

Abstract
Autism spectrum disorders (ASD) display both phenotypic and genetic heterogeneity, impeding the understanding of ASD and development of effective means of diagnosis and potential treatments. Genes affected by genomic variations for ASD converge in dozens of gene ontologies (GOs), but the relationship between the variations at the GO level have not been well elucidated. In the current study, multiple types of genomic variations were mapped to GOs and correlations among GOs were measured in ASD and control samples. Several ASD-unique GO correlations were found, suggesting the importance of co-occurrence of genomic variations in genes from different functional categories in ASD etiology. Combined with experimental data, several variations related to WNT signaling, neuron development, synapse morphology/function and organ morphogenesis were found to be important for ASD with macrocephaly, and novel co-occurrence patterns of them in ASD patients were found. Furthermore, we applied this gene ontology correlation analysis method to find genomic variations that contribute to ASD etiology in combination with changes in gene expression and transcription factor binding, providing novel insights into ASD with macrocephaly and a new methodology for the analysis of genomic variation.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disease that consists of social interaction abnormalities and repetitive behaviors (1). Beyond these two core symptoms, ASD patients may exhibit additional behaviors and comorbidities (2,3) such as seizures, aggressive behavior, intellectual disability and brain development abnormalities. Thus, ASD patients manifest substantial phenotypic heterogeneity. About 25% of ASD patients display early brain overgrowth (macrocephaly) as a comorbidity (4,5). Previous studies demonstrated that this overgrowth begins in mid-gestational fetal development and persists postnatally (4). Although ASD is often diagnosed by 3 years of age on the basis of the presence of core symptoms, the later age of onset makes it difficult to investigate prenatal pathophysiology. As the brain overgrowth abnormalities precede the behavioral abnormality, understanding the genetic mechanisms of ASD associated with macrocephaly could facilitate an earlier diagnosis and potential therapeutic targets of ASD.
The genetic heterogeneity of ASD is also extensive and has been broadly accepted. Many de novo, rare inherited and common genomic variations have been previously reported (6–8). ASD-related variations cover multiple categories, including single nucleotide variants (SNVs), insertions–deletions (INDELs) and structural variations (SVs) such as larger deletions or duplications (7,9–11). These variations affect ˃700 genes (1) in dozens of gene ontologies (12) (GOs), underscoring the substantial role of genetics in phenotypic heterogeneity. We believe by focusing on ASD with a single phenotypic attribute, namely ASD with macrocephaly, we can minimize the complexity because of ASD phenotypic and genetic heterogeneity, thereby simplifying the discovery of genetic mechanisms responsible for this subset of individuals with ASD.
Traditional GO analysis may not be adequate to identify genes responsible for ASD etiology. First, as a result of population history, evolution or special function, some regions/genes display high rates of variation. For example, genes functionally related to biological/cell adhesion display high rates of evolution in the human genome (13,14). Variations of these genes can be found in both ASD patients and normal individuals. Second, rare variations, or variations on genes with ‘house-keeping’ functions that may be important for the phenotype, may not cause significant results in GO analysis, as the number of these genes is relatively small.
Although previous analyses have concluded that genes dysregulated in different ASD patients vary greatly, they converge on similar sets of pathways (12,15). However, the combinatorial pattern of converged pathways for genomic variants that are key to ASD etiology remains unclear. It is important to explore the interaction among GOs enriched with genomic variations in ASD. A methodology for the analysis of pathway correlation has recently been developed. ClueGO (16) was developed as a Cytoscape (17) plug-in capable of grouping GO terms into modules on the basis of the similarity among GOs. This tool provided a network visualization of GOs overrepresented in gene lists uncovered in genetic or gene expression analysis. However, the connection between GOs identified by these methods was determined by the similarity of genes in GO terms, not the quantitative measurement of GOs affected by genomic variations.
Furthermore, it is important to integrate the analysis of different types of genetic variations, given that each type accounts for a small proportion of ASD risk (7,18). Most ASD genomic studies focused on a single type of genomic variation (9–11). In several recent review articles, the combinatorial effect of SNVs and CNVs was identified (18), but the studies lacked efficient quantitative models. Although most quantitative models were originally designed to integrate variants of a single type, they may be repurposed for the integration of different types of data. For example, DAWN (19,20) and MAGI (21) were designed to combine de novo mutations into modules with gene expression and protein–protein interaction networks as supporting information. However, these models require transmission score or gene expression data, which could limit the application of these models in ASD research.
Several recent studies for ASD with macrocephaly consistently found that genes in functional groups encompassing ‘cell adhesion’ and ‘neuron development’ were enriched with genomic variations and transcriptionally dysregulated genes (22–24). Among ASD patients lacking macrocephaly as a comorbidity, several genes in close proximity to regions identified by GWAS of ASD were also in these functional groups (25,26). These results suggested that genes in these functional groups are important for ASD, although they are not limited to the subgroup of individuals with ASD and macrocephaly. On the other hand, several studies, using mouse and human iPSC models, demonstrated that WNT pathway activity was dysregulated in ASD with macrocephaly, whereas GO analysis for genes enriched with genomic variations in ASD seldom detected WNT-related GOs. Therefore, we hypothesize that the co-occurrence of variations on genes in different GOs may be critically important for the etiology of ASD, including ASD with macrocephaly. Evaluation of co-occurrence in gene expression datasets, such as weighted correlation network analysis (WGCNA) (27), has proven to illuminate linked pathways dysregulated in a variety of disorders (22,28).
In the current study, we exploit SNV/INDEL and SV (deletion) data from both the Simons Simplex Collection (SSC) dataset (https://www.nature.com/articles/nbt0416-364) from the Simons Foundation Autism Research Initiative (SFARI) (SFARISSC dataset) and sequencing data from previously published iPSC models for ASD with macrocephaly by our laboratory and collaborators (22) (validation dataset). Both datasets detected SNVs, INDELs and SVs enriched in ASD with macrocephaly and control individuals. Traditional GO analysis for genes with these variations identified a set of GOs that were highly consistent between the two datasets, including ‘cell-cell adhesion’ and ‘neuron differentiation’, among others. Using a pipeline we developed, the correlated GO (cGO) pairs specific to ASD with macrocephaly were identified, helping us to identify a set of variations that together contributed to the etiology of ASD with macrocephaly. With the same pipeline examining samples of ASD microcephaly patients and ASD with no brain size change, several GO pairs and a group of genomic variations were identified for each of these subtypes of ASD. These findings demonstrate the utility of using co-occurring variation to identify potential links among various genes participating in disparate GOs in the etiology of various subsets of ASD.
Results
Genomic variations of genes that function in neurogenesis, neuron development and cell adhesion were enriched in, but not unique to, ASD-macro probands
On the basis of ≥2 standard deviations (SD) larger/smaller than mean head circumference standard (4), 41 ASD-macro and 37 ASD-micro probands were selected from the SFARI-SSC database (Supplementary Material, Table S1A). Another 38 probands were selected that displayed head circumferences close to average (ASD-other, Fig. 1A). The significant increase in head circumference of the ASD-macro probands over their siblings confirmed our method of sample selection (Fig. 1B).
Figure 1.
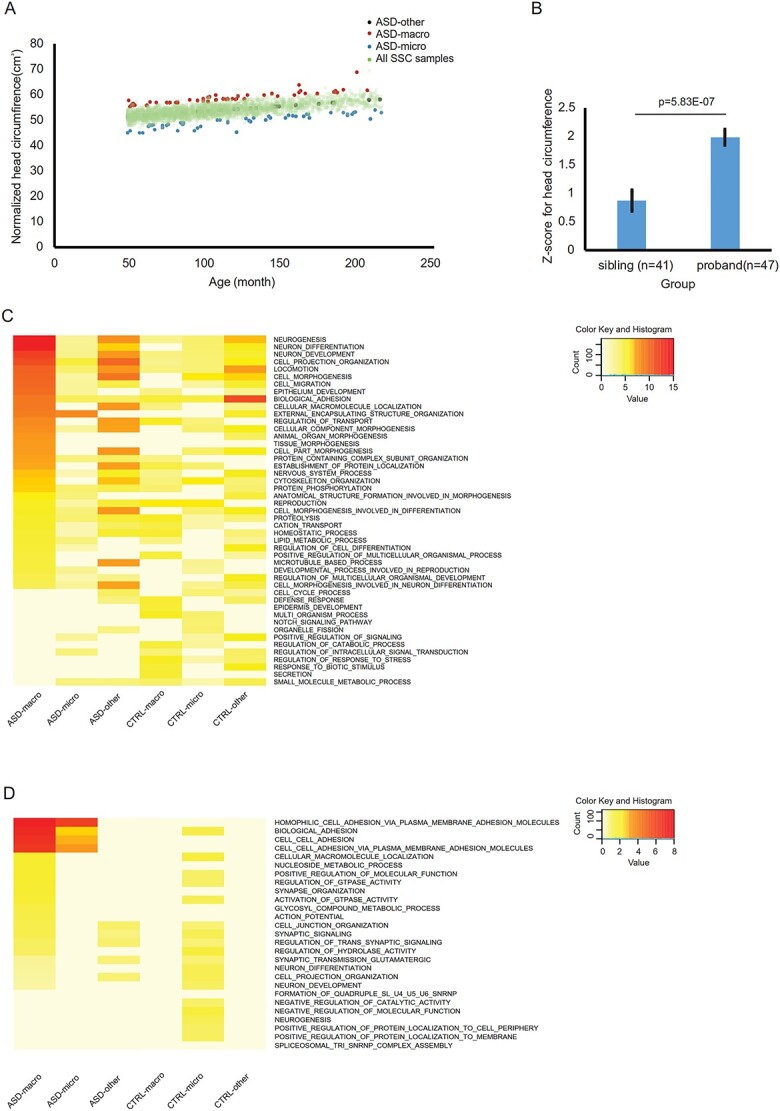
SNV, INDEL and deletions in ASD with macrocephaly and microcephaly from the SFARI Simplex Simons Collection (SFARI-SSC) dataset. (A) ASD with macrocephaly (red dots) and microcephaly (blue dots) individuals with head circumference 2 SD greater than or 2 SD less than the mean were selected from 2760 ASD probands in the SSC dataset, whereas ASD with no brain size phenotype (‘ASD-other’, black dots) were selected from the individuals with head circumference closest to the mean. All other ASD probands were plotted as green dots. (B) Head circumferences of probands with macrocephaly were significantly larger than their siblings (P = 5.8E-07). (C) The top 15 significant GOs for genes that displayed significant enrichment for probands over siblings on the basis of exome sequencing (SNV and INDEL) data were selected. Adjusted P-value for top 15 significant GOs from each of ASD-macro, ASD-micro and ASD-others list was plotted from most to least significant for ASD-macro from top to bottom. The color scale was proportional to the -log transformed adjust P-value. (D) GOs for genes intersected with deletions found in probands but not control individuals. Adjusted P-value for top 20 GOs from each of ASD-macro, ASD-micro and ASD-other list was plotted. The color scale was proportional to the -log transformed adjust P-value.
For genetic analysis, fathers of these 38 ASD-other probands were used as controls for ASD-macro and ASD-micro probands. Fathers from 37 ASD-micro families were used as controls for ASD-other probands. In total, 320 710 SNV/INDEL loci for these 191 individuals were retrieved from exome sequencing results of SFARI-SSC samples (Supplementary Material, Table S1A). After filtering for sample size, SNV/INDEL loci enriched in either ASDs or controls (enrich rate ≥0.1, see Materials and Methods) from each of the three groups (ASD-macro, ASD-micro and ASD-other) were selected. A total of 7373 loci for ASD-macro, 5233 loci for ASD-micro and 2458 loci for ASD-other were selected for subsequent analysis (Supplementary Material, Table S1B–D). Genes with these loci enriched in ASD probands were used as input for GO analysis (457 for ASD-macro, 411 for ASD-micro and 561 for ASD-other probands; 468, 480 and 491 for controls for these 3 groups, respectively) (Supplementary Material, Table S1E).
GO analysis demonstrated that, in ASD-macro samples, GO terms for neurogenesis, neuron development, organ morphogenesis and biological adhesion were overrepresented by genes with ASD-enriched loci (Fig. 1C, Supplementary Material, Table S2A). For ASD-micro samples, GO terms representing external encapsulating structure organization were most significant, together with GO terms related to neuron development and adhesion (Supplementary Material, Table S2B). The GO list for the ASD-other probands was similar to that of ASD-macro, with less significant levels for corresponding terms (Supplementary Material, Table S2C). These results suggested that the three types of ASD comorbidities could be characterized on the basis of enriched genomic variations. Specifically, genes related to neurodevelopment and adhesion were more represented in ASD-macro individuals.
The GO results for SV data confirmed the conclusion from the SNV/INDEL data. GO terms related to adhesion remained significant in ASD-macro and ASD-micro probands, whereas GO terms for neuron development and neuron differentiation were not the top terms in ASD-macro individuals, but were still significant (Fig. 1D, Supplementary Material, Table S1F–I).
Finding GOs associated with ASD comorbidities by gWGCNA pipeline
We hypothesized that the co-occurrence of genomic variations affecting genes with different but related biological functions would be important for the etiology of ASD and that different co-occurrence patterns of genomic variations would be present in ASD probands with different comorbidities. To determine this co-occurrence, we used WGCNA with genomic variation data as input (so called gWGCNA hereafter) and determined correlated ‘GO modules’ associated with brain size and ASD phenotype.
The 229 SFARI-SSC ASD and control samples were randomly assigned into two groups (ngroup1 = 115 and ngroup2 = 114). Genes with SV and SNV/INDEL loci (enrichment rate ≥0.1) were mapped to each GO for each sample (Supplementary Material, Table S3A). The gWGCNA pipeline was performed on each group with the same settings (nGO in module ≥30, module similarity ≤0.85). Performance of gWGCNA algorithm was similar in these two groups: the ‘similarity score’ was comparable for majority of Group 1 and Group 2 modules (Fig. 2A and B). The proportion of un-clustered GO module (gray module) was similar in the two groups. GO modules positively associated with ASD (ASD-only), with brain size (macrocephaly only) and with both (‘ASD-macro’) were detected in both Group 1 and Group 2 (Fig. 2C and D). Modules associated with ASD and negatively associated with brain size were considered ASD-micro modules. Other modules [except un-clustered GOs (gray module)] were labeled as ‘non-significant’ (Supplementary Material, Table S3C).
Figure 2.
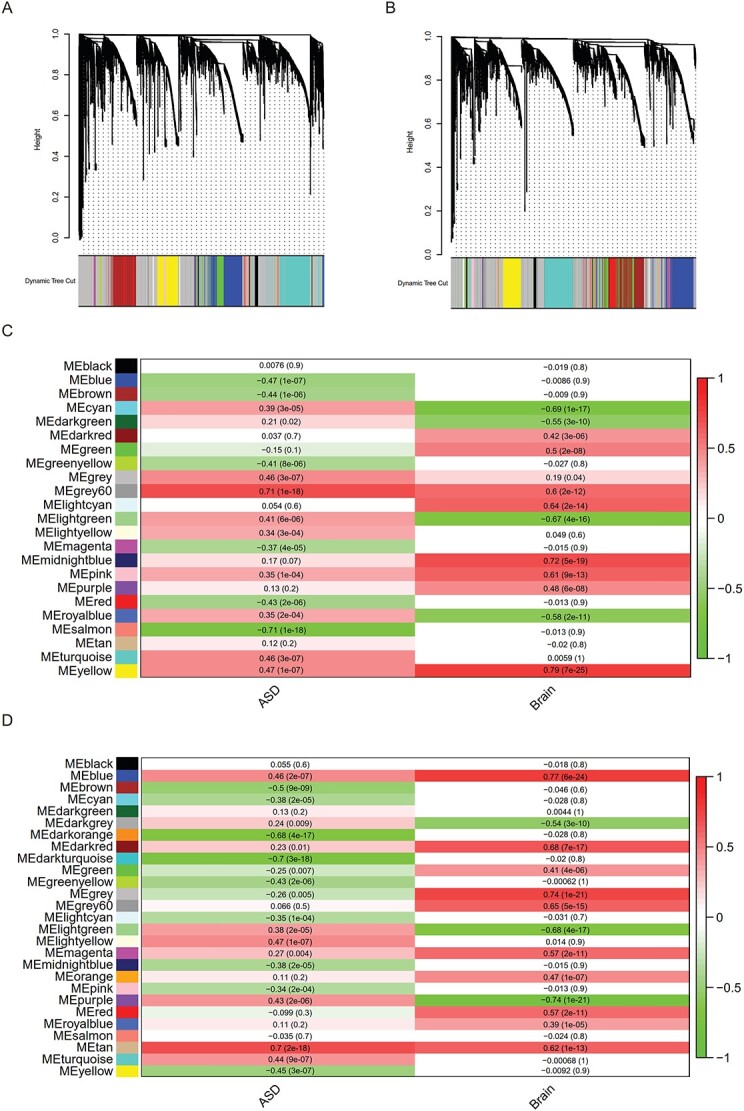
Find GO modules associated with ASD phenotype using WGCNA. (A) and (B) The GO dendrograms from WGCNA based on SNV/INDEL/SV data mapped to GOs from SFARI-SSC families in (A) Group 1 and (B) Group 2. (C) and (D) GO modules associated with ASD or brain size phenotype in (C) Group 1 and (D) Group 2. Pearson correlation (r) and P-value between phenotype (‘ASD’ or ‘Brain’) and eigenvalue of each GO module was shown in each grid, with the density of the red color representing positive correlation and the density of the green color representing negative correlation.
The GOs in corresponding module groups between Group 1 and Group 2 were conserved (Fig. 3A–E, Supplementary Material, Table S3D), with the highest proportion of overlap between Group 1 and 2 being for ‘ASD-only’ (73.9%, Fig. 3A), followed by ‘ASD-macro’ (65.5%, Fig. 3B), then ‘Macro-only’ (43.1%, Fig. 3C) and lowest for ‘ASD-micro’ (37.1%, Fig. 3D). These results suggested that this pipeline reliably detected GO modules associated with ASD and ASD-macro.
Figure 3.

GOs associated with ASD or brain size phenotype detected by the gWGCNA pipeline. The GOs within each of the significant modules associated with ASD and/or brain size in Figure 2 were collected. The numbers of GOs detected in Group 1 (blue circle), Group 2 (yellow circle) and overlap between the two groups (in red color) are displayed in a Venn diagram. Below each Venn diagram, the number of GOs in the GO module and representative GOs was listed. (A) GO modules that displayed significant positive association with ‘ASD’ and insignificant association with ‘brain’ were considered as ‘ASD-only’. (B) GO modules that displayed significant positive association with ‘ASD’ and significant positive association with ‘brain’ were considered ‘ASD-macro’. (C) GO modules that displayed insignificant association with ‘ASD’ and significant positive association with ‘brain’ were considered ‘macro-only’. (D) GO modules that displayed significant association with ‘ASD’ and significant negative association with ‘brain’ were considered ‘ASD-micro’. (E) All GO modules that displayed significant association with neither ‘ASD’ nor ‘brain’ were considered as ‘non-significant’.
GO groups for WNT, neuron morphology/function and organ morphogenesis were enriched in GOs associated with ASD macrocephaly
We next took the overlapped GOs for each module group as input to determine whether a few functionally similar GO groups (called ‘GO groups’ hereafter) were enriched. We first tested GOs overrepresented by genes with ASD-enriched CNVs (12), which included cell proliferation, GTPase/Ras activity and organ morphogenesis. In addition, WNT signaling was tested because WNT activity was decreased in ASD with macrocephaly (22). Furthermore, GOs presumably related to brain size change such as head/brain development and neuron morphology/function were examined.
A proportion test (29) demonstrated that WNT signaling, neuron function/morphology and organ morphogenesis-related GOs were enriched in GO modules associated with ASD-macro (Fig. 4A, Supplementary Material, Table S3E). Synapse-related GOs were enriched in those associated with ASD-only. These results are supported by previous publications that found the importance of WNT signaling for ASD with macrocephaly (22). GOs for neuron and organ morphogenesis were also significant and plausible GO categories found for brain size change. On the other hand, synapse function, especially vesicle release related GOs, was enriched in ASD-only associated GOs (Fig. 4A, Supplementary Material, Table S3E), suggesting that synapse function was dysregulated to affect neurological, behavioral and/or cognitive functions associated with ASD without brain size differences. That adhesion-related GOs was not enriched in the modules associated with ASD-macro may be because of the big number of GOs related with adhesion in non-significant GO modules. Therefore, even the number of adhesion-related GOs was big in GO modules significantly associated with ‘ASD’ or ‘Brain’, the proportion test result was not significant.
Figure 4.
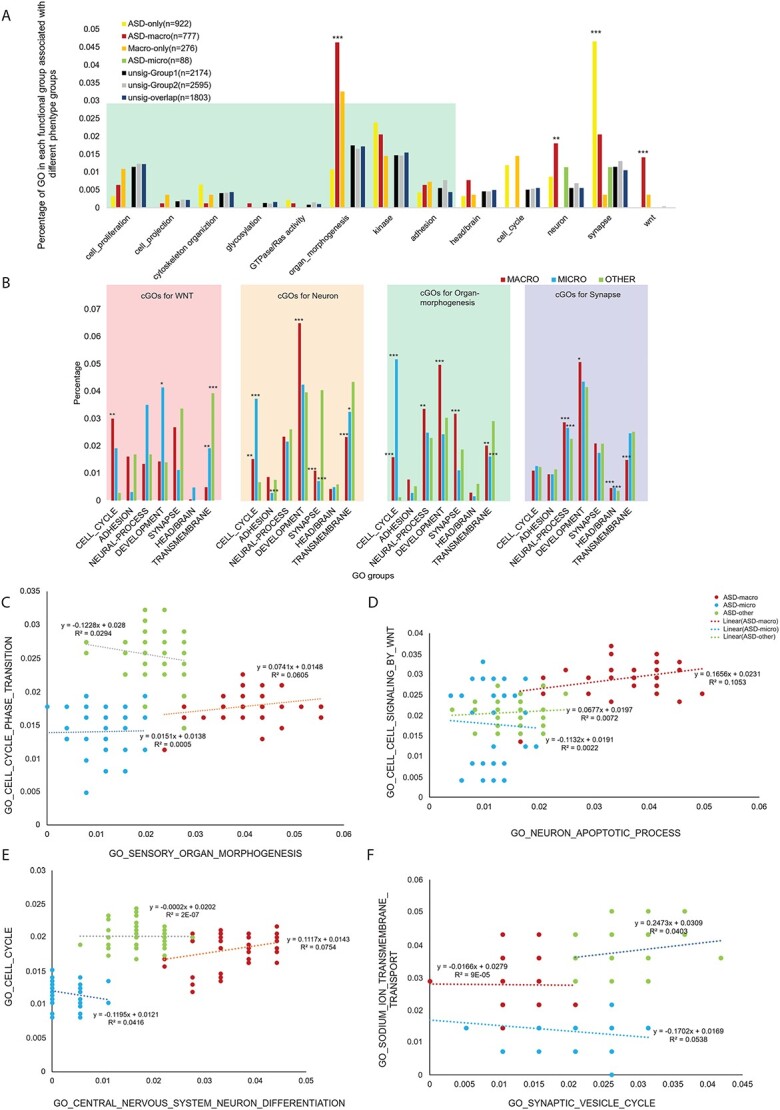
GO groups enriched in sGOs and cGOs. (A) The percentage of GOs related to specific functions (GO groups) was calculated for each of the five types of GO modules as described in Figure 3A–E. GO significantly enriched GO groups were indicated with associated P-values from the proportion test on the top of the bar. GO groups found to be significant from the analysis of CNVs from previously published results (12) were within the green shade. Other GO groups, such as ‘head/brain’, ‘cell cycle’, ‘neuron’, ‘synapse’ and ‘wnt’ were selected on the basis of our previously published data (22). (B) The percentage of selected GO groups (cGOs) for each of the four sGOs significant in (A) (WNT, neuron, organ morphology and synapse). Red bars represent the percentage of each of the GO groups from ASD-macro, blue bars represent the percentage of ASD microcephaly and green bars represent the percentage in ASD-other. Significance levels on top of the bars were suggested by asterisks (*P < 0.05; **P < 0.01; ***P < 0.001). (C)–(F) Example of GO–GO correlation. Genomic variation rate for each sGO was plotted on the x-axis and for each cGO was on the y-axis. Linear regression equation (y) and variance explained (R2) were on top of the regression line. Each dot represents one proband (ASD-macro in red, ASD microcephaly in blue and ASD-other in green). (C) GO central nervous system neuron differentiation versus GO cell cycle. (D) GO sensory organ morphogenesis versus GO cell cycle phase transition. (E) GO synaptic vesicle cycle versus GO sodium ion transmembrane transport. (F) GO neuron apoptotic process versus GO cell–cell signaling by WNT.
Several GOs, including cell cycle, were enriched in cGOs for ASD with different comorbidities
The four representative GO groups enriched in GOs associated with ASD phenotypes (WNT signaling activity, organ morphogenesis, neuron and synapse) were used as ‘seeding GOs’ (sGOs) for further analysis (Supplementary Material, Table S3F). With these sGOs, we calculated the Pearson correlation for each sGO versus all other (n = 8425) GOs in ASD-macro, ASD-micro and ASD-other (Supplementary Material, Table S3G–J). Consistent with our hypothesis that multiple genomic variations tend to affect genes with different but related functions and contribute to phenotype collaboratively in each individual, a set of ‘correlated GOs’ (so called cGOs hereafter) was found. In ASD-macro probands, there were 630 cGOs positively correlated with WNT sGOs, 2235 with neuron sGOs and 3011 with organ morphogenesis sGOs (Supplementary Material, Table S3G–I). In the ASD-other probands, 4322 cGOs were significantly correlated with synapse sGOs (Supplementary Material, Table S3J). As these positive correlations were unique to ASD probands (control samples showed either negative or insignificant correlation), variations on genes belonging to these GOs may contribute collaboratively to ASD etiology.
Similar to the enrichment test we performed for sGOs (Fig. 4A), we tested if specific GO groups were enriched in these cGOs (Fig. 4B). Cell cycle was enriched in cGOs correlated with all three sets of sGOs (sGO WNT, sGOs neuron and sGO organ_morphogenesis) for ASD-macro. On the other hand, the cell cycle was not enriched in cGOs correlated with sGO(synapse) for the ASD-other group. This finding suggested that cell cycle might be a very important biological process in brain size change in ASD. Interestingly, cell cycle-related GOs were significantly enriched in both cGOs found in ASD-macro and ASD-micro samples, except for the cGOs correlated with WNT sGOs, in which cell cycle-related GOs were only enriched in ASD-macro samples (Fig. 4B). These observations may together suggest that variations in WNT signaling could trigger changes in cell cycle to cause ASD with macrocephaly, but some other factor(s) may be responsible for changes in cell cycle in ASD with microcephaly. Two example plots further elucidate the correlation between cell cycle-related GOs and GOs related to neuron (Fig. 4C) or organ morphogenesis (Fig. 4D).
Adhesion-related GOs were never enriched in cGOs in either the ASD-macro or ASD-other group (Fig. 4B). Only the ASD-micro group showed deprivation of adhesion-related GOs compared with either ASD-macro or ASD-other in cGOs correlated with sGO ‘neuron’. These observations do not exclude the contribution of adhesion to ASD etiology; the suggested adhesion was not specific to any of these three ASD subgroups. In other words, adhesion-related variations may not account specifically for brain size comorbidity in ASD.
‘Neural process’-related GOs were all GOs related to the nervous system except for those with ‘neuron’ in the name. It(clarify) is not enriched in cGOs correlated with most sGOs for ASD-macro except for sGO ‘organ morphogenesis’. Nonetheless, in cGOs correlated with sGO for ASD-other (‘synapse’), ‘Neural process’-related cGOs were enriched in both the ASD-macro and ASD microcephaly group, which may suggest that this GO group may be too broad to be specifically linked with any sGO.
‘Development’-related GOs were enriched in cGOs correlated with sGOs for neuron and organ morphogenesis in the ASD-macro group (Fig. 4B). However, this enrichment was not observed in the ASD-micro group. Together, this finding could suggest that common variants on development-related genes may co-occur with variants affecting neuron/organ morphogenesis to cause ASD with macrocephaly.
‘Synapse’-related GOs were enriched in cGOs for sGO neuron in the ASD-other group, suggesting that the biological process related to neuron and synapse was correlated closer in the ASD-other group. Again, this endorsed the effect on the behavioral rather than brain morphological side of the synapse-related variations.
Surprisingly, the ‘head/brain’-related GOs were not enriched in cGOs correlated with most sGOs except for sGO synapse in the ASD-micro and ASD-macro group. Our interpretation was that variations for brain or head morphology changes may not often correlate with genes function on WNT/neuron/organ morphogenesis. Instead, they could function independently or tend to co-occur with variants with other functions such as synapse.
‘Transmembrane’-related GOs were enriched in cGOs for sGO neuron, sGO WNT and sGO organ morphogenesis in the ASD-other group, suggesting that the ‘transmembrane’-related GOs may ‘amplify’ the effect of variants on WNT, neuron and organ_morphogenesis-related genes in ASD without brain size changes. ‘Transmembrane’-related GOs were significantly enriched in cGOs correlated with sGO synapse in both the ASD microcephaly and ASD-other group. Considering that the enrichment of vesicle-related GO terms was enriched in sGO group synapse in the ASD-other group (11 of 43 sGOs for synapse, proportion test, P < 2.2e-16, Fig. 4A, Supplementary Material, Table S3F), the correlation between transmembrane process and vesicle-related synapse function was specific for the ASD-other group. Some examples may further consolidate this interpretation. In Figure 4E, the positive correlation between sGO Synaptic vesicle cycle and cGO sodium ion transmembrane was specific to the ASD-other group and the correlation in the ASD-macro and microcephaly groups was negative.
The enrichment of WNT-related GOs in cGOs correlated with different type of sGOs was not systematically tested but there were examples that endorsed the correlation between WNT signaling and neuron-related GOs in ASD-macro. For example, the sGO ‘neuron apoptotic process’ was positively associated with the cGO ‘cell cell signaling by WNT’ (Fig. 4F), the correlation was positive in both the ASD-macro and ASD-other groups and the correlation coefficient was much larger in the ASD-macro group, whereas the correlation was negative in the ASD-micro group.
Variations identified on sGOs from ASD with macrocephaly
With GOs directly (sGOs) and indirectly associated (cGOs) with ASD or brain size being identified, we next focused on identifying genomic variations important for phenotype in these GOs. Two factors were considered to prioritize the key variations: the number of the genes occurring in each of the sGOs or cGOs and the enrichment of specific gene loci in ASD over control samples. The product of both factors was defined as the ‘combined score’ and given a standardized value (z-score). Variation loci were then ranked based on this ‘combined enrichment score’, and only variations with a z-score > 0 were selected.
In sGOs for ASD-macro, 126 variations (26 SV and 100 SNV/INDEL loci) were on genes from the three groups of sGOs (GOs related to WNT, GOs related to organ morphogenesis and GOs related to neuron development/function, inner circle in Fig. 5A, left panel). The length of each color bar in both the inner and outer circles was proportional to the number of variants selected in each corresponding sGO.
Figure 5.

Genomic variations enriched in sGOs and cGOs. (A)–(C) Genomic variations detected in ASD-macro samples. (A) Summary of genomic variants on genes belonging to GOs directly associated with ASD phenotype (sGOs, inner circle) and on genes belonging to cGOs (outer circle). (B–C) Summary of genomic variants on sGOs (B) and cGOs (C). The number of SV, SNV/INDELs on each sGO/cGO group and representative genes/GOs was listed. Bold: SV, Red: Known ASD gene. (D) Summary of genomic variations detected in ASD-other samples.
About 50% (70 of 126) of variant loci were on genes belonging to all three types of sGOs (WNT, neuron and organ morphogenesis) for the ASD-macro (Group III in Fig. 5A, left panel). On the other hand ˃30% of variants selected were unique to either neuron- (Group VI) or organ morphogenesis- (Group I) related sGOs (Fig. 5B). These variants may affect specific biological process in ASD etiology. The number of variants on genes belonging(?) to both WNT- and morphogenesis-related sGOs (Group IV) or on genes belonging to both neuron- and morphogenesis-related sGOs (Group II) was low (Fig. 5B). The 126 variants on sGOs for ASD-macro affected in total 86 genes. These genes were enriched with genes known to relate to ASD (n = 19, proportion test, P = 7.146e – 15), including GRIN2B, PTEN, SMG6, WNT2B, etc. PTEN was known to be important for macrocephaly (30); another gene on PI3KAKT pathway (AKT3) further increased the confidence of the variants we found as new biomarkers for ASD macrocephaly.
Additional variations identified using sGO/cGO correlations from ASD macrocephaly
Next, we determined whether identifying cGOs with each of the sGOs would provide further insight into ASD with macrocephaly. In cGOs in ASD-macro group, 1476 genomic loci (91 SV and 1385 SNV/INDEL loci) were selected (outer circles in Fig. 5A, left panel).
Among these loci ˃50% (45 SV and 852 SNV/INDEL) were on cGOs (Group 3 in Fig. 5C, n = 375) correlated with all three types of sGOs (WNT, neuron and organ morphogenesis), suggesting that these variants could be ‘triggered’ by variants on genes in all these three major biological processes to cause ASD macrocephaly. Of these variants, 136 were on known ASD genes (proportion test, P < 2.2e-16), including NRXN1, RELN and SEMA5A, whereas 761 variations were on genes not yet related to ASD, including AKAP13, HTT, NAV2 and TET3. These identified variations are promising candidates for biomarkers of ASD with macrocephaly. On the other hand, there was a much smaller number of variants on cGOs correlated with single type of sGOs (Fig. 5C). For example, the number of cGOs correlated with sGO for morphogenesis was large (Group 1 in Fig. 5C, n = 1279), and variations selected from these cGOs were few (n = 154, 20 SVs, 134 SNV/INDELs). Similarly, from 528 cGOs correlated with sGO neuron (Group 7), only 25 variants passed the filter standard of the combined score. These suggest the combined score, which take both frequency of gene in cGOs and the locus level enrichment in the population into consideration, would largely suppress potential false positive rate in candidate loci detection from cGOs.
One exception was cGOs specifically correlated with sGO for WNT signaling (Group 5 in Fig. 5C). In 68 cGOs, 105 variations passed the combined score filter. This higher ratio of variants selected per cGO suggested that genes interacted with WNT signaling tend to be involved in multiple biological processes (GOs) and carry variants enriched in ASD.
Above average ratio of variants selected per cGO was also observed in the 106 cGOs correlated with both WNT and morphogenesis sGOs (Group 4 in Fig. 5C) and in 81 cGOs correlated with both neuron and WNT sGOs (Group 6). A total of 89 variants were selected in each group by combined score. These results together emphasized the variants, not only those on genes belonging to WNT signaling pathway but also those functionally interacting with WNT signaling, were important for ASD with macrocephaly, rendering them good biomarkers.
High consistency between SFARI-SSC and validation dataset results
We next determined whether any of our findings from the SFARI-SSC dataset could be replicated. Using the ASD and control fibroblast cell lines from a previous publication (22), we detected SNVs/INDELs using exome sequencing and detected SVs using matepair sequencing (These variations were called validation dataset hereafter). In detail, there were 333 846 SNV loci (Fig. 6A), corresponding to 837 180 SNV events (Fig. 6B) in 8 ASD and 5 control samples. There were 33 908 INDEL loci (Fig. 6C), corresponding to 69 031 INDEL events (Fig. 6D). Importantly, 80.82% of the 1924 selected genomic variation loci from SFARI-SSC ASD-macro samples had matches in the validation dataset (Fig. 6G, Supplementary Material, Tables S4A,S6A andS6E).
Figure 6.
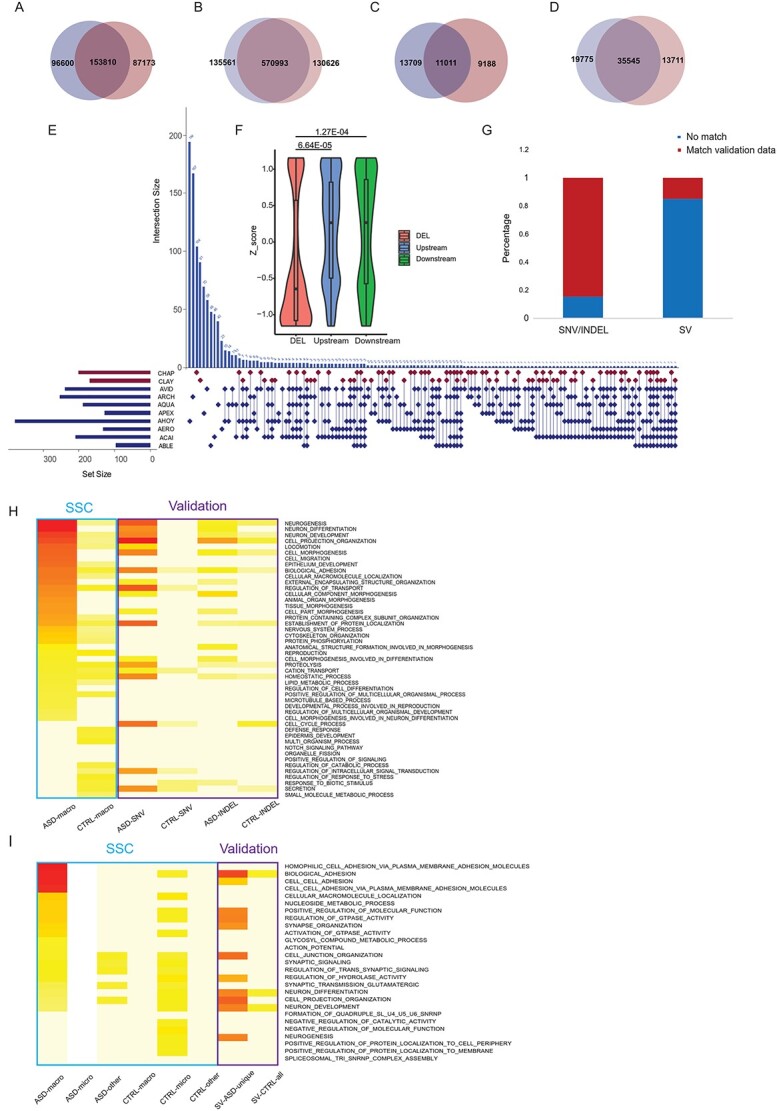
SNVs INDELs and SVs in validation dataset. (A)–(D) Venn diagrams of number of loci detected in the validation dataset for SV events (A), Blue: ASDs, Red: Controls. SNV events (B), INDEL loci (C) and INDEL events (D). Blue: ASDs, Red: Controls. (E) SV (Deletions) detected in eight ASD with macrocephaly and two control individuals. Horizontal bar beside each sample name represents the total number of deletions detected in that sample. Control samples were in red and ASD samples in blue. Each row of the dot matrix corresponds to one sample and each column corresponds to one set of deletion loci with the same distribution pattern among these samples. Each vertical histogram bar represents the recurrence of a specific distribution pattern. Also, the recurrence number for each combination was labeled on top of the histogram bar. (F) Z-score of reading depth of exonic regions overlapped with deletions in ASD samples (red) demonstrated a significant reduction compared with genomic regions 10 KB upstream (blue) and downstream (green) of the deletion event. (G) The proportion of selected genomic loci from the SFARI-SSC dataset (as in Fig. 5B–D, nASD_macro = 1514, n asd_other = 410) that overlapped with the validation dataset. (H) GOs for genes with SNV/INDELs enriched in ASD from the validation dataset correspondingto top 15 GOs for each SFARI-SSC group (as in Fig. 1C). Color scale was proportional to -log transformed adjusted P-value. The blue rectangle depicted SFARI-SSC results; the purple rectangle depicted validation results. (I) GOs for genes with ASD-specific SVs from validation dataset corresponding to top 20 GOs for each SFARI-SSC group (as in Fig. 1D).
In total, 761 deletions were detected in the validation dataset (Fig. 6E). Exome sequencing data showed exonic regions overlapped with deletions we detected have reduced reads compared with nearby up/downstream regions (Fig. 6F). Of the deletion loci selected using combined score from SFARI-SSC dataset, 16.4% could find matches with these deletions (Fig. 6G). The relatively low matching rate could be accounted for by the lower frequency of deletions compared with SNVs/INDELs in population and would tend to be missed in the small sample size validation dataset (Fig. 6G, Supplementary Material, Table S8A).
GOs overrepresented by genes enriched with SNVs/INDELs/SVs in ASDs over controls in the validation dataset largely overlapped with GOs overrepresented by genes with ASD-enriched variations in the SFARI-SSC dataset in the ASD-macrocephaly group (Fig. 6H and I). This GO pattern similarity confirmed that, although high individual heterogeneity exists at the gene level, the overall genetic machinery of ASD macrocephaly was conserved at the level of GOs and could be distinguished from those of control individuals and other ASD subgroups. In this sense, the RNASeq, ATAC-Seq and ChIPSeq data collected from the validation dataset could be related to genomic variations selected from the SFARI-SSC dataset with high confidence.
Interaction among genomic variations, gene expression and transcriptional regulation related to WNT/β-catenin in ASD macrocephaly
WNT signaling activity was significantly reduced in both mouse and human(?) neural progenitor cell (NPC) models of ASD with macrocephaly. For the 1514 loci we selected from the SFARI-SSC ASD-macro group, ChEA analysis showed that transcription factors (TFs) related to the WNT pathway (e.g. TCF4, SOX2, SMAD4, etc.) were overrepresented (Supplementary Material, Table S8G).
These SFARI-SSC ASD-macro results overlapped largely with the results from the ChEA analysis on genes overlapped with SV (deletion)s specific to ASD samples in validation dataset (Supplementary Material, Fig. S3, Supplementary Material, Table S8I). The overlap of ChEA results between SFARI-SSC ASD-other and the ASD-specific SV was much smaller (Supplementary Material, Fig. S3). Combined, these findings show that genomic variations affecting WNT signaling pathway-regulated transcriptional activity could contribute to the etiology of ASD with macrocephaly.
To further explore the effect of WNT-related transcription regulation on ASD with macrocephaly, we used publicly available ChIPSeq β-catenin binding data in human embryonic stem cells [hESCs; (31)], because β-catenin is the transcriptional effector of the canonical WNT pathway. Estaras et al. found 11 621 binding peaks distributed among various genomic regions (Fig. 7A) that bound a total of 4036 genes that were active in NPCs by ATAC-seq (Fig. 7B). A total of 227 β-catenin-bound genes overlapped with selected genomic variation loci from SFARI-SSC ASD-macro samples (Fig. 7B), which was much more frequent than the overlap (n = 74) with genes carrying loci selected from SFARI-SSC ASD-other samples (Fig. 7C). These results support the relationship between WNT/β-catenin signaling and ASD with macrocephaly.
Figure 7.
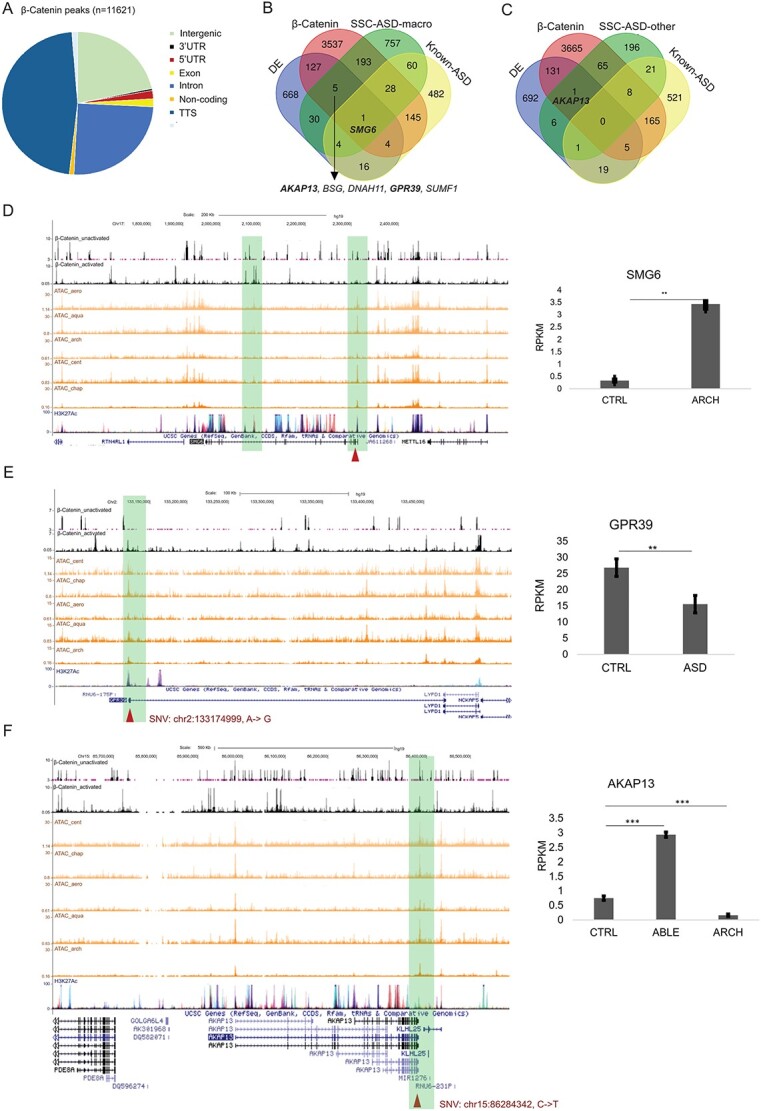
Possible effect of selected loci on gene expression and β-catenin transcription regulation. (A) Functional annotation of β-catenin binding peaks. (B) Selected genomic loci from SFARI-SSC ASD-macro samples that overlapped with β-catenin targets, DE genes and known ASD genes. Genes with loci selected by gWGCNA pipeline, showing differential expression and bound by β-catenin, were listed below the Venn diagram. SMG6, which meets all these criteria and is a known ASD gene, was labeled in the Venn diagram. Names of loci selected to be plotted individually were bold. (C) Selected genomic loci from SFARI-SSC ASD-other samples overlapped with β-catenin targets, DE genes and known ASD genes. (D) Example plot for SNV (Chr17:2203025, T- > G) on SMG6, black tracks: β-catenin ChIPSeq (upper: inactivated, lower: activated by WNT3A); orange tracks: ATACseq data; purple track: H3K27Ac ChIPSeq from UCSC. Right panel: RPKM for ASD line with this variation (ARCH) and CTRL NPC lines. (E) SNV (Chr2:133174999, A- > G) on GPR39. Right panel: RPKM for ASD and CTRL NPC lines. (F) SNV (chr15:86284342, C- > T) on AKAP13. Right panel: RPKM for ASD line with this variation (ABLE and ARCH) and CTRL NPC lines. Significance level of difference in gene expression between ASD and CTRL group was indicated using asterisks on top of the bar (*P < 0.05, **P < 0.01, ***P < 0.001).
We next examined whether any of the identified genes in the ASD loci displayed differences in gene expression in the NPCs from the iPSCs (define) from eight ASD and five control lines used in our validation dataset. Consistent with previous studies with these cell lines (22,32), the number of differentially expressed genes between ASD and control NPCs was small (n = 191, Supplementary Material, Table S5A). Among them, 40 overlapped with genomic variations selected from the SFARI-SSC ASD-macro samples (Fig. 7B) and eight overlapped with SFARI-SSC ASD-other samples (Fig. 7C). GO analysis of the 191 deferentially expressed (DE) genes did not yield any significant results. When we compared expression levels in each of the NPC lines with the average expression level of all four control lines, an additional 664 ‘individualized DE genes’ were detected (Fig. 7C and D, Supplementary Material, Table S5A).
The single gene identified that was differentially expressed, bound by β-catenin and was a known ASD gene was SMG6 (Fig. 7B). An SNV in the second exon of SMG6 (SMG6, Chr17: 2203025, T- > G) was enriched in both ASD-macro (by 17.5%) and ASD microcephaly probands (by 12.4%), but not in ASD-others from SFARI-SSC. This gene is a known ASD gene (33) and mainly functions in nonsense-mediated mRNA decay. This locus was also found in one ASD-macroc proband (ARCH) from the validation dataset. This locus is close to the TSS of SMG6 (Fig. 7D), which increased the possibility that this variation would affect TF binding. The β-catenin ChIPSeq peaks overlapped with SMG6 but not with this locus, so the function of this locus is unclear. We speculate that this locus could affect the interaction between β-catenin binding to the intron region of SMG6 and some other TFs that bind to TSS of SMG6. The RNASeq results demonstrated a significant decrease in SMG6 in the ASD NPC line (ARCH) compared with control lines.
One of the five genes that were differentially expressed and bound by β-catenin but not identified as known ASD gene, GPR39, contains an ASD-macro enriched SNV (Chr2:133174999, A- > G) (Fig. 7B and E). This gene encodes a rhodopsin-type G-protein-coupled receptor (GPCR) and is related to the pathophysiology of depression (34). It contains a binding target of β-catenin, and this variation (Chr2:133174999, A- > G) was in the ChIP binding area close to the TSS, which suggested that it could affect the binding of β-catenin to GPR39. The frequency of this allele in ASD in the SFARI-SSC ASD-macro samples was 18% higher than in control (Supplementary Material, Table S4A). This locus also displayed a 30% higher frequency in ASD than control in the validation dataset (Supplementary Material, Table S6A). RNASeq results demonstrated that GPR39 was downregulated in ASD (Fig. 7E). These findings combined suggest that this SNV in GPR39 may be a new biomarker for ASD with macrocephaly.
A second example within the five genes identified that was differentially expressed, bound by β-catenin and was not a known ASD gene, was AKAP13, which contains an SNV (chr15:86284342, C- > T, Fig. 7B, C and F). This gene encodes an A-kinase anchor protein, which is functionally related to both GPCR signaling and mTOR signaling (35). This SNV demonstrated a higher alternative allele frequency in ASD subjects than in controls in both the SFARI-SSC ASD-macro and SFARI-SSC ASD-micro samples (Supplementary Material, Table S6A). In contrast, the SFARI-SSC ASD-other samples did not demonstrate enrichment for this locus, suggesting that this locus might contribute to the brain size changes associated with ASD. The expression of AKAP13 was upregulated in one ASD NPC line (ABLE) but downregulated in another ASD sample (ARCH), which may be attributed to the stop gain mutation on CTNNB1 (coding for β-catenin) in the ARCH line. Considering the binding of β-catenin to both TSS and TTS region of AKAP13, this SNV loci could play a role linking WNT signaling and mTOR signaling to regulate brain development in the context of ASD.
Finally, we found TRIM2, which was bound by β-catenin and not a known ASD gene, overlapped with a deletion (chr4:154125517-154260572) that was enriched in ASD-macro samples (Fig. 7B, Supplementary Material, Fig. S3B). This gene plays a neuroprotective role and functions as an E3-ubiquitin ligase in proteasome-mediated degradation of target proteins. This ASD-macro enriched deletion covered the middle part to the 3′ end of the TRIM2. This deletion overlapped with both the ATAC-Seq peak and β-catenin binding peak within a HiC interval, and this interval looped with another HiC interval covering the 5′ end of the same gene. These results suggested that TRIM2 may be the transcriptional target of β-catenin, and the deletion (chr4:154125517-154260572) may be an important biomarker for ASD with macrocephaly. However, we did not find this deletion in our small validation dataset, so the functional effect of this deletion (e.g. effect on TRIM2 gene expression) could not be directly tested.
Discussion
Overall significance
ASD is characterized by phenotypic and genetic heterogeneity. The different comorbidities of ASD patients have been used to divide ASD patients into different phenotypic subgroups that may also reduce the genetic heterogeneity to facilitate the clarification of ASD genetic mechanism. However, one challenge of this approach is how to tease apart the genetic factors that mainly contribute to the comorbidities (macrocephaly, aggressive behavior, seizure, etc.) from those that mainly contribute to the defining behavioral abnormalities found in ASD. Here, we developed a new quantitative approach to detect the co-occurring genomic variations important for the development of ASD with and without macrocephaly, an important comorbidity in ⁓20–25% of ASD individuals (4). In detail, 160 genomic variations were identified through GOs directly associated with ASD and brain size phenotype, 1565 genomic variations were identified through GO–GO correlations and 104 of these 1715 variations were identified by both. These genes and variations, especially their combinatorial patterns, may provide novel biomarkers for different ASD subtypes. This pipeline could also be applied to analysis of other-omic data types, such as RNAseq, ChIPseq and ATACseq.
Dissection of ASD and brain size phenotype
In the current study, 922 GOs associated with ASD-only probands were identified. Synapse-related GOs were enriched in these seeding(?) or sGOs, which suggested that dysregulation at the synapse level might be specifically associated with behavioral abnormalities in ASD independent of brain size. For synapse-related sGOs, ‘transmembrane process’ related correlated or cGOs were significantly enriched. As transmembrane process was often functionally related to synapse development and function (e.g. synaptic vesicle recycling), this observation further supports the importance of synapse dysfunction in behavioral changes diagnostic for ASD (36).
On the other hand, WNT-, neuron- and organ morphogenesis-related GOs were enriched in 777 GOs associated with ASD-macro. This result is consistent with previous findings that increased neuron numbers in ASD with macrocephaly probands (4) and decreased WNT signaling pathway activity (22) are important for ASD with macrocephaly. For sGOs of WNT, neuron and organ morphogenesis, both ASD macro and micro probands showed enrichment of cell cycle-related cGOs, which is consistent with the conclusion on the basis of gene expression change in blood from ASD with macrocephaly probands (23). We validated the importance of the Wnt pathway correlation by performing RNA-seq, ChIP-seq for β-catenin and ATAC-seq on the ASD and control lines used as the validation dataset, which led to the identification of SMG6 as a Wnt-regulated ASD gene (Fig. 7D).
New biomarkers for ASD with macrocephaly
We identified six genes (AKAP13, BSG, DNAH11, GPR39, SMG6, SUMF1) as potential biomarkers for ASD with macrocephaly, as they were supported by our gWGCNA pipeline computation results from the SFARI-SSC dataset, β-catenin ChIPSeq and differential expression data from the validation dataset. Among these genes(?), SMG6 had already been reported to be related to the ASD gene (33), which increased the confidence that this gene was an ASD-macro biomarker. The other five genes were not previously linked to ASD, and our results suggest they could be new biomarkers for ASD-macro and require independent replication. In ASD-other samples from SFARI-SSC, AKAP13 was selected by our pipeline, confirmed by ChIPSeq and differential expression data from validation dataset. Considering it is one of the six candidates found in ASD-macro samples, it may be a new biomarker for ASD, not necessarily limited to ASD with macrocephaly.
We noticed a sharp decrease in the number of candidate loci selected by our gWGCNA pipeline and the number of loci validated by RNASeq, ChIPSeq data. Two factors could account for this decrease: first, the purpose of the gWGCNA pipeline was to determine the co-occurrence of genomic variations, and the expression level changes for these co-occurred variations in ASD NPC lines would be more complex than simply differential gene expression from control individuals at the single gene level. Second, we only have eight ASD-macro and five control cell lines in the validation dataset, which was ˃5-fold and less than the SFARI-SSC dataset sample size. Some low frequency variations could not be found in the validation dataset and, more importantly, many co-occurrence patterns could not be found in the rather small dataset. In the future, when larger expression/ChIPSeq datasets with genomic/clinical information available can be used as validation datasets, we believe a much higher proportion of our co-expression results could be determined and validated.
Cell cycle-related genes may affect both macro and microcephaly
We identified that cell cycle-related GOs were enriched in cGOs correlated with sGOs for WNT, neuron, organ-morphogenesis in ASD-macro and ASD-micro probands compared with ASD-other probands (Fig. 4B, Supplementary Material, Table S3G, I and J). This finding suggested that cell cycle is a biological process dysregulated in ASD-micro/macro probands, potentially affecting neural precursor cell/neuron proliferation/differentiation and altered neuron number in the brain, which is not surprising in an of itself. However, the correlation between WNT-related sGOs and cGOs for cell cycle suggested that variations in WNT signaling genes may be upstream of variations in cell cycle-related genes in ASD with macrocephaly genetic architecture, a novel observation. Similarly, variations in genes from sGOs for neuron and organ morphogenesis may be downstream of cell cycle to affect specific neuronal/developmental functions in ASD macro/microcephaly. In the 88 GOs associated with ASD microcephaly, WNT-related GOs were not overrepresented (Fig. 4A) but, using sGOs identified in ASD with macrocephaly, cell cycle-related cGOs were enriched in the ASD microcephaly samples (Fig. 4B). This result suggests that a WNT-cell cycle correlation exists in ASD with microcephaly, but this correlation is not as strong as that in ASD with macrocephaly. Importantly, this finding suggests that WNT signaling may be one of the upstream factors for cell cycle change in ASD microcephaly but is probably not the major factor.
WNT signaling for ASD with macrocephaly
Previous publications showed β-catenin/BRN2 transcriptional activity was decreased in human (22) and mouse models (37) for ASD with macrocephaly. Also, the gene expression level for WNT-related genes was downregulated in ASD with macrocephaly postmortem brain samples (38). Our results provide genomic level evidence of the dysregulation of WNT signaling in the context of ASD with macrocephaly. More importantly, our results elucidated how genomic variations in the WNT signaling pathway interact with other genomic variations in the context of ASD with macrocephaly. In detail, 62 WNT-related variations were identified on genes from sGOs, including 14 known ASD genes, such as DLG1, GRIN2B, WNT2, GPC6, TNN, etc. The other 48 genes may be potential new candidate loci for ASD with macrocephaly, including variations on HES1, FZD3, DKK1, GLI2, PRKN, AKT3, etc. Considering their potential effect on transcriptional regulation (Fig. 7B, Supplementary Material, Table S4A), TNN and GLI2 (Figs 5B and 7B) are good candidate biomarkers for ASD with macrocephaly. These loci were correlated with 1105 variations on cGOs (Supplementary Material, Table S4A), affecting genes including AKAP13, ESR1, CNTNAP2, GPR39, SEMA5A, etc. These results provide an example of how a small number of variations within WNT signaling co-occurred with a large number of variations on genes with a higher variation rate (e.g. neuron development) to cause complex disease.
Application of gWGCNA pipeline to analysis of RNASeq and ChIPSeq data
We have shown that the gWGCNA pipeline could integrate multiple types of genomic variations (SNV, INDEL, SV). This pipeline could also be applied to analysis of RNASeq and ChIPSeq data. It would be a promising next step to analyze HiC data with the gWGCNA pipeline in the context of complex human disease. The cGO (or gene, variation) pairs identified by gWGCNA and looping patterns detected by HiC could show how physically interacting deoxyribonucleic acid (DNA) regions and functionally related genes interplay to affect disease etiology. We believe the integration of different types of -omic level data would generate a more complete picture of ASD genetics.
Materials and Methods
Selection of samples from SFARI-SSC database
Brain circumferences of ASD probands from 2760 SSC families (1) were normalized by the age of the measurement. In detail, we calculated linear regression of head circumference versus age at measurement (in month) for SSC probands. Then the expected head circumference was compared with each real measurement, the difference between expected and measured value was divided by SD of the head circumferences (Supplementary Material, Table S1J). Probands with larger than 2 SD above average were labeled as ASD-macro (ASD probands with macrocephaly comorbidity) and those with 2 SD below average were labeled as ASD-micro (ASD with microcephaly). This standard resulted in the identification of 47 and 52 probands with macrocephaly and microcephaly, respectively. We next selected 52 probands from the same database with the smallest difference from average and labeled them as ‘ASD-other’ (probands with no brain size phenotype).
Genomic variation data from SSC dataset
SNV/INDEL data from the 41 ASD-macro, 37 ASD-micro and 38 ASD-other probands and their fathers were downloaded from the (SFARI-SSC database (https://www.sfari.org/resource/). Fathers from ASD-other families (n = 38) were used as controls for ASD-macro and ASD-micro probands. Fathers from the ASD-micro group (n = 37) were used as controls for ASD-other probands for subsequent genomic variation analysis (Supplementary Material, Table S1A).
In total 320 710 SNV/INDEL loci were downloaded. As the minimum sample number for each group for specific loci was required to be eight (nsample ≥8), a total of 60 617 loci were annotated using ANNOVAR. Variations in the intergenic, upstream or downstream regions [>10 kb distance from transcriptional start site (TSS) or transcription termination site (TTS)] were identified and excluded for further analysis. For each of the remaining loci, ALT allele frequencies were compared between each ASD group and controls. Loci with an increased ALT frequency of 10% or more in ASD than in CTRL were considered ‘ASD-enriched loci’. There were 7373, 5233 and 2458 ASD-enriched SNV/INDEL loci in ASD-macro, ASD-micro and ASD-other groups, respectively (Supplementary Material, Table S1B–D). These loci affected 457, 411 and 562 genes in each of the groups (Supplementary Material, Table S1E). These gene lists were the input for GO analysis using Gene Set Enrichment Analysis (GSEA) (39).
SV data were also obtained from the SFARI-SSC database. Only deletions were used for subsequent analysis. After filtering by nsample ≥8 in each group, 1044, 1330 and 1223 deletion loci were included for ASD-macro, ASD-micro and ASD-other, respectively (Supplementary Material, Table S1G–I). These loci were annotated by AnnotSV (40). The same standard identifying ASD-enriched loci as applied to SNV/INDEL data was used. GO analyses were performed for genes intersected with the ASD- or control-enriched deletions (Supplementary Material, Table S1F) using GSEA (see above).
Mapping genomic variations to GOs
The GO and gene lists were downloaded from a GSEA database (‘msigdb.v7.1.symbols.gmt’ under ‘All gene sets’). Genomic variation rate per GO was calculated based on these loci (in the formula listed below). Specifically, for each selected ASD or control individual from the SSC dataset, the number of ASD-enriched loci (increase of alternate (ALT) allele frequency in probands over controls ≥0.1) in each type of variation (SNV/INDELs and SVs) for each gene (genei) was summed for all genes in each GO (GOj) and normalized by the total number of genes in that GO to get variation_rateGOj.
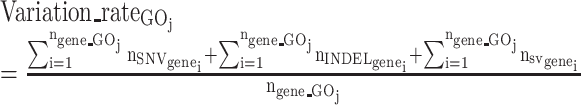 |
The variation-rateGO was calculated for all 10 131 GOs. GO with zero counts for all samples (n = 1845) was excluded from subsequent analysis. The individuals from each of the six groups (ASD-macro, ASD0micro and ASD-other, CTRL-macro, CTRL-micro and CTRL-other) were randomly divided into Group 1 and Group 2, with 115 and 114 samples, respectively (Supplementary Material, Table S3A).
gWGCNA analysis of genomic variation data
We employed a WGCNA, a data mining method used for studying biological networks on the basis of pairwise correlations between variables (27), to analyze GO correlations. With WGCNA default settings selected (soft threshold power = 6; minimum module size = 30), the sample tree was first generated for Group 1 on the basis of inter-sample Spearman correlation using variation_rateGO. Next, the WGCNA network was constructed with dissimilarities among modules to be at least 15%.
After the construction of the network, the eigengene for each module was associated with each of the two ‘phenotypes:’ ASD_vs_CTRL (labeled as ‘ASD’) (all ASD samples coded as 1 and controls as 0); and brain size (labeled as ‘Brain’) [all SSC probands with macrocephaly coded as 2, SSC proband with microcephaly as 0 and probands from ‘ASD-other’ group as 1, all control individuals (fathers) coded as 1] using Pearson correlation. Modules with significant correlation (P < 0.05) for either phenotype were detected for Group 1 samples. On the basis of the correlation with ASD and brain phenotypes, GO modules were defined as several ‘module groups’ such as ASD-macro, ASD-micro, etc. (Supplementary Material, Table S3B).
The same settings were then applied to network construction for Group 2 samples to detect GO modules associated with ASD and brain size. Venn diagrams were generated to find results from Group 1 that could be confirmed by those from Group 2 for each module group.
In the ‘confirmed’ GOs, if a few representative GO groups (e.g. GOs related to ‘WNT signaling’) were enriched in each module group, they were tested using a proportions test (29).
Correlation among GOs related to ASD
The enriched representative GOs detected above were used as sGOs. Pearson correlation among these sGOs and all other GOs for ASD and Control samples were calculated for each of three sets of samples: ASD-macro, ASD-micro and ASD-other. The correlation matrix was flattened using an R library ‘corrplot’, and the P-value for each GO pair was calculated. Using P < 0.05 as the cutoff, GO pairs that displayed positive and significant correlation in ASD samples and negative or insignificant correlation in control samples were considered ‘cGO’ for subsequent analysis.
Detection of loci enriched in seeding and cGOs
Total occurrence of each gene in each of the eight GO groups was calculated (4 sGO groups and 4 cGO groups, Supplementary Material, Table S3F–I) four sGO groups (GOs related to ‘neuron’, ‘WNT’, ‘organ morphogenesis’ and ‘synapse’, Fig. 5) and four groups of GOs correlated to each of the sGO. The occurrence value was standardized into a z-score.
The alternate allele frequency difference between ASD and CTRL (≥0.1, see above) was also standardized into a z-score. The ‘combined enrichment score’ for each locus was defined as the product of the z-score for gene frequency in a specific GO group and the z-score for alternate allele enrichment. Loci with positive combined enrichment score were selected.
Detection of SNV/INDEL loci enriched in ASD or controls in validation dataset
Whole genome DNA was extracted from fibroblast cell lines of eight ASD with macrocephaly and five gender/age-matched control individuals previously published with the clinical phenotype (22). Genomic variations detected in these cell lines were called ‘validation dataset’ in this paper. Exome libraries were produced in the Case Western Reserve University (CWRU) Genome Core using the Illumina ultra-sensitive exome library protocol. The libraries were sequenced on Illumina HiSeq 2500 at 8 libraries/run, which yielded ~100–150 million reads per library. The reads were aligned to the hg19 reference genome using BWA (41) with default settings (Gap open penalty = 6, Mismatch penalty = 4, etc.) and yielded about 20× coverage of the human genome in each library. Vcftools (42) were used to call variants. The SNV lists were generated after filtering out loci with low reading depth (<10), low number of reads in support of variant (n < 3), low alignment quality (q < 20) and low base quality (Q < 30). ANNOVAR (43) was used to annotate the SNVs. The ASD-enriched SNV/INDELs were those with significant P-value for ALT allele frequency difference between ASD and control [5000 times resampling using R, similar to previously reported method (44)]. All SNV/INDELs were compared with records in dbSNP. Filters used for calling INDELs were the same as those used for SNVs. The annotation of INDEL was performed using SeattleSeq Annotation 138 (45). GO analysis was performed on genes harboring selected SNV/INDELs using GSEA. SNVs under negative selection were found using Funseq (46).
Matepair sequencing and calling of deletions in validation dataset
‘Jumping libraries’ for matepair sequencing from eight ASD samples with macrocephaly and two control samples (aforementioned) were built using Illumina Nextera Mate Pair Sample Prep Kit. The libraries were sequenced in the CWRU sequencing core using an Illumina HiSeq 2500 machine with eight samples/run, which yield ~60 million reads per library. The reads were first trimmed off adapter sequences and reverse complemented using Nxtrim and then aligned to hg19 using BWA, resulting in an average physical coverage of about 2× and actual coverage of human genome of ~50×.
The bam files were used as input for DELLY (47) and LUMPY (48) (version 0.2.13) to call deletions. The start and end intervals of each structural variant (SV) from both algorithms were intersected using BEDTools (49). The matched intervals were used for the final inference of deletion breakpoints (Supplementary Material, Fig. S4). The deletions from all samples were collapsed if they overlapped. The frequency of deletions among ASD samples was calculated for these collapsed deletion events. The deletions from two control samples were inferred following the same process (‘control deletion list’). The deletions were classified as ‘ASD-Control-shared deletions’ if the ASD deletions overlapped with the control list or ‘ASD-specific deletions’ otherwise. Deletions were then annotated using AnnotSV (40). GO analysis for genes intersected with the deletions was performed using GSEA.
To test if the variations selected by our pipeline from SFARI-SSC samples were enriched by putative targets of any TFs, we applied CHEA analysis (50) for the genes that carried the 1514 loci selected from SFARI-SSC ASD-macro samples and 410 loci selected from SFARI-SSC ASD-other samples. Using the CHEA online (https://maayanlab.cloud/chea3/#top) function ‘ChIP-Seq- > Literature’ library. Similarly, genes affected by the 515 ASD-specific deletions in the validation dataset were tested with CHEA online tools. The results from the three input sets were compared using a Venn diagram (Supplementary Material, Fig. S5).
RNASeq for NPCs from validation dataset
Total ribonucleic acid (RNA) was extracted from NPCs from eight ASD-macro and five control lines as mentioned above. One control line (COVE) was excluded from subsequent analysis as its karyotype was abnormal. The RNA was purified and quantified, and samples of high quality (RIN ≥7.0) were used. The Illumina TruSeq Stranded Total RNA kit with Ribo Zero Gold (for rRNA removal by hybridization/bead capture) was used for library preparation. Optimized libraries were then loaded onto the HiSeq 2500 flowcell (8 libraries/lane) for 50 bp single-end sequencing.
Adapter sequences were trimmed and filtered using cutadapt (51). Reads that passed quality filter were aligned to hg38 using HISAT2 (52) and converted to sorted BAM files with samtools (53). Identification of differentially expressed genes and statistical analyses was performed using DESeq2 (54).
ChIPSeq for NPCs from validation dataset
ChIPSeq libraries for BRN2 were prepared using NPCs from three ASD lines and three control lines. About 10 million NPCs for each library were collected between Passages 6–9 at Day 3 (about 80% confluency) and cross linked using 4% formaldehyde for 10 min at room temperature. The cells were resuspended in lysis buffer, sonicated and incubated with antibody [POU3F2 (D2C1L) from Cell Signaling] linked DynaBeads Protein G overnight at 4°C. The DynaBeads were washed and then reverse crosslinked at 65° for 12 h. RNA and antibody were removed with RNase A (Ambion Cat # 2271) and proteinase K (Invitrogen, 25530-049). The pulled-down DNA was end-repaired, and a ploy-A tail was added, linked with adapter and PCR amplified. The PCR product was gel purified and fragments in the 250–400 bp were excised and purified with Qiagen MinElute Gel Extraction kit (Qiagen, 28606). The Bioanalyzer DNA 1000 assay (Agilent) was used to access the quality of the libraries. Seventy-five bp single-end reads were generated for high-quality libraries using the HiSeq 2500 (8 libraries/lane on flowcell) sequencing pipeline. Reads were adaptor trimmed with fastx_clipper, aligned with hg19 using BWA (41) and further processed using SamTools (53). Peaks were called by MACS14 (55) with default settings using sorted bam files with redundant reads removed. Called peaks were overlapped with published BRN2 on human NPC (56) and ATAC-Seq data from sample NPC lines (57) (see below).
Further bioinformatic processing of RNASeq, ChIPSeq and HiC data
The genes with selected genomic variation loci and differentially expressed between ASD and control NPCs were illustrated using Venn diagrams (https://bioinformatics.psb.ugent.be/webtools/Venn/). The ATAC-Seq data for three ASD and two control NPC lines (57) were obtained from the Gage laboratory through collaboration. These lines were a subset of the NPC lines on which we performed RNASeq and ChIPSeq experiments. Overlap between ATAC-seq peaks and BRN2 ChIPSeq were found using BedTools (49). Published BRN2 ChIPSeq data at NPC stage (with 2 samples) (56) were downloaded. To ascertain that the BRN2 binding positions were in actively transcriptional sites, BRN2 peaks from at least two control lines or published BRN2 peaks from both NPC samples needed to overlap with ATAC-Seq peak from both control lines.
Similarly, published β-catenin ChIPSeq from hESCs (31) were obtained from GEO and overlapped with control NPC ATAC-Seq data. The ATAC-Seq confirmed BRN2 and β-catenin ChIPSeq peaks were annotated using HOMER (58).
Published HiC data from human NPCs were obtained (59). Both sides of significantly associated HiC intervals were overlapped with selected genomic variations using BedTools.
Supplementary Material
Contributor Information
Chen Fu, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Justine Ngo, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Shanshan Zhang, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Leina Lu, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Alexander Miron, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Simon Schafer, The Salk Institute for Biological Studies, La Jolla, CA 92037, USA.
Fred H Gage, The Salk Institute for Biological Studies, La Jolla, CA 92037, USA.
Fulai Jin, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Fredrick R Schumacher, Department of Population and Quantitative Health Sciences, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Anthony Wynshaw-Boris, Department of Genetics and Genomic Science, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Conflict of Interest statement
None declared.
Funding
NIH R01MH113106 to A.W.B., and the JPB Foundation to F.H.G.
Author contributions
C.F and A.W.B. designed the study. C.F. performed matepair library preparation. C.F., J.N. and F.R.S. performed data analysis. L.L. performed HiC library preparation, S. Z. performed HiC data analysis. F.J. designed the HiC study. C.F., A.M., F.R.S. and A.W.B. contributed to manuscript preparation. All sequencing was performed in the Genomics Core of the Department of Genetics and Genomic Science, CWRU. S.S performed ATAC-Seq under supervision of F.G.
Data availability
All whole genome, whole exome, ChIP-seq and RNA-seq data are being uploaded to the NIMH Data Archive.
References
- 1. Abrahams, B.S., Arking, D.E., Campbell, D.B., Mefford, H.C., Morrow, E.M., Weiss, L.A., Menashe, I., Wadkins, T., Banerjee-Basu, S. and Packer, A. (2013) SFARI gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism, 4, 36. 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doshi-Velez, F., Ge, Y. and Kohane, I. (2014) Comorbidity clusters in autism spectrum disorders: an electronic health record time-series analysis. Pediatrics, 133, e54–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma, S.R., Gonda, X. and Tarazi, F.I. (2018) Autism spectrum disorder: classification, diagnosis and therapy. Pharmacol. Ther., 190, 91–104. [DOI] [PubMed] [Google Scholar]
- 4. Courchesne, E. (2004) Brain development in autism: early overgrowth followed by premature arrest of growth. Ment. Retard. Dev. Disabil. Res. Rev., 10, 106–111. [DOI] [PubMed] [Google Scholar]
- 5. Hewitson, L. (2013) Scientific challenges in developing biological markers for autism. OA Autism., 1. 10.13172/2052-7810-1-1-474. [DOI] [Google Scholar]
- 6. Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T.R., Correia, C., Abrahams, B.S., Sykes, N., Pagnamenta, A.T. et al. (2010) A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet., 19, 4072–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Devlin, B. and Scherer, S.W. (2012) Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev., 22, 229–237. [DOI] [PubMed] [Google Scholar]
- 8. Grove, J., Ripke, S., Als, T.D., Mattheisen, M., Walters, R.K., Won, H., Pallesen, J., Agerbo, E., Andreassen, O.A., Anney, R. et al. (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet., 51, 431–444.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brandler, W.M., Antaki, D., Gujral, M., Noor, A., Rosanio, G., Chapman, T.R., Barrera, D.J., Lin, G.N., Malhotra, D., Watts, A.C. et al. (2016) Frequency and complexity of De novo structural mutation in autism. Am. J. Hum. Genet., 98, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brandler, W.M., Antaki, D., Gujral, M., Kleiber, M.L., Whitney, J., Maile, M.S., Hong, O., Chapman, T.R., Tan, S., Tandon, P. et al. (2018) Paternally inherited cis-regulatory structural variants are associated with autism. Science (New York, N.Y.), 360, 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dong, S., Walker, M.F., Carriero, N.J., DiCola, M., Willsey, A.J., Ye, A.Y., Waqar, Z., Gonzalez, L.E., Overton, J.D., Frahm, S. et al. (2014) De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep., 9, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., Thiruvahindrapuram, B., Xu, X., Ziman, R., Wang, Z. et al. (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet., 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hynes, R.O. and Zhao, Q. (2000) The evolution of cell adhesion. J. Cell Biol., 150, F89–F96. [DOI] [PubMed] [Google Scholar]
- 14. Prabhakar, S., Noonan, J.P., Pääbo, S. and Rubin, E.M. (2006) Accelerated evolution of conserved noncoding sequences in humans. Science (New York, N.Y.), 314, 786. 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- 15. An, J.Y. and Claudianos, C. (2016) Genetic heterogeneity in autism: from single gene to a pathway perspective. Neurosci. Biobehav. Rev., 68, 442–453. [DOI] [PubMed] [Google Scholar]
- 16. Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., Fridman, W.H., Pagès, F., Trajanoski, Z. and Galon, J. (2009) ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics (Oxford, England), 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shannon, P., Markiel, A., Ozier, O., Baliga, N.S., Wang, J.T., Ramage, D., Amin, N., Schwikowski, B. and Ideker, T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iakoucheva, L.M., Muotri, A.R. and Sebat, J. (2019) Getting to the cores of autism. Cell, 178, 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Rubeis, S., He, X., Goldberg, A.P., Poultney, C.S., Samocha, K., Cicek, A.E., Kou, Y., Liu, L., Fromer, M., Walker, S. et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu, L., Lei, J., Sanders, S.J., Willsey, A.J., Kou, Y., Cicek, A.E., Klei, L., Lu, C., He, X., Li, M. et al. (2014) DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol. Autism, 5, 22. 10.1186/2040-2392-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hormozdiari, F., Penn, O., Borenstein, E. and Eichler, E.E. (2015) The discovery of integrated gene networks for autism and related disorders. Genome Res., 25, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marchetto, M.C., Belinson, H., Tian, Y., Freitas, B.C., Fu, C., Vadodaria, K., Beltrao-Braga, P., Trujillo, C.A., Mendes, A., Padmanabhan, K. et al. (2017) Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry, 22, 820–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pramparo, T., Lombardo, M.V., Campbell, K., Barnes, C.C., Marinero, S., Solso, S., Young, J., Mayo, M., Dale, A., Ahrens-Barbeau, C. et al. (2015) Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol. Syst. Biol., 11, 841. 10.15252/msb.20156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang, M., Wei, P.C., Lim, C.K., Gallina, I.S., Marshall, S., Marchetto, M.C., Alt, F.W. and Gage, F.H. (2020) Increased neural progenitor proliferation in a hiPSC model of autism induces replication stress-associated genome instability. Cell Stem Cell, 26, 221–233.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Glessner, J.T., Connolly, J.J. and Hakonarson, H. (2014) Genome-wide association studies of autism. Curr Behav Neurosci Rep, 1, 234–241. [Google Scholar]
- 26. Jiménez-Barrón, L.T., O'Rawe, J.A., Wu, Y., Yoon, M., Fang, H., Iossifov, I. and Lyon, G.J. (2015) Genome-wide variant analysis of simplex autism families with an integrative clinical-bioinformatics pipeline. Cold Spring Harb. Mol. Case Stud., 1, a000422. 10.1101/mcs.a000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gilabert-Juan, J., López-Campos, G., Sebastiá-Ortega, N., Guara-Ciurana, S., Ruso-Julve, F., Prieto, C., Crespo-Facorro, B., Sanjuán, J. and Moltó, M.D. (2019) Time dependent expression of the blood biomarkers EIF2D and TOX in patients with schizophrenia. Brain Behav. Immun., 80, 909–915. [DOI] [PubMed] [Google Scholar]
- 29. Newcombe, R.G. (1998) Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat. Med., 17, 857–872. [DOI] [PubMed] [Google Scholar]
- 30. Zahedi Abghari, F., Moradi, Y. and Akouchekian, M. (2019) PTEN gene mutations in patients with macrocephaly and classic autism: a systematic review. Med. J. Islam Repub. Iran, 33, 10. 10.34171/mjiri.33.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Estarás, C., Benner, C. and Jones, K.A. (2015) SMADs and YAP compete to control elongation of β-catenin: LEF-1-recruited RNAPII during hESC differentiation. Mol. Cell, 58, 780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoffman, G.E., Hartley, B.J., Flaherty, E., Ladran, I., Gochman, P., Ruderfer, D.M., Stahl, E.A., Rapoport, J., Sklar, P. and Brennand, K.J. (2017) Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat. Commun., 8, 2225. 10.1038/s41467-017-02330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nguyen, L.S., Kim, H.G., Rosenfeld, J.A., Shen, Y., Gusella, J.F., Lacassie, Y., Layman, L.C., Shaffer, L.G. and Gécz, J. (2013) Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet., 22, 1816–1825. [DOI] [PubMed] [Google Scholar]
- 34. Mlyniec, K., Budziszewska, B., Holst, B., Ostachowicz, B. and Nowak, G. (2014) GPR39 (zinc receptor) knockout mice exhibit depression-like behavior and CREB/BDNF down-regulation in the hippocampus. Int. J. Neuropsychopharmacol., 18, pyu002. 10.1093/ijnp/pyu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang, S., Wang, H., Melick, C.H., Jeong, M.H., Curukovic, A., Tiwary, S., Lama-Sherpa, T.D., Meng, D., Servage, K.A., James, N.G. and Jewell, J.L. (2021) AKAP13 couples GPCR signaling to mTORC1 inhibition. PLoS Genet., 17, e1009832. 10.1371/journal.pgen.1009832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masini, E., Loi, E., Vega-Benedetti, A.F., Carta, M., Doneddu, G., Fadda, R. and Zavattari, P. (2020) An overview of the main genetic, epigenetic and environmental factors involved in autism Spectrum disorder focusing on synaptic activity. Int. J. Mol. Sci., 21, 8290. 10.3390/ijms21218290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Belinson, H., Nakatani, J., Babineau, B.A., Birnbaum, R.Y., Ellegood, J., Bershteyn, M., McEvilly, R.J., Long, J.M., Willert, K., Klein, O.D. et al. (2016) Prenatal β-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol. Psychiatry, 21, 1417–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chow, M.L., Pramparo, T., Winn, M.E., Barnes, C.C., Li, H.R., Weiss, L., Fan, J.B., Murray, S., April, C., Belinson, H. et al. (2012) Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet., 8, e1002592. 10.1371/journal.pgen.1002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subramanian, A., Tamayo, P., Mootha, V.K., Mukherjee, S., Ebert, B.L., Gillette, M.A., Paulovich, A., Pomeroy, S.L., Golub, T.R., Lander, E.S. and Mesirov, J.P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geoffroy, V., Herenger, Y., Kress, A., Stoetzel, C., Piton, A., Dollfus, H. and Muller, J. (2018) AnnotSV: an integrated tool for structural variations annotation. Bioinformatics (Oxford, England), 34, 3572–3574. [DOI] [PubMed] [Google Scholar]
- 41. Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics (Oxford, England), 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Danecek, P., Auton, A., Abecasis, G., Albers, C.A., Banks, E., DePristo, M.A., Handsaker, R.E., Lunter, G., Marth, G.T., Sherry, S.T. et al. (2011) The variant call format and VCFtools. Bioinformatics (Oxford, England), 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang, K., Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids Res., 38, e164. 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Werling, D.M., Brand, H., An, J.Y., Stone, M.R., Zhu, L., Glessner, J.T., Collins, R.L., Dong, S., Layer, R.M., Markenscoff-Papadimitriou, E. et al. (2018) An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet., 50, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ng, S.B., Turner, E.H., Robertson, P.D., Flygare, S.D., Bigham, A.W., Lee, C., Shaffer, T., Wong, M., Bhattacharjee, A., Eichler, E.E. et al. (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature, 461, 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fu, Y., Liu, Z., Lou, S., Bedford, J., Mu, X.J., Yip, K.Y., Khurana, E. and Gerstein, M. (2014) FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol., 15, 480. 10.1186/s13059-014-0480-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rausch, T., Zichner, T., Schlattl, A., Stütz, A.M., Benes, V. and Korbel, J.O. (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics (Oxford, England), 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Layer, R.M., Chiang, C., Quinlan, A.R. and Hall, I.M. (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol., 15, R84. 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England), 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lachmann, A., Xu, H., Krishnan, J., Berger, S.I., Mazloom, A.R. and Ma'ayan, A. (2010) ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics (Oxford, England), 26, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin, M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J., 17, 10–12. [Google Scholar]
- 52. Pertea, M., Kim, D., Pertea, G.M., Leek, J.T. and Salzberg, S.L. (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT. StringTie and Ballgown. Nat. Protocols, 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England), 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Feng, J., Liu, T. and Zhang, Y. (2011) Using MACS to identify peaks from ChIP-Seq data. Curr. Protoc. Bioinformat., Chapter 2, Unit2.14–2.14. 10.1002/0471250953.bi0214s34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xue, Y., Qian, H., Hu, J., Zhou, B., Zhou, Y., Hu, X., Karakhanyan, A., Pang, Z. and Fu, X.D. (2016) Sequential regulatory loops as key gatekeepers for neuronal reprogramming in human cells. Nat. Neurosci., 19, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schafer, S.T., Paquola, A., Stern, S., Gosselin, D., Ku, M., Pena, M., Kuret, T., Liyanage, M., Mansour, A.A., Jaeger, B.N. et al. (2019) Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci., 22, 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y.C., Laslo, P., Cheng, J.X., Murre, C., Singh, H. and Glass, C.K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu, L., Liu, X., Huang, W.K., Giusti-Rodríguez, P., Cui, J., Zhang, S., Xu, W., Wen, Z., Ma, S., Rosen, J.D. et al. (2020) Robust hi-C maps of enhancer-promoter interactions reveal the function of non-coding genome in neural development and diseases. Mol. Cell, 79, 521–534.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All whole genome, whole exome, ChIP-seq and RNA-seq data are being uploaded to the NIMH Data Archive.