

Abstract
Objectives:
Iron oxide nanoparticles have been used to track the accumulation of chimeric antigen receptor (CAR) T-cells with magnetic resonance imaging (MRI). However, the only nanoparticle available for clinical applications to date, ferumoxytol, has caused rare but severe anaphylactic reactions. MegaPro nanoparticles (MegaPro-NP) provide an improved safety profile. We evaluated whether MegaPro-NP can be applied for in vivo tracking of CAR T-cells in a mouse model of glioblastoma multiforme (GBM).
Materials and Methods:
We labeled tumor-targeted CD70CAR (8R-70CAR) T-cells and non-tumor targeted controls with MegaPro-NP, followed by inductively coupled plasma optical emission spectroscopy (ICP-OES), Prussian blue (PB) staining and cell viability assays. Next, we treated forty-two NRG mice bearing U87-MG/eGFP-fLuc GBM xenografts with MegaPro-NP labeled/unlabeled CAR T-cells or labeled untargeted T-cells, and performed serial MRI, MPI and histology studies. The Kruskal-Wallis test was conducted to evaluate overall group differences, and the Mann-Whitney U test was applied to compare the pairs of groups.
Results:
MegaPro-NP-labeled CAR T-cells demonstrated significantly increased iron uptake compared to unlabeled controls (p<0.01). Cell viability, activation and exhaustion markers were not significantly different between the two groups (p>0.05). In vivo, tumor T2* relaxation times were significantly lower after treatment with MegaPro-NP-labeled CAR T-cells compared to untargeted T-cells (p<0.01). There is no significant difference in tumor growth inhibition between mice injected with labeled and unlabeled CAR T-cells.
Conclusions:
MegaPro-NP can be used for in vivo tracking of CAR T-cells. Since MegaPro-NP recently completed Phase II clinical trial investigation as an MRI contrast agent, MegaPro-NP is expected to applied to track CAR T-cells in cancer immunotherapy trials in the near future.
Keywords: MegaPro Iron Oxide Nanoparticles, Glioblastoma, CAR T-Cells, MRI, MPI
Introduction:
Patients with glioblastoma multiforme (GBM) have a 5-year survival rate of only 7.2%(1). Effective therapies are lacking. A new therapy approach involves genetic modification of chimeric antigen receptor (CAR) T-cell to express surface ligands that can recognize brain cancer cells (2). Recent studies reported that a combination therapy of CXCR2-receptor targeted CAR T-cell and ionizing radiation led to significant inhibition of GBM tumor growth in mouse models (3). However, the tumor biology in clinical trials is far more heterogenous in preclinical studies. Clinical experiences with other solid tumors have shown that the efficacy of CAR T-cell therapy varies substantially from patient to patient (4). A non-invasive and clinically translatable imaging technique could help to determine, who might benefit from new CAR T-cell immunotherapies.
We generated CAR T-cells targeting CD70, a tumor antigen that was found to correlate with tumor proliferation, migration and chemokine-mediated immune inhibition in GBM (3, 5, 6). To guide and improve T-cell trafficking, we modified the CD70CAR T cells(6), with an IL-8 receptor, CXCR2 (8R-70CAR). We demonstrate that 8R-70CAR markedly enhances the migration and persistence of modified T cells in tumors, induces complete tumor regression and long-lasting immunologic memory in preclinical tumor models, including GBM, ovarian and pancreatic cancer (3). Because of these findings, a phase-I trial using 8R-70CAR T cells for adults with newly diagnosed GBM will be initiated soon (NCT05353530). While our previous studies have shown that these tumor-targeted CAR T-cell achieved effective tumor growth inhibition in mouse models(3), a medical imaging technology that could visualize and quantify the tumor accumulation of the therapeutic cells in the tumor tissue would greatly enhance our ability to monitor and optimize combination therapies. Previous studies used PET imaging for in vivo CAR T-cell tracking (7). However, potential immune responses against genetically modified cells, cell death induced by radiolabeling, and the need to add PET scans to the clinical workflow represent barriers to this approach (8, 9). Since patients with GBM undergo serial MRIs as part of their routine clinical care, it would be easier from a practical point of view, if therapeutic cells could be visualized with MRI.
Preclinical iron oxide nanoparticles have been used to track the tumor accumulation of CAR T-cell using MRI(10, 11). However, the only nanoparticle currently available for clinical applications, ferumoxytol, demonstrates a limited r2 relaxivity and a risk for severe adverse reactions in some patients, such as hypersensitivity (e.g., pruritus, rash, urticaria, or wheezing), hypotension, and anaphylaxis or anaphylactoid reactions in 3.7%, 1.9% and 0.2% of subjects studied, respectively (12). Anaphylactic reactions are likely in response to ferumoxytol’s carboxymethyldextran coating. MegaPro nanoparticles (MegaPro-NP) are currently investigated in clinical trials (NCT03407495) and are coated with polyethyleneglycol (PEG), which may lead to an improved safety profile compared to Ferumoxytol (13).
Preclinically, we recently used a microfluidics device to mechanically label CAR T-cell with ferumoxytol and tracked the CAR T-cell accumulation in osteosarcomas with MRI (14). However, we did not yet track the accumulation of CAR T-cell in GBM models. Tracking CAR T-cell in the brain involves a variety of factors including the blood brain barrier (BBB) and central hypoxic/necrotic areas in GBM. These factors can negatively affect the tumor accumulation of CAR T-cell and requires sufficient iron loading of the therapeutic cells. Due to its larger hydrodynamic diameter, Megapro has demonstrated increased cellular uptake compared to Ferumoxytol. In addition, the T2 relaxivity of MegaPro (149.3mM-1S-1(0.47 T)) is higher compared to Ferumoxytol (89mM-1S-1(1.5 T, 37°C)(13), which leads to a higher signal-to-noise ratio (SNR) and higher sensitivity to detect CAR T-cell in brain tumors. The goal of our study was to evaluate if MegaPro-NP can track CAR T-cell accumulation in glioblastoma multiforme (GBM) in mouse models with MRI.
Materials and Methods:
CAR T-cell generation
8R-70CAR T cells were generated by transduction of donor T cells with a retroviral vector as previously described(3). Briefly, to produce retrovirus, GP2–293 cells were transfected with 2 μg pMD2.G (Addgene) and 2 μg transfer plasmid using 10 μl Lipofectamine 2000 (ThermoFisher Scientific) in 600 μl OPTI-MEM (ThermoFisher Scientific). Two days later, the supernatants were harvested for CAR transduction. Human peripheral blood mononuclear cells (PBMCs) from healthy donors or GBM patients were obtained. T cells from PBMCs were activated with anti-CD3/anti-CD28 Dynabeads (ThermoFisher Scientific) (cell-to-bead ratio = 1:3) 72 h before the transduction. After PBMC activation with DynaBeads (Day 1) and retroviral transduction (Day 4), 8R-70CAR T cells were harvested and cryopreserved on day 14.”
MegaPro-NP labeling of CAR T-cell
U87-MG/eGFP-fLuc cells will be mentioned as U87MG cells below. MegaPro-NP have a hydrodynamic diameter of 48 nm, a polydispersity index of 0.2 and an r2 relaxivity of 149.3 mM−1s−1 at 0.47 Tesla and 37°C(13). Triplicate samples of 2×106 T cells were incubated for 1, 2, 3 and 4 hours (h), with increasing concentrations of MegaPro-NP (20 μg/mL, 100 μg/mL, 500 μg/mL) in one ml of serum-free AIM-V media with added heparin (2 IU/mL) and protamine (60 μg/mL). Cellular iron was determined by ICP-OES and PB staining (Sigma-Aldrich, HT-20).
In vitro Magnetic Resonance Imaging (MRI) and Magnetic Particle Imaging (MPI)
Triplicate samples of 2 × 106 T cells at 1h and 1, 2, 3 and 4 weeks after labeling underwent MRI on a 7T MRI scanner (Bruker Biospin, Billerica, MA) using T2-weighted fast spin echo (FSE; repetition time TR= 2,400 ms, echo time TE=33 ms) and T2*-weighted multi-echo gradient echo sequences (MGE, flip angle=80°, TR=1,000 ms, TE=3–42 ms). Afterwards, cell samples underwent MPI on a Momentum MPI scanner (Magnetic Insight Inc., Alameda, CA). Image analysis was conducted with VivoQuant software (InviCRO, Boston, MA).
Cell viability and cell function assays
The viability of MegaPro labeled and unlabeled CAR T-cells was measured with a fluorometric assay (Abcam, ab112122). In addition, 2 × 104 U87-MG cells were cocultured with labeled or unlabeled CAR T-cells and cytotoxicity was measured with the bright-GloTM luciferase assay kit (Promega, E2610) using a SynergyH1 Hybrid Reader (BioTeK, Winooski, VT, USA). For cytokine release assays, 1 × 105 labeled or unlabeled CAR T-cells were cocultured with 1 × 105 U87-MG cells and the supernatant was collected on the second day for cytokine release assays using a Luminex human cytokine panel (EMD-Millipore). To measure exhaustion and activation markers, the cells were labeled with CD279 (PD-1), CD57,CD69 and CD25 antibodies (BD Biosciences) and evaluated on a BD Biosciences LSR-II flow cytometer, using FlowJo software (TreeStar, Ashland, OR).
Animal model
All animal procedures were approved by the Stanford Administrative Panel on Laboratory Animal Care (APLAC-12040) and were performed under isoflurane anesthesia. Forty-two eight-week-old female NRG mice (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ, the Jackson Laboratory) received stereotactic injections of 2 × 105 U87-MG cells. Nine additional mice without treatment served as controls for histology. The tumor growth was monitored by BLI, using an IVIS Spectrum (Caliper Life Science) and intraperitoneal injections of D-luciferin (firefly luciferase; 90 mg/kg). When the tumors reached a luminescent flux of 5 × 108 photons/second, animals received two fractionated irradiations at 4.5 Gy/day for two consecutive days. of the whole brain using a PXi X-Rad SmART cabinet irradiator (Precision X-Ray Inc., North Branford, CT) and a 10 mm collimator (15). Seven days after the irradiation, three groups of 14 mice each received intracardiac injections of 12 × 106 MegaPro-NP labeled or unlabeled CAR T-cells or labeled untargeted T-cells. See Figure 1 for the timeline of in vivo animal study.
Figure 1. In vivo study timeline.
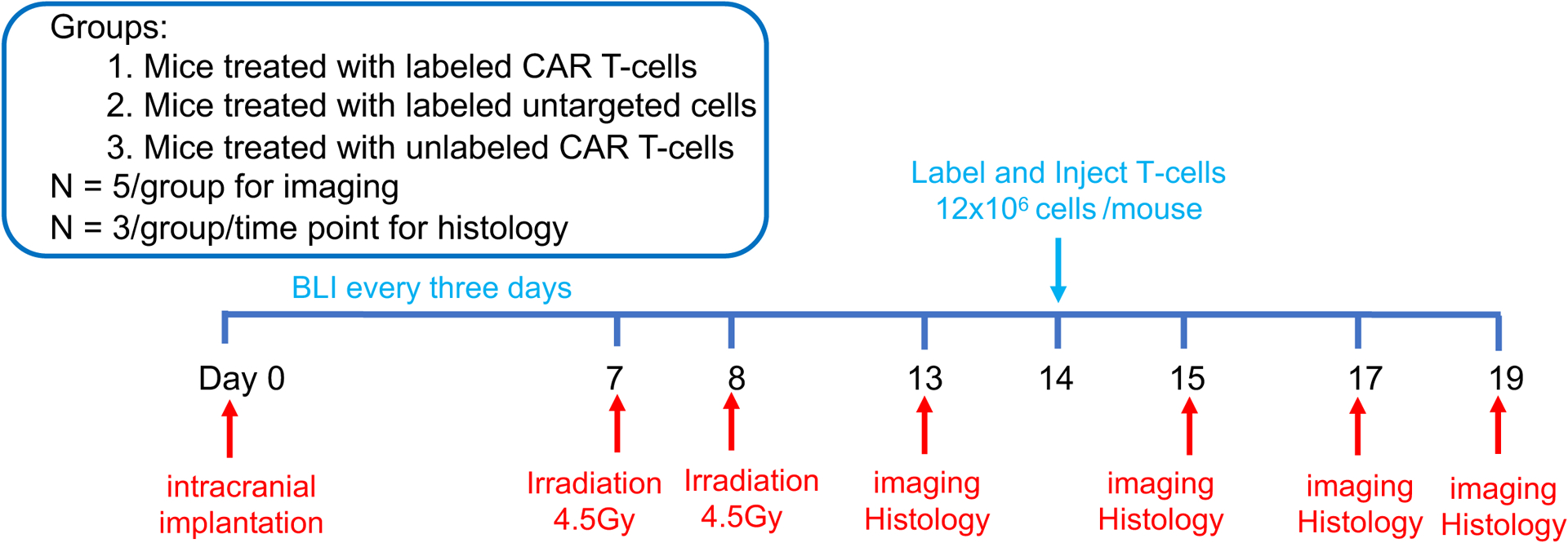
Forty-two NRG mice received two fractionated irradiation doses (2 × 4.5Gy), followed by intra-cardiac injection of 12 × 106 CAR T-cells or untargeted T-cells. The mice underwent BLI, MRI and MPI before, on D1, D3 and D5 after T-cell treatment (n=5). 3 mice/group/time point were sacrificed for histology (Nine additional mice without treatment served as controls). (Created using BioRender.com).
In vivo MRI and MPI
In vivo MRI was performed prior to and 1, 3 and 5 days after CAR T-cell therapy, using a 7T MRI scanner, a Millipede RF coil (ExtendMR LLC, Milpitas, CA) and the following sequences: T2-weighted FSE (TR=3,476 ms, TE=33 ms) and T2*-weighted MGE (TR=1,107 ms, TE=3–42 ms, α=30°). Tumor T2* maps were generated using the Bruker’s ParaVision 6.0.1 software and mean T2* relaxation times were measured using Osirix software (Pixmeo SARL, Bernex, Switzerland).
MPI was performed on a Momentum MPI scanner before and 1, 3 and 5 days after CAR T-cell therapy. Two-dimensional coronal projection images of the animals were acquired (FOV, 6×10 cm) using the standard scan mode. Images were analyzed with VivoQuant software (InviCRO. Boston, MA).
Histology
Three mice per experimental group were sacrificed on days 1, 3 and 5 after treatment and the brains were processed for PB staining (16). Tissue sections were evaluated with a NanoZoomer S60 Digital slide scanner (Hamamatsu Corporation, Bridgewater, NJ). Immunocytochemistry was conducted as previously described (15). Briefly, paraffin embedded tissue was sectioned (5 μm thick) and mounted on superfrost slides. After rehydration, the tissue was permeabilized with 0.1% Triton X-100 and was blocked in blocking buffer (Cell Signaling Technology, 12727S) for 1h. Subsequently, the tissue was incubated with CD45 primary antibody (1:500, Thermo Fisher Scientific, MA517687) at 4°C overnight and incubated with a goat anti-rat IgG (H+L) Alexa Fluor 594 secondary antibody (ThermoFisher scientific, A11007) for 1 hour at room temperature. The tissue was mounted with mounting medium with DAPI (Invitrogen, 00495952) and imaged using a BZ-X710 fluorescence microscope (Keyence, Cupertino, CA). The number of iron or CD45 positive cells was counted with Image-J.
Statistical analyses
The two analyzers were nanoparticle researchers with 5 and 6 years of experience, respectively, in MRI of mouse models of cancer. Data analyzers were blinded with regards to the experimental group of cells or animals. All in vitro work was repeated at least three times in independent experiments. Data are presented as mean ± SD. To exclude the limitation of sample size and normality assumptions, the Kruskal-Wallis test and Mann-Whitney U test were applied for statistical analyses. The Kruskal-Wallis test was conducted to evaluate the overall group difference, and if any group difference was detected (p<0.05), then the Mann-Whitney U test was applied to compare the pairs of groups. As there were 4–5 groups comparisons due to different time points, to correct for multiple comparisons, the threshold for significance level was set to be 0.01 in Mann-Whitney U test, p<0.01 was considered statistically significant results.
Results:
MegaPro-NP labeling of CAR T-cells
Cells labeled with different concentrations of MegaPro-NP for various times showed a significantly different iron content (Kruskal Wallis test; p<0.05; Figure 2A, Figure S1). Specifically, incubation of CAR-T-cells with 500 μg/mL of MegaPro-NP for 4 h led to an intracellular iron content of 2 ± 0.13 pg/cell (Figure 2A, Figure S1), which was significantly higher compared to unlabeled controls (0.07 ± 0.05 pg iron per cell; Mann-Whitney U test: p<0.01). Follow up studies showed that the iron content of the cells decreased over time (Figure 2B). The iron content of MegaPro-NP labeled cells was 0.36 ± 0.06 pg/cell at week 1 after labeling, which was still significantly higher compared to unlabeled controls (Mann-Whitney U test: p<0.01) and reached baseline values at week 4 (Figure 2C).
Figure 2. Evaluating the in vitro uptake of MegaPro-NP by CAR T-cells.
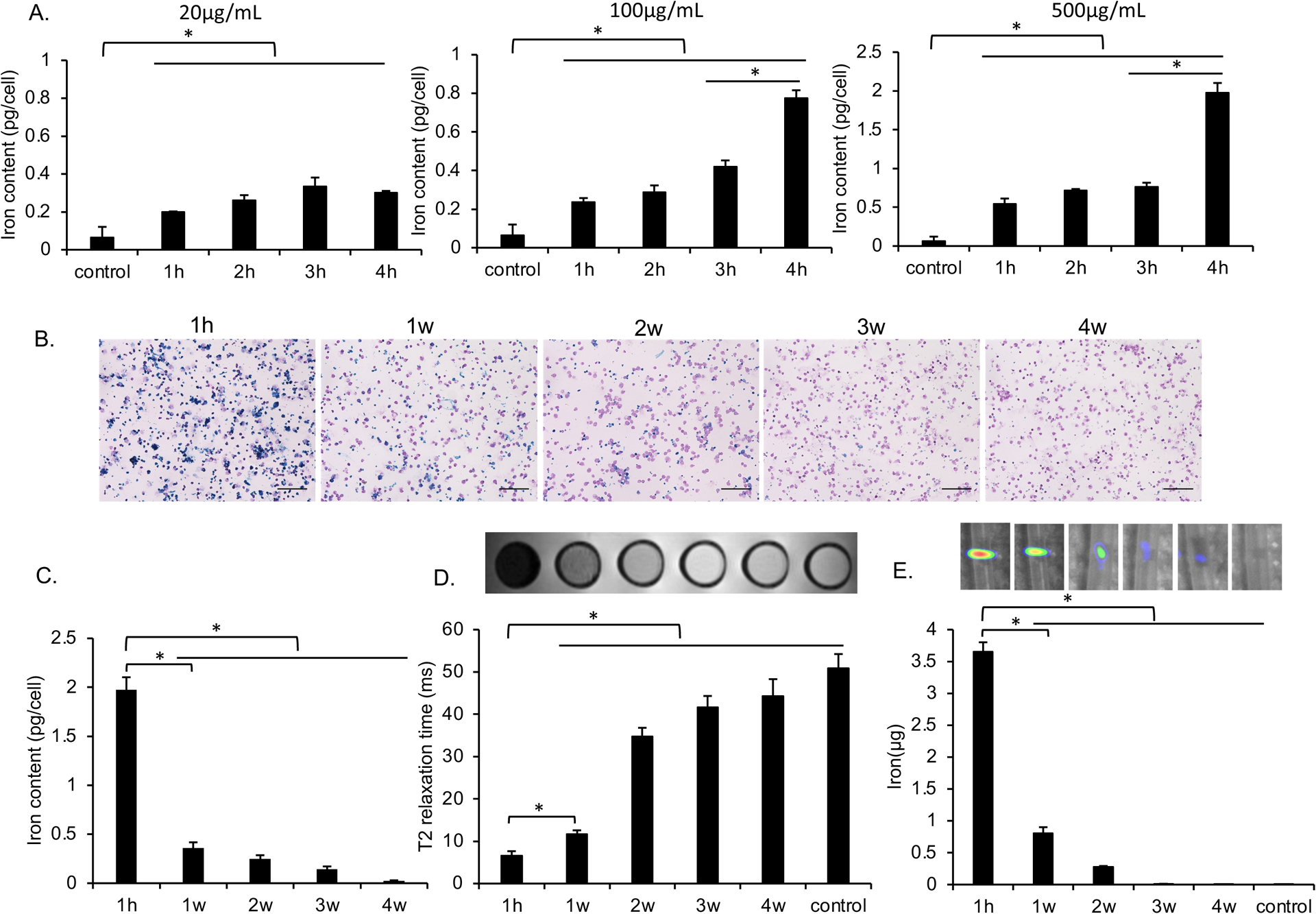
(A) Intracellular iron content of CAR T-cells, as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) after incubation with increasing concentrations of MegaPro-NP for increasing time periods. Data are displayed as means and standard deviations of triplicate experiments for each group. (B) Representative prussian blue stains of CAR T-cells at 1 hour as well as 1, 2, 3 and 4 weeks after labeling with MegaPro-NP at a concentration of 500 μg/mL for 4h. (scale bars, 100 μm) C) Corresponding intracellular iron content of CAR T-cells, as determined by ICP-OES. (D) Corresponding axial T2-weighted MRI images and T2* relaxation times of 2 million CAR T-cells at different time points after labeling with MegaPro-NP at a concentration of 500 μg/mL for 4 hours. (E) Corresponding MPI images of 2 million CAR T-cells at different time points after labeling with MegaPro-NP at a concentration of 500 μg/mL for 4 hours. The Kruskal–Wallis test was used to test the statistical difference among different time points, a p < 0.05 was considered statistically significant. The Kruskal-Wallis test was conducted to evaluate the overall group difference, and if any group difference was detected (p<0.05), then the Mann-Whitney U test was applied to compare the pairs of groups. A value of p < 0.01 was considered statistically significant and denoted with an asterisk.
The MegaPro-NP-labeled cells demonstrated marked hypointense signal on MRI after the labeling procedure and significantly lower T2* relaxation times compared to unlabeled cells (p < 0.01). Follow up MRI demonstrated slowly increasing T2* relaxation times over time (Figure 2D). Similarly, the MegaPro-NP-labeled cells demonstrated significantly higher MPI signal compared to unlabeled cells on day 1 (p < 0.01), which decreased over time (Figure 2E). MegaPro-NP-labeled and unlabeled cells demonstrated significantly different MRI and MPI signal at 1h compared to subsequent time points (Mann-Whitney U test; p<0.01).
The viability and function of the MegaPro-NP-labeled cells were not significantly different from unlabeled cells. When compared with unlabeled controls, MegaPro-NP labeled CAR T-cells demonstrated no significant difference in U87-MG cell killing ability (p>0.05; Figure 3A and 3B). Similarly, there were no significant differences in the secretion of key cytokines (p>0.05; Figure 3C and Figure S2C) or the expression of T-cell markers (exhaustion marker PD-1, senescence marker CD57 and activation marker CD25/CD69) between MegaPro-NP labeled and unlabeled CAR T-cells (p>0.05; Figure 3D). Serial cell viability assays over 5 days did not reveal any significant differences between labeled and unlabeled cells (p>0.05; Figure 3E). The iron is localized in the lysome of the cells after labeling from transmission electron microscopy (Figure S3). The total iron content within all labeled CAR T-cell decreased over an observation time of 4 weeks after labeling while cell numbers increased (Figure S4A and S4B).
Figure 3. Functional in vitro evaluation of CAR T-cells after MegaPro-NP labeling.
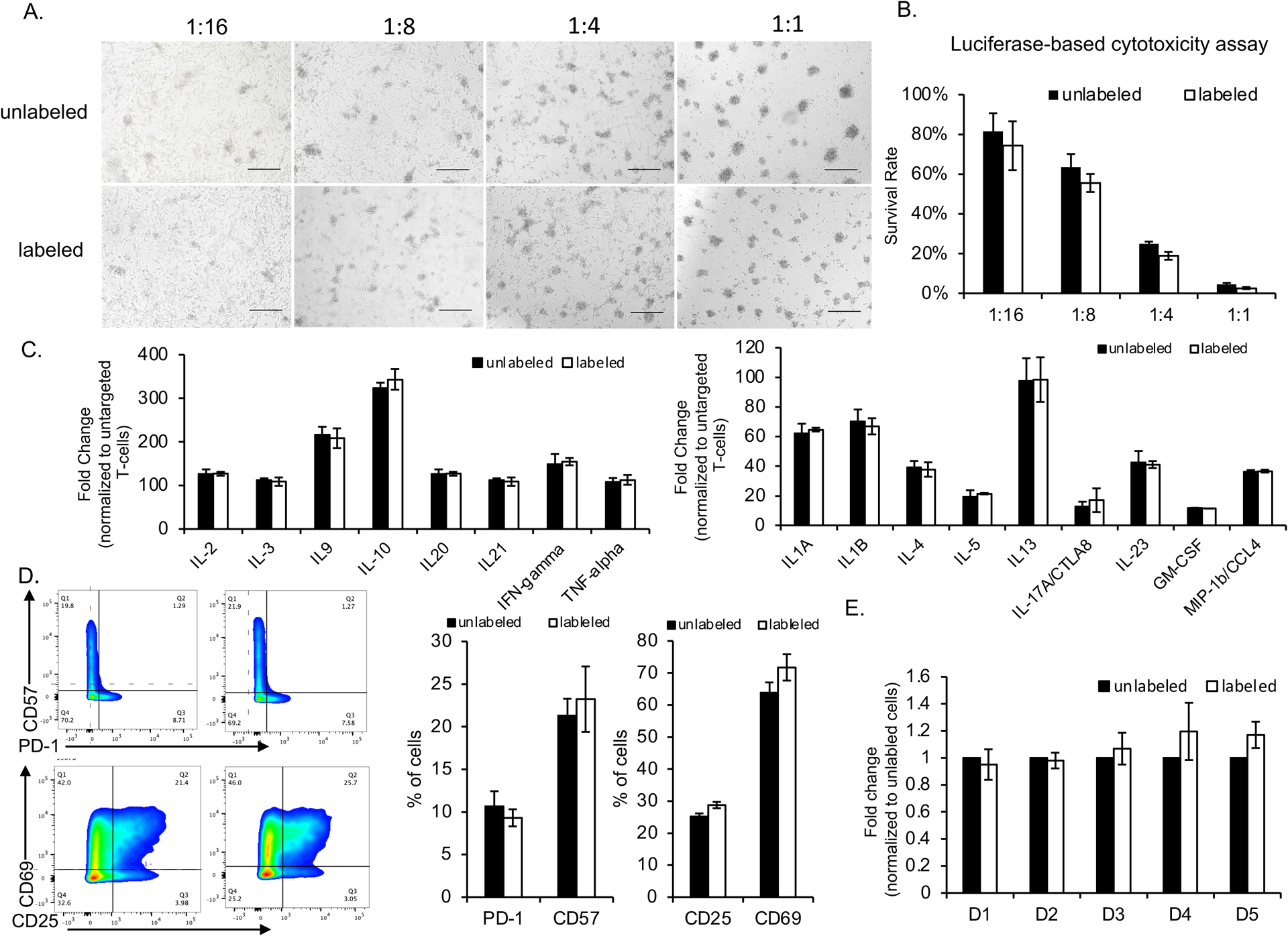
U87-MG cells (2 × 104 cells/well) were co-cultured with CAR T-cells at the indicated effector-to-target ratios and analyzed for various factors. (A) The morphologies of the cells were observed under a light microscope (scale bars, 100 μm). (B) Cancer cell viabilities, as quantified with a Luciferase-based cytotoxicity assay, after co-incubation with CAR T-cells at the indicated effector-to-target ratios. (C) Fold change of expression of various cytokines by labeled and unlabeled CAR T-cells, normalized to untargeted T-cells. (D) Relative number of labeled and unlabeled CAR T-cells which express exhaustion and activation markers. (E) Relative change in proliferation of labeled and unlabeled CAR T-cells at different time points after labeling (D = day). Comparisons between labeled and unlabeled cells were performed using Mann–Whitney U test (Figure 3B, 3C and 3D). For comparisons of three or more groups (Figure E), the values were analyzed with the Kruskal–Wallis test. There were no statistically significant differences between labeled and unlabeled CAR T-cells for any of the data shown.
Monitoring tumor growth by BLI
The BLI signal of GBM treated with MegaPro-NP labeled CAR T-cells increased on day 1 after treatment and then decreased significantly on day 3 and day 5 (Figure 4A and 4B). GBMs treated with untargeted T-cells displayed no such decline; the BLI intensity increased significantly over time (Figure 4A and 4B). Accordingly, tumors treated with MegaPro-NP-labeled CAR T-cells and untargeted T-cells demonstrated significant changes in tumor size on MRI over time (Figure 5A and 5C, Kruskal Wallis test; p<0.05). Tumor size measurements were significantly different between both groups at each time point after therapeutic cell administration (Mann-Whitney U test; p<0.01).
Figure 4. Bioluminescence Imaging (BLI) before and after treatment with CAR T-cells or untargeted controls.
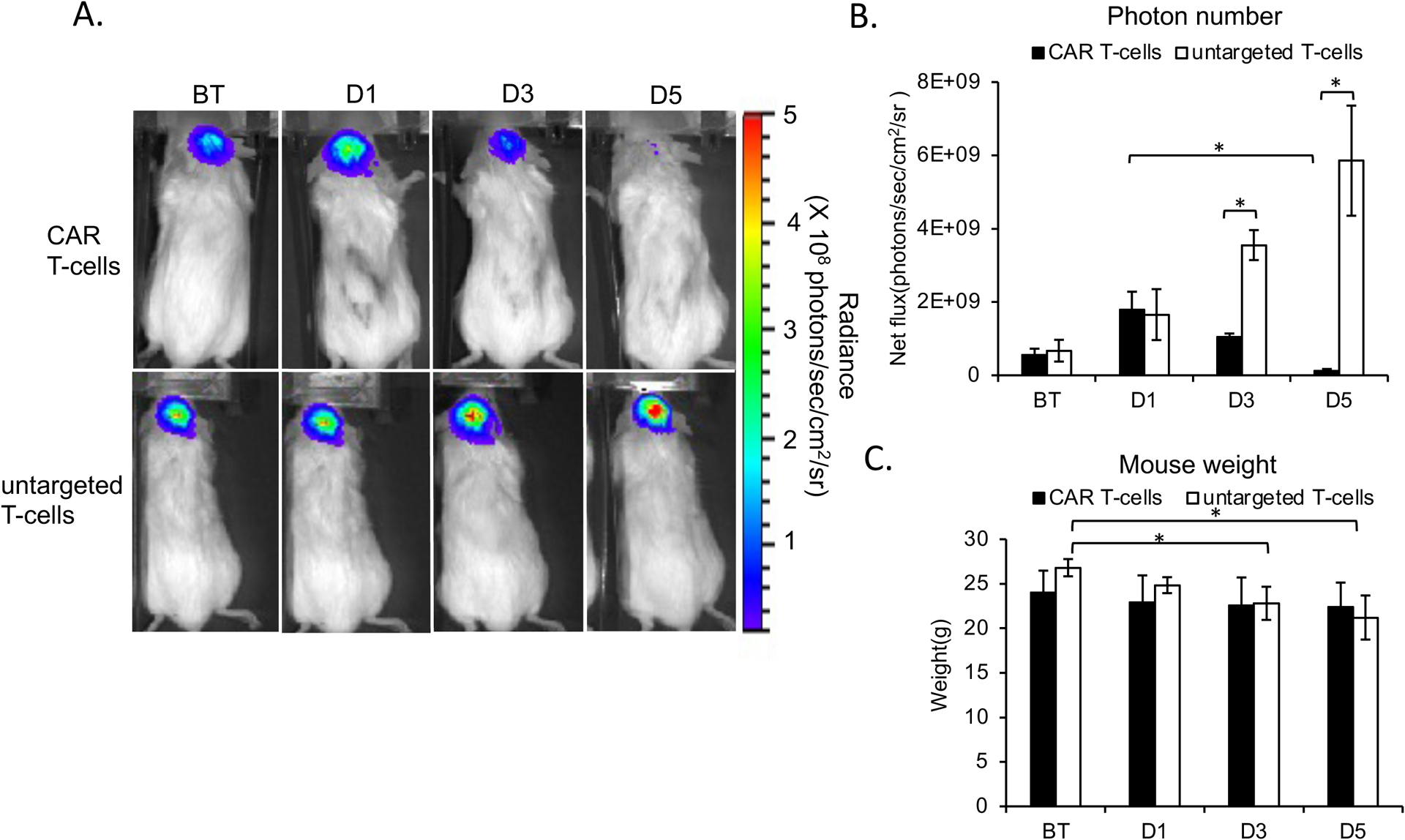
(A) Top row: BLI signal of a representative mouse infused with MegaPro-NP-labeled CAR T-cells increases on day 1 (D1) and decreases on day 3 and 5. Lower row: By contrast, BLI signal of a representative mouse infused with untargeted CAR T-cells increases continuously. (B) Corresponding flux of the tumor tissue demonstrates a significant decrease in tumor burden at day 3 and 5 after CAR T-cell therapy, while the tumor burden continuously increases after treatment with untargeted T-cells. (C) The body weight of mice infused with labeled CAR T-cells remained unchanged while the body weight of mice infused with labeled untargeted T-cells decreased over time. The Kruskal-Wallis test was conducted to evaluate the overall group difference, and if any group difference was detected (p<0.05), then the Mann-Whitney U test was applied to compare the pairs of groups. A value of p < 0.01 was considered statistically significant and denoted with an asterisk.
Figure 5. In Vivo detection of MegaPro-NP labeled CAR T-cells using MRI and MPI.
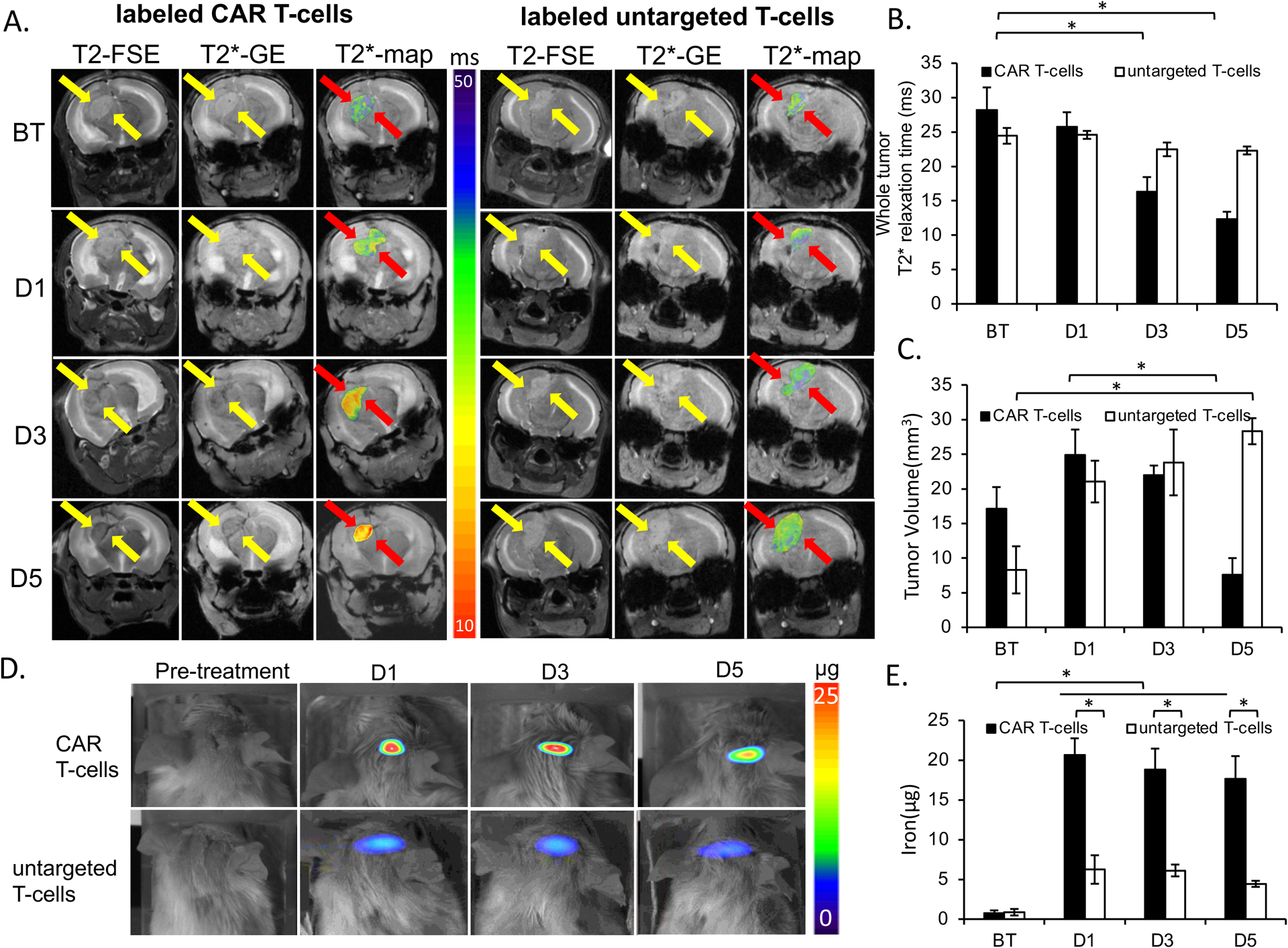
(A) T2-weighted FSE- and T2*-MGE image shows hyperintense U87-MG tumor before therapy (BT). Left: After infusion of MegaPro-NP-labeled CAR T-cells, the tumor demonstrates a signal decline at day 1 (D1), 3 (D3) and 5 (D5). T2* GE scan with superimposed T2* color map demonstrate T2* shortening of the tumor tissue. Right: After infusion of MegaPro-NP-labeled untargeted T-cells, a representative control tumor shows no signal change on T2- and T2*- weighted MRI scans or T2* color maps. (B) Corresponding mean T2* relaxation times of tumors treated with MegaPro-NP-labeled CAR T-cells or MegaPro-NP-labeled untargeted T-cells. Tumors treated with MegaPro-NP-labeled CAR T-cells demonstrated a significant decline in T2* relaxation times. (C) Mean tumor volume of mice treated with MegaPro-NP-labeled CAR T-cells or MegaPro-NP-labeled untargeted T-cells. (D) MPI images of mice treated with MegaPro-NP-labeled CAR T-cells demonstrate marked nanoparticle signal, while MPI images of mice treated with MegaPro-NP-labeled untargeted T-cells demonstrate minor MPI signal. (E) Corresponding quantitative MPI signal of tumors treated with MegaPro-NP-labeled CAR T-cells or MegaPro-NP-labeled untargeted T-cells. D = day. The Kruskal-Wallis test was conducted to evaluate the overall group difference, and if any group difference was detected (p<0.05), then the Mann-Whitney U test was applied to compare the pairs of groups. A value of p < 0.01 was considered statistically significant and denoted with an asterisk.
Mice injected with unlabeled CAR T-cells demonstrated the same tumor BLI signal kinetics compared to MegaPro-NP labeled CAR T-cells. The tumor BLI signal decreased significantly on D5 after treatment (p<0.01; Figure S4C and S4D). The weight of mice treated with untargeted T-cells decreased significantly over time (p<0.05), whereas mice treated with MegaPro-NP labeled and unlabeled CAR T-cells demonstrated no significant change in body weight before and after treatment (p>0.05; Figure 4C and Figure S4E).
In vivo tracking of MegaPro-NP labeled CAR T-cells using MRI and MPI
After intracardiac injection of 12 × 106 MegaPro-NP labeled CAR T-cells, the tumor MRI signal decreased on day 1 and kept decreasing on days 3 and 5 after treatment. By comparison, treatment with MegaPro-NP labeled untargeted T-cells did not lead to visible changes in tumor signal on day 1 after treatment and only slightly decreased tumor signal on days 3 and 5, which displayed no statistical significance compared with baseline values (Figure 5A). Control mice treated with unlabeled CAR T-cells did not show any tumor MRI enhancement (Figure S5A).
Accordingly, tumors treated with MegaPro-NP labeled CAR T-cells demonstrated significantly shorter tumor T2* values on day 3 (p<0.01) and day 5 (p<0.01) after infusion of iron-labeled CAR T-cells compared with baseline values. Conversely, tumors treated with labeled untargeted T-cells and unlabeled CAR T-cells did not show significant changes in T2* relaxation times over time (Kruskal Wallis test; p>0.05; Figure 5B, Figure S5B).
Tumor size: GBM treated with MegaPro-NP labeled CAR T-cells demonstrated an initial increase in tumor size on day 1 and day 3 when compared to baseline. However, the tumor size significantly decreased on day 5 (Mann-Whitney U test: p < 0.01; Figure 5C). By contrast, the tumor size of mice treated with untargeted T-cells increased rapidly at each time point after treatment (Mann-Whitney U test: p < 0.01; Figure 5C). The tumor size of mice was not significantly different between mice injected with labeled and unlabeled CAR T-cells at each of these times (p>0.5; Figure S5C).
Compared to baseline signal, we observed intense tumor MPI signal on day 1 after labeled CAR T-cells treatment, which decreased over time. The tumor MPI signal was significantly different at baseline compared to each time point after infusion of MegaPro-NP labeled CAR T-cells (Mann-Whitney U test; p < 0.01). The MPI signal of tumors treated with MegaPro-NP labeled untargeted T-cells was significantly lower compared to tumors treated with labeled CAR T-cells at each time point of observation (p < 0.01), suggesting lower tumor infiltration by non-tumor-targeted T-cells (Figure 5D and 5E). We did not observe MPI signal in mice injected with unlabeled CAR T-cells (Figure S5D and S5E).
Verifying CAR T-cells tumor accumulation on histology
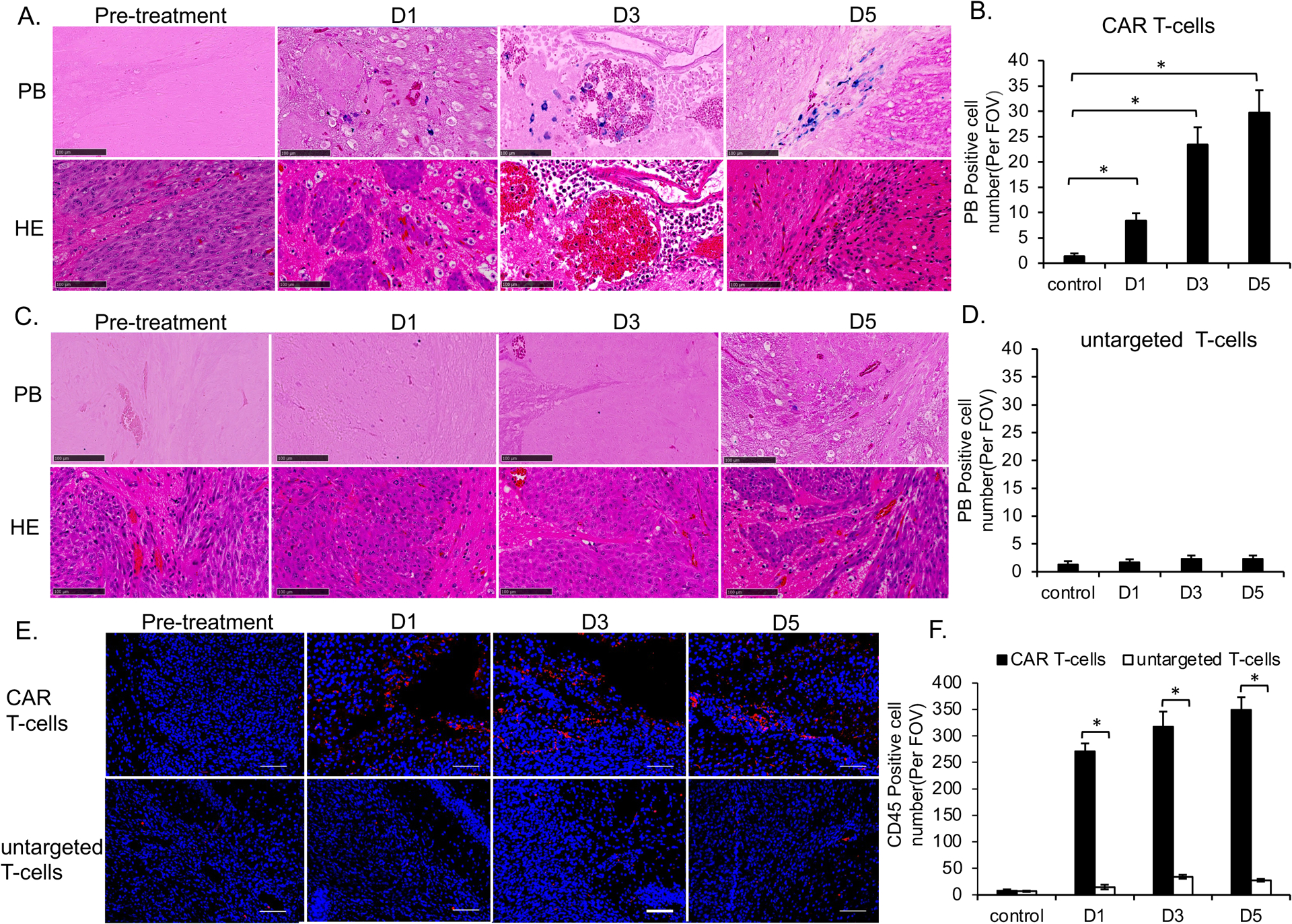
For GBM treated with MegaPro-NP labeled CAR T-cells, PB staining demonstrated an iron accumulation in the tumor periphery on day 1 (p < 0.01), which started to accumulate in the vascular niche on day 3 (p < 0.01) and increased in number on day 5, with development of a central necrosis (p < 0.01; Figure 6A and 6B). While lower quantities of PB positive iron nanoparticles were found in the tumor periphery in labeled untargeted T-cells group (Figure 6C and 6D). PB stains were negative in unlabeled CAR T-cells group (Figure S6A and S6B).
Figure 6. Histology demonstrated the accumulation of MegaPro labeled CAR T-cells in GBM.
(A) Tumor specimen before and after treatment with MegaPro-NP-labeled CAR T-cells. Prussian blue staining (upper row) showed iron containing cells. Hematoxylin and eosin (H&E) staining (lower row) showed the tumor morphology and vascular niches (scale bars, 100 μm). (B) Corresponding quantitative data show increasing quantities of prussian-blue positive cells with increasing time after infusion of MegaPro-NP-labeled CAR T-cells. (C) Tumor specimen before and after treatment with MegaPro-NP-labeled untargeted T-cells. Prussian blue staining (upper row) showed no iron containing cells. Hematoxylin and eosin (H&E) staining (lower row) showed the tumor morphology (scale bars, 100 μm). (D) Corresponding quantitative data show few prussian-blue positive cells. (E) CD45 immunostaining of representative brain tumors at different time points before and after infusion of either MegaPro-NP-labeled CAR T-cells (upper row) or MegaPro-NP-labeled untargeted T-cells (lower row). (F) Corresponding quantitative data show significantly more CD45 positive cells (red) in tumors treated with MegaPro-NP-labeled CAR T-cells than MegaPro-NP-labeled untargeted T-cells (scale bars, 100 μm). The Kruskal-Wallis test was conducted to evaluate the overall group difference, and if any group difference was detected (p<0.05), then the Mann-Whitney U test was applied to compare the pairs of groups. A value of p < 0.01 was considered statistically significant and denoted with an asterisk.
We found a significantly higher number of CD45-positive cells in tumors treated with labeled CAR T-cells versus untargeted T-cells at each time point (p<0.01; Figure 6E and 6F). We observed no significant difference in CD45-positive T-cells in tumors treated with MegaPro-NP labeled versus unlabeled CAR T-cells at each time point (p > 0.05; Figure S6C and S6D).
Discussion:
Our data showed that MegaPro-NP can be used to track the in vivo accumulation of CAR T-cells in GBM with MRI. Since MegaPro recently completed a phase II clinical trial, MegaPro is expected to become available for tracking CAR T-cells in ongoing cancer immunotherapy trials in the near future. Real-time in vivo cell tracking could provide objective information on CAR T-cells distribution in the tumor and normal brain tissue, which could inform early interventions to improve anti-tumor efficacy or reduce side effects (17). Our imaging technique could provide insights on CAR T-cells penetration into tumors and visualize potential off-target effects (2, 18).
Various types of iron oxide nanoparticles have been designed to label cells for biomedical applications (19). For example, highly aminated cross-linked iron oxide nanoworms with a size of 120 nm have been utilized to label CAR T-cells (10). However, these larger nanoparticles cause polarization of CAR T-cells populations toward CD4+ subtypes, and 5–20% of CAR T- cells underwent apoptosis during the labeling process. Another study compared co-precipitated and acid-treated iron oxide nanoparticles coated with 3-aminopropyl-triethoxysilane (APS)-, dimercaptosuccinic acid or dextran for improving their cell labeling efficiency; APS coating yielded a final size of 82 nm and displayed the highest labeling rate (20). However, the coated iron oxide nanoparticles slightly impaired cell migration in response to CXCL12 chemokines. This impaired function of the labeled cells could become a concern for clinical use. Conversely, our data show that the small amounts of iron oxide nanoparticles delivered by our approach have no effect on the in vitro or in vivo function of CAR T-cells. MegaPro-NP labeled CAR T-cells demonstrated similar viability and killing ability as unlabeled CAR T-cell both in in vitro (Fig 3A) and in vivo models (Figure 4B and Figure S4C), while their proliferation was slightly, although not significantly increased (Figure 3E). This is in accordance with previous reports by Berg et, al. who found that iron deprivation can cause growth arrest of T-cells and restored upon addition of exogenous iron (21). Cortes et al. reported that ferritin upregulation may support the survival of inflammatory cells in the microenvironment (22). Additionally, it exhibited comparable cytokine release, activation and exhaustion marker levels in MegaPro-NP labeled and unlabeled CAR T-cell (Figure 3C and 3D). Moreover, MRI and MPI signals indicated the accumulation of CAR T-cell in the tumor region (Figure 5A and 5D), which is verified by Prussian blue staining and CD45 immunofluorescent staining (Figure 6A and 6E). MegaPro-NP demonstrated a favorable safety profile in early clinical trials (NCT03407495) and did not impair CAR T-cell function or viability. While anaphylactic reactions to PEG coatings can occur, they are rare and significantly less frequent compared with adverse reactions against dextran- or carboxymethyldextran-coated nanoparticles (23).
We observed significant MRI signal in both in vitro (Figure 2D) and in vivo models with MegaPro-labeled CAR T-cell (Figure 5A). Previous imaging studies showed the presence of SPIO labeled T cells in rats models reflected by hypointense areas in the sacral part of the spinal cord 3–4 days after transfer (24). Similarly, localized hypointense areas were observed in rats with transplanted hearts and lungs 4 and 5, 24-, and 48-hour post- IOPC-NH2−labeled T-cell infusion, respectively (25). Our previous study also showed significant MRI signal changes after intravenous injection of ferumoxytol labeled CAR T-cells in an osteosarcoma model (14). Megpro-labeled CAR T-cell showed stronger MRI signal effects compared with ferumoxytol-labeled CAR T-cell, which might because MegaPro-NP have significantly higher r2 and r2* relaxivities compared with other nanoparticle compounds currently being applied in clinical applications (26, 27). Additionally, we also observed strong signal of MegaPro-NP in MPI. MPI is an emerging medical imaging technique which aims for high-sensitivity, and real-time imaging. It is reported that the detection limit of MPI is as low as ~ 200 cells (5.4 ng Fe) in a voxel (28), which is more sensitive than MRI. Additionally, MPI provides a sum image of the entire tumor. MRI provides multiple 2D slices of the tumor, which explains the significant signal in MPI at D1 after treatment but not on the MRI images in our study. MPI has not been extensively used to track T cells, one study was able to track T cells in a GBM model after intracerebroventricular administration of ferucarbotran-labeled T cells (29). The number of PB positive CAR T-cells increased from Day 1 to Day 5 while the MPI signal decreased (Figure 5D, Figure 6B). This could be due to a combined effect of iron dilution in proliferating CAR T-cell and iron metabolism within the cells. The total iron content decreased in CAR T-cells over an observation time of 4 weeks after labeling while cell numbers increased, this can be further explained by metabolization of the iron oxide nanoparticles by the cells over time (Figure S4A and S4B).
Of note, our CAR T-cells iron loading procedure only requires the addition of nanoparticles with clinically applicable protamine and heparin to the cell expansion media (30). And, otherwise, would not require any changes to current protocols for CAR T-cell culture or administration. Thus, oncologists could add the labeling solution to their expansion media, while radiologists could focus on the imaging part of cell tracking procedures. The described nanoparticle labeling approach could be readily used for many other cell therapies, beyond our own focus of GBM. However, we recognize several limitations of our work: We tested a specific type of CAR T cells that were engineered to target glioblastoma. Future studies will have to show the value of our technique for tracking therapeutic cells that are targeted to other brain tumors. Detecting iron-labeled CAR T-cell in the brain requires an iron-sensitive pulse sequence and is facilitated in the presence of high intrinsic T2-signal of the underlying tumor tissue at baseline, high vascularity of the tumor to ensure delivery of a high quantity of cells and effective tumor penetration by CAR T-cell. Susceptibility artifacts at the skull base could create challenges for tumors in this location. However, the brain is a favorable target organ for the detection of low quantities of iron due to the intrinsic low iron content, relatively high intrinsic T2* values of malignant brain tumors and clinically established iron sensitive MRI sequences for iron detection. Applications in other target organs might require additional pulse sequence development and optimization. For example, detection of iron-loaded therapeutic cells in organs of the reticuloendothelial system, such as liver, spleen and bone marrow would be considered much more challenging due to the intrinsic iron content of these organs. We infused a relatively high quantity of CAR T-cell in our mouse model, following previously reported protocols that led to tumor regression in this model (3). In humans, typically 1–10 × 106/kg cells are being infused(31). Clinical-translational applications have to show, if the accumulation of these quantities of Mega-Pro labeled cells in human brain tumors can reach MRI detection limits in human patients. MRI is likely not suited for biodistribution studies in human patients due to its limited sensitivity in certain target organs, as outlined above, and due to time constraints. Currently ongoing developments of clinical-translational MPI scanners could address this problem: As shown by our data and confirmed by others (14, 32, 33), the MPI technique is more sensitive than MRI and it selectively depicts nanoparticles and not endogenous iron.
In summary, we developed a clinically translatable CAR T-cell tracking technique using MegaPro-NP. Since MegaPro recently completed a phase II clinical trial and is expected to become available to cancer patients in the near future.
Supplementary Material
Conflicts of Interest and Source of Funding:
The authors acknowledge support from the ReMission Alliance against Brain Tumors, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NIH/NICHD, grant number R01HD103638). Dr. Jianping Huang was supported by the Department of Defense (DOD grant W81XWH-20-1-0726, J.H.) for developing tumor-targeted CAR-T cells. For the remaining authors none were declared.
References:
- 1.Ostrom QT, Patil N, Cioffi G, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(12 Suppl 2):iv1–iv96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 2019;10:2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin L, Tao H, Karachi A, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nabors LB, Ammirati M, Bierman PJ, et al. Central nervous system cancers. J Natl Compr Canc Netw. 2013;11(9):1114–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ge H, Mu L, Jin L, et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int J Cancer. 2017;141(7):1434–44. [DOI] [PubMed] [Google Scholar]
- 6.Jin L, Ge H, Long Y, et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol. 2018;20(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keu KV, Witney TH, Yaghoubi S, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017;9(373). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhirde A, Xie J, Swierczewska M, Chen X. Nanoparticles for cell labeling. Nanoscale. 2011;3(1):142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli F, Varani M, Lauri C, et al. Immune cell labelling and tracking: implications for adoptive cell transfer therapies. EJNMMI Radiopharm Chem. 2021;6(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Gaikwad H, Groman EV, et al. Highly aminated iron oxide nanoworms for simultaneous manufacturing and labeling of chimeric antigen receptor T cells. J Magn Magn Mater. 2022;541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balakrishnan PB, Sweeney EE. Nanoparticles for Enhanced Adoptive T Cell Therapies and Future Perspectives for CNS Tumors. Front Immunol. 2021;12:600659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85(5):315–9. [DOI] [PubMed] [Google Scholar]
- 13.Chen CL, Zhang H, Ye Q, et al. A new nano-sized iron oxide particle with high sensitivity for cellular magnetic resonance imaging. Mol Imaging Biol. 2011;13(5):825–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiru L, Zlitni A, Tousley AM, et al. In vivo imaging of nanoparticle-labeled CAR T cells. Proc Natl Acad Sci U S A. 2022;119(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu W, Klockow JL, Mohanty S, et al. Theranostic nanoparticles enhance the response of glioblastomas to radiation. Nanotheranostics. 2019;3(4):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nejadnik H, Jung KO, Theruvath AJ, et al. Instant labeling of therapeutic cells for multimodality imaging. Theranostics. 2020;10(13):6024–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivastava S, Riddell SR. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J Immunol. 2018;200(2):459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidts A, Maus MV. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol. 2018;9:2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arias LS, Pessan JP, Vieira APM, et al. Iron Oxide Nanoparticles for Biomedical Applications: A Perspective on Synthesis, Drugs, Antimicrobial Activity, and Toxicity. Antibiotics (Basel). 2018;7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanz-Ortega L, Rojas JM, Marcos A, et al. T cells loaded with magnetic nanoparticles are retained in peripheral lymph nodes by the application of a magnetic field. J Nanobiotechnology. 2019;17(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg V, Modak M, Brell J, et al. Iron Deprivation in Human T Cells Induces Nonproliferating Accessory Helper Cells. Immunohorizons. 2020;4(4):165–77. [DOI] [PubMed] [Google Scholar]
- 22.Cortes DF, Sha W, Hower V, et al. Differential gene expression in normal and transformed human mammary epithelial cells in response to oxidative stress. Free Radic Biol Med. 2011;50(11):1565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wenande E, Garvey LH. Immediate-type hypersensitivity to polyethylene glycols: a review. Clin Exp Allergy. 2016;46(7):907–22. [DOI] [PubMed] [Google Scholar]
- 24.Baeten K, Adriaensens P, Hendriks J, et al. Tracking of myelin-reactive T cells in experimental autoimmune encephalomyelitis (EAE) animals using small particles of iron oxide and MRI. NMR Biomed. 2010;23(6):601–9. [DOI] [PubMed] [Google Scholar]
- 25.Liu L, Ye Q, Wu Y, et al. Tracking T-cells in vivo with a new nano-sized MRI contrast agent. Nanomedicine. 2012;8(8):1345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nejadnik H, Pandit P, Lenkov O, et al. Ferumoxytol Can Be Used for Quantitative Magnetic Particle Imaging of Transplanted Stem Cells. Mol Imaging Biol. 2019;21(3):465–72. [Google Scholar]
- 27.Blockley NP, Jiang L, Gardener AG, et al. Field strength dependence of R1 and R2* relaxivities of human whole blood to ProHance, Vasovist, and deoxyhemoglobin. Magn Reson Med. 2008;60(6):1313–20. [DOI] [PubMed] [Google Scholar]
- 28.Zheng B, Vazin T, Goodwill PW, et al. Magnetic Particle Imaging tracks the long-term fate of in vivo neural cell implants with high image contrast. Sci Rep. 2015;5:14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera-Rodriguez A, Hoang-Minh LB, Chiu-Lam A, et al. Tracking adoptive T cell immunotherapy using magnetic particle imaging. Nanotheranostics. 2021;5(4):431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thu MS, Bryant LH, Coppola T, et al. Self-assembling nanocomplexes by combining ferumoxytol, heparin and protamine for cell tracking by magnetic resonance imaging. Nat Med. 2012;18(3):463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei YH, He YZ, Lin XY, et al. Regional Injection of CAR-T Cells for the Treatment of Refractory and Recurrent Diffuse Large B Cell Lymphoma: A Case Report. Front Cell Dev Biol. 2020;8:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindemann A, Ludtke-Buzug K, Fraderich BM, et al. Biological impact of superparamagnetic iron oxide nanoparticles for magnetic particle imaging of head and neck cancer cells. Int J Nanomedicine. 2014;9:5025–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tay ZW, Chandrasekharan P, Fellows BD, et al. Magnetic Particle Imaging: An Emerging Modality with Prospects in Diagnosis, Targeting and Therapy of Cancer. Cancers (Basel). 2021;13(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.