

Abstract
Gene regulation via chemically induced dimerization (CID) is useful for biomedical research. However, the number, type, versatility, and in vivo applications of CID tools are limited. Here, we demonstrate proteolysis targeting chimeras-based scalable CID (PROTAC-CID) platforms, by systematically engineering the available PROTAC systems for inducible gene regulation and gene editing. Further, we develop orthogonal PROTAC-CIDs that can fine-tune gene expression at gradient levels or multiplex biological signals with different logic gating operations. Coupling the PROTAC-CID platform with genetic circuits, we achieve digitally inducible expression of DNA recombinases, base- and prime editors for transient genome manipulation. Finally, we package a compact PROTAC-CID system into adeno-associated viral vectors for inducible and reversible gene activation in vivo. This work provides a versatile molecular toolbox that expands the scope of chemically inducible gene regulation in human cells and mice.
Keywords: PROTAC, CID, inducible gene regulation, mammal, genome editing, AAV delivery
Introduction
The ability to regulate gene expression with precise spatiotemporal control is essential for basic research and therapeutic development1–2. Small molecule inducers enable spatial, temporal, and quantitative gene regulations3–4. The chemically induced dimerization (CID) system comprises two proteins that can be dimerized by a small molecule, which can be used for inducible gene regulations by fusing the DNA binding domain and transcriptional regulation domain to each of the two CID proteins, respectively2. CID-based gene regulation systems have been used for the mining of diverse transactivation domains5, CRISPR-based gene activation6, and tailored modification of antibody N-glycosylation7. In addition to gene regulation, CID systems are also applied to mediate protein degradation8, cell therapy9, and programmable 3D genome positioning10–11.
Current CID systems mainly use small molecules from bacteria or plants. However, only a limited number of such systems are available. For example, rapamycin is a widely-used CID inducer yet possesses undesirable immunosuppressive and autophagy-inducing effects12–13. Other CID inducers, such as abscisic acid (ABA)14 and gibberellic acid analog (GA3)15, are inefficient because they require high concentrations for ideal protein dimerization. Prior efforts to expand CID toolboxes include designing or mining more small molecules16–19, identifying new protein partners through screening nanobody/antibody libraries20–21, or computation-assisted protein design22–23. However, the number and type of highly efficient CIDs remain limited, preventing more sophisticated applications in mammalian cells or organisms.
To address this unmet need, we leverage the proteolysis targeting chimeras (PROTACs), a rapidly growing group of small molecules that can harness the ubiquitin-proteasome system for proximity-induced degradation of the targeted proteins (Figure 1a)24–25. PROTACs are composed of a warhead that binds to the target protein, an anchor ligand that binds to an E3 ubiquitin ligase, and a linker that ties these two units together25. Over 1600 PROTAC small molecules have been developed, acting on more than 100 human protein targets through different E3 ubiquitin ligases, mainly as an effective strategy for cancer therapy26–27. In addition, because PROTAC and CID systems share similar characteristics, it is possible to repurpose PROTAC systems for CID-based applications.
Figure 1.
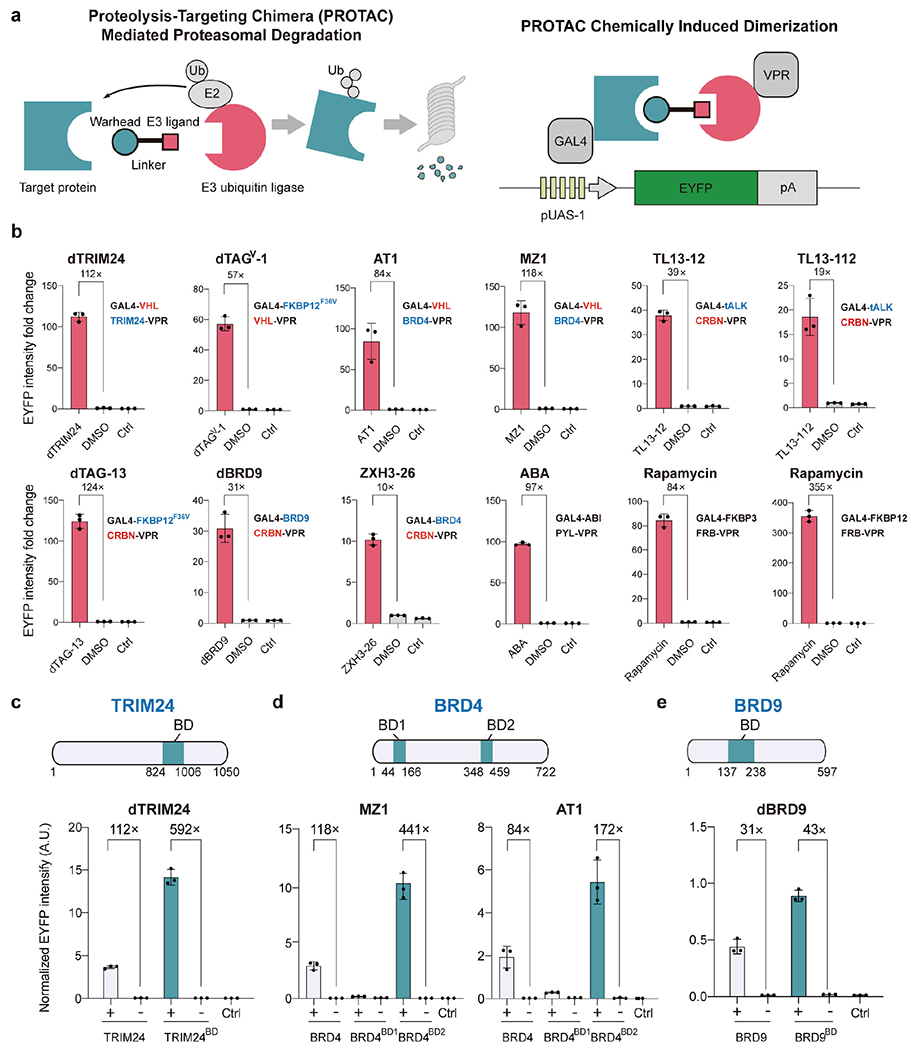
Repurposing PROTACs for inducible gene activation. (a) Schematic of the PROTAC system for the degradation of target proteins (left). E2, E2 ubiquitin ligase. Ub, Ubiquitin. Schematic of the repurposed PROTAC-CID system for inducible gene expression (right). A PROTAC target protein or an E3 ubiquitin ligase fused with the DNA-binding domain GAL4 or the transactivation domain VPR. GAL4 binds to the cognate upstream activation sequence promoter (pUAS-1). PROTACs recruit the VPR domain to the pUAS-1 for enhanced yellow fluorescent (EYFP) protein expression. pA, polyA signal. (b) The relative EYFP fluorescence intensity was measured by flow cytometry in response to PROTACs or solvent Dimethylsulfoxide (DMSO) after 2 days of induction. The concentration for each small molecule and the protein fusion strategy are listed in Table S1. The target protein is shown in blue and E3 ubiquitin ligase is shown in red. (c-e), EYFP signal intensity in the presence of 5 μM dTRIM24 (c), 100 nM MZ1 or 1 μM AT1 (d), or 1 μM dBRD9 (e) or DMSO (shown as “-”) with truncated TRIM24, BRD4, and BRD9, respectively, as indicated. The same data are shown Figure 1b–e with full-length TRIM24, BRD4, and BRD9 for comparison, (b-e) Data are shown in n = 3 biologically independent samples with an error bar of standard deviation (SD). AU, arbitrary units. See Materials and Methods for the EYFP intensity normalization calculation. HEK293T cells transfected with the reporter plasmid encoding pUAS-1 driven EYFP, with no PROTAC or DMSO added, as the blank control group (Ctrl).
Here, we established the PROTAC-based scalable CID (PROTAC-CID) platform by systematically repurposing PROTAC systems for highly efficient inducible transcriptional gene activation. As a result, many PROTACs interacting with different targets are now readily available for orthogonal, multiplexed, and graded gene regulation with different logic gating operations. Further, we demonstrate that our PROTAC-CIDs can be coupled with multi-layer genetic circuits to enable low-basal level inducible DNA manipulations, including site-specific DNA recombination, base- and prime-editing. Finally, we demonstrate the in vivo application potential of the PROTAC-CID system by using adeno-associated virus (AAV) delivery to create ON/OFF and reversible genetic switches in mice. The PROTAC-CIDs toolkits developed in this work could be impactful for biomedical research and applications.
Results
Establish PROTAC-CIDs for inducible gene expression in mammalian cells
We first fused either the GAL4 DNA binding domain or the VP64-p65-Rta (VPR) transactivation domain28 to each PROTAC interacting protein partner. The dimerization of the target protein and the E3 ubiquitin ligase induced by a PROTAC small molecule will bring GAL4 and VPR into proximity to drive the expression of a downstream reporter gene (EYFP) (Figure 1a). Out of the nine commercially available PROTACs we chose (Table S1), four (dTRIM2429, dTAGV-130, AT131, and MZ132) were used to conjugate the Von Hippel-Lindau E3 ubiquitin ligase (VHL) with various target proteins (TRIM24, FKBP12F36V, or BRD4); while the remaining five (TL13-1233, TL13-11233, dTAG-1334, dBRD935, and ZXH3-2636) were implemented to dimerize the Cereblon E3 ubiquitin ligase (CRBN) with different target proteins (truncated ALK (tALK), BRD9, FKBP12F36V or BRD4). We employed the full-length sequence of the PROTAC target proteins and the E3 ubiquitin ligases for initial tests, except that the small-molecule binding kinase domain (tALK) was truncated from the membrane-bound target protein ALK based on its crystal structure to facilitate nucleus translocation37. We co-transfected plasmids encoding these fusion genes into HEK293T cells, together with a plasmid encoding the EYFP reporter driven by the GAL4 cognate pUAS promoter (Figure S1a, b). After two days of induction, all nine PROTAC-CID systems induced EYFP expression compared with the control samples using DMSO (Figure 1b). Notably, three of our tested PROTACs, dTRIM24, dTAG-13, and MZ1, showed more than a 100-fold increase in EYFP expression at 5 μM, 100 nM, and 100 nM, respectively, is more efficient than the existing ABA-based CID system for gene activation (Figure 1b)14. Further, we performed additional analysis to clarify the basic leaky effect of the PROTAC-CID system. Among all the groups, ABA, dBRD9 and TL13-12 based CID showed nearly no leaky effect, while the rest of groups showed significant leaky effect (Figure S2). Molecular glue Lenalidomide is a compound previously identified to degrade IKAROS Family Zinc Finger 1 (IKZF1) or IKZF3 by recruiting CRBN38. To compare the activity of PROTACs with Lenalidomide for inducible gene activation, we designed a similar gene activation platform by fusing CRBN with VPR and GAL4 with either IKZF1 or IKZF3. However, we found that Lenalidomide is significantly less efficient than the dTAG-13 PROTAC-CID system that also uses fusions with CRBN or VPR (Figure S1c–e). These results highlight the advantages of modular PROTACs for CID-based gene activation.
We reasoned that the relatively large sizes of some target proteins, e.g., BRD9, BRD4, and TRIM24 (67, 80, and 117 kDa, respectively), could impose conformational constraints and limit the accessibility of PROTACs to form stable heterodimers31. We crafted new designs that relied on previous reports that the bromodomains (BDs) alone in TRIM2429 and BRD935 can bind to the dTRIM24 and dBRD9 PROTACs, while the BD1 and BD2 in BRD431 are capable of binding MZ1 and AT1. Fusion proteins of VPR with the BDs from BRD4 and TRIM24 (BRD4BD2-VPR and TRIM24BD-VPR) showed significant enhancement in EYFP activation when co-transfected with GAL4-VHL (Figure 1c–d). Compared to the DMSO-treated controls, TRIM24BD-VPR achieved a 592-fold increase in EYFP expression, and BRD4BD2-VPR displayed 441-fold EYFP induction, which exceeded that of rapamycin-based gene activation (355-fold) using FRB and FKBP12 (Figure 1b–d). The truncated GAL4-BRD9BD also displayed increased EYFP expression compared with GAL4-BRD9 (Figure 1e). Thus, our results indicated that truncation of the target proteins could further enhance PROTAC-CID platforms, possibly due to the enhanced PROTAC accessibility to the interaction domains. We also tested two truncated CRBN variants with no ubiquitin ligase function by deleting the 7-α-helical bundle domain in the structure. As a system for targeting proteins for degradation, this endogenous activity may potentially be counterproductive by degrading our fused platform proteins. However, both resulting variants retained high gene activation ability upon deletion, demonstrating the possibility of decreasing the undesired effects raised by overexpressing E3 ubiquitin ligases (Figure S3).
To characterize the sensitivity of the engineered PROTAC-CID systems, we profiled the dose-response of PROTACs. We observed dTAG-13, MZ1 and dTAGV-1 showing EC50 values of 53 nM, 32 nM and 228 nM, respectively, which are more sensitive than ABA (EC50 763 nM) for gene activation (Figure S4a). However, the EC50 values of all the tested PROTACs are higher than that of rapamycin (6 nM), indicating that the rapamycin-based systems are still relatively more sensitive than PROTAC-CID systems. Although we achieved more than 500-fold change in gene activation by using 5μM dTRIM24, high EC50 (6.3 μM) was observed (Figure S4a), which might be explained by its poor cell permeability39. To evaluate the modularity of the protein fusions in our PROTAC-CID systems, we tested different domain organizations of the PROTAC target proteins, E3 ubiquitin ligases, GAL4, and VPR for EYFP activations, by using the dTAG-13 and dBRD9 systems. All protein fusions successfully activated EYFP with differing expression levels (43- to 290-fold). These studies corroborated the feasibility and robustness of the PROTAC-CID systems for high-fold activation and sensitive inducible gene expression in mammalian cells for transgene activation (Figure S4b, c and Table S4).
Multiplexing and gradient gene regulation enabled by PROTAC-CID systems
Since several PROTAC small molecules interact with one or two of the same protein partners (Table S1), we tested the orthogonality of these PROTAC-CID systems in triggering gene activation with cognate or non-cognate protein pairs. Each small molecule (eight of our PROTACs and rapamycin as a standard for comparison) was added to the HEK293T cells transfected with plasmids encoding each combination of protein partners (seven different pairs in total). The successful dimerization of two protein pairs will drive the Firefly luciferase gene (Fluc) expression, enabling high-throughput semi-quantitative readouts. As predicted, high-induction levels of Fluc (62- to 1396-fold) were only observed under the correct cognate combinations after small molecule administration (Figure 2a and Figure S5). For example, dTAG-13 only activated Fluc expression in the cells transfected with plasmids containing the GAL4-FKBP12F36V and CRBN-VPR coding regions, while all other samples with non-cognate protein partners failed to show Fluc induction levels after being supplemented with dTAG-13(Figure 2a and Figure S5). MZ1/AT1 and TL13-12/TL-112 likewise interact with the same protein pairs as expected. These results demonstrated the orthogonality of the PROTAC-CID systems for high-level inducible gene expression, paving the road for expanding these platforms for multiplexing gene regulations.
Figure 2.
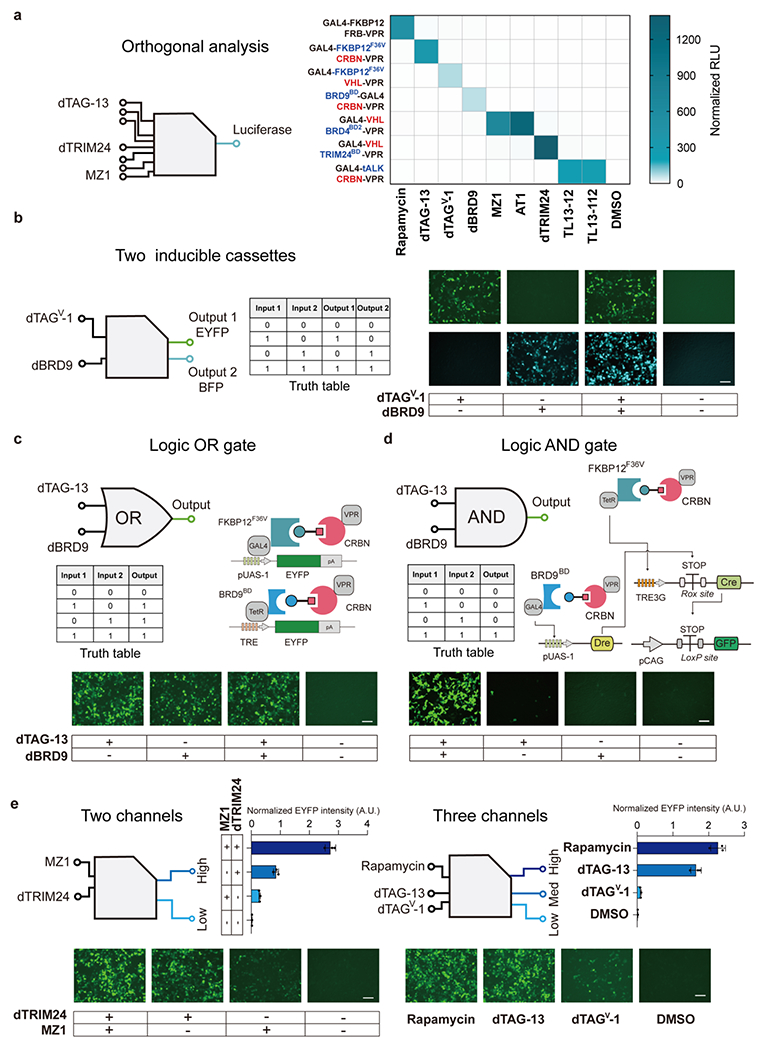
Multiplexing and gradient gene expression regulation by PROTAC-CIDs. (a), Orthogonality analysis of the PROTAC-CID systems. HEK293T cells were transfected with plasmids encoding each PROTAC-CID and the Fluc reporter, followed by treatment with indicated PROTACs or rapamycin (all cognate and noncognate pairs) for 2 days. Cells were lysed and assayed for the bioluminescence intensity. RLU, relative light units. n = 3 biologically independent repeats with the mean shown in the heatmap. (b), The diagram of a dual orthogonal inducible expression system simultaneously (left). Representative imaging of EYFP and BFP expression after dTAGV-1 or dBRD9 treatment (1 μM) (right). (c), Representative images of EYFP intensity of logic OR gate circuit in HEK293T cells. (b and c) Scale bar, 125 μm. (d), Representative images of the logic AND gate system based on inducible DNA recombinases in HEK293T cells. Scale bar, 100 μm. Pre-mature STOP transcriptional signal flanked with LoxP sites was placed between constitutive pCAG promoter and Gfp gene. Only two PROTAC inducers drive the Cre expression by inducing the TRE3G promoter and removing the STOP signal by induced Dre, the GFP signal will be observed. (c-d) HEK293T cells were induced by 1 μM dBRD9 and 100 nM dTAG-13 for 2 days. (e) Quantitative measurement and microscopy observation of EYFP intensity of the multiple-channel gene regulation system. dTRIM24 5 μM, MZ1 100 nM, Rapamycin 1 μM, dTAG-13 1 μM, dTAGV-1 1 μM. Scale bar, 125 μm. DMSO treated samples are shown as “-”. Data are shown in n = 3 biologically independent samples. Error bar represents SD.
We next tested the feasibility of dual PROTAC-based inducible gene cassettes (Figure S6). In the first cassette, GAL4 was fused with FKBP12F36V, and VHL was ligated with VPR to drive EYFP expression in response to dTAGV-1. In the second cassette, another DNA binding domain TetR was fused with BRD9BD, and CRBN was fused to VPR to drive the expression of BFP gene in the presence of dBRD9. We observed EYFP or BFP expression only in the presence of its corresponding inducer dTAGV-1 or dBRD9, while activation of both EYFP and BFP could be achieved by administering these two PROTACs simultaneously (Figure 2b and Figure S7a). Similarly, the MZ1 and dTAG-13 PROTAC-CID systems showed single gene activation with one small molecule and dual-gene activation using both PROTACs (Figure S7b). Since the strategy of biological computation relies on the protein or DNA to execute Boolean logic gate operation within a living organism to allow for cell discrimination and disease diagnosis4, 9, 40, we next explored the possibility of PROTAC-CID-enabled logic gating operations. By placing EYFP under the control of TRE (Tetracycline response element) or the pUAS-1 promoter, we observed strong EYFP expression by using one or two PROTACs, achieving clear logical OR gate responses (Figure 2c and Figure S6c). To develop a more sophisticated logical AND gate, we took advantage of two orthogonal site-specific DNA recombinases (Cre and Dre) for biological computation41. A polyA pre-stop transcription signal (STOP) flanked by either Dre or Cre DNA recombination sites (Rox-STOP-Rox or LoxP-STOP-LoxP, respectively) was put upstream of Cre or GFP to prevent gene expression. Two of our PROTAC-CID gene activation systems (dTAG-13 and dBRD9) were designed to drive the Cre and Dre expression, respectively. In our designed gene circuit, only when both dTAG-13 and dBRD9 inducers were added could Dre and Cre be expressed to remove their respective “STOP” signals, resulting in the eventual expression of GFP functioning as a clear logical AND readout (Figure 2d).
One of the limitations of a single inducer-controlled gene expression system is the existence of only one input, which restricts the programmability of gene activation22. We hypothesized that a multi-state transcriptional control system with differing gradients of gene activation could be achieved by combining different PROTAC-CID systems. Notably, some of our PROTACs can bind to the same E3 ubiquitin ligases, e.g., dTRIM24 and MZ1 both conjugate VHL, but bind to different target proteins (TRIM24 and BRD4 respectively). When HEK293T cells were transfected with GAL4-VHL, TRIM24BD-VPR, BRD4BD2-VPR, and the reporter plasmid, we observed three grades of EYFP intensity, 13-fold, 37-fold, or 120-fold, with MZ1, dTRIM24, or both these two PROTACs, respectively. Likewise, rapamycin, dTAG-13, and dTAGV-1 share the same target protein FKBP12F36V but recruit three different cognate partners (FRB, CRBN, and VHL) with various affinities. We also achieved three grades of activation by administration of rapamycin (136-fold), dTAG-13 (99-fold), and dTAGV-1 (6-fold) (Figure 2e and Figure S7d) in HEK293T cells transfected with all related constructs. Thus, the PROTAC-CID systems enable graded gene regulation.
Application of PROTAC-CIDs to regulate genome editing
Site-specific recombinases or integrases41, base editors (BEs)42, and prime editors (PE)43 are powerful molecular tools that enable manipulation of genomes in living cells. However, effective approaches that can control the functions of those tools, in a chemically inducible manner, are still lacking. To test whether PROTAC-CIDs can be used to induce Cre-based site-specific DNA recombination, we designed a “two-layer” genetic circuit and transfected plasmids encoding the dTAG-13 or dBRD9 PROTAC-CID system to drive the Cre expression in HEK293T cells. The loxP-STOP-LoxP cassette was placed upstream of the Gfp gene, where Cre protein can be recruited to remove the “STOP” signal for Cre-mediated GFP expression. We observed a strong GFP signal in the presence of 100 nM dTAG-13 or 1 μM dBRD9 (Figure 3a and Figure S8). However, leaky expression of GFP was still observed in both cases before the administration of the PROTACs, which was similar to previous reports regarding leakage within tetracycline and rapamycin inducible systems44–46. To address if the PROTAC-CID system could facilitate tighter control of Cre expression as digital outputs, we subsequently designed a “three-layer” genetic circuit by adding the orthogonal DNA recombinase, Dre. To eliminate the leaky expression of the un-stimulated state, we put a Rox-STOP-Rox site between the TRE3G promoter and Cre DNA coding region. Upon addition of dTAG-13, the PROTAC-CID system induces Dre expression to remove the “STOP” signal in front of the Cre gene. Downstream Cre expression then removes the “STOP” between the LoxP sites and leads to the eventual expression of GFP (Figure 3b). With the “three-layer” gene circuit, we observed robust GFP expression only in the presence of dTAG-13, indicating that the PROTAC-CID system could be combined with other synthetic genetic circuits to enable tight and digital gene regulation (Figure 3b).
Figure 3.
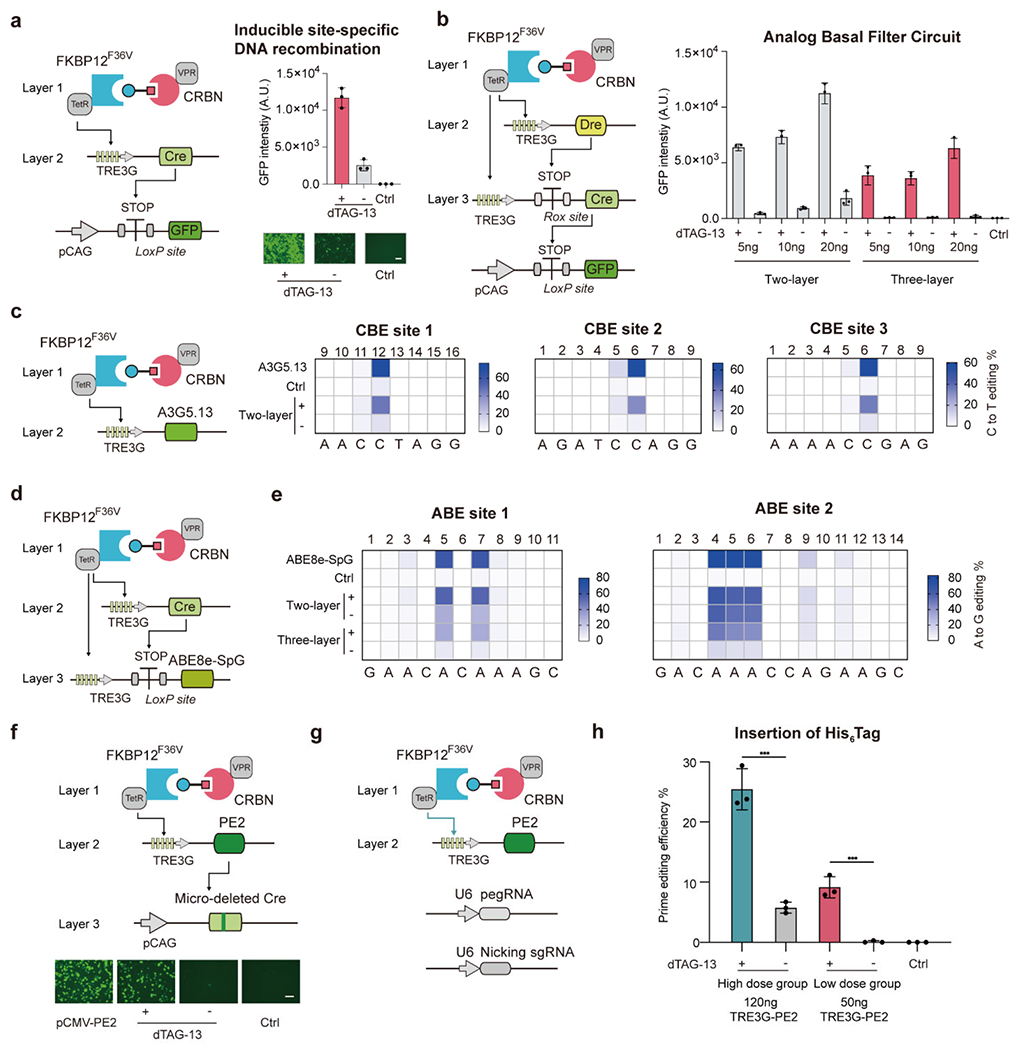
High-induction and low-basal level gene regulation for transient genome editing. (a) GFP expression in HEK293T cells was measured by flow cytometry or microscopy to report the expression level of Cre gene in the presence of 100 nM dTAG-13 or DMSO. (b) Schematic of “three-layer” genetic circuits for tightly controlled digital output (left). Quantitative GFP intensity for measuring the Cre expression in HEK293T cells (right). (a) and (b) HEK293T cells transfected with LoxP-STOP-LoxP-GFP reporter plasmid were used as the control group (Ctrl). (c-e), Quantification of base editing efficiency by PROTAC-CID based inducible base editing tools in HEK293T cells driven by 100 nM dTAG-13 for 3 days. (c) C to T editing by inducible A3G5.13-SpCas9 (d) and (e) A to G editing by TRE3G driven or “three-layer” circuit driven PAM-expanded ABE base editor by fusing ABE8e with the SpG Cas9 variant (ABE8e-SpG). (f) Schematic of the PROTAC-CID based inducible prime editing reporter platform (above). Representative images of the GFP intensity induced by dTAG-13 or DMSO after 48h induction (below). HEK293T cells were induced in the presence of 100 nM dTAG-13 or DMSO. HEK293T cells transfected with the LoxP-STOP-LoxP-GFP reporter plasmid and the pCAG driven mutated Cre as the Control group (Ctrl). (g) Schematic of PROTAC-CID based inducible prime editing system for endogenous genome editing. (h) Quantification of His6 Tag insertion efficiency in HEK293T cells after 3 days of induction by 100 nM dTAG-13 or DMSO. The pegRNA and nicking sgRNA sequences are listed in Table S3. ***, p < 0.001. n = 3 biologically independent repeats for all experiments. Error bar represents SD. DMSO is shown as “-” Scale bar, 125 μm.
Next, we aimed to use PROTAC-CID systems to control base editing in mammalian cells. Two main classes of DNA-modifying BEs have been developed, cytosine BEs (CBEs) and adenine BEs (ABEs), capable of converting base pairs of C•G to T•A and A•T to G•C, respectively. To enable PROTAC-based inducible base editing, we first integrated our previously developed CBE A3G5.1347 with the dTAG-13 PROTAC-CID system. We observed efficient 34.7-46.3% C-to-T editing across three different genomic sites in the presence of 100 nM dTAG-13. Only low levels of editing (4.0-10.7%) were detected without adding PROTACs (Figure 3c). However, when the inducible ABE8e48 was tested within our PROTAC-CID architecture, a high background of A-to-G editing (27.0% to 59.7%) was observed without dTAG-13 induction at two genomic sites. Since the engineered ABE8e is highly active, we speculated that even the low level of the leaky ABE expression could result in significant genome editing outcomes. Therefore, we coupled the Cre DNA recombinase with the PROTAC-CID system to decrease the basal level of ABE8e activity. By placing the LoxP-STOP-LoxP cassette between the TRE3G promoter and the region encoding the ABE8e, we observed high A-to-G editing (30.0% to 49.3%) in the presence of dTAG-13, while significantly reduced editing efficiency was observed in this condition without induction (7.3% to 15.3%), demonstrating the feasibility of PROTAC-CID-enabled genetic circuits to temper basal level induction of potent genome editors to tolerable levels (Figure 3d, e).
Finally, we sought to test whether the PROTAC-CID system can be used to develop inducible prime editors (PEs)43. We engineered a mutated Cre gene with 2-bp micro-deletion, which has no site-specific DNA recombinase activity due to the frameshift. Correct insertion of the missing 2-bp by PE will restore Cre activity by allowing it to remove the STOP signal for GFP expression, enabling an efficient proxy of successful PE activity. While constitutionally expressed PE2 elicited higher GFP intensity, the dTAG-13 induced a more than 25-fold increase in GFP expression without any leaky baseline editing, showing that the PE system can also be integrated into the “three-layer” genetic circuit and enable tight gene regulation (Figure 3f and Figure S9). When we applied the inducible PE system to insert a His6 Tag into an endogenous HEK3 locus in the HEK293T cells with a high TRE3G-driven PE2 plasmid dose (120 ng per well) (Figure 3g, h), more than 20% prime editing efficiency was observed in the presence of dTAG-13, although 5.8% of editing was in the control condition without PROTACs. When a lower PE plasmid dose (50 ng per well) was used, no observable leaky His6 Tag insertion was found in the unstimulated group and 9.2% of PE efficiency was achieved in the PROTAC-induced group (Figure 3g, h). These results demonstrate that the PROTAC-CID system could be used for inducible genome editing to overcome long-term safety concerns.
In vivo PROTAC-CID based inducible gene activation through AAV delivery
Coupling PROTAC-CID with AAV vectors could allow precise dosage and spatiotemporal control of gene expression in vivo, potentially valuable for toxicity management or personalized gene therapy. To test the PROTAC-CID system for such in vivo applications, we designed a compact PROTAC-CID system to pack into AAV vectors (Figure 4a and Figure S10). Since the gene fragments encoding BRD4BD2 and VHL are smaller than 700 bp and MZ1 displayed a low EC50 (Figure S3a), we selected the MZ1 PROTAC-CID systems by placing the GAL4-VHL and BRD4BD2-VP64-p65 in one AAV vector (Figure 4a; Virus a, AAV serotype 8). We also constructed another AAV vector expressing the Fluc gene driven by the pUAS-2 promoter (Figure 4a; Virus b, AAV serotype 8). After co-infecting the HEK293T cells, we treated the infected cells with 100 nM MZ1 for 3 days and observed a more than 60-fold increase in luciferase bioluminescence intensity (Figure 4b).
Figure 4.
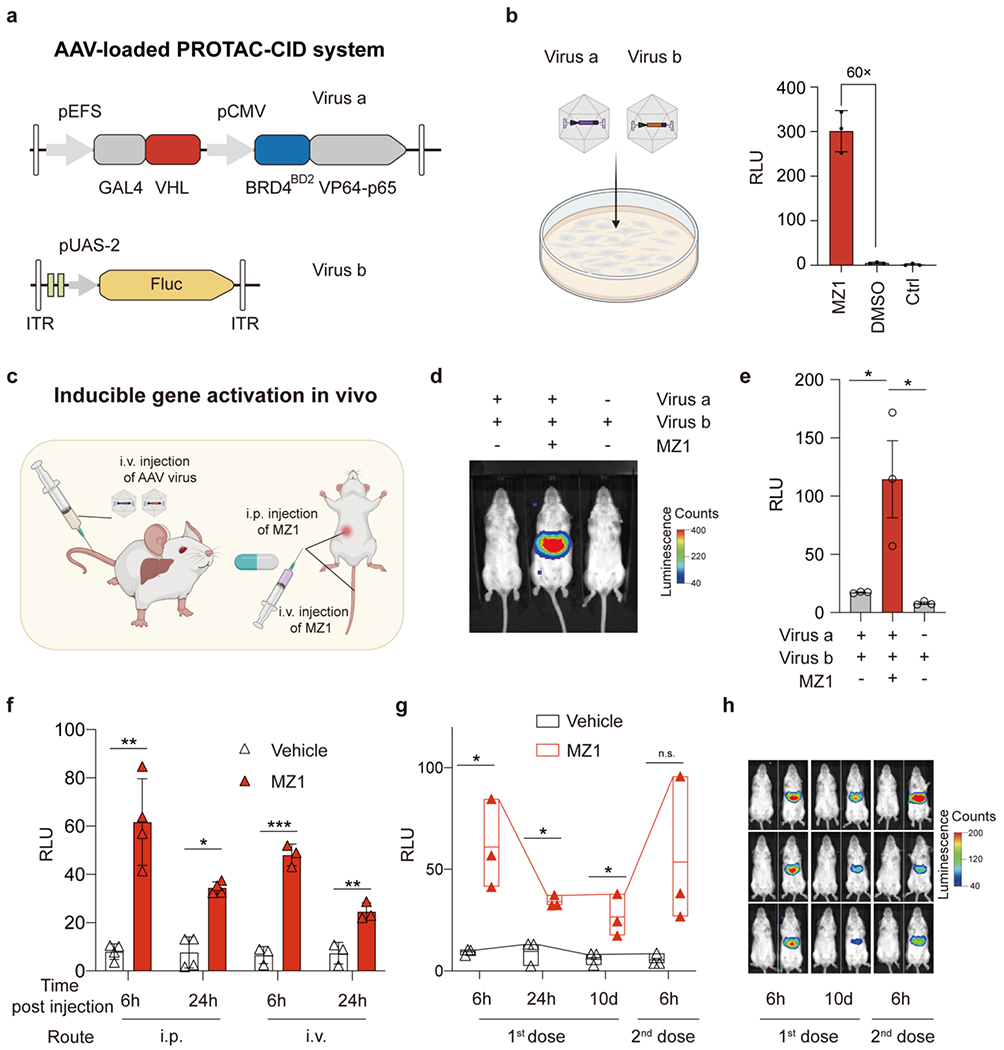
In vivo gene activation by PROTAC-CID. (a) Schematic of AAV-loaded PROTAC-CID system to induce Fluc gene expression. (b) Infecting HEK293T cells by Virus a and Virus b treated by 100 nM MZ1 or DMSO. HEK293T cells without treatment were used as the control (Ctrl). Cells were lysed at 3 days post-infection. (c) Schematic of AAV delivery and MZ1 administration routes. (d) Representative bioluminescence images of mice infected with AAV virus and treated with 10 mg/kg MZ1 or vehicle solution at 6 h post-MZ1 injection. (e) Quantification of the bioluminescence signals in (d). RLU, relative light units. n = 3 mice. The data are mean with standard error of the mean (SEM). (f) Bioluminescence signals after MZ1 injection intraperitoneally (i.p. 50 mg/kg, n = 4 mice) or intravenously (i.v. 10 mg/kg, n = 3 mice). The data shown are the mean with SD. (g-h) Floating bar plot of bioluminescence signals (min to max) with a line at mean and polyline linking the max point at different time points, and representative images of mice after repeated i.p. injection of 50 mg/kg MZ1 (n=3 mice). Asterisks indicate statistical significance using an unpaired two-tailed t-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., non-significant.
To validate the ability of PROTACs for in vivo inducible gene activation via AAV delivery (Figure 4c), we intravenously injected 8-week-old adult FVB/NCrl (FVB) mice with both Virus a and Virus b (Figure S11). 25 days after the AAV injection, we treated mice with 10 mg/kg MZ1 intraperitoneally and performed the bioluminescence imaging at 6 h post-MZ1 injection (Figure 4d). We observed a significant increase (~7-fold) of the luciferase expression in the liver compared with the group without the MZ1 treatment (Figure 4e). The basal bioluminescence intensity from the vehicle control group is comparable to the group only treated by Virus b, demonstrating a low leaky expression in the absence of MZ1 in vivo. Administrating MZ1 intravenously can also increase bioluminescence levels (~ 8-fold), suggesting PROTAC MZ1 is compatible with multiple administration routes to induce gene expression (Figure 4f).
The potential of PROTAC CID systems as distinct from traditional CRISPR-based gene editing lies in the fact that while a single dose of genome editors makes a permanent modification for life-long benefits, more appropriate gene therapy might require transient or reversible activation of transgene expression to the alignment of a daily rhythm (e.g., the rhythm of insulin) or according to disease progression. Therefore, the ability to modulate the transgene expression levels is a desirable feature for future gene regulation systems49–50. To test the possibility of activating gene expression repeatedly, we provided a second MZ1 injection 10 days after the first MZ1 treatment and observed an elevated expression of Fluc, demonstrating the relative ability for repeatable and reversible induction of the transgene expression (Figure 4g, h). Interestingly, even 10 days post-1st injection, we still observed a basal luciferase signal in MZ1 treated mice (Figure 4g, h), which could be caused by in vivo heritable epigenetic modification in the pUAS-2 promoter after the 1st treatment51. We did not observe a significant change in the expression of long isoform BRD4 (BRD4L), and short isoform BRD4 (BRD4S) was undetectable in both treated and untreated mice liver tissue, suggesting that the dose of MZ1 was not sufficient to degrade the endogenous target protein in the healthy liver (Figure 4 and Figures S12 and S16). Similarly, in cultured HEK293T cells, we only observed the degradation of BRD4S, not the conventional BRD4L in the presence of MZ1 (20 nM and 100 nM) (Figures S13 and S17). Additionally, we observed no abnormality or body weight change after MZ1 administration, indicating the relative safety of MZ1 and the PROTAC-CID system (Figure S14). In summary, the compact PROTAC-CID system in AAV enables inducible and repeated gene expression regulation in vivo.
Discussion
In this study, we establish the PROTAC-based scalable CID platforms by systematically engineering PROTACs for inducible transcriptional activation, enabling orthogonal, multiplexing, and digital gene regulation in human cells and mice. Given the rapid development of PROTACs26, the CID toolbox can be readily expanded. PROTAC protein partners are derived from human sources and could mitigate immune responses compared to ABA and other CID inducers. At least 13 PROTACs are being tested in clinical trials, while two PROTACs, ARV-110 and ARV-471, have passed phase I clinical trials with validated safety profiles and characterized pharmacological properties52. The established safety profiles of PROTACs make them potentially suitable for inducible gene or cell therapy. As a research tool, the effect of the repurposed PROTAC on gene expression regulation can be concurrent with the degradation of its endogenous substrate. Therefore, it is crucial to include the negative control with the same PROTAC treatment to correctly attribute the observed biological effects to gene expression regulation rather than degradation of the endogenous substrate of the PROTAC. There are many ways to minimize the interference of PROTAC-CID with the endogenous cellular process. For example, dTAG-13 and dTAGV-1 work with the FKBP12F36V protein partner and do not degrade wild-type FKBP1230, 34. Furthermore, our engineered overexpressed compact PROTAC interacting domain with higher affinity without affecting endogenous target proteins, as the endogenous BRD4 expression was not influenced using our PROTAC-CID in mice.
The limitation of the current PROTAC-CID platform is that the in vivo (in mice) activation ability of the PROTAC-CID system is lower than observed in vitro (HEK293T cells). The chemical compound applied in vivo typically undergoes a multilayered process of absorption, distribution, metabolism, and excretion, which can rapidly break down the chemical compound and excrete it through the urine tract to render it loss-of-function. However, PROTACs added in the cell culture medium can maintain a high concentration even after a long period of time, because the HEK293T cell itself can hardly decompose the chemical compound and no excretion process is available in vitro. Thus, the low concentration and short duration of the MZ1 in vivo could explain the observed poor activation ability of the relevant PROTAC-CID. To improve the in vivo performance of PROTAC-CID, we reason further efforts by increasing the concentration or dose of MZ1 for mouse injection. Alternatively, according to a recent study demonstrating a type of PROTACs with much extended pharmacokinetics53, it is promising to use such PROTAC-CID tools to increase the amount of induced protein product for in vivo applications.
Additionally, we administered a low AAV dosage, which might not be quite enough for high-fold-change gene activation. Specifically, our tested mice were infected with AAV virus (Figure 4c–h) at a dose of 2×1010 genome copies (GC) per mouse, however, in recent gene therapy studies54–55, AAV was administered at a dose from 1011 to 1012 GC per mouse. Thus, to further improve the in vivo performance of PROTAC-CID, we reason to administer a higher dose of AAV to increase the amount of PROTAC-CID in the delivered tissues and therefore give rise to a higher response. The PROTAC-CID gene activation platform can be further optimized. For example, the number of DNA binding sites in the pUAS-2 promoter can be increased to recruit more gene activation domains, as evidenced by recent research showing that synthetic promoters with more binding sites displayed dramatically increased reporter gene expression56. Therefore, an inducible promoter with more DNA binding sites could be engineered to increase the capability of the PROTAC-CID gene activation platform. In addition, recent bioengineered AAV capsids have exhibited much improved transduction in vivo including hepatocyte57, muscle tissue58 and brain tissue59. These engineered capsids can be used to replace the AAV8 capsid to further enhance the delivery efficiency. The VP64 and p65 transcriptional activators used in our manuscript are less effective as compared to VPR and SAM systems60–62. We reason that using VPR or SAM systems would further improve the gene activation fold-change of PROTAC-CID tools.
Conclusion
Here, via systematic engineering of available PROTAC systems, we built a variety of PROTAC-CID platforms that enable versatile inducible genetic manipulations. We demonstrate that PROTAC-CID platforms can be readily used for orthogonal, multiplexed, spatiotemporal and graded gene regulations by different logic gating operations. By coupling PROTAC-CID with multi-layer genetic circuits, we achieve low-basal level inducible DNA manipulations. Finally, using AAV delivery, we demonstrate the in vivo application potential of the PROTAC-CID platform. While we mainly focused on the applications of PROTAC-CID for transcriptional regulation, PROTAC-CID tools could be applied to more CID-related research. Some applications include controlling post-translational protein levels by dimerizing the split CRISPR/Cas effector proteins for inducible endogenous gene activation, base editing, or prime editing28, 43, 63–64. We envision future efforts will emerge to upgrade our versatile PROTAC-CID platforms with new functionalities and capabilities for a wider range of biomedical research and applications.
MATERIALS AND METHODS
Cell lines
HEK293T cells (American Type Culture Collection, no. CRL-3216) were cultured with high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific no. 10569044) with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific no. 10437028) and 1× penicillin-streptomycin (Thermo Fisher Scientific no. 15140122) at 37°C with 5% CO2.
Mouse model
FVB female mice were maintained and handled following laboratory animal treatments approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine (BCM). FVB mice were purchased from the Jackson Laboratory. All mice were kept on the 2920X Teklad Global Extruded Rodent Diet (Soy Protein-Free; Harlan Laboratories). 3-5 mice were housed in each cage in a 12 h light/12 h dark (LD, 7 am light-on, 7 pm light-off) condition with free access to water and food for all experiments.
Fluorescence assay using flow cytometry analysis
Cells were treated with 0.05% Trypsin EDTA (Thermo Fisher Scientific no. 25300054) or TrypLE (Thermo Fisher Scientific no. 12605028) after transfection and centrifuged at 3000 g for 5 min. The supernatant was removed and the cell pellet was resuspended with 200 μL phosphate-buffered saline (PBS) without calcium and magnesium (Thermo Fisher Scientific no. 10010023). Cells were transferred into 12×75 mm flow tubes (Global Scientific no. 110410). Cells were analyzed by MA900 flow cytometry (SONY). 405 nm laser (FL6 filter) was used for testing the BFP channel. 488 nm (FL1 filter) was used for testing the EYFP and GFP channels. Automated alignment using the Automatic Setup Beads kit (SONY no. LE-B3001) was performed before running samples. In Figure 3a, 3b, and Figure S8, experiments were performed under 2% FSC, 17% BSC, and 24% FL1. In Figure S9, experiments were performed under 2% FSC, 17% BSC, and 27% FL1. All the other experiments were performed under 2% FSC, 17% BSC, 24% FL1, and 27% FL6. Flow cytometry data were processed and analyzed by FlowJo 10. Events were first gated with forwarding scatter-Area (FSC-A) and side scatter-Area (SSC-A) to select viable cells and remove the debris and dead cells. Cells were further gated with FSC-A and FSC-Height (FSC-H) for selecting single cells. The example gating strategy was shown in Figure S15. The mean intensity of the reporter fluorescent protein (BFP and EYFP) in viable and single-cell populations were calculated by FlowJo 10 as the mean of FL6-A and FL1-A. When calculating the fold change of relative fluorescence units (RFU), the relative fluorescence units of EYFP were calculated by dividing the mean of fluorescence intensity of EYFP by the mean of BFP fluorescence intensity for normalization as the following formula:
Next, the mean of the normalized relative fluorescence units of the induced group was divided by the value of the control group for calculating the fold change as the following formula:
The mean of the normalized relative fluorescence units of the DMSO treated group in Figure S2 was divided by the value of the control group for calculating the fold change as the following formula:
Plasmid construction
Nucleotide oligos were synthesized by Integrated DNA Technologies (IDT). DNA fragments were amplified by 2× Phanta Max Master Mix (Vazyme no. p515) and assembled by golden gate strategy using T4 DNA ligase (New England Biolabs no. M0202), BsaI-HFv2 (New England Biolabs no. R3733), or Esp3I (Thermo Fisher Scientific no. ER0451). Golden gate reactions were performed in 10 μL reaction volume with 1 μL T4 DNA ligase reaction buffer (New England Biolabs no. B0202S), 0.5 μL BsaI-HFv2 (200 U) or 0.5 μL Esp3I (200 U) and proper volume of fragments and plasmids. Golden gate reactions were performed in a thermocycler by the following program: 37°C for 10 min, 16°C for 10 min for 10 cycles, 50°C for 10 min, and 80°C for 10 min. CRBN fragment was amplified from plenti-UbcP-HA-CRBN-pGK-HYG plasmid, a gift from William Kaelin (Addgene plasmid # 107378). FKBP12F36V fragment was amplified from pLEX305_FKBP12F36V-SHOC2, a gift from Andrew Aguirre (Addgene plasmid # 134522). VHL fragment was amplified from pDONR223_VHL_WT, a gift from Jesse Boehm & William Hahn & David Root (Addgene plasmid # 81874). BRD4 was amplified from GFP-BRD4, a gift from Kyle Miller (Addgene plasmid # 65378). ALK was amplified from pDONR223-ALK, a gift from William Hahn & David Root (Addgene plasmid # 23917). TRIM24 was amplified from Flag-TRIM24, a gift from Michelle Barton (Addgene plasmid # 28138). Dre was amplified from pCAG-NLS-HA-Dre, a gift from Pawel Pelczar (Addgene plasmid # 51272). IKZF3 was amplified from pIRIGF-IKZF3, a gift from William Kaelin (Addgene plasmid # 69046). IKZF1 was amplified from pFUW-tetO-IKZF1 a gift from Filipe Pereira (Addgene plasmid # 139807). SpG was amplified from pCAG-CBE4max-SpG-P2A-EGFP (RTW4552), a gift from Benjamin Kleinstiver (Addgene plasmid # 139998). pCAG-loxPSTOPloxP-ZsGreen was a gift from Pawel Pelczar (Addgene plasmid # 51269). PE2 was amplified from pCMV-PE2, a gift from David Liu (Addgene plasmid # 132775). gRNA was constructed into the scaffold plasmid lentiGuide-Puro, a gift from Feng Zhang (Addgene plasmid # 52963). For protein engineering, the 3D protein/small molecule complex or protein complex structure was visualized by UCSF Chimera v1.16. The concentration of plasmids was measured by Nanodrop Spectrophotometer (Thermo Fisher Scientific). Plasmids were sequenced by Genewiz. The protein sequences and primers were listed in Sequences S1 and S2.
Transfection and microscopy
Except for the data in Figure 3c, 3e, and 3h, cells were plated into 96 well plates (Corning no. 3598) and transfected by Polyethyleneimine Max (PEI Max) (Polysciences no. 24765-1). When the cell confluency reached 50% confluency, 1.5 μL PEI Max (1 mg/mL, pH = 7.1) and 100 μl DMEM were mixed with plasmids to be slowly dropped into the cell medium after 30 min of incubation. The medium was changed 12 h post-transfection. The inductive small molecules, including Rapamycin (STEM CELL technologies no. 73362), dTAG-13 (TOCRIS no. 6605/5), dTAGV-1 (TOCRIS no. 6744-5), dBRD9 (TOCRIS no. 6606/5), dTRIM24 (TOCRIS no. 6607/5), MZ1 (TOCRIS no. 6154/5), AT1 (TOCRIS no. 6356/5), TL13-12 (TOCRIS no. 6744/5), TL13-112 (TOCRIS no. 6745/5), dTAG-7 (TOCRIS no. 69125), ZXH3-26 (TOCRIS no. 6713/5) and ABA (GoldBio no. A-050-100), dissolved in Dimethyl sulfoxide (DMSO) (Sigma, no. D8418) were added. For the microscopy observation, the images were taken by Microscopy EVOS (Life Technologies) and processed by ImageJ65. If not otherwise noted, 60 ng of each plasmid were used for each sample. The transfection plasmid configuration was listed in Table S5.
Luciferase luminescence intensity measurement
Before transfection, HEK293T cells were seeded in the clear bottom 96 well assay plates (Corning no. 3610) with 100 μL DMEM with 10% FBS and 1× penicillin-streptomycin at 37 °C with 5% CO2. When cells reached 50 % confluency, 1.5 μL PEI Max (1 mg/mL, pH=7.1) and 100 μL DMEM were mixed with 60 ng of each plasmid (GAL4 and VPR fused target protein or E3 ubiquitin ligase, pUAS-2-Fluc) for 30 min at room temperature. The mixture was transferred into cells gently. 12 h after transfection, the medium was changed, and the inductive small molecules were supplied. 2 days post-induction, 120 μL of medium was aspirated from each well containing 200 μL of medium, leaving 80 μL of medium per well, then treated with 40 μL of lysis buffer (500 mM DTT (Sigma no. D0632), 10 mM coenzyme A (RPI no. C95275), 100 mM ATP (Thermo Fisher Scientific no. R1441), 80 mg/mL D-luciferin (GoldBio no. LUCNA-100), Triton lysis buffer (0.1082 M Tris-HCl, 0.0419 M Tris-Base, 75 mM NaCl, 3 mM MgCl2)). The plate was shaken with 20 s in “Orbital” mode with a frequency of 432 rpm and amplitude of 1 mm to lysis the cells. Recording the luminescence by plate reader Infinite M200 (TECAN) with 1000 ms integration time using Magellan v1.7 (TECAN). The mean of luciferase luminescence relative light units (RLU) was calculated by the average of three biological replications. Normalized RLU was calculated by dividing the RLU of the tested group by the mean of the RLU of the DMSO treated control group for normalization.
Base editing measurement
HEK293T cells were plated into 96 well plates (Corning no. 3598) for transfection when reaching 20 % confluency. 0.5 μL Lipofectamine 2000 (Life Technology no. 11661089) was mixed with 25 μL DMEM for 5 min incubation. 80 ng to 210 ng Plasmids (See Table S5 for plasmid dosage used in each condition) were then mixed with 25 μL DMEM and added into the Lipofectamine 2000 and DMEM mixture for 20 min incubation at the room temperature. The mixture was added to the cells gently. After 12 h, supernatants were changed with fresh 10% FBS (Thermo Fisher Scientific no. 10437028) DMEM medium (Thermo Fisher Scientific no. 10569044), Puromycin (10 μg/mL Thermo Fisher Scientific no. A1113803), and supplied with 100 nM dTAG-13 (TOCRIS no. 6605/5) or DMSO (Sigma, no. D8418). After 3 days, the cell medium was removed and cells were treated with 100 μL lysis buffer (10 mM Tris-HCl (pH=7.5), 0.05% SDS, and proteinase K (25 μg/mL, Thermo Fisher Scientific no.01169965)) followed by 37°C 1 h, 58°C 30 min and inactivated at 95°C for 20 min. The cell lysis was amplified by 2× Phanta Max Master Mix (Vazyme no. p515) following the program: 95°C 3 min, 95°C 15 s, and 58°C 15 s with 72°C for 35 cycles, and 72°C 5 min. The guide RNA sequences and primers were listed in Table S2. The editing efficiency was measured by Sanger sequencing and analyzed by EditR (https://moriaritylab.shinyapps.io/editr_v10/)66.
Prime editing measurement
HEK293T cells were seeded into 96 well plates (Corning no. 3598). When the cells reach 20% confluency, 265 ng to 290 ng plasmids (See Table S5 for plasmid dosage used in each condition) were firstly mixed with 25 μL DMEM. 0.5 μL Lipofectamine 2000 (Life Technology no. 11661089) was incubated with 25 μL DMEM for 5 min. Next, the plasmid solution was mixed with the Lipofectamine 2000 solution for 20 min and added to the cells gently. After 12 h, the supernatants were changed with fresh 10% FBS (Thermo Fisher Scientific no. 10437028) DMEM medium (Thermo Fisher Scientific no. 10569044) supplied with Puromycin (10 μg/mL Thermo Fisher Scientific no. A1113803) and 100 nM dTAG-13 (TOCRIS no. 6605/5) or DMSO (Sigma, no. D8418). 3 days post-induction, the supernatant was removed and supplied with 100 μL lysis buffer (10 mM Tris-HCl (pH=7.5), 0.05% SDS, and proteinase K (25 μg/mL, Thermo Fisher Scientific no.01169965)) followed by 37°C 1 h, 58°C 30 min and inactivated at 95°C for 20 min. To design primers to amplify the editing region, the gRNA target sequences as the inquiry by the BLAT Search Genome tool (https://genome.ucsc.edu/cgi-bin/hgBlat). The 2000 base pair (bp) flanking genomic DNA sequences were downloaded, and the primers were designed by Geneious Prime 3 (Biomatter). 0.5 μL cell lysis was amplified with DNA primers (listed in Table S3) by 2× Phanta Max Master Mix (Vazyme no. p515) following the program: 95°C 3 min, 95°C 15 s, and 58°C 15 s with 72°C for 35 cycles, and 72°C 5 min. The fragments were cleaned by PB buffer (Qiagen no. 166021045). 10 ng PCR products were used for Sanger sequencing (Genewiz). The insertion efficiency was analyzed online in TIDE (https://tide.nki.nl/)67 with the setting (left boundary=100, Decomposition window (115-685 bp), Indel size range (28 bp), P-value threshold=0.001).
Immunoblots and RT-qPCR
Mice liver tissues or culture cells were lysed in RIPA buffer (Abcam no. ab156034) supplemented with phosphatase inhibitor (Thermo Fisher Scientific no. PIA32957) and protease inhibitors (Thermo Fishers scientific no. A32965). Lysates were resolved by 10% Tris-glycine SDS-PAGE, transferred to PVDF membrane (Bio-Rad no. 1620177), and blotted with antibodies BRD4 (Abcam, ab128874), GAPDH (Cell Signaling Technology, 2118L), luciferase (ABclonal, requested), Ran (ABclonal, A0976), Hsp90 (Cell Signaling Technology, 4874). Images were acquired using LumiQuant AC600 (Acuronbio Technology Inc), and quantification analysis was processed by ImageJ software. Trizol reagent (Sigma no. T9424) was used to extract total RNA from the liver. RNAs were purified using the RNeasy Mini kit (QIAGEN no. 4106). The quality and concentration of total RNA were checked on NanoDrop™ 2000/2000c Spectrophotometers (Thermo Fisher Scientific no. ND2000LAPTOP). Reverse transcription of total RNA was performed using Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific no. 4368813) and qPCR was conducted with SYBR Green Master Mix (Abclonal no. RK21203) on QuantStudio 6 Real-time PCR system (Thermo Fisher Scientific).
AAV production and Mouse for in vivo delivery
High titer and purity AAV viruses were produced by the Neuroconnectivity Core of Baylor College of Medicine with 10 plates scale. The titer of AAV viruses was then measured by real-time qPCR. 8-week-old FVB female mice were infected with either Virus a or Virus a and Virus b (Figure 4c–h) at a dose of 2×1010 genome copies (GC) per mouse in saline (100 μL) via tail vein injection. 25 days after the virus injection, all mice were administered with MZ1 at the concentration of 10 mg/kg through intraperitoneal injection (i.p.). To compare the route of administration, we treated the mice with 50 mg/kg MZ1 by i.p. or 10 mg/kg by intravenous injection (i.v.). After ten days, to measure the repeatable activation, we treated mice with 50 mg/kg MZ1 by i.p. injection and observed the luciferase bioluminescence.
AAV infection on HEK293T cells
Before infection, HEK293T cells were seeded into 96 well plates (Corning no. 3610). When the confluency reached 50 %, 1 μL of each purified AAV virus (Virus a and Virus b in Figure 4) were added to the supernatant of HEK293T cells. 100 nM of MZ1 or DMSO was added to the supernatant. 3 days post-infection, cells with 80 μL medium were treated with 40 μL lysis buffer (500 mM DTT, 10 mM coenzyme A, 100 mM ATP, 80 mg/mL D-luciferin, Triton lysis buffer (0.1082 M Tris-HCl, 0.0419 M Tris-Base, 75 mM NaCl, 3 mM MgCl2)). To lysis the cells, Plate was shaken by plate reader Infinite M200 (TECAN) with 20 s in “Orbital” mode with a frequency of 432 rpm and Amplitude of 1 mm. Relative light units of luminescence were recorded by plate reader Infinite M200 (TECAN) with 1000 ms integration time using Magellan v1.7 (TECAN).
IVIS imaging system and quantification
Luciferase fluorescence intensity was measured by the IVIS imaging system (PerkinElmer). Mice were anesthetized with a mixture of isoflurane and oxygen, and then intraperitoneally (i.p.) injected with D-luciferin (15 mg/ml, GoldBio no. LUCNA-100). 5 min after the D-luciferin injection, mice were imaged with IVIS imaging system. Quantitative analysis of imaging signals (luminescence counts) was processed by Living Imaging software (PerkinElmer).
Statistical analysis
The number of independent experiments performed in parallel was represented by n in the figure legend. Two-tailed Student’s t-test was used for comparison shown in the figure legend. * P< 0.05; ** P< 0.01; *** P< 0.001. No statistical methods were used to predetermine sample size. Most data in this research were represented by bar graphs with the mean and individual points. Unless otherwise indicated, representative images were from three biologically independent repeats. No data were excluded for analysis. For in vivo experiments, different biological repeats represented different mice. We calculated the means and standard deviation (SD) with N=3 biological repeats unless stated otherwise. Prism 9 (GraphPad) was used to generate the bar plots and heatmap.
Supplementary Material
Acknowledgments
We thank Harshavardhan Deshmukh for the flow cytometry assistance. The figures were prepared with BioRender.com. This work was supported by the National Science Foundation CAREER AWARD (CBET-2143626 to X.G.); Robert A. Welch Foundation grant (C-1952 to X.G.); National Institutes of Health grants (HL157714 to X.G.; HL153320, DK111436, AG069966, and ES027544 to Z.S.); John S. Dunn Foundation, Mrs. Clifford Elder White Graham Endowed Research Fund, Cardiovascular Research Institute at Baylor College of Medicine, the DLDCCC (P30CA125123), the Specialized Programs of Research Excellence (SPORE) program (P50CA126752), the Gulf Coast Center for Precision Environmental Health (P30ES030285), and the Texas Medical Center Digestive Diseases Center (P30DK056338).
Footnotes
X.G. and D.M. are co-inventors on a provisional patent application filed by Rice University related to this work. The other authors declare no competing interests.
REFERENCES
- 1.Stanton BZ; Chory EJ; Crabtree GR, Chemically induced proximity in biology and medicine. Science 2018, 359 (6380), 1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fegan A; White B; Carlson JC; Wagner CR, Chemically controlled protein assembly: techniques and applications. Chem Rev 2010, 110 (6), 3315–3336. [DOI] [PubMed] [Google Scholar]
- 3.Jaeger MG; Winter GE, Fast-acting chemical tools to delineate causality in transcriptional control. Mol Cell 2021, 81 (8), 1617–1630. [DOI] [PubMed] [Google Scholar]
- 4.Kitada T; DiAndreth B; Teague B; Weiss R, Programming gene and engineered-cell therapies with synthetic biology. Science 2018, 359 (6376), 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alerasool N; Leng H; Lin ZY; Gingras AC; Taipale M, Identification and functional characterization of transcriptional activators in human cells. Mol Cell 2022, 82 (3), 677–695 e7. [DOI] [PubMed] [Google Scholar]
- 6.Gao Y; Xiong X; Wong S; Charles EJ; Lim WA; Qi LS, Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat Methods 2016, 13 (12), 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang MM; Gaidukov L; Jung G; Tseng WA; Scarcelli JJ; Cornell R; Marshall JK; Lyles JL; Sakorafas P; Chu AA; Cote K; Tzvetkova B; Dolatshahi S; Sumit M; Mulukutla BC; Lauffenburger DA; Figueroa B Jr.; Summers NM; Lu TK; Weiss R, Small-molecule control of antibody N-glycosylation in engineered mammalian cells. Nat Chem Biol 2019, 15 (7), 730–736. [DOI] [PubMed] [Google Scholar]
- 8.Deng W; Bates JA; Wei H; Bartoschek MD; Conradt B; Leonhardt H, Tunable light and drug induced depletion of target proteins. Nature Communications 2020, 11 (1), 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu CY; Roybal KT; Puchner EM; Onuffer J; Lim WA, Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350 (6258), aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H; Xu X; Nguyen CM; Liu Y; Gao Y; Lin X; Daley T; Kipniss NH; La Russa M; Qi LS, CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 2018, 175 (5), 1405–1417 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Y; Han M; Shang S; Wang H; Qi LS, Interrogation of the dynamic properties of higher-order heterochromatin using CRISPR-dCas9. Mol Cell 2021, 81 (20), 4287–4299 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown EJ; Schreiber SL, A Signaling Pathway to Translational Control. Cell 1996, 86 (4), 517–520. [DOI] [PubMed] [Google Scholar]
- 13.Brown EJ; Albers MW; Shin TB; Ichikawa K; Keith CT; Lane WAS; Schreiber SL, A mammalian protein targeted by G1-arresting rapamycie receptor complex. Nature 1994, 369, 756–758. [DOI] [PubMed] [Google Scholar]
- 14.Liang FS; Ho WQ; Crabtree GR, Engineering the ABA plant stress pathway for regulation of induced proximity. Sci Signal 2011, 4 (164), rs2 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto T; DeRose R; Suarez A; Ueno T; Chen M; Sun TP; Wolfgang MJ; Mukherjee C; Meyers DJ; Inoue T, Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat Chem Biol 2012, 8 (5), 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziegler MJ; Yserentant K; Dunsing V; Middel V; Gralak AJ; Pakari K; Bargstedt J; Kern C; Petrich A; Chiantia S; Strahle U; Herten DP; Wombacher R, Mandipropamid as a chemical inducer of proximity for in vivo applications. Nat Chem Biol 2022, 18 (1), 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jan M; Scarfo I; Larson RC; Walker A; Schmidts A; Guirguis AA; Gasser JA; Slabicki M; Bouffard AA; Castano AP; Kann MC; Cabral ML; Tepper A; Grinshpun DE; Sperling AS; Kyung T; Sievers QL; Birnbaum ME; Maus MV; Ebert BL, Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci TranslMed 2021, 13 (575), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayle JH; Grimley JS; Stankunas K; Gestwicki JE; Wandless TJ; Crabtree GR, Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem Biol 2006, 13 (1), 99–107. [DOI] [PubMed] [Google Scholar]
- 19.Liu P; Calderon A; Konstantinidis G; Hou J; Voss S; Chen X; Li F; Banerjee S; Hoffmann JE; Theiss C; Dehmelt L; Wu YW, A bioorthogonal small-molecule-switch system for controlling protein function in live cells. Angew Chem Int Ed Engl 2014, 53 (38), 10049–10055. [DOI] [PubMed] [Google Scholar]
- 20.Kang S; Davidsen K; Gomez-Castillo L; Jiang H; Fu X; Li Z; Liang Y; Jahn M; Moussa M; DiMaio F; Gu L, COMBINES-CID: An Efficient Method for De Novo Engineering of Highly Specific Chemically Induced Protein Dimerization Systems. J Am Chem Soc 2019, 141 (28), 10948–10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill ZB; Martinko AJ; Nguyen DP; Wells JA, Human antibody-based chemically induced dimerizers for cell therapeutic applications. Nat Chem Biol 2018, 14 (2), 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foight GW; Wang Z; Wei CT; Jr Greisen P; Warner KM; Cunningham-Bryant D; Park K; Brunette TJ; Sheffler W; Baker D; Maly DJ, Multi-input chemical control of protein dimerization for programming graded cellular responses. Nat Biotechnol 2019, 37 (10), 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shui S; Gainza P; Scheller L; Yang C; Kurumida Y; Rosset S; Georgeon S; Di Roberto RB; Castellanos-Rueda R; Reddy ST; Correia BE, A rational blueprint for the design of chemically-controlled protein switches. Nat Commun 2021, 12 (1), 5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schapira M; Calabrese MF; Bullock AN; Crews CM, Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 2019, 18 (12), 949–963. [DOI] [PubMed] [Google Scholar]
- 25.Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y, PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 2019, 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weng G; Shen C; Cao D; Gao J; Dong X; He Q; Yang B; Li D; Wu J; Hou T, PROTAC-DB: an online database of PROTACs. Nucleic Acids Res 2021, 49 (D1), D1381–D1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullard A, First targeted protein degrader hits the clinic. Nat Rev Drug Discov 2019, 237–239. [DOI] [PubMed] [Google Scholar]
- 28.Chavez A; Scheiman J; Vora S; Pruitt BW; Tuttle M; E PRI; Lin S; Kiani S; Guzman CD; Wiegand DJ; Ter-Ovanesyan D; Braff JL; Davidsohn N; Housden BE; Perrimon N; Weiss R; Aach J; Collins JJ; Church GM, Highly efficient Cas9-mediated transcriptional programming. Nat Methods 2015, 12 (4), 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gechijian LN; Buckley DL; Lawlor MA; Reyes JM; Paulk J; Ott CJ; Winter GE; Erb MA; Scott TG; Xu M; Seo HS; Dhe-Paganon S; Kwiatkowski NP; Perry JA; Qi J; Gray NS; Bradner JE, Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol 2018, 14 (4), 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nabet B; Ferguson FM; Seong BKA; Kuljanin M; Leggett AL; Mohardt ML; Robichaud A; Conway AS; Buckley DL; Mancias JD; Bradner JE; Stegmaier K; Gray NS, Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun 2020, 11 (1), 4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadd MS; Testa A; Lucas X; Chan KH; Chen W; Lamont DJ; Zengerle M; Ciulli A, Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 2017, 13 (5), 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zengerle M; Chan KH; Ciulli A, Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 2015, 10 (8), 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell CE; Gao Y; Tan L; Donovan KA; Nowak RP; Loehr A; Bahcall M; Fischer ES; Janne PA; George RE; Gray NS, Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J Med Chem 2018, 61 (9), 4249–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nabet B; Roberts JM; Buckley DL; Paulk J; Dastjerdi S; Yang A; Leggett AL; Erb MA; Lawlor MA; Souza A; Scott TG; Vittori S; Perry JA; Qi J; Winter GE; Wong KK; Gray NS; Bradner JE, The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 2018, 14 (5), 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Remillard D; Buckley DL; Paulk J; Brien GL; Sonnett M; Seo HS; Dastjerdi S; Wuhr M; Dhe-Paganon S; Armstrong SA; Bradner JE, Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chem Int Ed Engl 2017, 56 (21), 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowak RP; DeAngelo SL; Buckley D; He Z; Donovan KA; An J; Safaee N; Jedrychowski MP; Ponthier CM; Ishoey M; Zhang T; Mancias JD; Gray NS; Bradner JE; Fischer ES, Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol 2018, 14 (7), 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Epstein LF; Chen H; Emkey R; Whittington DA, The R1275Q neuroblastoma mutant and certain ATP-competitive inhibitors stabilize alternative activation loop conformations of anaplastic lymphoma kinase. J Biol Chem 2012, 287 (44), 37447–37457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kronke J; Fink EC; Hollenbach PW; MacBeth KJ; Hurst SN; Udeshi ND; Chamberlain PP; Mani DR; Man HW; Gandhi AK; Svinkina T; Schneider RK; McConkey M; Jaras M; Griffiths E; Wetzler M; Bullinger L; Cathers BE; Carr SA; Chopra R; Ebert BL, Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523 (7559), 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao H; Sun X; Rao Y, PROTAC Technology: Opportunities and Challenges. ACS Med Chem Lett 2020, 11 (3), 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z; Kibler RD; Hunt A; Busch F; Pearl J; Jia M; VanAernum ZL; Wicky BIM; Dods G; Liao H; Wilken MS; Ciarlo C; Green S; El-Samad H; Stamatoyannopoulos J; Wysocki VH; Jewett MC; Boyken SE; Baker D, De novo design of protein logic gates. Science 2020, 368 (6486), 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nern A; Pfeiffer BD; Svoboda K; Rubin GM, Multiple new site-specific recombinases for use in manipulating animal genomes. Proc Natl Acad Sci U S A 2011, 108 (34), 14198–14203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anzalone AV; Koblan LW; Liu DR, Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 2020, 38 (7), 824–844. [DOI] [PubMed] [Google Scholar]
- 43.Anzalone AV; Randolph PB; Davis JR; Sousa AA; Koblan LW; Levy JM; Chen PJ; Wilson C; Newby GA; Raguram A; Liu DR, Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576 (7785), 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agha-Mohammadi S; O’Malley M; Etemad A; Wang Z; Xiao X; Lotze MT, Second-generation tetracycline-regulatable promoter: repositioned tet operator elements optimize transactivator synergy while shorter minimal promoter offers tight basal leakiness. J Gene Med 2004, 6 (7), 817–828. [DOI] [PubMed] [Google Scholar]
- 45.Costello A; Lao NT; Gallagher C; Capella Roca B; Julius LAN; Suda S; Ducree J; King D; Wagner R; Barron N; Clynes M, Leaky Expression of the TET-On System Hinders Control of Endogenous miRNA Abundance. Biotechnol J 2019, 14 (3), e1800219. [DOI] [PubMed] [Google Scholar]
- 46.Liberles SD; Diver ST; Austin DJ; Schreiber SL, Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc Natl Acad Sci U S A 1997, 94 (15), 7825–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S; Ding N; Sun Y; Yuan T; Li J; Yuan Q; Liu L; Yang J; Wang Q; Kolomeisky AB; Hilton IB; Zuo E; Gao X, Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Sci Adv 2020, 6 (29), eaba1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richter MF; Zhao KT; Eton E; Lapinaite A; Newby GA; Thuronyi BW; Wilson C; Koblan LW; Zeng J; Bauer DE; Doudna JA; Liu DR, Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 2020, 38 (7), 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monteys AM; Hundley AA; Ranum PT; Tecedor L; Muehlmatt A; Lim E; Lukashev D; Sivasankaran R; Davidson BL, Regulated control of gene therapies by drug- induced splicing. Nature 2021, 596 (7871), 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye H; Daoud-El Baba M; Peng RW; Fussenegger M, A synthetic optogenetic transcription device enhances blood-glucose homeostasis in mice. Science 2011, 332 (6037), 1565–1568. [DOI] [PubMed] [Google Scholar]
- 51.Nunez JK; Chen J; Pommier GC; Cogan JZ; Replogle JM; Adriaens C; Ramadoss GN; Shi Q; Hung KL; Samelson AJ; Pogson AN; Kim JYS; Chung A; Leonetti MD; Chang HY; Kampmann M; Bernstein BE; Hovestadt V; Gilbert LA; Weissman JS, Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184 (9), 2503–2519 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mullard A, Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov 2021, 20 (4), 247–250. [DOI] [PubMed] [Google Scholar]
- 53.Mares A; Miah AH; Smith IED; Rackham M; Thawani AR; Cryan J; Haile PA; Votta BJ; Beal AM; Capriotti C; Reilly MA; Fisher DT; Zinn N; Bantscheff M; MacDonald TT; Vossenkamper A; Dace P; Churcher I; Benowitz AB; Watt G; Denyer J; Scott-Stevens P; Harling JD, Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Communications Biology 2020, 3 (1), 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis JR; Wang X; Witte IP; Huang TP; Levy JM; Raguram A; Banskota S; Seidah NG; Musunuru K; Liu DR, Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nature Biomedical Engineering 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nelson CE; Wu Y; Gemberling MP; Oliver ML; Waller MA; Bohning JD; Robinson-Hamm JN; Bulaklak K; Castellanos Rivera RM; Collier JH; Asokan A; Gersbach CA, Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nature Medicine 2019, 25 (3), 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen WCW; Gaidukov L; Lai Y; Wu M-R; Cao J; Gutbrod MJ; Choi GCG; Utomo RP; Chen Y-C; Wroblewska L; Kellis M; Zhang L; Weiss R; Lu TK, A synthetic transcription platform for programmable gene expression in mammalian cells. Nature Communications 2022, 13 (1), 6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paulk NK; Pekrun K; Zhu E; Nygaard S; Li B; Xu J; Chu K; Leborgne C; Dane AP; Haft A; Zhang Y; Zhang F; Morton C; Valentine MB; Davidoff AM; Nathwani AC; Mingozzi F; Grompe M; Alexander IE; Lisowski L; Kay MA, Bioengineered AAV Capsids with Combined High Human Liver Transduction In Vivo and Unique Humoral Seroreactivity. Molecular Therapy 2018, 26 (1), 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tabebordbar M; Lagerborg KA; Stanton A; King EM; Ye S; Tellez L; Krunnfusz A; Tavakoli S; Widrick JJ; Messemer KA; Troiano EC; Moghadaszadeh B; Peacker BL; Leacock KA; Horwitz N; Beggs AH; Wagers AJ; Sabeti PC, Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184 (19), 4919–4938.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goertsen D; Flytzanis NC; Goeden N; Chuapoco MR; Cummins A; Chen Y; Fan Y; Zhang Q; Sharma J; Duan Y; Wang L; Feng G; Chen Y; Ip NY; Pickel J; Gradinaru V, AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nature Neuroscience 2022, 25 (1), 106–115. [DOI] [PubMed] [Google Scholar]
- 60.Chavez A; Scheiman J; Vora S; Pruitt BW; Tuttle M; P R Iyer E; Lin S; Kiani S; Guzman CD; Wiegand DJ; Ter-Ovanesyan D; Braff JL; Davidsohn N; Housden BE; Perrimon N; Weiss R; Aach J; Collins JJ; Church GM, Highly efficient Cas9- mediated transcriptional programming. Nature Methods 2015, 12 (4), 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Konermann S; Brigham MD; Trevino AE; Joung J; Abudayyeh OO; Barcena C; Hsu PD; Habib N; Gootenberg JS; Nishimasu H; Nureki O; Zhang F, Genome- scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517 (7536), 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chavez A; Tuttle M; Pruitt BW; Ewen-Campen B; Chari R; Ter-Ovanesyan D; Haque SJ; Cecchi RJ; Kowal EJK; Buchthal J; Housden BE; Perrimon N; Collins JJ; Church G, Comparison of Cas9 activators in multiple species. Nature Methods 2016, 13 (7), 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berrios KN; Evitt NH; DeWeerd RA; Ren D; Luo M; Barka A; Wang T; Bartman CR; Lan Y; Green AM; Shi J; Kohli RM, Controllable genome editing with split-engineered base editors. Nat Chem Biol 2021, 17 (12), 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zetsche B; Volz SE; Zhang F, A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 2015, 33 (2), 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider CA; Rasband WS; Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012, 9 (7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kluesner MG; Nedveck DA; Lahr WS; Garbe JR; Abrahante JE; Webber BR; Moriarity BS, EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J 2018, 1, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brinkman EK; Kousholt AN; Harmsen T; Leemans C; Chen T; Jonkers J; van Steensel B, Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res 2018, 46 (10), e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.