

Abstract
Epidemiological studies indicate that chronic obstructive pulmonary disease (COPD) is associated with the incidence of changes in intestinal health. Cigarette smoking, as one of the major causes of COPD, can have an impact on the gastrointestinal system and promotes intestinal diseases. This points to the existence of gut–lung interactions, but an overview of the underlying mechanisms of the bidirectional connection between the lungs and the gut in COPD is lacking. The interaction between the lungs and the gut can occur through circulating inflammatory cells and mediators. Moreover, gut microbiota dysbiosis, observed in both COPD and intestinal disorders, can lead to a disturbed mucosal environment, including the intestinal barrier and immune system, and hence may negatively affect both the gut and the lungs. Furthermore, systemic hypoxia and oxidative stress that occur in COPD may also be involved in intestinal dysfunction and play a role in the gut–lung axis. In this review, we summarize data from clinical research, animal models, and in vitro studies that may explain the possible mechanisms of gut–lung interactions associated with COPD. Interesting observations on the possibility of promising future add-on therapies for intestinal dysfunction in patients with COPD are highlighted.
Keywords: gut–lung axis, lung diseases, systemic inflammation, hypoxia, microbiota
Chronic obstructive pulmonary disease (COPD) is characterized by chronic inflammation and chronic airway obstruction, leading to a progressive and irreversible decline in lung function (1). COPD is associated with exacerbations and comorbidities, resulting in a significant social and economic burden (2). There are several risk factors for COPD, including cigarette smoking, genetic factors, environmental pollution, and infections (3, 4).
A retrospective analysis of 1,228 patients with COPD showed that the majority of patients with COPD have at least one gastrointestinal symptom (5). Intestinal diseases, such as inflammatory bowel disease (IBD) and irritable bowel syndrome, are commonly observed in patients with COPD (6). The incidence of IBD is higher in patients with COPD compared with healthy subjects (7). Patients with COPD have intestinal microbiome dysbiosis and increased intestinal permeability, which is associated with inflammatory cell infiltration (8). Moreover, enterocyte damage in addition to intestinal hyperpermeability in patients with COPD, but not in control subjects, indicates functional changes in the gastrointestinal tract (9). Although the gut and lungs are separate organs with different functions and environments, they have structural similarities and interact in health and disease. There is growing interest in how lung health affects the function of the intestinal system and vice versa (10).
Cigarette smoke, as one of the leading causes of COPD, has detrimental effects on respiratory and gastrointestinal mucosal health. Cigarette smoke can induce inflammatory responses in the lung, which might “spill” into the systemic circulation, causing systemic inflammation and leading to (low-grade) inflammatory responses in the intestine (11). Cigarette smoke can cause oxidative stress and hypoxia in airway epithelium (12), while the impaired gas exchange induced by cigarette smoke in patients with COPD also leads to intestinal hypoxia and enterocyte damage and hence can result in an enhancement of intestinal permeability, disturbed immune function, and microbiome dysbiosis (8, 13, 14). In addition, soluble cigarette smoke particles may directly affect the gastrointestinal tract by entering the bloodstream after inhalation, or these particles may reach the gastrointestinal tract because of mucociliary clearance in the airways or direct swallowing of cigarette smoke (15).
In this review, we discuss recent advances in understanding the interactions between the lungs and the gut in COPD. Systemic inflammation, epithelial barrier dysfunction, oxidative stress, hypoxia, and gut microbiome dysbiosis are considered to play a major role in gut–lung interactions. Clinical studies and preclinical in vivo and in vitro experiments related to the gut–lung axis in COPD are discussed, as well as potential interventions and their modulation via the gut–lung axis aimed at treating or preventing the progression of COPD and its intestinal comorbidities.
Gastrointestinal Tract Disorders Associated with COPD
There is an association between gastrointestinal disorders and COPD. Gastrointestinal diseases are more prevalent in patients with COPD than in healthy subjects. A population-based retrospective cohort study using the administrative health databases of Québec showed that the incidence of IBD (i.e., Crohn’s disease [CD] and ulcerative colitis [UC]) was 55% and 30% higher in patients with COPD than in the general population, respectively. The highest incidence rate of IBD among patients with COPD was at 50–59 years of age (7). There is also evidence that up to 60% of patients with IBD have some degree of subclinical lung disease (16). In addition, both CD and UC were associated with an increased mortality in patients with asthma-related COPD (patients with COPD with mixed features of asthma and COPD), while UC also increased mortality among patients with COPD (17). Shared environmental risks (i.e., smoking and air pollution) are unlikely to fully explain this association, because cigarette smoke may have a protective role in UC (18). A meta-analysis presented that the risk of developing UC is significantly lower in smokers compared with people who have never smoked (19), whereas early smoking is associated with an increased risk for CD (20). However, the benefits of nicotine treatments in patients with UC in the form of transdermal patches, chewing gum, and nicotine-based enemas are not yet conclusive (21–23). Moreover, IBD is associated not only with COPD but also with other airway manifestations, such as bronchiectasis, characterized by airway inflammation and high volumes of sputum production (24).
A large U.S. population-based study (1997–2003) revealed that the prevalence of gastrointestinal ulcer disease in current and former smokers (11.4% and 11.5%, respectively) is almost doubled compared with never-smokers (6.0%). In addition, smokers who have a large daily intake of tobacco have higher risk of developing peptic ulcers compared with never-smokers (25).
A case–control study using data from the Ewha Womans University School of Medicine, Mokdong Hospital (Seoul, Republic of Korea), the database showed that the prevalence of colorectal adenomatous polyps was 66% (54 of 82) in the COPD group and 39% (98 of 251) in the non-COPD group matched for both age and sex. This indicated that the risk of colorectal malignant potential in the COPD group was higher than in the non-COPD group (26).
Although COPD is a well-known risk factor for lung cancer, a retrospective analysis of the National Health Insurance Service–National Sample Cohort in Korea indicated that COPD may also be an independent risk factor for cancers developing outside the lungs, such as colorectal and liver cancers, irrespective of smoking status (27). A clinical trial of 666 subjects (256 patients with liver diseases and 410 patients without liver diseases) showed that the prevalence of COPD was higher in patients with liver diseases (28). Furthermore, the prevalence of aseptic inflammation of the gallbladder is associated with COPD exacerbations (29).
Gastroesophageal reflux disease (GERD) was one of the most common comorbidities of COPD and was associated with exacerbations in patients with COPD in a national cross-sectional cohort study that included data from 141,057 patients with COPD using the Korean National Health Insurance Database (30). A longitudinal cohort study in the United Kingdom, following two groups of patients for 5 years, revealed that the relative risk of COPD diagnosis in patients with GERD was 1.17, and the relative risk of GERD diagnosis in patients with COPD was 1.46 (31). Cigarette smoking is a risk factor for both GERD and COPD in the general population (32, 33). Nicotine may affect esophageal sphincter tone and esophageal clearance and induce relaxation of the muscle of the esophageal sphincter, possibly increasing the duration of acid exposure and frequency of reflux events (34, 35). The rate of COPD exacerbations is twice as high in patients with GERD symptoms in comparison with those without GERD symptoms (36). A potential pathogenic mechanism for these COPD exacerbations might be related to the microaspiration of gastric contents and/or vagal irritation from gastroesophageal reflux, which may constitute airway irritants (37).
There is also an association between COPD and periodontitis. A meta-analysis demonstrated that patients with COPD have worse periodontal health status than patients without COPD, evidenced by more clinical attachment loss, gingival tissue inflammation, and bleeding (21). On the other hand, poor oral health is considered to be a contributing factor to the development of respiratory diseases (38).
In the next section, we discuss the interactions between the respiratory and gastrointestinal systems and their possible mechanisms of involvement in COPD, including changes in immunological and systemic inflammation, epithelial barrier function, hypoxia-related and oxidative stress processes, and the composition of the microbiota (Figure 1) (39). In addition, clinical evidence for the existence of the gut–lung axis in COPD is summarized in Table 1 (clinical part).
Figure 1.
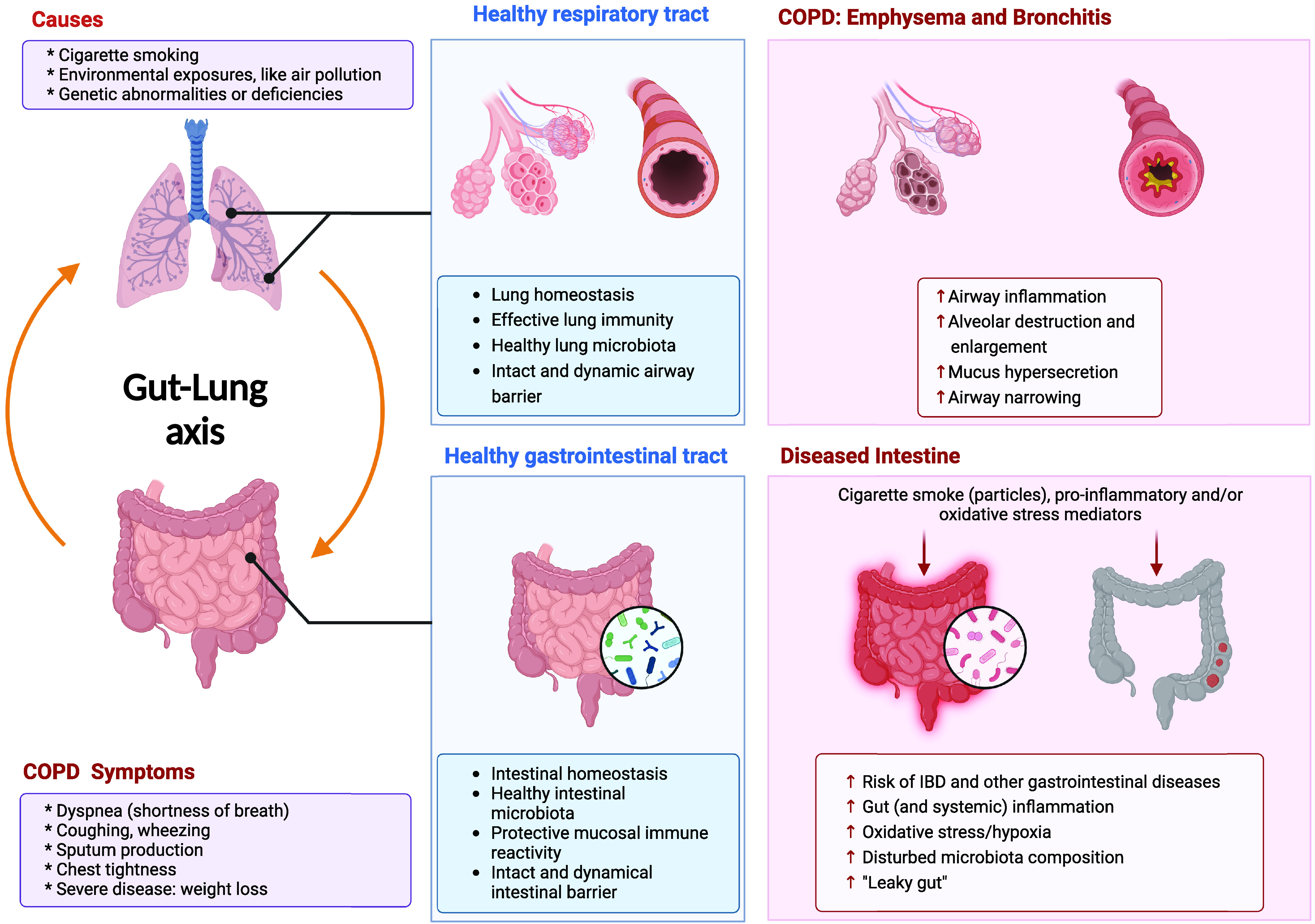
Background of chronic obstructive pulmonary disease (COPD) and the gut–lung axis in COPD. COPD, including emphysema and bronchitis, causes shortness of breath, coughing, and sputum production and is triggered mainly by cigarette smoking. COPD is associated with gastrointestinal symptoms. External (e.g., cigarette smoking) and internal (e.g., proinflammatory and oxidative stress mediators) danger signals can induce gastrointestinal dysfunction, including IBD, and other gastrointestinal diseases, such as colorectal cancer, gastesophageal reflux disease, periodontitis, and gallbladder disease. IBD = inflammatory bowel disease. Created with BioRender.com (39).
Table 1.
Clinical Evidence for Gut–Lung Axis in Chronic Obstructive Pulmonary Disease
| Target Subjects | Study Design | Effects on Intestine/Mortality | References |
|---|---|---|---|
| Patients with COPD Non-COPD control subjects | Population of Québec, 2001–2006 | The incidence of CD and ulcerative colitis was 55% and 30% higher in patients with COPD than in the general population, respectively The highest incidence rate for COPD cohort was at ages 50–59 yr |
7 |
| COPD Asthma–COPD Non-COPD control subjects | Health databases in Québec, age ⩾ 65 yr, without a private drug plan as control Residents received one or more prescriptions for a specific respiratory medication from 1990 to 2007, age ⩾ 41 Newly developed IBD in patients with COPD/asthma–COPD |
In patients with asthma–COPD, IBD increased the risk of mortality from respiratory conditions In patients with COPD, IBD increased the risk of death of digestive conditions |
17 |
| Patients with COPD Non-COPD control subjects | National Health Insurance Database of Korea Patients received prescriptions for COPD medications at least twice in 2009 |
The prevalence of GERD in patients with COPD was 28% GERD increased the risk of hospitalization in patients with COPD |
30 |
| Patients with COPD Non-COPD control subjects | Smokers, former smokers, and nonsmokers of the Korean population in 2002; ages 40–79 yr | Higher incidence of colorectal cancer in patients with COPD regardless of smoking status | 27 |
| Patients with COPD Non-COPD control subjects | Database from the Ewha Womans University School of Medicine, Mokdong Hospital (Seoul, Republic of Korea), January 2003 to April 2012 Subjects ⩾40 yr of age who underwent postbronchodilator spirometry Age and sex matched between groups |
The prevalence of colorectal adenomatous polyps was 39% (98 of 251) in the non-COPD group and 66% (54 of 82) in the COPD group Among 54 patients with adenomatous polyps in the COPD group, 47 had tubular adenoma, and 7 had villous adenoma |
26 |
| Patients with COPD Non-COPD control subjects | Meta-analysis from PubMed and Embase | Patients with COPD had worse periodontal health status, including deeper periodontal pockets, high degrees of clinical attachment loss, worse oral hygiene, more inflammation and bleeding in the gingival tissue, and a lower number of remaining teeth | 55 |
| Patients with chronic diseases | No diagnosis of respiratory diseases; ⩾40 yr of age | High COPD prevalence in patients with liver disease
|
28 |
| Patients with COPD Healthy control subjects | Moderate COPD (mean FEV1, 55 ± 3% predicted) | Functional alterations in the gastrointestinal tract
|
9 |
| Patients with CD Healthy control subjects | Current smokers, former smokers, and never-smokers with CD Age and sex matched between groups |
Current and former smokers with CD had higher systemic hypoxia and colonic remodeling than nonsmoker patients with CD
|
14 |
| Patients with COPD Healthy controls | Patients with COPD with GOLD stages 1 and 2 severity Patients with COPD with GOLD stages 3 and 4 severity |
Altered microbial diversity and composition
Lower concentrations of short-chain fatty acids |
72 |
| Patients with COPD Healthy controls | Patients with COPD with FEV1 < 80% predicted and FEV1:FVC ratio < 0.7; age > 40 yr with history of smoking Healthy control subjects with FEV1 > 80% predicted and FEV1:FVC ratio > 0.7; age > 40 yr without history of cardiac or respiratory disease and with normal lung function |
Fecal microbiome and metabolome of patients with COPD were distinct from those of healthy individuals
|
74 |
Definition of abbreviations: CD = Crohn’s disease; CI = confidence interval; COPD = chronic obstructive pulmonary disease; GERD = gastroesophageal reflux disease; GOLD = Global Initiative for Chronic Obstructive Lung Disease; IBD = inflammatory bowel disease; S. = Streptococcus; VEGF = vascular endothelial growth factor.
Systemic Inflammation
Keely and colleagues showed that exaggerated innate immune responses, characterized by increases in systemic proinflammatory mediators such as IL-6 and TNF-α (tumor necrosis factor-α), can contribute to the development of COPD (13). There is clear evidence that immune dysfunction precedes symptoms of intestinal disorders (40). The circulating proinflammatory mediators in patients with COPD may drive cross-organ inflammation; for example, systemic IL-6 together with pulmonary TGF-β (transforming growth factor-β) can trigger T-helper cell type 17 (Th17)–polarized inflammatory responses in the intestine (13). A prospective case–control study showed that circulating concentrations of IL-6 and C-reactive protein (CRP) before diagnosis were associated with the risk of CD and UC (40). In addition, raised plasma concentrations of CRP and IL-6 are present in patients with COPD (41). TNF-α is believed to play a central role in the pathophysiology of COPD and IBD (13, 42). TNF-α inhibitors, such as infliximab, adalimumab, and etanercept, are currently in use to achieve remission, to avoid disease progression, and to minimize surgical resections in patients with IBD (43–45). Elevated concentrations of TNF-α are observed in peripheral blood, bronchial biopsy samples, induced sputum, and bronchoalveolar lavage fluid (BALF) of patients with COPD (42). Unfortunately, contradictory results have been observed with anti-TNF therapy in COPD (46, 47).
Ahn and colleagues hypothesized that the spillover of exaggerated inflammation in COPD could lead to systemic consequences in other organs, such as carcinogenesis in the intestines (27). There is an intimate relationship between malignant cells and their inflammatory microenvironment (48). Cigarette smoke–induced chronic inflammation is associated with various proinflammatory cytokines, such as TNF-α, IL-1, and IL-6, and chemokines, such as CXCL8 (C-X-C motif chemokine ligand 8), which are capable of promoting tumor growth, tumor adhesion, and invasion (49). Cigarette smoke ingredients, such as nicotine, can directly activate the nicotinic acetylcholine receptors (nAChRs) on cancer cells and induce the release of growth factors, such as VEGF (vascular endothelial growth factor) and IL-1β, into the tumor microenvironment, which can increase tumor angiogenesis and therefore promote tumor growth (49). These activated inflammatory mediators, like TNF-α and IL-1, are also observed in peptic ulceration induced by increased numbers of Helicobacter pylori bacteria, which may enhance COPD progression (50).
Viglino and colleagues suggested that systemic inflammation, including TNF-α, also seems to play a role in the development of nonalcoholic fatty liver disease in patients with COPD (51). Moreover, IL-8 serum concentrations are significantly increased in patients with chronic liver disease (52) as well as in patients with COPD and smokers (53). IL-8 upregulation in bronchial biopsy samples from patients with COPD is associated with severe exacerbations and increased neutrophil recruitment (54).
Currently, there are no proper plasma or serum biomarkers to investigate the systemic effects in COPD-induced GERD (50).
Dysfunctional immune cells and inflammatory hyperreactivity in patients with COPD may prevent the host from effectively killing bacteria in the periodontium and thereby increase periodontal inflammation (55). In addition, COPD and periodontitis share common risk factors, including cigarette smoking (33). Zhou and colleagues recently found that periodontal treatment in patients with COPD improved lung function and decreased the frequency of COPD exacerbations (56). We assume that a disruption in the composition of the oral microbiota after periodontal treatment may play a role in the remission of COPD exacerbations (56, 57).
It can be hypothesized that excessive proinflammatory mediators produced in the lungs of patients with COPD affect the intestine through the systemic circulation, contributing to intestinal disease. Correspondingly, continuous intestinal inflammation can also exacerbate lung diseases by the presence of inflammatory mediators in the systemic circulation. During inflammation, the gut-associated lymphoid tissue regulates lymphocyte trafficking from intestinal tissue through the systemic circulation, which reflects the bronchus-associated lymphoid tissue, and these lymphocytes from the intestine and the lungs migrate to other mucosal sites as part of their shared mucosal immune system (13). Moreover, inflammatory mediators, such as circulating TNF-α, have been strongly implicated in comorbidities associated with COPD (58) and have been implicated in the pathogenesis of both COPD and IBD (16).
In addition, the concentrations of acrolein and other aldehydes, harmful volatile constituents of cigarette smoke, in saliva and exhaled breath condensate of heavy smokers are about 10-fold higher compared with those in healthy persons (59, 60). Therefore, soluble cigarette smoke constituents may directly reach the gastrointestinal tract after swallowing or via the systemic circulation, which can cause gastrointestinal mucosal injury and a delay in ulcer repair (61). Many cytotoxic substances of tobacco smoke, such as nicotine, enter the bloodstream and affect the intestine. Lindell and colleagues showed that the nicotine concentration in gastric fluid was >50 times higher than in plasma on smoking days (62).
Epithelial Barrier Dysfunction, Oxidative Stress, and Hypoxia
The integrity of the epithelial barrier is essential for maintaining respiratory and intestinal mucosal health. Cigarette smoke impairs the airway epithelial barrier function and induces increased permeability, which was more common in smokers with COPD compared with smokers with normal lung function (12, 63). Further downregulation of apical junctional complex gene expression in airway epithelium was observed in smokers with COPD compared with healthy smokers (63). Cigarette smoke can promote the production of endogenous reactive oxygen species (ROS) by airway epithelial cells and pulmonary immune cells (12). The ongoing production of ROS during cigarette smoke exposure and exacerbations will result in a substantial reduction in the general antioxidant capacity in patients with COPD (64), which may affect intestinal disorders.
The unmet metabolic demand (hypoxia) in patients with COPD during daily activities results in an increase in intestinal permeability and causes enterocyte damage (9). It has been described that chronic cigarette smoking changes microcirculation and significantly diminishes blood flow to the gastrointestinal mucosa, suggesting smoking-associated intestinal hypoxia, which may stimulate the development of inflammatory diseases (14, 20).
Elevated serum concentrations of VEGF, a marker of systemic hypoxia, have been reported in smokers as well as in patients with CD (65, 66). Fricker and colleagues showed the effects of smoking on increasing serum VEGF in patients with CD, which supports the hypothesis that smokers with impaired gas exchange and systemic hypoxia have a higher risk of developing CD (14).
In addition, patients with chronic hepatitis C who smoke may have more hepatic fibrosis, whereas cigarette smoke–induced hypoxia and associated increased VEGF and VEGF-D concentrations may be involved in the molecular mechanisms of fibrogenesis (67). Furthermore, smokers appear to be at higher risk of becoming infected with H. pylori, a major factor in the pathogenesis of peptic ulcer disease. This increased risk may be related to the adverse effects of smoking on antioxidants or the immune system (68).
COPD-related systemic hypoxemia also negatively affects the status of patients with esophageal cancer by accelerating inflammation, undernutrition, and angiogenesis (69). Hypoxia stabilizes HIF-1α (hypoxia-inducible factor-1α), leading to the expression of genes that are involved in tumor vascularization, metastasis and migration, cell survival, and chemoresistance, as observed in colorectal cancer (70).
Overall, we hypothesize that the toxic effects of cigarette smoke, inhaled ROS, and/or systemic hypoxia may extend systemically to the intestinal epithelium, resulting in intestinal epithelial dysfunction.
Changes in Microbiota Composition
Gut microbiota dysbiosis has been observed in patients with COPD compared with healthy individuals (71). A clinical study with stool samples from a cohort of 73 healthy humans and 99 patients with COPD showed that patients with COPD had a relatively lower proportion of Bacteroidetes and a higher proportion of Firmicutes compared with healthy control subjects. At the family level, there was a difference in the relative abundances for Fusobacteriaceae, Prevotellaceae, and Bacteroidaceae (72). Prevotella were also enriched in the stool of patients with COPD. Fecal matter from these patients with COPD and healthy control subjects was homogenized for fecal microbiota transplantation in male C57BL/6 recipients and in C57BL/6 mice exposed to smoke from biomass fuel to induce COPD-like changes. Interestingly, mice receiving fecal microbiota from patients with stages 3 and 4 COPD showed reductions in body weight, higher pulmonary inflammation, and increases in IL-1β and TNF-α concentrations in plasma compared with control mice. In addition, a higher number of T lymphocytes and a lower number of B lymphocytes in blood were observed in mice that received fecal microbiota from patients with COPD. Moreover, accelerated declines in lung function and emphysematous changes were observed in the mouse recipients of fecal microbiota from patients with COPD during biomass fuel smoke exposure. In conclusion, the altered gut microbiota of patients with COPD accelerated the development of COPD in mice (72).
Recently, a large multicenter study showed that the severity of COPD is associated with a decreased abundance of Prevotella and an increased abundance of Moraxella in the lung microbiome in concert with downregulation of genes promoting epithelial defense and upregulation of proinflammatory pathway responses (73). In addition, the abundance of Streptococcus in the airways was higher in patients with mild to moderate COPD than in healthy individuals (73).
The increased abundance of several Streptococcus species, including S. parasanguinis_B and S. salivarius, was also found in the intestine in patients with COPD (74). However, there were no differences between the gut microbiome composition of nonsmokers with COPD and smokers with COPD, supporting that this is a disease-associated phenotype rather than a phenotype driven by the influence of cigarette smoking on the gut microbiome (74).
Translocation of gut microbiota may play a role in the gut–lung cross-talk in patients with COPD. Dysbiosis of the intestinal microbiota has been shown to disturb the composition of the respiratory microbiota through changes in circulating inflammatory cytokines and via translocation of intestinal microbiota to the airways (16). Translocation of gut microbiota to the airways might be related to the leaky gut induced by cigarette smoke, although this has not yet been confirmed in patients with COPD (16). Pathogens frequently colonize the airways, which is associated with exacerbations in patients with COPD, promoting COPD development by amplifying lung and systemic inflammation. We speculate that the disruption of the lung epithelial barrier function may lead to the translocation of pathogenic bacteria. Translocated pathogens and pathogen-associated virulence factors may also enter the gastrointestinal tract, causing intestinal inflammation and related diseases.
In addition, the coordination of ventilation and swallowing may be subverted in patients with COPD (75), possibly resulting in the simultaneous microaspiration of the pharyngeal or oral microbiota into the respiratory and gastrointestinal tract. Another possible speculation is that the same embryonic origin and the structural similarities of gastrointestinal and respiratory tract may lead to similar responses and changes of the microbiota after systemic inflammation and immune responses caused by COPD (6).
Changes in Microbial Metabolites
Components of the lung microbiome and metabolome have potential collaborative roles in COPD pathogenesis (76). Accumulating evidence indicates that short-chain fatty acids (SCFAs), derived from intestinal microbial fermentation of indigestible foods, are the most prominent immunomodulatory metabolites and well-studied bacterial metabolites in the gastrointestinal tract that exert their functions along the gut–lung axis (77). The effects of microbiota-derived SCFAs are not limited to the intestinal compartment, and SCFAs disseminate from the gut into the bloodstream, reaching the bone marrow to promote bone marrow hematopoiesis, leading to the resolution of airway inflammation and maintenance of healthy homeostasis in the lung (78). Moreover, as SCFAs produced in the gut can enter the circulation, these metabolites may have direct effects on the lungs or may even be produced by the lung microbiota (79). Patients with COPD have lower concentrations of fecal SCFAs compared with healthy individuals, and patients’ COPD severity correlated with reductions in SCFA concentrations, including acetic acid, isobutyric acid, and isovaleric acid (72), which might be a subsequent event of a disrupted gut microbiota. Increasing SCFA concentrations and the abundance of beneficial gut microbiota by dietary fiber intake might be one of the solutions to mitigate COPD and its related comorbidities.
Limitations
Although there is some clinical evidence for a connection between the lungs and the gut in COPD, additional clinical information regarding the severity of disease, controls, active smoking/passive smoking, and smoking rate and duration would improve the quality of trials in the future.
Animal Studies: Gut–Lung Axis in COPD Models
Immunological Changes and Systemic Inflammation
Besides the clinical evidence showing an interaction between the gut and the lungs in COPD, numerous in vivo studies have been undertaken to unravel the underlying mechanisms of this bidirectional connection.
Female Balb/c mice exposed to mainstream cigarette smoke for 72 days demonstrated gastrointestinal changes, including changes in intestinal histomorphology and immune network for IgA production (80). Another study showed that female C57BL/6J mice exposed to cigarette smoke for 2 months and treated with dextran sodium sulfate (DSS) exhibit exaggerated intestinal inflammation, with elevated Th17 cells, type 3 innate lymphoid cells, and neutrophil responses in the intestine. This might be due to the increased systemic susceptibility to inflammation by a higher number of Th17 cells and neutrophils in the circulation after cigarette smoke exposure (81). Moreover, there is an accumulation in the number of CD11b+ (cluster of differentiation 11b) dendritic cells (DCs) in the intestinal Peyer’s patches of male C57BL/6 mice after 24 weeks of cigarette smoke exposure (82). Interestingly, lung DCs, like CD103+ mesenteric lymph node DCs, can stimulate the translocation of T cells into the gastrointestinal tract, probably via upregulating the gut-homing integrin α4β7 in vivo (83). In contrast, cigarette smoke seems to play a protective role in UC, as smoke exposure for 4 weeks reversed the CD4+/CD8+ ratio in blood and colon and increased B cells in the colon of C57BL6/cmdb mice with DSS-induced colitis (84). It has been described that cigarette smoke exposure for 27 days can reduce DSS-induced colitis severity in male C57BL/6 mice and modulates DSS-induced dysbiosis of specific bacterial genera, such as Akkermansia, Intestinimonas, and Lactobacillus, which may contribute to resolving intestinal inflammation or accelerate recovery. In addition, the activation of the cholinergic antiinflammatory pathway by nicotine might be one of the explanations for the protective role of cigarette smoke in UC (85). Qin and colleagues indicated that nicotine (subcutaneous injection of nicotine for 7 consecutive days) exerts its protective action in murine DSS colitis by upregulating microRNA-124 expression and inhibiting STAT3 (signal transducer and activator of transcription 3) activation (86).
Possibly, these contradictory observations regarding the effect of cigarette smoke exposure on murine colitis might be related to the different procedure/duration of cigarette smoke exposure and DSS treatment.
Moreover, the controversial effects of nicotine appear to be site specific, as nicotine treatment through drinking water for 2 weeks exacerbated jejunum damage but reduced colonic inflammation in male C57/BL10 IL-10−/− mice (87).
Elevated systemic mediators, such as chemokines/cytokines, might play a role in the lung–gut cross-talk in COPD. Verschuere and colleagues showed that the upregulation of CCL9 (C-C motif chemokine ligand 9) and CCL20 induced by cigarette smoke exposure may play a role in the increased epithelial apoptosis in the Peyer’s patches and the accumulation of CD11b+ DCs and CD4+ and CD8+ T cells in the Peyer’s patches in the ileum (82). These results suggest that cigarette smoking causes alterations in the epithelium lining of the Peyer’s patches, leading to changes in the composition of the underlying immune cell population, possibly via the CCL9–CCR1 (C-C motif chemokine receptor 1) and CCL20–CCR6 pathways.
Epithelial Barrier Dysfunction, Oxidative Stress, and Hypoxia
Animal studies have demonstrated that cigarette smoke–induced intestinal barrier dysfunction and subsequent inflammation of the intestinal mucosa may be one of the mechanisms of intestinal damage in COPD (88, 89). Dysfunctional and structural changes in the intestinal mucosal barrier, including neutrophil infiltration, epithelial shedding, reduced tight junction protein expression (occludin and ZO-1 [zonula occludens 1]), and increased intestinal expression concentrations of TNF-α, IFN-γ, and IL-8, were observed in Sprague-Dawley rats with emphysema after 6 months of side-stream cigarette smoke exposure (88). Male C57BL/6 mice exhibited elevated intestinal inflammation and reduced intestinal Paneth cell integrity after intragastric administration of cigarette smoke condensate (three times per week for 2 wk), resulting in reduced antimicrobial peptide production and bactericidal capacity (90). Mainstream cigarette smoke exposure for 24 weeks induced intestinal inflammation in male C57BL/6 mice, which was related to altered epithelial mucus profiles, including increased mRNA expression of Muc2 (mucin 2, oligomeric mucus/gel-forming) and Muc3 in the ileum and Muc4 in the distal colon (91).
Impaired gas exchange (marked reduction in diffusion factor for carbon monoxide) induced by 8 weeks of mainstream cigarette smoke exposure caused systemic hypoxia, as measured by serum VEGF and intestinal hypoxia throughout the mucosal layer in female C57BL/6 mice, which drives angiogenesis and intestinal epithelial barrier dysfunction, resulting in increased risk and severity of CD (14). Systemic and local ischemia might be caused by oxygen-free radicals present in inhaled smoke, as described in the previous part (Gastrointestinal Tract Disorders Associated with COPD). In addition, 14 weeks of whole-body cigarette smoke exposure elevated HIF-1α expression in the male Wistar rat small intestine, which was associated with increased oxidative stress and apoptosis, leading to the destruction of intestinal tight junctions (92).
Increased ROS generation and HIF-1α concentrations are also related to the development of colorectal cancer (93). Recently, a stress-activated mitochondrial protease (OMA1 [OMA1 zinc metallopeptidase]) has been found to promote colorectal cancer development by increasing ROS generation and stabilizing HIF-1α under hypoxia. OMA1 knockout suppresses colorectal cancer development in murine azoxymethane (AOM)/DSS and xenograft models of colorectal cancer (94).
Moreover, exposure to cigarette smoke for 2 months caused decreased colonic diameter and an increased frequency of colonic contractions in male Balb/c mice; it is possible that nicotine contributed to these changes in gut function by acting on enteric neurons expressing nAChR to contract intestinal smooth muscle (95).
Antioxidants might have therapeutical potentials in gastrointestinal dysfunction in patients with COPD. The antioxidant ebselen prevents the reduction in colonic diameter in mice exposed to cigarette smoke for 2 months (95).
Changes in Microbiota Composition
Papoutsopoulou and colleagues reviewed the deleterious influence of smoking on the human gut microbiota (96), but information is scarce in animal species. Cigarette smoke exposure for 4 weeks reduces the relative abundance of Bacteroidales but increases the presence of Firmicutes in C57BL/6 mouse fecal microbiota, while a high-fat diet exacerbates the cigarette smoke–induced reduction in gut microbiota diversity (97). Interestingly, fecal microbiota from control, antibiotic-treated, ampicillin-treated, or vancomycin-treated mice reversed emphysema features of cigarette smoke–recipient C57BL/6 mice, in which the commensal bacterium Parabacteroides goldsteinii might play a key role. The LPS derived from P. goldsteinii exhibits antiinflammatory properties, which can ameliorate experimental emphysema by acting as an antagonist of the TLR4 (Toll-like receptor 4) signaling pathway (98).
An increased abundance of gut Lachnospiraceae species was found in male C57BL/6 mice exposed to cigarette smoke for 24 weeks, which might be one of the contributing factors leading to aberrant inflammation and reduction of cellular physiological activities in the colon (91, 98). Consistently, an increased abundance of Lachnospiraceae species was also reported in other chronic inflammatory diseases, such as IBD, irritable bowel syndrome with diarrhea, and obesity (74, 98). Fecal microbiota transplantation from healthy control mice or mice receiving a high-fiber diet attenuated emphysema development in C57BL/6 mice via local and systemic inhibition of inflammation and changes in gut microbiota composition, including the increased amounts of Bacteroidaceae and Lachnospiraceae (99).
Intraoral cigarette smoke exposure for 3 months reduced the abundance of potentially beneficial bacteria (i.e., Clostridium, Turicibacter) and increased the abundance of potentially harmful bacteria (i.e., Desulfovibrio, Bilophila) in rats (100). Clostridium and Turicibacter are important commensal bacteria, which have the ability to regulate the gut barrier and inflammatory/immune responses related to intestinal diseases, such as IBD (100–102).
The genera Desulfovibrio and Bilophila likely participate in the development of colorectal cancer by producing hydrogen sulfide and promoting chronic inflammation (103). Cigarette smoke exposure for 28 weeks can promote colon tumorigenesis by inducing gut microbiota dysbiosis in a carcinogen AOM-induced mouse model of colorectal cancer. This gut microbiota dysbiosis was characterized by enrichment of Eggerthella lenta and depletion of Parabacteroides distasonis and Lactobacillus species and increased taurodeoxycholic acid, a bile acid metabolite, in the colon, resulting in gut barrier dysfunction and activation of oncogenic and proinflammatory signaling pathways. Moreover, transplanting stool from smoke-exposed mice into germ-free mice leads to enrichments in E. lenta and taurodeoxycholic acid, impaired colonic epithelium, and increased activation of proinflammatory pathways in recipient mice (104). Manipulation of the gut microbiota might represent a promising prophylactic strategy against intestinal disorders in patients with COPD.
Changes in Microbial Metabolites
Cigarette smoke exposure for 4 weeks significantly decreased SCFA concentrations, including acetic acid, propionic acid, butyric acid, and valeric acid, in the cecal content of Wistar rats (105), whereas 3 months of smoking exposure reduced propionate as well as amino acid and carbohydrate metabolism in the cecal and colonic contents of Wistar rats (100).
Gut microbiota dysbiosis induced by cigarette smoke plays a protumorigenic role in colorectal cancer. AOM-treated C57BL/6 mice were exposed to cigarette smoke for 28 weeks, and the fecal metabolic profile in smoke-exposed mice was significantly different from that of control mice. Bile acid biosynthesis was the top enriched pathway in cigarette smoke–exposed mice compared with smoke-free mice. This study showed that smoke-induced gut microbiota dysbiosis altered gut metabolite profiles and impaired gut barrier function, which might activate oncogenic MAPK (mitogen-activated protein kinase)/ERK (extracellular signal-regulated kinase) signaling in colonic epithelium (104).
A whey peptide–based enteral diet inhibited elastase-induced emphysema and lowered the total cell counts in BALF of female C57BL/6 mice, which might be related to the increase in cecal SCFAs (106). Interestingly, a mixture of nondigestible oligosaccharides prevented the emphysema-like changes induced by LPS and decreased the LPS-induced neutrophil influx by >60% in Balb/c mice (107). Discontinuous feeding with a fiber-free diet accelerated emphysematous lesions after 8 weeks of cigarette smoke in spontaneous hypertension male rats, as observed by enlarged and disrupted alveolar walls compared with rats fed the control diet. Moreover, the cigarette smoke–related decrease in total organic acid and acetic acid concentrations was also accelerated by discontinuous feeding with a fiber-free diet (108).
These studies confirm that shaping the gut microbiota composition and altering the production of microbial metabolites with specific dietary supplementation might have the potential for the treatment or prevention of COPD and its (intestinal) comorbidities.
Limitations of Animal Models
Issues that are difficult or impossible to study in humans for ethical, safety, or logistical reasons can be addressed by using in vivo models. A major limitation of cigarette smoke exposure models is that these models exhibit relatively mild pathological changes of COPD, and none of the models shows the progressive disease observed in humans with Global Initiative for Chronic Obstructive Lung Disease stage 3 or 4 COPD (109). In addition, because of the differences in lung anatomy and physiology between mice and humans, it is difficult to capture both chronic bronchitis–related and emphysematous changes in a single model. An ideal animal model of COPD would reproduce a combination of the principal features of the human disease, namely, chronic bronchitis, mucus hypersecretion, small airway remodeling, emphysema, impaired lung function, and systemic comorbidities. Unfortunately, all of the described animal models meet only some of these criteria, and in the majority of studies described in this review, animal models of cigarette smoke–induced emphysema were used (110). Therefore, we must be careful with the translation of preclinical research to clinical practice.
Lessons Learned from In Vitro Studies
Although it is difficult to integrate several organs (gut and lungs) in an in vitro system to mimic the physiological function of the human body, several in vitro studies have contributed to the knowledge of the underlying mechanisms of intestinal complications in COPD.
Human colorectal cancer (Caco-2) cells exposed to cigarette smoke extract for 3 hours showed reductions in cell necrosis, claudin-1, and E-cadherin expression and increases in microRNA-21 (a diagnostic marker for colorectal carcinoma), cytoskeleton rearrangement, cell motility, and invasiveness, which may contribute to colorectal cancer progression (111). Moreover, cigarette smoke condensate reduced cell viability and upregulated total ROS, mitochondrial ROS, and superoxide radicals in HCT-15 and HT-29 colon cells within 48 hours (112).
Increased oxidative stress also contributes to reduced gene and protein expression of tight and adherens junctions in human bronchial epithelial cells, which may increase susceptibility to inhaled pathogens and environmental stressors (12, 113). The presence of exogenous ROS in cigarette smoke can induce the fragmentation of hyaluronic acid in human tracheobronchial epithelial cells, leading to impaired airway barrier integrity by binding to the hyaluronan receptor layilin and mediating the RhoA (Ras homolog family member A)/Rho kinase–dependent decrease in E-cadherin expression (114).
Papoutsopoulou and colleagues reviewed the deleterious consequences of smoking on various cell types and physiological processes within the intestinal epithelium, including the direct and indirect effects of cigarette smoking on various parameters, such as oxidative damage, impairment of intestinal barrier, and immune cell function (96).
The biology of the oral epithelium is also altered by cigarette smoking, as observed by a cigarette smoke–induced reduction in the number of early apoptotic keratinocytes and an increase in the number of late apoptotic cells (115).
Among all the components described, nicotine as the major and highly addictive component of cigarette smoke is of great interest in several in vitro studies. Nicotine-derived nitrosamine ketone, a highly carcinogenic tobacco-specific nitrosamine, stimulates colon cancer HT-29 cell proliferation through β-adrenoceptors, and the effect was abolished by β1- or β2-adrenoceptor antagonists (116). Moreover, nicotine was found to reduce TNF-α release by LPS-stimulated human leukemia (THP-1) cells in vitro (117) and to inhibit the ex vivo production of IL-2 and TNF-α by human peripheral blood lymphocytes stimulated with phytohemagglutinin (118). An in vitro experiment using HT-29 cells showed that nicotine can inhibit TNF-α–induced IL-8 release, possibly via binding to the α7 subunit of the nAChR (119). Nicotine may bind to nAChR and prevents NF-κB (nuclear factor-κB) signaling, thereby preventing the release of HMGB1 (high-mobility group box 1) and inhibiting the release of proinflammatory cytokines such as TNF-α (120). The protective effect of nicotine on epithelium may partly explain the protective effect of cigarette smoking on UC, as observed in clinical studies reviewed by Berkowitz and colleagues (20).
In vitro and ex vivo studies showed that SCFAs have the ability to improve the regulation of inflammation, carcinogenesis, intestinal barrier function, and oxidative stress (121). SCFAs are known to strengthen gut barrier function (122) and to regulate inflammation and oxidative stress via SCFA signaling mechanisms, such as the promotion of histone acetylation and activation of G protein–coupled receptors (78). The SCFA butyrate enhances the intestinal barrier by facilitating a tight junction in human colonic Caco-2 cells (123, 124) and small intestine porcine IPEC-J2 cells when exposed to inflammatory conditions (125). SCFAs even showed prophylactic and restorative effects on airway epithelial barrier function, which might be related to increased ZO-1 expression (126).
All preclinical (in vivo and in vitro) evidence for the connection between the lungs and the gut in COPD is summarized in Table 2, and relevant studies regarding the effects of intestinal metabolites and dietary fiber in COPD are summarized in Table 3.
Table 2.
Preclinical Evidence for Gut–Lung Axis Using In Vivo and In Vitro Studies
|
In Vivo Studies | |||
|---|---|---|---|
| Target Subjects | Experimental Design | Effects on Intestine/Intestinal Cells | References |
| Male Sprague-Dawley rats 8 wk old | Air/CS exposure 2 h/d 5 d/wk 6 mo |
Dysfunctional and structural changes in the intestinal mucosal barrier
Intestinal inflammation
|
88 |
| Male C57BL/6 mice 7–8 wk old | Intragastric CS condensate or PBS 3 times/wk 2 wk |
Intestinal inflammation
|
90 |
| Male C57BL/6 mice 8–9 wk old | Air/CS exposure 5 cigarettes/round 4 times/d 5 d/wk 24 wk |
Intestinal inflammation
Altered bacterial activity and community structure
|
91 |
| Female C57BL/6 mice | Air/CS exposure 12 cigarettes/round Twice/day 5 times/wk 12 wk |
Systemic and intestinal hypoxia Epithelial barrier dysfunction |
14 |
| Male Wistar rats 6 wk old | Air/CS exposure 5 cigarettes Twice/day 7 d/wk 14 wk |
Hypoxia and oxidative stress
Increased intestinal permeability
Apoptosis |
92 |
| Female C57BL/6J mice 6–8 wk of age | Air/CS exposure 3 cigarettes/d 5 d/wk 2 mo Plus DSS challenge |
Enhanced intestinal pathological innate and adaptive immune responses
|
81 |
| Female Balb/c mice 11–13 wk old | Air/CS exposure 14 cigarettes/d Once/day 7 d/wk 72 d |
Changes in proximal intestinal histomorphology Changes in intestinal immune network for IgA production in distal small intestine Increased fecal sIgA concentrations and enlargement of Peyer’s patches |
80 |
| Male C57BL/6 mice 8–9 wk old | Air/CS exposure 5 cigarettes/round 4 times/d 5 d/wk 24 wk |
Epithelial apoptosis Intestinal inflammation
|
82 |
| Male C57BL/6 mice 6 wk old | Air/CS exposure 15-min CS exposure Twice/day 4 wk Plus normal-food diet/high-fat diet |
CS exposure increased the accumulation of total cholesterol in liver Insulin resistance in the liver of mice with high-fat diet CS exposure altered the microbiota composition
|
97 |
| Female C57BL/6 mice 8–10 wk old | Air/CS exposure 12 cigarettes Twice/day 5 d/wk 12 wk Plus fecal microbiota transplantation (∼1 × 108 cfu in 100 μl PBS) Oral gavage 12 wk |
Altered microbiota composition
Fecal microbiota transplantation restored COPD pathogenesis
|
98 |
| Female and male Wistar rats 5 wk old | Air/CS intraoral exposure 20-min exposure/round Twice/day 3 mo |
Altered microbiota composition
Active smoking might impair amino acid, lipid, and propanoate metabolism function |
100 |
| Male C57BL6/cmdb mice 8 wk old | Colitis induction by DSS Plus air/CS exposure 20 cigarettes/round Once/day 5 d/wk 4 wk |
Decreased severity of DSS-induced colitis
|
84 |
| Male C57Bl/6 mice 8–10 wk old | Air/CS exposure for 27 d Plus colitis induction by DSS |
CS attenuated DSS-induced dysbiosis of specific bacterial genera CS resolved the inflammation and accelerated recovery |
85 |
| Male specific pathogen–free C57BL/6 mice 8–10 wk old | Air/smoke (produced by smoldering China fir sawdust) 40 g/exposure, 3-h periods Twice/day 5 d/wk 20 wk Plus fecal transplantation from patients with COPD Single oral administration of 100 μl per mouse twice/week 20 wk |
Accelerated declines in lung function Severe emphysematous changes Airway remodeling Mucus hypersecretion |
72 |
|
In Vitro Studies | |||
|---|---|---|---|
| Cell Line | Exposures | Effects | References |
| Caco-2 cells | CSE (5% and 10%) Different time points (3 and 24 h) |
Cell necrosis ↓ Claudin-1 and E-cadherin expression ↓ Cell permeability ↑ |
111 |
| HT-29 cells | TNF-α (50 ng/ml) exposure plus nicotine (10−11–10−6 M) | TNF-α–induced IL-8 release ↓ | 119 |
Definition of abbreviations: CCL = C-C motif chemokine ligand; CD = cluster of differentiation; COPD = chronic obstructive pulmonary disease; CS = cigarette smoke; CSE = cigarette smoke extract; Cxcl2 = C-X-C motif chemokine ligand 2; DC = dendritic cell; DSS = dextran sodium sulfate; HIF-1α = hypoxia-inducible factor-1α; ILC3 = type 3 innate lymphoid; Muc2 = mucin 2, oligomeric mucus/gel-forming; PBS = phosphate-buffered saline; sIgA = secretory IgA; Tgf-β = transforming growth factor-β; Th17 = T-helper cell type 17; TJ = tight junction; TNF-α = tumor necrosis factor-α; ZO-1 = zonula occludens 1.
Table 3.
The Effects of Intestinal Metabolites and Dietary Fiber in Chronic Obstructive Pulmonary Disease
| Subjects | Treatment/Diet | Effect in the Lung | References |
|---|---|---|---|
| Patients with COPD 1,557 cases from the Swedish National Patient Register during follow-up (2002–2014) |
Dietary fiber intake was assessed in 1987 and 1997 using a food frequency questionnaire | Long-term high dietary fiber intake (⩾26.5 vs. <17.6 g/d) was associated with a 30% lower risk of COPD Current and ex-smokers with low long-term total fiber intake had a 33-fold or 10-fold higher risk of COPD compared with never-smokers |
131 |
| 63 patients with COPD of GOLD stages 1 and 2 severity 30 patients with COPD of GOLD stages 3 and 4 severity 60 healthy control subjects |
— | Total SCFA concentrations were significantly lower in stool samples of the COPD stages 3 and 4 group compared with healthy subjects and those with COPD stages 1 and 2 Acetic acid, isobutyric acid, and isovaleric acid concentrations were significantly lower in the subjects with COPD stages 3 and 4 |
72 |
| Female C57BL/6 mice 8 wk old | Cigarette smoke exposure 5 d/wk for 4 wk Plus 50 μg (1 μg/μl) of poly(I:C) via nasal aspiration twice a week at 3 and 4 wk High-fiber diet performed with a 4-wk study period |
Mice receiving a high-fiber diet presented less severe emphysema
|
99 |
| Male albino rats of Wistar Kyoto 10 wk old | Cigarette smoke exposure for 4 wk 30 cigarettes for 20 min 5 d/wk |
Cecal concentrations of organic acids, such as acetic acid, propionic acid, butyric acid, and valeric acid, significantly decreased in the cigarette smoke–exposed mice | 105 |
| Wistar rats 5 wk old | Intraoral smoking exposure for 3 mo 20 min per time in the morning and afternoon |
Impaired amino acid, lipid, propanoate metabolism function Microbiota associated with metabolism pathways for some antioxidants and vitamins, such as glutathione, lipoic acid, retinol, taurine, hypotaurine, riboflavin, and cofactors, was enriched in the active smoking group |
100 |
| Female C57BL/6 mice 6 wk old | Elastase-induced emphysema | The whey peptide–based enteral diet group exhibited protective effects in the lung
|
106 |
| Male Balb/c by Jico mice 6 wk old | LPS-induced emphysema Diet with nondigestible oligosaccharides |
Nondigestible oligosaccharides diminished disease progression
|
107 |
Definition of abbreviations: BALF = bronchoalveolar lavage fluid; COPD = chronic obstructive pulmonary disease; GOLD = Global Initiative for Chronic Obstructive Lung Disease; poly(I:C) = polyinosinic:polycytidylic acid; SCFA = short-chain fatty acid.
Conclusions
COPD is a complex disease and is often associated with comorbidities such as intestinal diseases. The concept of the “gut–lung axis” has gained attention with the discovery of the influence of gut health on lung immunity (127). Advances in understanding the potential mechanisms of gut–lung cross-talk have highlighted the bidirectional cross-talk between the gut and the lungs, showing the impact of the lungs on intestinal health and the importance of the intestine in maintaining immune homeostasis. In this translational review, this bidirectional cross-talk between the gut and the lungs in COPD is verified on the basis of clinical, in vivo, and in vitro studies. Overall, the findings of these studies support one another and demonstrate that COPD and/or cigarette smoking can cause intestinal inflammation, barrier disruption, hypoxia, and oxidative stress, as well as microbiota dysbiosis and altered microbial metabolite production. The possible connections between the gut and the lungs and postulated mechanisms for how the gut–lung axis affects COPD are described in Figure 2. Systemic inflammation is considered one of the major causes of gut–lung cross-talk in COPD. Excessive inflammatory mediators generated from the lung can affect gastrointestinal health through the systemic and lymphatic systems. Correspondingly, inflammation in the gastrointestinal tract can also potentiate the development of lung diseases. Moreover, impaired gas exchange capacity in the lung and systemic hypoxia in COPD can lead to gastrointestinal epithelial integrity damage. In addition, gut microbiota dysbiosis in COPD can lead to disruption of the intestinal barrier integrity and can activate local and systemic immune responses, thereby aggravating the immunity and pathology in the lung. SCFAs are produced mainly by the gut microbiota through fermentation of dietary fibers and are known to beneficially influence host homeostasis (128). Although the function of intestinal microbiota in modulating local and systemic immune responses has been extensively studied, the impact of the lung microbiota and its products in regulating immunity has just begun, and further research is needed to better understand host immune–lung microbiome interactions.
Figure 2.
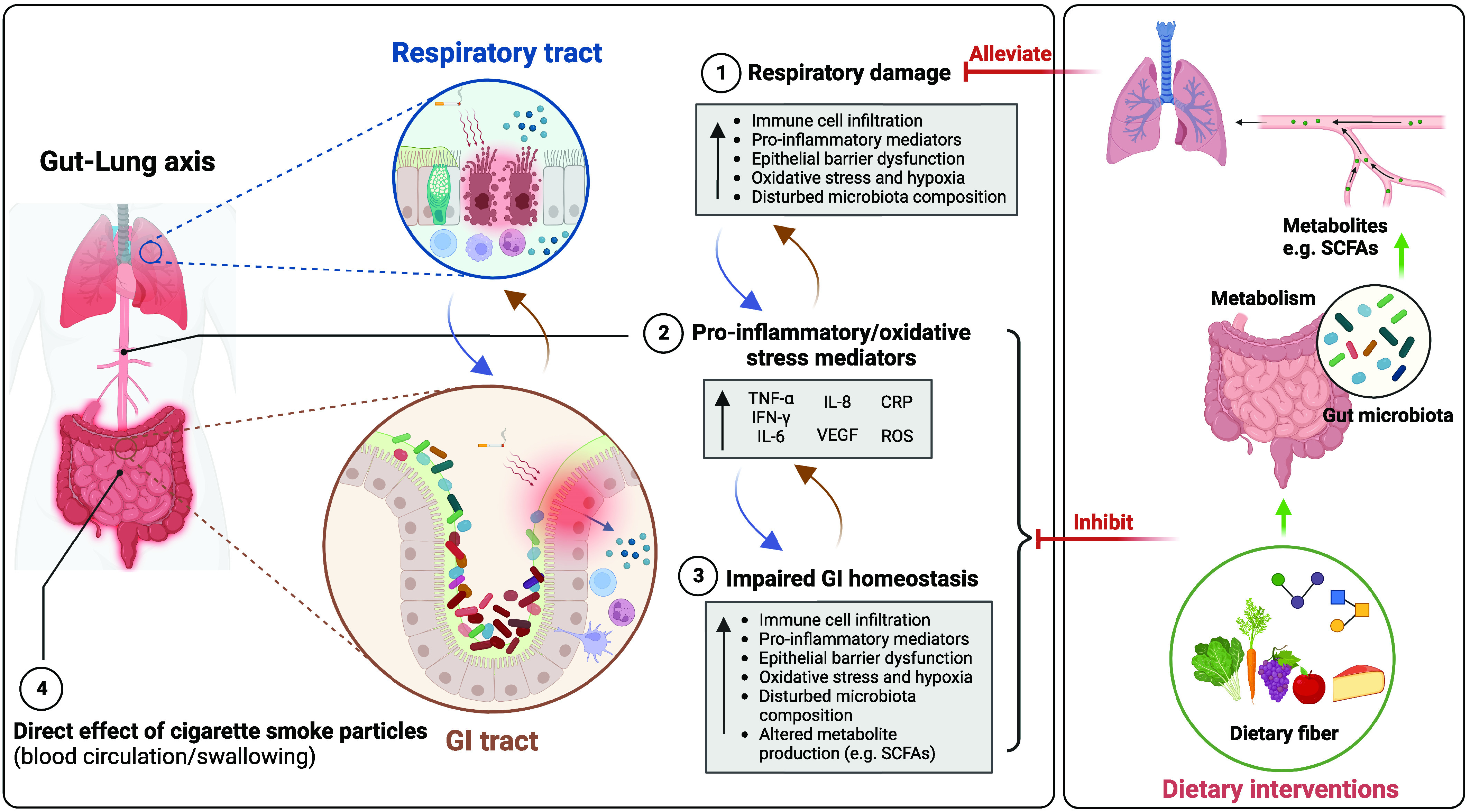
Proposed connections between the gut and lungs in chronic obstructive pulmonary disease (COPD). COPD is characterized by immune cell infiltration, proinflammatory mediators in the lungs, impaired epithelial barrier function, oxidative stress, and hypoxia in the lungs. (1) Damaged and activated lung cells further stimulate the innate immune response through elevated proinflammatory mediators (e.g., TNF-α, IFN-γ, IL-6, IL-8, CRP, ROS) (2). Those proinflammatory mediators (and cigarette smoke particles) can migrate to the gastrointestinal tract, (partly) via the systemic circulation, exacerbating intestinal impairments, including increased inflammatory immune infiltration, epithelial barrier damage, oxidative stress, and hypoxia in the gastrointestinal tract. Moreover, (long-term) cigarette smoking and inflammatory immune cell infiltration in the gut can change the microbiota composition, leading to decreased abundance of health-promoting commensal bacteria (3). In addition, soluble cigarette smoke particles may directly affect the gastrointestinal tract by entering the bloodstream after inhalation, or these particles may reach the gastrointestinal tract due to mucociliary clearance in the airways or direct swallowing of cigarette smoke (4). Impaired gut function not only increases the production and entry of proinflammatory mediators into the systemic circulation but also attenuates nutrient absorption, antioxidant capacity, and protection from pathogens and other environmental stimuli, further exacerbating COPD. Dietary intervention, such as fibers, can manipulate the gut microbiota composition to promote host health, increasing the abundance of health-promoting commensal bacteria, decreasing the permeability of intestinal mucosa, and enhancing the bacterial synthesis of immune-modulating compounds (e.g., SCFAs). This may decrease proinflammatory mediators and alleviate the symptoms in the lung. CRP = C-reactive protein; GI = gastrointestinal; ROS = reactive oxygen species; SCFA = short-chain fatty acid; TNF-α = tumor necrosis factor α; VEGF = vascular endothelial growth factor. Created with BioRender.com (39).
The current management of COPD is focused mainly on suppressing airway inflammation and decreasing symptoms. The purpose of this translational review is not only to emphasize the interaction between the gut and the lung in COPD but also to highlight the possibility of targeting the intestine to improve clinical outcomes in patients with COPD. There is an urgent need for research into the primary prevention of COPD and deceleration of disease progression. The intestine might be a promising target, through bidirectional gut and lung cross-talk, to improve lung health and minimize COPD and related comorbidities. An understudied area of COPD management might be nutritional interventions. On the basis of the literature presented in this review, understanding the importance of the gut–lung axis in COPD, it can be suggested that antiinflammatory and/or nutritional intervention strategies targeting the intestine might be used as a new add-on therapy to suppress the development of COPD and its comorbidities.
Future Perspectives
The existence of the gut–lung axis in COPD has become increasingly clear. The gut microbiome plays an important role in profiling the intestinal and systemic immune system, thereby regulating lung health. Therefore, therapies focusing on gut microbiota manipulation might be considered in patients with COPD (and associated intestinal comorbidities). Dietary fibers, with their prebiotic and immunomodulatory properties, are also capable of increasing SCFA-producing bacteria (129) and exerting antiinflammatory effects in respiratory diseases, such as asthma (101). SCFAs are also potent antiinflammatory and barrier-protective molecules, which may reach the airways via the systemic circulation (130). Moreover, on the basis of a murine study showing that healthy fecal microbiota transplantation attenuates emphysema development via local and systemic inhibition of inflammation and changes in gut microbiota composition (99), fecal microbiota transplantation might represent a new paradigm for the treatment of COPD, and further research in this area is certainly needed.
Footnotes
Supported by Chinese Scholarship Council grant 201706170055 (L.W.) and Fundamental Research Funds for the Central Universities grant 1124007125 (Y.C.).
Originally Published in Press as DOI: 10.1164/rccm.202206-1066TR on March 8, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis . 2018;13:3341–3348. doi: 10.2147/COPD.S176122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaughan A, Frazer ZA, Hansbro PM, Yang IA. COPD and the gut–lung axis: the therapeutic potential of fibre. J Thorac Dis . 2019;11:S2173–S2180. doi: 10.21037/jtd.2019.10.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet . 2012;379:1341–1351. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ritchie AI, Baker JR, Parekh TM, Allinson JP, Bhatt SP, Donnelly LE, et al. Update in chronic obstructive pulmonary disease 2020. Am J Respir Crit Care Med . 2021;204:14–22. doi: 10.1164/rccm.202102-0253UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rutten EP, Spruit MA, Franssen FM, Buurman WA, Wouters EF, Lenaerts K. GI symptoms in patients with COPD. Chest . 2014;145:1437–1438. doi: 10.1378/chest.14-0285. [DOI] [PubMed] [Google Scholar]
- 6. Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut–lung axis. Nat Rev Microbiol . 2017;15:55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 7. Brassard P, Vutcovici M, Ernst P, Patenaude V, Sewitch M, Suissa S, et al. Increased incidence of inflammatory bowel disease in Québec residents with airway diseases. Eur Respir J . 2015;45:962–968. doi: 10.1183/09031936.00079414. [DOI] [PubMed] [Google Scholar]
- 8. Keely S, Hansbro PM. Lung–gut cross talk: a potential mechanism for intestinal dysfunction in patients with COPD. Chest . 2014;145:199–200. doi: 10.1378/chest.13-2077. [DOI] [PubMed] [Google Scholar]
- 9. Rutten EPA, Lenaerts K, Buurman WA, Wouters EFM. Disturbed intestinal integrity in patients with COPD: effects of activities of daily living. Chest . 2014;145:245–252. doi: 10.1378/chest.13-0584. [DOI] [PubMed] [Google Scholar]
- 10. Willis KA, Stewart JD, Ambalavanan N. Recent advances in understanding the ecology of the lung microbiota and deciphering the gut–lung axis. Am J Physiol Lung Cell Mol Physiol . 2020;319:L710–L716. doi: 10.1152/ajplung.00360.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beloborodova EI, Akimova LA, Burkovskaia BA, Asanova AV, Semenenko EV. Activity of systemic inflammatory reaction in patients with chronic obstructive pulmonary disease in regard to small intestinal absorption function [in Russian] Ter Arkh . 2009;81:19–23. [PubMed] [Google Scholar]
- 12. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol . 2018;58:157–169. doi: 10.1165/rcmb.2017-0200TR. [DOI] [PubMed] [Google Scholar]
- 13. Keely S, Talley NJ, Hansbro PM. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal Immunol . 2012;5:7–18. doi: 10.1038/mi.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fricker M, Goggins BJ, Mateer S, Jones B, Kim RY, Gellatly SL, et al. Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction. JCI Insight . 2018;3:e94040. doi: 10.1172/jci.insight.94040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai Z, Zhang H, Wei Y, Cong F. Hyaluronan-inorganic nanohybrid materials for biomedical applications. Biomacromolecules . 2017;18:1677–1696. doi: 10.1021/acs.biomac.7b00424. [DOI] [PubMed] [Google Scholar]
- 16. Raftery AL, Tsantikos E, Harris NL, Hibbs ML. Links between inflammatory bowel disease and chronic obstructive pulmonary disease. Front Immunol . 2020;11:2144. doi: 10.3389/fimmu.2020.02144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vutcovici M, Bitton A, Ernst P, Kezouh A, Suissa S, Brassard P. Inflammatory bowel disease and risk of mortality in COPD. Eur Respir J . 2016;47:1357–1364. doi: 10.1183/13993003.01945-2015. [DOI] [PubMed] [Google Scholar]
- 18. Birrenbach T, Böcker U. Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm Bowel Dis . 2004;10:848–859. doi: 10.1097/00054725-200411000-00019. [DOI] [PubMed] [Google Scholar]
- 19. Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc . 2006;81:1462–1471. doi: 10.4065/81.11.1462. [DOI] [PubMed] [Google Scholar]
- 20. Berkowitz L, Schultz BM, Salazar GA, Pardo-Roa C, Sebastián VP, Álvarez-Lobos MM, et al. Impact of cigarette smoking on the gastrointestinal tract inflammation: opposing effects in Crohn’s disease and ulcerative colitis. Front Immunol . 2018;9:74. doi: 10.3389/fimmu.2018.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ingram JR, Thomas GA, Rhodes J, Green JT, Hawkes ND, Swift JL, et al. A randomized trial of nicotine enemas for active ulcerative colitis. Clin Gastroenterol Hepatol . 2005;3:1107–1114. doi: 10.1016/s1542-3565(05)00849-9. [DOI] [PubMed] [Google Scholar]
- 22. Lashner BA, Hanauer SB, Silverstein MD. Testing nicotine gum for ulcerative colitis patients: experience with single-patient trials. Dig Dis Sci . 1990;35:827–832. doi: 10.1007/BF01536795. [DOI] [PubMed] [Google Scholar]
- 23. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, et al. Transdermal nicotine for active ulcerative colitis. N Engl J Med . 1994;330:811–815. doi: 10.1056/NEJM199403243301202. [DOI] [PubMed] [Google Scholar]
- 24. Ji XQ, Wang LX, Lu DG. Pulmonary manifestations of inflammatory bowel disease. World J Gastroenterol . 2014;20:13501–13511. doi: 10.3748/wjg.v20.i37.13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garrow D, Delegge MH. Risk factors for gastrointestinal ulcer disease in the US population. Dig Dis Sci . 2010;55:66–72. doi: 10.1007/s10620-008-0708-x. [DOI] [PubMed] [Google Scholar]
- 26. Chun EM, Kim SW, Lim SY. Prevalence of colorectal adenomatous polyps in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis . 2015;10:955–960. doi: 10.2147/COPD.S83341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahn SV, Lee E, Park B, Jung JH, Park JE, Sheen SS, et al. Cancer development in patients with COPD: a retrospective analysis of the National Health Insurance Service–National Sample Cohort in Korea. BMC Pulm Med . 2020;20:170. doi: 10.1186/s12890-020-01194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Minakata Y, Ueda H, Akamatsu K, Kanda M, Yanagisawa S, Ichikawa T, et al. High COPD prevalence in patients with liver disease. Intern Med . 2010;49:2687–2691. doi: 10.2169/internalmedicine.49.3948. [DOI] [PubMed] [Google Scholar]
- 29.Dudka T, Dudka I, Pavlyuk VO. In: Scientific progress of medicine and pharmacy of the EU countries. Antonyuk LV, Prokofiev MV, Klimovich MM, Afanasiuk OI, Shmaliy VI, Yakovets OO, et al., editors. Czestochowa: Baltija Publishing; 2021. Changes in the characteristics of the gallbladder in patients with chronic cholecystitis and COPD; pp. 46–48. [Google Scholar]
- 30. Kim J, Lee JH, Kim Y, Kim K, Oh YM, Yoo KH, et al. Association between chronic obstructive pulmonary disease and gastroesophageal reflux disease: a national cross-sectional cohort study. BMC Pulm Med . 2013;13:51. doi: 10.1186/1471-2466-13-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. García Rodríguez LA, Ruigómez A, Martín-Merino E, Johansson S, Wallander MA. Relationship between gastroesophageal reflux disease and COPD in UK primary care. Chest . 2008;134:1223–1230. doi: 10.1378/chest.08-0902. [DOI] [PubMed] [Google Scholar]
- 32. El-Serag H, Hill C, Jones R. Systematic review: the epidemiology of gastro-oesophageal reflux disease in primary care, using the UK General Practice Research Database. Aliment Pharmacol Ther . 2009;29:470–480. doi: 10.1111/j.1365-2036.2008.03901.x. [DOI] [PubMed] [Google Scholar]
- 33. Hyman JJ, Reid BC. Cigarette smoking, periodontal disease: and chronic obstructive pulmonary disease. J Periodontol . 2004;75:9–15. doi: 10.1902/jop.2004.75.1.9. [DOI] [PubMed] [Google Scholar]
- 34. Pandolfino JE, Kahrilas PJ. Smoking and gastro-oesophageal reflux disease. Eur J Gastroenterol Hepatol . 2000;12:837–842. doi: 10.1097/00042737-200012080-00002. [DOI] [PubMed] [Google Scholar]
- 35. Kadakia SC, Kikendall JW, Maydonovitch C, Johnson LF. Effect of cigarette smoking on gastroesophageal reflux measured by 24-h ambulatory esophageal pH monitoring. Am J Gastroenterol . 1995;90:1785–1790. [PubMed] [Google Scholar]
- 36. Rascon-Aguilar IE, Pamer M, Wludyka P, Cury J, Coultas D, Lambiase LR, et al. Role of gastroesophageal reflux symptoms in exacerbations of COPD. Chest . 2006;130:1096–1101. doi: 10.1378/chest.130.4.1096. [DOI] [PubMed] [Google Scholar]
- 37. Gaude GS. Pulmonary manifestations of gastroesophageal reflux disease. Ann Thorac Med . 2009;4:115–123. doi: 10.4103/1817-1737.53347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gomes-Filho IS, Cruz SSD, Trindade SC, Passos-Soares JS, Carvalho-Filho PC, Figueiredo ACMG, et al. Periodontitis and respiratory diseases: a systematic review with meta-analysis. Oral Dis . 2020;26:439–446. doi: 10.1111/odi.13228. [DOI] [PubMed] [Google Scholar]
- 39.Biorender. 2023. BioRender.comhttps://app.biorender.com/
- 40. Lochhead P, Khalili H, Ananthakrishnan AN, Richter JM, Chan AT. Association between circulating levels of C-reactive protein and interleukin-6 and risk of inflammatory bowel disease. Clin Gastroenterol Hepatol . 2016;14:818–824.e6. doi: 10.1016/j.cgh.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yanbaeva DG, Dentener MA, Spruit MA, Houwing-Duistermaat JJ, Kotz D, Passos VL, et al. IL6 and CRP haplotypes are associated with COPD risk and systemic inflammation: a case-control study. BMC Med Genet . 2009;10:23. doi: 10.1186/1471-2350-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matera MG, Calzetta L, Cazzola M. TNF-alpha inhibitors in asthma and COPD: we must not throw the baby out with the bath water. Pulm Pharmacol Ther . 2010;23:121–128. doi: 10.1016/j.pupt.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Ringerike T, Elvsaas IKO, Coll P, Lundin KEA, Movik E, Gjertsen MK.2008. [PubMed]
- 44. Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P. Anti-TNF therapy in Crohn’s disease. Int J Mol Sci . 2018;19:2244. doi: 10.3390/ijms19082244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pugliese D, Felice C, Papa A, Gasbarrini A, Rapaccini GL, Guidi L, et al. Anti TNF-α therapy for ulcerative colitis: current status and prospects for the future. Expert Rev Clin Immunol . 2017;13:223–233. doi: 10.1080/1744666X.2017.1243468. [DOI] [PubMed] [Google Scholar]
- 46. Suissa S, Ernst P, Hudson M. TNF-alpha antagonists and the prevention of hospitalisation for chronic obstructive pulmonary disease. Pulm Pharmacol Ther . 2008;21:234–238. doi: 10.1016/j.pupt.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 47. Barnes PJ. Unexpected failure of anti-tumor necrosis factor therapy in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2007;175:866–867. doi: 10.1164/rccm.200702-253ED. [DOI] [PubMed] [Google Scholar]
- 48. Peek RM, Jr, Mohla S, DuBois RN. Inflammation in the genesis and perpetuation of cancer: summary and recommendations from a national cancer institute-sponsored meeting. Cancer Res . 2005;65:8583–8586. doi: 10.1158/0008-5472.CAN-05-1777. [DOI] [PubMed] [Google Scholar]
- 49. Li LF, Chan RL, Lu L, Shen J, Zhang L, Wu WK, et al. Cigarette smoking and gastrointestinal diseases: the causal relationship and underlying molecular mechanisms (review) Int J Mol Med . 2014;34:372–380. doi: 10.3892/ijmm.2014.1786. [DOI] [PubMed] [Google Scholar]
- 50. MacNee W. Systemic inflammatory biomarkers and co-morbidities of chronic obstructive pulmonary disease. Ann Med . 2013;45:291–300. doi: 10.3109/07853890.2012.732703. [DOI] [PubMed] [Google Scholar]
- 51. Viglino D, Jullian-Desayes I, Minoves M, Aron-Wisnewsky J, Leroy V, Zarski JP, et al. Nonalcoholic fatty liver disease in chronic obstructive pulmonary disease. Eur Respir J . 2017;49:1601923. doi: 10.1183/13993003.01923-2016. [DOI] [PubMed] [Google Scholar]
- 52. Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C, et al. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One . 2011;6:e21381. doi: 10.1371/journal.pone.0021381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yadav RS, Kant S, Tripathi PM, Pathak AK, Mahdi AA. Transcription factor NF-κB, interleukin-1β, and interleukin-8 expression and its association with tobacco smoking and severity in chronic obstructive pulmonary disease. Gene Rep . 2022;26:101453. [Google Scholar]
- 54. Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, et al. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2003;168:968–975. doi: 10.1164/rccm.200208-794OC. [DOI] [PubMed] [Google Scholar]
- 55. Shi Q, Zhang B, Xing H, Yang S, Xu J, Liu H. Patients with chronic obstructive pulmonary disease suffer from worse periodontal health—evidence from a meta-analysis. Front Physiol . 2018;9:33. doi: 10.3389/fphys.2018.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou X, Han J, Liu Z, Song Y, Wang Z, Sun Z. Effects of periodontal treatment on lung function and exacerbation frequency in patients with chronic obstructive pulmonary disease and chronic periodontitis: a 2-year pilot randomized controlled trial. J Clin Periodontol . 2014;41:564–572. doi: 10.1111/jcpe.12247. [DOI] [PubMed] [Google Scholar]
- 57. Mammen MJ, Scannapieco FA, Sethi S. Oral-lung microbiome interactions in lung diseases. Periodontol 2000 . 2020;83:234–241. doi: 10.1111/prd.12301. [DOI] [PubMed] [Google Scholar]
- 58. Sevenoaks MJ, Stockley RA. Chronic obstructive pulmonary disease, inflammation and co-morbidity—a common inflammatory phenotype? Respir Res . 2006;7:70. doi: 10.1186/1465-9921-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andreoli R, Manini P, Corradi M, Mutti A, Niessen WM. Determination of patterns of biologically relevant aldehydes in exhaled breath condensate of healthy subjects by liquid chromatography/atmospheric chemical ionization tandem mass spectrometry. Rapid Commun Mass Spectrom . 2003;17:637–645. doi: 10.1002/rcm.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Annovazzi L, Cattaneo V, Viglio S, Perani E, Zanone C, Rota C, et al. High-performance liquid chromatography and capillary electrophoresis: methodological challenges for the determination of biologically relevant low-aliphatic aldehydes in human saliva. Electrophoresis . 2004;25:1255–1263. doi: 10.1002/elps.200305843. [DOI] [PubMed] [Google Scholar]
- 61. Zhang L, Ren JW, Wong CC, Wu WK, Ren SX, Shen J, et al. Effects of cigarette smoke and its active components on ulcer formation and healing in the gastrointestinal mucosa. Curr Med Chem . 2012;19:63–69. doi: 10.2174/092986712803413926. [DOI] [PubMed] [Google Scholar]
- 62. Lindell G, Farnebo LO, Chen D, Nexø E, Rask Madsen J, Bukhave K, et al. Acute effects of smoking during modified sham feeding in duodenal ulcer patients: an analysis of nicotine, acid secretion, gastrin, catecholamines, epidermal growth factor, prostaglandin E2, and bile acids. Scand J Gastroenterol . 1993;28:487–494. doi: 10.3109/00365529309098254. [DOI] [PubMed] [Google Scholar]
- 63. Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci . 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest . 2013;144:266–273. doi: 10.1378/chest.12-2664. [DOI] [PubMed] [Google Scholar]
- 65. Park HY, Hahm CR, Jeon K, Koh WJ, Suh GY, Chung MP, et al. Serum vascular endothelial growth factor and angiopoietin-2 are associated with the severity of systemic inflammation rather than the presence of hemoptysis in patients with inflammatory lung disease. Yonsei Med J . 2012;53:369–376. doi: 10.3349/ymj.2012.53.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Griga T, Tromm A, Spranger J, May B. Increased serum levels of vascular endothelial growth factor in patients with inflammatory bowel disease. Scand J Gastroenterol . 1998;33:504–508. doi: 10.1080/00365529850172070. [DOI] [PubMed] [Google Scholar]
- 67. Dev A, Patel K, Conrad A, Blatt LM, McHutchison JG. Relationship of smoking and fibrosis in patients with chronic hepatitis C. Clin Gastroenterol Hepatol . 2006;4:797–801. doi: 10.1016/j.cgh.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 68. Parasher G, Eastwood GL. Smoking and peptic ulcer in the Helicobacter pylori era. Eur J Gastroenterol Hepatol . 2000;12:843–853. doi: 10.1097/00042737-200012080-00003. [DOI] [PubMed] [Google Scholar]
- 69. Krzystek-Korpacka M, Matusiewicz M, Diakowska D, Grabowski K, Neubauer K, Kustrzeba-Wojcicka I, et al. Respiratory insufficiency related to COPD accelerates systemic inflammation, under-nutrition, and angiogenesis in esophageal malignancies. Exp Oncol . 2008;30:75–80. [PubMed] [Google Scholar]
- 70. Nagaraju GP, Bramhachari PV, Raghu G, El-Rayes BF. Hypoxia inducible factor-1α: its role in colorectal carcinogenesis and metastasis. Cancer Lett . 2015;366:11–18. doi: 10.1016/j.canlet.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 71. Marsland BJ, Trompette A, Gollwitzer ES. The gut–lung axis in respiratory disease. Ann Am Thorac Soc . 2015;12:S150–S156. doi: 10.1513/AnnalsATS.201503-133AW. [DOI] [PubMed] [Google Scholar]
- 72. Li N, Dai Z, Wang Z, Deng Z, Zhang J, Pu J, et al. Gut microbiota dysbiosis contributes to the development of chronic obstructive pulmonary disease. Respir Res . 2021;22:274. doi: 10.1186/s12931-021-01872-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ramsheh MY, Haldar K, Esteve-Codina A, Purser LF, Richardson M, Müller-Quernheim J, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancet Microbe . 2021;2:e300–e310. doi: 10.1016/S2666-5247(21)00035-5. [DOI] [PubMed] [Google Scholar]
- 74. Bowerman KL, Rehman SF, Vaughan A, Lachner N, Budden KF, Kim RY, et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat Commun . 2020;11:5886. doi: 10.1038/s41467-020-19701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cvejic L, Harding R, Churchward T, Turton A, Finlay P, Massey D, et al. Laryngeal penetration and aspiration in individuals with stable COPD. Respirology . 2011;16:269–275. doi: 10.1111/j.1440-1843.2010.01875.x. [DOI] [PubMed] [Google Scholar]
- 76. Madapoosi SS, Cruickshank-Quinn C, Opron K, Erb-Downward JR, Begley LA, Li G, et al. SPIROMICS Research Group Lung microbiota and metabolites collectively associate with clinical outcomes in milder stage chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2022;206:427–439. doi: 10.1164/rccm.202110-2241OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Qu L, Cheng Q, Wang Y, Mu H, Zhang Y. COPD and gut–lung axis: how microbiota and host inflammasome influence COPD and related therapeutics. Front Microbiol . 2022;13:868086. doi: 10.3389/fmicb.2022.868086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dang AT, Marsland BJ. Microbes, metabolites, and the gut–lung axis. Mucosal Immunol . 2019;12:843–850. doi: 10.1038/s41385-019-0160-6. [DOI] [PubMed] [Google Scholar]
- 79. Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)–mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol . 2019;10:277. doi: 10.3389/fimmu.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang L, Pelgrim CE, Peralta Marzal LN, Korver S, van Ark I, Leusink-Muis T, et al. Changes in intestinal homeostasis and immunity in a cigarette smoke- and LPS-induced murine model for COPD: the lung–gut axis. Am J Physiol Lung Cell Mol Physiol . 2022;323:L266–L280. doi: 10.1152/ajplung.00486.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim M, Gu B, Madison MC, Song HW, Norwood K, Hill AA, et al. Cigarette smoke induces intestinal inflammation via a Th17 cell–neutrophil axis. Front Immunol . 2019;10:75. doi: 10.3389/fimmu.2019.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Verschuere S, Bracke KR, Demoor T, Plantinga M, Verbrugghe P, Ferdinande L, et al. Cigarette smoking alters epithelial apoptosis and immune composition in murine GALT. Lab Invest . 2011;91:1056–1067. doi: 10.1038/labinvest.2011.74. [DOI] [PubMed] [Google Scholar]
- 83. Ruane D, Brane L, Reis BS, Cheong C, Poles J, Do Y, et al. Lung dendritic cells induce migration of protective T cells to the gastrointestinal tract. J Exp Med . 2013;210:1871–1888. doi: 10.1084/jem.20122762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Daniluk J, Daniluk U, Reszec J, Rusak M, Dabrowska M, Dabrowski A. Protective effect of cigarette smoke on the course of dextran sulfate sodium-induced colitis is accompanied by lymphocyte subpopulation changes in the blood and colon. Int J Colorectal Dis . 2017;32:1551–1559. doi: 10.1007/s00384-017-2882-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lo Sasso G, Phillips BW, Sewer A, Battey JND, Kondylis A, Talikka M, et al. The reduction of DSS-induced colitis severity in mice exposed to cigarette smoke is linked to immune modulation and microbial shifts. Sci Rep . 2020;10:3829. doi: 10.1038/s41598-020-60175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Qin Z, Wan JJ, Sun Y, Wu T, Wang PY, Du P, et al. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J Mol Med (Berl) . 2017;95:221–233. doi: 10.1007/s00109-016-1473-5. [DOI] [PubMed] [Google Scholar]
- 87. Eliakim R, Fan QX, Babyatsky MW. Chronic nicotine administration differentially alters jejunal and colonic inflammation in interleukin-10 deficient mice. Eur J Gastroenterol Hepatol . 2002;14:607–614. doi: 10.1097/00042737-200206000-00005. [DOI] [PubMed] [Google Scholar]
- 88. Xin X, Dai W, Wu J, Fang L, Zhao M, Zhang P, et al. Mechanism of intestinal mucosal barrier dysfunction in a rat model of chronic obstructive pulmonary disease: an observational study. Exp Ther Med . 2016;12:1331–1336. doi: 10.3892/etm.2016.3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zuo L, Li Y, Wang H, Wu R, Zhu W, Zhang W, et al. Cigarette smoking is associated with intestinal barrier dysfunction in the small intestine but not in the large intestine of mice. J Crohn’s Colitis . 2014;8:1710–1722. doi: 10.1016/j.crohns.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 90. Berkowitz L, Pardo-Roa C, Salazar GA, Salazar-Echegarai F, Miranda JP, Ramírez G, et al. Mucosal exposure to cigarette components induces intestinal inflammation and alters antimicrobial response in mice. Front Immunol . 2019;10:2289. doi: 10.3389/fimmu.2019.02289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Allais L, Kerckhof FM, Verschuere S, Bracke KR, De Smet R, Laukens D, et al. Chronic cigarette smoke exposure induces microbial and inflammatory shifts and mucin changes in the murine gut. Environ Microbiol . 2016;18:1352–1363. doi: 10.1111/1462-2920.12934. [DOI] [PubMed] [Google Scholar]
- 92. Li H, Wu Q, Xu L, Li X, Duan J, Zhan J, et al. Increased oxidative stress and disrupted small intestinal tight junctions in cigarette smoke–exposed rats. Mol Med Rep . 2015;11:4639–4644. doi: 10.3892/mmr.2015.3234. [DOI] [PubMed] [Google Scholar]
- 93. Basak D, Uddin MN, Hancock J. The role of oxidative stress and its counteractive utility in colorectal cancer (CRC) Cancers (Basel) . 2020;12:3336. doi: 10.3390/cancers12113336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wu Z, Zuo M, Zeng L, Cui K, Liu B, Yan C, et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep . 2021;22:e50827. doi: 10.15252/embr.202050827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Balasuriya GK, Mohsenipour M, Brassington K, Dobric A, De Luca SN, Mou K, et al. Ebselen prevents cigarette smoke–induced gastrointestinal dysfunction in mice. Clin Sci (Lond) . 2020;134:2943–2957. doi: 10.1042/CS20200886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Papoutsopoulou S, Satsangi J, Campbell BJ, Probert CS. Review article: impact of cigarette smoking on intestinal inflammation-direct and indirect mechanisms. Aliment Pharmacol Ther . 2020;51:1268–1285. doi: 10.1111/apt.15774. [DOI] [PubMed] [Google Scholar]
- 97. Yang Y, Yang C, Lei Z, Rong H, Yu S, Wu H, et al. Cigarette smoking exposure breaks the homeostasis of cholesterol and bile acid metabolism and induces gut microbiota dysbiosis in mice with different diets. Toxicology . 2021;450:152678. doi: 10.1016/j.tox.2021.152678. [DOI] [PubMed] [Google Scholar]
- 98. Lai HC, Lin TL, Chen TW, Kuo YL, Chang CJ, Wu TR, et al. Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut . 2022;71:309–321. doi: 10.1136/gutjnl-2020-322599. [DOI] [PubMed] [Google Scholar]
- 99. Jang YO, Lee SH, Choi JJ, Kim DH, Choi JM, Kang MJ, et al. Fecal microbial transplantation and a high fiber diet attenuates emphysema development by suppressing inflammation and apoptosis. Exp Mol Med . 2020;52:1128–1139. doi: 10.1038/s12276-020-0469-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang X, Ye P, Fang L, Ge S, Huang F, Polverini PJ, et al. Active smoking induces aberrations in digestive tract microbiota of rats. Front Cell Infect Microbiol . 2021;11:737204. doi: 10.3389/fcimb.2021.737204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cai Y, Folkerts J, Folkerts G, Maurer M, Braber S. Microbiota-dependent and -independent effects of dietary fibre on human health. Br J Pharmacol . 2020;177:1363–1381. doi: 10.1111/bph.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhong Y, Nyman M, Fåk F. Modulation of gut microbiota in rats fed high-fat diets by processing whole-grain barley to barley malt. Mol Nutr Food Res . 2015;59:2066–2076. doi: 10.1002/mnfr.201500187. [DOI] [PubMed] [Google Scholar]
- 103. Ai D, Pan H, Li X, Gao Y, Liu G, Xia LC. Identifying gut microbiota associated with colorectal cancer using a zero-inflated lognormal model. Front Microbiol . 2019;10:826. doi: 10.3389/fmicb.2019.00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bai X, Wei H, Liu W, Coker OO, Gou H, Liu C, et al. Cigarette smoke promotes colorectal cancer through modulation of gut microbiota and related metabolites. Gut . 2022;71:2439–2450. doi: 10.1136/gutjnl-2021-325021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tomoda K, Kubo K, Asahara T, Andoh A, Nomoto K, Nishii Y, et al. Cigarette smoke decreases organic acids levels and population of bifidobacterium in the caecum of rats. J Toxicol Sci . 2011;36:261–266. doi: 10.2131/jts.36.261. [DOI] [PubMed] [Google Scholar]
- 106. Tomoda K, Kubo K, Dairiki K, Yamaji T, Yamamoto Y, Nishii Y, et al. Whey peptide-based enteral diet attenuated elastase-induced emphysema with increase in short chain fatty acids in mice. BMC Pulm Med . 2015;15:64. doi: 10.1186/s12890-015-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Janbazacyabar H, van Bergenhenegouwen J, Verheijden KAT, Leusink-Muis T, van Helvoort A, Garssen J, et al. Non-digestible oligosaccharides partially prevent the development of LPS-induced lung emphysema in mice. PharmaNutrition . 2019;10:100163. [Google Scholar]
- 108. Tomoda K, Kubo K, Yamamoto Y, Kimura H. Alteration in gut environment accelerates emphysematous lesions by cigarette smoke in rats discontinuously fed with fiber-free diet [abstract] Am J Respir Crit Care Med . 2014;189:A3000. [Google Scholar]
- 109. Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol . 2008;295:L1–L15. doi: 10.1152/ajplung.90200.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tanner L, Single AB. Animal models reflecting chronic obstructive pulmonary disease and related respiratory disorders: translating pre-clinical data into clinical relevance. J Innate Immun . 2020;12:203–225. doi: 10.1159/000502489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dino P, D’Anna C, Sangiorgi C, Di Sano C, Di Vincenzo S, Ferraro M, et al. Cigarette smoke extract modulates E-cadherin, claudin-1 and miR-21 and promotes cancer invasiveness in human colorectal adenocarcinoma cells. Toxicol Lett . 2019;317:102–109. doi: 10.1016/j.toxlet.2019.09.020. [DOI] [PubMed] [Google Scholar]
- 112. Sharma S, Kumar DS, Dhakar B, Avti P, Khanduja K. PO-239 Benzo(a)pyrene, an active product of cigarette smoke, role in pla2 isoforms activation in colon cancer. ESMO Open . 2018;3:A113–A114. [Google Scholar]
- 113. Heijink IH, Brandenburg SM, Postma DS, van Oosterhout AJ. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur Respir J . 2012;39:419–428. doi: 10.1183/09031936.00193810. [DOI] [PubMed] [Google Scholar]
- 114. Forteza RM, Casalino-Matsuda SM, Falcon NS, Valencia Gattas M, Monzon ME. Hyaluronan and layilin mediate loss of airway epithelial barrier function induced by cigarette smoke by decreasing E-cadherin. J Biol Chem . 2012;287:42288–42298. doi: 10.1074/jbc.M112.387795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Michcik A, Cichorek M, Daca A, Chomik P, Wojcik S, Zawrocki A, et al. Tobacco smoking alters the number of oral epithelial cells with apoptotic features. Folia Histochem Cytobiol . 2014;52:60–68. doi: 10.5603/FHC.2014.0007. [DOI] [PubMed] [Google Scholar]
- 116. Wu WK, Wong HP, Luo SW, Chan K, Huang FY, Hui MK, et al. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone from cigarette smoke stimulates colon cancer growth via beta-adrenoceptors. Cancer Res . 2005;65:5272–5277. doi: 10.1158/0008-5472.CAN-05-0205. [DOI] [PubMed] [Google Scholar]
- 117. Sykes AP, Brampton C, Klee S, Chander CL, Whelan C, Parsons ME. An investigation into the effect and mechanisms of action of nicotine in inflammatory bowel disease. Inflamm Res . 2000;49:311–319. doi: 10.1007/s000110050597. [DOI] [PubMed] [Google Scholar]
- 118. Madretsma GS, Donze GJ, van Dijk AP, Tak CJ, Wilson JH, Zijlstra FJ. Nicotine inhibits the in vitro production of interleukin 2 and tumour necrosis factor-alpha by human mononuclear cells. Immunopharmacology . 1996;35:47–51. doi: 10.1016/0162-3109(96)00122-1. [DOI] [PubMed] [Google Scholar]
- 119. Summers AE, Whelan CJ, Parsons ME. Nicotinic acetylcholine receptor subunits and receptor activity in the epithelial cell line HT29. Life Sci . 2003;72:2091–2094. doi: 10.1016/s0024-3205(03)00089-4. [DOI] [PubMed] [Google Scholar]
- 120. McGilligan VE, Wallace JM, Heavey PM, Ridley DL, Rowland IR. Hypothesis about mechanisms through which nicotine might exert its effect on the interdependence of inflammation and gut barrier function in ulcerative colitis. Inflamm Bowel Dis . 2007;13:108–115. doi: 10.1002/ibd.20020. [DOI] [PubMed] [Google Scholar]
- 121. Liu P, Wang Y, Yang G, Zhang Q, Meng L, Xin Y, et al. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol Res . 2021;165:105420. doi: 10.1016/j.phrs.2021.105420. [DOI] [PubMed] [Google Scholar]
- 122. Suzuki T, Yoshida S, Hara H. Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Br J Nutr . 2008;100:297–305. doi: 10.1017/S0007114508888733. [DOI] [PubMed] [Google Scholar]
- 123. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr . 2009;139:1619–1625. doi: 10.3945/jn.109.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Miao W, Wu X, Wang K, Wang W, Wang Y, Li Z, et al. Sodium butyrate promotes reassembly of tight junctions in Caco-2 monolayers involving inhibition of MLCK/MLC2 pathway and phosphorylation of PKCβ2. Int J Mol Sci . 2016;17:1696. doi: 10.3390/ijms17101696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yan H, Ajuwon KM. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS One . 2017;12:e0179586. doi: 10.1371/journal.pone.0179586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Richards LB, Li M, Folkerts G, Henricks PAJ, Garssen J, van Esch BCAM. Butyrate and propionate restore the cytokine and house dust mite compromised barrier function of human bronchial airway epithelial cells. Int J Mol Sci . 2020;22:65. doi: 10.3390/ijms22010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bulanda E, Wypych TP. Bypassing the gut–lung axis via microbial metabolites: implications for chronic respiratory diseases. Front Microbiol . 2022;13:857418. doi: 10.3389/fmicb.2022.857418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ashique S, De Rubis G, Sirohi E, Mishra N, Rihan M, Garg A, et al. Short chain fatty acids: fundamental mediators of the gut–lung axis and their involvement in pulmonary diseases. Chem Biol Interact . 2022;368:110231. doi: 10.1016/j.cbi.2022.110231. [DOI] [PubMed] [Google Scholar]
- 129. Fu Y, Moscoso DI, Porter J, Krishnareddy S, Abrams JA, Seres D, et al. Relationship between dietary fiber intake and short-chain fatty acid-producing bacteria during critical illness: a prospective cohort study. JPEN J Parenter Enteral Nutr . 2020;44:463–471. doi: 10.1002/jpen.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunology . 2016;5:e73. doi: 10.1038/cti.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Szmidt MK, Kaluza J, Harris HR, Linden A, Wolk A. Long-term dietary fiber intake and risk of chronic obstructive pulmonary disease: a prospective cohort study of women. Eur J Nutr . 2020;59:1869–1879. doi: 10.1007/s00394-019-02038-w. [DOI] [PMC free article] [PubMed] [Google Scholar]