efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet 29, 1796–1803 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bukowska-Olech E et al. Hereditary multiple exostoses—a review of the molecular background, diagnostics, and potential therapeutic strategies. Front. Genet 12, 759129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clement N & Porter D Hereditary multiple exostoses: anatomical distribution and burden of exostoses is dependent upon genotype and gender. Scott. Med. J 59, 35–44 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Cook A et al. Genetic heterogeneity in families with hereditary multiple exostoses. Am. J. Hum. Genet 53, 71–79 (1993). [PMC free article] [PubMed] [Google Scholar]
- 30.Stenson PD et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet 136, 665–677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bovee JV et al. EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am. J. Hum. Genet 65, 689–698 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raskind WH et al. Evaluation of locus heterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum. Mutat 11, 231–239 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Wuyts W et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet 62, 346–354 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Philippe C et al. Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am. J. Hum. Genet 61, 520–528 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morimoto K et al. Transgenic expression of the EXT2 gene in developing chondrocytes enhances the synthesis of heparan sulfate and bone formation in mice. Biochem. Biophys. Res. Commun 292, 999–1009 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Li Z et al. Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1. Nat. Chem. Biol 13, 757–763 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Lira-Navarrete E & Hurtado-Guerrero R A perspective on structural and mechanistic aspects of protein O-fucosylation. Acta Crystallogr F Struct. Biol. Commun 74, 443–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lira-Navarrete E et al. Structural insights into the mechanism of protein O-fucosylation. PLoS ONE 6, e25365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urbanowicz BR et al. Structural, mutagenic and in silico studies of xyloglucan fucosylation in Arabidopsis thaliana suggest a water-mediated mechanism. Plant J 91, 931–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negishi M, Dong J, Darden TA, Pedersen LG & Pedersen LC Glucosaminylglycan biosynthesis: what we can learn from the X-ray crystal structures of glycosyltransferases GlcAT1 and EXTL2. Biochem. Biophys. Res. Commun 303, 393–398 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Albesa-Jove D, Sainz-Polo MA, Marina A & Guerin ME Structural snapshots of α-1,3-galactosyltransferase with native substrates: insight into the catalytic mechanism of retaining glycosyltransferases. Angew. Chem. Int. Ed. Engl 56, 14853–14857 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Yu H et al. Notch-modifying xylosyltransferase structures support an SNi-like retaining mechanism. Nat. Chem. Biol 11, 847–854 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kreuger J & Kjellen L Heparan sulfate biosynthesis: regulation and variability. J. Histochem. Cytochem 60, 898–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitagawa H, Shimakawa H & Sugahara K The tumor suppressor EXT-like gene EXTL2 encodes an α1, 4-N-acetylhexosaminyltransferase that transfers N-acetylgalactosamine and N-acetylglucosamine to the common glycosaminoglycan-protein linkage region. The key enzyme for the chain initiation of heparan sulfate. J. Biol. Chem 274, 13933–13937 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Wilson LFL et al. The structure of EXTL3 helps to explain the different roles of bi-domain exostosins in heparan sulfate synthesis. Nat. Commun 13, 3314 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim BT et al. Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode α1,4-N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/heparin biosynthesis. Proc. Natl Acad. Sci. USA 98, 7176–7181 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osawa T et al. Crystal structure of chondroitin polymerase from Escherichia coli K4. Biochem. Biophys. Res. Commun 378, 10–14 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Katz M & Diskin R The homodimeric structure of the LARGE1 dual glycosyltransferase Preprint at 10.1101/2022.05.11.491581v1.full (2022). [DOI] [PMC free article] [PubMed]
- 49.Joseph S et al. Structure and mechanism of LARGE1 matriglycan polymerase Preprint at 10.1101/2022.05.12.491222v1 (2022). [DOI]
- 50.Maloney FP et al. Structure, substrate recognition and initiation of hyaluronan synthase. Nature 604, 195–201 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 51.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohou A & Grigorieff N CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchan DWA & Jones DT The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 47, W402–W407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goddard TD et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams CJ et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robert X & Gouet P Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miteva MA, Guyon F & Tuffery P Frog2: efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res 38, W622–W627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM 3D maps of human exostosin-1 and exostosin-2 heterodimer in the apo form (3.1 Å), bound to a 4-mer acceptor substrate (3.0 Å), bound to a 7-mer acceptor substrate (2.8 Å), complexed with UDP-GlcNAc (3.3 Å) or complexed with UDP-GlcA (3.0 Å), have been deposited in the Electron Microscopy Data Bank under accession codes EMD-25035, EMD-25036, EMD-25037, EMD-26701 and EMD-26702, respectively. The associated atomic models have been deposited in the Protein Data Bank (PDB) under accession codes 7SCH, 7SCJ, 7SCK, 7UQX and 7UQY, respectively. The crystal structure of the mouse EXTL2 (PDB accession code 1ON6) was used for comparison with human EXT1–2 structure. These data are available from the authors upon reasonable request. Source data are provided with this paper.