

Abstract
Background & Aims:
Vinyl chloride (VC) monomer is a volatile organic compound commonly used in industry. At high exposure levels, VC causes liver cancer and toxicant-associated steatohepatitis. However, lower exposure levels (i.e., sub-regulatory exposure limits) that do not directly damage the liver, enhance injury caused by Western diet (WD). It is still unknown if the long-term impact of transient low-concentration VC enhances the risk of liver cancer development. This is especially a concern given that fatty liver disease is in and of itself a risk factor for the development of liver cancer.
Methods:
C57Bl/6J mice were fed WD or control diet (CD) for 1 year. During the first 12 weeks of feeding only, mice were also exposed to VC via inhalation at sub-regulatory limit concentrations (<1 ppm) or air for 6 hours/day, 5 days/week.
Results:
Feeding WD for 1 year caused significant hepatic injury, which was exacerbated by VC. Additionally, VC increased the number of tumors which ranged from moderately to poorly differentiated hepatocellular carcinoma (HCC). Transcriptomic analysis demonstrated VC-induced changes in metabolic but also ribosomal processes. Epitranscriptomic analysis showed a VC-induced shift of the modification pattern that has been associated with metabolic disease, mitochondrial dysfunction, and cancer.
Conclusions:
These data indicate that VC sensitizes the liver to other stressors (e.g., WD), resulting in enhanced tumorigenesis. These data raise concerns about potential interactions between VC exposure and WD. It also emphasizes that current safety restrictions may be insufficient to account for other factors that can influence hepatotoxicity.
Keywords: liver disease, chloroethylene, volatile organic compound, hepatocellular cancer, exposure
Introduction
The global burden of chronic liver disease (CLD) has been steadily increasing.(Asrani et al., 2019) CLD is not a single pathological manifestation but rather a spectrum with various stages of severity, ranging from steatosis to steatohepatitis and subsequently to the more severe fibrosis and cirrhosis (Sayiner et al., 2016). The incidence of HCC has also been increasing in parallel with that of CLD,(Villanueva, 2019) as ~90% of HCC occurs on the background of advanced CLD, especially cirrhosis (Farazi and DePinho, 2006). HCC is the 6th most common cancer worldwide (Villanueva, 2019). However, as treatment options for advanced HCC are limited, the 5-year survival rate for HCC is low, and it is consequently the 2nd most lethal solid cancer (Jemal et al., 2017). Unlike most cancers, the mortality rate of HCC has only improved incrementally in recent decades (Llovet et al., 2018).
Liver diseases are well-known to show categorical health disparities within a given population (Nguyen and Thuluvath, 2008). One of the components of consideration in local health disparities is differential exposure to environmental chemicals (Smith and Laribi, 2021). Indeed, disparities in exposure to environmental hazards often overlap with other sociodemographic inequities (Johnston and Cushing, 2020). Although severe CLD is the primary risk factor for the development of HCC, developing studies indicate that overall risk is modified by environmental factors. The prevailing hypothesis is that these risk modifiers act by increasing the mutation frequency in the damaged liver and/or by favoring clonal expansion and progression of precancerous lesions. Moreover, exposure to numerous chemicals has been linked with an increase in HCC incidence, including vinyl chloride (VC) (Beier and Arteel, 2021).
VC is a ubiquitous environmental pollutant. Its global production was recently estimated at 27 million metric tons annually. VC has been identified as a solvent degradation product at waste sites, in the groundwater near military installations and natural gas fracking sites (Kielhorn et al., 2000; Carpenter, 2016). Major environmental exposure risk stems from ambient air (concentrations of VC near production sites can be quite high - PPM range), or from contaminated groundwater (Dimmick, 1981). VC readily volatilizes in buildings located above contaminated groundwater, where these vapors then accumulate ((U.S.EPA), 2000). Owing to its widespread presence and its known potential human risk, VC is ranked #4 on the ATSDR Hazardous Substance Priority List (ATSDR, 2006).
Exposure to some anthropogenic chemicals is associated with increased HCC risk, including VC (2012). However, their prevalence is inadequately quantified, and their epidemiology limited. Moreover, whether this is a direct effect on HCC development or an indirect effect (via causing CLD) is not clear and the diagnosis of liver injury caused by chemicals is challenging, one of exclusion and often requires an interdisciplinary approach (2019). Most research has focused on high occupational exposure to VC (>1 ppm) (Cave et al., 2010). However, recent data demonstrate that VC, at concentrations that are not hepatotoxic per se, exacerbates experimental NAFLD (Lang and Beier, 2018; Lang et al., 2018). Moreover, VC is an independent risk factor for liver cancer in patients with CLD from other causes (e.g., alcohol, HCV, etc), suggesting that VC exposure may enhance HCC risk (Lotti, 2017). Therefore, the major goal of the study was to prove this hypothesis using this newly developed animal model of exposure to low levels of VC in combination with a Western diet.
Materials and methods
Animals and Treatments.
Six-week-old male C57Bl/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in a pathogen-free barrier accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and procedures were approved by the local Institutional Animal Care and Use Committee. Food and tap water were provided ad libitum. Mice were fed either a control diet (CD, Teklad diets, # TD.120336) or a high-fat/high-carbohydrate diet, aka Western diet (WD, 42% fat, 43% carbohydrate, Teklad diets, # TD.07511) for 12 weeks, as previously described (Lang et al., 2018; Lang et al., 2020). Mice were exposed to VC (Kin-tek, La Marque, TX) at ~0.85 ± 0.1 ppm, or air, in inhalation chambers for 6 hours per day, 5 days per week for 12 weeks (Lang et al., 2018; Lang et al., 2020). Body weight and food consumption were monitored throughout the exposure regimen. Animals were sacrificed either immediately following 12 weeks of exposure or following an additional 9 months of housing (±WD feeding). At sacrifice, mice were fasted for 4 hours and anesthetized with ketamine/xylazine (100/15 mg/kg, i.p.). Blood was collected from the vena cava, and citrated plasma was stored at −80°C. Liver tissue was snap-frozen in liquid nitrogen, embedded in frozen specimen medium (Sakura Finetek, Torrance, CA), or was fixed in 10% neutral buffered formalin.
Biochemical analyses and histology.
Plasma levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined using standard kits (see Supplemental Table 1). Formalin fixed, paraffin embedded liver sections were stained with hematoxylin & eosin (H&E) for general morphology. Pathology and tumor burden were assessed by a trained pathologist (S. Monga) in a blinded fashion. Paraffin-embedded sections of the liver were stained with hematoxylin & eosin (H&E) to assess the overall hepatic structure. Cellular proliferation was visualized via PCNA (biotinylated, 1:300, #M0879, DAKO, Carpenteria, CA) and Ki-67 antibodies (1:5000, # ab15580, Abcam, Boston, MA). Cell cycle progression (per 1,000 hepatocytes) was estimated using PCNA staining patterns and cell morphology as described previously (Wang et al., 2000; von Montfort et al., 2010; Beier et al., 2016). Tumor immune microenvironment was assessed via CD45 immunohistochemistry (#98819, Cell Signalling) Immunohistochemistry for angiogenesis using a CD31 antibody (1:50, #550274, BD Pharmingen). Oxidative stress was determined by visualizing 4-hydroxynonenal (4-HNE) adducts via immunohistochemistry using a rabbit anti-mouse primary antibody (1:500, #HNE11-S, Alpha Diagnostics, San Antonio, TX). Frozen samples kept in OCT compound were cut and used for Oil Red O staining (for neutral lipids) and fibrin immunostaining. Sections were stained with Oil Red O (#O-9755, Sigma, St. Louis, MO) solution for 10–12 minutes and then rinsed in distilled water before staining with hematoxylin and mounting with aqueous media. Glutamine synthetase was visualized by indirect immunohistochemistry (1:50, #sc-74430, Santa Cruz Biotechnology, Dallas, TX). Type 3 (reticulin) and type 1 collagen formation was assessed by visualization of silver and Sirius Red (SR) stained liver sections, respectively. Image analysis was performed using Metamorph Image Analysis Software (Molecular devices, Sunnyvale, CA) and expressed as positive staining in % of microscope field (Lang et al., 2019). The sample size for all the immunohistochemistry and staining was between 4–5. For analysis, 10 randomly selected images were taken from each sample and analyzed using the Metamorph Image Analysis Software.
RNA-Seq and data analysis.
Transcriptomic analysis of RNA from whole liver tissue (bulk RNA-Seq) was performed as described previously (Tao et al., 2021). Briefly, total RNA was extracted using Trizol and treated with DNAse1. Ribosomal RNA was removed from the samples using RIBO-ZeroTM Magnetic kit (Epicentre, Madison, WI). RNA was reversetranscribed to cDNA and amplified using TruSeq™ RNA Sample Prep Kit v2 from Illumina, Inc (San Diego, CA). The cDNA library was similarly sequenced in Illumina HiSeq2500 sequencer, see Supplemental Table 2 for number of reads. Quality control on raw RNA-Seq data was performed using the tool FastQC and low-quality reads and adapter sequences were filtered out using the tool Trimmomatic. The surviving reads were then aligned to the mouse reference genome mm10 by STAR aligner, and the genes were quantified using the tool HTSeq. Then differential expression analyses, using R package DESeq2, were performed to compare the pairwise experimental groups. Differentially expressed genes (DEGs) were defined by FDR=5% and fold-change equal to or greater than 1.5. These DEGs were further used as input for ingenuity pathway analysis (IPA) to detect significantly-enriched pathways and common upstream regulators (see Supplemental Table 3). Pathway analyses on Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and JASPAR databases were also performed using the DEGs. Significant pathways were defined by FDR=5%.
The Cancer Genome Atlas (TCGA) data mining.
To explore the TCGA database, gene expression profiles for 374 tumors and 50 normal (or adjacent tumor) tissues with HCC and RNA sequencing data were collected from Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/) as previously described (Tao et al., 2021). Differential expression analysis comparing normal and tumor samples were performed by R package DESeq2. The potential association between RNA expression patterns and TCGA clinical features of HCC patients was compared, which included age of first diagnosis, gender, race, risk factors, fibrosis score (Ishak scale), tumor stage, tumor grade and vital status. The frequency of missing data at baseline was 0% for sex, 0.2% for Age, 2.4% for race, 5.1% for Risk Factors, 42.5% for fibrosis score, 6.1% for tumor stage, 0.8%. Vital status for 8.8% patients was lost to follow-up.
In order to compare the mouse study with human TCGA cohort, DEGs comparing the experimental groups in the mouse model were recruited as biomarkers. The mouse genes were first mapped to human homology genes via the MGI database. Then these markers were applied to the TCGA liver hepatocellular carcinoma (LIHC) cohort. Hierarchical clustering was then performed on the tumor cases to group all the HCC patients into subtypes based to the mouse signatures selected in our study. The resulting marker genes were grouped into clusters as well. Kaplan-Meier survival curves were plotted on the two groups clustered by these gene markers and p-value was calculated by the log rank test. To further explore the consistency between mouse and human studies, common differentially expressed genes in both our mouse model and the TCGA cohort were detected. Pearson correlation and Spearman’s correlation were calculated on these common DEGs.
Epitranscriptomic analysis.
Total RNA was extracted from whole liver. RNA extracts were denatured at 90 °C for 3 min and then chilled in an ice-water for 3 min. To detect modifications of RNA on nucleosides level, 15 μg RNAs were first mixed with 240 units nuclease S1 and 2 μL of reaction buffer with pH= 4.5 (500 mM sodium acetate, 2.8 M NaCl, and 45 mM ZnSO4). The mixture was incubated at 37°C for 4 h. After removing nuclease S1 using a microcon centrifugal filter (Microcon YM-10), the pH of the solution was adjusted to basic using 2.5 μL of a mixture with pH = 9.3 (1 mM MgCl2, 0.1 mM ZnCl2, 1 mM spermidine, 50 mM Tris-HCl) and 0.5 units of phosphodiesterase I was added. The solution was incubated at 37 °C overnight, and phosphodiesterase I was then removed using Microcon YM-10. Finally, 3 units of antarctic phosphatase were added. The solution was incubated at 37 °C for 4 h and antarctic phosphatase was removed using Microcon YM-3.
All samples were analyzed randomly on a Thermo Q Exactive HF Hybrid Quadrupole-Orbitrap Mass Spectrometer coupled with a Thermo DIONEX UltiMate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) using the method developed previously.(He et al., 2019) To obtain full MS data, every sample was analyzed by LC-MS in positive mode (+). For metabolite identification, one pooled sample for each group was analyzed by LC-MS/MS in positive mode to acquire MS/MS spectra at three collision energies (20, 40, and 60 eV).
For LC-MS data analysis, XCMS software was used for spectrum deconvolution, and MetSign software was used for cross-sample peak list alignment, normalization, and statistical analysis. To identify nucleosides, the LC-MS/MS data of the pooled samples were matched to the MS/MS spectra of 94 nucleoside standards recorded in Compound Discoverer (v3.2, Thermo Scientific) that contains parent ion m/z value, retention time, and MS/MS spectra. Chemspider and mzCloud were used for the identification of unknown compounds. The threshold for the spectral similarity of the MS/MS spectra of a nucleoside standard and a spectrum of the pooled sample was set as ≥ 40 with a maximum score of 100, while the thresholds of the retention time difference and m/z variation window were set as ≤ 0.15 min and ≤ 5 ppm, respectively.
Statistics.
Summary data represent means ± SEM (n = 4–5). ANOVA with Bonferroni’s post-hoc test or the Mann-Whitney rank sum test was used for the determination of statistical significance among treatment groups, as appropriate. A p-value < 0.05 was selected before the study as the level of significance.
Results
VC increases WD-induced tumor formation.
Recent studies by this group with a novel mouse model determined the impact of VC inhalation at levels below the current OSHA limit (<1 ppm) on the development NAFLD to identify potential mechanisms of VC induced liver injury (Lang et al., 2018; Lang et al., 2020). In that model, VC exposure alone caused no overt liver injury in mice fed CD. However, in experimental NAFLD caused by WD feeding, 12 weeks of VC significantly increased liver damage as shown by elevated serum transaminases and steatosis (Figures 1B–D). Moreover, VC caused metabolic dyshomeostasis and enhanced WD-induced oxidative and endoplasmic reticulum stress (Chen et al., 2019). Here, the long-term impact of this transient VC exposure on later development of liver cancer was determined. Specifically, following the 12-week VC exposure, mice were returned to room air and continued to be fed WD (or CD) for 9 months (while not exposed to VC; timeline, Figure 1A).
Figure 1: VC enhances WD-induced tumorigenesis.
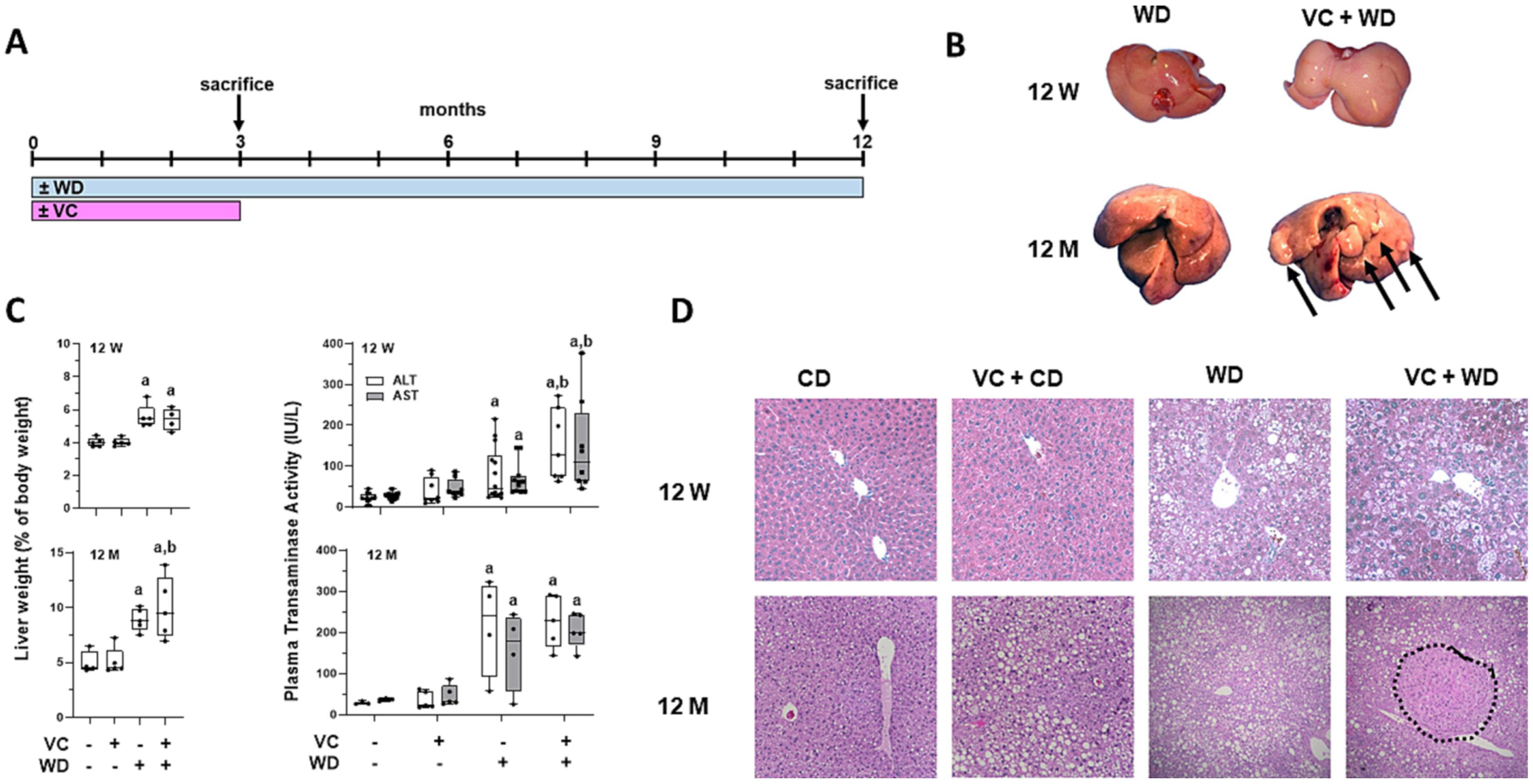
A: Timeline of exposure, feeding and sacrifice regimen. B: Gross liver comparison of WD vs. VC+WD at 12 weeks (12 W) and 12 months (12 M). Visible tumors at 12 M in the VC + WD group only (denoted via arrows). C: VC significantly increased LW:BW ratio, tumor number/size in the WD group only at the 12-month time point. D: VC enhanced WD-induced elevation in transaminases (ALT and AST) after 12 W. There was no difference in transaminase levels 9 months after cessation of VC exposure in the CD or WD group. E: H&E stain at 12 W and 12 M; HCC outlined; 4 × magnification. a, p<0.05 compared to control; b, p<0.05 compared to no VC. n=5 per group.
The were no changes in overall body weight (BW) gain caused by VC (BW in g: CD: 40.4±3.28, CD+VC: 39.9±1.43, WD: 52.3±2.05, WD+VC: 52.0±2.55). CD livers were normal for aged mice, with mild pericentral and midzonal steatosis that was predominantly macro-vesicular (Figures 1D and 2A). Quantitative lipid analysis demonstrated no significant effect of VC in mice fed CD (Figure 2 A, F and G). WD feeding for 1 year caused lobular steatosis (macro- and micro-vesicular), while WD+VC also caused well circumscribed neoplastic foci that contained no or modest steatosis (Figures 1D and 2A). In the WD group, 20% of the livers showed the presence of neoplastic foci, none of which showed the histologic features of HCC. In contrast, in the VC+WD group, 100% of the livers showed tumors, most of which were validated to be HCC by pathologic assessment and IHC (e.g. H&E, glutamine synthetase, CD45, Ki-67, PCNA Figures 1B, 1D and 2.
Figure 2: VC enhances WD-induced tumor number and proliferation after 12 months.
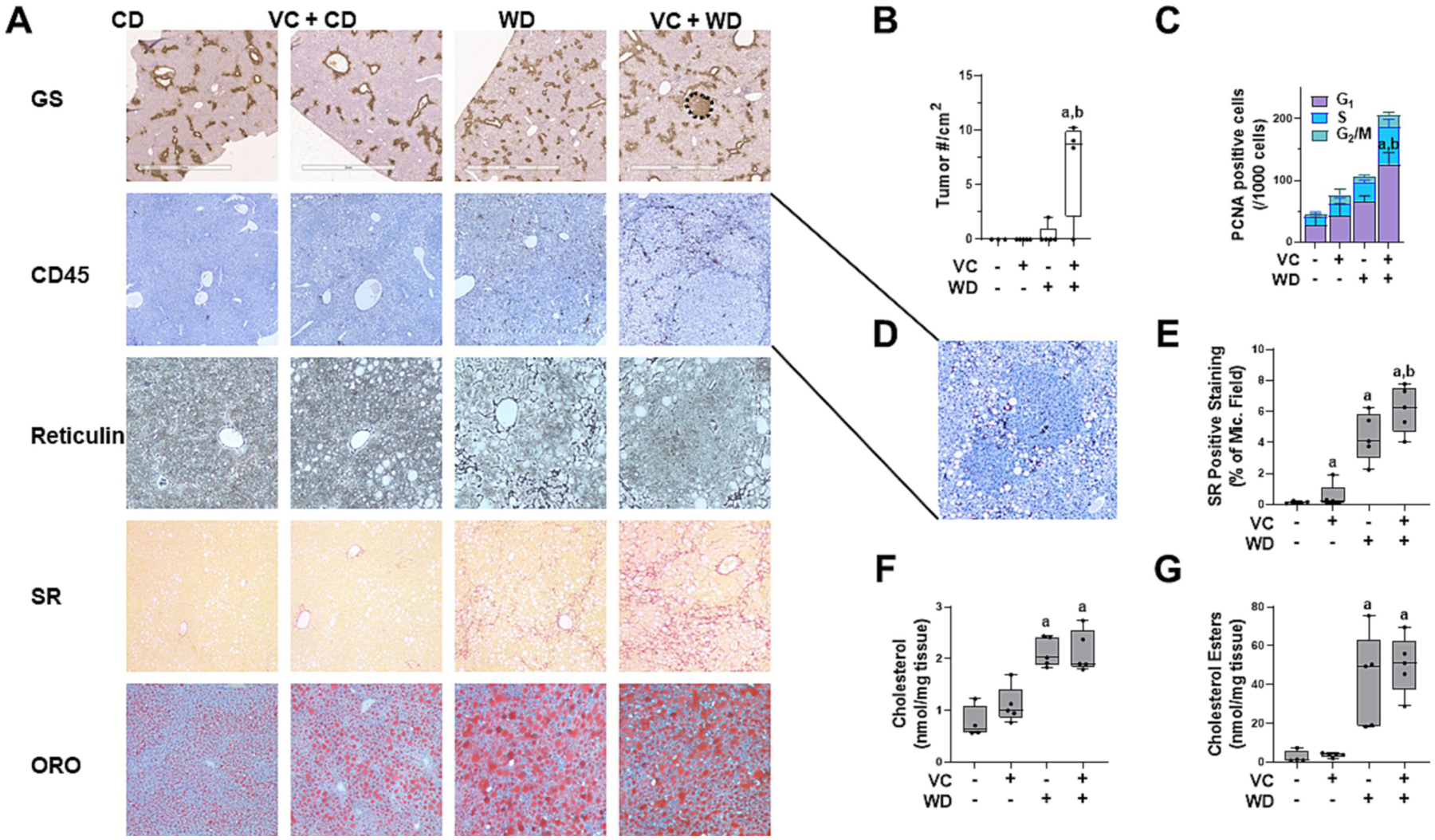
A: Immunohistochemistry was performed for glutamine synthetase(nodule outlined, bar represents 2 mm), and CD45 (4 × magnification). Liver sections were stained for reticulin (20 × magnification), Sirius Red (SR, collagen; 10 × magnification) and Oil Red O (ORO, lipids; 10 × magnification). B: VC significantly increased tumor number/cm2 and C: number of PCNA+ cells in the WD group. PCNA+ cells were counted and graphed as positive cells according to their stage in the cell cycle (G1 – gap1, S - synthesis, G2 – gap2/M - mitosis). D: Inset of 8x magnification of tumor nodules with CD45+ cells in the VC + WD group. E: SR positive staining was expressed as % of microscope field. F: Cholesterol and G: cholesterol esters were measured in hepatic lipid extracts. a, p<0.05 compared to control; b, p<0.05 compared to no VC. n=5 per group.
Tumor burden is depicted in liver weight (LW):body weight (BW) ratios and tumor number/cm2 (Figures 1C and 2B). The tumors from WD+VC were visible as well-circumscribed lesions with Ki67+ (Supplemental Figure 1) and PCNA+ cells (Figure 2C). Pathologic assessment demonstrated tumors ranging from moderately- to poorly-differentiated HCC with clear basophilic cytoplasm, moderate to high nuclear atypia, loss of the reticulin framework, and modest to high peritumor and moderate intratumor CD45+ inflammatory cell infiltration (Figure 2A and 2D). While no fibrosis was evident in CD or VC+CD groups, there was evidence of fibrosis in WD regardless of VC exposure, but more extensive fibrosis was visible in VC+WD as shown by Sirius red staining (Figure 2). Thus, 12 weeks transient exposure to VC at the beginning of WD feeding clearly exacerbated the normal tissue damage, increased fibrosis and inflammation creating a micro-environment permissive to tumor initiation and growth that led to development of HCC.
The enhancement of HCC by VC exposure leads to a unique transcriptomic phenotype.
To shed light on the potential mechanisms by which VC exposure enhanced HCC development caused by WD, transcriptomic analysis (bulk RNA-Seq) was performed. VC exposure (in contrast to air controls) significantly changed the expression of several genes at 12 months in WD-fed mice (DEGs up: 201, down: 168; Figure 3A), a robust increase in changes compared to the 12-week time point in the WD+VC group (DEGs up: 70, down: 60; Supplemental Figure 2). Importantly, RNA-Seq result confirmed that VC enhanced the development of WD-induced HCC, as demonstrated by ‘top significant diseases and disorders’ (Supplemental Figure 4). Based on the DEGs, GO, KEGG and JASPAR pathway analyses were performed, demonstrating 62 significantly changed pathways, including several involved in metabolic processes such as lipid metabolism that were robustly induced at 12 months by the combination of transient VC exposure and WD (Figure 3B and Supplemental Figure 3). More specifically, these included GO terms for fatty acid metabolic process, cholesterol metabolic process, and sterol metabolic process. In addition, Ingenuity Pathway Analysis (IPA) was performed on the DEGs. Employing the IPA Upstream Regulator analytic, PPARα signaling (Z-score=−2.0; p=8.5×10−13) was predicted to be downregulated in WD-fed mice transiently exposed to VC vs. air. Despite the robust increase in cholesterol metabolic processes, there was no difference in oil red-o (ORO) positive staining and cholesterol or cholesterol esters between WD and WD with transient VC exposure (Figure 2).
Figure 3: RNA-Seq analysis of the effects of transient VC and WD at the 12-month time-point.
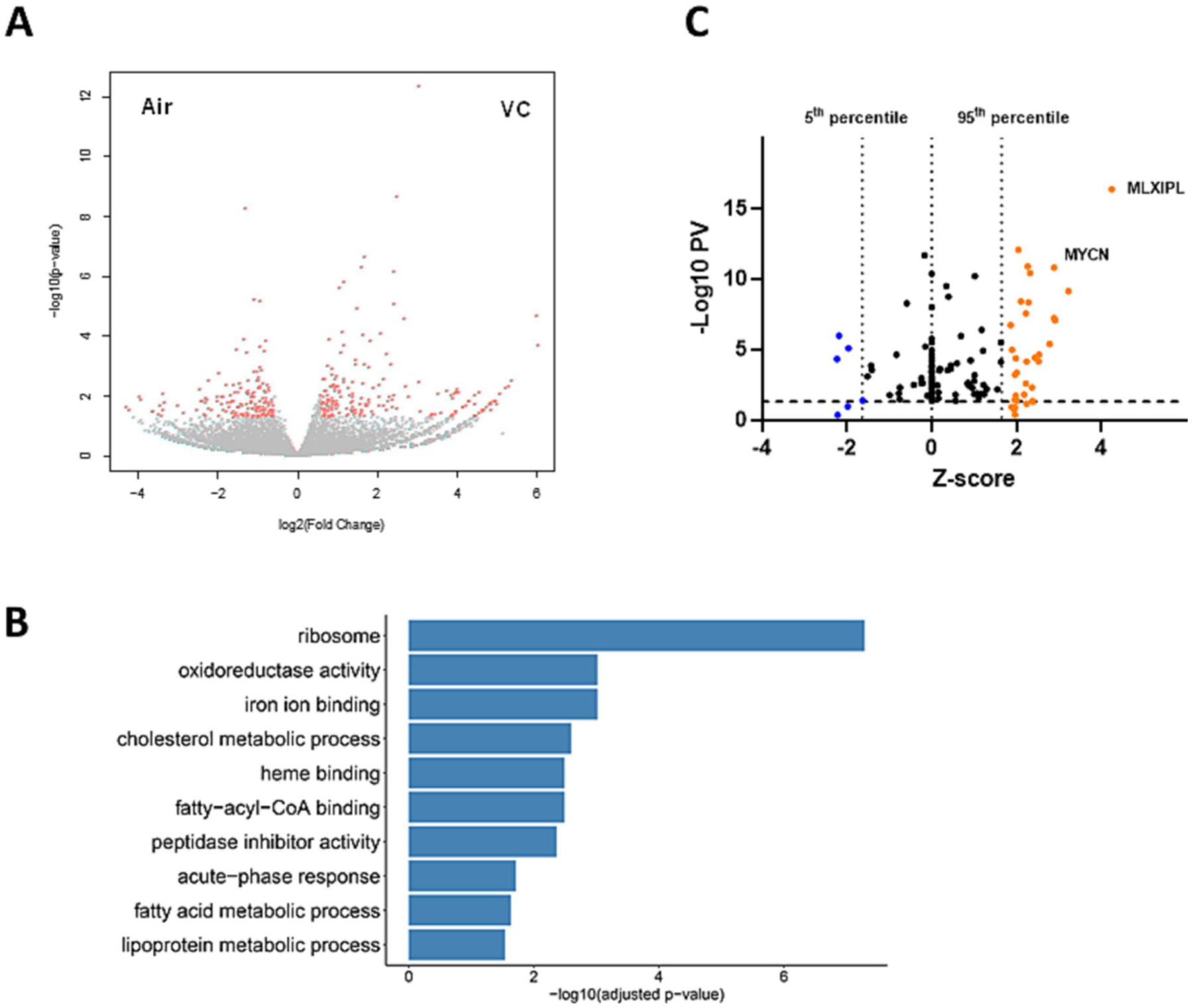
Livers of mice from the 12-month time point were analyzed by bulk RNA-Seq. n=4 per group. A: volcano plot comparing the expression pattern in mice exposed to VC (versus air) at the 12-month time point. Each dot represents a gene, with red dots highlighting significantly up- or down-regulated genes. B: top GO terms identified changed by pathway enrichment analysis. C: modified volcano plot based on IPA upstream regulator analysis, using significance (−Log10PV) and number of standard deviations (Z-score). Each dot represents the transcription factors on livers of mice exposed to WD vs. VC+WD at 12 months. The p-value for significance is plotted as a function of the Z-score. MLXIPL: (ChREBP) promotes HCC proliferation. MYCN: (N-Myc) proto-oncogene; associated with HCC.
In addition to the changes in metabolic processes, VC also caused major changes in ribosomal processes and RNA processing at the 12-month time point in mice fed a WD (Figure 3B and Supplemental Figure 3). While this pathway for ribosome also came up in the CD group, the p-value was much lower (KEGG P-value: 5; Supplemental Figure 5) compared to the WD group in which it is the top significant pathway (KEGG P-value > 20; Supplemental Figure 3). Employing the IPA Upstream Regulator analytic, VC significantly increased (compared to WD) key regulators of metabolic pathways and ribosomal processes, including MLXIPL (up; carbohydrate-responsive element binding protein, ChREBP), MYCN (up; N-Myc: regulates ribosome biogenesis and mitochondrial gene expression programs; Figure 3C). When the expression pattern caused by VC exposure (VC+WD 12 months vs. 12 weeks; Figure 4) was analyzed (DEGs up: 213, down: 267; 26 significant pathways, Figure 4B), employing the IPA Upstream Regulator analytic, dramatic changes in key regulators of metabolic pathways and ribosomal processes, including MLXIPL and MYCN (both up; analogus to data from the 12 month time-point, Figure 3C), ACOX (down; involved in β-oxidation) and LARP1 (down; involved in ribosomal biogenesis) (Figure 4C). This pattern was also reflected in the ‘top significant pathways’ (Figure 4D), highlighting the EIF2 signaling pathway, which is involved in translation, initiation, and assembly of the ribosomal subunits (see below).
Figure 4: RNA-Seq pathway analysis of the effects of transient VC.
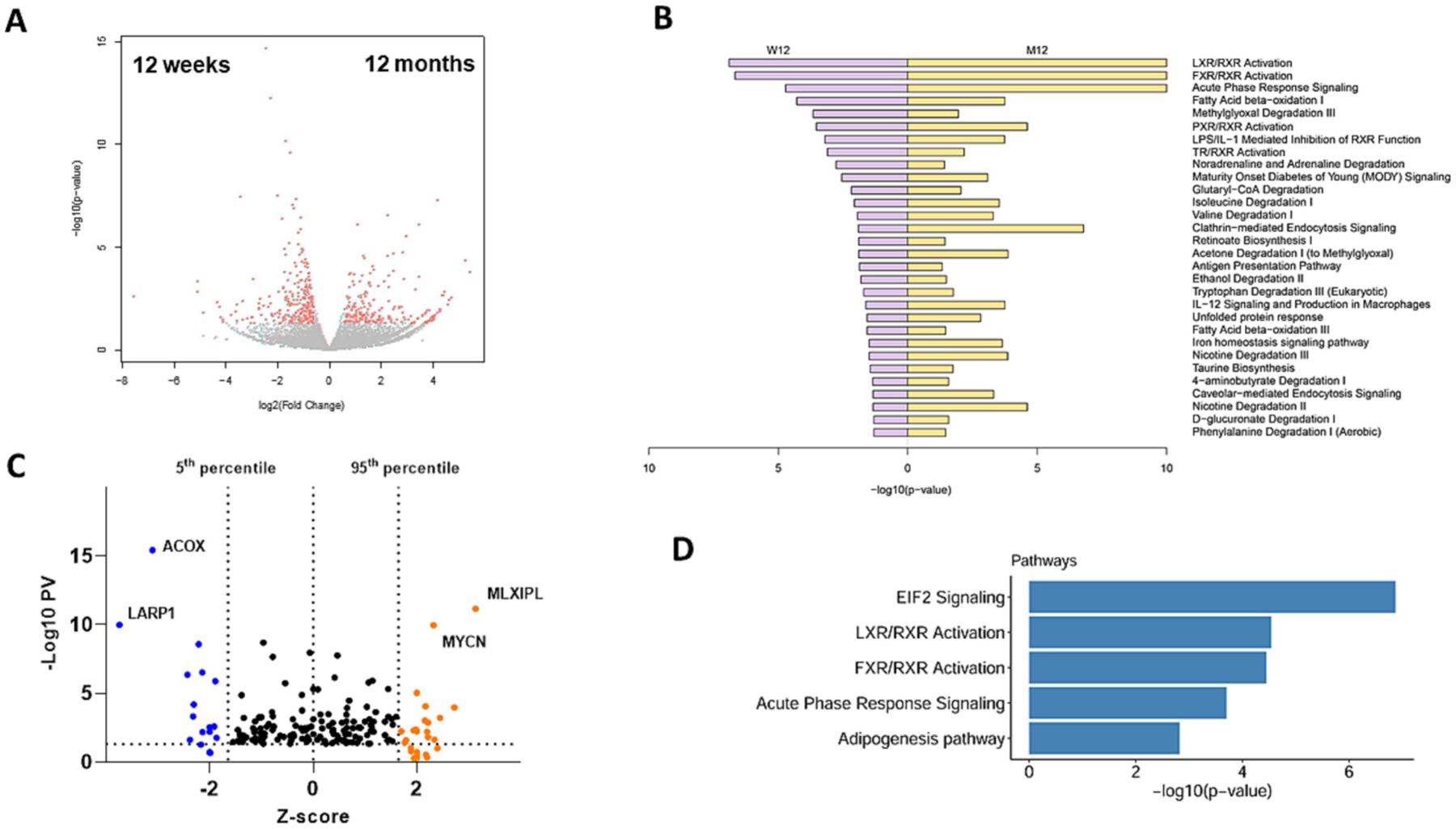
Livers of mice exposed to WD+VC from both time points were analyzed by bulk RNA-Seq. n=4 per group. A: volcano plot comparing the expression patterns at 12-months vs. 12-weeks. Each dot represents a gene, with red dots highlighting significantly up- or down-regulated genes. B: top GO pathways identified changed by pathway enrichment analysis, comparing VC+WD at 12 weeks vs. 12 months. C: modified volcano plot based on IPA upstream regulator analysis, using significance (-Log10PV) and number of standard deviations (Z-score). Each dot represents the transcription factors on livers of mice exposed to VC+WD at 12 weeks vs. 12 months. The p-value for significance is plotted as a function of the Z-score. D: Significant pathways detected by IPA. ACOX: first enzyme in β-oxidation pathway; decrease associated with poor outcome in HCC. LARP1: RNA-binding protein; ribosome biogenesis. MLXIPL: (ChREBP) promotes HCC proliferation. MYCN: (N-Myc) proto-oncogene; associated with HCC. EIF2: stress-related master controller of RNA processing.
Biomarkers in mouse model provide insight in human HCC cohort.
The RNA-Seq results provide weight-of-evidence that transient VC exposure can enhance the development of HCC caused by WD, as demonstrated by ‘top significant diseases and disorders’ (Supplemental Figure 4), via mechanisms likely involving altered ribosomal processes (Figure 4 and Supplemental Figure 4). However, whether these results have relevance to human HCC remained unclear. To address this, all changed genes that are involved in ribosomal processes (KEGG: 03010; see Figure 3 and Supplemental Figure 3) were applied to human ortholog expression in the Liver Hepatocellular Carcinoma (LIHC) sequencing results in the Cancer Genome Atlas (TCGA). These signature genes clustered all the TCGA tumor samples into 5 hierarchical clusters (Figure 5A). To determine if the tumors occurring in WD-fed mice exposed to transient VC share molecular signatures with HCC patients, the degree of similarity between the human differential expression analysis of HCC cases and the VC mouse model were assessed. First, differentially expressed genes (DEGs) from the TCGA cohort comparing normal vs tumor samples and from the mouse model were selected. Next, mouse DEGs were mapped to their human orthologs according to the MGI mouse-human ortholog database (http://www.informatics.jax.org/). Common up- and down-regulated genes in the cohorts were shown in Figure 5B. This analysis revealed a significantly high correlation between the sets of changes in two cohorts (groups 3 and 4; Figure 5A), with a Spearman’s correlation of 0.71 with a p-value of 2.2×10−16 and with a Pearson correlation of 0.681 with a p-value of 9.741×10−13 (Figure 5B). Taken together, these data indicate that the key transcription changes observed in our mouse model of VC exposure have strong expression concordance with a subset of human HCC.
Figure 5: TCGA data analysis.
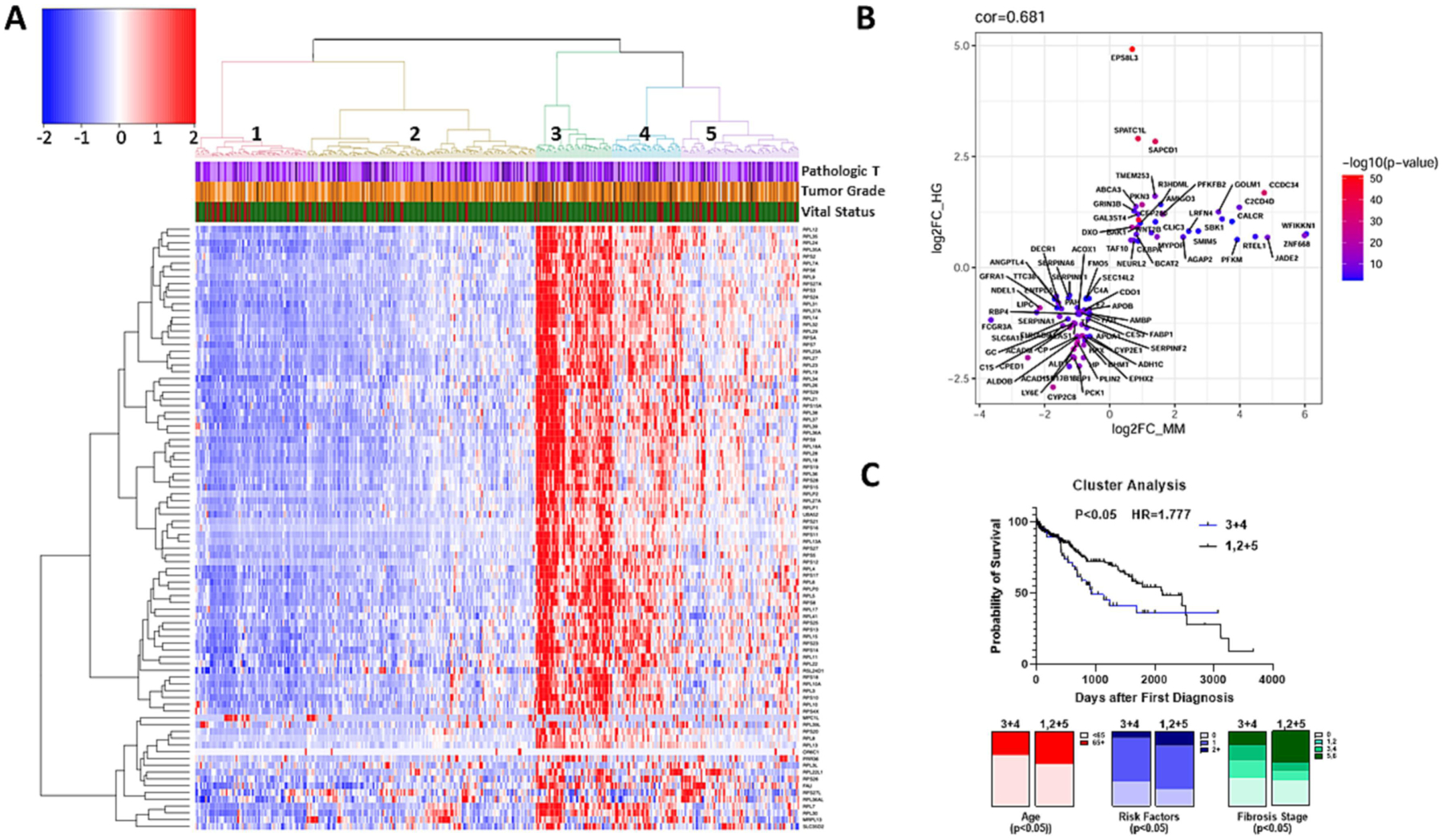
A unique gene signature of VC-induced tumors correlates with a subset of human HCC. A: Heatmap of the human TCGA samples divided based on the 89 genes involved in ribosomal pathways (KEGG: 03010) and changed by VC exposure after 12 months (see Figures 3 and 4). Cancer grade (Pathologic T and Tumor Grade) and mortality (vitality; green=alive) are also shown for individual HCC cases. B: A differential gene expression analysis of the human HCC subset comparing normal liver vs. tumor was performed and the extent to which these gene changes were recapitulated in the mouse model was assessed. Common up- and down-regulated in both human and mouse models are highly correlated with a Spearman correlation of 0.71 with a p-value of 2.2×10−16 and with a Pearson correlation of 0.681 with a p-value of 9.741×10−13. C: The TCGA cohort was separated by enrichment for high expression of KEGG:03010 (clusters 3 and 4) and low expression and compared with survival probability (Kaplan-Meier survival analysis) and clinical features, (e.g., age of first diagnosis, risk factors for liver disease, and Ishak fibrosis score (see also Table 1).
The clinical variables from the TCGA database associated with the 5 hierarchical clusters were then analyzed. The 2 clusters associated with an elevated expression pattern of ribosomal proteins cluster 3 (green) and cluster 4 (blue) were compared with the other clusters. Univariate Chi-square analysis indicated that patients in Clusters 3 and 4 were more evenly distributed by sex, younger, with fewer primary risk factors for HCC (e.g., alcohol, NAFLD, HCV, etc.), and had less severe CLD (Ishak scale; Figure 5C and Table 1). In fact, the number of patients with cirrhosis (Ishak 5 and 6) was ~50% lower in clusters 3 and 4 compared with the other clusters. Although these factors would be expected to correlate with better HCC survival, total mortality tended to be ~40% higher (p=0.0767), and Kaplan-Meier assessment of the predicted survival indicated that these groups actually had a steeper rate of mortality than the other 3 clusters by unadjusted Cox Proportional Hazard analysis (Figure 5C; HR=1.65, 95% CI 1.04–2.63, p=0.035). This Hazard Ratio was not increased by multivariate analysis of known risk factors for HCC mortality (e.g., age, pathologic T, tumor grade, fibrosis score and number of HCC risk factors; not shown).
Table 1: Association between enrichment of genes for the KEGG term ribosomal processes (03010) and patients’ clinicopathological features.
The Cancer Genome Atlas (TCGA) cohort was separated by enrichment for high expression of KEGG:03010 (clusters 3 and 4; see Figure 5) and low expression and compared with clinical features, which included age of first diagnosis, gender, race, risk factors, fibrosis score (Ishak scale), tumor stage, tumor grade and vital status. The unadjusted hazard ratio (HR) was calculated from the Kaplan-Meier survival analyses (see Figure 5).
| Measure | Category | Clusters 3+4 (n=90) | Clusters 1,2+5 (n=284) | p-value |
|---|---|---|---|---|
| Age at first | Years (±SD) | 57.6±13.9 | 60.0±13.3 | 0.1237 |
| Diagnosis | <65 (%) | 68.9 | 56.5 | 0.0376 |
| 65+ (%) | 31.1 | 43.5 | ||
| Sex | Male (%) | 60.0 | 70.0 | 0.0376 |
| Female (%) | 40.0 | 30.0 | ||
| Race | White (%) | 41.1 | 52.3 | 0.1549 |
| Black (%) | 3.3 | 4.9 | ||
| Asian (%) | 52.2 | 39.9 | ||
| Am. Indian (%) | 1.1 | 0.4 | ||
| Risk Factors | None (%) | 33.3 | 23.2 | 0.0324 |
| 1 (%) | 58.3 | 57.9 | ||
| 2+ (%) | 8.3 | 18.9 | ||
| Fibrosis Score | 0 (%) | 38.5 | 34.1 | 0.0220 |
| Ishak Scale | 1,2 (%) | 23.1 | 12.5 | |
| 3,4 (%) | 20.5 | 11.4 | ||
| 5,6 (%) | 17.5 | 42.0 | ||
| Tumor Stage | I (%) | 40.7 | 52.3 | 0.2383 |
| II (%) | 26.7 | 24.4 | ||
| III (%) | 30.2 | 22.3 | ||
| IV (%) | 2.3 | 1.1 | ||
| Tumor Grade | T1 (%) | 40.0 | 52.3 | 0.1917 |
| pT | T2 (%) | 27.8 | 24.9 | |
| T3 (%) | 27.8 | 19.6 | ||
| T4 (%) | 4.4 | 3.2 | ||
| Deaths | (%) | 34.25 | 23.97 | 0.0767 |
| Unadjusted HR | HR (95% CI) | 1.65 (1.04–2.63) | 0.035 | |
The enhancement of HCC by VC exposure leads to a unique epitranscriptomic phenotype.
RNA-Seq analysis identified several pathways (e.g. EIF2 pathway, RNA processing – both playing important roles in hepatic injury and cancer) affected by transient VC in combination with WD. However, while these results here were highly significant, the overall amount of DEGs was rather low (see above and Figures 4 and 5). Emerging evidence indicates that the biological impact of RNAs is not solely regulated by their presence or absence. Specifically, these processes are also regulated by secondary RNA structure (i.e., folding) and chemical modifications of RNA bases, which is similar in concept to DNA (i.e., epigenetics). These interdependent non-sequence features of RNA (i.e., epitranscriptomics) exert direct control over the transcriptome and thereby influence many aspects of cell function independent of RNA abundance. Importantly, RNA modifications may also alter RNA-protein interactions during gene expression. Therefore, an LC-MS/MS analysis of total hepatic nucleoside and nucleotides was performed on samples from the 12-week (Supplemental Figure 6 and Supplemental Table 4) and the 12-month time points (Figure 6A and B). An analysis of changes over time (WD+VC, 12 months vs. 12 weeks, Figure 6C and Supplemental Table 4) was also performed. VC induced significant changes to the modification pattern (Figure 6) that have been associated with cancer (e.g., m3C, m6A, m1A, and Gm), but also with mitochondrial dysfunction and metabolic disease (e.g., m4C, m6A, and m1A).
Figure 6: VC changes the pattern of total hepatic nucleosides/nucleotides.
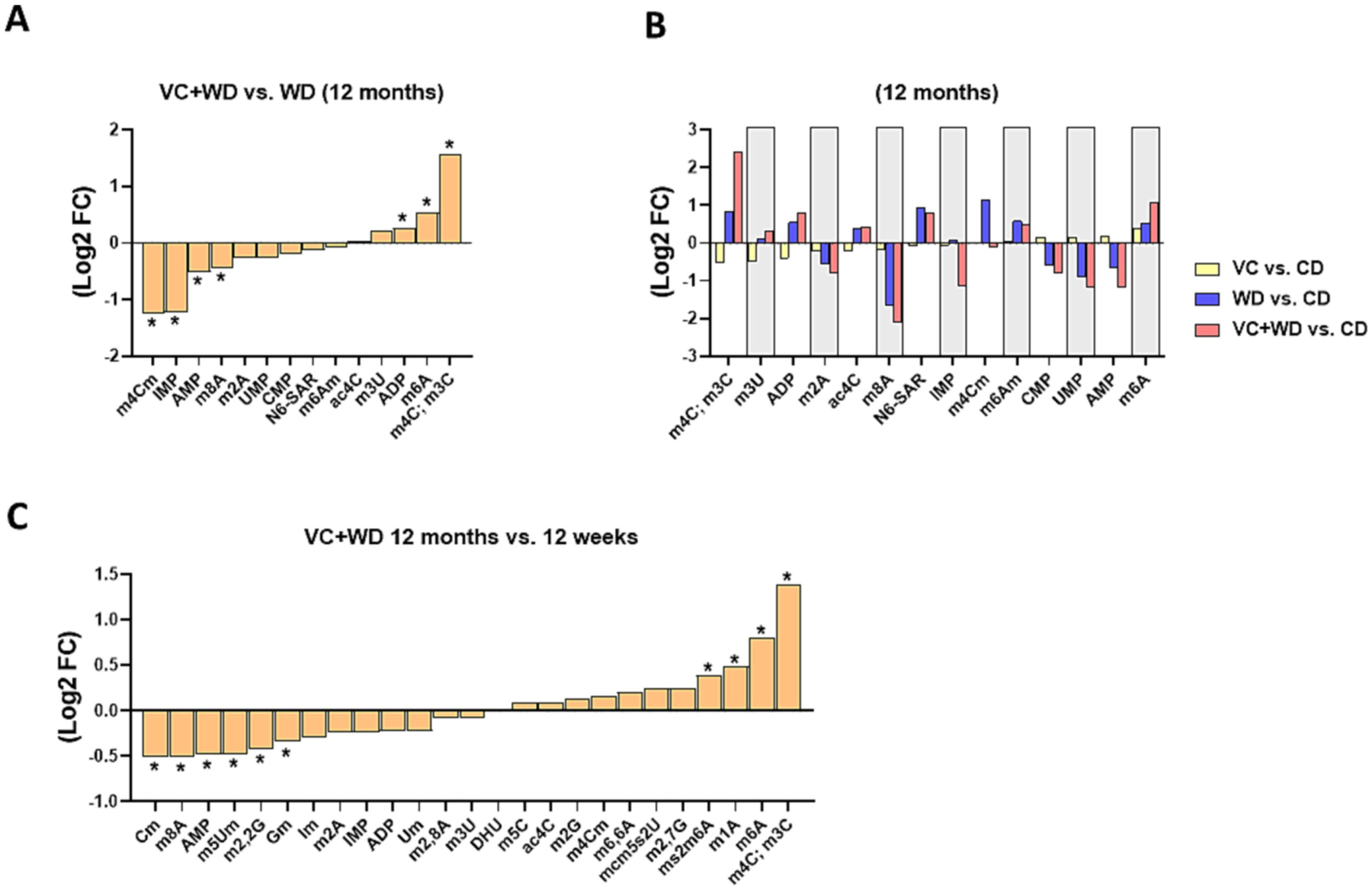
LC-MS/MS epitranscriptomic analysis of total hepatic nucleosides and nucleotides (Log2FC) for A: VC+WD vs. WD at 12 months. B: VC vs. CD, WD vs CD and VC+WD vs. CD at 12 months. C: VC+WD 12 months vs. 12 weeks. *, p<0.05 (FDR) log2 fold change (FC). n=5 per group.
Discussion
HCC occurs predominantly in the context of CLD, where cycles of hepatocyte injury, inflammation and regeneration trigger survival and proliferation of hepatocytes harboring oncogenic mutations. Therefore, the incidence of HCC has not surprisingly paralleled the increase in CLD in recent years. Although advanced therapeutic options have recently improved survival for most cancers, the low survival rate for HCC has not changed for decades, although the advent of immunotherapy is showing promise in a subset of HCC cases. These factors taken together explain why HCC-related deaths continue to rise (Llovet et al., 2015). It has been demonstrated that multiple parallel hits drive not only CLD pathogenesis, but also the development of HCC on the background of CLD (Kim et al., 2021). Although environmental exposure is increasingly identified as a potential risk factor for CLD (Beier and Arteel, 2021), few studies have investigated the impact of environmental exposure on the risk of developing HCC. The purpose of this study is to begin to address this gap in our knowledge.
High occupational VC exposure is well-known to be associated with the development of hemangiosarcoma and HCC (2008; 2012; Fedeli et al., 2019), which were largely eradicated with a lowering of exposure limits in the 1970’s. However, these safety guidelines are potentially outdated, considering the significant increase in average BMI and NAFLD/NASH across the global population in the subsequent years (Younossi et al., 2016). Therefore, it has been unclear if the interaction between VC exposure and underlying liver disease enhances the risk for the development of liver cancer at exposure levels lower than the current safety limits. The primary goal of the current study was to investigate the process underlying the development of HCC with a newly developed animal model of transient exposure to low levels of VC coupled with prolonged WD feeding. The present study’s findings demonstrated that transient (12 weeks) VC exposure accelerates and enhances tumorigenesis caused by experimental NAFLD in mice (Supplemental Figure 4). This enhancement of HCC appears not to be simply via a worsening of the underlying CLD, but rather a direct effect on HCC pathogenesis, as most indices of liver damage were not significantly altered between the air- and VC-exposed cohorts (Figures 1–2).
Transient VC exposure altered several metabolic processes (Figure 3), and the expression pattern of key regulators of lipid synthesis, including ChREBP (MLXIPL) in animals fed WD for 12 months (Figures 3 and 4). These findings are in line with previous work showing that VC exposure alters metabolism in precancerous models (Lang et al., 2018; Chen et al., 2019; Schnegelberger et al., 2021). Although altered metabolism is an established key factor in the development of CLD, more recent work has identified that it also may contribute to cancer biology and the development of HCC (Meyer et al., 2002; Chen et al., 2018b; Sangineto et al., 2020). Moreover, ChREBP activation has been previously shown to promote HCC proliferation and has been associated with poor outcome in patients with HCC (Ribback et al., 2018). Taken together, these data suggest that VC exposure causes prolonged alterations in metabolism that exacerbate HCC development.
Another set of pathways was associated with increases in ribosomal processes, including ribosomal biogenesis, structural constitution, and RNA processing. Moreover, the EIF2 signaling pathway, a major mediator of translation, initiation, and assembly of ribosomal subunits, was strongly induced. These findings are in line with previous work in humans, which has identified t-RNA aminoacylation, the initial step in translation initiation, as the top canonical pathway affected by occupational VC exposure (Guardiola et al., 2016). It is well established that altered ribosomal processes and ribosome biogenesis play an important role in cancer, as any quantitative change in ribosome concentration may impact translation patterns and favor expression of specific mRNAs to the detriment of others (Penzo et al., 2019). In cancer cells, disruption of ribosome biogenesis and protein synthesis is associated with altered expression of key genes encoding translation initiation factors and proto-oncogenes such as MYC (Bastide and David, 2018). Indeed, here transient VC not only induced major changes in expression of ChREBP (see above), but also strongly induced n-MYC (MYCN). Moreover, MYC and ChREBP transcription factors have been shown to cooperatively regulate normal and neoplastic hepatocyte proliferation in mice (Wang et al., 2018). These novel findings were not observed in previous studies of VC exposure in precancerous models and were not prevalent at earlier (12 weeks) sacrifices in this study (Supplemental Figure 2). These results suggest that the alteration of ribosomal processes is not directly related to the liver injury caused by VC exposure, per se, but rather develops after VC exposure is completed. Given the fact that VC and its metabolites have a very short half-life in mammalian species (hours) and do not bioaccumulate, these findings cannot be explained by residual chemicals present in the animals.
The mechanisms by which ribosomal processes and RNA processing were induced under these conditions remain unclear. These changes may be a direct result of the metabolic changes caused by VC exposure. For example, previous studies have shown that activated ChREBP pathways can lead to upregulation of ribosomal processes (Wang et al., 2018). However, recent work has indicated that direct modifications of ribosomes and/or RNAs may also influence these processes. This potential effect is especially important, as about 2/3 of the ribosome is comprised of RNA. Cellular RNAs are subject to multiple chemical modifications, and the study of these chemical modifications (i.e. epitranscriptomics) has a growing research interest in disease and dysfunction (Delaunay and Frye, 2019). The epitranscriptome is shaped through the activity of evolutionarily conserved factors that encode (writers), decode (readers) and remove (erasers) various chemical modifications of RNA bases. The work to date indicates that epitranscriptomic alterations play important roles in regulating RNA metabolism, translation, localization, stability, turnover, as well as in binding to proteins or other RNAs, which ultimately affects metabolism, disease progression, and carcinogenesis.
While qualitative RNA modifications can dictate oncogenic potential, the knowledge about specific modifications and their role in carcinogenesis is still limited. However, N6-methyladenosine (m6A), regarded as the most abundant internal modification, has been associated with poor prognosis in HCC, clinically (Chen et al., 2018a; Zhao et al., 2020). Functionally, in HCC m6A has been demonstrated to enhance cell proliferation, and migration via inhibiting tumor suppressor expression (Chen et al., 2018a). Moreover, m6A methylation sites have been shown to be enriched in processes associated with lipid metabolism (Zhao et al., 2020). Importantly, VC strongly increased m6A abundance at the 12-month time point, in line with the RNA-Seq data suggesting a potential role for m6A in the metabolic and tumorigenic changes caused by VC (Figure 6 and Supplemental Table 4). Moreover, it has been shown that together with the EIF2 signaling pathway, which was also strongly induced here (Figure 4), m6A mRNA methylation reconfigures the cellular adaptation to stress conditions (Zhou et al., 2018), a critical factor in cancer initiation and progression.
In addition to m6A, the 3-methylcytosine (m3C) modification, which is present in both tRNA and mRNA and displays diverse roles in developing diseases through the regulation of tRNA fate, was strongly increased (Xu et al., 2017; Cui et al., 2021). It has been shown recently that methylation in tRNA enhanced the proliferative activity of hepatocellular carcinoma (HCC) (Ignatova et al., 2020). However, several of the RNA modifications observed in this study (e.g., m4C, Cm, m8A. etc.) have never been associated with tumorigenesis or more specifically HCC (Supplemental Table 4). For example, currently not much information is known about the 4-methylcytosine (m4C) modification and its impact on human disease; however, m4C on mitochondrial rRNA has been associated with mitochondrial function. It should be noted that one limitation of this study is that while LC-MS/MS analysis employed here is highly sensitive and quantitative, it cannot distinguish between m3C and m4C nor determine which RNA sites are modified (Cui et al., 2021). Furthermore, changes to whole liver nucleoside/nucleotide modifications were measured here. Future studies will need to determine the location of the modification and RNA species. The field of epitranscriptomics is developing and these results may therefore point to new epitranscriptomic profiles that may be relevant for cancer development in general and HCC in particular.
The increase in ribosomal processes may also have an impact on HCC development and outcome. Two clusters of human HCC, identified from the TCGA database, were enriched for increases in expression of genes associated with ribosomal processes. Interestingly, HCC in these groups more frequently occurred on the background of less severe CLD and with fewer known risk factors. This group also had a higher Hazard Ratio for mortality. This administrative and retrospective analysis does not allow identification of other causes that may drive HCC. However, there was strong concordance in the overall expression patterns of this human HCC and the murine orthologs from WD-fed mice exposed to VC. It is therefore distinctly possible that unidentified environmental exposure such as to volatile organic compounds (e.g. VC) may be a hidden factor that contributed to the development of HCC in this cohort. Since several environmental chemicals like VC may impart similar effects on the target organ (Beier and Arteel, 2021), this potential environmental exposure may be representative of more than VC per se, and by extension, may represent exposures that also cause epitranscriptomic changes. These results bolster the need for more comprehensive documentation of exposure (i.e., the “exposome”) (Cheung et al., 2020).
CLD and HCC have been demonstrated to be sexually dimorphic with a higher incidence in women of certain populations (Table 1). Therefore, a limitation of the current study is that only male mice were used. However, this group has previously demonstrated that female mice were protected from liver injury at 12 weeks (Wahlang et al., 2020). Future studies should therefore address dimorphism during tumorigenesis caused by VC in mice. Prior chronic animal model investigations, particularly in the nutritional area, have found a delay in a tumor endpoint rather than a complete absence of tumor initiation. Additional time points may be necessary in the future to elucidate the full pathologic profile. However, the current proof-of concept model clearly demonstrates that lower environmental toxicant exposures may be an underestimated risk modifier in the development of HCC.
Conclusion:
This is the first study to demonstrate VC-induced tumorigenesis at concentrations that are currently considered safe. The data indicate that transient VC exposure sensitizes the liver to other stressors (e.g., WD), involving dysregulation of metabolic pathways, an enrichment of genes for ribosomal processes, and changes to the epitranscriptomic pattern. These changes result in enhanced tumorigenesis and correlate with a subset of human HCC that tended to be younger with fewer primary risk factors for HCC. These data raise concerns about potential for overlap between hypercaloric diets and exposure to lower concentrations of VC, as well as the health implications of this co-exposure for humans that may be underappreciated with our current knowledgebase. It also emphasizes that current safety restrictions may be insufficient to account for other factors that can influence hepatotoxicity.
Supplementary Material
Highlights:
Transient VC exposure enhances tumorigenesis caused by Western diet
Transient VC alters metabolic and ribosomal processes
Transient VC changes epitranscriptomic regulation
A unique gene signature of VC-induced tumors correlates with a subset of human HCC
Funding:
This study was funded by awards from the National Institutes of Health: K01 DK096042, R03 DK107912, R21 ES031531, R01 DK133454 to Juliane Beier; and P30DK120531 to Satdarshan (Paul) S. Monga. This research was also supported, in part, by the National Institute of Environmental Health Sciences (R35ES028373, R01ES032189, P42ES023716, P30ES030283, R21ES031510); the National Institute of General Medical Sciences (P20GM113226); the National Institute on Alcohol Abuse and Alcoholism (P50AA024337). This research was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
Silvia Liu: Methodology, Visualization, Investigation, Validation, Data curation, Statistical Analysis, Writing- Original draft preparation; Liqing He: Conceptualization, Methodology, Software, Statistical Analysis, Writing- Original draft preparation; Olivia B Bannister: Visualization, Investigation, Validation, Formal Analysis, Writing- Original draft preparation Jiang Li: Visualization, Investigation, Validation; Regina D Schnegelberger: Visualization, Investigation, Validation, Formal Analysis; Charis-Marie Vanderpuye: Visualization, Investigation, Validation; Andrew D Althouse: Data curation, Statistical Analysis; Francisco J Schopfer: Methodology, Visualization, Investigation, Validation; Banrida Wahlang: Visualization, Investigation, Validation; Matthew C Cave: Conceptualization; Satdarshan P Monga: Conceptualization, Visualization, Investigation, Validation, Reviewing and Editing; Xiang Zhang: Conceptualization, Reviewing and Editing; Gavin E Arteel: Conceptualization, Reviewing and Editing; and Juliane I Beier: Project Administration, Visualization, Conceptualization, Methodology, Writing- Reviewing and Editing, Funding acquisition, Resources.
Data Availability
Transcriptome sequencing data generated in this study were deposited into NCBI Gene Expression Omnibus (GEO), with accession ID GSE197038. Raw sequencing reads and gene quantification profile can be downloaded at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197038.
References
- 2012. Chemical agents and related occupations. IARC Monogr Eval Carcinog Risks Hum 100, 9–562. [PMC free article] [PubMed] [Google Scholar]
- 2019. EASL Clinical Practice Guideline: Occupational liver diseases. J Hepatol 71, 1022–1037. [DOI] [PubMed] [Google Scholar]
- (U.S.EPA), U.S.E.P.A., 2000. Toxicological review of vinyl chloride in support of summary information on the Integrated Risk Information System (IRIS), pp.
- Asrani SK, Devarbhavi H, Eaton J, Kamath PS, 2019. Burden of liver diseases in the world. J Hepatol 70, 151–171. [DOI] [PubMed] [Google Scholar]
- ATSDR, 2006. Agency for Toxic Substances and Disease Registry (ATSDR): Toxicological profile for Vinyl Chloride, pp. [PubMed] [Google Scholar]
- Bastide A, David A, 2018. The ribosome, (slow) beating heart of cancer (stem) cell. Oncogenesis 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier JI, Arteel GE, 2021. Environmental exposure as a risk-modifying factor in liver diseases: Knowns and unknowns. Acta Pharm Sin B 11, 3768–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier JI, Guo L, Ritzenthaler JD, Joshi-Barve S, Roman J, Arteel GE, 2016. Fibrin-mediated integrin signaling plays a critical role in hepatic regeneration after partial hepatectomy in mice. Ann Hepatol 15, 762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DO, 2016. Hydraulic fracturing for natural gas: impact on health and environment. Rev Environ Health 31, 47–51. [DOI] [PubMed] [Google Scholar]
- Cave M, Falkner KC, Ray M, Joshi-Barve S, Brock G, Khan R, Bon Homme M, McClain CJ, 2010. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology 51, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lang AL, Poff GD, Ding WX, Beier JI, 2019. Vinyl chloride-induced interaction of nonalcoholic and toxicant-associated steatohepatitis: Protection by the ALDH2 activator Alda-1. Redox Biol 24, 101205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, Wong CC, Ng IO, Wong CM, 2018a. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 67, 2254–2270. [DOI] [PubMed] [Google Scholar]
- Chen XF, Tian MX, Sun RQ, Zhang ML, Zhou LS, Jin L, Chen LL, Zhou WJ, Duan KL, Chen YJ, Gao C, Cheng ZL, Wang F, Zhang JY, Sun YP, Yu HX, Zhao YZ, Yang Y, Liu WR, Shi YH, Xiong Y, Guan KL, Ye D, 2018b. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AC, Walker DI, Juran BD, Miller GW, Lazaridis KN, 2020. Studying the Exposome to Understand the Environmental Determinants of Complex Liver Diseases. Hepatology 71, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Liu Q, Sendinc E, Shi Y, Gregory RI, 2021. Nucleotide resolution profiling of m3C RNA modification by HAC-seq. Nucleic Acids Res 49, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay S, Frye M, 2019. RNA modifications regulating cell fate in cancer. Nat Cell Biol 21, 552–559. [DOI] [PubMed] [Google Scholar]
- Dimmick WF, 1981. EPA programs of vinyl chloride monitoring in ambient air. Environ Health Perspect 41, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi PA, DePinho RA, 2006. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 6, 674–687. [DOI] [PubMed] [Google Scholar]
- Fedeli U, Girardi P, Gardiman G, Zara D, Scoizzato L, Ballarin MN, Baccini M, Pirastu R, Comba P, Mastrangelo G, 2019. Mortality from liver angiosarcoma, hepatocellular carcinoma, and cirrhosis among vinyl chloride workers. American journal of industrial medicine 62, 14–20. [DOI] [PubMed] [Google Scholar]
- Guardiola JJ, Beier JI, Falkner KC, Wheeler B, McClain CJ, Cave M, 2016. Occupational exposures at a polyvinyl chloride production facility are associated with significant changes to the plasma metabolome. Toxicol Appl Pharmacol 313, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wei X, Ma X, Yin X, Song M, Donninger H, Yaddanapudi K, McClain CJ, Zhang X, 2019. Simultaneous Quantification of Nucleosides and Nucleotides from Biological Samples. J Am Soc Mass Spectrom 30, 987–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humans, I.W.G.o.t.E.o.C.R.t., 2012. Chemical agents and related occupations. IARC Monogr Eval Carcinog Risks Hum 100, 9–562. [PMC free article] [PubMed] [Google Scholar]
- IARC, 2008. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 97. 1,3-butadiene, ethylene oxide and vinyl halides (vinyl fluoride, vinyl chloride and vinyl bromide), IARC Monogr Eval Carcinog Risks Hum, pp. 3–471. [PMC free article] [PubMed] [Google Scholar]
- Ignatova VV, Kaiser S, Ho JSY, Bing X, Stolz P, Tan YX, Lee CL, Gay FPH, Lastres PR, Gerlini R, Rathkolb B, Aguilar-Pimentel A, Sanz-Moreno A, Klein-Rodewald T, Calzada-Wack J, Ibragimov E, Valenta M, Lukauskas S, Pavesi A, Marschall S, Leuchtenberger S, Fuchs H, Gailus-Durner V, de Angelis MH, Bultmann S, Rando OJ, Guccione E, Kellner SM, Schneider R, 2020. METTL6 is a tRNA m(3)C methyltransferase that regulates pluripotency and tumor cell growth. Sci Adv 6, eaaz4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL, Anderson RN, Henley SJ, Kohler BA, Penberthy L, Feuer EJ, Weir HK, 2017. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J Natl Cancer Inst 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Cushing L, 2020. Chemical Exposures, Health, and Environmental Justice in Communities Living on the Fenceline of Industry. Current environmental health reports 7, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielhorn J, Melber C, Wahnschaffe U, Aitio A, Mangelsdorf I, 2000. Vinyl chloride: still a cause for concern. Environ Health Perspect 108, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, He F, Karin M, 2021. From Liver Fat to Cancer: Perils of the Western Diet. Cancers (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AL, Beier JI, 2018. Interaction of volatile organic compounds and underlying liver disease: a new paradigm for risk. Biol Chem 399, 1237–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AL, Chen L, Poff GD, Ding WX, Barnett RA, Arteel GE, Beier JI, 2018. Vinyl chloride dysregulates metabolic homeostasis and enhances diet-induced liver injury in mice. Hepatol Commun 2, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AL, Goldsmith WT, Schnegelberger RD, Arteel GE, Beier JI, 2020. Vinyl Chloride and High-Fat Diet as a Model of Environment and Obesity Interaction. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AL, Krueger AM, Schnegelberger RD, Kaelin BR, Rakutt MJ, Chen L, Arteel GE, Beier JI, 2019. Rapamycin attenuates liver injury caused by vinyl chloride metabolite chloroethanol and lipopolysaccharide in mice. Toxicol Appl Pharmacol 382, 114745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Montal R, Sia D, Finn RS, 2018. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 15, 599–616. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Villanueva A, Lachenmayer A, Finn RS, 2015. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 12, 408–424. [DOI] [PubMed] [Google Scholar]
- Lotti M, 2017. Do occupational exposures to vinyl chloride cause hepatocellular carcinoma and cirrhosis? Liver Int 37, 630–633. [DOI] [PubMed] [Google Scholar]
- Meyer K, Jia Y, Cao WQ, Kashireddy P, Rao MS, 2002. Expression of peroxisome proliferator-activated receptor alpha, and PPARalpha regulated genes in spontaneously developed hepatocellular carcinomas in fatty acyl-CoA oxidase null mice. Int J Oncol 21, 1175–1180. [PubMed] [Google Scholar]
- Nguyen GC, Thuluvath PJ, 2008. Racial disparity in liver disease: Biological, cultural, or socioeconomic factors. Hepatology 47, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Penzo M, Montanaro L, Treré D, Derenzini M, 2019. The Ribosome Biogenesis-Cancer Connection. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribback S, Che L, Pilo MG, Cigliano A, Latte G, Pes GM, Porcu A, Pascale RM, Li L, Qiao Y, Dombrowski F, Chen X, Calvisi DF, 2018. Oncogene-dependent addiction to carbohydrate-responsive element binding protein in hepatocellular carcinoma. Cell Cycle 17, 1496–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangineto M, Villani R, Cavallone F, Romano A, Loizzi D, Serviddio G, 2020. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayiner M, Koenig A, Henry L, Younossi ZM, 2016. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin Liver Dis 20, 205–214. [DOI] [PubMed] [Google Scholar]
- Schnegelberger RD, Lang AL, Arteel GE, Beier JI, 2021. Environmental toxicant-induced maladaptive mitochondrial changes: A potential unifying mechanism in fatty liver disease? Acta Pharm Sin B 11, 3756–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Laribi O, 2021. Environmental Justice in the American Public Health Context: Trends in the Scientific Literature at the Intersection Between Health, Environment, and Social Status. J Racial Ethn Health Disparities. [DOI] [PubMed] [Google Scholar]
- Tao J, Krutsenko Y, Moghe A, Singh S, Poddar M, Bell A, Oertel M, Singhi AD, Geller D, Chen X, Lujambio A, Liu S, Monga SP, 2021. Nuclear factor erythroid 2-related factor 2 and β-Catenin Coactivation in Hepatocellular Cancer: Biological and Therapeutic Implications. Hepatology 74, 741–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A, 2019. Hepatocellular Carcinoma. N Engl J Med 380, 1450–1462. [DOI] [PubMed] [Google Scholar]
- von Montfort C, Beier JI, Kaiser JP, Guo L, Joshi-Barve S, Pritchard MT, States JC, Arteel GE, 2010. PAI-1 plays a protective role in CCl4-induced hepatic fibrosis in mice: role of hepatocyte division. Am J Physiol Gastrointest Liver Physiol 298, G657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Hardesty JE, Head KZ, Jin J, Falkner KC, Prough RA, Cave MC, Beier JI, 2020. Hepatic Injury Caused by the Environmental Toxicant Vinyl Chloride is Sex-Dependent in Mice. Toxicol Sci 174, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Dolezal JM, Kulkarni S, Lu J, Mandel J, Jackson LE, Alencastro F, Duncan AW, Prochownik EV, 2018. Myc and ChREBP transcription factors cooperatively regulate normal and neoplastic hepatocyte proliferation in mice. J Biol Chem 293, 14740–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Fontenot RD, Soni MG, Bucci TJ, Mehendale HM, 2000. Enhanced hepatotoxicity and toxic outcome of thioacetamide in streptozotocin-induced diabetic rats. Toxicol Appl Pharmacol 166, 92–100. [DOI] [PubMed] [Google Scholar]
- Xu L, Liu X, Sheng N, Oo KS, Liang J, Chionh YH, Xu J, Ye F, Gao YG, Dedon PC, Fu XY, 2017. Three distinct 3-methylcytidine (m(3)C) methyltransferases modify tRNA and mRNA in mice and humans. J Biol Chem 292, 14695–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M, 2016. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Meng J, Su R, Zhang J, Chen J, Ma X, Xia Q, 2020. Epitranscriptomics in liver disease: Basic concepts and therapeutic potential. J Hepatol 73, 664–679. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wan J, Shu XE, Mao Y, Liu XM, Yuan X, Zhang X, Hess ME, Brüning JC, Qian SB, 2018. N(6)-Methyladenosine Guides mRNA Alternative Translation during Integrated Stress Response. Mol Cell 69, 636–647.e637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptome sequencing data generated in this study were deposited into NCBI Gene Expression Omnibus (GEO), with accession ID GSE197038. Raw sequencing reads and gene quantification profile can be downloaded at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197038.