

Abstract
The antiphospholipid syndrome (APS) is the most common cause of acquired immune-mediated thrombophilia. This syndrome is broadly defined by the presence of arterial or venous thrombosis, or pregnancy morbidity, in the presence of high levels of antiphospholipid antibodies. Despite recognition of this disorder more than 50 years ago, a fundamental unifying pathogenesis has not been determined. Due to this, mechanism-based therapies for APS are not available, and current management following thrombotic events suggests anticoagulation of indeterminate duration, or for obstetric complications, heparin/low molecular weight heparin and aspirin. However, APS is an autoimmune disorder, and several approaches focused on modulating the immune response or its effectors have been employed. Those which have been most extensively studied include hydroxychloroquine, rituximab and eculizumab, an inhibitor of complement C5. In this report, we review in depth, and critique, key clinical studies of these agents. Since all of these studies are small, our conclusions are qualified. However, it appears that hydroxychloroquine may enhance the anticoagulant efficacy of vitamin K antagonists in APS patients, and that rituximab may ameliorate some of the “non-criteria” manifestations of APS. The catastrophic antiphospholipid syndrome (CAPS) is associated with diffuse thrombosis, multi-organ dysfunction, and ~30% mortality. A high incidence of complement regulatory gene mutations, and compelling data concerning the efficacy of eculizumab in CAPS, suggests an important role for complement in this disorder. However, additional work is needed to clarify the role of complement in non-catastrophic APS, though emerging data suggests that complement inhibition may be effective in preventing thrombosis in these patients as well.
Keywords: Antiphospholipid, Lupus, Complement, Eculizumab, Plasma exchange, Thrombophilia
Introduction
The antiphospholipid syndrome (APS) is an autoimmune disorder characterized by arterial or venous thrombosis and/or recurrent obstetrical morbidity accompanied by persistently high levels of antiphospholipid antibodies (aPL) [1]. aPL are autoantibodies with specificity primarily toward phospholipid binding proteins [2] and include the lupus anticoagulant (LAC), anti-beta2-glycoprotein-I (B2GPI), anti-cardiolipin (aCL), and anti-prothrombin antibodies (IgG or IgM), [1] among others. Cofactor-independent aPL have also been described [3,4]. APS is the most common acquired thrombophilia [5] and results in both venous and arterial thrombosis, most commonly deep vein thromboses (DVT) and stroke, respectively [6]. APS is also associated with significant obstetric morbidity [6]. A comprehensive pathogenic model that can inform effective therapies remains to be established. The cornerstone of management following a thrombotic event is indefinite anticoagulation with a vitamin K antagonist (VKA), or aspirin and heparin for obstetric manifestations [7]. Several novel treatment approaches that target immune components of the syndrome have been described [8,9]. Agents that have been most extensively studied include hydroxychloroquine, the B-cell depleting monoclonal antibody rituximab, and eculizumab, a monoclonal antibody to complement C5. In the discussion that follows, we review the relevant pathophysiology, and the strength of the evidence supporting a role for these therapies in APS management.
Diagnosis of APS
The revised Sapporo criteria [10], originally developed to standardize enrollment of APS patients into clinical studies rather than to diagnose APS, are nevertheless widely used in clinical practice for APS diagnosis. These criteria include both clinical and laboratory parameters. Clinical criteria include objectively confirmed venous, arterial, or small vessel thrombosis, or obstetrical morbidity (unexplained death of a morphologically normal fetus at or beyond the 10th week of gestation, the premature birth of a neonate before the 34th week of gestation, and/or 3 or more unexplained, consecutive pregnancy losses before the 10th week of gestation) [1]. Laboratory criteria include a lupus anticoagulant, defined by International Society of Thrombosis and Hemostasis (ISTH) guidelines [11], and/or the presence of IgG or IgM anticardiolipin or anti-β2GPI antibodies at levels exceeding the 99th percentile of normal controls (ELISA) [11]. As aPL can be transient, the laboratory criteria must be met on 2 separate occasions at least 12 weeks apart. A subset of APS patients (approximately 1%) [6] present with the highly thrombotic phenotype referred to as catastrophic APS (CAPS). CAPS presents with simultaneous or rapidly successive thromboses in 3 or more organ systems occurring in less than 1 week in the presence of aPL [12]. Thrombosis in CAPS may pre-dominately affect microvascular beds, although large vessel venous or arterial events occur [12]. CAPS is highly morbid [13], may be refractory to standard treatment with anticoagulation, glucocorticoids and plasma exchange, and is associated mortality of approximately 30% [6]. Additional clinical features not formally included in the revised Sapporo criteria, but nonetheless recognized to occur in APS have been termed “non-criteria” manifestations, and include superficial venous thrombosis, thrombocytopenia, cardiac valvular disease, skin ulcers and livedo reticularis, aPL-associated nephropathy, migraine headaches, and cognitive dysfunction, among others [14].
In practice, the Sapporo criteria have several shortcomings. For example, there is limited guidance as to whether patients who develop thrombosis or pregnancy morbidity in the presence of low or intermediate aPL levels should be considered to have APS [11]. Similarly, patients presenting with a convincing clinical picture but with transient, or persistently negative aPL titers, are sometimes diagnosed with “seronegative APS” though there is a lack of consensus around this entity [15,16]. In contrast, some patients may have persistently high levels of aPL without experiencing thrombotic or obstetrical events. Laboratory analytical issues such as interassay and interlaboratory variability present additional challenges in APS diagnosis [7,11,17]. Hence, despite recent advances, the ability to risk stratify aPL patients remains limited, reflecting incomplete information concerning disease mechanisms in individual patients.
Pathophysiology
aPL are primarily directed against phospholipid-binding proteins such as prothrombin and B2GPI [2,18], although prothrombotic, cofactor-independent aPL have also been described [3,4]. The majority of published work in APS has focused on anti-β2GPI antibodies [2,19,20], and these studies generally demonstrate a significant association of anti-β2GPI antibodies with clinical events [2]. A “two hit” hypothesis proposes that aPL prime the intravascular space and a “second hit” (eg, infection, vessel injury, surgery) triggers a thrombotic event [2,7]. This hypothesis is supported by the observation that aPL positivity alone may be insufficient to drive the disease manifestations. For example, in a murine model, affinity purified IgG antibodies against B2GP1 from APS patients caused thrombosis in mice treated with a proinflammatory factor (LPS) in a complement-dependent manner, but did not cause thrombosis alone [20]. Passive infusion of human aPL IgG or affinity-purified anti-β2GPI antibodies into mice has been shown to potentiate thrombosis induced by a triggering event, such as vessel injury [20–22]. aPL generate the prothrombotic milieu through multiple mechanisms, including platelet, endothelial cell, and monocyte activation, in concert with inhibition of natural anticoagulant and fibrinolytic systems [2,7,22]. Activated endothelial cells increase the expression of adhesion molecules and tissue factor, and the release of von Willebrand factor, proinflammatory cytokines and extracellular vesicles, while decreasing endothelial cell-derived nitric oxide, in response to anti-β2GPI antibodies [7,23]. Anti-β2GP1 antibodies also stimulate monocyte tissue factor expression and release of inflammatory cytokines [24], as well as the release of neutrophil extracellular traps (NETs). [25] aPL associated with pregnancy loss may have additional mechanisms, including impairment of trophoblast migration leading to defective placentation [2].
There is compelling evidence for a role of complement in the pathogenesis of APS [2,22,26,27]. The complement system consists of numerous membrane-bound and soluble proteins that play a central role in innate immunity [28]. Activation of complement results in opsonization of pathogens but requires precise regulation to avoid damage to normal tissues; dysregulation of complement plays a critical role in disorders such as paroxysmal nocturnal hemoglobinuria (PNH) and complement-mediated hemolytic uremic syndrome (CM-HUS) [28]. There is significant interplay between the complement and coagulation systems [29,30] and some evidence suggests that activated complement contributes to aPL-associated thrombotic events (Figure 1) [26,31–33]. In preclinical models, enhancement of thrombosis and/or fetal loss that follows passive infusion of aPL into mice is ameliorated in mice deficient in C3, C5 or C6 [32,33], as well as in mice pretreated with the C5 inhibitor, coversine [34]. Moreover, in a murine model of fetal loss, complement activation led to local generation of C5a, which stimulated migration of tissue factor-expressing neutrophils into the placental bed [35]. Studies in humans, however, are not as clear cut and further study is needed. Patients with APS have elevated levels of circulating complement activation products C3a and C4a, demonstrating ongoing complement activation in many [36] (Figure 2); however, these do not appear to clearly distinguish patients with thrombosis from those without (Figure 3) [37]. This may reflect the fact that relevant complement effects occur on cell surfaces rather than in plasma.
Fig. 1.
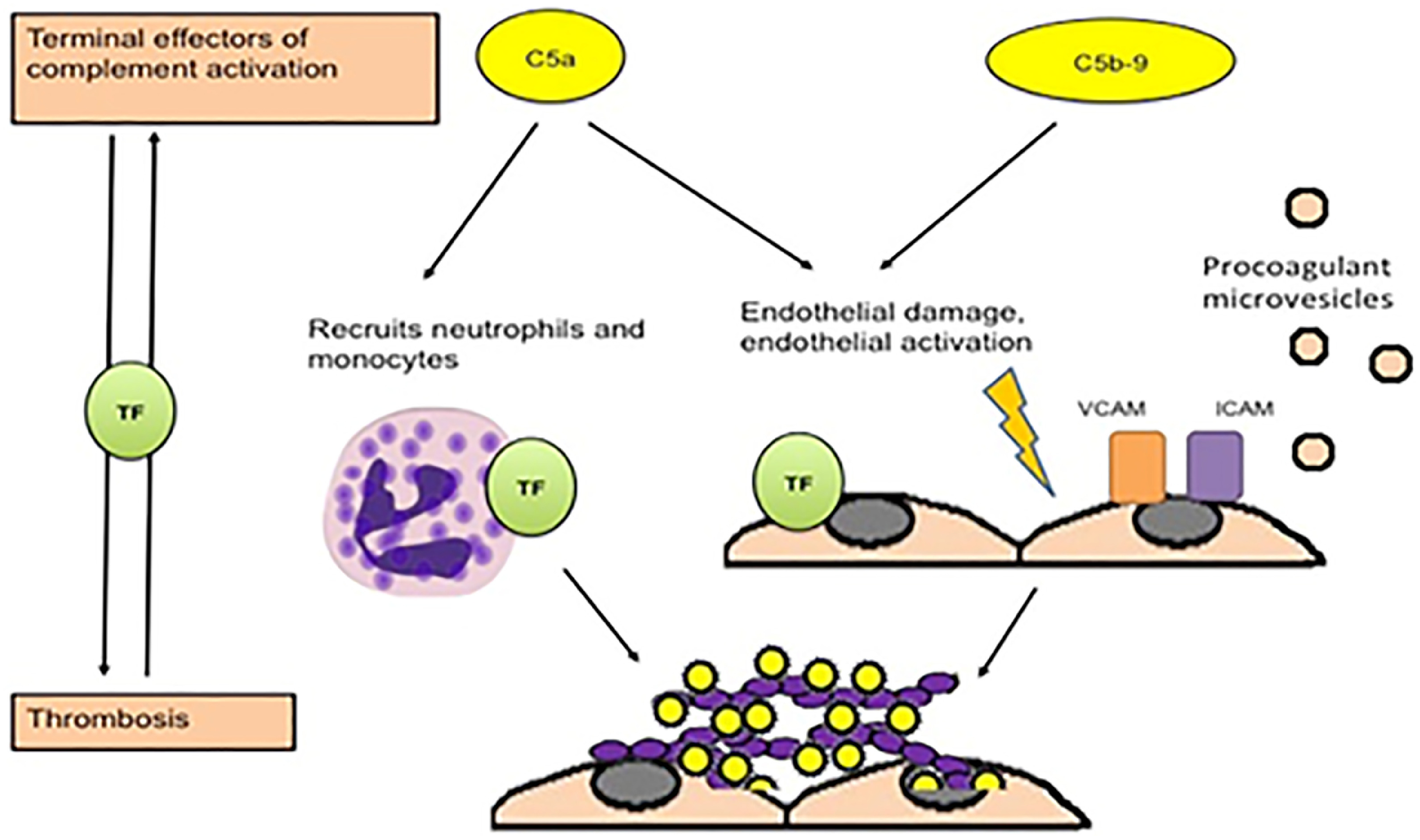
Potential mechanisms of thrombosis induced by aPL induced by complement, from Chaturvedi, Brodsky and McCrae26.
Fig. 2.
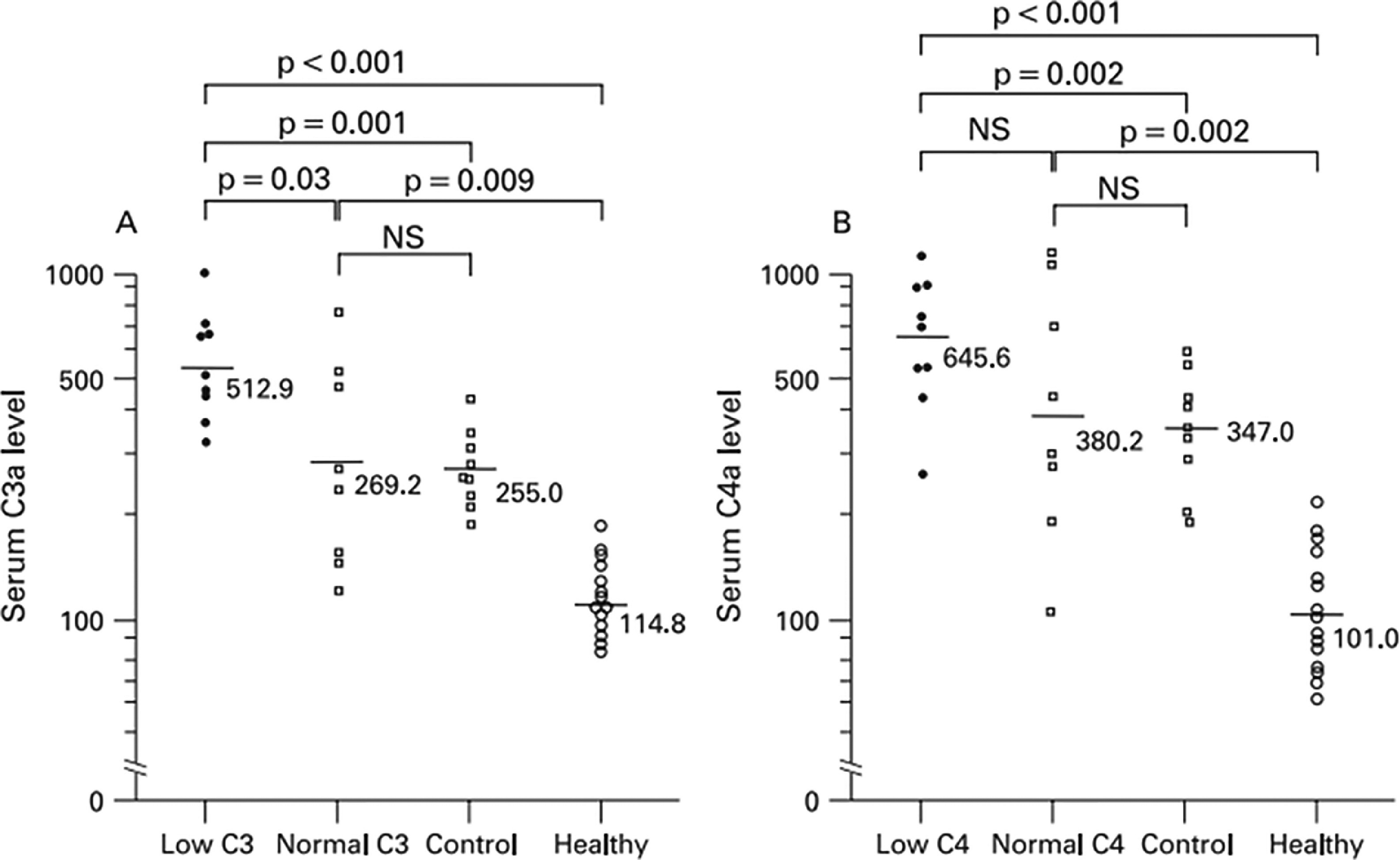
Serum C3a and C4a levels in 17 patients with primary APS, 9 patients with non-systemic SLE connective tissue diseases and 15 healthy volunteers, as reported by Oku et al.36 Low C3/C4 and normal C3/C4 refers to the levels of C3 of C4 in the plasmas being assessed, since some patients with APS were found to have hypocomplementemia.
Fig. 3.
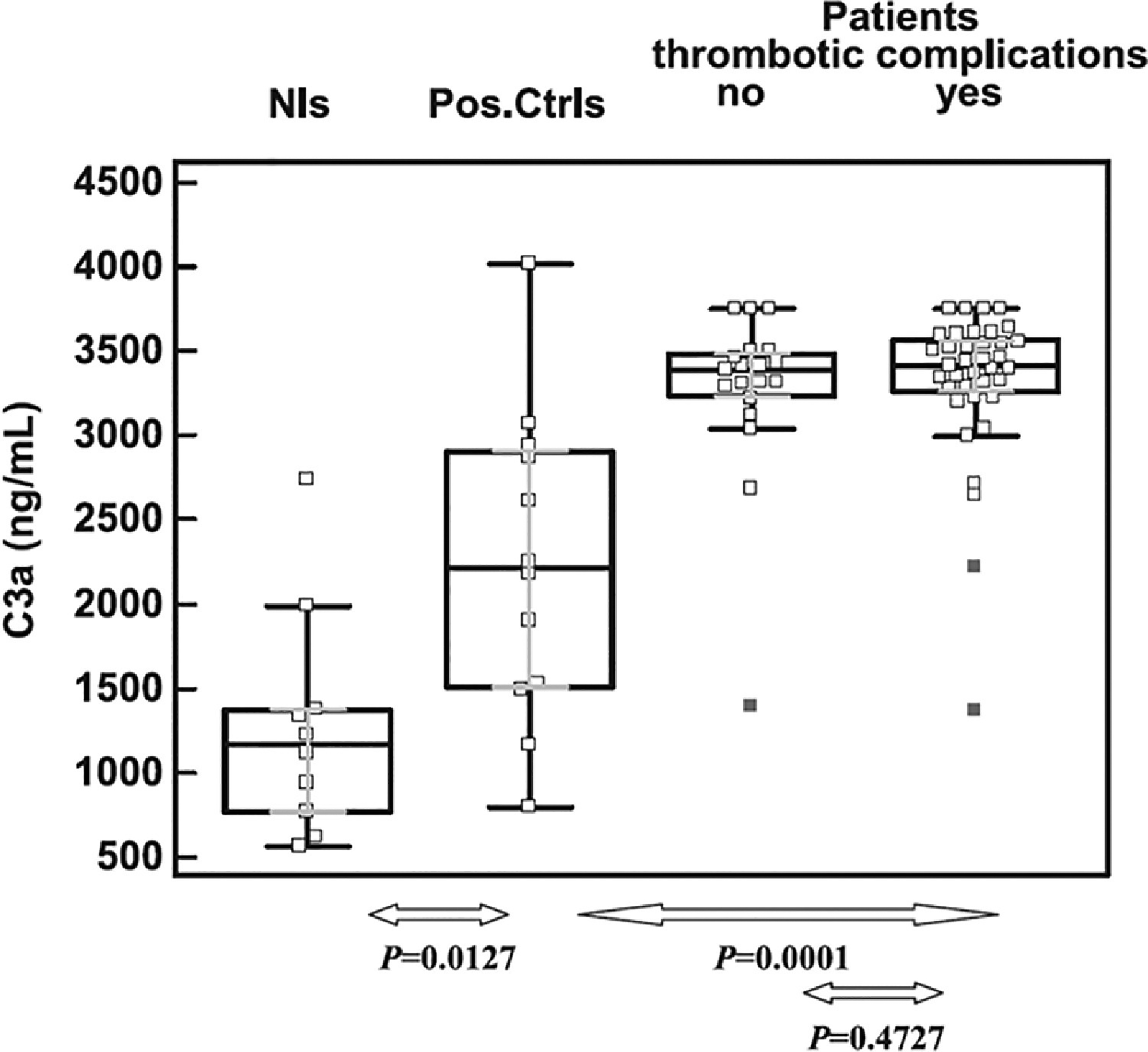
Relation between complement activation and aPL-associated thrombosis, from Devreese and Hoylaerts37.
Despite some uncertainty in “standard” APS, evidence of unregulated complement activation is present in patients who develop CAPS and is likely to play a role in pathogenesis. A high incidence of complement regulatory gene mutations, similar to that observed in CM-HUS, has been reported in these individuals, and supports the premise of complement activation as a driver of the disease process [31]. As the low incidence of CAPS precludes randomized trials of complement inhibitors, complement inhibition therefore has become an emerging target in management of CAPS and refractory APS. In the discussion that follows we review several reports of use of the complement C5 inhibitor eculizumab in these settings.
Complement Inhibitors in CAPS and Refractory Antiphospholipid Syndrome
Current management of CAPs is empiric. Guidelines from an expert panel that attempted to develop an evidence-based approach to APS concluded that all recommendations were based on very low certainty of evidence, with the majority being conditional [38]. CAPS is initially treated using a combination of anticoagulation and high dose steroids, with most experts also recommending plasma exchange; of these approaches, however, only anticoagulation was considered to be a strong recommendation. Intravenous immunoglobulin (IVIg) has been used in some cases, particularly in patients who cannot tolerate plasma exchange [5,12]. Rituximab may be added in cases refractory to first line therapy [39], and cyclophosphamide or other immunosuppression has been used in patients with systemic lupus erythematosus or secondary APS/CAPS, though neither has shown convincing benefit [5,12].
Eculizumab is a humanized monoclonal antibody with specificity for complement C5. Binding inhibits cleavage of C5, preventing the generation of the terminal complement C5b9 complex [40]. Several case reports, case series, and systematic reviews [41–43] have described responses to eculizumab in CAPS (Table 1). The majority of reported cases describe favorable responses, though whether these reports truly reflect efficacy or publication bias to highlight positive outcomes is unknown. Two small retrospective cohort studies provide insight. Yelnik et al [44] conducted a retrospective cohort study of 11 patients presenting with severe CAPS. Despite standard management with anticoagulation, glucocorticoids, and plasma exchange, these patients experienced a deteriorating course, and received eculizumab a median (range) of 25 days (3–100) after episode onset. Five of 11 patients responded (45%), but 6 had no response; 4 of the non-responders died of CAPS complications. This was the first study to report inconsistent responses of patients with CAPS to eculizumab. The authors suggested likely publication bias in prior case reports, but also questioned whether earlier initiation of eculizumab might have salvaged additional patients. In response to the question of time to treatment, Faguer and Ribes [45] published results from a retrospective study of 5 patients (6 total episodes of CAPS) refractory to corticosteroids, anticoagulation and plasma exchange, as well as rituximab (given in 5/6 episodes), who were ultimately treated with eculizumab. Eculizumab was given relatively soon, a median (range) of 7 (2–15) days after episode onset. Four patients (5 episodes) demonstrated rapid and sustained responses to eculizumab. One patient died of refractory CAPS and associated hemophagocytic lymphohistiocytosis. No increase in infectious complications was observed with early administration of eculizumab.
Table 1.
Summary of reports of eculizumab in patients with antiphospholipid syndrome.
| – | Patient | Prior therapies | Eculizumab dose/duration | Outcome |
|---|---|---|---|---|
| Shapira [55] | 28 M with SLE, APS with PE, arterial thrombosis leading to leg amputation, and recurrent CAPS | Heparin, argatroban, fondaparinux, cyclophosphamide (CYC), steroids, IVIG, lepirudin, bivalirudin, aspirin, clopidogrel, plasma exchange (PEX) | Eculizumab 900 mg once, then 1200 mg biweekly for 1 y | Normalization of cytopenias and resolution of thrombotic events |
| Strakhan [43] | 36 F with HTN, acute renal failure, stroke, acute coronary syndrome, and MAHA | PEX, steroids | Eculizumab 900mg weekly for 4 wk then 1200 mg biweekly | Gradual improvement of MAHA, continued dialysis |
| Appenzeller [56] | 30 F with ITP and primary APS who developed CAPS after pregnancy | Hydroxychloroquine (HCQ), heparin, steroids, rituximab, PEX, immunoadsorption, dialysis | Eculizumab for 3 mo, mycophenylate, steroids | Resolution of thrombocytopenia and MAHA, with late partial relapse, dialysis dependent |
| Lonze [57] | 2 patients with prior CAPS, undergoing renal transplant | Prednisone, rituximab, anticoagulation | Eculizumab, 900mg the day after transplant, then 1200 mg biweekly | Both with functioning allografts and no recurrence of thrombotic events, follow up 4 mo −4 y |
| Zikos [58] | 47 M with primary APS, then CAPS with thrombocytopenia, multifocal thrombi including renal and hepatic infarcts | Heparin, PEX, IVIG, steroids, argatroban, heparin | Eculizumab 900mg weekly for 2 wk, then 1,200 mg q 7–10 d | Gradual improvement in cytopenias, ascites, and splenomegaly, no further thrombotic events for 16 mo of follow up, continued dialysis |
| Gustavsen [59] | 22 F with arterial thrombosis and ischemic ulcerations during pregnancy | Warfarin, LMWH, aspirin | Eculizumab 600mg weekly for 2 wk in anticipation of Cesarean section | No further thrombosis, improvement in ischemic pain, no adverse fetal effects |
| Hussain [47] | 18 F with triple positive APS with recurrent thrombosis, hepativ infarction, PE | enoxaparin, fondaparinux, apixaban, rivaroxaban, warfarin, aspirin and clopidogrel, HCQ, steroids, rituximab, PEX | Eculizumab 600mg weekly x4, then 900mg biweekly plus fondaparinux, aspirin, clopidogrel, hydroxychloroquine | No recurrence of thrombosis with 27 mo of follow up |
| Tinti [41] | 54 F with primary APS with intestinal infarction, then CAPS with ischemic limb, acute bilateral effusions, acute decompensated heart failure, acute kidney injury | HCQ, steroids, warfarin, steroids, IVIG, PEX | Eculizumab 600mg weekly for 1 mo with steroids, warfarin, hydroxychloroquine | Rapid improvement in respiratory involvement, progressive improvement in thrombocytopenia, anemia, and serum creatinine, and normalization of C3 and C4 serum levels. No recurrence of thrombosis for 1 y |
| Chitalia [60] | Three cases of CAPS | All received steroids and anticoagulation, one additionally treated with rituximab, and one with PEX | Eculizumab dosing and regimen details not available | One with initital improvement in skin mottling and no further thrombosis but died during initial hospitalization (after 4 doses of eculizumab) due to septic shock; 1 discharged with maintenance eculizumab, length of follow up not available; 1 with resolution of CAPS and continues on maintenance eculizumab, length of follow up N/A |
| Guillot [42] | 78 F with history of PE presents with CAPS with renal injury, hypertension and cerebral vascular lesions | Steroids, anticoagulation, PEX | Eculizumab 900mg weekly x 4, then 1200mg biweekly for 2 mo with steroids, anticoagulation and HCQ | No recurrence of CAPS in 12 mo of follow up |
| Chidharla [61] | 64 F with triple positive APS develops acute encephalopathy and COVID associated CAPS with acute venous thrombosis, new ischemic stroke, adrenal hemorrhage | Dexamethasone, remdesivir, PEX, rituximab, IVIG | Eculizumab 900mg weekly x2 | No recurrent thrombotic events at 1 mo follow up |
| Skoczynska [62] | 35 F with SLE and CAPS with severe TMA and respiratory, circulatory and renal failure | PEX, steroids, anticoagulation, dialysis | Eculizumab 900mg weekly for 1 mo, then 1200mg biweekly for 8 mo | Improvement in neurological and circulatory function, ongoing dialysis dependence, remission of CAPS at 9 mo follow up |
| Ruffatti [63] | 32 F with pregnancy-associated CAPS with renal failure, respiratory and circulatory failure, ischemic digits | Anticoagulation, steroids, PEX, IVIG | 1200mg weekly x3 then 900mg weekly x 6 | Rapid remission of CAPS symptoms |
| Nauseef [64] | 54 M with APS and Factor II G20210A carrier, and steroid-refractory ITP, develops CAPS with bilateral adrenal hemorrhage, acute thrombocytopenia, IVC thrombus and PE, and ear cartilage thrombosis | Steroids, IVIG, rituximab, oseltamivir, romiplostim, anticoagulation, HCQ | Eculizumab 900mg weekly x 4, then 1200mg biweekly x 6 mo | Found to have heterozygous deletion in CFHR3-CFHR1, no recurrence of thrombosis 1 y after cessation of eculizumab |
| Kello [65] | Case series of 9 patients with SLE and/or APS; 6 APS and 7 SLE, 4 SLE and APS | All APS patients received steroids and anticoagulation, 5 additionally received PEX, 2 cyclophosphamide, 1 mycophenolate mofetil, and 1 rituximab | Eculizumab duration ranged 3–60 mo | No recurrences of TMA, variable disease-free follow up after discontinuation from 1–30 mo, 100% survival at 3 mo, 2 subsequent deaths related to discontuation of dialysis |
| Rovere-Querini [66] | 33 F with FVL, primary triple positive APS, prior PE and 2 miscarriages develops CAPS with acute hemorrage at 30 + 6 wk gestation with hemolytic anemia, thrombocytopenia, acute kidney injury | Warfarin replaced by LMWH during pregnancy, aspirin, HCQ, rituximab | Eculizumab 600mg hospitalization day 7 and 14, cesarean section day 9 due to thrombocytopenia. | Mild transient thrombocytopenia and transient inhibition of CH50 with low C3 in the newborn, with no adverse effects and spontaneous normalization |
| Kronbichler [67] | 30 F with IgA deficiency, ITP, splenectomy, APS, SLE with pregnancy-associated CAPS with cerebral, myocardial, renal and pulmonary involvement | Steroids, rituximab, PEX (severe intolerance), immunoadsorption, dialysis | Eculizumab 900mg weekly x4, then 1200mg biweekly for 3 mo | Resolution of TMA, lupus flare while on eculizumab |
| Wig [68] | 43 F with primary APS with thrombotic and pregnancy morbidity, with subsequent CAPS with HTN, intracranial hemorrhage, and recurrent thrombosis despite closely monitored anticoagulation | steroids, CYC, PEX, rituximab, IVIG, anticoagulation | Eculizumab 900mg once then 1200mg weekly x 6 until resolution of thrombocytopenia, then tapered 1200mg every other week, 900mg every other week, 900mg monthly | Improvement in thrombocytopenia, recovered of renal function and cessation of dialysis, reduction in steroids, functional improvement following strokes and living independently |
The most comprehensive data on eculizumab use in CAPS to date comes from Lopez-Benjume [46] who published a descriptive analysis from the “CAPS Registry.” Of the 584 patients in this registry, 39 (6.7%) were treated with eculizumab. Seventy-seven percent of the treated patients had primary APS. Twenty-nine (74%) patients recovered from the episode of CAPS, 4 showed a partial response, 9 (23%) had progressive symptoms and 5 patients ultimately died. Responses were sustained with only 1 relapse during a median follow up of 10.7 months. Most patients were treated with 900 mg of eculizumab weekly for 4 weeks, followed by 1200 mg every 2 weeks for variable durations.
Taken together, these data suggest a potentially important role for eculizumab in treatment of CAPS. However, these reports are retrospective, and reporting bias may exist, thus additional studies are needed to answer multiple outstanding questions and guide the most efficient and effective use of this expensive agent. The presence of mutations in complement regulatory genes in these patients, that in some cases resemble those seen in CM-HUS, suggest an important role for the alternative pathway of complement. However, whether APS patients with specific complement regulatory gene mutations will respond better to complement inhibition has not been assessed. The optimal timing of administration, and duration of eculizumab in CAPS is unknown, and how to incorporate eculizumab into a multiagent regimen that includes plasma exchange has also not been determined. In most reports, treatment with eculizumab was continued for weeks to more than a year, but whether prolonged treatment is needed likely varies among patients and at this point there are no biomarkers to suggest the safety of treatment discontinuation. Given the rarity of CAPS, prospective randomized trials of eculizumab will be difficult, and answers to these questions may need to be derived from databases of real-world experience with this agent.
Occasional patients with APS, while not meeting criteria for CAPS, follow an aggressive course and develop recurrent thrombotic events despite optimal anticoagulation. Others may experience small thrombi, resembling vasculitis. While there is no clear treatment path for such patients, a recent case report suggests potential benefit of eculizumab in refractory APS [47]. This report describes an 18-year old female with persistently triple-positive APS, who developed spontaneous thrombosis in multiple areas, including the inferior vena cava, brachiocephalic and axillary veins. She was treated with multiple anticoagulant regimens over the course of several years, including enoxaparin, fondaparinux, apixaban, rivaroxaban, and warfarin, both alone and in combination with antiplatelet agents (aspirin, clopidogrel). Despite these measures, as well as the use of hydroxychloroquine (HCQ), corticosteroids, rituximab, and even extensive plasma exchange (that was ineffective in lowering her aPL levels), she continued to develop recurrent thrombi. Ultimately, after developing hepatic infarction and another episode of pulmonary embolism, the patient was initiated on eculizumab at a dose of 600 mg/wk for 4 weeks, followed by 900 mg every 2 weeks thereafter. Fondaparinux, clopidogrel, HCQ and clopidogrel were continued. On this regimen, she remained thrombosis free for more than 27 months, with follow up ongoing (Figure 4). Though only a single case, much can be learned from such experiences, and this remarkable outcome suggests that complement activation may have been substantially contributing to this patient’s refractory thrombosis. Though the cost of eculizumab precludes its use in all but the most severely affected APS patients, several orally available complement inhibitors are in development and may offer a promising new approach to APS. We are unaware of any reports concerning the long acting C5 inhibitor raviluzumab, or the newly-approved C3 inhibitor, pegcetoplan, in APS. Further work is needed to better define APS patients likely to benefit from these and other emerging agents.
Fig. 4.
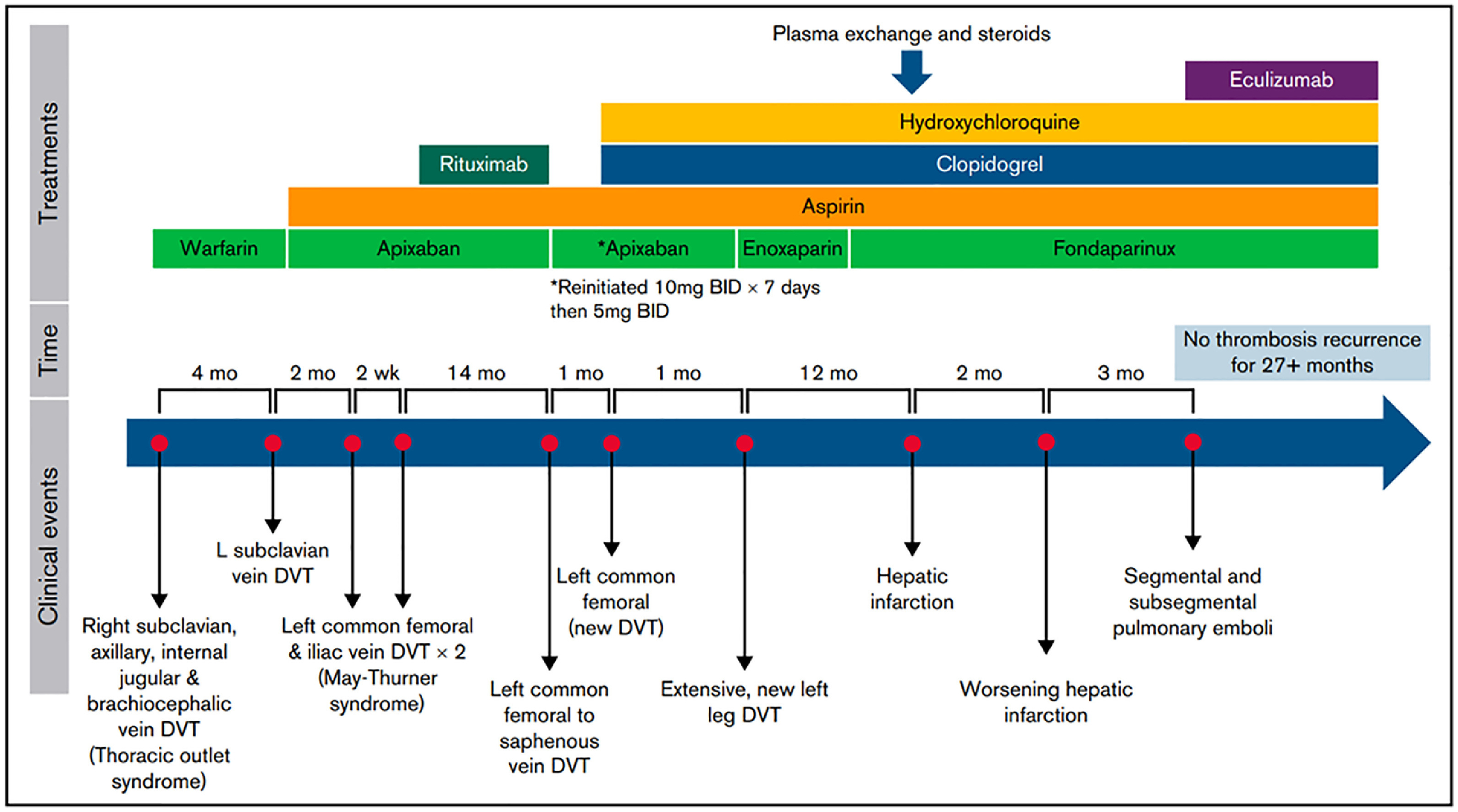
Clinical course and timeline of a case of refractory APS that ultimately responded to eculizumab, as reported by Hussain et al.47
Antithrombotic Effects of Hydroxychloroquine in Primary Antiphospholipid Syndrome
Hydroxychloroquine (HCQ) has been shown to reverse aPL-mediated disruption of annexin A5 crystal structures that shield phospholipid, with inhibition of coagulation reactions and implications for thrombosis and pregnancy complication [48]. Other studies have suggested an immunomodulatory action of HCQ mediated by decreasing antigen presentation and reducing production of proinflammatory cytokines. Anti-thrombotic effects may also reflect altered platelet adhesion and engagement with clotting factors [49,50].
A prospective study by Schmidt-Tanguy et al [49]. examined the role of hydroxychloroquine as secondary prophylaxis for recurrent thrombosis in primary APS. This small prospective, non-randomized trial enrolled 40 patients with primary APS and a history of VTE (arterial and obstetric morbidity were excluded). Patients were screened for traditional risk factors for thrombosis and genetic thrombophilia’s and underwent baseline venous Doppler and D-dimer assessment. Twenty patients received HCQ (400 mg daily) in addition to a VKA (fluinidione), while the other 20 received VKA alone. The baseline characteristics for the 2 cohorts were similar and, importantly, were comparable in terms of the aPL profile, single or double aPL positivity, and aPL evolution over time. At the end of the 36-month trial, there were 2 recurrent VTE (30%) in patients receiving anticoagulation alone vs none in patients receiving HCQ and VKA. The percentage of time in the therapeutic INR range was high in all cases. Two patients (10%) in each cohort experienced minor bleeding. Though this was a small, non-randomized study with few thrombotic events, it was the first prospective trial to assess the role HCQ in addition to anticoagulation, and suggested a possible ancillary role for HCQ in secondary prevention of thrombosis.
Building upon this, Kravvariti et al [51] reported a pilot open-label prospective randomized clinical trial to evaluate the effect of HCQ on thrombosis prevention and aPL levels in patients with primary APS. [51] Patients were followed prospectively for up to 3 years for development of thrombosis and assessment of aPL titers. All patients were continued on their existing anticoagulation regimens without modification. While initiation of immunosuppressive treatment was not allowed, patients on steroids at the time of enrollment were allowed to continue. Thrombotic events occurred in 7 of 50 (14%) patients in the trial – 6 in the standard of care cohort (4 patients on VKA and 2 patients on VKA plus low dose aspirin), and 1 in the HCQ plus standard of care arm (this patient later revealed they had discontinued HCQ). In the intention-to-treat analysis, HCQ use was associated with 85% reduction in the incidence rate of thrombosis (0.001 vs 0.008 for usual care, log-rank P = .048). In the Cox model, stratified by triple-positive aPL status and adjusted for other traditional risk factors for thrombosis, HCQ use was associated with a multivariate hazard ratio of 0.09 (95% CI = 0.011.−26, P = .074). Strengths of this trial include the prospective randomized design, a well-defined patient population inclusive of obstetric APS, and efforts taken to control for potential confounding and time-varying effects.
The ongoing HIBISCUS trial [50] is a multicenter, international double-blind RCT to evaluate the safety and effectiveness of HCQ in secondary prevention of obstetrical and thrombotic events in primary APS. This trial should further contribute to our understanding of the role of HCQ in this setting.
Rituximab for Non-Criteria Manifestations of Antiphospholipid Syndrome
Rituximab is a chimeric monoclonal antibody that binds to CD20 on B lymphocytes and results in their depletion. In mouse models of SLE-associated APS, B cell blockade prevented disease onset [52,53] and prolonged survival [53]. Case reports and small case series suggest a potential role for B-cell directed therapy in recurrent thrombotic APS [39], though rituximab has not been systematically studied for this indication.
Erkan et al [54] reported the results of the RITAPS trial (Pilot Study of Rituximab for the Anticoagulation Resistant Manifestations of Antiphospholipid Syndrome), a single-center phase II trial of rituximab for non-criteria manifestations of APS. The RITAPS trial was designed with the primary objective of evaluating the safety of rituximab in aPL-positive patients, and secondarily to assess the effect on non-criteria manifestations and the aPL profile. Of note, only approximately half of the patients in this trial met the revised Sapporo criteria. The non-criteria manifestations of enrolled patients included thrombocytopenia, cardiac valvular disease, skin ulceration, APS nephropathy, and/or cognitive dysfunction. Eligible patients received 1000 mg of rituximab on days 1 and 15, and were subsequently followed for up to 1 year. Nineteen patients were enrolled in the trial and 11 (58%) met criteria for primary APS. Twelve patients (63%) were triple positive for aPL. Rituximab did not significantly alter the aPL profile of patients: all patients with positive results of LAC, aCL, and/or anti-β2GPI IgG antibody tests at baseline had positive results at 24 weeks and 52 weeks. With regard to clinical end points, 2 patients had improvement in thrombocytopenia, while 2 had no response. No patients with cardiac valvular disease improved during the established follow up period. All 4 patients with skin ulcerations experienced a complete response, though 1 developed a recurrence. Three patients with cognitive dysfunction experienced remission with normalization of the cognitive impairment index, and one had a partial response with >50% improvement. The authors conclude with appropriate caution given the small cohort that rituximab may be effective at controlling some of the non-criteria manifestations of APS.
The exploratory aims of the trial offer insights into the biological effects of rituximab in APS patients. Rituximab treatment predictably resulted in rapid B cell depletion within 2 weeks of the first infusion in all but 1 subject. B cell levels returned to baseline or normal levels, with significant inter-subject variability, over an interval from 6 to 24 months. One patient had a positive baseline human anti-chimeric antibody (HACA) test. At 36 weeks 9 (56%) of 16 patients had developed HACAs. Four of 6 (67%) patients with no response and 5 of the 11 (45%) patients with partial response, complete response, or recurrence demonstrated HACAs.
This trial remains one of the few prospective trials to systematically evaluate rituximab in APS. Although limited due to the small sample size, the trial offers a few key insights. First, the study demonstrated the safety of rituximab in APS. However, the response of aPL levels to rituximab was disappointing, with no substantial change in aPL profiles during 12 months of follow up; this suggests minimal or no effect on long-lived plasma cells or memory B cells. As the authors hypothesize, B cell effector function independent of antibody production may explain the interesting observation of some patients experiencing a clinical response in non-criteria manifestations without a substantial change in aPL profile. On the other hand, APS patients may display a heterogeneous antibody profile, and improvement in thrombocytopenia in this cohort may reflect reduction in levels of antibodies to “non-aPL” targets such as platelet glycoproteins; these responses are consistent with the 60% response rate to rituximab observed in immune thrombocytopenia (ITP). The most interesting responses were seen in skin ulceration where all patients had resolution of their lesions, and importantly, cognitive function where improvements were observed in aspects of attention, visuomotor speed, and flexibility.
Conclusions
Traditional management of APS remains anticoagulation. However, better and more durable treatment of this disorder will require a better understanding of the underlying immune dysregulation, the role of complement, and many other facets of the disorder that remain incompletely understood. The emerging treatment landscape in APS is in its infancy and the heterogeneity of both the patient population and the serologic characteristics of APS create challenges for the prospective study of novel treatment strategies. Further large-scale collaborative trials are needed to translate insights from preclinical and early clinical research into standard of care practice.
Funding
This work was supported by grant HL143402 (to KRM). AH is supported by grant U2CDK129330 (to John R Sedor and Walter F Boron).
Footnotes
Conflicts of Interest
The authors have no conflict of interest to disclose in relation to the submitted review.
References
- [1].Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- [2].Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat Rev Rheumatol 2011;7:330–9. doi: 10.1038/nrrheum.2011.52. [DOI] [PubMed] [Google Scholar]
- [3].Manukyan D, Müller-Calleja N, Jäckel S, Luchmann K, Mönnikes R, Kiouptsi K, et al. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J Thromb Haemost 2016;14:1011–20. doi: 10.1111/jth.13263. [DOI] [PubMed] [Google Scholar]
- [4].Müller-Calleja N, Ritter S, Hollerbach A, Falter T, Lackner KJ, Ruf W. Complement C5 but not C3 is expendable for tissue factor activation by cofactor-independent antiphospholipid antibodies. Blood Adv 2018;2:979–86. doi: 10.1182/bloodadvances.2018017095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ortel TL, Erkan D, Kitchens CS. How I treat catastrophic thrombotic syndromes. Blood 2015;126:1285–93. doi: 10.1182/BLOOD-2014-09-551978. [DOI] [PubMed] [Google Scholar]
- [6].Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, De Ramón E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis 2015;74:1011–18. doi: 10.1136/ANNRHEUMDIS-2013-204838. [DOI] [PubMed] [Google Scholar]
- [7].Chaturvedi S, McCrae KR. Diagnosis and management of the antiphospholipid syndrome. Blood Rev 2017;31:406. doi: 10.1016/J.BLRE.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rodziewicz M, D’Cruz DP. An update on the management of antiphospholipid syndrome. Ther Adv Musculoskelet Dis 2020;12. doi: 10.1177/1759720X20910855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cohen H, Cuadrado MJ, Erkan D, Duarte-Garcia A, Isenberg DA, Knight JS, et al. 16th International congress on antiphospholipid antibodies task force report on antiphospholipid syndrome treatment trends. Lupus 2020;29:1571–93. doi: 10.1177/0961203320950461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum 1999;42:1309–11. doi: 10.1002/1529-0131(199907)42:71309::AID-ANR13.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- [11].Devreese KMJ, Ortel TL, Pengo V, de Laat B. Laboratory criteria for antiphospholipid syndrome: communication from the SSC of the ISTH. J Thromb Haemost 2018;16:809–13. doi: 10.1111/JTH.13976. [DOI] [PubMed] [Google Scholar]
- [12].Cervera R, Rodríguez-Pintó I, Colafrancesco S, Conti F, Valesini G, Rosário C, et al. 14th international congress on antiphospholipid antibodies task force report on catastrophic antiphospholipid syndrome. Autoimmun Rev 2014;13:699–707. doi: 10.1016/j.autrev.2014.03.002. [DOI] [PubMed] [Google Scholar]
- [13].Cervera R, Espinosa G. Update on the catastrophic antiphospholipid syndrome and the “cAPS registryo. Semin Thromb Hemost 2012;38:333–8. doi: 10.1055/S-0032-1304718. [DOI] [PubMed] [Google Scholar]
- [14].Erkan D, Lockshin MD. Non-criteria manifestations of antiphospholipid syndrome. Lupus 2010;19:424–7. doi: 10.1177/0961203309360545. [DOI] [PubMed] [Google Scholar]
- [15].Hughes GRV, Khamashta MA. Seronegative antiphospholipid syndrome. Ann Rheum Dis 2003;62:1127. doi: 10.1136/ard.2003.006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nayfe R, Uthman I, Aoun J, Aldin ES, Merashli M, Khamashta MA. Seronegative antiphospholipid syndrome. Rheumatol (United Kingdom) 2013;52:1358–67. doi: 10.1093/rheumatology/ket126. [DOI] [PubMed] [Google Scholar]
- [17].Willis R, Lakos G, Harris EN. Standardization of Antiphospholipid antibody testing-historical perspectives and ongoing initiatives. Semin Thromb Hemost 2014;40:172–7. doi: 10.1055/S-0033-1364207/ID/JR02041-31. [DOI] [PubMed] [Google Scholar]
- [18].Galli M, Borrelli G, Jacobsen EM, Marfisi RM, Finazzi G, Marchioli R, et al. Clinical significance of different antiphospholipid antibodies in the WAPS (warfarin in the antiphospholipid syndrome) study. Blood 2007;110:1178–83. doi: 10.1182/BLOOD-2007-01-066043. [DOI] [PubMed] [Google Scholar]
- [19].Pengo V, Ruffatti A, Legnani C, Gresele P, Barcellona D, Erba N, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010;8:237–42. doi: 10.1111/j.1538-7836.2009.03674.x. [DOI] [PubMed] [Google Scholar]
- [20].Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to β2-glycoprotein I is complement dependent and requires a priming factor. Blood 2005;106:2340–6. doi: 10.1182/blood-2005-03-1319. [DOI] [PubMed] [Google Scholar]
- [21].Jankowski M, Vreys I, Wittevrongel C, Boon D, Vermylen J, Hoylaerts MF, et al. Thrombogenicity of β2-glycoprotein I-dependent antiphospholipid antibodies in a photochemically induced thrombosis model in the hamster. Blood 2003;101:157–62. doi: 10.1182/BLOOD-2002-05-1310. [DOI] [PubMed] [Google Scholar]
- [22].Willis R, Pierangeli SS. Pathophysiology of the antiphospholipid antibody syndrome. Autoimmun Highlights 2011;2:35–52. doi: 10.1007/s13317-011-0017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Williams FM, Parmar K, Hughes GR HB. Systemic endothelial cell markers in primary antiphospholipid syndrome. Thromb Haemost 2000;84:742–6. [PubMed] [Google Scholar]
- [24].Yasuda S, Bohgaki M, Atsumi T, Koike T. Pathogenesis of antiphospholipid antibodies: impairment of fibrinolysis and monocyte activation via the p38 mitogen-activated protein kinase pathway. Immunobiology 2005;210:775–80. doi: 10.1016/j.imbio.2005.10.009. [DOI] [PubMed] [Google Scholar]
- [25].Collison J Preventing NETosis to reduce thrombosis. Nat Rev Rheumatol 2019;15:317. doi: 10.1038/s41584-019-0234-6. [DOI] [PubMed] [Google Scholar]
- [26].Chaturvedi S, Brodsky RA, McCrae KR. Complement in the pathophysiology of the antiphospholipid syndrome. Front Immunol 2019;10:449. doi: 10.3389/fimmu.2019.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oku K, Nakamura H, Kono M, Ohmura K, Kato M, Bohgaki T, et al. Complement and thrombosis in the antiphospholipid syndrome. Autoimmun Rev 2016;15:1001–4. doi: 10.1016/j.autrev.2016.07.020. [DOI] [PubMed] [Google Scholar]
- [28].Sarma JV, Ward PA. The complement system. Cell Tissue Res 2011;343:227–35. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res 2016;118:1392–408. doi: 10.1161/CIRCRESAHA.116.306853. [DOI] [PubMed] [Google Scholar]
- [30].Dzik S Complement and coagulation: cross talk through time. Transfus Med Rev 2019;33:199–206. doi: 10.1016/J.TMRV.2019.08.004. [DOI] [PubMed] [Google Scholar]
- [31].Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 2020;135:239–51. doi: 10.1182/blood.2019003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Carrera-Marín AL, Romay-Penabad Z, Papalardo E, Reyes-Maldonado E, García-Latorre E, Vargas G, et al. C6 knock-out mice are protected from thrombophilia mediated by antiphospholipid antibodies. Lupus 2012;21:1497–505. doi: 10.1177/0961203312458839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum 2005;52:2120–4. doi: 10.1002/ART.21157. [DOI] [PubMed] [Google Scholar]
- [34].Romay-Penabad Z, Carrera Marin AL, Willis R, Weston-Davies W, Machin S, Cohen H, et al. Complement C5-inhibitor rEV576 (coversin) ameliorates in-vivo effects of antiphospholipid antibodies. Lupus 2014;23:1324–6. doi: 10.1177/0961203314546022. [DOI] [PubMed] [Google Scholar]
- [35].Seshan SV, Franzke CW, Redecha P, Monestier M, Mackman N, Girardi G. Role of tissue factor in a mouse model of thrombotic microangiopathy induced by antiphospholipid antibodies. Blood 2009;114:1675–83. doi: 10.1182/blood-2009-01-199117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, et al. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis 2009;68:1030–5. doi: 10.1136/ARD.2008.090670. [DOI] [PubMed] [Google Scholar]
- [37].Devreese KMJ, Hoylaerts MF. Is there an association between complement activation and antiphospholipid antibody-related thrombosis? Thromb Haemost 2010;104:1279–81. doi: 10.1160/TH10-06-0410/ID/JR0410-11. [DOI] [PubMed] [Google Scholar]
- [38].Legault K, Schunemann H, Hillis C, Yeung C, Akl EA, Carrier M, et al. Mc-Master RARE-Bestpractices clinical practice guideline on diagnosis and management of the catastrophic antiphospholipid syndrome. J Thromb Haemost 2018;16:1656–64. doi: 10.1111/jth.14192. [DOI] [PubMed] [Google Scholar]
- [39].Berman H, Rodríguez-Pintó I, Cervera R, Morel N, Costedoat-Chalumeau N, Erkan D, et al. Rituximab use in the catastrophic antiphospholipid syndrome: Descriptive analysis of the CAPS registry patients receiving rituximab. Autoimmun Rev 2013;12:1085–90. doi: 10.1016/J.AUTREV.2013.05.004. [DOI] [PubMed] [Google Scholar]
- [40].Dubois EA, Eculizumab Cohen AF. Br J Clin Pharmacol 2009;68:318–19. doi: 10.1111/J.1365-2125.2009.03491.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tinti MG, Carnevale V, Inglese M, Molinaro F, Bernal M, Migliore A, et al. Eculizumab in refractory catastrophic antiphospholipid syndrome: a case report and systematic review of the literature. Clin Exp Med 2019;19:281–8. doi: 10.1007/S10238-019-00565-8. [DOI] [PubMed] [Google Scholar]
- [42].Guillot M, Rafat C, Buob D, Coppo P, Jamme M, Rondeau E, et al. Eculizumab for catastrophic antiphospholipid syndrome—a case report and literature review. Rheumatology 2018;57:2055–7. doi: 10.1093/RHEUMATOLOGY/KEY228. [DOI] [PubMed] [Google Scholar]
- [43].Strakhan M, Hurtado-Sbordoni M, Galeas N, Bakirhan K, Alexis K, Elrafei T. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol 2014;2014:1–7. doi: 10.1155/2014/704371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yelnik CM, Miranda S, Mékinian A, Lazaro E, Quéméneur T, Provot F, et al. Patients with refractory catastrophic antiphospholipid syndrome respond inconsistently to eculizumab. Blood 2020;136:2473–7. doi: 10.1182/BLOOD.2020007499. [DOI] [PubMed] [Google Scholar]
- [45].Faguer S, Ribes D. Early use of eculizumab for catastrophic antiphospholipid syndrome. Br J Haematol 2022;196:e12–14. doi: 10.1111/bjh.17783. [DOI] [PubMed] [Google Scholar]
- [46].López-Benjume B, Rodríguez-Pintó I, Amigo MC, Erkan D, Shoenfeld Y, Cervera R, et al. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the “CAPS Registry. Autoimmun Rev 2022;21:103055. doi: 10.1016/J.AUTREV.2022.103055. [DOI] [PubMed] [Google Scholar]
- [47].Hussain H, Tarantino MD, Chaturvedi S, McCrae KR, Roberts JC. Eculizumab for refractory thrombosis in antiphospholipid syndrome. Blood Adv 2022;6:1271–7. doi: 10.1182/BLOODADVANCES.2021005657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rand JH, Wu X-X, Quinn AS, Taatjes DJ. The annexin A5-mediated pathogenic mechanism in the antiphospholipid syndrome: role in pregnancy losses and thrombosis. Lupus 2010;19:460–9. doi: 10.1177/0961203310361485. [DOI] [PubMed] [Google Scholar]
- [49].Schmidt-Tanguy A, Voswinkel J, Henrion D, Subra JF, Loufrani L, Rohmer V, et al. Antithrombotic effects of hydroxychloroquine in primary antiphospholipid syndrome patients. J Thromb Haemost 2013;11:1927–9. doi: 10.1111/JTH.12363. [DOI] [PubMed] [Google Scholar]
- [50].Belizna C, Pregnolato F, Abad S, Alijotas-Reig J, Amital H, Amoura Z, et al. HIBISCUS: Hydroxychloroquine for the secondary prevention of thrombotic and obstetrical events in primary antiphospholipid syndrome. Autoimmun Rev 2018;17:1153–68. doi: 10.1016/J.AUTREV.2018.05.012. [DOI] [PubMed] [Google Scholar]
- [51].Kravvariti E, Koutsogianni A, Samoli E, Sfikakis PP, Tektonidou MG. The effect of hydroxychloroquine on thrombosis prevention and antiphospholipid antibody levels in primary antiphospholipid syndrome: A pilot open label randomized prospective study. Autoimmun Rev 2020;19. doi: 10.1016/J.AUTREV.2020.102491. [DOI] [PubMed] [Google Scholar]
- [52].Akkerman A, Huang W, Wang X, Ramanujam M, Schiffer L, Madaio M, et al. CTLA4Ig Prevents initiation but not evolution of anti-phospholipid syndrome in NZW/BXSB Mice. Autoimmunity 2004;37:445. doi: 10.1080/08916930400008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kahn P, Ramanujam M, Bethunaickan R, Huang W, Tao H, Madaio MP, et al. Prevention of murine antiphospholipid syndrome by BAFF blockade. Arthritis Rheum 2008;58:2824–34. doi: 10.1002/ART.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Erkan D, Vega J, Ramón G, Kozora E, Lockshin MD. A pilot open-label phase II trial of rituximab for non-criteria manifestations of antiphospholipid syndrome. ARTHRITIS Rheum 2013;65:464–71. doi: 10.1002/art.37759. [DOI] [PubMed] [Google Scholar]
- [55].Shapira I, Andrade D, Allen SL, Salmon JE. Induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. ARTHRITIS Rheum 2012;64:2719–23. doi: 10.1002/art.34440. [DOI] [PubMed] [Google Scholar]
- [56].Appenzeller S, Souza FHC, Wagner Silva de Souza A, Shoenfeld Y, de Carvalho JF. HELLP syndrome and its relationship with antiphospholipid syndrome and antiphospholipid antibodies. Semin Arthritis Rheum 2011;41:517–23. doi: 10.1016/j.semarthrit.2011.05.007. [DOI] [PubMed] [Google Scholar]
- [57].Lonze BE, Singer AL, Montgomery RA. Eculizumab and renal transplantation in a patient with CAPS. N Engl J Med 2010;362:1744–5. doi: 10.1056/nejmc0910965. [DOI] [PubMed] [Google Scholar]
- [58].Zikos TA, Sokolove J, Ahuja N, Berube C. Eculizumab induces sustained remission in a patient with refractory primary catastrophic antiphospholipid syndrome. J Clin Rheumatol 2015;21:311–13. doi: 10.1097/RHU.0000000000000290. [DOI] [PubMed] [Google Scholar]
- [59].Gustavsen A, Skattum L, Bergseth G, Lorentzen B, Floisand Y, Bosnes V, et al. Effect on mother and child of eculizumab given before caesarean section in a patient with severe antiphospholipid syndrome. Med (United States) 2017;96. doi: 10.1097/MD.0000000000006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chitalia A, Swoboda D, Broome C. Management of catastrophic antiphosphopholipid syndrome with eculizumab. Blood 2016;128:2603. doi: 10.1182/BLOOD.V128.22.2603.2603. [DOI] [Google Scholar]
- [61].Chidharla A, Syed SB, Chatterjee T, Tarantino MD. A case report of covid-associated catastrophic antiphospholipid syndrome successfully treated with eculizumab. J Blood Med 2021;12:929–33. doi: 10.2147/JBM.S324873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Skoczynska M, Crowther MA, Chowaniec M, Ponikowska M, Chaturvedi S, Legault K. Thrombotic microangiopathy in the course of catastrophic antiphospholipid syndrome successfully treated with eculizumab: case report and systematic review of the literature. Lupus 2020;29:631–9. doi: 10.1177/0961203320917460. [DOI] [PubMed] [Google Scholar]
- [63].Ruffatti A, Tarzia V, Fedrigo M, Calligaro A, Favaro M, Macor P, et al. Evidence of complement activation in the thrombotic small vessels of a patient with catastrophic antiphospholipid syndrome treated with eculizumab. Autoimmun Rev 2019;18:561–3. doi: 10.1016/J.AUTREV.2019.03.015. [DOI] [PubMed] [Google Scholar]
- [64].Nauseef JT, Lim HI, DeSancho MT. Successful outcome with eculizumab treatment in a patient with antiphospholipid syndrome presenting with an unusual thrombotic storm. J Thromb Thrombolysis 2021;52:597–600. doi: 10.1007/s11239-020-02343-w. [DOI] [PubMed] [Google Scholar]
- [65].Kello N, El Khoury L, Marder G, Furie R, Zapantis E, Horowitz DL. Secondary thrombotic microangiopathy in systemic lupus erythematosus and antiphospholipid syndrome, the role of complement and use of eculizumab: Case series and review of literature. Semin Arthritis Rheum 2019;49:74–83. doi: 10.1016/j.semarthrit.2018.11.005. [DOI] [PubMed] [Google Scholar]
- [66].Rovere-Querini P, Canti V, Erra R, Bianchi E, Slaviero G, D’Angelo A, et al. Eculizumab in a pregnant patient with laboratory onset of catastrophic antiphospholipid syndrome A case report. Med (United States) 2018;97. doi: 10.1097/MD.0000000000012584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kronbichler A, Frank R, Kirschfink M, Szilágyi Á, Csuka D, Prohászka Z, et al. Efficacy of eculizumab in a patient with immunoadsorption- dependent catastrophic antiphospholipid syndrome: A case report. Med (United States) 2014;93. doi: 10.1097/MD.0000000000000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wig S, Chan M, Thachil J, Bruce I, Barnes T. A case of relapsing and refractory catastrophic anti-phospholipid syndrome successfully managed with eculizumab, a complement 5 inhibitor. Rheumatol (United Kingdom) 2015;55:382–4. doi: 10.1093/rheumatology/kev371. [DOI] [PubMed] [Google Scholar]