

Abstract
It has been over twenty years since the linear scaling of reaction intermediate adsorption energies started to coin the fields of heterogeneous and electrocatalysis as a blessing and a curse at the same time. It has established the possibility to construct activity volcano plots as a function of a single or two readily accessible adsorption energies as descriptors, but also limited the maximal catalytic conversion rate. In this work, it is found that these established adsorption energy-based descriptor spaces are not applicable to electrochemistry, because they are lacking an important additional dimension, the potential of zero charge. This extra dimension arises from the interaction of the electric double layer with reaction intermediates which does not scale with adsorption energies. At the example of the electrochemical reduction of CO2 it is shown that the addition of this descriptor breaks the scaling relations, opening up a huge chemical space that is readily accessible via potential of zero charge-based material design. The potential of zero charge also explains product selectivity trends of electrochemical CO2 reduction in close agreement with reported experimental data highlighting its importance for electrocatalyst design.
Subject terms: Electrocatalysis, Catalytic mechanisms, Density functional theory, Materials for energy and catalysis
The electric double layer controls the product selectivity and conversion rate of electrochemical processes. Here the author presents a double layer-corrected computational electrocatalyst screening approach using CO2 reduction as an example that highlights the importance of the potential of zero charge descriptor.
Introduction
Over the last years, computational, first-principles-based simulations have become a key tool in the global thrive towards a sustainable energy landscape. Density Functional Theory (DFT) has become in particular in heterogeneous surface catalysis a valuable means to explore reaction pathways and evaluate the catalytic ability of specific materials and active sites1. Unfortunately, complex reaction pathways often occur which require the computation of a large number of intermediate’s formation energies and reaction barriers, making it hard to utilize this approach for high-throughput screening of catalysts. The discovery of an often linear correlation of intermediate’s adsorption energies2,3, known as scaling relations4 has been thus a breakthrough in reducing the dimensionality of the catalytic activity space. In combination with Brønsted-Evans-Polanyi (BEP) relations5–8 which resemble the often linear correlations of activation energies with intermediate adsorption energies, this results in activity curves as a function of a single or two adsorption energies. According to the Sabatier principle9,10 these take the form of activity volcanos with the best catalyst binding the descriptor intermediate neither too weak nor too strong11. This has allowed the high-throughput screening of catalysts12–15 and a simplified discussion of experimental findings by a single or two adsorption energies16. On the downside, the scaling relations have made it impossible to independently vary the kinetic barriers of all elementary steps which would be needed to achieve the optimal catalyst2,3. The breaking of scaling relations to go beyond the dimensionality and thus limiting activity shown in the activity volcano has therefore been a key obstacle for the development of more active solid catalysts17,18.
In contrast to thermocatalysis, electrocatalysis involves the transfer of electrons and protons which electrochemical potentials needs to be calculated. The Computational Hydrogen Electrode (CHE) approach considers entirely proton-coupled electron transfer (PCET) steps and derives these electrochemical potentials simply from reference to the Standard Hydrogen Electrode (SHE) at which these are in equilibrium with Hydrogen gas19,20. By again considering scaling and BEP relations for PCET steps21, the full potential and pH dependence of all intermediate’s formation energies is accessible from often only a single or two formation energies22. Again, this descriptor-based approach has been used from high-throughput screening using machine learning23–25 or interpretation of experimental findings22.
Due to the origination of this approach from surface science, the formation energies of reaction intermediates even in electrochemistry were calculated on simple solid surface-vacuum model systems. Recently, this simplistic picture has been increasingly doubted, mostly due to the neglect of the electric double layer26,27. At applied potentials away from the Potential of Zero Charge (PZC), all metal electrodes build up surface charge density which is compensated by counter charge in the solution creating a significant interfacial electric double layer field. This surface charge density and the corresponding electric field have been shown to strongly affect the stability of key reaction intermediates and with that the catalyst’s performance and product selectivity28–35. The electric double layer effect is particularly critical in the electrochemical reduction of CO2 (CO2RR). Here, the formation of intermediates like *CO2 and *OCCO is rate-limiting which gives rise to a significant electric dipole that is stabilized by the surrounding double layer field28–30,32. Despite this recently discovered significance of the electric double layer, a correspondingly corrected descriptor-picture or activity volcano has not been derived so far. Instead, the community has mostly relied on the surface chemical approach resulting in one-dimensional (e.g. CO adsorption energy in CO2RR to CO36) or two-dimensional (e.g. C and CO adsorption energies for CO2RR to higher reduced products37,38) activity volcano plots.
In this work, I develop a descriptor-based activity model for CO2RR to CO with an explicit account for the electrochemical interface. The focus on CO2RR is due to two reasons. First, previous studies have revealed significant electric double-layer effects, stressing the urgency of this extension for the reliable screening of electrocatalysts. Secondly, electrochemical CO2RR has recently emerged as an attractive prospect for a sustainable carbon cycle and energy society. The focus on CO is due to the economical relevance of this process39 and the simple and well-established reaction mechanism allowing for a more reliable kinetic model. From this, it is revealed that the PZC and CO formation energies as the relevant descriptors and also discover that the same descriptors can be also quite generally used to discuss product selectivity of electrochemical CO2RR. Thus the descriptor space might be used to new CO2RR electrocatalysts for a defined product. This work makes a significant step forward in the screening of electrocatalysts and opens design avenues based on the PZC of materials that have been neglected so far.
Results
Development of descriptor space
Figure 1 schematically illustrates the workflow for deriving an electric double layer-corrected CO2RR to CO activity volcano, in this work exemplarily for metallic electrocatalysts. I briefly summarize the procedure here and direct the interested reader to the Methods section A for more details. The electrochemical reduction of CO2 to CO is considered to follow the well-established mechanism 30,40,41:
| 1 |
The formation/adsorption energy ΔiΩ of each reaction intermediate i is calculated as (see Methods section A for a derivation):
| 2a |
| 2b |
with the surface charge density σ, the electrode potential vs. SHE U, the zero-point energy (ZPE) and finite temperature correction term ΔiET,S,ZPE,∘, the gap or Helmholtz capacitance Cgap and the potential of zero charge (PZC) UPZC. ΔiE(σ(U)) is the DFT calculated expression, the surface-charge dependent adsorption energy of a specific reaction intermediate relative to the bare slab and CO2(g) (in the case of kinetic modeling or reference to the *COOH and *CO2 adsorption energies) or CO(g) (in the case of reference to the *CO adsorption energy).
Fig. 1. Schematic illustration of the derivation of electrochemical activity volcanos.

DFT calculations provide adsorption energies on a chosen facet as a function of surface charge density which can be interpolated. As a result, adsorption energies are obtained at zero surface charge density and the charge sensitivity parameter, and the process is repeated for various facets to generate a database. Then, the data is used to search for correlations among the properties to reduce the dimensionality of descriptors needed to define all adsorption energies and charge sensitivity parameters and thus the full free energy diagram. After this, the determined descriptors CO adsorption energy and PZC are used to build up a two-dimensional TOF figure. For this, the micro-kinetic rate equations are solved on a grid of points on this two-dimensional figure where each point defines a descriptor pair that uniquely links to a full free energy diagram via the determined correlations. To get the potential- and pH dependence of the free energy diagram at a given point, the CHE model is used together with the surface charging relation which eliminates the surface charge density from the expression. Kinetics are described by Brønsted-Evans-Polanyi (BEP) scaling relations with empirically chosen parameters.
Within DFT, the effect of surface charge is included by means of an implicit solvation model and planar counter charge approach, and a quadratic fit was considered first29,30,42. The results for adsorption of the three reaction intermediates on various metallic facets are summarized in Supplementary Materials Table S1. The quadratic contribution to ΔiE related to the quadratic coefficient Δib turns out to be at least an order of magnitude smaller than the linear contribution related to the coefficient Δia (cf. Supplementary Materials Fig. S5), making ΔiE(σ) linear to a good approximation (corresponding to a constant capacitance during adsorption43). The quadratic term is thus dropped in the following. From a physical point of view, the charge sensitivity parameter Δia is proportional to the electric dipole moment change during adsorption:
| 3 |
with the vacuum permittivity ε0, the dielectric permittivity of the Helmholtz layer εgap and the change of the electric dipole moment during adsorption Δiμ. Changes in the electric dipole moment are triggered by charge transfer between adsorbate and surface26,43–46. The charge sensitivity parameter for CO2 adsorption, and thus the generated dipole moment during adsorption, is a factor of ≈3–10 larger than the coefficients for *COOH and *CO formation (Supplementary Materials Table S1). This is due to the strong interfacial dipole moment created during CO2 adsorption which is stabilized by the surrounding electric double-layer field29,30. The remaining potential- and pH terms in Eq. (2a) stem from the CHE expression for the thermodynamic energy of a PCET 19,30.
Finally, Eq. (2b) denotes the surface charging relation or equation of state, for which again a plate capacitor was assumed. In this expression, the gap capacitance is taken to be constant across metals (≈20 μF/cm2)47–49 and the PZC which is well known to be proportional to the work function or Fermi level26,50, is derived from the calculated Fermi level of each surface plus a constant offset of 4.92 V obtained from fitting the Fermi level to the experimental PZC data (cf. Supplementary Materials Fig. S1). From the energy expression above, all formation energies are determined by three material-specific parameters for each intermediate, the formation energy at the PZC, ΔiE0, the parameters measuring the sensitivity of the intermediate with respect to surface charging Δia, and the PZC UPZC. The ZPE and finite temperature corrections are considered roughly independent of the surface51.
To further reduce the nine-dimensional descriptor space of the three intermediates which determine the free energy landscape, linear correlations are searched for within this space. First, all properties are correlated with only the CO adsorption energy ΔCOE0. In agreement with the literature33,52 a good linear scaling with the *COOH formation energy ΔCOOHE0 (R2 = 0.90) is obtained (Supplementary Materials Fig. S2). The prediction of the *CO2 adsorption energy in contrast is significantly worse with a reduced R2 = 0.72. Most critically, however, the remaining charge sensitivity parameter Δia turns out to be not predictable using the CO adsorption energies since no linear correlation can be identified. In addition, the charge sensitivity parameters of *CO and *COOH are found to be uncorrelated (Supplementary Materials Fig. S3) which according to Eq. (2a) means that ΔCOΩ and ΔCOOHΩ are also not correlated. This suggests a breaking of the well-established *COOH-*CO scaling relation33,52, meaning that even without the presence of the *CO2 intermediate deviations from a one-dimensional activity volcano for CO2 reduction to CO are to be expected due to surface charging.
As explained above, Δia relates to the amount of charge transferred from the metal surface to the adsorbate during the adsorption process. The charge is transferred from the Fermi level of the metallic slab to the adsorbate. Due to this, the Fermi level seems like an intuitive choice for predicting the qualitative trends of charge transfer and thus Δia across metals. Indeed, the work function which correlates with the PZC trends across metals50,53, has been already been discussed as an important parameter for CO2RR to CO54 and the hydrogen evolution reaction (HER)55,56. Intriguingly, the inclusion of this descriptor in a two-dimensional multi-regression model happens to increase all R2 values of adsorption energies and charge sensitivity parameters significantly to 0.9–1.0, with only the charge sensitivity parameter of *CO2 being not perfectly scalable at R2 = 0.76 (Supplementary Materials Fig. S2, S4 and Table S2). The larger uncertainty is likely related to the instability of the *CO2 state on many surfaces which requires extrapolating the energy to smaller charges using a fixed high charge geometry29. This extrapolation procedure bears some numerical error which might actually be canceled out to some degree by the linear scaling model. I also note that a correlation between adsorption dipole (and thus Δia) and PZC has been noticed by a different work recently46. Δia has, however, also a strong dependence on the adsorption site as well29, which might be in parts included in the other CO adsorption energy descriptor, as suggested by the strong linear partial correlation of the charging parameter of CO with the CO adsorption energy (cf. Supplementary Materials Fig. S4 and Table S2).
In sum, the two-dimensional descriptor space spanned by CO adsorption energy and Fermi level gives a reliable representation of the full-dimensional free energy landscape for all considered metal surfaces. Note that this includes both planar surface facets, but also stepped once, highlighting the generality of this approach. The interested reader is also referred to the Supplementary Materials Section S1 for a practical example of how to use this scheme to derive all formation energies starting from a given descriptor pair.
The derived multi-linear relations ΔiE0(U,pH; ΔCOE0, UPZC) and Δia(U,pH; ΔCOE0, UPZC) given in Supplementary Materials Table S2 fully define the thermodynamic free energy landscape at a given pair of descriptor values. To also define kinetic barriers, the approach of Hansen et al. is adapted which first fixes the activation barrier at the equilibrium, reversible potential of each elementary step to a defined value36. This idea is extended to include the effect of surface charging on the reversible potential following Eqs. (2a) and (2b). Potential dependence is then included via the BEP approach for which previous theoretical studies have indicated the validity21. The resulting linear scaling of the change of the thermodynamic driving force ΔiΩ across elementary steps using a charge transfer coefficient βi leads then to a Butler-Volmer-like equation (see Methods section B for details). This gives rise to two more parameters for the two electrochemical barriers, the fixed barrier at the reversible potential and the parameter βi defining the initial/final state-likeliness (both in terms of energetics and field-dependence) of the transition state and thus also its potential dependence. Instead of calculating these two parameters, I rationalize their values from previous experimental studies30. The reader is referred to Methods section B for a detailed discussion of this procedure and also the reasons for neglecting additional kinetic barriers for CO desorption and CO2 adsorption.
CO activity volcano
Starting from activation energies of all elementary steps, a mean-field micro-kinetic model (MKM) for the reaction mechanism presented in Eq. (1) is constructed and solved in the steady-state approximation using the CatMAP program package11. Fig. 2 shows the activity volcano plot of the turnover frequency (TOF) of CO formation obtained from the MKM model as a function of the two descriptors, CO adsorption energy, and PZC, at −1.0 V vs. SHE, the voltage at which CO is primarily formed on most CO forming metals29,33. In the following, this two-dimensional model co2-2d is compared to the conventional CO2RR to CO kinetic model (co2-1d)36 which does not consider the presence of the *CO2 intermediate nor the presence of surface charging.
Fig. 2. Charge-corrected descriptor-based volcano for the CO2RR to CO activity at -1 V vs. SHE (pH 6.8).

Contour lines depict the turnover frequency for CO2 to CO conversion from the descriptor-based kinetic model as a function of PZC and CO adsorption energy (relative to CO gas and without ZPE, free energy and gas corrections). Solid contour lines represent iso-lines of the absolute value of the hydrogen adsorption free energy, predicted as a function of the two descriptors. The right panel shows the conventional CO2RR to CO activity volcano which does not consider the CO2 intermediate or surface charging. It was generated using the kinetic parameters in this work (black) and the original kinetic model (red)36 taking adsorption energies at the potential of zero charge to be correlated with the CO adsorption energy only. Source data are provided as a Source Data file.
By means of a degree of rate control analysis, it is first found that the CO activity volcano in the co2-2d model decomposes into four distinct regions with different rate-limiting steps (shown in Fig. 2). The co2-1d model becomes instead a one-dimensional activity volcano as a function of the *CO adsorption energy36 and shows only two different rate-limiting steps, *COOH to *CO for the weakly CO-binding, and *CO desorption for the strongly CO-binding leg. For very weakly CO-binding materials, the co2-2d model shows that the *COOH to *CO step is only rate-limiting for materials with a positive PZC, and thus those which have significant surface charge density at −1 V vs. SHE (cf. Eq. (2b)), stabilizing the adsorption of *CO2. At more negative PZCs, however, CO2 adsorption is always rate-limiting. This can be explained by the stronger adsorption dipole of *CO2 which results in at least one order of magnitude larger Δia (cf. ref. 29 and Supplementary Materials Table S1) that destabilizes *CO2 significantly more compared to *COOH when moving to more negative PZC values, and thus reduced negative surface charge density.
At highly negatively charged electrodes with a very positive PZC, another transition can be found at intermediate CO binding strengths for which the *CO2 to *COOH step becomes limiting. As seen in Supplementary Materials Table S2, both *CO2 and *COOH adsorption energies have a similar *CO adsorption energy scaling of around ≈0.6 which makes them become less stabilized compared to *CO when going to more strongly *CO binding metals. Thus, the *COOH to *CO step becomes increasingly likely while the previous step becomes rate-limiting. As expected, the *COOH to *CO region enlarges in both PZC and CO adsorption energy direction when increasing the reversible potential activation barrier of the *COOH to *CO step (Supplementary Materials Fig. S6). As explained in the Methods section B, this barrier is chosen based on experimental results on Au, and affects only the rate-limiting step regions, not the actual value of the TOF (Supplementary Materials Fig. S6).
Since the *CO intermediate is the one that is most strongly stabilized when moving to strongly CO-binding metals, CO desorption becomes rate-limiting inevitably (Supplementary Materials Table S2). It is notable though that the transition line marking this region has a strong curvature resulting from the PZC dependence of the CO activity in the co2-2d model. This distinguishes the volcano substantially from the co2-1d model. The curvature of the volcano indicates that catalysts with the same CO adsorption energy can exhibit significantly different activity and thus also free energy diagrams depending on their PZC. As explained above, *CO2 is significantly more sensitive to the PZC compared to the other intermediates, being strongly stabilized when going to more positive PZC values (cf. Table S2). Therefore, at a fixed *CO adsorption energy and relatively strong *CO binding, an increasing PZC will at some point always make the *CO2 to *COOH step rate-limiting. On the other hand, for a very negative PZC, CO2 is drastically destabilized which will make CO2 adsorption rate-limiting at some point. The result is a defined PZC region in which *CO desorption is rate-limiting which includes all the transition metals that were investigated in this work.
All metals adsorbing CO weaker or equal to Cu lie in the region where CO2 adsorption is rate-limiting. In this region (with the exception of Cu), a negligible coverage of all reaction intermediates is found (cf. Supplementary Materials Fig. S8) in line with experimental works on Au57. If CO2 adsorption is rate-limiting, no pH-dependence is expected of the CO partial current on an absolute potential scale, even under acidic conditions, since no proton is involved in the rate-limiting step and all coverages are small so that the pH-dependence of the number of free active sites is negligible. This prediction from our model has been indeed recently verified on Au30, Ag, Sn and In41. Previous experimental studies have shown that at −0.7 V vs. SHE, the pH-dependent formation of *COOH becomes rate-limiting on Au30. Since I used this experimental result to parametrize the kinetic model (cf. Methods section B), our model reproduces this transition quantitatively, and the rate-limiting step region of *COOH to *CO moves to more negative PZCs so that it is located exactly over Au at this potential (cf. Supplementary Materials Fig. S10). More importantly, however, the co2-2d model can also reproduce that on Ag, a corresponding switch of the rate-limiting step associated with a reduction of the Tafel slope from 120–140 mV/dec33,58 to around 80 mV/dec58 is observed already at around −0.4 V vs. SHE (Supplementary Materials Fig. S10). Our model reveals that the physical reason for this earlier switch to CO2 adsorption being limiting is the less positive PZC of Ag reducing the interfacial electric field and thus the driving force for CO2 adsorption. The agreement with these trends increases the trust in the transferability of our kinetic model and the developed descriptor space.
An additional indication that our model is an improvement to the conventional model is the comparison between Au and Ag. The right panel of Fig. 2 shows the conventional activity volcano constructed from the CO adsorption energy as the mere descriptor without any account for the *CO2 intermediate nor for surface charging (adsorption energies from ΔiE0). Kinetic parameters are either taken to be the same as in the two-dimensional volcano (black line) or from the original published model (red line)36. By taking the (100) facet as an example, the CO TOF increases by 0.7 orders of magnitude for the original co2-1d model, and 1.4 orders of magnitude for the updated co2-1d model. From analyzing previous experimental data in Supplementary Materials Fig. S730,33,59,60 on various planar Au and Ag surfaces in H-cells, it becomes, however, clear that for the same facet, Au surfaces are commonly up to three orders of magnitude more active than Ag in the lower kinetic potential region (which is not limited by mass transport30). By comparing to Fig. 2, the co2-2d model captures this order of magnitude and shows that the difference to the co2-1d model arises from the non-horizontal TOF contour lines in the region where CO2 adsorption is rate-limiting. These arise due to the CO2 intermediate which is increasingly stabilized when moving to more positive PZC values due to the increased negative surface charge density explaining the much higher activity of Au compared to Ag.
The fact that CO2 adsorption is rate-limiting on all strongly CO-producing metals suggests that rather than the pH, the surface charge density might be a much more effective parameter to tune the CO production rate (cf. also Fig. S9 showing a minor pH effect). In addition, since Au under-binds CO, the addition of a strongly CO-binding metal like Pd might be moving the system into the more active region, as recently experimentally demonstrated for Au-Pd nanoparticle catalysts61 and depicted here by the Pd3Au alloy catalyst in Fig. 2. Another interesting observation is that Al(111) is predicted to be reasonably active. Although thermodynamically, Al is likely to be at least partially oxidized under electrochemical CO2RR conditions62, the co2-2d model suggests that the remaining Al sites might be very active and indeed Al-based bimetallics have been already reported as promising catalysts 24.
For all considered strongly CO-adsorbing metals, CO desorption is rate-limiting, and the surface is fully covered with CO (cf. Supplementary Materials Fig. S8). According to the co2-2d model, Pd is a catalyst with impressively high activity comparable to Ag. Pd is indeed also experimentally known to produce CO63–65, albeit it is rarely discussed as a promising CO2RR catalyst due to the prevailing HER. To visualize the regions of high HER rate, the hydrogen adsorption energy (without accounting for the field interaction which is a small effect32) is correlated with the two descriptors revealing a high R2 = 0.90. This correlation originates mostly from the correlation with the CO adsorption energy (R2 = 0.87), leading to almost horizontal isosurfaces of the H adsorption energy in Fig. 2, with the stronger binding CO metals showing increased HER activity. The high CO coverage, however, can to some degree suppress HER by poisoning active sites66. Therefore if means to suppress HER can be employed and improved, such electrocatalysts might become highly promising candidates67,68. Finally, I also mention that due to saturation of the CO coverage (cf. Supplementary Materials Fig. S8), the CO formation rate is independent of pH also in this region, suggesting again a minor role of the pH in enhancing the CO production rate (cf. also Supplementary Materials Fig. S9 showing the pH effect on the activity volcano).
The most interesting part of the activity volcano is the central high CO activity region, where the rate-limiting step switches from CO2 adsorption to CO desorption. Cu lies according to our model closest to this region with the binding of CO being neither too weak nor too strong leading to a partially CO-covered surface (Supplementary Materials Fig. S8 and ref. 69). The three to four orders of magnitude higher activity of Cu vs. Au stands in contrast to the previously published model36 (shown in the right panel of Fig. 2, red line) suggesting Au has comparable or even higher TOF (looking at the (211) facet). Using our kinetic parameters, however, the co2-1d model gives a comparable order of magnitude change from Au to Cu (right panel in Fig. 2, black line). Both co2-1d models, however, locate the maximum of the activity volcano at a too-weak CO binding, as well as not give an account for the strong curvature with PZC. The latter is important, as it can easily move a catalyst outside the active region, despite a perfectly matching CO adsorption energy. Experimentally, further reduced products like C1 (methane) and C2 (ethylene) products are usually observed on Cu, depending on the potential region and pH70,71. Our calculations suggest, however, that Cu is actually a very excellent catalyst for producing CO, indicating that a rather significant amount of higher reduced products might be formed by re-adsorption of CO. Also the combination with a higher PZC and stronger CO binding metal like Pd might be promising as it moves the system into the higher activity region, and corresponding alloys showing high CO selectivity have been recently reported72.
Tuning bimetallic alloys for CO production
The maximum of the co2-2d volcano lies in a region of space where no bare transition metal surface is located. In principle, transition metals might be combined for example in the form of alloy compounds, resulting in hybrid properties between the two materials making it possible to navigate through the descriptor space. Multi-metallic catalysts could be formed by doping a substrate with a different metal e.g. in the form of single atoms73, forming an overlayer74, forming homogeneously mixed high entropy alloys75, pure alloy crystal phases, nano-particles, etc76. In Fig. 3 these possibilities are evaluated at the example of Au-Cu bimetallic alloys to discuss how such structural motifs could help to navigate in the here reported descriptor space. CO will always adsorb on the most strongly binding site and thus the presence of Cu sites on the surface will always set the CO adsorption energy descriptor to a value close to Cu. Variations of this adsorption energy across the bimetallics are rather small, only about ± 0.1 eV. This suggests that the tuning of material properties based on adsorption energies may not be particularly successful in actually designing better catalysts. I stress that this simplified treatment does not take into account coverage distributions across active sites, a saturation of the Cu sites, etc. However, in contrast to the comparably difficult modulation of the CO adsorption energy, the PZC can be much more easily tuned over a wide range. Looking at this descriptor in more detail, one can first note by comparing analogous surface configurations that the less noble metal dominates the PZC. In order to increase the PZC of Cu to make it more active for CO formation, the most efficient way is to dope Cu into an Au substrate. Also having small Cu clusters on an Au substrate can be beneficial up to a full monolayer of Cu. In contrast, high entropy alloy or single crystalline crystals promise much less tunability of the PZC, fixing the value to the less noble metal. The importance of the substrate in tuning the PZC of a catalytically active site on the surface has been so far relatively rarely discussed35 but could indeed play a much more important role than previously expected. By comparing to Fig. 2, increases of the PZC relative to Cu would activate the catalyst for the formation of CO. CO producing Cu-Au alloy catalysts where Cu and Au are mixed homogeneously in the surface have been reported in the literature 77.
Fig. 3. Effect of Cu-Au bimetallic composition and structure in the here reported descriptor space.
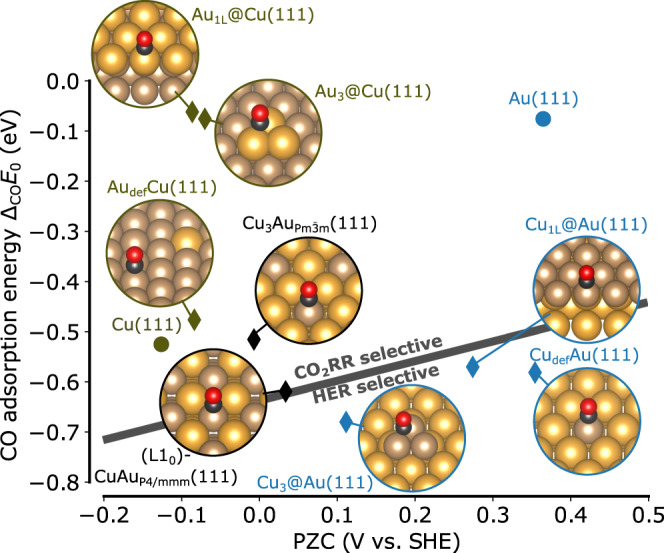
The gray line indicates a transition from CO2RR to HER selectivity. Circles refer to the data points which were actually used for the training our descriptor model, and diamonds for those where only the descriptor values were evaluated. Source data are provided as a Source Data file.
Description of experimental product selectivity trends
So far, the introduced two-dimensional descriptor space was mainly discussed based on the CO2 to CO conversion rate landscape. However, a large part of the metals presented in the figures so far is not actually selectively forming CO during CO2RR, but instead formate/formic acid, C1- or C2- products or even performing the HER. The construction of a kinetic model for all of these processes has been recently attempted including suggestions of descriptors like the *CO, *OH, *C and *H adsorption energies37,38,78,79, but these models still lack in their ability to explain all selectivity trends78. An interesting outcome from previous studies was the discovered correlation of the adsorption energies of all intermediates in the CO2RR process on Cu to C1- or C2- products with the CO adsorption energy80. This suggests that the same descriptor pair that I suggest here might be again valid also to describe the activity for other products and thus also the selectivity trends. In addition, the formation of C2 products is limited by the field-driven coupling of two *CO intermediates28,29,69,81–87 which could suggest the necessity of the PZC descriptor to explain selectivity trends.
If the discovered descriptors would be useful also for the description of other product pathways, a plot of the experimentally revealed dominant product selectivity as a function of these descriptors should clearly separate into selectivity regions88. Figure 4a shows the experimentally determined dominant product of electrochemical CO2RR at −1.4 V vs. SHE and pH = 6.8 from studies using planar electrodes (details in Supplementary Materials Section S3). As seen from this picture, the descriptor space clearly separates into defined regions of specific product selectivity. While a large part of the descriptor map is relatively unexplored, such as for example Rh, this suggests that the descriptors may also be suitable to screen catalysts for a desired product of choice. Particularly notable is also again the different impact of the two descriptors. The CO adsorption energy distinguishes the CO2RR- from the HER-selective region. This can be understood by the fact that stronger CO binding surfaces are generally poisoned by *CO66, but in turn have an almost optimal *H adsorption energy (cf. the H-adsorption energy lines in Fig. 2). CO2RR activity is only expected if the CO adsorption energy is in the intermediate to weak region (< −1 eV) where *H adsorption is also significantly weaker so that CO2RR becomes competitive. Further reduced products compared to formate and CO are only formed if *CO can be more stabilized on the surface so that it can further react, as in the case of intermediately strongly CO-binding metals such as Cu. The PZC determines the product selectivity of CO2RR. At weaker CO binding, the PZC distinguishes formate vs. CO selectivity. An interesting fact, since it has been noted before35 that CO formation might be a field-sensitive process, driven at positive PZC values, while formate is produced via CO2 reacting directly with *H89, a field insensitive process35. The latter becomes the dominating path if the CO2 adsorption step is very uphill in energy, which is the case at more negative PZC values, blocking the CO-forming pathway. This might explain why the PZC is effective in distinguishing formate from CO selectivity.
Fig. 4. CO2RR product selectivity as a function of the here-developed descriptors.

a Main experimental product selectivity30,33,59,64,90,134–137 for electrochemical CO2RR as a function of the developed descriptor space. Red markers: No selectivity data available. The red-boxed Cu region is discussed in detail in (b and c). b Ethylene/methane selectivity on a log scale as a function of the here-developed descriptors. The data was taken from constant-current experiments by Hori et al90. and interpolated via a triangular, linear fit. c Ethylene/methane selectivity on a log scale as a function of the here-developed descriptors. The data is obtained from extrapolating constant potential data83,90,138 to the same potential of −1.4 V vs. SHE. d Product selectivity for C2+ products vs. products beyond formate and CO ( > 2e−) as extracted from ref. 91 at −1.3 V vs. SHE plotted vs. the PZC. Source data are provided as a Source Data file.
To gain more insights into the region of higher reduced products, meaning the formation of methane, ethylene, ethanol, etc., the Cu region is enlarged, and experimental data is analyzed in detail using the here-developed descriptors. Hori et al. recorded the ethylene vs. methane selectivity at a constant current on a variety of Cu single crystalline facets90, and this is shown in Fig. 4 b. The data shows an interesting volcano-like trend with a maximum selectivity for ethylene at a PZC of −0.4 V vs. SHE. When looking at these trends, it is also important to consider typical experimental error bars, which can change the selectivity easily by 0.130. Therefore, the selectivity-descriptor relation is refit by a Gaussian process regression with regularization accounting for systematic noise. As Supplementary Materials Fig. S11 shows, this treatment smooths out the variations in the direction of the CO adsorption energy resulting in a simple one-dimensional selectivity volcano as a function of the PZC. Unfortunately, the data of Hori et al. was recorded at constant current, and the selectivity drastically changes with potential37. To explore this point in more detail, potential-dependent CO2RR data on Cu single crystals is extracted from literature and plotted in the same figure (Supplementary Materials Fig. S12). The data is then interpolated to estimate the selectivity at a constant potential (Supplementary Materials Fig. S13). From this much sparser data set, a similar dependence on PZC can be found, as seen in Fig. 4c with a maximum at a similar position for −1.4 V vs. SHE which, however, shifts with potential (Supplementary Materials Fig. S14). While the sparsity of the experimental data makes it hard to make final conclusions about the location of the selectivity maximum, it is interesting to see, how well the two descriptors can define the selectivity space, once the relevant experimental data is present.
To explore the generality of the volcano-like PZC trend of the ethylene/methane selectivity, the PZC descriptor is also applied to predict the C2 vs. > 2e− reduction (including all products but CO and formate) selectivity on metal-doped Cu catalysts from recent experimental data91(Fig. 4b). Interestingly, the descriptor correlates nicely with the selectivity data with the exception of the experimental data on Sn which has been also noted as a special case in the original paper91. The enhancement of C2 product selectivity with increased PZC can be explained by increased surface charge density (Eq. (2b)) and the resulting increased electric field. It has been previously shown that the coupling of two *CO intermediates that is rate-limiting for C2 product formation has a strong field-dependence and correspondingly large Δia-coefficients28,29,69,81–86. Due to this, a more positive PZC usually increases the C2 over C1 product selectivity. This is in line with the low-PZC branch of the selectivity volcano predicted from analyzing the experimental Cu facets data in Figs. 4b and c. This also agrees with recent experimental data on organic molecule-functionalized Cu surfaces showing higher surface charge density to enhance the C2 product selectivity92. At even higher PZC values, the selectivity is predicted by this analysis to go down at some point, likely due to an over-stabilization of *OCCO and the next step becoming rate-limiting.
I also compared our descriptor space to the one more conventionally discussed in the community consisting of CO and H adsorption energies88. Supplementary Materials Fig. S15 shows the selectivity map which reveals important differences to our map. First, as I discussed above, the H adsorption energy correlates much more closely with the CO adsorption energy than the PZC leading to selectivity separation lines lying on the diagonal instead of vertical and horizontal. Secondly, the H adsorption energy cannot efficiently separate the different product selectivity regions, like formate and CO, or the trend from C1 to C2 products. Another problem with using this descriptor is the assignment of the correct adsorption site for H. In addition to the general problem of assigning an active site, some arbitrariness arises for example in the question if this should be the most stable site on a chosen facet or the CO2RR active site. These findings indicate, that the PZC – which depends on the facet, but is roughly independent of local variations in the field, and thus particular active site environments – might be a more useful addition to the CO adsorption energy descriptor to rationalize experimental selectivity trends.
Discussion
The results of this work highlight the importance of the PZC as an electrochemical design parameter. It is noted that this study is not the first to notice the significance of the PZC. In particular, in the HER community, activity-work function and activity-PZC trends have been discovered93–95, but until today not fully understood. Possible explanations have been suggested, e.g. that a more positive PZC leads to a more negatively charged electrode and thus enthalpic stabilization of the charged transition state which is partially compensated by entropic changes due to proton accumulation94,96. Here, I presented a fully enthalpic explanation. While the adsorbed *H is charge-neutral at first sight, it still transfers charge and thus exhibits a small32, but significant97 interfacial dipole which can be stabilized by e.g. going to more positive PZC values. Without making any claims on the possibly significant entropic contribution, this work suggests a fundamental, enthalpic explanation for the work function or PZC dependence of electrochemical processes and demonstrated the high significance, especially for electrochemical CO2 reduction.
To prepare materials with optimal PZC, structural design concepts need to be developed. Such concepts are relatively well known for the work function: Halide adsorption98 or the adsorption of organic molecules92,99 are just a few examples, of how the work function can be tuned in addition to the substrate effect presented above. Employing work function design strategies the future task should thus be to find materials with the optimal CO adsorption energy and PZC. For this, high-throughput computational screening of materials should be performed which can be facilitated by machine learning methods that utilize geometric and physical features. The prediction of the adsorption energy is thereby tricky, as it requires knowledge about the active site which is not always obvious from experimental setups. Besides that, the development of material features for learning CO adsorption energies100 or work functions101,102 is in process and can be utilized in this plan.
Moreover, I comment on the PZC that I used throughout this manuscript and its suitability as a descriptor for high-throughput material screening. The PZC that I used is ultimately an ideal, theoretical value that is comparable roughly to the experimental PZC in the absence of specific adsorption and reconstruction (Supplementary Materials Fig. S1). It can be theoretically directly obtained from the DFT calculation of the slab system from the Fermi level and the relation presented in Supplementary Materials Fig. S1 (practically, a new relationship might need to be obtained to reference vs. true vacuum). Experimentally, the PZC, or as it is also called the potential of zero free charge (PZFC) is prone to additional environmental effects such as the type of anions or cations, or the pH due to specific adsorption and reconstruction27,103 and is generally measured with a larger uncertainty, although recently it has been even assigned to grains of a catalyst surface93. Therefore, for comparison of the presented theory with experimental systems, it might be also useful to make use of the advances of vacuum techniques to measure the more accurately measurable work function55 instead and use the relation with the PZC50, such as that presented in Supplementary Materials Fig. S1 to derive the theoretical PZC that is used for all plots presented above. I stress that it is the ideal, theoretical PZC or work function that explains the selectivity trends e.g. in Fig. 4, rather than the true PZC. Of course, for more complex systems which are prone to reconstruction or specific adsorption, the model should be revised again using theoretical methods to resolve the double layer in atomic detail.
Lastly, I also comment on the importance of cation effects for the activity and selectivity trends presented here. Cation concentration and identity have been shown to affect both HER104–106 and CO2RR29,30,32,35,106–110 conversion efficiency and product selectivity. For CO2 reduction, cations affect the selectivity mostly be stabilizing key intermediates by direct bonding and field stabilization29,107,108. Thus, our presented activity trends could shift substantially under different electrolyte environments, a point which should be explored in future studies. According to our previous work, mono-valent cation effects could be incorporated by changing the surface charging relation used in this work to one including cation-size effects29.
In this work, I introduced the PZC as a so-far underrated parameter to describe activity and selectivity trends in electrochemical energy conversion. The descriptor arises from the electric double layer field which influences the stability of certain reaction intermediates which give rise to a strong interfacial dipole, such as e.g. the *CO2 intermediate in the electrochemical reduction of CO2. To show this, I performed DFT calculations on metallic catalysts accounting for the electric double layer and found that the full free energy diagram can be mapped onto a two-dimensional descriptor space composed of CO adsorption energy and PZC. From micro-kinetic modeling, I found the PZC to effectively break the scaling of intermediate formation energies opening up new material design spaces. Due to the global nature of the PZC, this design space is much more flexibly accessible via the PZC compared to adsorption energies. Using the here-developed descriptor space, I explained a variety of experimentally observed trends in CO2 reduction, shedding much trust in the model. An important outcome is for example a volcano-like dependence of the C2 vs. C1 product formation rate on the PZC. Our results indicate that the PZC represents a so far less recognized, but important parameter for the discovery of electrocatalysts and the explanation of experimental trends and findings.
Methods
A: DFT calculations
Conceptual details
The adsorption energy (or grand potential26,44) of any reaction intermediate i is defined as:
| 4 |
with U denoting the electrochemical potential of electrons in the electrode at which the energies of the adsorbed and empty slab state are evaluated vs. the SHE reference electrode, i.e. the electrode potential on a SHE scale. Ni denotes the number of each adsorbate element, where the elemental reference chemical potentials μ are given as:
| 5 |
In the microkinetic modeling framework, we always use the CO2(g) reference, but in the plots shown in the main paper, the CO reference is used to define the adsorption energy of CO. The electrochemical potentials of protons and electrons are defined via the CHE approach which assumes electrochemical equilibrium at the SHE reference electrode allowing for re-reference of the electrochemical potentials19,20. We further defined for convenience:
| 6 |
where we integrated the DFT-calculated properties into a single term ΔiEU. The corrections for finite pressure (standard conditions) and temperature and zero-point energy were moved into the term ΔiET,S,ZPE,∘. To obtain ΔiEU, a standard approach is to utilize charge equilibration to match the Fermi levels (= electron’s electrochemical potentials) of the initial and final state (grand-canonical constant potential approach) and then obtain the potential-dependence by varying the excess charge44,111. Here, we disregard the often small charge equilibration contribution and approximate the energy difference as a constant charge energy difference ΔiEσ30,44,111, for simplicity here denoted as ΔiE:
| 7 |
With that, the adsorption energy becomes:
| 8 |
The surface charge density is related to the potential by a simple plate-capacitor equation:
| 9 |
with the Helmholtz or gap capacitance Cgap and the potential of zero charge (PZC) vs. SHE UPZC. This is valid because, under electrochemical conditions, the diffuse layer contributions to the surface charge density are small29. The PZC is accessible from the simulations via the Fermi level ϵF. In our simulations, we use a small dielectric permittivity of εb = 6 (see computational details discussion) defining the potential reference. The Fermi level or work function is in our system thus measured relative to almost vacuum (εb = 6). We found a fairly linear correlation of the experimental PZC vs. SHE and the theoretical PZC − ϵF/e vs. εb = 6 from a series of metallic facets (Supplementary Materials Fig. S1), related merely by a constant shift of 4.92 V:
| 10 |
The constant shift represents the absolute SHE potential within the implicit solvent model applied26. For the gap capacitance we took the approximately metal-independent value of around 20 μF/cm247–49. The advantage of this formulation compared to the constant potential formulation is that it avoids the problem of implicit solvation models not accurately producing the capacitance value across several metals using a single implicit solvation model parameter set112. We note that the real picture of the electric double layer might be more complex than described here, with specific adsorption, effects of the diffuse layer, ionic correlations, etc. but in many cases, even a simple plate-capacitor motivated model can correctly describe trends across materials26. This is also since the capacitive response of most metal electrodes under highly polarizing CO2RR conditions of −1 to −1.5 V vs. SHE is dominated by the Helmholtz response with only small contributions of the diffuse layer26,29.
To evaluate ΔiE(σ), we employed an implicit solvent model and a planar counter charge representation of the mobile counter-charges. From this, we found an approximately linear dependence, so that:
| 11 |
We note that this approximation will generally break if the capacitance changes during the adsorption event (the 2nd-order term). In our case, we found this term, however, to be relatively minor and similar to previous studies43,45. The charge sensitivity parameters Δia are related to the change of the PZC during the adsorption event, or in other words, the partial charge transferred during the adsorption event, or electrosorption valence26,43,44. The partial charge transferred generates an electric dipole moment, which modifies the PZC and interacts with the interfacial electric field. In summary, in our approach, the potential-dependent free energy diagram is completely defined from three formation energies ΔiE0, the field sensitivity parameters Δia, the PZC of each material UPZC and the finite temperature and zero-point energy correction term ΔiET,S,ZPE,∘.
Technical details
Density functional theory calculations of reaction energetics were carried out with a periodic plane-wave implementation and ultrasoft pseudopotentials using QUANTUM ESPRESSO version 6.1113 interfaced with the Atomistic Simulation Environment (ASE)114. We applied ultra-soft pseudopotentials and the BEEF-vdW functional, which provides a reasonable description of van der Waals forces while maintaining an accurate prediction of chemisorption energies115. The bulk lattice constant of the face-centered cubic (fcc) materials was optimized within QUANTUM ESPRESSO using spin-polarized DFT, plane-wave, and density cutoffs of 900 and 9000 eV, respectively, 1 × 1 unit cells, and 13 × 13 × 13 Monkhorst-Pack k-point grids116, until forces were converged to less than 0.05 eV/Å.
Following the bulk relaxation, surfaces were cut using ASE114, CatKIT117 and Pymatgen118. For the face-centered cubic (fcc) (111) and (100) surfaces, large 4 × 4 orthogonal supercells were constructed to minimize adsorbate-adsorbate interactions and approach the limit where constant charge and constant potential calculations approach each other43 and a 3 × 3 × 1 Monkhorst-Pack k-point grid used119, which led to converged energies and Fermi level. Correspondingly, orthogonal 3 × 4 supercells and 4 × 3 × 1 Monkhorst-Pack k-point grids were used for the fcc(211) surfaces. For all other surfaces, the separation of surface adsorbates was attempted to be chosen similarly to the other surfaces and the k-point grid density was kept as similar as possible. Adsorption energies were evaluated using asymmetric (adsorbates on one side of the slab) three-layered supercells with the lowest layer constrained and 20 Å separation of the surface slabs. Spin-unpolarized calculations were performed using plane-wave and density cutoffs of 700 and 7000 eV, respectively, as well as a Fermi-level smearing width of 0.1 eV. The reported Fermi energies in the figures have been, however, mostly obtained using lower plane-wave and density cutoffs of 500 and 5000 eV, respectively.
The SCCS implicit solvation model as implemented in the Environ QUANTUM ESPRESSO module120 was used to model the presence of implicit water in all surface slab calculations. We employed the “fitg03” (in a.u.: ) solvation parameter set which has been optimized for neutral molecule solvation energies120. The bulk relative dielectric permittivity was set to εb = 6 corresponding to highly constrained water that has been rationalized in our previous study29. Cavitation and repulsive energy terms are included by introducing an energy term proportional to the cavity surface area as described in ref. 120 and we here apply the parameter (α + γ) = 11.5 dyn/cm from the fitg03 parameter set. Dispersion interactions are ignored since they depend on the cavity volume which is an ill-defined property in surface slab calculations. The surface charge density was modulated in order to simulate the response of adsorbate-free energies to the presence of an electric double-layer field. A planar counter charge with a slab separation of 5 Å was applied to neutralize the simulation cell. A parabolic correction was applied in the Environ calculations to decouple the electrostatic interaction between the periodically repeated slabs.
All structures were relaxed at the corresponding surface charge density using a BFGS line search algorithm until force components were less than 0.05 eV/Å. *CO2 requires for many metallic surfaces the presence of negative excess surface charge to be stable. In line with our previous work29,30, we, therefore, extrapolated the formation energy for smaller surface charge densities by performing single point calculations using the optimized geometry at the least negative surface charge where the adsorbate still adsorbed. In general, the most stable adsorption sites were searched for by testing various adsorption sites, despite the (111) facets, where the most stable sites are well known (fcc or hcp hollow for *H, top for *CO, bridge bonded configuration for *COOH, see e.g.33).
Zero-point energy and finite temperature corrections in the harmonic oscillator approximation were taken from our previous work on gold30, and it was assumed that these values do not change much between different metals and facets. We applied an energy correction of 0.33 eV to the gas phase energy calculation of CO2, which was determined from experimental gas-phase reaction energies115. This results in an equilibrium potential for CO2 reduction to CO of −0.04 V vs. SHE (pH = 0) which is close to the experimental value of −0.10 V vs. SHE70. Further, double bond corrections of +0.25 eV were applied to *COOH and *CO269, and a hydrogen bonding solvation correction of −0.25 eV to the energy of *COOH121, due to the underestimation of this contribution by implicit solvation techniques122. The corrections were the same as in ref. 30.
B: Micro-kinetic modeling
Conceptual details
Micro-kinetic modeling was performed using the CatMAP package11. CatMAP solves the mean-field rate equations (all implemented as reversible) in the steady-state assumption. Within CatMAP, Butler-Volmer kinetics123 for proton-coupled electron transfers (PCETs) are implemented for each elementary step36. We extended this formulation to also include surface charging by the electric double layer. Then the activation energy for a PCET step becomes:
| 12 |
where is the activation energy at the reversible potential of the elementary step i to i + 1 and the ∘ indicates standard conditions (activities of 1, pH = 0). ΔiRΩ∘ is the difference in formation energies of two reaction intermediates i and i + 1, i.e. the reaction energy of that particular elementary step (the subscript R is used to differentiate it from the formation/adsorption energy which is defined relative to the empty slab). The meaning of βi becomes clear when the reversible potential of each step is related at which:
| 13 |
and therefore following Eq. (8):
| 14 |
And therefore:
| 15 |
where in the last term, we introduced again the grand potential which energetic contribution needs to be shifted to the right potential. With this expression, we can rewrite Eq. (12):
| 16 |
The first βi-term is the known term from reversible electrochemical kinetics formulated for each PCET step36. The last term depicts the charge stabilization of the transition state relative to the initial state. In experiment, usually an effective βeff is observed, which can be defined by rewriting as30:
| 17 |
with the effective βeff containing both contributions from the transferred electron and the double layer stabilization:
| 18 |
We further used a pre-exponential factor of 1013 s−1 that was chosen based on the entropy-free pre-factor kBT/h (Boltzmann constant kB) that appears in transition state theory124 which was used by various kinetic CO2R studies36,69.
The above summarizes the calculation of barriers within CatMAP under standard conditions. Under non-standard conditions, e.g. pH > 0, the chemical potential of the initial state decreases, reducing the driving force ΔiRΩ by 0.0592 eV ⋅ pH. Assuming that the chemical potential of the transition state cannot be tuned by the pH125, this means that the activation energy changes by the same amount:
| 19 |
Rationalization of kinetic barriers and model
In principle, kinetic parameters can be estimated e.g. via grand-canonical DFT126,127 or extrapolation methods128,129 using e.g. the nudged elastic band (NEB) method for localizing transition states. In practice, however, these simulations rely on a frozen electrolyte structure model from which activation energies on the 0K potential energy landscape are calculated. This generally neglects the dynamic nature of the electric double layer which can significantly contribute to the kinetic barrier. In addition, the use of hybrid implicit-explicit solvation methods in the grand-canonical schemes might lead to problems in capacitances and strong dependence on the interfacial water structure43. Due to these reasons, we decided instead to follow a semi-empirical approach to derive the kinetic parameters which we describe as follows.
In our previous work on CO2 reduction at the Au(211) facet, we have found from constant potential NEB calculations that the transition state of the CO2 adsorption and CO2 to *COOH steps lies very close to the final state for all potentials30. Following Eq. (12), this means that βi ≈ 1 for these two steps. In addition, we found in the same work that the difference between activation energy and reaction energy, i.e. at all voltages, so that . For simplicity, we assumed that these conclusions hold also for all other considered metal surfaces, an approximation that has to be verified by future simulations. In line with our settings, a recent work has suggested that the transition state of the *CO2 to *COOH step lies close to *COOH with no additional kinetic barrier, but proposed an additional barrier for CO2 adsorption which we did not consider here130.
For the *COOH to *CO step, it is expected that the transition state is in the center of the initial and final states, due to the Tafel slope from previous experiments being 42 mV/dec. A value of 40 mV/dec is expected if the 2nd PCET is rate-limiting and 40. This corresponds to a value of βi = 0.40 for Au(211) derived from Eq. (18) and the charging parameters that we calculated. For other metals, there can be significant changes in the Tafel slope, since high coverages of CO are expected on the strongly CO-adsorbing metals, which will change the kinetic equation that determines the Tafel slope. The charge-transfer coefficient is in principle more likely to be similar, but still, significant changes could occur. For simplicity in this work, we, however, ignored such variations in line with other previous works36. With the availability of more accurate means for electrochemical barriers, these assumptions should be tested again in future works.
The remaining free parameter is the activation energy at the reversible potential of the *COOH to *CO step. According to our previous collaborative experimental-theoretical work, this step becomes rate-limiting on Au at pH 6.8 at potentials larger than −0.7 V vs. SHE. At all potentials more negative than that CO2 adsorption is rate-limiting. We thus decided to tune this parameter to match the voltage at which the rate-limiting step changes with our model and by that obtained a value of eV. Choosing this value gives exactly the correct rate-limiting step transition voltage. See also Supplementary Materials Fig. S6 for the effect of this barrier on the rate-limiting step regions. Again, we assume that this value is constant across all metals again following the previous works36. This value might be at the low end of the error bar, since the *CO2 to *COOH step starts playing a role with this setting for the Au surfaces (cf. Supplementary Materials Fig. S10) which has not been observed experimentally30. With a slightly higher value, the importance of this step reduces (cf. Supplementary Materials Fig. S6). Also recent theoretical studies suggest a bit higher barrier130.
Lastly, the activation barrier for CO desorption was set to zero, following previous studies which generally found small kinetic barriers on Ru and Pt131,132.
Pressures
All micro-kinetic models were evaluated for a CO2 pressure of 1 bar and CO pressure of 0. Further, we set the H2O pressure to the vapor pressure in equilibrium with the liquid phase Pvap = 3.5 kPa, roughly the experimental value at room temperature133 to convert the gas phase chemical potential into a liquid phase chemical potential 30.
Supplementary information
Description of additional supplementary files
Acknowledgements
S.R. acknowledges the financial support provided by the National Research Foundation of Korea (Grant No. NRF-2021R1C1C1008776) and the support of the National Supercomputing Center for supercomputing resources, including technical support (No. KSC-2021-CRE-0410). Finally, as special thanks goes to Prof. Hyungjun Kim at the Chemistry department of KAIST for insightful discussions and support. Additionally, financial support from “The Institute of Basic Science, College of Science, Korea University” is acknowledged.
Source data
Author contributions
S.R. conceived the research plan, carried out all calculations and wrote the manuscript.
Peer review
Peer review information
Nature Communications thanks Ryosuke Jinnouchi and the other anonymous, reviewer(s) for their contribution to the peer review of this work
Data availability
All other data is available on request from the author. Source data are provided with this paper.
Competing interests
The author declares no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-37929-4.
References
- 1.Reuter K. Ab initio thermodynamics and First-Principles microkinetics for surface catalysis. Catal. Lett. 2016;146:541–563. doi: 10.1007/s10562-015-1684-3. [DOI] [Google Scholar]
- 2.Nørskov JK, Bligaard T, Kleis J. Chemistry. rate control and reaction engineering. Science. 2009;324:1655–1656. doi: 10.1126/science.1174885. [DOI] [PubMed] [Google Scholar]
- 3.Avanesian T, Christopher P. Scaled degree of rate control: identifying elementary steps that control differences in performance of transition-metal catalysts. Acs Catalysis. 2016;6:5268–5272. doi: 10.1021/acscatal.6b01547. [DOI] [Google Scholar]
- 4.Abild-Pedersen F, et al. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys. Rev. Lett. 2007;99:016105. doi: 10.1103/PhysRevLett.99.016105. [DOI] [PubMed] [Google Scholar]
- 5.Evans MG, Polanyi M. Further considerations on the thermodynamics of chemical equilibria and reaction rates. T. Faraday. Soc. 1936;32:1333. doi: 10.1039/tf9363201333. [DOI] [Google Scholar]
- 6.Brønsted JN, Pedersen KJ. Stöchiometrie und verwandtschaftslehre. Z. Phys. Chem. 1924;108:185–235. [Google Scholar]
- 7.Wang S, et al. Universal Brønsted-Evans-Polanyi relations for C–C, C–O, C–N, N–O, N–N, and O–O dissociation reactions. Catal. Letters. 2011;141:370–373. doi: 10.1007/s10562-010-0477-y. [DOI] [Google Scholar]
- 8.Cheng J, et al. Brønsted-Evans-Polanyi relation of multistep reactions and volcano curve in heterogeneous catalysis. J. Phys. Chem. C. 2008;112:1308–1311. doi: 10.1021/jp711191j. [DOI] [Google Scholar]
- 9.Sabatier, P. La catalyse en chimie organique, librairie polytechnique Ch. Beranger, Editeur. Paris, page 180, (1913).
- 10.Che M. Nobel prize in chemistry 1912 to sabatier: organic chemistry or catalysis? Catal. Today. 2013;218-219:162–171. doi: 10.1016/j.cattod.2013.07.006. [DOI] [Google Scholar]
- 11.Medford AJ, et al. From the sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015;328:36–42. doi: 10.1016/j.jcat.2014.12.033. [DOI] [Google Scholar]
- 12.Mamun O, Winther KT, Boes JR, Bligaard T. High-throughput calculations of catalytic properties of bimetallic alloy surfaces. Sci. Data. 2019;6:76. doi: 10.1038/s41597-019-0080-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoyt RA, et al. Machine learning prediction of H adsorption energies on ag alloys. J. Chem. Inf. Model. 2019;59:1357–1365. doi: 10.1021/acs.jcim.8b00657. [DOI] [PubMed] [Google Scholar]
- 14.Nayak S, Bhattacharjee S, Choi J-H, Lee SC. Machine learning and scaling laws for prediction of accurate adsorption energy. J. Phys. Chem. A. 2020;124:247–254. doi: 10.1021/acs.jpca.9b07569. [DOI] [PubMed] [Google Scholar]
- 15.Li Z, Ma X, Xin H. Feature engineering of machine-learning chemisorption models for catalyst design. Catal. Today. 2017;280:232–238. doi: 10.1016/j.cattod.2016.04.013. [DOI] [Google Scholar]
- 16.Nørskov, J. K., Scheffler, M. & Toulhoat, H. Density functional theory in surface science and heterogeneous catalysis. MRS Bull. 31, 669–674 (2006).
- 17.Khorshidi A, Violet J, Hashemi J, Peterson AA. How strain can break the scaling relations of catalysis. Nat. Catal. 2018;1:263–268. doi: 10.1038/s41929-018-0054-0. [DOI] [Google Scholar]
- 18.Huang Z-F, et al. Strategies to break the scaling relation toward enhanced oxygen electrocatalysis. Matter. 2019;1:1494–1518. doi: 10.1016/j.matt.2019.09.011. [DOI] [Google Scholar]
- 19.Nørskov JK, et al. Origin of the overpotential for oxygen reduction at a Fuel-Cell cathode. J. Phys. Chem. B. 2004;108:17886–17892. doi: 10.1021/jp047349j. [DOI] [Google Scholar]
- 20.Rossmeisl J, Logadottir A, Nørskov JK. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005;319:178–184. doi: 10.1016/j.chemphys.2005.05.038. [DOI] [Google Scholar]
- 21.Chun H-J, Zeng Z, Greeley J. Direct demonstration of unified Brønsted-Evans-Polanyi relationships for proton-coupled electron transfer reactions on transition metal surfaces. J. Electrochem. Soc. 2020;167:166516. doi: 10.1149/1945-7111/abd3bc. [DOI] [Google Scholar]
- 22.Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science355, eaad4998 (2017). [DOI] [PubMed]
- 23.Back S, Tran K, Ulissi ZW. Toward a design of active oxygen evolution catalysts: insights from automated density functional theory calculations and machine learning. ACS Catal. 2019;9:7651–7659. doi: 10.1021/acscatal.9b02416. [DOI] [Google Scholar]
- 24.Zhong M, et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature. 2020;581:178–183. doi: 10.1038/s41586-020-2242-8. [DOI] [PubMed] [Google Scholar]
- 25.Liu, J. et al. Toward excellence of electrocatalyst design by emerging descriptor-oriented machine learning Adv. Funct. Mater. 32, 2110748 (2022).
- 26.Ringe, S., Hörmann, N. G., Oberhofer, H. & Reuter, K. Implicit solvation methods for catalysis at electrified interfaces arXiv preprint arXiv:2108.02461, (2021). [DOI] [PMC free article] [PubMed]
- 27.Sebastián-Pascual, P., Shao-Horn, Y. & Escudero-Escribano, M. Toward understanding the role of the electric double layer structure and electrolyte effects on well-defined interfaces for electrocatalysis. Curr. Opin. Electrochem.32, 100918 (2021).
- 28.Sandberg RB, Montoya JH, Chan K, Nørskov JK. CO-CO coupling on cu facets: coverage, strain and field effects. Surf. Sci. 2016;654:56–62. doi: 10.1016/j.susc.2016.08.006. [DOI] [Google Scholar]
- 29.Ringe S, et al. Understanding cation effects in electrochemical CO2 reduction. Energy Environ. Sci. 2019;12:3001–3014. doi: 10.1039/C9EE01341E. [DOI] [Google Scholar]
- 30.Ringe S, et al. Double layer charging driven carbon dioxide adsorption limits the rate of electrochemical carbon dioxide reduction on gold. Nat. Commun. 2020;11:33. doi: 10.1038/s41467-019-13777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen LD, Urushihara M, Chan K, Nørskov JK. Electric field effects in electrochemical CO2 reduction. ACS Catal. 2016;6:7133–7139. doi: 10.1021/acscatal.6b02299. [DOI] [Google Scholar]
- 32.Resasco J, et al. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J. Am. Chem. Soc. 2017;139:11277–11287. doi: 10.1021/jacs.7b06765. [DOI] [PubMed] [Google Scholar]
- 33.Clark, E. L. et al. Influence of atomic surface structure on the activity of ag for the electrochemical reduction of CO2 to CO. ACS Catal. 9, 4006–4014 (2019).
- 34.Kim DH, et al. Selective electrochemical reduction of nitric oxide to hydroxylamine by atomically dispersed iron catalyst. Nat. Commun. 2021;12:1–11. doi: 10.1038/s41467-021-22147-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee M-Y, Ringe S, Kim H, Kang S, Kwon Y. Electric field mediated selectivity switching of electrochemical CO2 reduction from formate to CO on carbon supported sn. ACS Energy Lett. 2020;5:2987–2994. doi: 10.1021/acsenergylett.0c01387. [DOI] [Google Scholar]
- 36.Hansen HA, Varley JB, Peterson AA, Nørskov JK. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 2013;4:388–392. doi: 10.1021/jz3021155. [DOI] [PubMed] [Google Scholar]
- 37.Peng H, et al. The role of atomic carbon in directing electrochemical CO(2) reduction to multicarbon products. Energy Environ. Sci. 2021;14:473–482. doi: 10.1039/D0EE02826F. [DOI] [Google Scholar]
- 38.Tang MT, Peng H, Lamoureux PS, Bajdich M, Abild-Pedersen F. From electricity to fuels: descriptors for C1 selectivity in electrochemical CO2 reduction. Appl. Catal. B. 2020;279:119384. doi: 10.1016/j.apcatb.2020.119384. [DOI] [Google Scholar]
- 39.Jouny M, Luc W, Jiao F. General techno-economic analysis of CO2 electrolysis systems. Ind. Eng. Chem. Res. 2018;57:2165–2177. doi: 10.1021/acs.iecr.7b03514. [DOI] [Google Scholar]
- 40.Dunwell M, Luc W, Yan Y, Jiao F, Xu B. Understanding surface-mediated electrochemical reactions: CO2 reduction and beyond. ACS Catal. 2018;8:8121–8129. doi: 10.1021/acscatal.8b02181. [DOI] [Google Scholar]
- 41.Deng W, Zhang P, Seger B, Gong J. Unraveling the rate-limiting step of two-electron transfer electrochemical reduction of carbon dioxide. Nat. Commun. 2022;13:803. doi: 10.1038/s41467-022-28436-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nattino F, Truscott M, Marzari N, Andreussi O. Continuum models of the electrochemical diffuse layer in electronic-structure calculations. J. Chem. Phys. 2019;150:041722. doi: 10.1063/1.5054588. [DOI] [PubMed] [Google Scholar]
- 43.Gauthier JA, et al. Unified approach to implicit and explicit solvent simulations of electrochemical reaction energetics. J. Chem. Theory Comput. 2019;15:6895–6906. doi: 10.1021/acs.jctc.9b00717. [DOI] [PubMed] [Google Scholar]
- 44.Hörmann NG, Marzari N, Reuter K. Electrosorption at metal surfaces from first principles. NPJ Comput. Mater. 2020;6:1–10. doi: 10.1038/s41524-020-00394-4. [DOI] [Google Scholar]
- 45.Gauthier JA, Dickens CF, Ringe S, Chan K. Practical considerations for continuum models applied to surface electrochemistry. Chem. Phys. Chem. 2019;20:3074–3080. doi: 10.1002/cphc.201900536. [DOI] [PubMed] [Google Scholar]
- 46.Beinlich SD, Hörmann NG, Reuter K. Field effects at protruding defect sites in electrocatalysis at metal electrodes? ACS Catal. 2022;12:6143–6148. doi: 10.1021/acscatal.2c00997. [DOI] [Google Scholar]
- 47.Valette G. Double layer on silver single-crystal electrodes in contact with electrolytes having anions which present a slight specific adsorption: Part i. the (110) face. J. Electroanal. Chem. Interfacial Electrochem. 1981;122:285–297. doi: 10.1016/S0022-0728(81)80159-3. [DOI] [Google Scholar]
- 48.Valette G. Double layer on silver single crystal electrodes in contact with electrolytes having anions which are slightly specifically adsorbed: Part II. the (100) face. J. Electroanal. Chem. Interfacial Electrochem. 1982;138:37–54. doi: 10.1016/0022-0728(82)87126-X. [DOI] [Google Scholar]
- 49.Valette G. Double layer on silver single crystal electrodes in contact with electrolytes having anions which are slightly specifically adsorbed: Part III. the (111) face. J. Electroanal. Chem. Interfacial Electrochem. 1989;269:191–203. doi: 10.1016/0022-0728(89)80112-3. [DOI] [Google Scholar]
- 50.Trasatti S. Work function, electronegativity, and electrochemical behaviour of metals: II. potentials of zero charge and “electrochemical” work functions. J. Electroanal. Chem. Interfacial Electrochem. 1971;33:358. doi: 10.1016/S0022-0728(71)80123-7. [DOI] [Google Scholar]
- 51.Wellendorff J, et al. A benchmark database for adsorption bond energies to transition metal surfaces and comparison to selected DFT functionals. Surf. Sci. 2015;640:36–44. doi: 10.1016/j.susc.2015.03.023. [DOI] [Google Scholar]
- 52.Shi C, Hansen HA, Lausche AC, Nørskov JK. Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 2014;16:4720–4727. doi: 10.1039/c3cp54822h. [DOI] [PubMed] [Google Scholar]
- 53.Letchworth-Weaver K, Arias TA. Joint density functional theory of the electrode-electrolyte interface: Application to fixed electrode potentials, interfacial capacitances, and potentials of zero charge. Phys. Rev. B. 2012;86:075140. doi: 10.1103/PhysRevB.86.075140. [DOI] [Google Scholar]
- 54.Liu T, et al. Improving interfacial electron transfer via tuning work function of electrodes for electrocatalysis: from theory to experiment. J. Phys. Chem. C. 2019;123:28319–28326. doi: 10.1021/acs.jpcc.9b09875. [DOI] [Google Scholar]
- 55.Zeradjanin AR, et al. Balanced work function as a driver for facile hydrogen evolution reaction - comprehension and experimental assessment of interfacial catalytic descriptor. Phys. Chem. Chem. Phys. 2017;19:17019–17027. doi: 10.1039/C7CP03081A. [DOI] [PubMed] [Google Scholar]
- 56.Ledezma-Yanez I, et al. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat. Energy. 2017;2:17031. doi: 10.1038/nenergy.2017.31. [DOI] [Google Scholar]
- 57.Katayama, Y. et al. An in situ Surface-Enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-Bound and O-Bound reaction intermediates J. Phys. Chem. C123, 5951–5963 (2018).
- 58.Ma M, Trześniewski BJ, Xie J, Smith WA. Selective and efficient reduction of carbon dioxide to carbon monoxide on Oxide-Derived nanostructured silver electrocatalysts. Angew. Chem. Int. Ed. 2016;55:9748–9752. doi: 10.1002/anie.201604654. [DOI] [PubMed] [Google Scholar]
- 59.Mezzavilla, S., Horch, S., Stephens, I. E. L., Seger, B. & Chorkendorff, I. Structure sensitivity in the electrocatalytic reduction of CO2 with gold catalysts. Angew. Chem. Int. Ed Engl. 58, 3774–3778 (2019). [DOI] [PubMed]
- 60.Hoshi N, Kato M, Hori Y. Electrochemical reduction of CO2 on single crystal electrodes of silver ag(111), ag(100) and ag(110) J. Electroanal. Chem. 1997;440:283–286. doi: 10.1016/S0022-0728(97)00447-6. [DOI] [Google Scholar]
- 61.Bellini M, et al. A Gold–Palladium nanoparticle alloy catalyst for CO production from CO 2 electroreduction. Energy Technol. 2019;7:1800859. doi: 10.1002/ente.201800859. [DOI] [Google Scholar]
- 62.O’Keeke, T. J. Pourbaix diagrams. In K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer, S. Mahajan, & P. Veyssière, editors, Encyclopedia of Materials. Sci. Technol. 7774–7781. Elsevier, Oxford (2001).
- 63.Ohkawa K, Hashimoto K, Fujishima A. Part 1 . the effect of hydrogen absorption on the electrochemical behavior on palladium electrodes. J. Electroanal. Chem. 1993;345:445–456. doi: 10.1016/0022-0728(93)80495-4. [DOI] [Google Scholar]
- 64.Noda H, et al. Electrochemical reduction of carbon dioxide at various metal electrodes in aqueous potassium hydrogen carbonate solution. Bull. Chem. Soc. Jpn. 1990;63:2459–2462. doi: 10.1246/bcsj.63.2459. [DOI] [Google Scholar]
- 65.Guo R-H, Liu C-F, Wei T-C, Hu C-C. Electrochemical behavior of CO2 reduction on palladium nanoparticles: Dependence of adsorbed CO on electrode potential. Electrochem. Commun. 2017;80:24–28. doi: 10.1016/j.elecom.2017.05.005. [DOI] [Google Scholar]
- 66.Song H, Tan YC, Kim B, Ringe S, Oh J. Tunable product selectivity in electrochemical CO2 reduction on Well-Mixed Ni-Cu alloys. ACS Appl. Mater. Interfaces. 2021;13:55272–55280. doi: 10.1021/acsami.1c19224. [DOI] [PubMed] [Google Scholar]
- 67.Huang JE, et al. CO2 electrolysis to multicarbon products in strong acid. Science. 2021;372:1074–1078. doi: 10.1126/science.abg6582. [DOI] [PubMed] [Google Scholar]
- 68.Bondue CJ, Graf M, Goyal A, Koper MTM. Suppression of hydrogen evolution in acidic electrolytes by electrochemical CO2 reduction. J. Am. Chem. Soc. 2021;143:279–285. doi: 10.1021/jacs.0c10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, et al. ph effects on the electrochemical reduction of CO(2) towards C2 products on stepped copper. Nat. Commun. 2019;10:32. doi: 10.1038/s41467-018-07970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019). [DOI] [PubMed]
- 71.Hori Y, Takahashi I, Koga O, Hoshi N. Selective formation of C2 compounds from electrochemical reduction of CO2 at a series of copper single crystal electrodes. J. Phys. Chem. B. 2002;106:15–17. doi: 10.1021/jp013478d. [DOI] [Google Scholar]
- 72.Yin Z, et al. Highly selective palladium-copper bimetallic electrocatalysts for the electrochemical reduction of CO2 to CO. Nano Energy. 2016;27:35–43. doi: 10.1016/j.nanoen.2016.06.035. [DOI] [Google Scholar]
- 73.Schumann, J. et al. Periodic trends in adsorption energies around Single-Atom alloy active sites. J. Phys. Chem. Lett. 12, 10060–10067 (2021). [DOI] [PubMed]
- 74.Groß A. Tailoring the reactivity of bimetallic overlayer and surface alloy systems. J. Phys. Condens. Matter. 2009;21:084205. doi: 10.1088/0953-8984/21/8/084205. [DOI] [PubMed] [Google Scholar]
- 75.George EP, Raabe D, Ritchie RO. High-entropy alloys. Nat. Rev. Mater. 2019;4:515–534. doi: 10.1038/s41578-019-0121-4. [DOI] [Google Scholar]
- 76.Furukawa S, Komatsu T, Shimizu K-I. Catalyst design concept based on a variety of alloy materials: a personal account and relevant studies. J. Mater. Chem. A Mater. Energy Sustain. 2020;8:15620–15645. doi: 10.1039/D0TA03733H. [DOI] [Google Scholar]
- 77.Christophe J, Doneux T, Buess-Herman C. Electroreduction of carbon dioxide on copper-based electrodes: activity of copper single crystals and Copper–Gold alloys. Electrocatalysis. 2012;3:139–146. doi: 10.1007/s12678-012-0095-0. [DOI] [Google Scholar]
- 78.Bagger A, Christensen O, Ivaništšev V, Rossmeisl J. Catalytic CO2/CO reduction: gas, aqueous, and aprotic phases. ACS Catal. 2022;12:2561–2568. doi: 10.1021/acscatal.1c05358. [DOI] [Google Scholar]
- 79.Peng H-J, Tang MT, Halldin Stenlid J, Liu X, Abild-Pedersen F. Trends in oxygenate/hydrocarbon selectivity for electrochemical CO(2) reduction to C2 products. Nat. Commun. 2022;13:1399. doi: 10.1038/s41467-022-29140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu X, et al. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 2017;8:15438. doi: 10.1038/ncomms15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Varela AS, Kroschel M, Reier T, Strasser P. Controlling the selectivity of CO2 electroreduction on copper: the effect of the electrolyte concentration and the importance of the local ph. Catal. Today. 2016;260:8–13. doi: 10.1016/j.cattod.2015.06.009. [DOI] [Google Scholar]
- 82.Cheng T, Xiao H, Goddard WA. 3rd Full atomistic reaction mechanism with kinetics for CO reduction on cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc. Natl. Acad. Sci. USA. 2017;114:1795–1800. doi: 10.1073/pnas.1612106114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang Y, Handoko AD, Hirunsit P, Yeo BS. Electrochemical reduction of CO2 using copper Single-Crystal surfaces: Effects of CO* coverage on the selective formation of ethylene. ACS Catal. 2017;7:1749–1756. doi: 10.1021/acscatal.6b03147. [DOI] [Google Scholar]
- 84.Tsang CF, et al. Potential-Dependent adsorption of CO and its Low-Overpotential reduction to CH3CH2OH on cu(511) surface reconstructed from cu(pc): Operando studies by seriatim STM-EQCN-DEMS. J. Electrochem. Soc. 2018;165:J3350–J3354. doi: 10.1149/2.0451815jes. [DOI] [Google Scholar]
- 85.Gauthier, J. A., Lin, Z., Head-Gordon, M. and Bell, A. T. Pathways for the formation of c2+ products under alkaline conditions during the electrochemical reduction of CO2.ACS Energy Lett. 7, 1679–1686 (2022).
- 86.Kastlunger G, et al. Using ph dependence to understand mechanisms in electrochemical CO reduction. ACS Catal. 2022;12:4344–4357. doi: 10.1021/acscatal.1c05520. [DOI] [Google Scholar]
- 87.Deng, W. et al. Unraveling the rate-determine step of c2+ products during electrochemical co reduction. 10.21203/rs.3.rs-2376669/v1 (2023).
- 88.Bagger A, Ju W, Varela AS, Strasser P, Rossmeisl J. Electrochemical CO2 reduction: a classification problem. Chemphyschem. 2017;18:3266–3273. doi: 10.1002/cphc.201700736. [DOI] [PubMed] [Google Scholar]
- 89.Cheng T, Xiao H, Goddard WA. Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the cu(100) surface at 298K from quantum mechanics free energy calculations with explicit water. J. Am. Chem. Soc. 2016;138:13802–13805. doi: 10.1021/jacs.6b08534. [DOI] [PubMed] [Google Scholar]
- 90.Hori Y, Takahashi I, Koga O, Hoshi N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A Chem. 2003;199(May):39–47. doi: 10.1016/S1381-1169(03)00016-5. [DOI] [Google Scholar]
- 91.Nishimura, Y. F. et al. Guiding the catalytic properties of copper for electrochemical CO2 reduction by metal atom decoration. ACS Appl. Mater. Interfaces. 13, 52044–52054 (2021). [DOI] [PubMed]
- 92.Lim CYJ, et al. Surface charge as activity descriptors for electrochemical CO2 reduction to multi-carbon products on organic-functionalised cu. Nat. Commun. 2023;14:335. doi: 10.1038/s41467-023-35912-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Gordon E, Ren H. Mapping the potential of zero charge and electrocatalytic activity of metal-electrolyte interface via a grain-by-grain approach. Anal. Chem. 2020;92:2859–2865. doi: 10.1021/acs.analchem.9b05502. [DOI] [PubMed] [Google Scholar]
- 94.Dubouis N, Grimaud A. The hydrogen evolution reaction: from material to interfacial descriptors. Chem. Sci. 2019;10:9165–9181. doi: 10.1039/C9SC03831K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trasatti S. Work function, electronegativity, and electrochemical behaviour of metals: III. electrolytic hydrogen evolution in acid solutions. J. Electroanal. Chem. Interfacial Electrochem. 1972;39:163–184. doi: 10.1016/S0022-0728(72)80485-6. [DOI] [Google Scholar]
- 96.Rossmeisl J, Chan K, Skúlason E, Björketun ME, Tripkovic V. On the pH dependence of electrochemical proton transfer barriers. Catal. Today. 2016;262:36–40. doi: 10.1016/j.cattod.2015.08.016. [DOI] [Google Scholar]
- 97.Wu Y, et al. A Two-Dimensional MoS2 catalysis transistor by solid-state ion gating manipulation and adjustment (SIGMA) Nano Lett. 2019;19:7293–7300. doi: 10.1021/acs.nanolett.9b02888. [DOI] [PubMed] [Google Scholar]
- 98.Gossenberger F, Roman T, Forster-Tonigold K, Groß A. Change of the work function of platinum electrodes induced by halide adsorption. Beilstein J. Nanotechnol. 2014;5:152–161. doi: 10.3762/bjnano.5.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang Y, et al. Tuning the work function of stepped metal surfaces by adsorption of organic molecules. J. Phys. Condens. Matter. 2017;29:204001. doi: 10.1088/1361-648X/aa693e. [DOI] [PubMed] [Google Scholar]
- 100.Tran K, Ulissi ZW. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 2018;1:696–703. doi: 10.1038/s41929-018-0142-1. [DOI] [Google Scholar]
- 101.Hashimoto W, Tsuji Y, Yoshizawa K. Optimization of work function via bayesian machine learning combined with first-principles calculation. J. Phys. Chem. C. 2020;124:9958–9970. doi: 10.1021/acs.jpcc.0c01106. [DOI] [Google Scholar]
- 102.Sasikumar K, Chan H, Narayanan B, Sankaranarayanan SKRS. Machine learning applied to a variable charge atomistic model for Cu/Hf binary alloy oxide heterostructures. Chem. Mater. 2019;31:3089–3102. doi: 10.1021/acs.chemmater.8b03969. [DOI] [Google Scholar]
- 103.Ojha K, Doblhoff-Dier K, Koper MTM. Double-layer structure of the pt(111)-aqueous electrolyte interface. Proc. Natl. Acad. Sci. USA. 2022;119:e2116016119. doi: 10.1073/pnas.2116016119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Monteiro MCO, Goyal A, Moerland P, Koper MTM. Understanding cation trends for hydrogen evolution on platinum and gold electrodes in alkaline media. ACS Catal. 2021;11:14328–14335. doi: 10.1021/acscatal.1c04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goyal A, Koper MTM. The interrelated effect of cations and electrolyte ph on the hydrogen evolution reaction on gold electrodes in alkaline media. Angew. Chem. Weinheim Bergstr. Ger. 2021;133:13564–13574. doi: 10.1002/ange.202102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monteiro MCO, Dattila F, López N, Koper MTM. The role of cation acidity on the competition between hydrogen evolution and CO2 reduction on gold electrodes. J. Am. Chem. Soc. 2022;144:1589–1602. doi: 10.1021/jacs.1c10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shin S-J, et al. A unifying mechanism for cation effect modulating C1 and C2 productions from CO2 electroreduction. Nat. Commun. 2022;13:5482. doi: 10.1038/s41467-022-33199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Monteiro MCO, et al. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 2021;4:654–662. doi: 10.1038/s41929-021-00655-5. [DOI] [Google Scholar]
- 109.Marcandalli, G., Monteiro, M. C., Goyal, A. & Koper, M. T. Electrolyte effects on CO2 electrochemical reduction to CO.Acc. Chem. Res. 55, 1900–1911 (2022). [DOI] [PMC free article] [PubMed]
- 110.Kim, M. K. et al. Design of less than 1 nm scale spaces on SnO 2 nanoparticles for high-performance electrochemical CO 2 reduction. Adv. Funct. Mater. 32, 2107349 (2021).
- 111.Gauthier JA, Chen LD, Bajdich M, Chan K. Implications of the fractional charge of hydroxide at the electrochemical interface. Phys. Chem. Chem. Phys. 2020;22:6964–6969. doi: 10.1039/C9CP05952K. [DOI] [PubMed] [Google Scholar]
- 112.Hörmann NG, Andreussi O, Marzari N. Grand canonical simulations of electrochemical interfaces in implicit solvation models. J. Chem. Phys. 2019;150:041730. doi: 10.1063/1.5054580. [DOI] [PubMed] [Google Scholar]
- 113.Giannozzi P, et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009;21:395502. doi: 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- 114.Hjorth Larsen A, et al. The atomic simulation environment—a python library for working with atoms. J. Phys. Condens. Matter. 2017;29:273002. doi: 10.1088/1361-648X/aa680e. [DOI] [PubMed] [Google Scholar]
- 115.Studt F, Abild-Pedersen F, Varley JB, Nørskov JK. CO and CO2 hydrogenation to methanol calculated using the BEEF-vdW functional. Catal. Lett. 2013;143:71–73. doi: 10.1007/s10562-012-0947-5. [DOI] [Google Scholar]
- 116.Monkhorst HJ, Pack JD. Special points for brillouin-zone integrations. Phys. Rev. B Condens. Matter. 1976;13:5188–5192. doi: 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- 117.Boes, J. R., Mamun, O., Winther, K. & Bligaard, T. Graph theory approach to High-Throughput surface adsorption structure generation J. Phys. Chem. A123, 2281–2285 (2019). [DOI] [PubMed]
- 118.Kirklin S, et al. The open quantum materials database (oqmd): assessing the accuracy of dft formation energies. NPJ Computational Materials. 2015;1:1–15. doi: 10.1038/npjcompumats.2015.10. [DOI] [Google Scholar]
- 119.Monkhorst HJ, Pack JD. Special points for brillouin-zone integrations. Phys. Rev. B Condens. Matter. 1976;13:5188–5192. doi: 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- 120.Andreussi O, Dabo I, Marzari N. Revised self-consistent continuum solvation in electronic-structure calculations. J. Chem. Phys. 2012;136:064102. doi: 10.1063/1.3676407. [DOI] [PubMed] [Google Scholar]
- 121.Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci.3, 1311–1315 (2010).
- 122.Cramer CJ, Truhlar DG. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem. Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 123.Bazant MZ. Theory of chemical kinetics and charge transfer based on nonequilibrium thermodynamics. Acc. Chem. Res. 2013;46:1144–1160. doi: 10.1021/ar300145c. [DOI] [PubMed] [Google Scholar]
- 124.Nørskov, J. K., Studt, F., Abild-Pedersen, F. & Bligaard, T. Fundamental Concepts in Heterogeneous Catalysis. p. 47 (Wiley, Harvard, 2015).
- 125.Wang L, et al. Electrochemical carbon monoxide reduction on polycrystalline copper: effects of potential, pressure, and ph on selectivity toward multicarbon and oxygenated products. ACS Catal. 2018;8:7445–7454. doi: 10.1021/acscatal.8b01200. [DOI] [Google Scholar]
- 126.Gao, G. & Wang, L.-W. A potential and ph inclusive microkinetic model for hydrogen reactions on pt surface. Chem. Catal.1, 1331–1345 (2021).
- 127.Choi C, Gu GH, Noh J, Park HS, Jung Y. Understanding potential-dependent competition between electrocatalytic dinitrogen and proton reduction reactions. Nat. Commun. 2021;12:4353. doi: 10.1038/s41467-021-24539-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chan K, Nørskov JK. Electrochemical barriers made simple. J. Phys. Chem. Lett. 2015;6:2663–2668. doi: 10.1021/acs.jpclett.5b01043. [DOI] [PubMed] [Google Scholar]
- 129.Chan K, Nørskov JK. Potential dependence of electrochemical barriers from ab initio calculations. J. Phys. Chem. Lett. 2016;7:1686–1690. doi: 10.1021/acs.jpclett.6b00382. [DOI] [PubMed] [Google Scholar]
- 130.Qin X, Vegge T, Hansen HA. Cation-coordinated inner-sphere CO2 electroreduction at au-water interfaces. J. Am. Chem. Soc. 2023;145:1897–1905. doi: 10.1021/jacs.2c11643. [DOI] [PubMed] [Google Scholar]
- 131.Stampfl C, Scheffler M. Energy barriers and chemical properties in the coadsorption of carbon monoxide and oxygen on ru(0001) Phys. Rev. B Condens. Matter. 2002;65:155417. doi: 10.1103/PhysRevB.65.155417. [DOI] [Google Scholar]
- 132.Guo C, Wang Z, Wang D, Wang H-F, Hu P. First-Principles determination of CO adsorption and desorption on pt(111) in the free energy landscape. J. Phys. Chem. C. 2018;122:21478–21483. doi: 10.1021/acs.jpcc.8b06782. [DOI] [Google Scholar]
- 133.Lide, D. R. CRC Handbook of Chemistry and Physics, 85th Edition CRC Press, June (2004).
- 134.Hori, Y., Wakebe, H., Tsukamoto, T. & Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media (1994).
- 135.Kuhl KP, et al. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014;136:14107–14113. doi: 10.1021/ja505791r. [DOI] [PubMed] [Google Scholar]
- 136.Yang Z, Oropeza FE, Zhang KHL. P-block metal-based (sn, in, bi, pb) electrocatalysts for selective reduction of CO2 to formate. APL Mater. 2020;8:060901. doi: 10.1063/5.0004194. [DOI] [Google Scholar]
- 137.Li J, et al. Two-Dimensional SnO2 nanosheets for efficient carbon dioxide electroreduction to formate. ACS Sustainable Chem. Eng. 2020;8:4975–4982. doi: 10.1021/acssuschemeng.0c01070. [DOI] [Google Scholar]
- 138.Hahn C, et al. Engineering cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons. Proc. Natl. Acad. Sci. USA. 2017;114:5918–5923. doi: 10.1073/pnas.1618935114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of additional supplementary files
Data Availability Statement
All other data is available on request from the author. Source data are provided with this paper.