

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease caused by more than sixty genes identified through classic linkage analysis and new sequencing methods. Yet no clear mechanism of onset, cure, or effective treatment is known. Popular discourse classifies the proteins encoded from ALS-related genes into four disrupted processes: proteostasis, mitochondrial function and ROS, nucleic acid regulation, and cytoskeletal dynamics. Surprisingly, the mechanisms detailing the contribution of the neuronal cytoskeletal in ALS are the least explored, despite involvement in these cell processes. Eight genes directly regulate properties of cytoskeleton function and are essential for the health and survival of motor neurons, including: TUBA4A, SPAST KIF5A, DCTN1, NF, PRPH, ALS2, and PFN1. Here we review the properties and studies exploring the contribution of each of these genes to ALS.
Keywords: Microtubules, Actin, Cytoskeleton, Amyotrophic lateral sclerosis (ALS)
1. Introduction: the cytoskeleton is a convergence point in ALS
Amyotrophic lateral sclerosis (ALS) is a rare and fatal disease (Lasiene and Yamanaka, 2011; Phatnani et al., 2013; Ragagnin et al., 2019). Although close to 95% of all ALS cases are sporadic, genetic studies have identified that mutations in SOD1, C9ORF72, FUS, and TDP43 genes may account for ~60% of all inherited cases, albeit with large variations in the disease penetrance and severity (Renton et al., 2014; Gregory et al., 2020; Masrori and Van Damme, 2020; Nguyen et al., 2018). ALS pathogenesis is characterized by the degeneration of motor neurons and disturbances to glia that further impair mechanisms of glia-motor neuron interaction and trophic support (Philips and Rothstein, 2014). Many different mechanisms are thought to explain disease onset including impairments to proteostasis, DNA repair, RNA metabolism, neuronal excitability, and intracellular transport. Notably, the neuronal cytoskeleton (e.g., actin filaments, intermediate filaments, and microtubules) is a convergence point required to execute each of these functions, and is clearly disrupted in mammalian cell models recapitulating ALS. SOD1 mutations gain affinity for actin and tubulin, which impairs neuronal growth cones and mitochondria fission (Osking et al., 2019; Muñoz-Lasso et al., 2020). C9orf72 acts as a roadblock to kinesin and dynein motor-based transport, and also disrupts actin filament disassembly (Fumagalli et al., 2021; Shiota et al., 2022; Webster et al., 2018; Sivadasan et al., 2016). Cytoplasmic inclusions of FUS prevent ribonucleotide cargos from reaching their normal locations by aberrantly sequestering motor proteins and influencing the post-translational states of microtubule tracks (Yasuda et al., 2017). Finally, the presence of pathological TDP-43 in the cytoplasm of motor neurons is correlated with diminished axon length, complexity, and mitochondrial transport (Herzog et al., 2017; Oberstadt et al., 2018a; Briese et al., 2020).
Disease-induced disturbances to axonal transport may be the most obvious connection between ALS and the cytoskeleton, because it conveniently explains several disparate phenotypes including: the aberrant localization of diverse proteins, lipids, and organelles; the accumulation of misfolded proteins or toxic aggregates from reduced clearance; and the decreased transmission of neuronal signals (Julien and Beaulieu, 2000; Castellanos-Montiel et al., 2020; Theunissen et al., 2021; Millecamps and Julien, 2013). All these scenarios may arise through deficits in functional tracks, motor proteins, cargo, or the adapter proteins used to connect cargos to the motors walking on the tracks (Fig. 1A) (Kapitein and Hoogenraad, 2015; Coles and Bradke, 2015; Tas et al., 2017; Saxton and Hollenbeck, 2012; Leterrier, 2021; Hammer and Wagner, 2013; Chua et al., 2012; Lewis et al., 2009). Normally, neuronal cargos are transported far distances (>1 m) along microtubules and move at different speeds and directions depending on the cargo and the linking motor (Nirschl et al., 2017). For perspective, the fastest kinesin motors take >4 days to traverse microtubules spanning the length of the longest motor neurons! To reach their final destinations, some ALS-relevant cargos are transferred from microtubules to actin filament tracks (Fig. 1B) (Coles and Bradke, 2015; Nirschl et al., 2017; Ally et al., 2009; Dogterom and Koenderink, 2019; Pimm and Henty-Ridilla, 2021). Microtubule or actin filament highways that become dilapidated, disassembled, or otherwise unpassable prevent neurons from transmitting cellular distress signals or receiving essential maintenance factors. Thus, even small impediments to or accumulated inefficiencies in this system explain an important component of symptomatic onset.
Fig. 1.
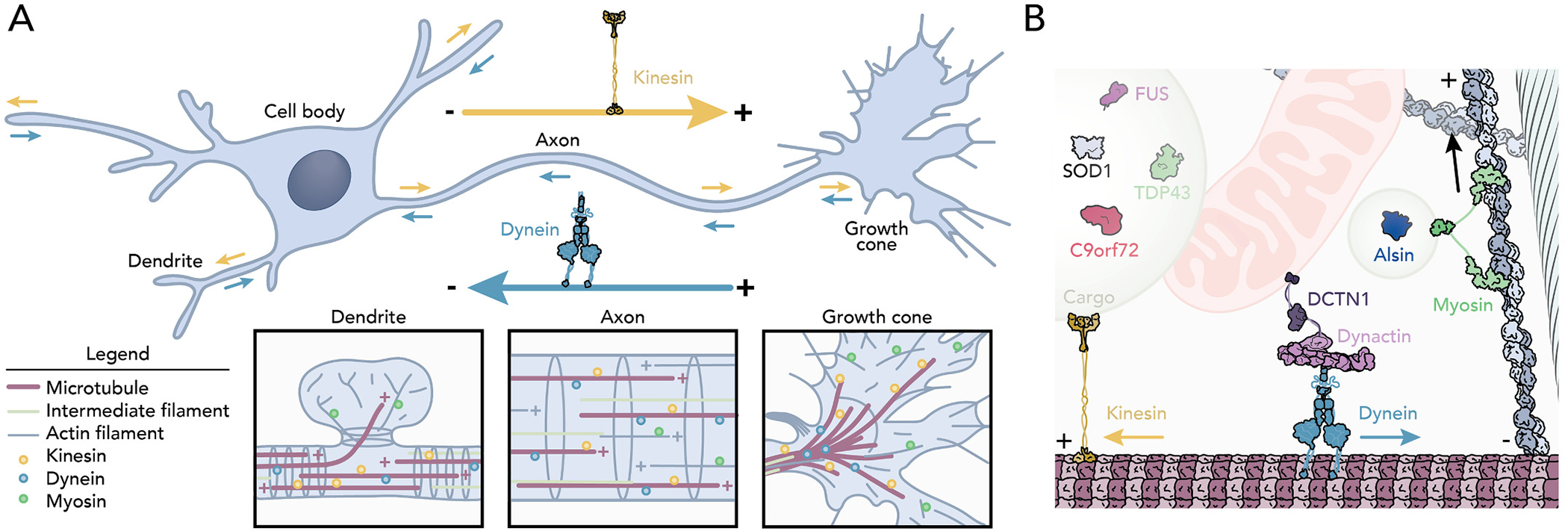
The neuronal cytoskeleton as tracks for intracellular transport. (A) Cartoon highlighting the functionally/structurally different features found in motor neurons including: a dendrite, cell body, axon, and growth cone. Kinesin motors (yellow) mediate anterograde transport. Dynein motors (teal) facilitate retrograde transport. Insets: localization of the transport components in (left) dendritic spines, in (middle) the axon, and in (right) growth cones. (B) Cytoskeleton-directed transport. Kinesin, dynein-dynactin complex, and myosin motors move vesicles, mitochondria, intermediate filaments, proteins, and many ALS-related proteins in cells.
Although more than sixty ALS-causative genes have been identified, only eight encode key cytoskeletal proteins, including: α-tubulin (TUBA4A), spastin (SPAST), kinesin (KIF5A), a subunit of the dynein-dynactin complex (DCTN1), neurofilament (NF), peripherin (PRPH), alsin (ALS2), and profilin (PFN1) (Cirulli et al., 2015; Wu et al., 2012; Liu et al., 2017; Smith et al., 2014; Zhang et al., 2019). Each is linked to transport mechanisms, is required for normal development, is present in cells besides those affected by ALS, and with the exception of ALS2, is inherited in a dominant manner (Table 1) (Millecamps and Julien, 2013; Yang et al., 2001; Topp et al., 2005). Pathogenic variants of these genes only explain <5% of all ALS cases (Table 1) (Cirulli et al., 2015; Wu et al., 2012; Liu et al., 2017; Smith et al., 2014; Zhang et al., 2019), and disruptions to intracellular transport alone do not completely explain the nuance of disease onset. The timing of ALS onset remains a major open question, as most patients do not present noticeable symptoms until middle age, when numerous cell activities fail. Changes in the expression of some cytoskeleton-related genes coincide with symptomatic onset, including an increase in intermediate filament proteins (e.g., NF and PRPH) and the reduced expression of tubulin, syntaxin1B, dynactin subunits, and glutamate receptors (Julien et al., 1995; Robertson et al., 2003; Poesen and Van Damme, 2019; Zucchi et al., 2020; Wang et al., 2021; Yadav et al., 2022). Nevertheless, such studies assessing the contribution of cytoskeleton-related genes to ALS onset are further compounded by the absence of these genes from available expression surveys (Yadav et al., 2022; Jiang et al., 2005). Here we discuss the contribution of cytoskeleton genes in ALS including and beyond functions as an intracellular shipping system. Understanding these mechanisms may offer insight into neuronal function and additional considerations for emerging therapies.
Table 1.
Summary of mutations identified for ALS-associated cytoskeleton genes.
| Gene | Abbrev. | ALS-associated variants | Genetics | Prevalence | Refs. |
|---|---|---|---|---|---|
|
| |||||
| Alsin | ALS2 | V261A; K1548G | AR | 0.007% | (Yang et al., 2001; Hadano et al., 2001) |
| Dynactin subunit-1 | DCTN1 | E34Q; G59S; D63Y; M571T; R785W; R1101K; T1249I | AD | 0.4% | (Puls et al., 2003; Münch et al., 2004; Stockmann et al., 2013; Münch et al., 2005) |
| Kinesin family member 5A | KIF5A | K29R; P46S; S291G; R297Q; E413G; Q474H; H542N; G577S; P986L; T1001Q; R1007K; Δ544–1032† | HM & SV | 0.2% | (Nicolas et al., 2018; He et al., 2020) |
| Neurofilament | NF | A380T; deletions: Δ528–561; Δ655–662; Δ663–668; Δ663–677; Δ743–748; Δ790; insertion: 714‡ | DLOF & IM | 0.18% | (Cirulli et al., 2015; Corbo and Hays, 1992; Hirano, 1991) |
| Peripherin | PRPH | R133P; D141Y‡ | DLOF | 0.04% | (Gros-Louis et al., 2004; Leung et al., 2006; Corrado et al., 2011) |
| Profilin-1 | PFN1 | A20T; C71G; T109M; M114T; E117G; G118V; R136W; Q139L | AD | 2.5% | (Wu et al., 2012; Ingre et al., 2013; Tiloca et al., 2013) |
| Spastin | SPAST | S44L§; R65H§; N542G | AD | 0.02% | (Münch et al., 2008; Meyer et al., 2005; Brugman et al., 2009) |
| α-Tubulin 4A | TUBA4A | V7I; G43V§, T145P; R215C; R320C; R320H; A383T; W407X; D438N | AD & NS | 1.4% | (Smith et al., 2014; Pensato et al., 2015) |
Abbreviations: AR, autosomal recessive; AD, autosomal dominant, HM, heterozygous missense; SV, splicing variant; DLOF, dominant loss of function; IM, insertion mutant; NS, nonsense mutation.
Fig. 3B illustrates a general kinesin structure, however the motor domain used is from KIF5A (PDBID:3KIN).
No experimental structural information is available.
Residue not present in the structure used in Fig. 3D.
2. Microtubule highways: main tracks for transport
Intracellular trafficking defects are linked to nearly all forms of ALS, regardless of gene variant (Castellanos-Montiel et al., 2020; Burk and Pasterkamp, 2019). Are these disruptions a cause or are they caused indirectly through the cacophony of other dysfunctional cell functions impinging on the cytoskeleton? Neuronal transport requires a specific arrangement of actin filaments and microtubules for normal function (Fig. 1). Although not perfectly uniform, most neuronal microtubules are oriented with growing plus-ends toward terminal actin-rich growth cones, and stable minus-ends pointing toward the cell body (Fig. 1A) (Coles and Bradke, 2015; Tas et al., 2017; Leterrier, 2021).This arrangement effectively allows plus-end directed kinesin motors to move cargos toward the cell periphery (e.g., anterograde transport) and minus-end directed dynein motors to shuttle cargos inward toward the cell body (e.g., retrograde transport) (Fig. 1A). Intermediate filaments reinforce actin and microtubule paths but do not serve as tracks for transport (Bott and Winckler, 2020). No direct evidence (i.e., ALS-causative mutations in actin or myosin motors) suggests actin filament tracks are compromised or defective in ALS (Sundaramoorthy et al., 2015). Thus, transport relevant to ALS is predominately microtubule-based (Fig. 1B). Whether disease mutations build compromised tracks or alter microtubule dynamics remains an important open question.
There are multiple isoforms of tubulin, and each assemble into microtubules in a concentration-dependent process where tubulin dimers build protofilaments from templates (e.g., ɣ-TuRc) to form microtubule tracks (Fig. 3A) (Kapitein and Hoogenraad, 2015; Buscaglia et al., 2020). 1.4% of ALS cases are explained by nine autosomal dominant mutations present in a single isotype of tubulin, TUBA4A (V7I, G43V, T145P, R215C, R320C, R320H, A383T, W407X, and D438N) (Table 1; Fig. 2A) (Smith et al., 2014; Pensato et al., 2015; Van Damme et al., 2017). TUBA4A is present but not the most abundant α-tubulin in neurons, and becomes down-regulated before disease onset (Yadav et al., 2022; Jiang et al., 2005; Buscaglia et al., 2020). Whether TUBA4A mutations compromise the overall structure or motor protein functions is not clear. At least two ALS-related residues are located adjacent to the β-tubulin binding surface (T145 and W407) (Fig. 2A). These mutations may limit the association of β-tubulin and disrupt heterodimer formation (Fig. 3A). Further, while no ALS-related residues appear on the surface that mediates interactions with motor proteins, disruptions to protofilament assembly could alter the binding or movement of microtubule motors. Testing these ideas directly will require specific systems that use only the TUBA4A isotype (Davis et al., 1993; Sackett et al., 2010; Ti et al., 2020). However, experiments from COS7 cells overexpressing TUBA4A (W407X) suggest that both microtubule tracks and polymerization are compromised (Fig. 3B) (Smith et al., 2014).
Fig. 3.
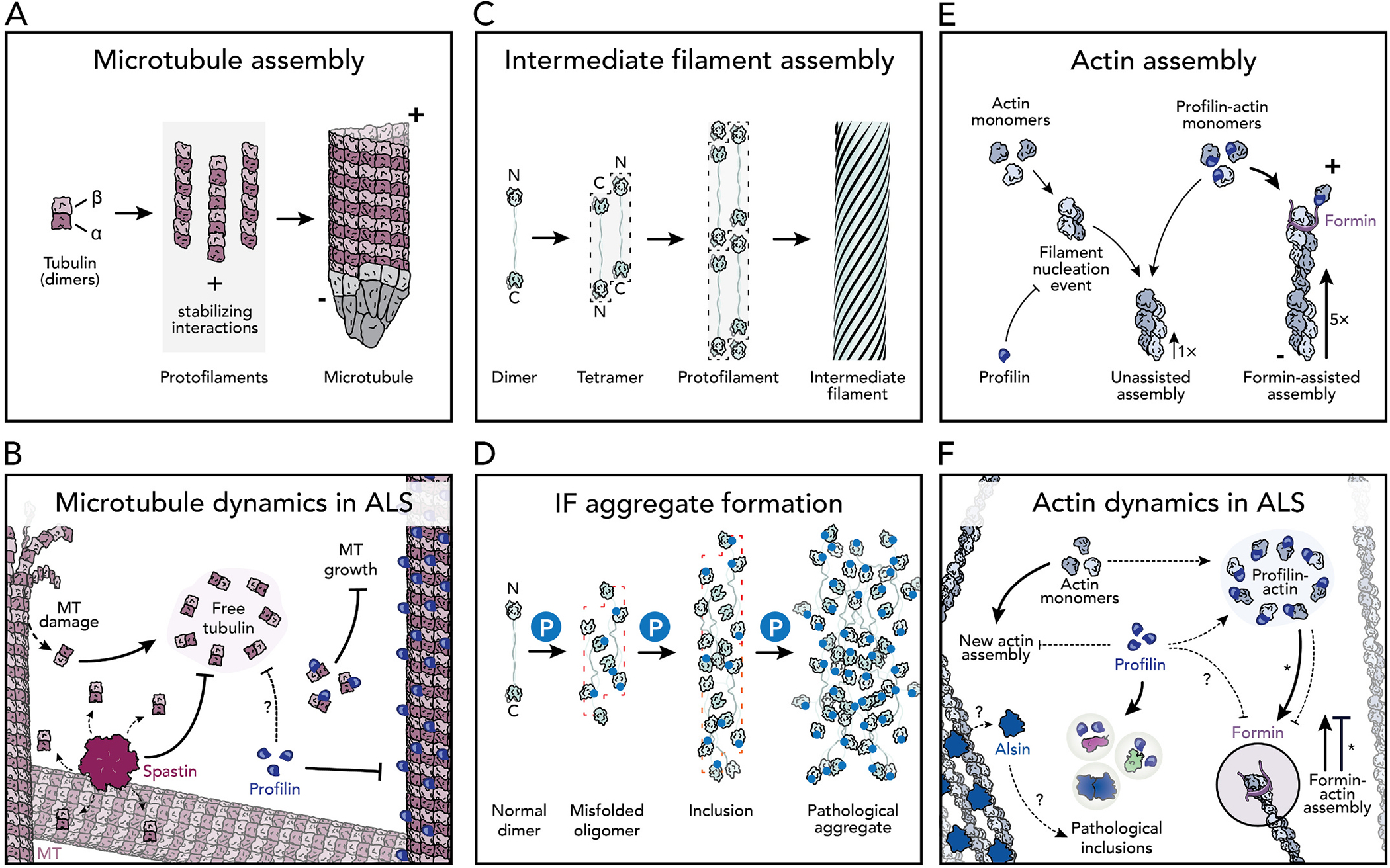
Mechanisms of cytoskeletal dynamics in the normal and predicted ALS-states. (A) Microtubule assembly. Dimers of α- and β-tubulin self-assemble to form protofilaments. Protofilaments are stabilized by ɣ-TuRC. Polarity: +, plus-end; −, minus-end. (B) Spastin (pink) disassembles microtubules excising subunits from the microtubule lattice. Profilin variants (dark blue) lose the affinity to bind to microtubules and possibly tubulin dimers, thereby destabilizing microtubule dynamics in ALS. (C) Intermediate filaments assembly. (D) Neurofilament and peripherin intermediate filaments become hyperphosphorylated (blue P), triggering the formation of pathological aggregates. (E) Actin filament assembly. PFN1 inhibits filament nucleation, but not the elongation rate. Formin proteins further stimulate actin elongation with PFN1. Polarity: +, plus-end; −, slower minus-end. (F) Profilin-mediated actin assembly mechanisms are disrupted in ALS including: reduced affinity for actin monomers and reduced formin-actin assembly. Asterisks note that the M114T and G118V profilin mutations enhance, rather than hinder, formin activities (Liu et al., 2022; Schmidt et al., 2021). Alsin (ALS2) association with actin is thought to be lost in ALS.
Fig. 2.
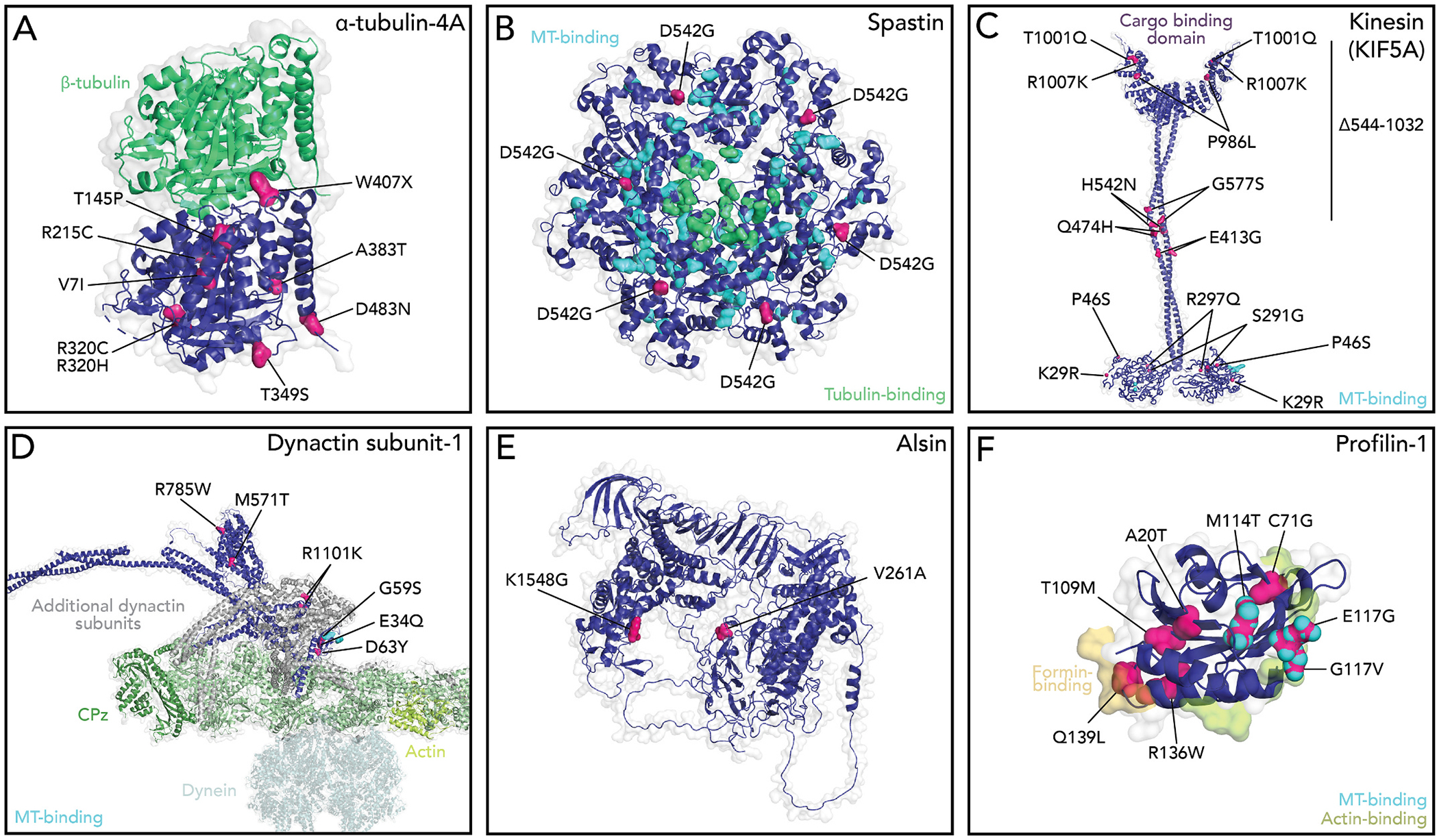
Positions of ALS-linked mutations in structures of cytoskeleton associated proteins. Cytoskeleton regulation proteins (dark blue) highlighting the positions of ALS-linked residues (pink). (A) α-tubulin relative to β-tubulin in the α/β heterodimer are shown (green). PDBID: 5KMG. (B) ALS residues in the spastin homohexamer. S44L and R65H residues are not present in the available structure. Microtubule (teal)- and tubulin-binding (green) residues are highlighted. PDBID: 6PEN. (C) General features of kinesin with the KIF5A motor domain. Microtubule binding residues (teal) are highlighted. Modeled after (Vale, 2003). PDBIDs: 4UXY, 1GK4, 3NF1. (D) View dynactin subunit-1 (DCTN1), with microtubule binding residues highlighted(teal). PDBIDs: 5NVU, 6ZNL, 2COY, 4RFX, 3E2U; and Alpha-fold ID: Q14203 (Jumper et al., 2021). (E) Alsin (ALS2). Alpha-fold ID: Q96Q42. (F) Profilin-1 and relevant binding surfaces to actin (lime green), formin proteins (yellow), and microtubules (teal) highlighted. PDBID: 2PAV.
In addition to mechanisms involving tubulin alone, hundreds of other proteins regulate the dynamics of neuronal microtubules. For example, SPAST disassembles microtubules by pulling on the C-terminal tail of α- and β-tubulin dimers to excise individual tubulin subunits from the microtubule lattice (Vemu et al., 2017; Sandate et al., 2019; Han et al., 2020). This mechanism generates pieces of microtubules that may serve as templates for new microtubule assembly (Roll-Mecak and Vale, 2008; Wood et al., 2006), or contribute to the loss of functions seen with the TUBA4A(W407X) nonsense mutation that lacks the α-tubulin tail (above). While SPAST is the primary cause of hereditary spastic paraplegia (HSP), three unrelated mutations (S44L, R65H, and N542G) also cause ALS (Table 1; Fig. 2B) (Cirulli et al., 2015; Münch et al., 2008; Meyer et al., 2005; Brugman et al., 2009; Tremolizzo et al., 2014). These mutations eliminate SPAST’s microtubule binding and disassembly activities, leading to undesirable microtubule growth (Fig. 3B). Furthermore, the loss of functional SPAST increases microtubule polyglutamylation, which in turn reduces the microtubule affinity of kinesin motors (particularly KIF5), and suppresses overall anterograde transport (Lopes et al., 2020; Solowska et al., 2008). In contrast to SPAST, PFN1 stimulates assembly by binding tubulin dimers and microtubules (Henty-Ridilla et al., 2017; Pimm et al., 2022). Some variants eliminate these functions and redistribute cellular PFN1 pools, disrupting the delicate coordination of microtubules and actin filaments at transport terminals, discussed further below (Henty-Ridilla et al., 2017; Pinto-Costa et al., 2020). Thus, compromised structural integrity of microtubule tracks through mutations in tubulin building blocks or essential regulatory proteins have devastating consequences for intracellular transport and overall neuronal health (Liang et al., 2016; Dumont et al., 2015; Kabir et al., 2020; Morikawa et al., 2015; Triclin et al., 2021; Thery and Blanchoin, 2021; Budaitis et al., 2021).
3. Tethering cargos to tracks: motors and cargo-linking proteins
Kinesin-1 motors (e.g., KIF5A) navigate microtubule tracks to mediate the anterograde transport of mitochondria, vesicles, and signaling molecules (Fig. 1) (Hirokawa and Takemura, 2005). KIF5A is exclusively expressed in motor neurons, essential for development and function, and the only kinesin with reported mutations that cause ALS (Nicolas et al., 2018; Niclas et al., 1994; Fagerberg et al., 2014; Brenner et al., 2018; He et al., 2020; Baron et al., 2022). The twelve variants (0.2% prevalence) linked to ALS or the Δ544–1032 truncation, localize to several conserved functional domains including: the motor (K29R, P46S, S291G, and R297Q), the coiled-coil (E413G, Q474G, H542N, and G577S), and a C-terminal cargo binding region (P986L, T1001Q, R1007K, and Δ544–1032) (Table 1; Fig. 2C) (Zhang et al., 2019; Nicolas et al., 2018). Regardless of variant, the mechanism of impaired long-range cargo transport is most likely failed motor locomotion or aberrant cargo unions that either unlink normal ligands or load new disease-relevant ones onto tracks. In zebrafish or rat hippocampal neurons the reduction of KIF5A results in hyperexcitability, axonal degradation, and defective mitochondrial transport that cannot be restored with other KIF5 isoforms or kinesin motors (Campbell et al., 2014; Zhao et al., 2020; Xia et al., 2003). Overexpression of a disease relevant fragment of KIF5A’s cargo binding domain releases autoinhibition and hyper-activates axonal transport and aberrant interactions with RNA (Baron et al., 2022). In contrast, modulating KIF5A had no observable effect on anterograde transport in SOD1 disease models (Shi et al., 2010), implying that additional motors interact or interfere with neuronal cargos in ALS.
In contrast to kinesin, dynein motors cruise along microtubules and require the dynactin complex as a cofactor to execute retrograde transport (Fig. 1) (Schroer, 2004). Twelve ALS-relevant mutations manifest in only DCTN1, which directly links cargos to the motor and improves dynein’s processivity (E34Q, G59S, D63Y, M571T, R785W, R1101K, and T1249I) (Table 1; Fig. 2D) (Nicolas et al., 2018; Lau et al., 2021; Waterman-Storer et al., 1995; Vaughan et al., 2002; Holzbaur and Vallee, 1994; Puls et al., 2003). ALS-related disruptions through DCTN1 (G59S) prevent the linkage of essential cargo to dynein-based transport systems, which in turn impairs motor neuron development and synaptic function (Liu et al., 2017; Bercier et al., 2019; Kuźma-Kozakiewicz et al., 2013; Levy et al., 2006; Moore et al., 2009; Lai et al., 2007). DCTN1 is the only cytoskeleton-related gene associated with polygenic manifestations of ALS, found in combination with variants of dynein-transported cargos, specifically SETX, ATXN2, FIG. 4, and C9ORF72 (Cady et al., 2015). Further, even without DCTN1 variants, all observed polygenic ALS cases display disease-related impairments to retrograde transport (Shi et al., 2010; Kuźma-Kozakiewicz et al., 2013; Maimon et al., 2021). Many mechanisms may explain the transport defects for these commonly misfolded proteins including that dynein-linkage sights become obscured (Cristofani et al., 2017; Licata et al., 2022). Regardless of the exact mechanism detailing the engagement between microtubules, motors, or cargos, defects in axonal transport are an established feature of ALS and other neurodegenerative diseases.
4. Intermediate filament aggregation is a pathological hallmark of ALS
Intermediate filaments (e.g., NF and PRPH) are formed from a dynamic self-assembly process, but unlike actin or microtubules the final polymers lack polarity and do not act as tracks for intracellular transport (Fig. 3C) (Xiao et al., 2006; Yuan et al., 2017). Instead, NF and PRPH influence transport on actin and microtubules by structurally reinforcing those filaments (Xiao et al., 2006; Yuan et al., 2017). Aggregates of either NF or PRPH intermediate filament proteins are a considered early pathological hallmarks of ALS, and found in nearly all patients, directly causing 0.22% of all familial ALS (Table 1) (Cirulli et al., 2015; Gros-Louis et al., 2004; Leung et al., 2006; Corrado et al., 2011; Corbo and Hays, 1992). However, the metaphorical road to pathological onset through intermediate filaments is bumpy and confusing. Disease-relevant hyperphosphorylation of NF and PRPH is thought to occur before noticeable defects in axonal transport because motor neurons in disease models often degenerate before aggregates form; this could trigger intermediate filament aggregation and also slowed transport (Fig. 3D) (Robertson et al., 2003; Beaulieu et al., 2000). However, this model does not consider whether pathological inclusions are present but below the detection readouts used to visualize aggregate formation, or changes to axonal transport, how NF and PRPH copolymers may contribute to pathological onset, or models that suggest intermediate filament compilations act as protective agents against the disease (Corbo and Hays, 1992; Hirano, 1991; Ho et al., 1998; Julien and Mushynski, 1998; Beaulieu and Julien, 2003). Unfortunately, neither NF or PRPH can be produced in conventional purification and expression systems for use in biochemical assays that to directly address some of these ideas or further decipher the mechanisms of intermediate filament quality control (Bott and Winckler, 2020; Didonna and Opal, 2019).
5. ALS2 and PFN1: direct links to disease-related actin dynamics?
Actin filament regulation is essential for all cells, but particularly critical for neurons, regulating neuronal outgrowth, organelle biogenesis, the stability of axons, and synaptic function (Coles and Bradke, 2015; Leterrier, 2021; Konietzny et al., 2017; Venkatesh et al., 2020). Similar to microtubules, the spontaneous ordered assembly of actin filaments, is concentration-dependent and imparts structural polarity to polymers important for mechanisms of intracellular transport and force generation (Fig. 3E) (Pollard, 2016). ALS2 and PFN1 are almost exclusively studied in the context of actin polymerization in ALS, because direct links to actin dynamics to ALS have been difficult to isolate from other critical cell processes, and because both are also linked to neuronal microtubules and other important regulatory processes.
ALS2 regulates actin-based neurite outgrowth as a prominent guanine nucleotide exchange factor (GEF) for the small GTPase Rab5 (Yang et al., 2001; Topp et al., 2005; Hadano et al., 2001; Cai and Yang, 2016; Tudor et al., 2005; Devon et al., 2006). Together these proteins regulate actin polymerization associated with early endosome dynamics. ALS2 mutations (A261A and K1548G) and deletions produce a very rare (0.007%), autosomal recessive form of juvenile ALS (Table 1; Fig. 2E) (Cirulli et al., 2015; Hamida et al., 1990; Hentati et al., 1994; Alsultan et al., 2016). The pathomechanism of these mutations likely involves unlinking or disturbing the connections between ALS2 and Rab5, which reduces actin-based intracellular trafficking and glutamate signaling at synapses (Lai et al., 2009). However, ALS2 colocalizes at sites of actin and microtubule overlap and is also capable of phase-separation mediated through a region containing A261, but we do not know if these observations are relevant to ALS (Topp et al., 2005; Tudor et al., 2005; Gautam et al., 2016; Sato et al., 2018; Kunita et al., 2007; Millecamps et al., 2005; Hsu et al., 2018). Indeed, neurons devoid of ALS2 display a significant increase in glutamate receptors, autophagy, and sensitivity to oxidative stress, which are each regulated by actin filament assembly and other mechanisms (Fig. 3F) (Gautam et al., 2016; Cai, 2005; Millecamps et al., 2010).
PFN1 variants (A20T, C71G, T109M, M114T, E117G, G118V, R136W, and Q139L) cause an autosomal dominant form of ALS that disturbs actin dynamics in two opposing ways (Table 1; Fig. 2F) (Wu et al., 2012; Alsultan et al., 2016; Ingre et al., 2013; Tiloca et al., 2013; Boopathy et al., 2015). First, PFN1 directly binds to actin monomers, preventing actin filament assembly by blocking the site of new monomer addition (Fig. 3E and F) (Boopathy et al., 2015; Skruber et al., 2018; Pimm et al., 2020; Davey and Moens, 2020; Suarez and Kovar, 2016; Liu et al., 2022; Ferron et al., 2007). Second, PFN1 enhances actin assembly through interactions with formin proteins (Fig. 3E and F) (Suarez and Kovar, 2016; Liu et al., 2022; Giampetruzzi et al., 2019; Schmidt et al., 2021; Skruber et al., 2019). Combined these properties could directly regulate the shape and function of neuronal growth cones and synapses (Pinto-Costa et al., 2020; Witke et al., 1998; Michaelsen-Preusse et al., 2016; Grintsevich et al., 2021). However, other roles beyond actin regulation also deserve consideration. The interaction between PFN1 and components of the dynactin complex are synthetic lethal in yeast (Figley et al., 2014). PFN1 also potently stimulates microtubule assembly mediated through ALS-relevant mutations, specifically M114T, E117G, and G118V (Henty-Ridilla et al., 2017; Pinto-Costa et al., 2020; Liu et al., 2022). Some PFN1 variants localize to microtubules in cells (Witke et al., 1998; Michaelsen-Preusse et al., 2016). Combined these observations strongly suggest that PFN1 contributes to ALS by regulating the microtubule tracks and transport machinery (e.g. kinesins, dynein, or spastin) (Pimm et al., 2022; Pinto-Costa et al., 2020; Liu et al., 2022; Figley et al., 2014). PFN1 also directly associates with aggregation prone variants of TDP-43, FUS, and UBQLN2 (Rutherford et al., 2013; Kawaguchi et al., 2020; Oberstadt et al., 2018b; Tanaka and Hasegawa, 2016; Matsukawa et al., 2016) and may be related to a protein misfolding or sequestration mechanism that prohibits normal cytoskeletal dynamics or autophagy related processes (Fig. 3F) (Boopathy et al., 2015; Liu et al., 2022; Schmidt et al., 2021; Del Poggetto et al., 2016; Kiaei et al., 2018; Lee et al., 2010). Therefore, additional studies are required to truly decipher whether the cytoskeleton is disrupted due to the presence of cytotoxic aggregates or vice versa.
6. Final thoughts and conclusions
The idea that the cytoskeleton’s role in ALS pathology sums to disrupted tracks for transport is certainly a logical and attractive notion, reinforced by the discovery of disease-causing mutations in bona fide microtubule-based machinery. Axonal transport is essential for the homoeostasis of neurons facilitating synaptic communications within and between cells, efficiently clearing disease-relevant protein aggregates, and a common convergence point interrelated to many pathological mechanisms that become compromised in neurodegenerative disease. However, these mechanisms alone do not differentiate ALS from other forms of neurodegeneration or other aggregopathies. While it may not fully explain what is happening to patients suffering from the disease, understanding the basic mechanisms that regulate cytoskeletal proteins is integral to developing specific drug targets for the treatment of ALS. Future studies that combine cell and biochemical approaches will help to dissect these complex questions and the secrets of the tracks and machines inside.
Acknowledgements
We are grateful to Marc Ridilla (Foundation for Prader-Willi Research), members of the Henty-Ridilla lab, and anonymous reviewers for helpful comments that improved this work. Research in the Henty-Ridilla laboratory is supported by a Sinsheimer Scholar Award, ALSA starter grant (20-IIP-506), and NIH R35 award GM133485.
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
References
- Ally S, Larson AG, Barlan K, et al. , 2009. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J. Cell Biol. 187, 1071–1082. 10.1083/jcb.200908075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsultan A, Waller R, Heath P, Kirby J, 2016. The genetics of amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis 6, 49–64. 10.2147/DNND.S84956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron DM, Fenton AR, Saez-Atienzar S, et al. , 2022. ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 39 (1), 110598 10.1016/j.celrep.2022.110598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J-M, Julien J-P, 2003. Peripherin-mediated death of motor neurons rescued by overexpression of neurofilament NF-H proteins. J. Neurochem. 85 (1), 248–256. 10.1046/j.1471-4159.2003.01653.x. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Jacomy H, Julien J-P, 2000. Formation of intermediate filament protein aggregates with disparate effects in two transgenic mouse models lacking the neurofilament light subunit. J. Neurosci. 20 (14), 5321–5328. 10.1523/JNEUROSCI.20-14-05321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercier V, Hubbard JM, Fidelin K, et al. , 2019. Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol. Neurodegener. 14 (1), 27. 10.1186/s13024-019-0327-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathy S, Silvas TV, Tischbein M, et al. , 2015. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc. Natl. Acad. Sci. U. S. A. 112 (26), 7984–7989. 10.1073/pnas.1424108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott CJ, Winckler B, 2020. Intermediate filaments in developing neurons: beyond structure. Cytoskeleton 77 (3–4), 110–128. 10.1002/cm.21597. [DOI] [PubMed] [Google Scholar]
- Brenner D, Yilmaz R, Müller K, et al. , 2018. Hot-spot KIF5A mutations cause familial ALS. Brain 141 (3), 688–697. 10.1093/brain/awx370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese M, Saal-Bauernschubert L, Lüningschrör P, et al. , 2020. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol Commun 8, 116. 10.1186/s40478-020-00987-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugman F, Veldink JH, Franssen H, et al. , 2009. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndromes. Arch. Neurol. 66 (4), 509–514. 10.1001/archneurol.2009.19. [DOI] [PubMed] [Google Scholar]
- Budaitis BG, Badieyan S, Yue Y, et al. , 2021. A kinesin-1 variant reveals motor-induced microtubule damage in cells. bioRxiv. 10.1101/2021.10.19.464974, 2021.10.19.464974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk K, Pasterkamp RJ, 2019. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 137, 859–877. 10.1007/s00401-019-01964-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscaglia G, Northington KR, Moore JK, Bates EA, 2020. Reduced TUBA1A tubulin causes defects in trafficking and impaired adult motor behavior. eNeuro 7 (2). 10.1523/ENEURO.0045-20.2020. ENEURO.0045–20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady J, Allred P, Bali T, et al. , 2015. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 77 (1), 100–113. 10.1002/ana.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, 2005. Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J. Neurosci. 25 (33), 7567–7574. 10.1523/JNEUROSCI.1645-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Yang EJ, 2016. Ginsenoside reattenuates neuroinflammation in a symptomatic ALS animal model. Am J Chin Med 44 (2), 401–413. 10.1142/S0192415X16500233. [DOI] [PubMed] [Google Scholar]
- Campbell PD, Shen K, Sapio MR, et al. , 2014. Unique function of kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 34 (44), 14717–14732. 10.1523/JNEUROSCI.2770-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos-Montiel MJ, Chaineau M, Durcan TM, 2020. The neglected genes of ALS: cytoskeletal dynamics impact synaptic degeneration in ALS. Front. Cell. Neurosci. 14, 594975 10.3389/fncel.2020.594975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JJE, Butkevich E, Worseck JM, et al. , 2012. Phosphorylation-regulated axonal dependent transport of syntaxin 1 is mediated by a Kinesin-1 adapter. Proc. Natl. Acad. Sci. U. S. A. 109 (15), 5862–5867. 10.1073/pnas.1113819109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, et al. , 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CH, Bradke F, 2015. Coordinating neuronal actin-microtubule dynamics. Curr. Biol. 25, R677–R691. 10.1016/j.cub.2015.06.020. [DOI] [PubMed] [Google Scholar]
- Corbo M, Hays AP, 1992. Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J. Neuropathol. Exp. Neurol. 51 (5), 531–537. 10.1097/00005072-199209000-00008. [DOI] [PubMed] [Google Scholar]
- Corrado L, Carlomagno Y, Falasco L, et al. , 2011. A novel peripherin gene (PRPH) mutation identified in one sporadic amyotrophic lateral sclerosis patient. Neurobiol. Aging 32 (3), 552.e1–552.e6. 10.1016/j.neurobiolaging.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Cristofani R, Crippa V, Rusmini P, et al. , 2017. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 13 (8), 1280–1303. 10.1080/15548627.2017.1308985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey RJ, Moens PD, 2020. Profilin: many facets of a small protein. Biophys. Rev. 12 (4), 827–849. 10.1007/s12551-020-00723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A, Sage CR, Wilson L, Farrell KW, 1993. Purification and biochemical characterization of tubulin from the budding yeast Saccharomyces cerevisiae. Biochemistry 32, 8823–8835. 10.1021/bi00085a013. [DOI] [PubMed] [Google Scholar]
- Del Poggetto E, Gori L, Chiti F, 2016. Biophysical analysis of three novel profilin-1 variants associated with amyotrophic lateral sclerosis indicates a correlation between their aggregation propensity and the structural features of their globular state. Biol. Chem. 397 (9), 927–937. 10.1515/hsz-2016-0154. [DOI] [PubMed] [Google Scholar]
- Devon RS, Orban PC, Gerrow K, et al. , 2006. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. U. S. A. 103 (25), 9595–9600. 10.1073/pnas.0510197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didonna A, Opal P, 2019. The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol. Neurodegener. 14 (1), 19. 10.1186/s13024-019-0318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogterom M, Koenderink GH, 2019. Actin-microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 20, 38–54. 10.1038/s41580-018-0067-1. [DOI] [PubMed] [Google Scholar]
- Dumont ELP, Do C, Hess H, 2015. Molecular wear of microtubules propelled by surface-adhered kinesins. Nat. Nanotechnol. 10 (2), 166–169. 10.1038/nnano.2014.334. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, et al. , 2014. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13 (2), 397–406. 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F, Rebowski G, Lee SH, Dominguez R, 2007. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 26 (21), 4597–4606. 10.1038/sj.emboj.7601874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley MD, Bieri G, Kolaitis R-M, et al. , 2014. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 34 (24), 8083–8097. 10.1523/JNEUROSCI.0543-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli L, Young FL, Boeynaems S, et al. , 2021. C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 7 (15) 10.1126/sciadv.abg3013 eabg3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Jara JH, Sekerkova G, et al. , 2016. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum. Mol. Genet. 25 (6), 1074–1087. 10.1093/hmg/ddv631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampetruzzi A, Danielson EW, Gumina V, et al. , 2019. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 10 (1), 3827. 10.1038/s41467-019-11837-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JM, Fagegaltier D, Phatnani H, Harms MB, 2020. Genetics of amyotrophic lateral sclerosis. Curr Genet Med Rep 8, 121–131. 10.1007/s40142-020-00194-8. [DOI] [Google Scholar]
- Grintsevich EE, Ahmed G, Ginosyan AA, et al. , 2021. Profilin and Mical combine to impair F-actin assembly and promote disassembly and remodeling. Nat. Commun. 12 (1), 5542. 10.1038/s41467-021-25781-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros-Louis F, Lariviere R, Gowing G, et al. , 2004. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J. Biol. Chem. 279 (44), 45951–45956. 10.1074/jbc.M408139200. [DOI] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, et al. , 2001. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29 (2), 166–173. 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- Hamida MB, Hentati F, Hamida CB, 1990. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis): conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 113 (2), 347–363. 10.1093/brain/113.2.347. [DOI] [PubMed] [Google Scholar]
- Hammer JA, Wagner W, 2013. Functions of class V Myosins in neurons. J. Biol. Chem. 288 (40), 28428–28434. 10.1074/jbc.R113.514497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Schubert HL, McCullough J, et al. , 2020. Structure of spastin bound to a glutamate-rich peptide implies a hand-over-hand mechanism of substrate translocation. J. Biol. Chem. 295 (2), 435–443. 10.1074/jbc.AC119.009890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Liu X, Tang L, et al. , 2020. Whole-exome sequencing identified novel KIF5A mutations in Chinese patients with amyotrophic lateral sclerosis and charcot-marie-tooth type 2. J. Neurol. Neurosurg. Psychiatry 91 (3), 326–328. 10.1136/jnnp-2019-320483. [DOI] [PubMed] [Google Scholar]
- Hentati A, Bejaoui K, Pericak-Vance MA, et al. , 1994. Linkage of recessive familial amyotrophic lateral sclerosis to chromosome 2q33–q35. Nat. Genet. 7 (3), 425–428. 10.1038/ng0794-425. [DOI] [PubMed] [Google Scholar]
- Henty-Ridilla JL, Juanes MA, Goode BL, 2017. Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr. Biol. 27 (22), 3535–3543.e4. 10.1016/j.cub.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog JJ, Deshpande M, Shapiro L, et al. , 2017. TDP-43 misexpression causes defects in dendritic growth. Sci. Rep. 7, 15656. 10.1038/s41598-017-15914-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, 1991. Cytopathology of amyotrophic lateral sclerosis. Adv. Neurol. 56, 91–101. https://pubmed.ncbi.nlm.nih.gov/1649547/. [PubMed] [Google Scholar]
- Hirokawa N, Takemura R, 2005. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6 (3), 201–214. 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Ho CL, Martys JL, Mikhailov A, et al. , 1998. Novel features of intermediate filament dynamics revealed by green fluorescent protein chimeras. J. Cell Sci. 111 (13), 1767–1778. 10.1242/jcs.111.13.1767. [DOI] [PubMed] [Google Scholar]
- Holzbaur ELF, Vallee RB, 1994. Dyneins: molecular structure and cellular function. Annu. Rev. Cell Biol. 10, 339–372. 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Hsu F, Spannl S, Ferguson C, et al. , 2018. Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. ELife 7, e32282. 10.7554/eLife.32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingre C, Landers JE, Rizik N, et al. , 2013. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol. Aging 34 (6). 10.1016/j.neurobiolaging.2012.10.009, 1708.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y-M, Yamamoto M, Kobayashi Y, et al. , 2005. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251. 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- Julien J-P, Beaulieu J-M, 2000. Cytoskeletal abnormalities in amyotrophic lateral sclerosis: beneficial or detrimental effects? J. Neurol. Sci. 180, 7–14. 10.1016/S0022-510X(00)00422-6. [DOI] [PubMed] [Google Scholar]
- Julien J-P, Mushynski WE, 1998. Neurofilaments in health and aisease. Prog. Nucleic Acid Res. Mol. Biol. 16, 1–23. 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- Julien J-P, Côté F, Collard J-F, 1995. Mice overexpressing the human neurofilament heavy gene as a model of ALS. Neurobiol. Aging 16, 487–490. 10.1016/0197-4580(94)00169-2. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, et al. , 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir AMR, Sada K, Kakugo A, 2020. Breaking of buckled microtubules is mediated by kinesins. Biochem. Biophys. Res. Commun. 524 (1), 249–254. 10.1016/j.bbrc.2020.01.082. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC, 2015. Building the neuronal microtubule cytoskeleton. Neuron 87, 492–506. 10.1016/j.neuron.2015.05.046. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Rollins MG, Moinpour M, et al. , 2020. Changes to the TDP-43 and FUS interactomes induced by DNA damage. J. Proteome Res. 19 (1), 360–370. 10.1021/acs.jproteome.9b00575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Balasubramaniam M, Govind Kumar V, et al. , 2018. ALS-causing mutations in profilin-1 alter its conformational dynamics: a computational approach to explain propensity for aggregation. Sci. Rep. 8 (1), 13102. 10.1038/s41598-018-31199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konietzny A, Bär J, Mikhaylova M, 2017. Dendritic actin cytoskeleton: structure, functions, and regulations. Front. Cell. Neurosci. 11, 147. 10.3389/fncel.2017.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunita R, Otomo A, Mizumura H, et al. , 2007. The Rab5 activator ALS2/alsin acts as a novel Rac1 effector through Rac1-activated endocytosis. J. Biol. Chem. 282 (22), 16599–16611. 10.1074/jbc.M610682200. [DOI] [PubMed] [Google Scholar]
- Kuźma-Kozakiewicz M, Chudy A, Kaźmierczak B, et al. , 2013. Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem. Res. 38 (12), 2463–2473. 10.1007/s11064-013-1160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Lin X, Chandran J, et al. , 2007. The G59S mutation in p150glued causes dysfunction of dynactin in mice. J. Neurosci. 27 (51), 13982–13990. 10.1523/JNEUROSCI.4226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Xie C, Shim H, et al. , 2009. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol Brain 2, 23. 10.1186/1756-6606-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasiene J, Yamanaka K, 2011. Glial cells in amyotrophic lateral sclerosis. Neurol Res Int 2011, 1–7. 10.1155/2011/718987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CK, O’Reilly FJ, Santhanam B, et al. , 2021. Cryo-EM reveals the complex architecture of dynactin’s shoulder region and pointed end. EMBO J. 40 (8), e106164 10.15252/embj.2020106164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Koga H, Kawaguchi Y, et al. , 2010. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29 (5), 969–980. 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, 2021. A pictorial history of the neuronal cytoskeleton. J. Neurosci. 41, 11–27. 10.1523/JNEUROSCI.2872-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CL, He CZ, Kaufmann P, et al. , 2006. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol. 14 (3), 290–296. 10.1111/j.1750-3639.2004.tb00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JR, Sumner CJ, Caviston JP, et al. , 2006. A motor neuron disease–associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 172 (5), 733–745. 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL, Mao T, Svoboda K, Arnold DB, 2009. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat. Neurosci. 12 (5), 568–576. 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WH, Li Q, Rifat Faysal KM, et al. , 2016. Microtubule defects influence kinesin-based transport in vitro. Biophys. J. 110 (10), 2229–2240. 10.1016/j.bpj.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata NV, Cristofani R, Salomonsson S, et al. , 2022. C9orf72 ALS/FTD dipeptide repeat protein levels are reduced by small molecules that inhibit PKA or enhance protein degradation. EMBO J. 41 (1), e105026 10.15252/embj.2020105026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yang L, Tang L, et al. , 2017. DCTN1 gene analysis in Chinese patients with sporadic amyotrophic lateral sclerosis. PLoS One 12, e0182572. 10.1371/journal.pone.0182572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Pimm ML, Haarer B, et al. , 2022. Biochemical characterization of actin assembly mechanisms with ALS-associated profilin variants. Eur. J. Cell Biol. 101 (2), 151212 10.1016/j.ejcb.2022.151212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes AT, Hausrat TJ, Heisler FF, et al. , 2020. Spastin depletion increases tubulin polyglutamylation and impairs kinesin-mediated neuronal transport, leading to working and associative memory deficits. PLoS Biol. 18, e3000820 10.1371/journal.pbio.3000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimon R, Ankol L, Gradus Pery T, et al. , 2021. A CRMP4-dependent retrograde axon-to-soma death signal in amyotrophic lateral sclerosis. EMBO J. 40 (17), e107586 10.15252/embj.2020107586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masrori P, Van Damme P, 2020. Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27 (10), 1918–1929. 10.1111/ene.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa K, Hashimoto T, Matsumoto T, et al. , 2016. Familial ALS-linked mutations in profilin 1 exacerbate TDP-43-induced degeneration in the retina of Drosophila melanogaster through an increase in the cytoplasmic localization of TDP43. J. Biol. Chem. 291 (45), 23464–23476. 10.1074/jbc.M116.729152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Schwan A, Dullinger JS, et al. , 2005. Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology 65, 141–143. 10.1212/01.wnl.0000167130.31618.0a. [DOI] [PubMed] [Google Scholar]
- Michaelsen-Preusse K, Zessin S, Grigoryan G, et al. , 2016. Neuronal profilins in health and disease: relevance for spine plasticity and fragile X syndrome. Proc. Natl. Acad. Sci. U. S. A. 113 (12), 3365–3370. 10.1073/pnas.1516697113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien J-P, 2013. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176. 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Millecamps S, Gentil BJ, Gros-Louis F, et al. , 2005. Alsin is partially associated with centrosome in human cells. Biochim. Biophys. Acta 1745 (1), 84–100. 10.1016/j.bbamcr.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Millecamps S, Salachas F, Cazeneuve C, et al. , 2010. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J. Med. Genet. 47 (8), 554–560. 10.1136/jmg.2010.077180. [DOI] [PubMed] [Google Scholar]
- Moore JK, Sept D, Cooper JA, 2009. Neurodegeneration mutations in dynactin impair dynein-dependent nuclear migration. Proc. Natl. Acad. Sci. U. S. A. 106, 5147–5152. 10.1073/pnas.0810828106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa M, Yajima H, Nitta R, et al. , 2015. X-ray and Cryo-EM structures reveal mutual conformational changes of kinesin and GTP-state microtubules upon binding. EMBO J. 34 (9), 1270–1286. 10.15252/embj.201490588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch C, Sedlmeier R, Meyer T, et al. , 2004. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63 (4), 724–726. 10.1212/01.WNL.0000134608.83927.B1. [DOI] [PubMed] [Google Scholar]
- Münch C, Rosenbohm A, Sperfeld A-D, et al. , 2005. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58 (5), 777–780. 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- Münch C, Rolfs A, Meyer T, 2008. Heterozygous S44L missense change of the spastin gene in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 9 (4), 251–253. 10.1080/17482960801900172. [DOI] [PubMed] [Google Scholar]
- Muñoz-Lasso DC, Romá-Mateo C, Pallardó FV, Gonzalez-Cabo P, 2020. Much more than a scaffold: cytoskeletal proteins in neurological disorders. Cells 9 (2), 358. 10.3390/cells9020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HP, Van Broeckhoven C, van der Zee J, 2018. ALS genes in the genomic era and their implications for FTD. Trends Genet. 34 (6), 404–423. 10.1016/j.tig.2018. [DOI] [PubMed] [Google Scholar]
- Niclas J, Navone F, Hom-Booker N, Vale RD, 1994. Cloning and localization of a conventional kinesin motor expressed exclusively in neurons. Neuron 12 (5), 1059–1072. 10.1016/0896-6273(94)90314-X. [DOI] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, et al. , 2018. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97 (6), 1268–1283.e6. 10.1016/j.neuron.2018.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirschl JJ, Ghiretti AE, Holzbaur ELF, 2017. The impact of cytoskeletal organization on the local regulation of neuronal transport. Nat. Rev. Neurosci. 18, 585–597. 10.1038/nrn.2017.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstadt M, Claßen J, Arendt T, Holzer M, 2018a. TDP-43 and cytoskeletal proteins in ALS. Mol. Neurobiol. 55, 3143–3151. 10.1007/s12035-017-0543-1. [DOI] [PubMed] [Google Scholar]
- Oberstadt M, Stieler J, Simpong DL, et al. , 2018b. TDP-43 self-interaction is modulated by redox-active compounds Auranofin Chelerythrine and Riluzole. Sci. Rep. 8 (1), 2248. 10.1038/s41598-018-20565-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osking Z, Ayers JI, Hildebrandt R, et al. , 2019. ALS-linked SOD1 mutants enhance neurite outgrowth and branching in adult motor neurons. iScience 11, 294–304. 10.1016/j.isci.2018.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensato V, Tiloca C, Corrado L, et al. , 2015. TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J. Neurol. 262 (5), 1376–1378. 10.1007/s00415-015-7739-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani HP, Guarnieri P, Friedman BA, et al. , 2013. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. U. S. A. 110 (8), E756–E765. 10.1073/pnas.1222361110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Rothstein JD, 2014. Glial cells in amyotrophic lateral sclerosis. Exp. Neurol. 262, 111–120. 10.1016/j.expneurol.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimm ML, Henty-Ridilla JL, 2021. New twists in actin-microtubule interactions. Mol. Biol. Cell 32, 211–217. 10.1091/mbc.E19-09-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimm ML, Hotaling J, Henty-Ridilla JL, 2020. Profilin choreographs actin and microtubules in cells and cancer. Int. Rev. Cell Mol. Biol. 10.1016/bs.ircmb.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimm ML, Liu X, Tuli F, et al. , 2022. Visualizing molecules of functional human profilin. eLife 11, e76485. 10.7554/eLife.76485, 2021.09.01.458557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Costa R, Sousa SC, Leite SC, et al. , 2020. Profilin 1 delivery tunes cytoskeletal dynamics toward CNS axon regeneration. J. Clin. Invest. 130 (4), 2024–2040. 10.1172/JCI125771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poesen K, Van Damme P, 2019. Diagnostic and prognostic performance of neurofilaments in ALS. Front. Neurol. 9, 1167. 10.3389/fneur.2018.01167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, 2016. Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 8 (8), a018226 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, et al. , 2003. Mutant dynactin in motor neuron disease. Nat. Genet. 33 (4), 455–456. 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Ragagnin AMG, Shadfar S, Vidal M, et al. , 2019. Motor neuron susceptibility in ALS/FTD. Front. Neurosci. 13, 532. 10.3389/fnins.2019.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chió A, Traynor BJ, 2014. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17 (1), 17–23. 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J, Doroudchi MM, Nguyen MD, et al. , 2003. A neurotoxic peripherin splice variant in a mouse model of ALS. J. Cell Biol. 160, 939–949. 10.1083/jcb.200205027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, Vale RD, 2008. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451 (7176), 363–367. 10.1038/nature06482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, Lewis J, Clippinger AK, et al. , 2013. Unbiased screen reveals ubiquilin-1 and −2 highly associated with huntingtin inclusions. Brain Res. 1524, 62–73. 10.1016/j.brainres.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackett DL, Werbovetz KA, Morrissette NS, 2010. Isolating tubulin from nonneural sources. Methods Cell Biol. 95, 17–32. 10.1016/S0091-679X(10)95002-4. [DOI] [PubMed] [Google Scholar]
- Sandate CR, Szyk A, Zehr EA, et al. , 2019. An allosteric network in spastin couples multiple activities required for microtubule severing. Nat. Struct. Mol. Biol. 26 (8), 671–678. 10.1038/s41594-019-0257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Otomo A, Ueda MT, et al. , 2018. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J. Biol. Chem. 293 (44), 17135–17153. 10.1074/jbc.RA118.003849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ, 2012. The axonal transport of mitochondria. J. Cell Sci. 118 (23), 5411–5419. 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EJ, Funes S, McKeon JE, et al. , 2021. ALS-linked PFN1 variants exhibit loss and gain of functions in the context of formin-induced actin polymerization. Proc. Natl. Acad. Sci. U. S. A. 118 (23), e2024605118 10.1073/pnas.2024605118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA, 2004. Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779. 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- Shi P, Ström A-L, Gal J, Zhu H, 2010. Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 1802 (9), 707–716. 10.1016/j.bbadis.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota T, Nagata R, Kikuchi S, et al. , 2022. C9orf72-derived proline:arginine poly-dipeptides modulate cytoskeleton and mechanical stress response. Front Cell Dev Biol 10, 750829. 10.3389/fcell.2022.750829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivadasan R, Hornburg D, Drepper C, et al. , 2016. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19 (12), 1610–1618. 10.1038/nn.4407. [DOI] [PubMed] [Google Scholar]
- Skruber K, Read T-A, Vitriol EA, 2018. Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell Sci. 131 (1) 10.1242/jcs.203760jcs203760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skruber K, Warp PV, Shklyarov R, et al. , 2019. Arp2/3 and Mena/VASP require profilin 1 for actin network assembly at the leading edge. Curr. Biol. 30 (14) 10.1016/j.cub.2020.04.085, 2651–2664.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN, Ticozzi N, Fallini C, et al. , 2014. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska JM, Morfini G, Falnikar A, et al. , 2008. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J. Neurosci. 28 (9), 2147–2157. 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann M, Meyer-Ohlendorf M, Achberger K, et al. , 2013. The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and parkinsonian syndromes. J. Neural Transm. 120 (5), 785–798. 10.1007/s00702-012-0910-z. [DOI] [PubMed] [Google Scholar]
- Suarez C, Kovar DR, 2016. Internetwork competition for monomers governs actin cytoskeleton organization. Nat. Rev. Mol. Cell Biol. 17 (12), 799–810. 10.1038/nrm.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy V, Sultana JM, Atkin JD, 2015. Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front. Neurosci. 9, 400. 10.3389/fnins.2015.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Hasegawa M, 2016. Profilin 1 mutants form aggregates that induce accumulation of prion-like TDP-43. Prion 10 (4), 283–289. 10.1080/19336896.2016.1207033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas RP, Chazeau A, Cloin BMC, et al. , 2017. Differentiation between oppositely oriented microtubules controls polarized neuronal transport. Neuron 96, 1264–1271.e5. 10.1016/j.neuron.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry M, Blanchoin L, 2021. Microtubule self-repair. Curr. Opin. Cell Biol. 68, 144–154. 10.1016/j.ceb.2020.10.012. [DOI] [PubMed] [Google Scholar]
- Theunissen F, West PK, Brennan S, et al. , 2021. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl Neurodegener 10, 46. 10.1186/s40035-021-00272-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ti S-C, Wieczorek M, Kapoor TM, 2020. Purification of affinity tag-free recombinant tubulin from insect cells. STAR Protoc 1, 100011. 10.1016/j.xpro.2019.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiloca C, Ticozzi N, Pensato V, et al. , 2013. Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol. Aging 34 (5), 1517.e9–1517.e10. 10.1016/j.neurobiolaging.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp JD, Carney DS, Horazdovsky BF, 2005. Biochemical Characterization of Alsin, a Rab5 and Rac1 Guanine Nucleotide Exchange Factor. Methods in Enzymology. Elsevier, pp. 261–276. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Sala G, Conti E, et al. , 2014. Valproate treatment in an ALS patient carrying a c.194G>a spastin mutation and SMN2 homozygous deletion. Case Rep Neurol Med 2014, 216094. 10.1155/2014/216094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triclin S, Inoue D, Gaillard J, et al. , 2021. Self-repair protects microtubules from destruction by molecular motors. Nat. Mater. 20 (6), 883–891. 10.1038/s41563-020-00905-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor EL, Perkinton MS, Schmidt A, et al. , 2005. ALS2/Alsin regulates Rac-PAK signaling and neurite outgrowth. J. Biol. Chem. 280 (41), 34735–34740. 10.1074/jbc.M506216200. [DOI] [PubMed] [Google Scholar]
- Vale RD, 2003. The molecular motor toolbox for intracellular transport. Cell 112 (4), 467–480. 10.1016/S0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W, Van Den Bosch L, 2017. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech. 10, 537–549. 10.1242/dmm.029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan PS, Miura P, Henderson M, et al. , 2002. A role for regulated binding of p150Glued to microtubule plus ends in organelle transport. J. Cell Biol. 158 (2), 305–319. 10.1083/jcb.200201029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemu A, Atherton J, Spector JO, et al. , 2017. Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 28 (25), 3564–3572. 10.1091/mbc.e17-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, Mathew A, Koushika SP, 2020. Role of actin in organelle trafficking in neurons. Cytoskeleton 77 (3–4), 97–109. 10.1002/cm.21580. [DOI] [PubMed] [Google Scholar]
- Wang JC, Ramaswami G, Geschwind DH, 2021. Gene co-expression network analysis in human spinal cord highlights mechanisms underlying amyotrophic lateral sclerosis susceptibility. Sci. Rep. 11, 5748. 10.1038/s41598-021-85061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki S, Holzbaur EL, 1995. The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc. Natl. Acad. Sci. U. S. A. 92 (5), 1634–1638. 10.1073/pnas.92.5.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Grierson AJ, De Vos KJ, 2018. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 9 (5), 399–408. 10.1080/21541248.2016.1240495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W, Podtelejnikov AV, Di Nardo A, et al. , 1998. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 17 (4), 967–976. 10.1093/emboj/17.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JD, Landers JA, Bingley M, et al. , 2006. The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet. 15 (18), 2763–2771. 10.1093/hmg/ddl212. [DOI] [PubMed] [Google Scholar]
- Wu C-H, Fallini C, Ticozzi N, et al. , 2012. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C-H, Roberts EA, Her L-S, et al. , 2003. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 161 (1), 55–66. 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, McLean J, Robertson J, 2006. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim. Biophys. Acta 1762 (11− 12), 1001–1012. 10.1016/j.bbadis.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Yadav A, Matson KJE, Li L, et al. , 2022. The human motoneuron expression signature is defined by ALS-related genes. bioRxiv. 10.1101/2022.03.25.485808, 2022.03.25.485808. [DOI] [Google Scholar]
- Yang Y, Hentati A, Deng H-X, et al. , 2001. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 29 (2), 160–165. 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Clatterbuck-Soper SF, Jackrel ME, et al. , 2017. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol. 216 (4), 1015–1034. 10.1083/jcb.201608022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Veeranna, Nixon RA, 2017. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 9 (4), a018309 10.1101/cshperspect.a018309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Liu Q, Shen D, et al. , 2019. Mutation analysis of KIF5A in Chinese amyotrophic lateral sclerosis patients. Neurobiol. Aging 73, 229.e1–229.e4. 10.1016/j.neurobiolaging.2018.08.006. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fok AHK, Fan R, et al. , 2020. Specific depletion of the motor protein KIF5B leads to deficits in dendritic transport, synaptic plasticity and memory. ELife 9, e53456. 10.7554/eLife.53456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchi E, Bonetto V, Sorarù G, et al. , 2020. Neurofilaments in motor neuron disorders: towards promising diagnostic and prognostic biomarkers. Mol. Neurodegener. 15, 58. 10.1186/s13024-020-00406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]