

Abstract
Regenerative medicine as a field has emerged as a new component of modern medicine and medical research that encompasses a wide range of products including cellular and acellular therapies. As this new field emerged, regulatory agencies like the Food and Drug Administration (FDA) rapidly adapted existing regulatory frameworks to address the transplantation, gene therapy, cell-based therapeutics, and acellular biologics that fall under the broader regenerative medicine umbrella. Where it has not been possible to modify existing regulation and processes, entirely new frameworks have been generated with the intention of providing flexible, forward-facing systems to regulate this rapidly growing field. This review discusses the current state of FDA regulatory affairs in the context of stem cells and extracellular vesicles by highlighting gaps in the current regulatory system and then discussing where regulatory science in regenerative medicine may be headed based on these gaps and the FDA’s historical ability to deal with emerging fields. Lastly, we utilize case studies in stem cell and acellular based treatments to demonstrate how regulatory science has evolved in regenerative medicine and highlight the ongoing clinical efforts and challenges of these therapies.
Keywords: extracellular vesicles, stem cells, translational research, nanomedicine, regenerative medicine
1. Introduction
In a review of FDA regulatory science and regulatory affairs, it is important to clearly define and delineate the two terms. The FDA defines regulatory science as “the science of developing new tools, standards, and approaches to assess the safety, efficacy, quality, and performance of all FDA-regulated products.”1 In contrast, regulatory affairs utilizes regulatory science to “protect consumers/patients and enhance public health by ensuring timely access to safe, quality FDA-regulated products.”2
Regenerative medicine as a field has emerged as a new component of modern medicine and medical research. Promising cell culture, animal model, and early clinical studies have demonstrated efficacy in several diseases and conditions.3–6 Transplantation, gene therapy, cell-based therapeutics, and acellular biologics all fall under the broader regenerative medicine umbrella. As each new modality has emerged, regulatory agencies like the Food and Drug Administration (FDA) have rapidly adapted existing regulatory frameworks to address these applications. These regulatory frameworks are the organization and implementation of enacting standard regulations for a particular product type, the goal of which is to protect consumers. Where it has not been possible to modify existing regulation and processes, entirely new frameworks have been generated with the intention of providing flexible, forward-facing systems to regulate the rapidly growing field of regenerative medicine.
Although the regenerative medicine field encompasses a wide range of products, we will focus on two main categories of regenerative products in this review: cellular and acellular therapies. Applications of cell-based therapies, especially stem cells (SCs), have been established and are being utilized in clinical applications (Figure 1). An analysis of the regulation of SCs demonstrates how current regulatory frameworks have affected and continue to affect current regenerative medicine products. Conversely, newer, acellular biologics like biogenic extracellular vesicles (EVs) are only in the beginning phases of regulation, with most products being in the basic science translational stage of production (Figure 1). Products that are in this early stage of the regulatory pipeline provide an opportunity for changes to be made to improve the process of translation of regenerative therapies.
Figure 1.
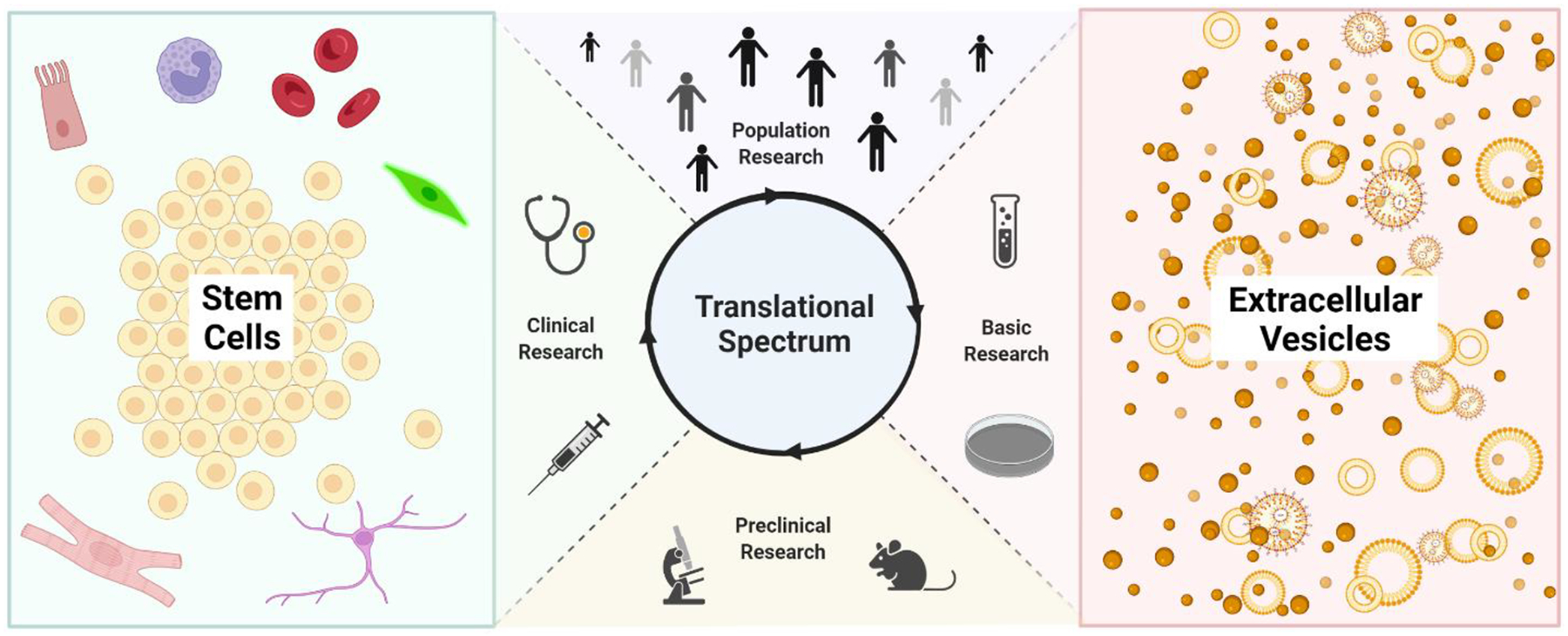
The Translational Stages of SC and EV Regenerative Therapies
We will begin our discussion of the current state of FDA regulation as it applies to regenerative medicine by discussing how the FDA applies and might apply its priority areas or goals for advancing regulatory science. It is important to lay this groundwork to understand not only the current state of regulation, but also gaps, and where we propose regulatory science in regenerative medicine may be headed. The following sections will discuss regulatory affairs in SC and EV based products (while we acknowledge that SC and EV based products do not comprise all regenerative therapies, we will use “regenerative therapies” in this review to refer to SC and EV based products). We will conclude by reiterating the major points discussed.
2. Regulation in the era of regenerative medicine
2.1. FDA efforts in regulatory science
In 2011, the FDA published a strategic plan for advancing regulatory science where it discussed plans to increase the speed of translation while maintaining critical evaluation of the safety and efficacy of products. In this document, the FDA identified eight priority areas in regulatory science necessary to its plan; a ninth priority area was added in 2013.7,8 The nine priority areas are depicted in Figure 2. The FDA also recently released a new document in 2021 outlining what it identified as “The Focus Areas of Regulatory Science” in which the FDA identifies 4 new Strategic Initiatives: 1. Public health preparedness and response, 2. Increasing choice and competition through innovation, 3. Unleashing the power of data, and 4. Empowering patients and consumers. Each strategic initiative is composed of several Focus Areas of Regulatory Science (FARS) which focus research on a unique aspect of the broader goal of the strategic initiative. The major purpose of the 2021 document was to identify areas of importance/ongoing research at the FDA because of the advancement of technology and medicine since 2011. Because this section discusses historical and ongoing FDA efforts for regulation, we will discuss FDA efforts and gaps in the context of six priority areas (i.e., priority areas 1, 2, 3, 4, 5, and 8) pertaining to SC and EV based products. Priority areas 6, 7, and 9 are tangential to SC and EV-based products and will not be discussed in detail in this review (e.g. for priority area 7, many medical treatments could be considered “medical counter measures”).
Figure 2.
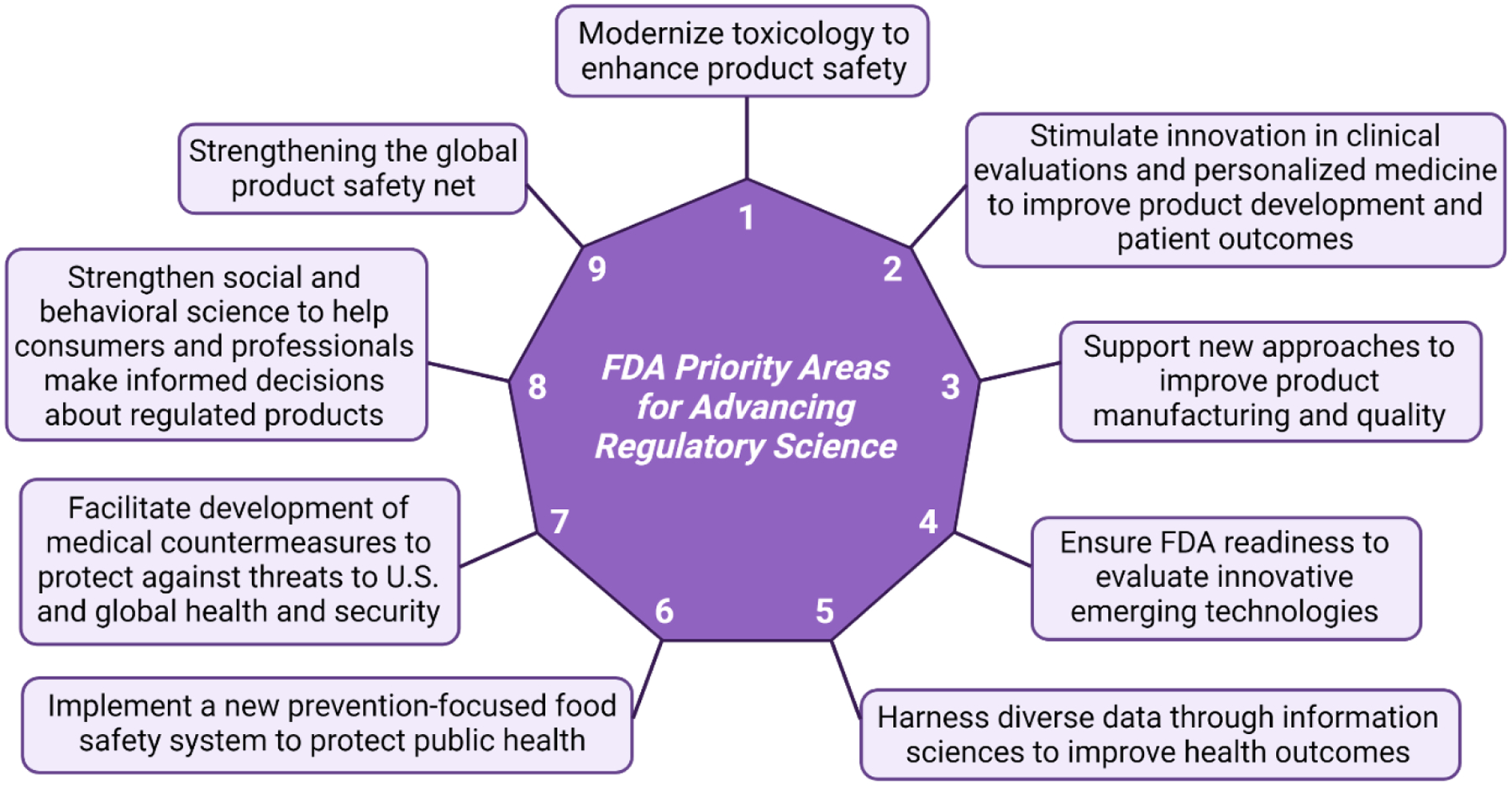
The Nine FDA Priority Areas for Advancing Regulatory Science7,8
2.2. Priority Areas 1: Modernize toxicology to enhance product safety and 2: Stimulate innovation in clinical evaluations and personalized medicine to improve product development and patient outcomes
The biologic complexity of regenerative therapies (compared to drugs and devices) in composition and mechanistic action, as well as inherent difficulties in quality control and manufacturing methods have presented challenges to the status quo evaluation of these products. For example, developing pure, homogeneous samples of biogenic EVs is technically difficult to achieve because of their natural heterogeneity and by the imprecision in which they are defined.9 Some purification methods used to isolate precise subsets of EVs (e.g. immunoprecipitation) may damage the product or render it biologically inactive and therapeutically useless.9 Because of this, understanding the composition, defining the broad population of EVs, and ensuring their safety profile as whole prior to evaluation of efficacy in humans may be an alternative to previously established methods of evaluating purity. Similar to the FDA regulation of SC based therapies, modernizing toxicology and stimulating innovation in the evaluation of complex or heterogeneous biological products are paramount to facilitating rapid and safe translation of emergent therapies (i.e., EVs) in regenerative medicine.7
2.3. Priority Area 3: Support new approaches to improve product manufacturing and quality
Since the 1990’s, the definition of what constitutes “regenerative medicine” has evolved and will continue to do so as the field acquires better understanding of the many sources and applications of regenerative products in medicine.10 The FDA regulates products based on how they primarily achieve therapeutic effect. As such, most EV- and SC-based therapies/products currently fall under the regulation of the FDA’s Center for Biologics Evaluation and Research (CBER), which regulates biologically derived products for human use.11 It is important to note that combination products, such as drug-loaded EVs or SC products that include some scaffold or device components, may be assigned to CBER or other centers such as the Center for Drug Evaluation and Research (CDER) and in unique cases the Center for Devices and Radiological Health (CDRH) depending on the product’s primary mode of action; thus in some cases, products may require evaluation from multiple centers.12
In 2011, the current good tissue practice (CGTP) FDA guidance was finalized with guidelines and standards for the use of SC and other cell-based product (this guidance was subsequently updated in July of 2020)13. Standardization for the generation of these biologics was a major step in the FDA helping to support modern manufacturing of biomedical products. EVs used in regenerative therapy can be collected from human tissue or manufactured by collecting them from cultured cells. However, there is currently no standardization in the collection/purification of EVs; the isolation of desired EVs can be accomplished by multiple methods such as: size-exclusion chromatography (SEC), dialysis or PEGylation with polyethylene-glycol. The various methods of isolation from the same sample source can change the population of EVs isolated, making standardization of isolation an important consideration.14 Sample storage methods can vary as well (lyophilization and reconstitution vs. frozen vs. freshly prepared) but ideal sample format may vary dependent on shipping requirements, short- and long-term storage availability, and how sample efficacy might vary with specific EV products. In 2019, the CBER Advanced Technologies Team (CATT) was established to improve communication between various stakeholders in the development and manufacturing of biologics and the FDA.15 CATT also funds research for work involving the development of methods to manufacture current or evolving biologics.16 Funding extramural research in this manner stimulates modernization of manufacturing products and falls under the FDA’s third priority area.
2.4. Priority Area 4: Ensure FDA readiness to evaluate innovative emerging technologies
Novel regenerative medicine products have created new market opportunities for “rogue” operators who prey on the public’s limited knowledge and understanding of science and medicine at large.9,17 There have been several instances in which the FDA has issued warning notices to facilities that tout curative properties of EV or SC injections that are given to patients without prior clinical trials or FDA approval18; these facilities recruit and target vulnerable patients that have not been able to find successful treatments for persistent and/or chronic ailments.17 Each year, FDA centers report Warning and Untitled Letters that are issued when (1) products/therapies violate federal regulations as a means of achieving voluntary compliance or (2) notify groups with official documentation that the FDA is aware of violations in the hope of achieving voluntary compliance.19 The CBER 2020 Fiscal Year Report from the Director indicated that the FDA issued five Warning Letters and four Untitled Letters for unapproved COVID-19 products including vaccine, exosome, adipose and umbilical cord products; four Warning Letters and nine Untitled Letters were also issued for similar unapproved regenerative medicine products unrelated to COVID-19.18
In 2017, the FDA published a comprehensive regenerative medicine policy framework indicating a period of enforcement discretion that ended in November of 2020.17,20 During this period, the FDA at its discretion could choose not to engage in enforcement activities to allow for product manufacturers to engage with the FDA to determine if they need regulatory approval and the best approach to secure adequate approval. The FDA used a risk-based approach to compliance which included sending letters when concerns regarding patient safety risks or violations arise17. When voluntary compliance is not achieved or victims of unregulated products are harmed, states may take litigatory action against the providers using those products.21,22 Additionally, the FDA can take action to enforce compliance including seizure of products, injunction, criminal prosecution, and fines up to $500,000 (depending on the specific details of each case)23. Though regenerative medicine provides unique opportunities for advances in biomedicine, it is essential that the products are properly tested to ensure safety of the general public as well as of vulnerable populations. In these efforts, the FDA demonstrates its ability to evaluate emerging technologies.
2.5. Priority Area 5: Harness diverse data through information sciences to improve health outcomes
Management of product safety has continuously improved since the FDA’s inception in 1906. Collection and utilization of early clinical data for SC and EV products (and newer products in general) is essential to improve patient outcomes. The Sentinel Initiative seeks to provide the FDA a means of performing effective safety monitoring on a large scale using data from three sources.24 Sentinel uses de-identified, real-world data obtained from medical records to perform algorithm-based risk analysis of post-market approved treatments via its Active Risk Identification and Analysis (ARIA) system.25 FDA-Catalyst is another monitoring system within Sentinel’s infrastructure and generates data by direct interaction with patients and/or healthcare providers (e.g. via safety monitoring surveys or phone applications for event reporting).24 The third source, CBER’s Biologics Effectiveness and Safety (BEST) System, resides outside of Sentinel’s infrastructure but provides information on the testing of biologics.26 The Sentinel Initiative ultimately seeks to develop tools for rapid and large-scale product safety monitoring to help guide risk assessment; by utilizing methods such as natural language processing and other advanced analytics, this system may play an important role in ensuring the safety of novel products – especially as innovation drives the need for rapid, real-world, data-driven assessment of products. Though EV therapies are earlier stage in the translational pipeline compared to SC therapies, collection and reporting of data from the limited number of EV clinical trials as well as future trials will maintain an important role in the development of EV treatments. These efforts fall under the FDA’s fifth priority area of harnessing diverse data to improve patient outcomes.
2.6. Priority Area 8: Strengthen social and behavioral science to help consumers and professionals make informed decisions about regulated products
Many regenerative medicine products are minimally manipulated. In July 2020, the FDA released an updated regulatory guidance for industry and other scientific stakeholders in which the criteria and definitions of minimal manipulation and homologous use are presented.13 Regulatory guidances are not “rule books” of required criteria for the products to which they refer; rather, they are suggestions or recommendations for basic safety and characterization of new products – the FDA reserves the right to request additional testing and safety endpoints in preclinical and clinical trials that may not be laid out in regulatory guidances. As previously mentioned, the novelty of the regenerative medicine field requires that FDA evaluation of products is flexible, based on the scientific community’s latest understanding of biological based therapeutics.7 The aforementioned 2020 guidance defines minimal manipulation for cells or nonstructural tissues as “processing that does not alter the relevant biological characteristics of cells or tissues” and states that when there is not available information on the criteria for processing of products which meet minimal manipulation requirements, the FDA considers such processing “more than minimal manipulation.”13 In the same guidance, homologous use is defined as “the repair, reconstruction, replacement, or supplementation of a recipient’s cells or tissue with a cellular and tissue-based product that performs the same basic function or functions in the recipient as in the donor,” and the FDA also takes into account the “labeling, advertising, and other indications of a manufacturer’s objective intent” to determine whether the definition of homologous use is applicable.13 Thus, the utilization of induced pluripotent stem cells (iPSCs), which often require lengthy treatment of cells in culture, would not meet the definition of minimal manipulation whereas autologous adult stem cell treatments could. However, the broad definitions presented in the regulatory guidance still allow room for scientific interpretation, which may allow some clinics’ assertions, attitudes, or beliefs that their unregulated, biologic-based treatments meet safety and use requirements. By issuing public warnings to these clinics and consumers, the FDA uses the eighth priority (Figure 2) to help the everyday consumer make educated choices when it comes to the use of regenerative products as therapies.
2.7. Considerations for regulatory science
Some have argued that downgrading regulation can in fact drive more rapid translation of products – but potentially at the cost of safety.27,28 On the one hand, regenerative therapies hold great promise for degenerative conditions over chemically-based therapies that have been ineffective at ameliorating some diseases; however, the new frontier of regenerative medicine therapeutics presents safety and efficacy challenges.28 South Korea was possibly the first to change its regulation of SC based products, giving priority for approval and assessment to meet the growing market demand. In 2012, South Korea had approved five times more SC products compared to Japan (between 2010 and 2012).27 This prompted changes to the Japanese requirements, which serves as an example of how reducing regulation might lead to faster translation. In Japan, regenerative therapies have their own unique regulatory designation and are allowed conditional approval after phase one clinical trials demonstrate safety.27,29 In contrast to Japan, the FDA focuses on product purity, safety, and efficacy – meaning that phase two trials demonstrating efficacy are required prior to product marketing. Since 2014 The United States and Japan have approved 12 regenerative medicine therapies each (3 duplicate therapies for allogeneic transplantation have been issued to 3 separate institutions since 2014 in the United States, making the total 14 prior to adjustment.30,31 Although the same number of therapies have been approved in both countries, an argument could be made that the Japanese system is more efficient, considering Japan has historically allocated less capital towards research compared to the United States via the National Institutes of Health.29
The landscape of regulatory systems is ever-evolving as scientific innovation in the biomedical sciences propels research into new frontiers. The field of regenerative medicine holds great promise for life-saving treatments, but the complexity of these products and how they are manufactured and tested pose ongoing challenges. The United States’ regulatory system takes into account the need for innovation in this area and its many programs in product approval and safety monitoring demonstrate its capability in areas of growing interest. The FDA’s Broad Agency Announcement (BAA) is a special contract mechanism that has been soliciting proposals from academia, industry, or other government agencies to advance regulatory science in the nine FDA priority areas.32 Additionally, FDA Centers of Excellence in Regulatory Science and Innovation (CERSIs) are institutions that collaborate with the FDA to advance regulatory science; the priority research areas for CERSI are identified by the FDA based on what it deems as “current unmet regulatory needs.”33 The key to safe, effective, and rapid translation requires balancing safety evaluation while supporting innovation. The next sections of this review will examine further details relating to the regulatory science specific to SC and EV along with discussion of ongoing clinical efforts and challenges.
3. Cellular regenerative medicine approaches
3.1. Emergence of Cellular Therapies
The commonly accepted history of cellular therapy dates to the 19th century, when French scientists investigated the anti-aging effects of injected primate testes extract, although others have highlighted blood transfusions in Ovid’s Metamorphosis as the earliest documented instance.34,35 Decades later in 1931, Paul Niehans began to make the unsubstantiated claim of treating various cancers by injecting bovine embryonic material. More moderns applications of cell therapies began in the 1950s, when research efforts by Billingham, Brent, and Medawar focused on preventing transplant rejection in animal models. This work culminated in 1968 with the first successful human bone marrow transplant.36 Over the next several decades, the scope of cell therapies began to expand, with each new therapy pushing the boundary of evidence-based medicine. These new therapies challenged the existing regulatory paradigm and individual US states responded on a case-by-case basis, largely driven by high profile events.37
This piecemeal approach was finally replaced in 1993 when the FDA started to categorize human cell therapies and products (HCT/Ps) as drugs or devices, and established a new system for handling, processing, marketing, documenting and recalling problematic interventions.38 In 1998 the FDA made a clear transition in its consideration of HCT/Ps that persists today, essentially carving out segments of HCT/Ps that do not require premarket approval. This thinking was crystalized in 2005 in the Federal Code of Regulations Title 21, part 1270 and 1271.38 The effect of the new regulations was to establish three tiers of risk for HCT/Ps with escalating levels of regulatory requirements. The concepts of “minimal manipulation” and “homologous use” became key in delineating those therapies that required full premarket approval and those that could be administered without the need for extensive clinical trials. As such, many of the advancements in the HCT/P space became heavily shaped by the interpretation of these terms. Guidance documents in 2013 and 2014 served to refine the types of therapies contained within each tier and to clarify how these decisions would be made.13,39 The 2016 passage of the 21st Century Cures Act charged the FDA to establish a program, the Regenerative Medicine Advanced Therapies Designation, to facilitate the rapid review of regenerative therapies.40–42 Public consultation surrounding the 21st Century Cures Act provided the impetus for the FDA to release four guidance documents in November of 2017 that further supplemented the existing regulatory landscape. The current regulatory documents for human cell therapies and products are detailed in Table 1.
Table 1.
Primary FDA Governing Documents For Human Cell Therapies and Products (HCT/Ps)a
| FDA Document | Year | Key Features |
|---|---|---|
| Federal Food, Drug, and Cosmetic Act43 | 1993 | Categorized HCT/Ps as drugs or devices, bringing them under the FDA’s regulation |
| Public Health Service Act, Section 35144 | 1998 | Established and regulates a category of biological products |
| Public Health Service Act, Section 36145 | 1998 | Regulations to control the spread of communicable disease, including the purity and release criteria for HCT/Ps |
| Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps) | 2009 | Established manufacturing standards for human cells, tissues, and HCT/Ps |
| 21st Century Cures Act40 | 2016 | Established new mechanisms for review, alternative approval endpoints, and increased funding for regenerative medicine research |
| Same Surgical Procedure Exemption under 21 CFR1271.15(b)46 | 2017 | Describes what procedures qualify as “same” and what can be done to process HCT/Ps prior to reutilization |
| Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue based products13 | 2017 | Defines “minimal manipulation” and “homologous use”, how to apply these definitions to HCT/Ps, and describes how the FDA will enforce these definitions |
| Evaluation of Devices used with Regenerative Medicine Advanced Therapies41 | 2017 | Details how the FDA will handle devices used in Regenerative Medicine Advanced Therapies (RMATs) |
| Expedited Programs for Regenerative Medicine Therapies for Serious Conditions42 | 2017 | Describes the new RMAT designation and additional effort to increase communication and collaboration between the FDA and investigators |
Abbreviations: CGTP, current good tissue practice; HCT/Ps, human cell therapies and products; RMATs, regenerative medicine advanced therapies
Taken together, the governing documents detailed in Table 1 create a regulatory landscape that categorizes HCT/Ps into a 3-tier system based on risk to the patient (Figure 3). The lowest tier, Tier 1, covers HCT/Ps harvested and readministered in the “same surgical procedure” and fertility treatments between intimate partners. Tier 2, which deals with minimally manipulated and homologous use HCT/Ps, is perhaps the most important tier, as it represents the border between needing a full premarket approval process (clinical trials, etc.) versus a lower regulatory bar. The final tier, Tier 3, can be thought of as any HCT/P that does not meet the requirements of either Tier 1 or 2.
Figure 3.
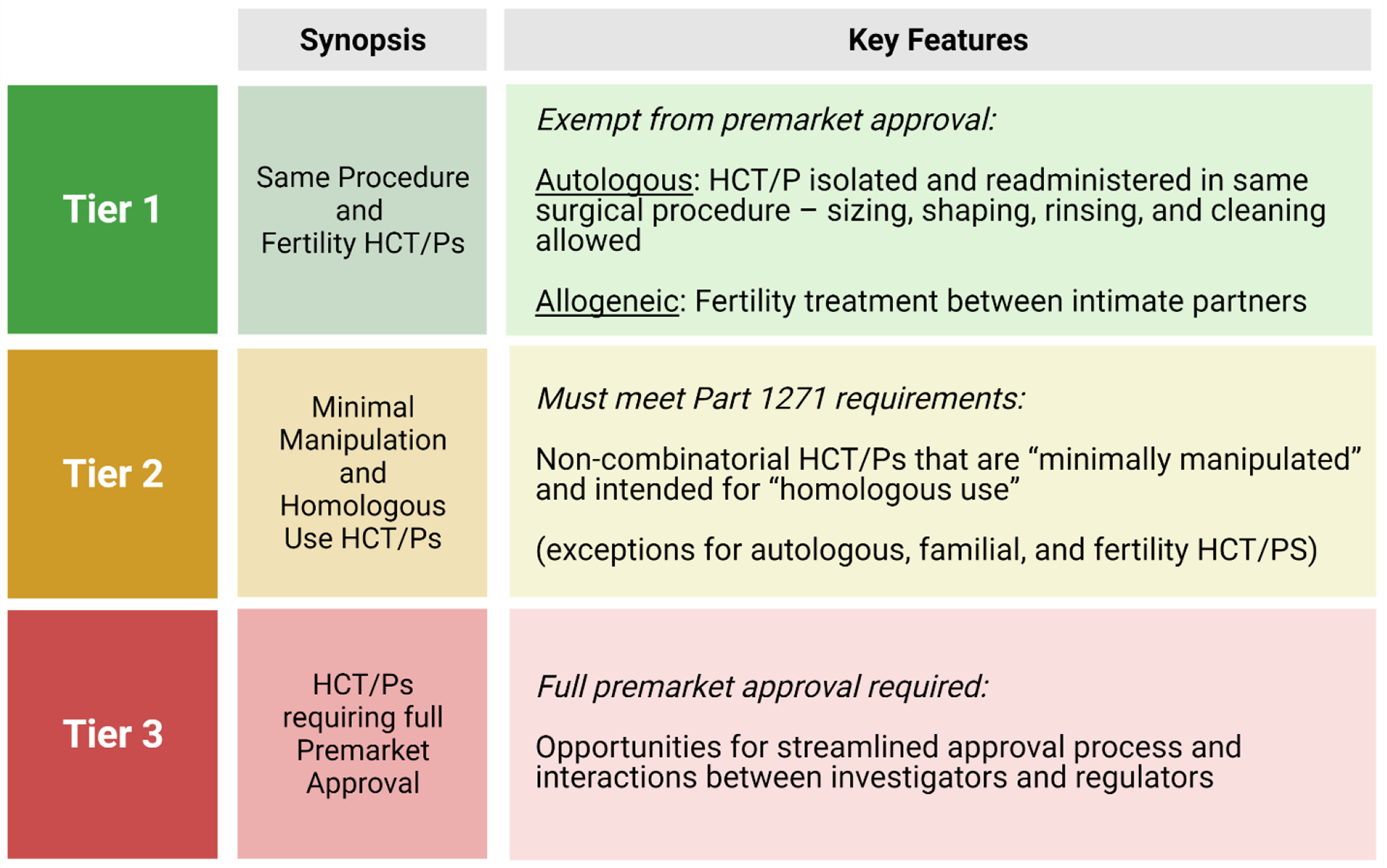
Regulatory Tiers of HCT/Ps
3.2. Trends in HCT/Ps
The tiered system of HCT/Ps regulation creates a clear incentive for commercial interests to position a given HCT/P within either the first or second tier, since the third tier requires extensive investment of both time and capital to satisfy clinical trial requirements. Many current regenerative human cell therapies involve the use of mesenchymal stem cells (MSCs). MSCs are generally thought to exert a transient effect on the body through paracrine signaling, extracellular vesicles, and cell-cell contact.47 However, certain therapeutics intend to utilize the capacity of MSCs to differentiate into more mature cell types.48 These two separate uses of MSCs provide a useful, although not always accurate, demarkation between Tier 2 and Tier 3 products. HCT/Ps with transient effects are more likely to fall into Tier 2 while HCT/Ps that involve long-term function are typically Tier 3.
While MSCs are a key player in emerging regenerative HCT/Ps, hematopoietic stem cells (HSCs) have historically been key drivers of the field and remain incredibly therapeutically useful.49 The vast majority of HSC based HCT/Ps are intended for leukemia patients, whose prognosis often necessitate rapid approval and administration of HCT/Ps. However, these pressures are not confined to leukemia as many of the diseases that regenerative medicine seeks to ameliorate place similar pressure on regulators to expedite the approval of potential therapeutics.
The emerging use of MSCs and historic use of HSCs highlight a trend in the regenerative medicine space – the utilization of HCT/Ps intended to persist within a patient and function for an indefinite period. Examples include bone marrow transplants, pancreatic islet cells, and dopaminergic neurons.50–53 While these HCT/Ps present the exciting possibility of improving chronic diseases for the lifespan of the recipient, they also create a series of unique regulatory challenges. Unlike transient HCT/Ps, long-term HCT/Ps that persist for up to the life of a patient are extremely challenging to administer safely, as the risk for complications extend beyond what can reasonably be investigated in preclinical studies.
To meet these regulatory challenges, each new category of long-term HCT/Ps requires a set of standardized criteria regarding characterization, purity, potency, and genetic landscape. Where clear markers exist, release criteria can be established using flow cytometry, immunohistochemistry, or other appropriate laboratory techniques. These criteria can be expanded to include the manner of sourcing and processing for the HCT/P. Furthermore, in instances where the HCT/P is intended to impart some biological effect on the recipient, efforts to establish standardized potency requirements should be established. For those HCT/Ps that utilize genetic modification, to correct or overexpress a particular gene, additional assurances would be required to ensure genetic stability. Taken together, these approaches could be used to establish a series of clear definitions for each category of HCT/P. These definitions would help to eliminate ambiguity from a regulatory perspective and ensure patients receive uniform treatments. The application of these suggestions need not represent a major barrier to innovation in the HCT/P space, instead, they can provide a unified series of criteria to evaluate broad categories of HCT/Ps.
3.3. Pancreatic β cells as a HCT/P case study
To use a contemporary therapy as a case study, these standardized criteria could readily be applied to VX-880, an HCT/P intended to ameliorate severe hypoglycemia and hypoglycemia unawareness in patients with type 1 diabetes which is currently under clinical trial investigation by the pharmaceutical company Vertex Pharmaceutical Incorporated.54,55 VX-880 is an allogenic, stem cell derived, pancreatic β cell product that is administered via the portal vein to the liver. Patients receiving VX-880 also receive immunosuppressant drugs to prevent rejection. This HCT/P draws on decades of existing literature that allows the suggested regulatory criteria to be neatly applied.56
The protocol used to generate the VX-880 cells involves a directed differentiation protocol to generate pancreatic β cells in a stepwise manner from human pluripotent stem cells. In these protocols, insulin and the transcription factor Nkx6.1 are commonly used as flow cytometry markers to help define the β cell population. Likewise, flow markers from previous stages of the differentiation (i.e., Oct4, Sox2, Nanog for pluripotent cells) can be used as flow markers to identify populations of cells that have not been successfully differentiated. These flow cytometry markers can further be validated with immunohistochemistry. Additionally, because the therapeutic mode of action for β cells is the secretion of insulin, it is important to characterize the potency of these cells. This can be achieved via a glucose-stimulated insulin secretion assay, a test where β cells are exposed to different concentrations of glucose and the amount of insulin they produce under each condition can be quantified and compared. However, the use of human pluripotent stem cells as the cell source of VX-880 creates the risk of teratoma formation once transplanted. Careful usage of the identity markers mentioned above can help to minimize this risk, by minimizing the number of non-terminally differentiated cells, but investigators must clearly establish the risk of teratoma formation by conducting long-term transplantation studies in translationally relevant animal models. Importantly, the transplantation of HCT/Ps intended to persist for an indefinite period, in this case VX-880, also necessitates actionable plans to monitor the status of the cells in vivo and remove them should the need arise.57
Taken together, these criteria provide a unified framework to evaluate not only VX-880 but all future pancreatic β cell based HCT/Ps that may follow. Applying this same lens to other long-term HCT/Ps can help to establish a system for standardizing characterization requirements that can reduce the risk of complications for recipients. This system can be broken down into identity, purity, function, and stability. As detailed above, existing protein and genetic markers can be used to establish the identity of a given HCT/P (Ex., Nkx6.1 and insulin for pancreatic beta cells). These same markers can be used to quantify the purity of a population, with bespoke purity standards applied to each therapy (Ex., 100% PDx1+/0% Oct4+ cells for a pancreatic beta cell HCT/P). Where functional standards exist, an HCT/P can be validated to ensure that it will perform its extended function in the body, in essence providing a measure of confidence in therapeutic potency (Ex., glucose stimulate insulin secretion for pancreatic beta cells). Finally, efforts should be made to establish the stability of a given long-term HCT/P (Ex., How long pancreatic beta cells retain their identity and the risk of teratoma formation). When applied to long-term HCT/Ps, this system can provide a common framework to compare emerging therapies and to ensure patient safety.
4. Cell-derived, acellular regenerative medicine approaches
4.1. Emergence of Cell-derived Therapies
More recently in regenerative medicine, a new generation of nanoscale, biomimetic products has emerged.58,59 Extracellular vesicles are a subcategory of the biogenic secretome and have widespread implications for the next phase of translational regenerative medicine.60 They are broadly defined as subcellular particles that function in cell-to-cell communication, have a lipid bilayer, and contain contents from their parent cell.59 The term EV has been adopted by leading nanoscientists as the default term for this broad group of particles; which include exosomes, microvesicles, apoptotic bodies, and more.59 While the sizes and subcellular origin of the entities of this group vary, they will be broadly discussed here as a collective, since subcategories are very challenging to fully delineate or characterize with today’s technology. This has been discussed at length elsewhere.14,59,61 As we will make clear, the emergence of nanoparticles has begun to revolutionize regenerative medicine, bringing both exciting new therapeutic opportunities and regulatory challenges.
Nanomedicine has long been an interest of scientists due to differing properties of nanoscale products versus microscale products.62 Nanoparticles have a combination of desirable traits: 1) Their small size allows better solubility. 2) Their large surface area to volume ratio increases potential sites of interaction per particle. 3) Their versatile cargo-carrying capacity allows them to be used as tools to carry therapies, diagnostic biomarkers, or therapeutics themselves.58,62 These distinct uses are relevant as they affect regulation of the end products, which include nanopharmaceuticals, theranostics, and nanoimaging products.63 These major categories of products (tools, biomarkers, or therapies) include many kinds of nanoparticles, both synthetic and biogenic, and combinations (like biogenic nanoparticles filled with synthetic drugs). When applied to regenerative medicine, nanoparticles seem to exhibit a strong influence on tissue regeneration and repair signaling and processes, regardless of the source.
Due to technological limitations, EVs, first theorized in 1947 and later confirmed in the 1960s, have only recently seen intensive study.63–65 This emerging interest in nanoparticles was sparked by the discovery that they could carry functional major histocompatibility receptors on their membrane and could stimulate T cells.66 Unfortunately, sudden interest in their functional contents and effects caused nomenclature and referral issues that are still experienced today. These issues sparked the formation of the International Society for Extracellular Vesicles (ISEV), which seeks to standardize the field.61,64 The nomenclature confusion causes regulatory issues as regulation of products relies on appropriate classification, which requires standardized characterization and nomenclature in the field. However, the ISEV has only had a handful of meetings over the last decade, and these field-wide issues are still affecting and will continue to affect regulation of these products.38
4.2. Trends in Nanomedicine
Nanoparticle use in the clinic is still in its infancy. The earliest introduction of these products as tools was in the 1990s, when the first FDA-approved nanodrug, Doxil, was approved.63,67,68 Doxil was a nanoformulation of the conventional chemotherapy doxorubicin, which allowed fewer side effects to patients from chemotherapy due to increased solubility of nanoparticles, which reduced the need for toxic solvents and thus decreased toxicity.69 The solubility advantage that nanoformulations could achieve over their microscale counterparts became a common reason for their use in chemotherapies in the following years, with chemotherapies like Abraxane exhibiting much better tolerance in patients at higher doses when used in a nanoformulation versus conventional formulations.63,68,70 These higher doses allow increased chemotherapy efficacy.63 Since 1995, over 30 nanotherapeutics have been approved worldwide for clinical use; however, the majority of these are simply nanoformulations of already approved products.14,63,71
EVs were first used as therapies for cancer, much like the vast majority of nanomedicines.65 They have also been used to treat a graft vs. host disease patient.65 It is currently thought that repair or restoration is possible via EVs because their functional contents, which can be any functional biomacromolecule (i.e., protein, DNA, RNA, lipids), can promote similar biological effects as their parent cells.59,72 Regardless of the origin of the EVs, whether exfoliation from plasma membrane or endocytic pathway, or the parent tissue of the vesicles, EVs seem to play important roles in tissue regeneration and repair signalling and processes in many different diseases often by decreasing inflammation and inflammation-associated damage.72
However, until 2020 there were no FDA approved EV-based or nanomedicine treatments for the general population, as prior approved, nanomedicine-based therapies were for very specific and typically for cancerous conditions.65,68 Until 2020, there have also been limited regulatory documents released that specifically focus on EVs. A growing number of clinics advertise unregulated EV products as a therapy, with some response from the FDA. In 2019, the FDA released a safety notification regarding EV products (called exosome products in the document) as well as a consumer alert on the regenerative medicine products discussed in this article: stem cells and extracellular vesicles (again referenced as exosomes in the alert).21,73
As an emerging field, there is no standardization for clinical grade production and quality control of therapeutic products.63 In many instances, the technique of isolating EVs is still being optimized, and thus quality control measures are yet to be validated or widely used, creating a large stumbling block to translation of these products to clinical trials and beyond.65 Currently, many argue that approval of EV products has been extremely limited compared to the investments made since the 1990s, but if the regulation of stem cell products can serve as a guide, regenerative discoveries with great potential often see several decades in lag time for translation to patients.63,74,75
At the beginning of 2020, although some EV products were trickling through the translational pipeline, most companies were more concerned with developing the infrastructure needed to support these products, both in the research and development phase (i.e., better imaging and analysis technology), as well as in the manufacturing stage, developing clinical grade, scalable, and efficient isolation filtering protocols and effective quality control systems.72
The worldwide SARS-CoV-2 pandemic was as revolutionary to the nanomedicine space as it was to most areas of science. The mRNA vaccines developed against SARS-CoV-2 used nanoformulations and were encapsulated by lipid nanoparticles, thus propelling the translation of nanomedicine at unimaginable speed. Not only were there several vaccines that used lipid nanoformulations (Pfizer, Moderna), this use allowed increased public awareness of nanomedicine in general, the vaccines were proven safe and effective, creating the world’s largest post market study population.14,71 The pandemic created the regulatory conditions that nanomedicine needed-a very large study population with proven safety and efficacy data to act as a control for comparisons for future translational nanomedicines.68 Now, more than sixty nanotherapeutics are in the clinical trialing stage of the translational pipeline, and with the success of the mRNA vaccines, lipid nanoparticle-based therapeutics are flooding the nanomedicine space.71
4.3. Characterization
From the FDA’s inception, thorough characterization and purity concerns for food and drugs have been of the utmost importance. However, biogenic EV products cannot currently be readily fully characterized, nor made completely “pure”. Even a complete isolation of uniform diameter EVs (currently impossible76) may contain a group of nanoparticles with differing contents, shapes, and membrane composition, all differences that affect function and efficacy of the product.63 With biogenic EV products isolated from tissue or body fluid, there is solid evidence of other, non-EV nanoparticles present, as current isolation methods are not EV specific.77 Liposomes, or globules of fat made soluble for triglyceride transport in the blood, are common “impurities” in these preparations. These purity issues, as well as the unknown nano-properties of these products are the main concern in relation to safety of these products.68,78 To further complicate matters, the overlapping divisions of EVs into their main subcategories of exosome, microvesicle, and apoptotic body are currently based mainly on size, but there is a growing push to also consider internal complexity and functional contents for subdivsions.65,76 Groups like the ISEV have created guidances on standardization practices but, to date, no single approach exists.65,77
Isolation of EVs can be achieved through several methodologies, each generating slightly different populations.65,72,79 Moreover, different sources, like different patients, body sources, sexes, disease or infection states, will have different populations. Even after isolation, there are no standardized storage protocols for these products, and storage conditions can also affect specific populations in distinct ways.65,72 This technological and standardization gap only adds to purity concerns and creates manufacturing limitations.67
4.4. Regulation of Nanomedicines
As is clear, biogenic nanoparticle EV products are very complex and poorly understood due to the many technical limitations in studying them. What the nanomedicine field does know is that there are several potential categories of these products from a regulatory standpoint. 1) Biogenic EVs can be isolated from unmodified tissues/cells/body fluid (naïve EVs from naïve sources). These EVs fall under CBER, much like cellular therapies, and need to follow the path of other biologics.65,80 2) Biogenic EVs can be isolated from a genetically modified source, but the genetic modification is not carried in the EVs themselves (i.e., naïve EVs from modified sources). For this situation, since the EV would be the main functional part of the product and that would not be manipulated, products of this type would be considered biologics.65,80 3) Biogenic EVs could be isolated from genetically manipulated sources and contain a gene therapy as well (modified EVs from modified sources). This category falls into the realm and regulation of gene therapy products, which still fall under CBER as biologics.65 4) Biogenic EVs could be isolated from unmodified sources, but synthetically filled with pharmaceuticals, from chemicals to biological molecules like miRNAs.65 This use, where the EV is a delivery tool as opposed to the therapeutic itself is regulated as a combination product, where the EV is not the primary acting agent, but changes drug distribution via vehicle-specific properties.65 5) Non-EV, biogenic nanoparticles include liposomes, which do not contain the same range of functional cargo as EVs, but are used as delivery tools to allow nanoformulations in a more biocompatible capsule.77 These nanoparticles are regulated differently mainly because of three major differences from EVs: their lack of natural functional contents, their passive accumulation in tissues versus the specific targeting ability demonstrated by EVs, and their longer half-lives than EVs.77 These represent a 5th category of biogenic particle products: liposomes, and similar to category 4, are regulated as delivery vehicles of biologics.77 As shown in Figure 4, the active agent of the nanoparticle product determines its categorization, but does not yet influence its regulation.77
Figure 4.
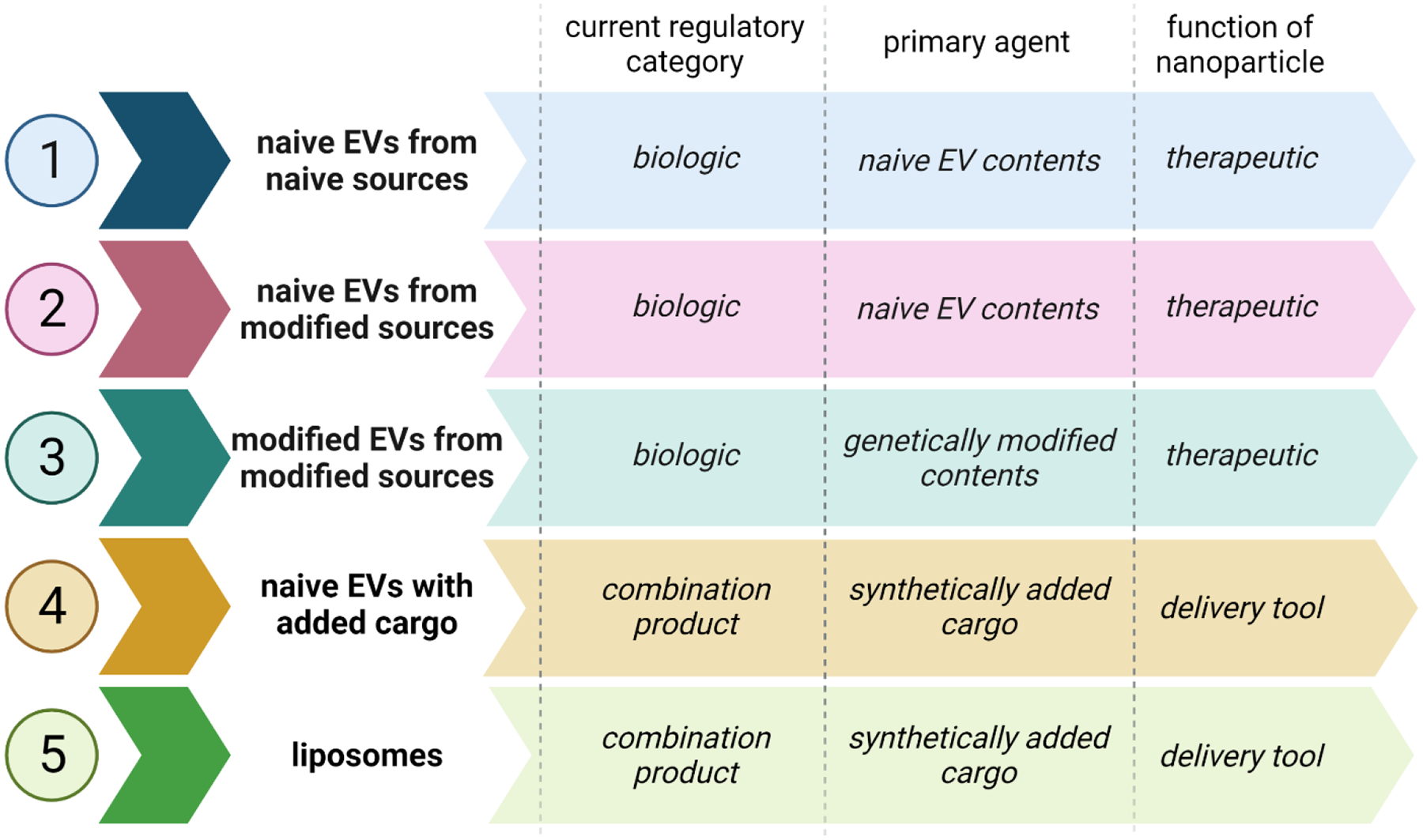
Potential Strata for Regulation of Biogenic Nanoparticles
All of these categories are of interest in the regenerative medicine space, however this article will not describe gene therapy products. All potential categories of biogenic nanoparticles fall under the wide regulatory umbrella of CBER as biologics or biologics in combination products (Figure 4), and would require the same translational pipeline and similar evaluation as other drugs and biologics, but this infrastructure does not account for some of the current limitations in characterization and evaluation of biogenic nanotherapeutics.80,81
In terms of specific regulation for nanomedicines, the FDA considers a product nanotechnology if it either contains nanoparticles or displays nanoscale properties.63 As previously mentioned, the FDA has published guidelines about characterization; however, there are no gold standards as of yet.63 Despite these specific guidelines, the regulatory process for nanomedicines is the same as for other biologics and follows the standard process of pre-IND submission, Phase 1–3 clinical trials, and IND application prior to marketing approval.63 This rather general process has garnered criticism, with some saying that there should be a regulatory pathway specific to nanomedicines, mainly because toxicity testing and characterization of these products is not foolproof.78,82
Furthermore, within nanomedicines, biogenic EVs are often heterogenous mixtures.83 As their contents and/or isolation methods can change their categorization, many call for robust physiochemical and functional profiling to be analyzed to assess their quality control, safety, and efficacy.59,65,72,83 Most agree that regulation of biogenic EV products, regenerative or otherwise, should clearly demonstrate a good risk to benefit ratio, have a known active substance, and known mechanism of action.59,84 Unfortunately, this trifecta is rarely possible, with most of these novel products having unknown mechanisms of action or primary acting agents. As EVs can contain a variety of functional biomolecules, all or some of which may work in concert to mediate their effect, and the products are heterogenous mixes, such determinations are difficult to obtain and basic scientists and clinicians are limited by available methods and technology.59,65 With these unknowns, others ask for increased regulation of these products, as they could potentially pose high risks to patients, but those that argue for heavy caution are met with the equally strong argument that safety profiles of these products, from all sources, are very good; for example, there is huge nanoparticle transfer during standard blood or plasma transfusions, but these are known to be low risk procedures.59,85–87 Although EVs are biogenic molecules and participate in almost all physiological cell-to-cell processes in the body, their biocompatibility must still be proved with each individual product.77
This push and pull for increased regulation while keeping a balance of progress with novel medicinal products is one very familiar to the regenerative medicine field. It is important to summarize here that there are no current regulatory requirements for nanomedicines of any kind and that regulation built around safety and efficacy testing of conventional thermotherapeutics may not be effective in testing nanoformulations, as their functional properties are different on all levels assessed by toxicity studies.59,65
As for biogenic EV use as biomarkers for diagnostics, drug delivery vehicles, or therapeutics themselves, these are goals with proposed products coming throughout the translational pipeline now.65,72,77,79 There are a variety of sources of biogenic EVs already being prepared for clinical use including mesenchymal stem cells, dendritic cells, adipose tissue, plasma, and more.77,88 As every known cell produces EVs for intercellular communication, only more applications and products are to come.14
5. Conclusion
As regenerative medicine continues to expand and mature as a field, its innovations are becoming translated into therapeutics. This creates a need to build a modular, flexible, and collaborative approach to regulation. As a critical part of the leading edge of science, regulatory agencies will need to fully utilize the breadth of expertise, including the research community, to create guidance based on the most current knowledge. Fortunately, precedents exist within the HCT/P sphere that can be used to inform the governance of future innovations. HCT/Ps are categorized on a risk-based system that relies on means of production and intended use. While we propose an additional framework to help guide governance of these products based on duration of therapeutic use (transient vs. long-term), salient lessons can be applied from the regulation of HCT/Ps to acellular (EV) products.
To a certain extent, EVs can be thought of as lagging HCT/Ps by a few decades in both technological innovations and therapeutic exploration. Given this lag, regulators have the unique opportunity to preemptively begin to shape a regulatory framework based on other, more mature, regenerative therapies. EVs can be regulated in many ways, as they have versatile uses in the clinic, from tools of drug delivery, to therapeutics themselves.65 Characterizing EVs remains a key roadblock to their regulation and translation. Without standardized characterization methods and manufacturing protocols for production, they will continue to remain at the bench where they show great promise at reducing inflammation and disease but challenge the current regulatory frameworks that are available, as these focus on clear definitions of biological products.59,63,77 As suggested for SCs, specific guidance for EVs and EV-based products may be needed to clarify the regulatory framework around these difficult-to-define nanoproducts.
The FDA’s regulatory system in relation to regenerative medicine, while somewhat slower than other systems such as Japan’s system, provides a good balance of modular approach to regulation. Arguments have been made for “downgrading regulation” of regenerative medicine products to shorten the time to translation; but it is imperative that the safety of products that may be used transitorily or perpetually be evaluated for safety and efficacy. The International Council for Harmonisation (IHC), established in 1990, provides a forum where regulatory agencies and industry representatives discuss and recommend harmonization in technical product requirements for medical treatments. The IHC provides resources to achieve its goal of regulatory harmonisation such as extensive recommended guidelines on safety, quality, and efficacy; these documents are similar to the FDA regulatory guidances, though they are not necessarily product-type focused like the FDA regulatory guidance.89 Collaborative tools like this are important for the development of regulatory science as requirements can be based on international scientific consensus. Science-driven evaluation of novel therapeutics with these pipelines in place may allow for faster time to translation without eschewing product safety concerns. As science and medicine delve into next-generation biologics via regenerative therapies, it is important that regulatory systems rise to meet new challenges – which the FDA is well poised to do.
Acknowledgements
All figures were created with use of Biorender.com.
Declaration of interest
The authors declare that there are no conflicts of interest. The work has been performed with support from National Institutes of Health (NIH) grant TL1 TR002380 and National Institute of Allergy and Infectious Disease (NIAID) grants R21 AI145356, R21 AI152318, R21 AI154927, American Heart Association grant 20TPA35490415, and Mayo Clinic Center for Regenerative Medicine grants.
Abbreviations
- ARIA
Active Risk Identification and Analysis
- BAA
Broad Agency Announcement
- BEST
Biologics Effectiveness and Safety
- CATT
CBER Advanced Technologies Team
- CBER
Center for Biologics Evaluation and Research
- CERSIs
Centers of Excellence in Regulatory Science and Innovation
- CDER
Center for Drug Evaluation and Research
- CGTP
current good tissue practice
- EVs
extracellular vesicles
- FARS
Focus Areas of Regulatory Science
- FDA
Food and Drug Administration
- HCT/Ps
human cell therapies and products
- HSCs
hematopoietic stem cells
- IHC
International Council for Harmonisation
- iPSCs
induced pluripotent stem cells
- ISEV
International Society for Extracellular Vesicles
- MSCs
mesenchymal stem cells
- SCs
stem cells
- SEC
size-exclusion chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.U.S. Food and Drug Administration. 2021. Advancing Regulatory Science. https://www.fda.gov/science-research/science-and-research-special-topics/advancing-regulatory-science
- 2.U.S. Food and Drug Administration. 2021. ORA Vision, Mission, and Values. https://www.fda.gov/about-fda/office-regulatory-affairs/ora-vision-mission-and-values
- 3.Ragni E, Perucca Orfei C, De Luca P, et al. Inflammatory priming enhances mesenchymal stromal cell secretome potential as a clinical product for regenerative medicine approaches through secreted factors and EV-miRNAs: the example of joint disease. Stem Cell Res Ther 2020;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mao AS, Mooney DJ. Regenerative medicine: Current therapies and future directions. Proc Natl Acad Sci U S A 2015;112:14452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keshtkar S, Azarpira N, Ghahremani MH. Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine. Stem Cell Res Ther 2018;9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvites R, Branquinho M, Sousa AC, Lopes B, Sousa P, Mauricio AC. Mesenchymal Stem/Stromal Cells and Their Paracrine Activity-Immunomodulation Mechanisms and How to Influence the Therapeutic Potential. Pharmaceutics 2022;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.U.S. Food and Drug Administration. 2011. Advancing Regulatory Science at FDA. https://www.fda.gov/media/81109/download
- 8.U.S. Food and Drug Administration. 2018. Priority Area 9: Strengthening the Global Product Safety Net. https://www.fda.gov/science-research/advancing-regulatory-science/priority-area-9-strengthening-global-product-safety-net
- 9.Muthu S, Bapat A, Jain R, Jeyaraman N, Jeyaraman M. Exosomal therapy-a new frontier in regenerative medicine. Stem Cell Investig 2021;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampogna G, Guraya SY, Forgione A. Regenerative medicine: Historical roots and potential strategies in modern medicine. J Microsc Ultrastruct 2015;3:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.U.S. Food and Druf Administration. 2021. Center for Biologics Evaluation and Research (CBER). https://www.fda.gov/about-fda/fda-organization/center-biologics-evaluation-and-research-cber
- 12.U.S. Food and Drug Administration. 2018. Transfer of Therapeutic Products to the Center for Drug Evaluation and Research (CDER). https://www.fda.gov/about-fda/center-biologics-evaluation-and-research-cber/transfer-therapeutic-products-center-drug-evaluation-and-research-cder
- 13.U.S. Department of Health and Human Services. 2020. Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use. https://www.fda.gov/media/109176/download
- 14.Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.U.S. Food and Drug Administration. 2021. CBER Advanced Technologies Program. https://www.fda.gov/vaccines-blood-biologics/industry-biologics/cber-advanced-technologies-program
- 16.U.S. Food and Drug Administration. 2021. CBER Advanced Technologies Program Extramural Research Funding. https://www.fda.gov/vaccines-blood-biologics/industry-biologics/cber-advanced-technologies-program-extramural-research-funding
- 17.Richardson E, Akkas F, Master Z. Evaluating the FDA regenerative medicine framework: opportunities for stakeholders. Regen Med 2020;15:1825–32. [DOI] [PubMed] [Google Scholar]
- 18.U.S. Food and Drug Administration. 2021. FY 2020 Report from the Director. https://www.fda.gov/vaccines-blood-biologics/fy-2020-report-director
- 19.U.S. Food and Drug Administration. 2021. Advisory Action Letters. https://www.fda.gov/animal-veterinary/compliance-enforcement/advisory-action-letters
- 20.U.S. Food and Drug Administration. 2019. Framework for the Regulation of Regenerative Medicine Products. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/framework-regulation-regenerative-medicine-products
- 21.U.S. Food and Drug Administration. 2019. Public Safety Notification on Exosome Products. https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/public-safety-notification-exosome-products
- 22.State of Nebraska v. Omaha Stem Cells, Regenerative Medicine and Anti-Aging Institute of Omaha, Stem Cell Centers of Alaska, Travis Autor and Emily Autor. The Distric Court of Douglas County, Nebraska; Filed 2020, not yet decided. [Google Scholar]
- 23.U.S. Food and Drug Administration. 2017. Types of FDA Enforcement Actions. https://www.fda.gov/animal-veterinary/resources-you/types-fda-enforcement-actions#:~:text=The%20range%20of%20enforcement%20activities,assure%20compliance%20with%20the%20law.
- 24.U.S. Food and Drug Administration. 2019. Sentinel System Five-Year Strategy. https://www.fda.gov/media/120333/download
- 25.Desai RJ, Matheny ME, Johnson K, et al. Broadening the reach of the FDA Sentinel system: A roadmap for integrating electronic health record data in a causal analysis framework. NPJ Digit Med 2021;4:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.U.S. Food and Drug Administration. 2020. CBER Biologics Effectiveness and Safety (BEST) System. https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/cber-biologics-effectiveness-and-safety-best-system
- 27.Sipp D, Sleeboom-Faulkner M. Downgrading of regulation in regenerative medicine. Science 2019;365:644–6. [DOI] [PubMed] [Google Scholar]
- 28.Marks P, Gottlieb S. Balancing Safety and Innovation for Cell-Based Regenerative Medicine. N Engl J Med 2018;378:954–9. [DOI] [PubMed] [Google Scholar]
- 29.Sleeboom-Faulkner M Regulatory brokerage: Competitive advantage and regulation in the field of regenerative medicine. Soc Stud Sci 2019;49:355–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.U.S. Food and Drug Administration. 2022. Approved Cellular and Gene Therapy Products. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products
- 31.Pharmaceuticals and Medical Devices Agency. 2022. Review Reports: Regenerative Medical Products. https://www.pmda.go.jp/english/review-services/reviews/approved-information/0004.html
- 32.U.S. Food and Drug Administration. 2021. Regulatory Science Extramural Research and Development Projects. https://www.fda.gov/science-research/advancing-regulatory-science/regulatory-science-extramural-research-and-development-projects
- 33.U.S. Food and Drug Administration. 2021. Centers of Excellence in Regulatory Science and Innovation. https://www.fda.gov/science-research/advancing-regulatory-science/centers-excellence-regulatory-science-and-innovation-cersis
- 34.Lefrere JJ, Berche P. [Doctor Brown-Sequard’s therapy]. Ann Endocrinol (Paris) 2010;71:69–75. [DOI] [PubMed] [Google Scholar]
- 35.Learoyd P The history of blood transfusion prior to the 20th century--part 1. Transfus Med 2012;22:308–14. [DOI] [PubMed] [Google Scholar]
- 36.Starzl TE. History of clinical transplantation. World J Surg 2000;24:759–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The Pew Charitable Trusts. 2019. FDA’s Framework for Regulating Regenerative Medicine Will Improve Oversight. https://www.pewtrusts.org/en/research-and-analysis/reports/2019/10/17/fdas-framework-for-regulating-regenerative-medicine-will-improve-oversight
- 38.U.S. Food and Drug Administration. 1997. 21 CFR 1270 - Human Tissue Intennded for Transplantation. https://www.govinfo.gov/app/details/CFR-2010-title21-vol8/CFR-2010-title21-vol8-part1270
- 39.Administration USFaD. Same Surgical Procedure Exception under 21 CFR 1271.15(b): Questions and Answers Regarding the Scope of the Exception. Center for Biologics Evaluation and Research: FDA; 2017. November 2017. [Google Scholar]
- 40.U.S. Food and Drug Administration. 2016. 21st Century Cures Act.https://www.congress.gov/114/plaws/publ255/PLAW-114publ255.pdf
- 41.U.S. Food and Drug Administration. 2019. Evaluation of Devices Used with Regenerative Medicine Advanced Therapies. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/evaluation-devices-used-regenerative-medicine-advanced-therapies
- 42.U.S. Food and Drug Administration. 2019. Expedited Programs for Regenerative Medicine Therapies for Serious Conditions. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-regenerative-medicine-therapies-serious-conditions
- 43.U.S. Government. 1906. Federal Food, Drug, and Cosmetic Act. http://uscode.house.gov/view.xhtml?path=/prelim@title21&edition=prelim
- 44.U.S. Government. 2022. Public Health Service Act, Section 351. https://uscode.house.gov/view.xhtml?req=(title:42%20section:262%20edition:prelim)
- 45.U.S. Government. 2022. Public Health Service Act, Section 361. https://uscode.house.gov/view.xhtml?req=(title:42%20section:264%20edition:prelim)
- 46.U.S. Food and Drug Administration. 2017. Same Surgical Procedure Exception under 21 CFR 1271.15(b): Questions and Answers Regarding the Scope of the Exception. https://www.fda.gov/media/89920/download
- 47.Fan XL, Zhang Y, Li X, Fu QL. Mechanisms underlying the protective effects of mesenchymal stem cell-based therapy. Cell Mol Life Sci 2020;77:2771–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller AM, Huppertz S, Henschler R. Hematopoietic Stem Cells in Regenerative Medicine: Astray or on the Path? Transfus Med Hemother 2016;43:247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernandez R, Jimenez-Luna C, Perales-Adan J, Perazzoli G, Melguizo C, Prados J. Differentiation of Human Mesenchymal Stem Cells towards Neuronal Lineage: Clinical Trials in Nervous System Disorders. Biomol Ther (Seoul) 2020;28:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velazco-Cruz L, Goedegebuure MM, Millman JR. Advances Toward Engineering Functionally Mature Human Pluripotent Stem Cell-Derived beta Cells. Front Bioeng Biotechnol 2020;8:786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leonard A, Bertaina A, Bonfim C, et al. Curative therapy for hemoglobinopathies: an International Society for Cell & Gene Therapy Stem Cell Engineering Committee review comparing outcomes, accessibility and cost of ex vivo stem cell gene therapy versus allogeneic hematopoietic stem cell transplantation. Cytotherapy 2021. [DOI] [PubMed] [Google Scholar]
- 52.Astori G, Soncin S, Lo Cicero V, et al. Bone marrow derived stem cells in regenerative medicine as advanced therapy medicinal products. Am J Transl Res 2010;2:285–95. [PMC free article] [PubMed] [Google Scholar]
- 53.Harris JP, Burrell JC, Struzyna LA, et al. Emerging regenerative medicine and tissue engineering strategies for Parkinson’s disease. NPJ Parkinsons Dis 2020;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.A Safety, Tolerability, and Efficacy Study of VX-880 in Participants With Type 1 Diabetes. National Library of Medicine, 2021. (Accessed 12/31/2021, 2021, at https://clinicaltrials.gov/ct2/show/NCT04786262?term=VX-880&cond=type+1+diabetes&draw=2&rank=1.) [Google Scholar]
- 55.VRTX-GEN. Vertex Announces Positive Day 90 Data for the First Patient in the Phase 1/2 Clinical Trial Dosed With VX-880, a Novel Investigational Stem Cell-Derived Therapy for the Treatment of Type 1 Diabetes. investors.vrtx.com: Vertex Pharmaceuticals Incorporated; 2021. [Google Scholar]
- 56.Pagliuca FW, Millman JR, Gurtler M, et al. Generation of functional human pancreatic beta cells in vitro. Cell 2014;159:428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jensen NK, Ingvorsen C, Petersen DR, et al. Characterization of the Nonendocrine Cell Populations in Human Embryonic Stem Cell-Derived (hESC) Islet-Like Clusters Posttransplantation. Toxicol Pathol 2021;49:1269–87. [DOI] [PubMed] [Google Scholar]
- 58.Friedrichs S, Bowman DM. COVID-19 may become nanomedicine’s finest hour yet. Nat Nanotechnol 2021;16:362–4. [DOI] [PubMed] [Google Scholar]
- 59.Silva AKA, Morille M, Piffoux M, et al. Development of extracellular vesicle-based medicinal products: A position paper of the group “Extracellular Vesicle translatiOn to clinicaL perspectiVEs - EVOLVE France”. Adv Drug Deliv Rev 2021;179:114001. [DOI] [PubMed] [Google Scholar]
- 60.Yanez-Mo M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 2015;4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witwer KW, Thery C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J Extracell Vesicles 2019;8:1648167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh P, Singh D, Sa P, Mohapatra P, Khuntia A, S KS. Insights from nanotechnology in COVID-19: prevention, detection, therapy and immunomodulation. Nanomedicine (Lond) 2021;16:1219–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ventola CL. Progress in Nanomedicine: Approved and Investigational Nanodrugs. P T 2017;42:742–55. [PMC free article] [PubMed] [Google Scholar]
- 64.Bazzan E, Tine M, Casara A, et al. Critical Review of the Evolution of Extracellular Vesicles’ Knowledge: From 1946 to Today. Int J Mol Sci 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lener T, Gimona M, Aigner L, et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Vesicles 2015;4:30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raposo G, Nijman HW, Stoorvogel W, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med 1996;183:1161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioeng Transl Med 2019;4:e10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milane L, Amiji M. Clinical approval of nanotechnology-based SARS-CoV-2 mRNA vaccines: impact on translational nanomedicine. Drug Deliv Transl Res 2021;11:1309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolfram J, Zhu M, Yang Y, et al. Safety of Nanoparticles in Medicine. Curr Drug Targets 2015;16:1671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caster JM, Patel AN, Zhang T, Wang A. Investigational nanomedicines in 2016: a review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2017;9. [DOI] [PubMed] [Google Scholar]
- 71.Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng Transl Med 2021:e10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fuster-Matanzo A, Gessler F, Leonardi T, Iraci N, Pluchino S. Acellular approaches for regenerative medicine: on the verge of clinical trials with extracellular membrane vesicles? Stem Cell Res Ther 2015;6:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.U.S. Food and Drug Administration. 2020. Consumer Alert on Regenerative Medicine Products Including Stem Cells and Exosomes. https://www.fda.gov/vaccines-blood-biologics/consumers-biologics/consumer-alert-regenerative-medicine-products-including-stem-cells-and-exosomes
- 74.Havel HA. Where Are the Nanodrugs? An Industry Perspective on Development of Drug Products Containing Nanomaterials. AAPS J 2016;18:1351–3. [DOI] [PubMed] [Google Scholar]
- 75.Weissig V, Guzman-Villanueva D. Nanopharmaceuticals (part 2): products in the pipeline. Int J Nanomedicine 2015;10:1245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles 2013;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol 2021;16:748–59. [DOI] [PubMed] [Google Scholar]
- 78.Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm Res 2016;33:2373–87. [DOI] [PubMed] [Google Scholar]
- 79.Allelein S, Medina-Perez P, Lopes ALH, et al. Potential and challenges of specifically isolating extracellular vesicles from heterogeneous populations. Sci Rep 2021;11:11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.U.S. Food and Drug Administration. 2022. Development & Approval Process (CBER). https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber
- 81.U.S. Food and Drug Administration. 2021. Cellular & Gene Therapy Products. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products
- 82.Ventola CL. The nanomedicine revolution: part 3: regulatory and safety challenges. P T 2012;37:631–9. [PMC free article] [PubMed] [Google Scholar]
- 83.Parliament E Commission Directive 2003/63/EC. Official Journal of the European Union 2003;159:46–94. [Google Scholar]
- 84.Havel H, Finch G, Strode P, et al. Nanomedicines: From Bench to Bedside and Beyond. AAPS J 2016;18:1373–8. [DOI] [PubMed] [Google Scholar]
- 85.Agency CfMPfHUEM. Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. European Medicines Agency; 2017:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Joyner MJ, Wright RS, Fairweather D, et al. Early safety indicators of COVID-19 convalescent plasma in 5000 patients. J Clin Invest 2020;130:4791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joyner MJ, Bruno KA, Klassen SA, et al. Safety Update: COVID-19 Convalescent Plasma in 20,000 Hospitalized Patients. Mayo Clin Proc 2020;95:1888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med 2005;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.International Council for Harmonisation. 2022. ICH Guidelines. https://www.ich.org/page/ich-guidelines