

Abstract
The shaker rat carries a naturally occurring mutation leading to progressive ataxia characterized by Purkinje cell (PC) loss. We previously reported on fine-mapping the shaker locus to the long arm of the rat X chromosome. In this work, we sought to identify the mutated gene underlying the shaker phenotype and confirm its identity by functional complementation. We fine-mapped the candidate region and analyzed cerebellar transcriptomes, identifying a XM_217630.9 (Slc9a6):c.[191_195delinsA] variant in the Slc9a6 gene that segregated with disease. We generated an adeno-associated virus (AAV) targeting Slc9a6 expression to PCs using the mouse L7–6 (L7) promoter. We administered the AAV prior to the onset of PC degeneration through intracerebroventricular injection and found that it reduced the shaker motor, molecular and cellular phenotypes. Therefore, Slc9a6 is mutated in shaker and AAV-based gene therapy may be a viable therapeutic strategy for Christianson syndrome, also caused by Slc9a6 mutation.
Introduction
Degenerative cerebellar ataxias comprise a large and heterogeneous group of disorders. Symptoms, age of onset, rate of progression and prevalence are highly variable. At least in part because of this heterogeneity, symptoms are difficult to treat, and most patients never experience more than symptomatic care. Animal models specific to individual forms of ataxias may ultimately prove important in generating new therapeutic strategies. Although there are a number of animal models of ataxia, particularly for the spinocerebellar ataxias (1–10), there are no models of most forms of ataxia. Whereas many ataxia rodent models have been generated by genetic engineering, a number of natural occurring ataxic mutant mice have been investigated (11–19), contributing to understanding cerebellar function and neurodegeneration, as well as to therapeutic strategies.
We and others have reported on the shaker rat, a spontaneous, X-linked, recessive model of tremor and ataxia characterized by Purkinje cell (PC) degeneration (20–25). Early work focused on characterizing the ataxia (20,26) and PC degeneration (21) present in the shaker rat, as well as developing an inbred Wistar Furth (WF) shaker rat strain (27). More recent work has focused on determining the precise cause of PC loss in shaker rats (24,25,28). We used the model to carry out preclinical studies of low-frequency deep brain stimulation of the deep cerebellum to reduce ataxia and tremor (22), which other groups have recently continued to develop (29–31). In these studies, we developed robust quantitative approaches for analyzing the shaker rat’s motor phenotype, some of which have been more recently further validated through deep neural network-based through markerless motion tracking approaches (32).
We previously mapped the shaker phenotype to the end of the long arm of rat chromosome X using F2 hybrid mapping crossing shaker rats on the WF background with wild-type (wt) Brown Norwegian (BN) rats. This was complemented by analysis of cerebellar transcriptomes seeking to identify X-chromosomal transcripts with reduced expression, as well as coding or mis-splicing events. These studies confirmed location of the shaker locus on rat chromosome X using polymorphic genetic markers. This analysis narrowed the candidate gene region to 26 Mbp of the telomeric region of the long arm and distal to marker DXRat21. We identified Atp2b3 encoding PMCA3 as containing a potentially disease-coding variant. However, functional studies in yeast complementation assays did not support an effect of the amino acid substitution on function of the Ca++ pump (23).
Subsequently, we identified genetic recombination events in symptomatic F2 shaker rats, definitively excluding the Atp2b3 variant as causative. We now report on additional studies combining viral, genetic, transcriptomic and motor analysis techniques to identify the shaker mutation, a deletion in Slc9a6 (solute carrier family 9, member A6) that encodes NHE6 (sodium-hydrogen exchanger 6). The deletion is predicted to lead to a truncated NHE6. Of note, SLC9A6 is mutated in Christianson syndrome (CS), a severe epileptic encephalopathy with ataxia and autistic features (33). We sought functionally confirmation of the pathogenicity of the Slc9a6 mutation, and we were motivated to perform functional complementation studies in part on the basis of clinical reports indicating that even the expression of a non-mutated SLC9A6 in 10% of transcripts is sufficient to yield a substantially attenuated CS phenotype (34). We restored NHE6 expression in PCs using viral transduction, reducing neurodegeneration and improving motor behavior, confirming Slc9a6 as causative.
Given that numerous aspects of the shaker phenotype have been reported on by multiple groups (20–28,32,35–37), we propose the shaker rat as beneficial for use in therapeutic development for CS. There exists a wealth of information regarding its degenerative processes (21,23–25,28,36), motor phenotyping (20,22,35) on the basis of widely used force plate actometer (38,39), and in this work, we investigate the molecular phenotype in detail. Coupled with the viral gene replacement methods explored herein, these provide excellent targets to evaluate in the future development of gene therapy for CS.
Results
Brief overview
We delineated the genetic region of interest and analyzed cerebellar and whole brain transcriptomes. We genotyped rats across the set of candidate genes and eliminated all candidate genes except Slc9a6 through recombination events. We administered an L7-Slc9a6-green fluorescent protein (GFP) adeno-associated virus (AAV) to shaker rats and found that driving expression of wt NHE6 to PCs in shaker rats reduced cerebellar gait ataxia. We proceeded to evaluate cerebellar samples and cerebellar histology and found that administration L7-Slc9a6-GFP AAV yielded improvements in PC-specific transcripts and proteins and decreased PC loss. Thus, we functionally demonstrated that loss-of-function mutation of Slc9a6 was causative of the shaker phenotype.
Shaker mapping
Using an F2 hybrid intercross, we mapped shaker to chromosome X distal to 133.45 (DxRat21) to qter. Our previous analysis of cerebellar transcriptomes revealed an amino substitution in PMCA3, encoded by Atp2b3, which we evaluated as a potentially disease-causing variant (23). Although the change occurred in a highly conserved amino acid, analysis in yeast did not reveal any change in calcium transporter function. Subsequent to our publication, we observed several shaker WF rats that did not carry this variant. Although this clearly excluded Atp2b3 as the causative gene, we were not able to discern whether shaker mapped proximal or distal to Atp2b3.
We developed a dual strategy to identify the shaker gene using genetic mapping of additional recombination events in our F2 WF/ BN hybrids, as well as more effective ribonucleic acid (RNA)-seq analysis with greater depth and longer read lengths. To improve genetic mapping, we conducted database searches in Rnor_6.0, and Medical College of Wisconsin Rat Genome Database, and we identified six single-nucleotide polymorphisms in the candidate region. Of these, only rs106120845 was informative between BN and WF and mapped the shaker gene distal to 144.59 (Fig. 1A).
Figure 1.
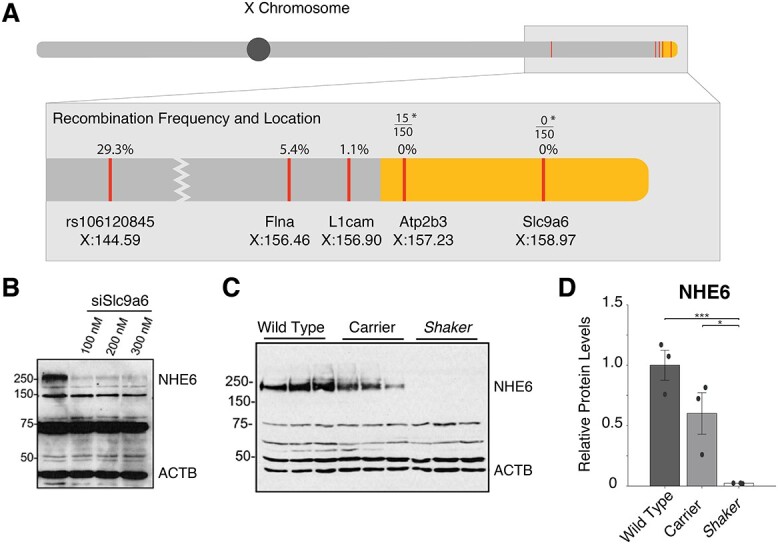
Linkage map of shaker and analysis of NHE6. (A) Locations of crucial recombination events in the WF/BN F2 intercross in 92 rats are shown. Yellow refers to our narrowed region of interest. *Of 150 shaker rats in the WF background, 15 rats were wt for ATP2b3, but none were wt for Slc9a6. (B) Western blots show that the band corresponding to glycosylated NHE6 is knocked down by siSlc9a6, validating the commercial antibody. (C) and (D) Less NHE6 expression is seen in obligate carriers than in wt; this is reduced further in shaker rats. Errors bars represent standard error of the mean; dots refer to individual animals.
To generate additional genetic markers and potentially identify the mutation itself, we used deep RNA-sequencing of cerebellar and hemispheric tissues of three obligate affected and three wt filial mates to identify coding variants, and we evaluated all variants and differentially expressed genes (DEGs). Utilizing the variant calling tool VarScan v2.3.3 (40), we found 15 changes predicted to be deleterious (3 high, 12 moderate) in eight genes on chromosome X. Of these, 4 were frameshift variants and 11 were missense variants. Once we limited variants to the new candidate linkage region distal to 144.59, variants in three genes remained: one missense variant and splice region variant in Flna (filamin A), one frameshift variant in L1cam (L1 cell adhesion molecule), and one frameshift variant in Slc9a6.
All variants in the candidate genes were amplified by polymerase chain reaction (PCR) and Sanger sequenced utilizing the WF and F2 WF/BN progeny cited in our previous publication (23). In addition, 55 affected and 10 aged (45-week old) asymptomatic rats were screened for the Atp2b3 and Slc9a6 variants. The respective recombination fractions are shown in Figure 1A, indicating smaller recombination fractions toward the telomere. Although variants in Flna and L1cam were found to have multiple recombination events, the frameshift variant in Slc9a6 showed no recombination events in 242 shaker rats.
Further confirmation for Slc9a6 as the shaker locus came from analyzing DEGs in cerebellar transcriptomes. The top DEGs on chromosome X were Gpr34 (G protein-coupled receptor 34), Tlr13 (toll-like receptor 13) and Slc9a6. Of these top DEGs on chromosome X, Gpr34 and Tlr13 were well outside the linkage region, further supporting Slc9a6 as the shaker allele.
Mutation
The Slc9a6 frameshift variant identified in shaker rats occurs at amino acid 64 and results in a predicted truncated protein. This frameshift was categorized as deletion–insertion variant, with base pairs 191–195 deleted, with one base pair inserted. The nomenclature is as follows: XM_217630.9(Slc9a6) c.[191_195delinsA], p.Ala64Glufs*23). Slc9a6 is abundantly expressed in brain (41), especially in PCs. In humans, mutation of human Slc9a6 causes CS, a recessive X-linked epileptic encephalopathy with prominent ataxia (42).
NHE6 loss of function in shaker rats
After identifying Slc9a6 as the likely shaker candidate gene, we tested whether the predicted NHE6 truncation would result in loss of NHE6 abundance. We evaluated several commercially available antibodies, including a rabbit polyclonal antibody that recognized several protein species at different molecular weights from 40 to 250 kDa. We sought to confirm the identity of these proteins, especially the ~250 kDa protein, which we predicted would correspond to the fully glycosylated, dimeric form of NHE6. Indeed, as the molecular weight fluctuates with glycosylation, the fully glycosylated, dimeric form of NHE6 has been reported as 200–250+ kDa (43,44). We treated HEK293 cells with ascending doses of SLC9A6 siRNA (45) and found that only the 250 kDa protein was reduced in intensity. This confirmed that the ~250 kDa protein responded to Slc9a6 knockdown and likely represented glycosylated NHE6 (Fig. 1B). Of note, the 75 kDa band, which would correspond to a non-modified NHE6 moiety, did not show any change in abundance and represents a non-specific protein band, as do the other bands below 250 kDa.
To examine NHE6 expression in wt and shaker rats, we performed western blotting of cerebellar extracts in age-matched wt, female heterozygote and male shaker rats (n = 3 for each at 5 weeks of age). We found gene-dosage dependent reduction of the ~250 kDa protein in that band intensity was reduced in heterozygotes to ~50% and to 0% in shaker rats. This was consistent with loss of function caused by the frameshift Slc9a6 mutation (Fig. 1C and D). Notably, a lower molecular weight band representing a truncated NHE6 was not observed. Differences in the number and size of non-specific western blot bands were attributed to the use of a commercial antibody not previously used in NHE6 mutant rodent models, as opposed to in-house custom antibodies, as well as strain and species differences from other NHE6 mutant models.
AAV vectors for PC targeting
To functionally test whether Slc9a6 causes shaker, we developed a viral vector targeting wt rat Slc9a6 expression to PCs and analyzed motor behavior, mRNA and protein expression and PC morphology. We previously reported that shaker rats exhibited PC loss by 7 weeks of age and that PCs appear to be the predominant cell type affected early in pathogenesis (23). Therefore, we generated a plasmid expressing a GFP-tagged rat wt Slc9a6 under control of the PC-specific optimized mouse L7–6 (L7) promoter (46). This plasmid was packaged in the PHP.eB capsid, ensuring widespread transduction in the central nervous system when injected into the lateral ventricle (47).
To test whether intracerebroventricular (ICV) delivery of the L7-Slc9a6 AAV would indeed result in expression in PCs, we generated a control vector expressing GFP under the control of the L7–6 promoter. We administered the L7-GFP AAV via ICV injection at three concentrations (5, 10 and 20 μl at a concentration of 2.26 × 10^11 vG/μl), as well as 10 μl of saline in four age-matched, wt rats. The 10 μl injection resulted in significant transduction of PCs (Supplementary Material, Fig. S2A), as imaged from 40 μm slices of cerebellum.
When we injected a comparable dose of Slc9a6-GFP AAV, the larger Slc9a6-GFP fusion protein did not generate detectable fluorescence, likely caused by relatively low translational efficiency of the substantially larger construct and, potentially, conformational changes affecting epitope recognition. Therefore, we performed western blotting using an anti-GFP antibody. We analyzed cerebellar extracts from 4 rats administered L7-Slc9a6-GFP AAV, at 25–35 weeks of age. Although a number of non-specific bands were detected, we observed a band that corresponds to the molecular weight of glycosylated NHE6 fused to GFP with a molecular weight of 250+ kDa. This band was not observed in uninjected shaker, carrier or wt rats, but was observed exclusively in shaker rats with motor improvement (see below). One shaker rat without motor improvement after injection also lacked a glycosylated NHE6-GFP band (Supplementary Material, Fig. S2B).
Motor performance is substantially improved in shaker rats treated with the L7-Slc9a6-GFP AAV
We sought functional evidence to demonstrate that mutation of Slc9a6 was causative of the shaker phenotype. We tested whether AAV-based expression of the wt Slc9a6 gene in shaker rats would prevent or reduce the motor phenotype. We injected animals with either L7-Slc9a6-GFP AAV or the control L7-GFP-AAV at 5 weeks of age and analyzed 30-min un-cued motor recordings taken in each rat at 7, 18 and 25 weeks of age: 7 weeks was chosen given that it corresponds to the typical onset of PC loss; 18 weeks was chosen given that it follows the typical completion of PC loss and represents the first time point after which motor ataxia no longer significantly worsened in untreated rats, both in this and previous work; finally, 25 weeks was the final and most statistically conservative time point of the study (22–24). Figure 2A shows representative traces of rapid movements from rats with different genotypes and treatments at 18 weeks of age. We computed straightness of gait by taking a ratio of total distance traveled versus displacement. At 8 weeks, there were no significant differences across groups (Fig. 2B). At 18 weeks, shaker rats administered the control L7-GFP AAV were severely ataxic, whereas those administered L7-GFP-Slc9a6 AAV had greatly improved motor performance (Fig. 2C). These improvements were maintained through the final time point of motor recordings, 25 weeks of age (Fig. 2D). At all time points, uninjected wt rats were insignificantly different in gait compared with wt rats injected with L7-GFP AAV. Notably, while the shaker rats administered the control AAV made almost exclusively uncoordinated movements at 18 and 25 weeks, several L7-Slc9a6-GFP AAV-treated shaker rats exhibited a strong motor therapeutic effect, being almost indistinguishable from wt rats, even at 25 weeks of age (Fig. 2E). Representative videos taken at 18 weeks of age demonstrating the efficacy of the L7-Slc9a6-GFP AAV in improving the gait of shaker rats are shown in Supplementary Material, Video S1.
Figure 2.
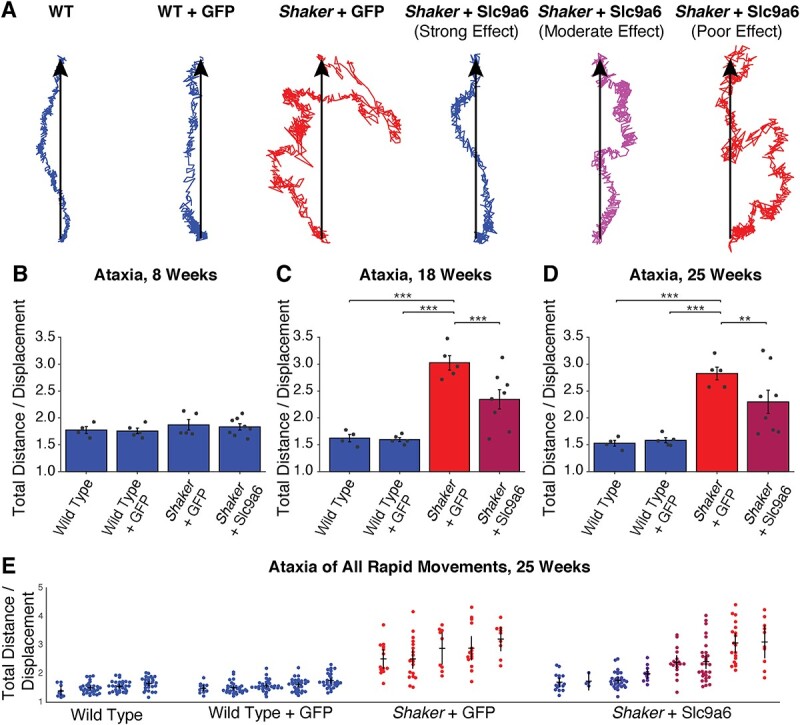
Administration of L7-Slc9a6-GFP AAV at 5 weeks of age reduces ataxic motor phenotype at later time points. (A) Center of mass traces from representative movements made at 18 weeks of age. Treated animals are grouped on the basis of treatment efficacy. Color scale refers to quantitative severity of ataxia. Blue refers to WT animals, red refers to untreated shakers (shaker + L7-GFP AAV). Percentage of severity from WT to untreated is represented by the shade of purple. (B)–(D) Straightness of gait is computed as a measure of motor coordination. Shown is a ratio of total distance traveled versus displacement, averaged across all rapid movements made in 30 min at each time point. Ratios are shown at (B) 8 weeks, shortly after the typical onset of PC loss, (C) 18 weeks, shortly after the typical completion of PC loss and (D) 25 weeks, at which point the motor symptom progression is typically complete. L7-GFP AAV had no impact on gait, but L7-Slc9a6-GFP AAV reduced motor dysfunction. (E) Straightness of gait ratios are shown for all rapid movements recorded in all rats at 25 weeks. Untreated shakers primarily make uncoordinated movements, whereas treated shakers with a strong effect primarily make coordinated movements.
Slc9a6 mRNA and NHE6 are partially restored in shaker rats treated with the L7-Slc9a6-GFP AAV
Cerebella from half of each control and treatment group were taken for molecular analyses. Notably, rats chosen from the treatment group for molecular analyses were representative of and spanned the group. Specifically, animals with the 1st, 3rd, 5th and 8th best motor treatment effect among the eight treated animals were those analyzed at the molecular level. We performed western blotting for NHE6, identifying glycosylated NHE6 in shaker rats treated with L7-Slc9a6-GFP AAV (Fig. 3A and B, Supplementary Material, Fig. S3). Quantification of NHE6 levels (Fig. 3C) showed significant NHE6 increases in shaker rats that were administered L7-Slc9a6-GFP AAV compared with those only administered L7-GFP AAV. We quantified cerebellar Slc9a6 mRNA levels by quantitative real-time PCR (qRT-PCR) and found that compared with wt or carrier, Slc9a6 expression was lower in shakers administered the control L7-GFP AAV, and this expression was partially restored by treatment with L7-Slc9a6-GFP AAV (Fig. 3D).
Figure 3.
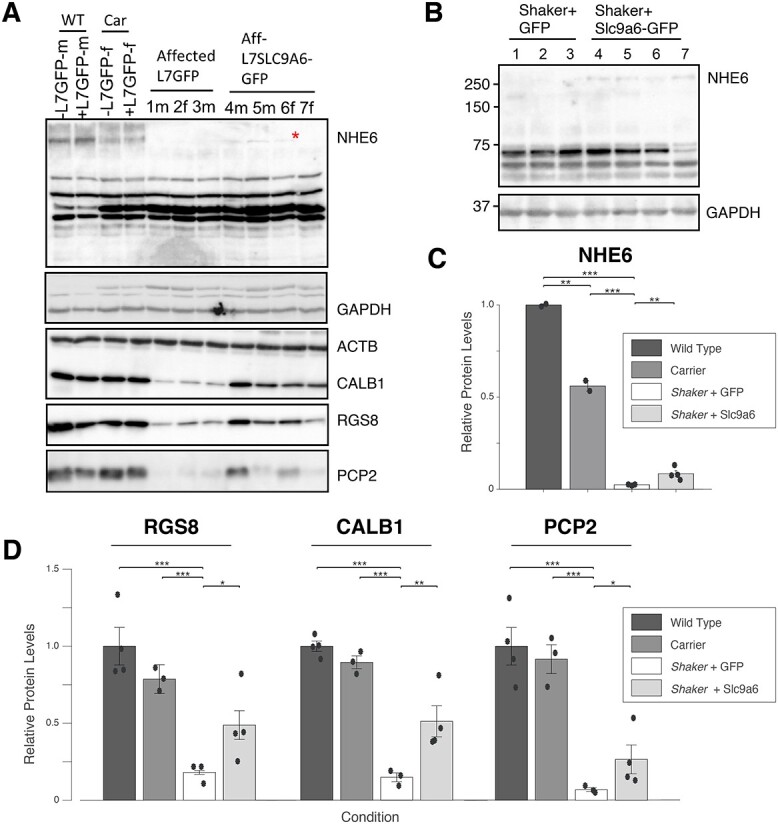
Administration of L7-Slc9a6-GFP AAV modestly, but significantly increases glycosylated NHE6 and Slc9a6 mRNA. (A) Western blotting of cerebellar extracts with Abcam antibody shows that the glycosylated NHE6 band is reduced in carriers, and fully reduced in shakers administered the control AAV. A faint band is seen in shaker rats treated with L7-Slc9a6-GFP AAV with ICV injection at 5 weeks of age. (B) Repeat of (A) with ThermoFisher antibody confirms lack of the glycosylated NHE6 band in shakers + L7-GFP AAV and presence in shakers + L7-Slc9a6-GFP AAV. (C) Quantified results from (A). L7-Slc9a6-GFP AAV significantly restores NHE6 expression in shaker rats compared with those administered the control AAV. (D) Slc9a6 mRNA expression is evaluated through qRT-PCR. Slc9a6 is reduced in shaker rats compared with wt or carrier rats, whereas administering L7-Slc9a6-GFP AAV increases expression.
PC-specific markers are partially restored in shaker rats treated with the L7-Slc9a6-GFP AAV
We quantified protein and mRNA expression for several markers associated with PC quantity and function established in prior detailed analyses of PC degeneration (3,48–53). First, we analyzed cerebellar protein extracts by quantitative western blotting of calbindin-1 (CALB1), Regulator of G protein signaling 8 (RGS8) and Purkinje Cell Protein 2 (PCP2). We found that levels of each PC-specific protein were reduced in shakers compared with wt and carriers at 25+ weeks of age. CALB1 and RGS8 levels were partially restored by L7-Slc9a6-GFP AAV (Fig. 4A and B), whereas PCP2 normalization trended toward significance (P = 0.062). Next, we quantified Calb1, Rgs8 and Pcp2 mRNA levels by qRT-PCR. We found that all 3 mRNAs were significantly reduced in shaker rats compared with wt and carriers. Upon transduction with L7-Slc9a6-GFP AAV, levels were significantly increased in shakers (Fig. 4C).
Figure 4.

Administration of L7-Slc9a6-GFP AAV improves levels of key PC-specific mRNAs and proteins. (A) Western blotting of cerebellar protein extracts shows reduced expression of CALB1, RGS8 and PCP2 in shaker rats administered the control AAV compared with wt and carriers. Shaker rats administered the L7-SlC9a6-GFP AAV show partially restored expression. (B) Quantification of (A). For each key PC-specific gene, administration of L7-Slc9a6-GFP AAV resulted in significantly increased expression in shaker rats compared with those administered the control L7-GFP AAV. (C) mRNA expression was evaluated for Rgs8, Calb1 and Pcp2 through qRT-PCR. Compared with wt, each marker was reduced significantly in carrier females and further in shaker rats administered the control L7-GFP AAV. Shaker rats administered the L7-Slc9a6-GFP AAV showed significant improvements in each marker compared with those administered the control L7-GFP AAV.
Finally, we sought to confirm these results by immunocytochemical analysis of PCs. To assess number of PC somata and extent of dendritic arborization we employed staining with an antibody to CALB1, a calcium binding protein that is highly enriched in PCs. We stained parasagittal slices from each animal sacrificed after completion of the motor experiments at 25 weeks of age with an anti-Calb1 antibody. We found that PCs in wt rats show strong CALB1 staining PC, with near total loss outside of a flocculonodular exclusion zone in shaker rats administered only the L7-GFP AAV. However, loss in other lobules was consistently partially prevented by treating shaker rats with the L7-Slc9a6-GFP AAV, matching mRNA and protein results. We show representative examples of each group in Figure 5.
Figure 5.
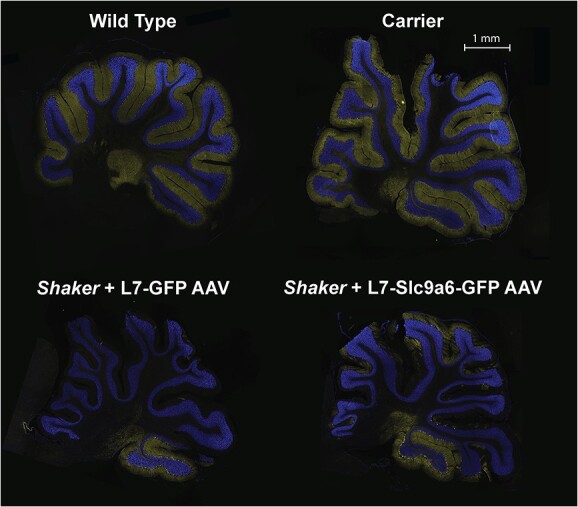
Representative 20 μm full cerebellar sagittal slices stained with Anti-Calb1 (yellow), counterstained with DAPI (blue). The L7-Slc9a6-GFP AAV prevented PC loss in shaker rats compared with those administered the L7-GFP control AAV. Shown are representative sagittal slices across groups. Notably, PCs tend to be preserved in the flocculonodular lobule 10. Even a relatively small degree of PC rescue led to substantial motor recovery (Fig. 2).
Discussion
General
Loss-of-function mutations in endosomal Na+/H+ exchanger 6 (NHE6) encoded by SLC9A6 cause the X-linked neurologic disorder CS (42). Patients exhibit symptoms associated with both neurodevelopmental and neurodegenerative abnormalities (54). NHE6 is mainly expressed in early and recycling endosomes and plays an important role in regulating endosomal pH (55). Loss of NHE6 function results in overacidification of the endosome (56), with decreased endosome-lysosome fusion in NHE6-null neurons (57).
We identified a frameshifting mutation in Slc9a6 that is predicted to result in an early frame shift at amino acid 64 resulting in a truncated protein of 87 amino acids. This is a spontaneous mutation that results in a tremor and ataxic phenotype, hence the designation of this strain as shaker. Pathologically, the phenotype is characterized by cerebellar volume loss and PC death that proceeds from anterior to posterior, generally with less severe cell loss in a flocculonodular part of the cerebellum (23–25) (Fig. 5). We previously verified abnormal cerebellar and PC pathology in shaker rats confirming a cerebellar ataxia-like phenotype (23).
The study objective was to identify the gene responsible for the shaker phenotype by linkage analysis combined with candidate gene analysis. In a previous study, we investigated Atp2b3, which mapped to the shaker candidate region. This was a promising candidate as Atp2b3 mutations are associated with congenital cerebellar ataxia in humans (58). However, we observed no difference in channel function by comparing wt Atp2b3 with Atp2b3R35C (23). Additionally, recombination events observed subsequent to our publication excluded Atp2b3. This also refined the region to 2.7 Mb, extending from 157.2 to 159.9 (Xq37-qter). Although the Slc9a6 variant did not emerge as a candidate in previous work, this current study employed both longer reads and greater depth of coverage. In addition, Slc9a6 mapped more proximal in the X chromosome in rat genomes prior to Rnor_6.0. Ultimately, on the basis of both linkage analysis and functional evidence, we concluded that a protein-truncating mutation found in the Slc9a6 gene is causative of the shaker phenotype.
The shaker rat as a model of Christianson syndrome
At least 54 different genes on the X chromosome are linked to mental retardation (59). One of these is SLC9A6, and mutations in SLC9A6 truncating the NHE6 protein cause CS (33,42,60), resulting in an Angelman-like, recessive, X-linked, progressive, neurodevelopmental syndrome. Symptom onset occurs in infancy, with patients exhibiting intellectual disability, autism, ataxia, lack of speech and seizures, among other symptoms (42,54).
Our work led to the discovery of a novel protein truncating variant (PTV) (p.A64Efs*23) in Slc9a6 in the shaker rat, making this the first rat model of CS. Other groups have worked with Slc9a6 knockout mice as well as Slc9a6 mutant mice (43,61,62) to demonstrate that Slc9a6 loss of function causes the relevant defects, including disrupted endosomal-lysosomal function, neurodevelopmental and neurodegenerative pathology, impaired plasticity and impoverished neuronal arborization (43,56,61,63–66). Further, Slc9a6 heterozygous knockout mice develop PC, motor and visuospatial abnormalities (67). Other Slc9a6 rodent models are not reported as recapitulating all components of the human disease, such as seizures, cognitive deficits or autistic behavior. Given that rats have a complex social repertoire, the shaker rat may be well suited for detailed dissection of the behavioral/cognitive components of CS.
NHE6 function in the context of the shaker rat
NHE6 is a membrane-bound protein functioning as a Na+/H+ exchanger, providing a leak pathway for protons and limiting endosomal acidification (68). NHE6 expression is high in PCs and coincides with PC loss in NHE6-null mice (65). NHE6 mutation leads to neuronal degeneration through apoptosis (43), which is the mechanism for PCs death in the shaker rat (24,25). However, NHE6 expression is substantially more widespread than just PCs, and loss of function could potentially impact the entire nervous system (64). Thus, future work in the shaker rat should include an evaluation of NHE6 deficits in cerebrum and spinal cord.
Functional evidence suggesting that Slc9a6 mutation underlies the shaker phenotype
In this work, we generated an AAV vector targeting expression of Slc9a6 to PCs using the L7 (Pcp2) promoter. AAV administration after P1–P2 in rodents can result in relatively low neuronal yield (69). Indeed, our ICV injection at 5 weeks of age provided relatively low levels of glycosylated NHE6 compared with levels in wt rats, ~10% of wt cerebellar NHE6 (Fig. 3). However, despite relatively low cerebellar NHE6, PC-specific proteins and mRNA levels, markers of PC health, improved substantially toward normalization (Fig. 4). It is interesting that key mRNAs and proteins decreased in SCA2 mouse models—CALB1, PCP2 and RGS8—are also decreased in shaker. Although it is known that these changes occur prior to PC cell loss or significant rarification of PC dendrites in SCA2 mouse models (48), future studies will be needed to examine transcriptomic changes across different time points.
Behavioral analyses that we performed used a previously described force plate paradigm that allows analysis of tremor and gait ataxia at a spatial and temporal resolution exceeding that of video-based analysis (22). Ataxic phenotypes significantly improved, with a subset of rats approaching near-normal motor function (Fig. 2). These functional data strongly support Slc9a6 as the gene responsible for the shaker phenotype.
The prospect of gene therapy for CS
Viral gene therapy is an attractive strategy for treating CS, supported by this study. However, therapeutic optimization of the viral construct will be required. First, a therapeutic construct would lack a GFP tag; given the large size of our construct with the GFP tag, it is likely that translational efficiency would be substantially better in this proposed construct, which would likely yield greater therapeutic benefit. Further, using a promoter driving expression beyond just PCs is almost certainly necessary to treat a number of other CS symptoms. With this in mind, using a pan-neuronal promoter such as CAG may be ideal (70). However, NHE6 gain of function has been reported as over-alkalizing endosomes, yielding neuronal atrophy (71). Further, off target effects may occur, particularly with improper dosing. Therefore, precise dose response studies will be of high importance for therapeutic development.
Notably, relatively minimal NHE6 expression may be necessary for substantial benefit. Our results indicate that ~10% cerebellar NHE6 expression may be sufficient to prevent the generation of ataxia in many cases. This is consistent with literature on human patients; whereas total loss of function universally creates a severe phenotype, even just the presence of 10% normal transcripts has been reported as yielding a substantially attenuated phenotype consisting of mild intellectual deficit, attention deficit disorder, speech difficulties, microcephaly and mild asymptomatic cerebellar atrophy (34).
Phenotypic variation across the shaker rat, NHE6-null rodents and humans with CS
Several engineered NHE6 null and mutant rodent models have contributed substantially to the study of CS and neurodegeneration. To date, mouse models have primarily been used (56,57,61,63,64,66,67,72–74), whereas an NHE6-null rat was recently reported (75). Reports of NHE6-null mice indicate a slower progression of PC degeneration, perhaps with less severe PC loss (65,66). Among the possible underlying mechanisms, one involves differential activity of NHE6 paralogs such as NHE9, which is also localized in recycling endosomes.
Rat models of NHE6 dysfunction may ultimately prove more useful than mouse models for the study of associated amyloid beta and tau deposition (75), whereas a diversity of models across species with loss of function created through mutation or knockout will be highly useful for therapeutic development for CS. Compared with the recently reported NHE6-null rat in the Sprague Dawley background (75), the WF shaker rat appears to have a later onset of PC loss and motor dysfunction. However, it should be noted that the NHE6-null rat’s motor phenotype was evaluated via digigait and rotorod, whereas we evaluated ataxia directly through actometry. Thus, direct comparisons of motor progression or severity are difficult to make. Further, it is notable that shaker first arose in Sprague Dawley rats before being crossed into the WF background (20,21,26,27), with similar phenotypic onset timing to the NHE6 null rat when still in a Sprague Dawley background. This indicates that strain differences are likely a major contributor to differences in phenotypic progression. Indeed, there exists a complex interplay between vesicular ATP-dependent proton pumps and the NHEs, and it is possible that any number of modifiers may yield important differences in endosomal pH and PC health. Taken together, the shaker rat, NHE6 null rat and NHE6 mouse create a powerful and diverse set of models from which to test future gene therapeutic strategies for CS. The substantial previous phenotyping work in the shaker rat (20–24, 26, 27, 32, 35–39) combined with the methods contained herein will enable the shaker rat to play an important role in the development of a gene therapy.
Potential caveats
NHE6 protein levels in treated shaker rats significantly increased but remained relatively low. We interpret this as being related to restored NHE6 expression only in PCs, but not astrocytes. Perhaps the use of a ubiquitous promoter would restore NHE6 closer to wt levels. Despite this, it is apparent that even some degree of restoration of NHE6 expression targeted to a specific neuronal type is sufficient for substantial protection of motor function.
Successful expression of the L7-Slc9a6-GFP AAV was determined through western blotting rather than histology. Although the L7-GFP AAV yielded strong histological signal (Supplementary Material, Fig. S2A), GFP expressed by the L7-Slc9a6-GFP AAV was not clearly detectable over background autofluorescence by fluorescent microscopy. This may have been caused by either low expression or misfolding of the fused GFP. Importantly, however, a specific band corresponding to the Slc9a6–GFP fusion protein was detected upon western blotting the cerebella of L7-Slc9a6-GFP AAV-treated shaker rats (Supplementary Material, Fig. S2B) and was exclusive to rats with therapeutic benefit. Further, expression was also validated through Slc9a6 mRNA and NHE6 protein expression, both of which were increased significantly in cerebellum. This yielded significant changes in numerous key PC proteins associated with very substantial improvement in motor performance and indicating that the L7–6 targeting remained intact in L7-Slc9a6-GFP. As we were approaching AAV packaging limits with our construct, this likely substantially impacted translational efficiency (76). Future work using the L7 promoter would benefit from a dual virus injection consisting of primarily L7-Slc9a6 AAV and a small amount of L7-GFP AAV to validate viral presence in cerebellum. This approach would enable both visualization of transduction and greater translational efficiency.
Conclusions
The cerebellar dysfunction and motor symptoms exhibited by the shaker rat are caused by loss-of-function mutation in the Slc9a6 gene. Thus, the shaker rat is a spontaneous model of CS. Given the shaker rat’s strong motor phenotype, it should be considered as a model for mechanistic study and therapeutic development for CS.
Materials and Methods
All animal work was done under an approved Institutional Animal Care and Use Committee protocol at the University of Utah. Rats were housed 2–5 per cage under a 12:12 h light cycle with food and water provided ad libitum. We maintain the shaker colony in the WF inbred background (Envigo RMS LLC).
Genetic mapping
The shaker rat used in these studies is a spontaneous X-linked recessive mutant that we have previously described (23). Once the previously identified candidate gene Atp2b3 was excluded, we refined to 2.7 Mb, extending from 157.2 to 159.9 (Xq37-qter) through an F2, WF/BN intercross. The F1 offspring were generated by breeding affected WF males and commercial wt BN female rats, and the F2 generation was obtained by breeding affected F1 males with obligate carrier females. An assumption was made that the WF alleles would segregate in close proximity to the causative shaker gene. An additional 55 WF affected males and ten 45-week old asymptomatic (non Slc9a6 variant positive) N6F0 WF were added to our previously reported cohort for a total 242 affected male rats: 150 WF and 92 F2 WF/BN.
RNA-Seq
We conducted deep RNA-seq using total RNA on whole cerebella and brain hemispheres of three wt and three obligate affected 5-week old WF males. We extracted total RNA with miRNAeasy minikit (Qiagen Inc., Valencia, CA), obtaining RNA quality using the Agilent RNA ScreenTape Assay (Agilent; Santa Clara, CA). Library preparation took place utilizing Illumina TruSeq Stranded RNA Kit with Ribo-Zero Gold (rat). We generated paired-end 125-bp reads on a Hiseq 2000 machine at the University of Utah Microarray and Genomic Analysis Shared Resource using Illumina HiSeq 125 cycle paired-end sequencing v4.
We used SAMTools Mpileup on the alignment files and VarScan v2.3.3 (40) for variant calling. Per our exclusion of Atp2b3 as a causative disease gene, we focused on variants found on ChrX: 157239000 to qter. In order to estimate variance–mean dependence and test for differential expression, we utilized DESeq2 (77).
We examined the relative expression of X chromosome genes to look for differential expression and logarithmic fold changes. We first sorted our data by Max DESeq2 AdjP values and then by Log2 Ratio. The Max DESeq2 AdjP values were then defined as the maximum observed -10Log10(AdjP) between any pairwise DESeq2 comparisons. All P-values and AdjP were phred transformed where the value is −10*Log10(AdjP or pval); thus, −10*Log10(0.05) = 13. Therefore, significant cutoffs are those of values equal to or greater than 13. The Log2 ratio is defined as log2 (T/R), where T is the gene expression level in affected rats and R is the gene expression level in the wt samples.
Mutation description
To generate standardized nomenclature for this PTV, we utilized the freely available Mutalyzer-Description extractor software to generate the Human Genome Variation Society variant description. Although the National Center for Biotechnology Information (NCBI) Reference sequence XM_217630.9 is more reflective of the Slc9a6 human transcript, the most recent Rat Genome release Rnor_6.0 has a transcript different at the 5′ end of the observed mutation adding an additional 20 amino acids (ENSRNOT00000066809.3) (Supplementary Material, Fig. S1). Because Mutalyzer-Description extractor software is only compatible with NCBI sequences, we used NCBI to generate the variant description and Rnor_6.0 to anchor the markers used for genetic mapping.
Genotyping methods
We isolated genomic deoxyribonucleic acid (DNA) from rat tail tips using Qiagen genomic DNA extraction kit (Qiagen Inc., USA). We performed genotyping via PCR; a 363 bp fragment was produced with the following primers: forward of 5′-AAGACATGGCTGTGGCTCGG-3′ and reverse of 5′-AGCTAGGGGACAGGGGTCCG-3′. Because of the minimal difference in fragment sizes (4 bp), we confirmed genotype via Sanger sequencing with the same forward primer.
AAV production
We derived rat cDNA sequences for Slc9a6 (XM_001053956) from the NCBI DNA database, which we used to design primers to PCR-amplify the coding sequences from a cDNA library made from wt rat cerebellar RNA. The primer set was as follows: Slc9a6-F: 5′-TTTATGGCTGTG GCTCGGCGCGGCTGG-3′ and Slc9a6-R: 5′-TTTCGGCTGGACTGTGTCTTGTGTCATC-3′. We cloned the amplified PCR product into topoisomerase I (TOPO) vector (TOPO™ TA Cloning™ Kit, ThermoFisher, Cat# K457502) and verified by sequencing. The adeno-associated virus, pAAV/L7-6-GFP-WPRE (woodchuck hepatitis post transcriptional regulatory element)-SV40, was a gift from Dr Hirokazu Hirai (46), and the pUCmini-iCAP-PHP.eB was designed by the Gradinaru group (Addgene, plasmid #103005) (47). For the PHP.eB AAV expression plasmid, the Slc9a6 cDNA was re-PCR amplified from TOPO cloning vector using AgeI restriction sites and subsequently cloned into pAAV/L7-6-GFP-WPRE plasmid at AgeI site, designated as pAAV/L7-6-Slc9a6-GFP. All constructs were verified by sequencing. We generated recombinant AAV particles in the Drug Discovery Core Facility, University of Utah. Briefly, HEK-293 T cells were co-transfected with the following plasmids: pAAV/L7-6-Slc9A6-GFP or pAAV/L7-6-GFP (control) with pHelper (Stratagene, La Jolla, CA, USA), and pUCmini-iCAP-PHP.eB followed by viral particles purification, concentration and genomic titer determination.
Surgical procedures
In total, 18 age-matched rats were administered either the L7-GFP control AAV or the L7-Slc9a6-GFP AAV, including three male wt rats, two female carriers, six males hemizygous and seven females homozygous for the candidate allele. We administered the L7-Slc9a6-GFP AAV to eight shakers, and the control L7-GFP AAV to five shakers and five wt or carriers. An additional four rats—two male wt and two female carriers—served as uninjected controls. Littermate effect was avoided by group randomization. The wt rats were all male given that our focus was on treatment of shaker rats and the fact that it is not possible to generate wt and shaker females in the same litter. Notably, we previously analyzed the gait of wt males and females and found no significant differences in straightness of gait between wt animals of both sexes (22). We anesthetized rats with 2% isoflurane at 35 +/− 2 days of age. We shaved and disinfected the surgical site and placed rats on an electronically controlled heating pad set to body temperature in a stereotactic frame. We administered 0.1 ml bupivacaine to the incision site and opened to the scalp and dried. We marked the craniotomy site for the injection of AAV to the right lateral ventricle with respect to bregma and used a burr to drill a hole in the marked location. We inserted a needle attached to a syringe (Hamilton Co., Reno, NV) into the lateral ventricle, waited for 3 min, then injected 10 μl of virus at < 2.0 μl/min. Five minutes after injection, the needle was withdrawn at a rate not exceeding 1.0 mm/min. We sutured the incision and provided 0.1 mg/kg carprofen subcutaneously daily for 3 days. Rats were then given a 12- to 16-day recovery period.
Motor analyses
We made 30-min force plate-based motor recordings in the aforementioned rats at 7, 18 and 25 weeks of age using previously described methods (22,38), collecting data through in-house software and a force plate actometer device, a widely used and described tool for motor and behavioral data collection (39). These timepoints chosen were on the basis of typical start of PC degeneration, after the end of the typical PC degeneration, and when motor symptom progression is complete (22). We tracked center of mass at 1000 Hz and analyzed straightness of gait during rapid movements as a measure of ataxia of gait. We computed a ratio of distance traveled to displacement during rapid movements, in which we previously showed that wt rats average 1.5–1.7 and severely ataxic rats average ~3.0 (22). Notably, we previously found that carrier rats have insignificantly different motor ataxia from wt rats. It is impossible to breed both affected females and wt females in the same litter. Thus, in the interest of litter matching across groups, motor results labeled as wt are a combination of wt and carrier.
Extraction of tissues for cellular and molecular analyses
Following experiments, we deeply anesthetized rats and extracted all cerebella. In 21 of 22 rats used in AAV experiments, we retained one of half of each brains for histological analyses, whereas one brain was not evaluated because of damage on removal. We took the other half of the 11 of the 22 cerebella—as well as two more uninjected, age-matched wt and carrier rats—for a total of 15 samples and flash froze at −80°C. These samples were homogenized, with half of each sample used for mRNA analyses and the other half used for protein analyses.
Preparation of protein lysates and western blotting
We prepared cellular extracts by a single-step lysis method (78). We suspended harvested cells in sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer [Laemmli sample buffer (Bio-Rad, Cat# 161-0737)] before boiling for 5 min. We used equal amounts of the extracts for western blot analyses. We prepared rat cerebellar protein extracts by homogenization of tissues in extraction buffer [25 mM Tris–HCl pH 7.6, 300 mM NaCl, 0.5% Nonidet P-40, 2 mM EDTA, 2 mM MgCl2, 0.5 M urea, and protease inhibitors (Sigma-Aldrich, P-8340)], followed by centrifugation at 4°C for 20 min at 14 000 rotations per minute (RPM), only using supernatants for western blotting. We resolved protein extracts by SDS-PAGE and transferred to Hybond P membranes (Amersham Bioscience) before processing for western blotting according to our previously published protocol (78). We used the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Cat# WBKLSO500) to visualize the signals and detected on the ChemiDoc MP imager (Bio-Rad). For some blots, we used a film developing system and quantified band intensities using ImageJ software after inversion of the images. Relative protein abundances were expressed as ratios to beta-actin (ACTB) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
We used the following antibodies and dilutions for western blotting: Anti-NHE6/Slc9a6 antibody [(1: 5000), Abcam, Cat# ab137185], Anti-NHE6/Slc9a6 antibody [(1:3000), ThermoFisher, Cat# PA5-36997], GFP antibody (B-2) [(1:2000), Santa Cruz, sc-9996], GAPDH (14C10) rabbit mAb [(1:5000), Cell Signaling, Cat# 2118], monoclonal anti-β-Actin−peroxidase antibody (clone AC-15) [(1:30 000), Sigma-Aldrich, A3854], monoclonal Anti-Calbindin-D-28 K antibody [(1:5000), Sigma-Aldrich, C9848], RGS8 antibody [(1:5000) (Novus Biologicals, NBP2-20153)], PCP-2 antibody (F-3) [(1:3000), Santa Cruz, sc-137 064], and GFP rabbit polyclonal antibody (FL) [(1:1000), Santa Cruz, sc-8334]. The secondary antibodies were peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H + L) antibody [(1:5000), Jackson ImmunoResearch Laboratories, Cat# 111-035-144] and Peroxidase-conjugated horse anti-mouse IgG (H + L) antibody [(1:5000) (Vector laboratories, PI-2000)]. Initial western blots were carried out using three wt rats of both genders, three carrier females and three shaker rats of both genders, whereas later western blots were made from the rats described below, as well as two additional uninjected, age-matched wt rats, one male and one female, and two additional carrier females.
siRNAs and reagents
For siRNA experiments, we used the following siRNAs: All Star Negative Control siRNA (Qiagen, Cat# 1027280), human siSLC9A6: 5′-GGA ACA GCA AUU UCU UGU UUC GUUA-3′ (45). siRNA oligonucleotides were synthesized by Invitrogen, USA. The oligonucleotides were deprotected and the complementary strands were annealed.
RNA analyses
Total RNA from flash frozen cerebellums was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The extracted RNA was then used to synthesize cDNA using ProtoScript® First Strand cDNA Synthesis Kit (New England Biolabs). Quantitative real-time PCR (qRT-PCR) was performed in an Applied Biosystems™ QuantStudio™ 12 K Flex Real-time PCR system using TaqMan™ Gene Expression Assays (FAM) (ThermoFisher, actin Rn00667869_m1, slc9a6 Rn01402034_m1, Calb1 Rn00583140_m1, fam107b Rn0165005_m1, pcp2 Rn01403231_g1, rgs8 Rn00571066_m1, gfap Rn01253033_m1) and TaqMan™ Fast Advanced Master Mix (ThermoFisher). PCR cycling parameters were set according to the manufacturer’s protocol for TaqMan™ Fast Advanced Master Mix and TaqMan™ Gene Expression Assays. The qPCR data was analyzed by the ΔΔCt method, using ΔΔCt values calculated by the QuantStudio™ 12 K Flex software.
Histological analyses
We incubated half of each cerebellum in 4% PFA for 24 h followed by 24 h at each of 10%, 20% and 30% sucrose. Tissue were cryoprotected in optimal cutting temperature blocks, flash frozen and then cut into 20 μm coronal slices with a Leica 5100S cryostat (Wetzlar, Germany), retaining one slice every 200 μm. In all 21 animals, we stained at least three slices with a mouse monoclonal anti-calbindin-D-28K (1:500) and a counterstain for DAPI.
Statistical analyses
Comparisons were made via two-sample Student’s t-tests with n = the number of animals unless otherwise noted. P-values are reported via significance bars for all mentioned comparisons as follows: ns refers to P > 0.05, whereas * refers to P < 0.05, ** refers to P < 0.01 and *** refers to P < 0.001.
Supplementary Material
Acknowledgements
The authors thank Christopher R. Nelson for his technical assistance with histological slice preparation. We acknowledge the Cell Imaging Core at the University of Utah for use of several microscopes and thank Dr Michael Bridge for his assistance.
Conflict of Interest statement. None declared.
Contributor Information
Karla P Figueroa, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA.
Collin J Anderson, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA; School of Medical Sciences, University of Sydney, Camperdown NSW 2006, Australia; School of Biomedical Engineering University of Sydney, Darlington NSW 2008, Australia.
Sharan Paul, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA.
Warunee Dansithong, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA.
Mandi Gandelman, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA.
Daniel R Scoles, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA.
Stefan M Pulst, Department of Neurology, University of Utah, Salt Lake City, UT 84132, USA.
Funding
National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (NINDS) (R37 NS033123) (Pulst), NIH NINDS (R21 NS104799) (Pulst), NIH NINDS (R21 NS079852) (Pulst), an RTW Charitable Foundation Grant (Pulst, Anderson, Paul), a National Ataxia Foundation Postdoctoral Fellowship (Anderson), a National Ataxia Foundation Junior Investigator Award (Anderson) and a Harrington Discovery Institute Rare Disease Scholar Award (Scoles).
Data Availability
Supporting data for this study are available from the corresponding author upon reasonable request.
References
- 1. Burright, E.N., Brent Clark, H., Servadio, A., Matilla, T., Feddersen, R.M., Yunis, W.S., Duvick, L.A., Zoghbi, H.Y. and Orr, H.T. (1995) SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell, 82, 937–948. [DOI] [PubMed] [Google Scholar]
- 2. Huynh, D.P., Figueroa, K., Hoang, N. and Pulst, S.-M. (2000) Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet., 26, 44–50. [DOI] [PubMed] [Google Scholar]
- 3. Dansithong, W., Paul, S., Figueroa, K.P., Rinehart, M.D., Wiest, S., Pflieger, L.T., Scoles, D.R. and Pulst, S.M. (2015) Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet., 11, e1005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colomer Gould, V.F. (2012) Mouse models of spinocerebellar ataxia type 3 (Machado-Joseph disease). Neurotherapeutics, 9, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watase, K., Barrett, C.F., Miyazaki, T., Ishiguro, T., Ishikawa, K., Hu, Y., Unno, T., Sun, Y., Kasai, S., Watanabe, M. et al. (2008) Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl. Acad. Sci., 105, 11987–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoo, S.Y., Pennesi, M.E., Weeber, E.J., Xu, B., Atkinson, R., Chen, S., Armstrong, D.L., Wu, S.M., Sweatt, J.D. and Zoghbi, H.Y. (2003) SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron, 37, 383–401. [DOI] [PubMed] [Google Scholar]
- 7. White, M., Xia, G., Gao, R., Wakamiya, M., Sarkar, P.S., McFarland, K. and Ashizawa, T. (2012) Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure — a toxic RNA gain-of-function model. J. Neurosci. Res., 90, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cendelin, J., Cvetanovic, M., Gandelman, M., Hirai, H., Orr, H.T., Pulst, S.M., Strupp, M., Tichanek, F., Tuma, J. and Manto, M. (2021) Consensus paper: strengths and weaknesses of animal models of spinocerebellar ataxias and their clinical implications. Cerebellum, 3, 452–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramani, B., Harris, G.M., Huang, R., Seki, T., Murphy, G.G., Costa, M., Fischer, S., Saunders, T.L., Xia, G., McEachin, R.C. et al. (2015) A knockin mouse model of spinocerebellar ataxia type 3 exhibits prominent aggregate pathology and aberrant splicing of the disease gene transcript. Hum. Mol. Genet., 24, 1211–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramani, B., Panwar, B., Moore, L.R., Wang, B., Huang, R., Guan, Y. and Paulson, H.L. (2017) Comparison of spinocerebellar ataxia type 3 mouse models identifies early gain-of-function, cell-autonomous transcriptional changes in oligodendrocytes. Hum. Mol. Genet., 26, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Phillips, R.J.S. (1960) ‘Lurcher’, a new gene in linkage group XI of the house mouse. J. Genet., 57, 35. [Google Scholar]
- 12. Lalouette, A., Guénet, J.L. and Vriz, S. (1998) Hotfoot mouse mutations affect the delta 2 glutamate receptor gene and are allelic to lurcher. Genomics, 50, 9–13. [DOI] [PubMed] [Google Scholar]
- 13. Sidman, R.L., Lane, P.W. and Dickie, M.M. (1962) Staggerer, a new mutation in the mouse affecting the cerebellum. Science, 137, 610–612. [DOI] [PubMed] [Google Scholar]
- 14. Hamilton, B.A., Frankel, W.N., Kerrebrock, A.W., Hawkins, T.L., FitzHugh, W., Kusumi, K., Russell, L.B., Mueller, K.L., van Berkel, V., Birren, B.W. et al. (1996) Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature, 379, 736–739. [DOI] [PubMed] [Google Scholar]
- 15. Falconer, D.S. (1951) Two new mutants, “trembler” and “reeler”, with neurological actions in the house mouse (Mus musculus L.). J. Genet., 50, 192–201. [DOI] [PubMed] [Google Scholar]
- 16. Patil, N., Cox, D.R., Bhat, D., Faham, M., Myers, R.M. and Peterson, A.S. (1995) A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat. Genet., 11, 126–129. [DOI] [PubMed] [Google Scholar]
- 17. Sweet, H.O., Bronson, R.T., Johnson, K.R., Cook, S.A. and Davisson, M.T. (1996) Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm. Genome Off. J. Int. Mamm. Genome Soc., 7, 798–802. [DOI] [PubMed] [Google Scholar]
- 18. Cendelin, J. (2014) From mice to men: lessons from mutant ataxic mice. Cerebellum Ataxias, 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manto, M. and Marmolino, D. (2009) Animal models of human cerebellar ataxias: a cornerstone for the therapies of the twenty-first century. Cerebellum, 8, 137–154. [DOI] [PubMed] [Google Scholar]
- 20. La Regina, M.C., Yates-Siilata, K., Woods, L. and Tolbert, D. (1992) Preliminary characterization of hereditary cerebellar ataxia in rats. Lab. Anim. Sci., 42, 19–26. [PubMed] [Google Scholar]
- 21. Tolbert, D.L., Ewald, M., Gutting, J. and La Regina, M.C. (1995) Spatial and temporal pattern of Purkinje cell degeneration in shaker mutant rats with hereditary cerebellar ataxia. J. Comp. Neurol., 355, 490–507. [DOI] [PubMed] [Google Scholar]
- 22. Anderson, C.J., Figueroa, K.P., Dorval, A.D. and Pulst, S.M. (2019) Deep cerebellar stimulation reduces ataxic motor symptoms in the shaker rat. Ann. Neurol., 85, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Figueroa, K.P., Paul, S., Calì, T., Lopreiato, R., Karan, S., Frizzarin, M., Ames, D., Zanni, G., Brini, M., Dansithong, W. et al. (2016) Spontaneous shaker rat mutant – a new model for X-linked tremor/ataxia. Dis. Model. Mech., 9, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Erekat, N.S. (2017) Cerebellar Purkinje cells die by apoptosis in the shaker mutant rat. Brain Res., 1657, 323–332. [DOI] [PubMed] [Google Scholar]
- 25. Erekat, N.S. (2018) Autophagy precedes apoptosis among at risk cerebellar Purkinje cells in the shaker mutant rat: an ultrastructural study. Ultrastruct. Pathol., 42, 162–169. [DOI] [PubMed] [Google Scholar]
- 26. Wolf, L.W., LaRegina, M.C. and Tolbert, D.L. (1996) A behavioral study of the development of hereditary cerebellar ataxia in the shaker rat mutant. Behav. Brain Res., 75, 67–81. [DOI] [PubMed] [Google Scholar]
- 27. Clark, B.R., LaRegina, M. and Tolbert, D.L. (2000) X-linked transmission of the shaker mutation in rats with hereditary Purkinje cell degeneration and ataxia. Brain Res., 858, 264–273. [DOI] [PubMed] [Google Scholar]
- 28. Erekat, N.S. (2019) Active caspase-3 upregulation is augmented in at-risk cerebellar Purkinje cells following inferior olive chemoablation in the shaker mutant rat: an immunofluorescence study. Neurol. Res., 41, 234–241. [DOI] [PubMed] [Google Scholar]
- 29. Miterko, L.N., Lin, T., Zhou, J., van der Heijden, M.E., Beckinghausen, J., White, J.J. and Sillitoe, R.V. (2021) Neuromodulation of the cerebellum rescues movement in a mouse model of ataxia. Nat. Commun., 12, 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cury, R.G., França, C., Silva, V., Barbosa, E.R., Capato, T.T.C., Lepski, G., Duarte, K.P., Teixeira, M.J. and Andrade, D.C. (2019) Effects of dentate nucleus stimulation in spinocerebellar ataxia type 3. Parkinsonism Relat. Disord., 69, 91–93. [DOI] [PubMed] [Google Scholar]
- 31. Cury, R.G., França, C., Duarte, K.P., Paraguay, I., Diniz, J.M., Cunha, P., Galhardoni, R., Silva, V., Iglesio, R., Bissoli, A.B. et al. (2021) Safety and outcomes of dentate nucleus deep brain stimulation for cerebellar ataxia. Cerebellum, 21, 861–865. [DOI] [PubMed] [Google Scholar]
- 32. Lang, J., Haas, E., Hübener-Schmid, J., Anderson, C.J., Pulst, S.M., Giese, M.A. and Ilg, W. (2020) Detecting and quantifying ataxia-related motor impairments in rodents using markerless motion tracking with deep neural networks. 2020 42nd Annual International Conference of the IEEE Engineering in Medicine Biology Society (EMBC), 3642–3648. [DOI] [PMC free article] [PubMed]
- 33. Schroer, R.J., Holden, K.R., Tarpey, P.S., Matheus, M.G., Griesemer, D.A., Friez, M.J., Fan, J.Z., Simensen, R.J., Strømme, P., Stevenson, R.E. et al. (2010) Natural history of Christianson syndrome. Am. J. Med. Genet. A, 0, 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Masurel-Paulet, A., Piton, A., Chancenotte, S., Redin, C., Thauvin-Robinet, C., Henrenger, Y., Minot, D., Creppy, A., Ruffier-Bourdet, M., Thevenon, J. et al. (2016) A new family with an SLC9A6 mutation expanding the phenotypic spectrum of Christianson syndrome. Am. J. Med. Genet. A, 170, 2103–2110. [DOI] [PubMed] [Google Scholar]
- 35. Kuo, S.-H., Louis, E.D., Faust, P.L., Handforth, A., Chang, S.-Y., Avlar, B., Lang, E.J., Pan, M.-K., Miterko, L.N., Brown, A.M. et al. (2019) Current opinions and consensus for studying tremor in animal models. Cerebellum Lond. Engl., 18, 1036–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Erekat, N.S. (2022) Programmed cell death in cerebellar Purkinje neurons. J. Integr. Neurosci., 21, 30. [DOI] [PubMed] [Google Scholar]
- 37. Erekat, N.S. (2010) Mechanisms Underlying Cerebellar Purkinje Cell Death in the Shaker Mutant Rat. Ph.D. In Saint Louis University, Missouri, USA. [Google Scholar]
- 38. Fowler, S.C., Birkestrand, B.R., Chen, R., Moss, S.J., Vorontsova, E., Wang, G. and Zarcone, T.J. (2001) A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J. Neurosci. Methods, 107, 107–124. [DOI] [PubMed] [Google Scholar]
- 39. Zarcone, T.J. (2022) Neuroscience and actometry: an example of the benefits of the precise measurement of behavior. Brain Res. Bull., 185, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koboldt, D.C., Zhang, Q., Larson, D.E., Shen, D., McLellan, M.D., Lin, L., Miller, C.A., Mardis, E.R., Ding, L. and Wilson, R.K. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res., 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gene Detail: Allen Brain Atlas: Mouse Brain. https://mouse.brain-map.org/gene/show/88236 (accessed Feb 7, 2022).
- 42. Gilfillan, G.D., Selmer, K.K., Roxrud, I., Smith, R., Kyllerman, M., Eiklid, K., Kroken, M., Mattingsdal, M., Egeland, T., Stenmark, H. et al. (2008) SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am. J. Hum. Genet., 82, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ilie, A., Gao, A.Y.L., Reid, J., Boucher, A., McEwan, C., Barrière, H., Lukacs, G.L., McKinney, R.A. and Orlowski, J. (2016) A Christianson syndrome-linked deletion mutation (∆287ES288) in SLC9A6 disrupts recycling endosomal function and elicits neurodegeneration and cell death. Mol. Neurodegener., 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ilie, A., Boucher, A., Park, J., Berghuis, A.M., McKinney, R.A. and Orlowski, J. (2020) Assorted dysfunctions of endosomal alkali cation/proton exchanger SLC9A6 variants linked to Christianson syndrome. J. Biol. Chem., 295, 7075–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ohgaki, R., Matsushita, M., Kanazawa, H., Ogihara, S., Hoekstra, D. and van IJzendoorn, S.C.D. (2010) The Na+/H+ exchanger NHE6 in the endosomal recycling system is involved in the development of apical bile Canalicular surface domains in HepG2 cells. Mol. Biol. Cell, 21, 1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nitta, K., Matsuzaki, Y., Konno, A. and Hirai, H. (2017) Minimal Purkinje cell-specific PCP2/L7 promoter virally available for rodents and non-human primates. Mol. Ther. Methods Clin. Dev., 6, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chan, K.Y., Jang, M.J., Yoo, B.B., Greenbaum, A., Ravi, N., Wu, W.-L., Sánchez-Guardado, L., Lois, C., Mazmanian, S.K., Deverman, B.E. et al. (2017) Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci., 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hansen, S.T., Meera, P., Otis, T.S. and Pulst, S.M. (2013) Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet., 22, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scoles, D.R., Meera, P., Schneider, M., Paul, S., Dansithong, W., Figueroa, K.P., Hung, G., Rigo, F., Bennett, C.F., Otis, T.S. et al. (2017) Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature, 544, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pflieger, L.T., Dansithong, W., Paul, S., Scoles, D.R., Figueroa, K.P., Meera, P., Otis, T.S., Facelli, J.C. and Pulst, S.M. (2017) Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum. Mol. Genet., 26, 3069–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu, Q.-W. and Kapfhammer, J.P. (2021) Modulation of increased mGluR1 Signaling by RGS8 protects Purkinje cells from dendritic reduction and could be a common mechanism in diverse forms of spinocerebellar ataxia. Front. Cell Dev. Biol., 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Niewiadomska-Cimicka, A., Doussau, F., Perot, J.-B., Roux, M.J., Keime, C., Hache, A., Piguet, F., Novati, A., Weber, C., Yalcin, B. et al. (2021) SCA7 mouse cerebellar pathology reveals preferential downregulation of key Purkinje cell-identity genes and shared disease signature with SCA1 and SCA2. J. Neurosci., 41, 4910–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Itoh, M., Odagiri, M., Abe, H. and Saitoh, O. (2001) RGS8 protein is distributed in dendrites and cell body of cerebellar Purkinje cell. Biochem. Biophys. Res. Commun., 287, 223–228. [DOI] [PubMed] [Google Scholar]
- 54. Christianson, A.L., Stevenson, R.E., van der Meyden, C.H., Pelser, J., Theron, F.W., van Rensburg, P.L., Chandler, M. and Schwartz, C.E. (1999) X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large south African kindred is localised to Xq24-q27. J. Med. Genet., 36, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brett, C.L., Wei, Y., Donowitz, M. and Rao, R. (2002) Human Na(+)/H(+) exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. Am. J. Physiol. Cell Physiol., 282, C1031–C1041. [DOI] [PubMed] [Google Scholar]
- 56. Ouyang, Q., Lizarraga, S.B., Schmidt, M., Yang, U., Gong, J., Ellisor, D., Kauer, J.A. and Morrow, E.M. (2013) Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron, 80, 97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pescosolido, M.F., Ouyang, Q., Liu, J.S. and Morrow, E.M. (2021) Loss of Christianson syndrome Na+/H+ exchanger 6 (NHE6) causes abnormal endosome maturation and trafficking underlying lysosome dysfunction in neurons. J. Neurosci., 41, 9235–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zanni, G., Calì, T., Kalscheuer, V.M., Ottolini, D., Barresi, S., Lebrun, N., Montecchi-Palazzi, L., Hu, H., Chelly, J., Bertini, E. et al. (2012) Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA, 109, 14514–14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. OMIM Phenotypic Series – PS309510 https://omim.org/phenotypicSeries/PS309510?sort=geneSymbols (accessed Aug 10, 2020).
- 60. Pescosolido, M.F., Stein, D.M., Schmidt, M., El Achkar, C.M., Sabbagh, M., Rogg, J.M., Tantravahi, U., McLean, R.L., Liu, J.S., Poduri, A. et al. (2014) Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann. Neurol., 76, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ouyang, Q., Joesch-Cohen, L., Mishra, S., Riaz, H.A., Schmidt, M. and Morrow, E.M. (2019) Functional assessment in vivo of the mouse homolog of the human Ala-9-Ser NHE6 variant. eNeuro, 6, ENEURO.0046–ENEU19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao, A.Y.L., Ilie, A., Chang, P.K.Y., Orlowski, J. and McKinney, R.A. (2019) A Christianson syndrome-linked deletion mutation (Δ287ES288) in SLC9A6 impairs hippocampal neuronal plasticity. Neurobiol. Dis., 130, 104490. [DOI] [PubMed] [Google Scholar]
- 63. Prasad, H. and Rao, R. (2018) Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. USA, 115, E6640–E6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kerner-Rossi, M., Gulinello, M., Walkley, S. and Dobrenis, K. (2019) Pathobiology of Christianson syndrome: linking disrupted endosomal-lysosomal function with intellectual disability and sensory impairments. Neurobiol. Learn. Mem., 165, 106867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Strømme, P., Dobrenis, K., Sillitoe, R.V., Gulinello, M., Ali, N.F., Davidson, C., Micsenyi, M.C., Stephney, G., Ellevog, L., Klungland, A. et al. (2011) X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal–lysosomal dysfunction. Brain, 134, 3369–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu, M., Ouyang, Q., Gong, J., Pescosolido, M.F., Pruett, B.S., Mishra, S., Schmidt, M., Jones, R.N., Gamsiz Uzun, E.D., Lizarraga, S.B. et al. (2017) Mixed neurodevelopmental and neurodegenerative pathology in Nhe6-null mouse model of Christianson syndrome. eNeuro, 4, ENEURO.0388-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sikora, J., Leddy, J., Gulinello, M. and Walkley, S.U. (2016) X-linked Christianson syndrome: heterozygous female Slc9a6 knockout mice develop mosaic neuropathological changes and related behavioral abnormalities. Dis. Model. Mech., 9, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Prasad, H. and Rao, R. (2015) The Na+/H+ exchanger NHE6 modulates endosomal pH to control processing of amyloid precursor protein in a cell culture model of Alzheimer disease. J. Biol. Chem., 290, 5311–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chakrabarty, P., Rosario, A., Cruz, P., Siemienski, Z., Ceballos-Diaz, C., Crosby, K., Jansen, K., Borchelt, D.R., Kim, J.-Y., Jankowsky, J.L. et al. (2013) Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS One, 8, e67680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lukashchuk, V., Lewis, K.E., Coldicott, I., Grierson, A.J. and Azzouz, M. (2016) AAV9-mediated central nervous system–targeted gene delivery via cisterna magna route in mice. Mol. Ther. Methods Clin. Dev., 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ilie, A., Gao, A.Y.L., Boucher, A., Park, J., Berghuis, A.M., Hoffer, M.J.V., Hilhorst-Hofstee, Y., McKinney, R.A. and Orlowski, J. (2019) A potential gain-of-function variant of SLC9A6 leads to endosomal alkalinization and neuronal atrophy associated with Christianson syndrome. Neurobiol. Dis., 121, 187–204. [DOI] [PubMed] [Google Scholar]
- 72. Petitjean, H., Fatima, T., Mouchbahani-Constance, S., Davidova, A., Ferland, C.E., Orlowski, J. and Sharif-Naeini, R. (2020) Loss of SLC9A6/NHE6 impairs nociception in a mouse model of Christianson syndrome. Pain, 161, 2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kucharava, K., Brand, Y., Albano, G., Sekulic-Jablanovic, M., Glutz, A., Xian, X., Herz, J., Bodmer, D., Fuster, D.G. and Petkovic, V. (2020) Sodium-hydrogen exchanger 6 (NHE6) deficiency leads to hearing loss, via reduced endosomal signalling through the BDNF/Trk pathway. Sci. Rep., 10, 3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schnyder, D., Albano, G., Kucharczyk, P., Dolder, S., Siegrist, M., Anderegg, M., Pathare, G., Hofstetter, W., Baron, R. and Fuster, D.G. (2021) Deletion of the sodium/hydrogen exchanger 6 causes low bone volume in adult mice. Bone, 153, 116178. [DOI] [PubMed] [Google Scholar]
- 75. Lee, Y., Miller, M.R., Fernandez, M.A., Berg, E.L., Prada, A.M., Ouyang, Q., Schmidt, M., Silverman, J.L., Young-Pearse, T.L. and Morrow, E.M. (2021) Early lysosome defects precede neurodegeneration with amyloid-β and tau aggregation in NHE6-null rat brain. Brain, 145, 3187–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dong, J.-Y., Fan, P.-D. and Frizzell, R.A. (1996) Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther., 7, 2101–2112. [DOI] [PubMed] [Google Scholar]
- 77. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Paul, S., Dansithong, W., Figueroa, K.P., Scoles, D.R. and Pulst, S.M. (2018) Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun., 9, 3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supporting data for this study are available from the corresponding author upon reasonable request.