

Abstract
Reduced Survival Motor Neuron (SMN) protein triggers the motor neuron disease, spinal muscular atrophy (SMA). Restoring SMN prevents disease but it is not known how neuromuscular function is preserved. We used model mice to map and identify an Hspa8G470R synaptic chaperone variant, which suppressed SMA. Expression of the variant in the severely affected mutant mice increased lifespan >10-fold, improved motor performance and mitigated neuromuscular pathology. Mechanistically, Hspa8G470R altered SMN2 splicing and simultaneously stimulated formation of a tripartite chaperone complex, critical for synaptic homeostasis, by augmenting its interaction with other complex members. Concomitantly, synaptic vesicular SNARE complex formation, which relies on chaperone activity for sustained neuromuscular synaptic transmission, and was found perturbed in SMA mice and patient-derived motor neurons, was restored in modified mutants. Identification of the Hspa8G470R SMA modifier implicates SMN in SNARE complex assembly and casts new light on how deficiency of the ubiquitous protein causes motor neuron disease.
Keywords: Spinal muscular atrophy, Survival motor neuron protein, modifiers, SNARE complex assembly, Hspa8
Graphical Abstract
eTOC Blurb
Why does paucity of the housekeeping SMN protein trigger selective motor neuron degeneration? Kim et al. probe this by identifying a genetic suppressor of neurodegeneration in spinal muscular atrophy model mice. The suppressor, a synaptic chaperone, reveals a role for SMN in assembling synaptic SNARE complexes specifically at neuromuscular junctions.
Introduction
Homozygous loss of the Survival Motor Neuron 1 (SMN1) gene triggers the neuromuscular disorder, spinal muscular atrophy (SMA).1–3 A paralogue, SMN2, is invariably present in patients, but fails to counter SMN1 loss owing to a splice-switching exon 7 transition that renders the copy gene unable to produce adequate SMN protein.4,5 Still, SMN2 is an intuitively appealing therapeutic target for the treatment of SMA. Indeed, nusinersen and risdiplam, two agents currently approved for SMA therapy, mitigate disease by inducing SMN2 to express greater amounts of intact SMN. A third agent, onasemnogene abeparvovec, restores SMN by means of an AAV9 vector. These therapies, if delivered in a timely manner effectively prevent disease onset.6 Yet, developing these therapeutic modalities did not address our understanding of how SMN governs neuromuscular health. SMA mechanisms have instead been gleaned primarily from studies that investigated SMN functions. These have implicated SMN in diverse roles including, notably, functions in RNA metabolism.7 However, a compelling link between such functions and the selective vulnerability of the neuromuscular system in SMA is yet to emerge.
One approach to defining disease-relevant mechanisms involves the identification of genetic modifiers.8,9 Indeed, such a strategy has been pursued for SMA.10,11 Yet, what has emerged, especially from genetic screens carried out in invertebrates, has rarely been confirmed in mammalian models. Besides, the search for SMA modifiers in worms, flies and even rodents is hampered by the absence of a true, SMN2-like hypomorph in these model systems. Attempts to identify SMA modifiers by studying families with discordantly affected sibs have been more fruitful.12–15 However, perplexingly, expressing the genes that emerged from these studies in humanized, SMN2-expressing mice failed to modify disease without first augmenting the mutants with an SMN-enhancing agent. Moreover, none of the putative modifiers was ever linked to a distinct genomic alteration.12–14 Consequently, discerning precisely why the identified modifiers, which are indistinguishable in modified and unmodified subjects, alter disease is difficult.
We previously generated a severely affected, humanized mouse model of SMA.16 Consistent with observations of discordant SMA sibs and the notion of disease modifiers, we found that distinct mouse strain backgrounds altered phenotypic severity in the mutants. Here we report the mapping, identification and functional significance of a novel SMA modifier associated with the altered phenotypes. The modifier, a G470R variant of the synaptic chaperone, Hspa8, dramatically suppresses severe disease. Investigations of the mechanism of action of Hspa8G470R led to the discovery that SNARE complex assembly, a process reliant on Hspa8 chaperone activity and crucial for neurotransmission, is impaired at the neuromuscular synapses of SMA mice but restored in mutants expressing the modifier. Our results suggest that SMN functions in a hitherto unrecognized manner – to facilitate assembly of synaptic complexes critical to neuromuscular function. Moreover, to our knowledge, this is the first report of an SMA modifier to have emerged from an unbiased genetic study conducted in an established, mammalian model of the human disease.
Results
Evidence of a genetic modifier of the mouse SMA phenotype
We previously developed severe (“Δ7”) SMA model mice.16 On a mixed (FVB/N × C57BL/6) background, median and maximum lifespans of the mutants (SMN2+/+;SMNΔ7+/+;Smn−/−) were ~11 days and 25 days respectively. These were reduced on the pure parental strains (Figure 1A). Consistent with their shortened lifespans, FVB/N SMA mutants were significantly smaller than were those on the mixed background (Figure 1B), suggestive of the existence of protective modifiers in one or both parental strains. To map the modifiers, a classic F2 intercross was employed to produce SMA mutants. Most mutants thus derived exhibited the typically severe SMA phenotype, succumbing to disease at approximately PND14. However, ~6% presented with an unexpectedly mild phenotype characterized by a maximum lifespan exceeding 300 days (Figure 1C). This dramatic extension of the two-week lifespan of typically severe SMA mice was accompanied by an increase in body weight (Figure 1B) and a complete recovery, as assessed in righting ability at PND14, of the paralytic SMA phenotype (Figure 1D). Overtly, the mice remained active into adulthood but developed necrotic tails. Still, the results strongly suggested the disease-modifying effects of an SMA suppressor.
Figure 1. Evidence and mapping of a C57BL/6-derived modifier of SMA in model mice.
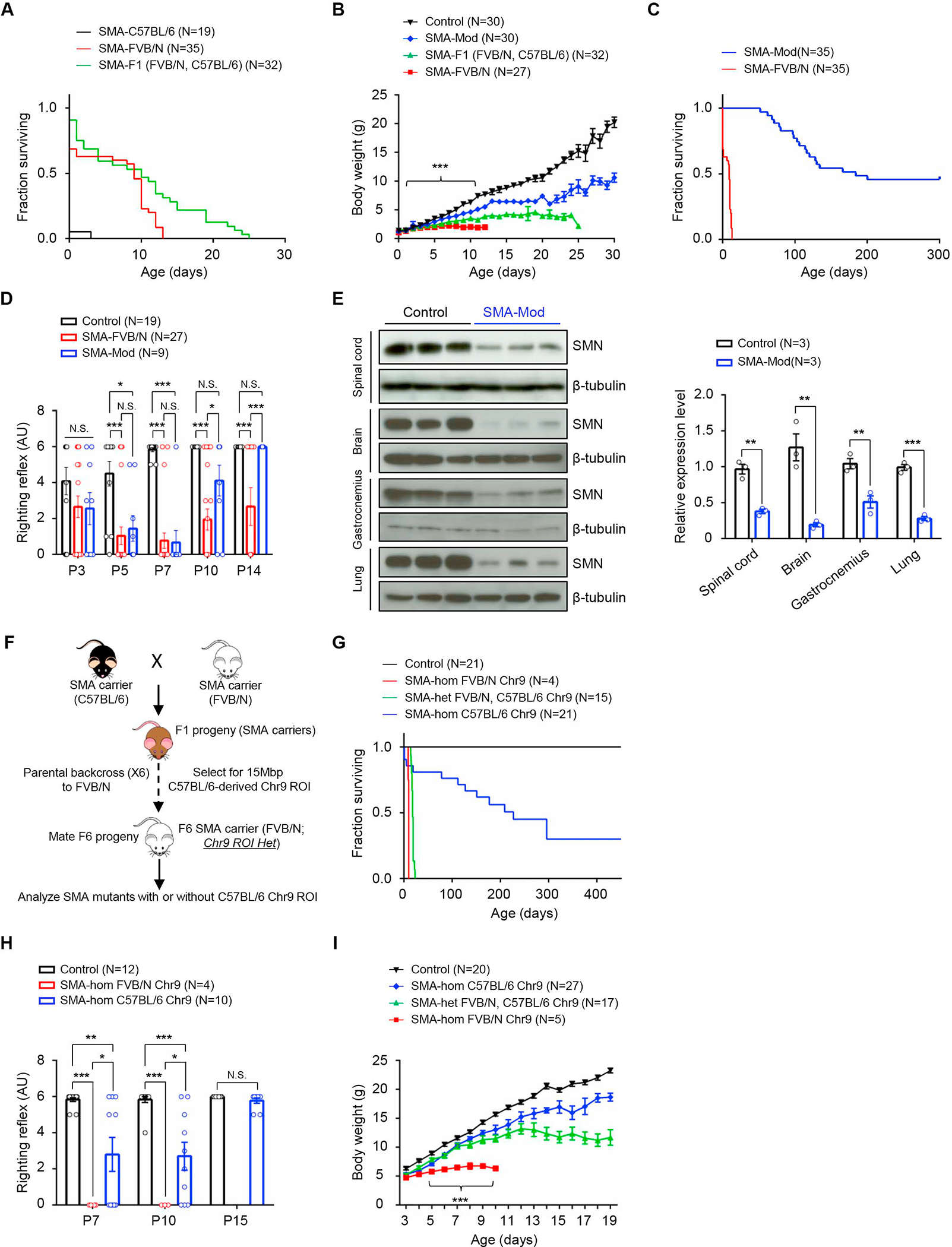
(A) Kaplan-Meier survival curves depicting altered lifespans of model mice on pure or hybrid strain backgrounds. P < 0.0001 between C57BL/6 and FVB/N mutants, log-rank test. (B) Weight curves of pure and hybrid strain SMA mutants. ***, P < 0.001, PND2 – PND12 between FVB/N and SMA-Mod mutants, t tests. Reduced disease severity in SMA-Mod mutants relative to FVB/N-derived mutants, as assessed by (C) increased lifespans and (D) improved motor performance. Note: P < 0.0001 for panel (C) log-rank test; *, **, ***, P < 0.05, P < 0.01 and P < 0.001 respectively, Kruskal-Wallis test (D). (E) Western blots and quantified results of blots depicting greatly reduced SMN in SMA-Mod mutants. **, ***, P < 0.01 and P < 0.001 respectively, t tests. (F) Diagrammatic representation of the strategy employed to map the SMA modifier to Chr.9. (G) Kaplan-Meier survival curves demonstrating that mutants harboring the C57BL/6-derived Chr.9 ROI are more mildly affected than those homozygous FVB/N for this genomic region. P < 0.0001 between SMA-hom C57BL/6 and SMA-hom FVB/N mutants, log-rank test. SMA-hom C57BL/6 Chr.9 mutants were also less severely affected based on (H) the righting reflex assay and (I) their tendency to gain more weight than their SMA-hom FVB/N Chr.9 counterparts. Note: *, **, P < 0.05 and P < 0.01, Kruskal-Wallis test for panel (H); ***, P < 0.001 between SMA-hom C57BL/6 and SMA-hom FVB/N mutants in panel (I). N.S. – not significant. SMA-Mod – SMN2+/+;SMNΔ7+/+;Hspa8G470R;Smn−/−, Controls – SMN2+/+;SMNΔ7+/+;Smn+/−. Data: mean ± SEM. See also Video S1, Table S1 and Figs. S1–S3.
We examined the modified mutants for SMN levels and evidence of neuromuscular pathology classically seen in SMA.17–19 At PND70, the mutants continued to express acutely low SMN protein (Figure 1E). Yet, morphometric counts of spinal motor neurons revealed no difference between the modified mutants and healthy controls (Figure S1A) and, consistent with this finding, there was neither a change in mutant spinal motor neuron soma size nor evidence of deafferentiation (Figure S1B – S1D). An examination of two muscles, the gastrocnemius and triceps, did not uncover any group atrophy of the fibers but, congruent with the smaller size of the mutants, did reveal a uniform drop in fiber size (Figure S1E – S1G). Δ7 SMA mice have defective neuromuscular junctions (NMJs) characterized by small, immature endplates bereft of the perforations observed in wild-type (WT) NMJs.20–22 These defects were partially rescued in the modified mutants; the endplates remained smaller than in controls but appeared perforated and fully mature (Figure S1H – S1J). Moreover, intracellular recordings to gauge NMJ function indicated that neurotransmission in the EDL muscle was normalized in the modified mutants (Figure S1K – S1M). These observations suggested that the modifier responsible for extending lifespan in our mutants also mitigated the neuromuscular defects typical of SMA.
Δ7 mutants are known to suffer cardiac abnormalities.23 We investigated if such dysfunction was also resolved in our F2 SMA modified mutants. Echocardiography indicated that cardiac function was indeed normalized in the mutants (Figure S2A – S2K). Consistent with this observation, gross cardiac structure and heart to body mass ratios were normal (Figure S2L and S2M). Collectively, these preliminary findings suggested that disease modification was systemic, involving cardiac function too.
The SMA modifier is mapped to mouse chromosome 9
To reveal the chromosomal location of the SMA modifier, a cohort of F2 mutants exhibiting either the severe or the mild phenotype was subjected to a genome-wide scan using ~180 and ~730 single nucleotide polymorphic (SNP) markers. We found linkage between the modified phenotype and a region of chromosome 9 (Chr.9) deriving from C57BL/6 and extending from 34Mbp to 49Mbp (Tables S1A and S1B). To ascertain if this region of Chr.9 from the C57BL/6 strain was sufficient to produce the mild phenotype, we introduced it via successive backcrosses and SNP-based selection into SMA carriers deriving the rest of their genomes from the FVB/N strain. Such carriers, heterozygous for the region of interest (ROI), were then bred to produce mutants (Figure. 1F). As expected, mutants homozygous FVB/N for the ROI were very severely affected and perished on or before PND10. In contrast, mutants heterozygous at the ROI exhibited a median survival of 18 days, and mice deriving both copies of the Chr.9 ROI from C57BL/6 lived much longer (Figure 1G). This suggested that the region of Chr. 9 being investigated did indeed harbor an SMA suppressor and, furthermore, that its disease-mitigating effects were dose dependent. Consistent with the longer lifespans of mutants harboring the ROI, we found that motor performance in the mice was improved and body weight enhanced relative to severely affected SMA mice (Figure 1H and 1I). Investigations of muscle fiber morphology and spinal motor neuron counts confirmed the modifying effects of the Chr.9 ROI. At PND7, myofiber area frequency distribution analysis showed that fibers of mutants homozygous for the modifying ROI were closer in size to that of controls (Figure S3A), and this was borne out when average fiber size was quantified. Fibers from mutants with the modifying ROI were significantly larger than those from mutants devoid of it, although they remained smaller than fibers in controls (Figure S3B). By 6 months, differences in fiber size between modified mutants and controls were no longer detectable and, morphologically, the mutant muscles appeared healthy (Figure S3C and S3D). Motor neuron counts at this stage failed to detect significant difference between controls and mutants homozygous for the ROI (Figure S3E and S3F).
A G470R variant in the synaptic chaperone Hspa8 is the SMA modifier
To ascertain the precise identity of the SMA modifier, a second cohort of F2 mutants, similar to those employed to localize it chromosomally, was subjected to whole genome sequencing and variants specifically in the Chr.9 ROI examined in detail. Our rough localization studies predicted that fully modified mutants, defined as those with lifespans of > 50 days, would be homozygous for the C57BL/6 version of the modifier, while typically severe mutants, succumbing to disease before PND14, would inherit both modifier alleles from the FVB/N strain. Mutants heterozygous for the modifier were expected to constitute a third group with an intermediate phenotype. We furthermore focused on non-synonymous changes in protein coding genes. A genome-wide association study (GWAS) revealed approximately 4.9 million total SNP variants, 0.8% of which were in protein coding regions (Figure 2A, 2B and Table S2A). After filtering for stringency, 4742 significant SNPs emerged from the GWAS. The preponderance of these mapped to the Chr.9 ROI (Figure 2C – 2E and Table S2B) and were non-exonic; only 2 were non-synonymous variants. One of these resulted in a T1309M alteration in IGSF9b, a protein expressed in synapses.24 However, the residue is not evolutionarily conserved and was rejected. The second variant of interest, a G→C nucleotide change, caused a G470R substitution in Hspa8 and piqued our curiosity for several reasons. First, the transversion is not evident in the ancestral C57BL/6 strain, suggesting that it arose de novo in our C57BL/6 colony. Second, Hspa8 is a chaperone, important for synaptic proteostasis and neurotransmission25,26, functions disrupted in SMA. Finally, the glycine at position 470 is a constituent of a 10 amino acid IPPAPRGVPQ motif, perfectly conserved from worms to humans suggesting an important function.
Figure 2. GWAS links an Hspa8 variant to SMA disease suppression.
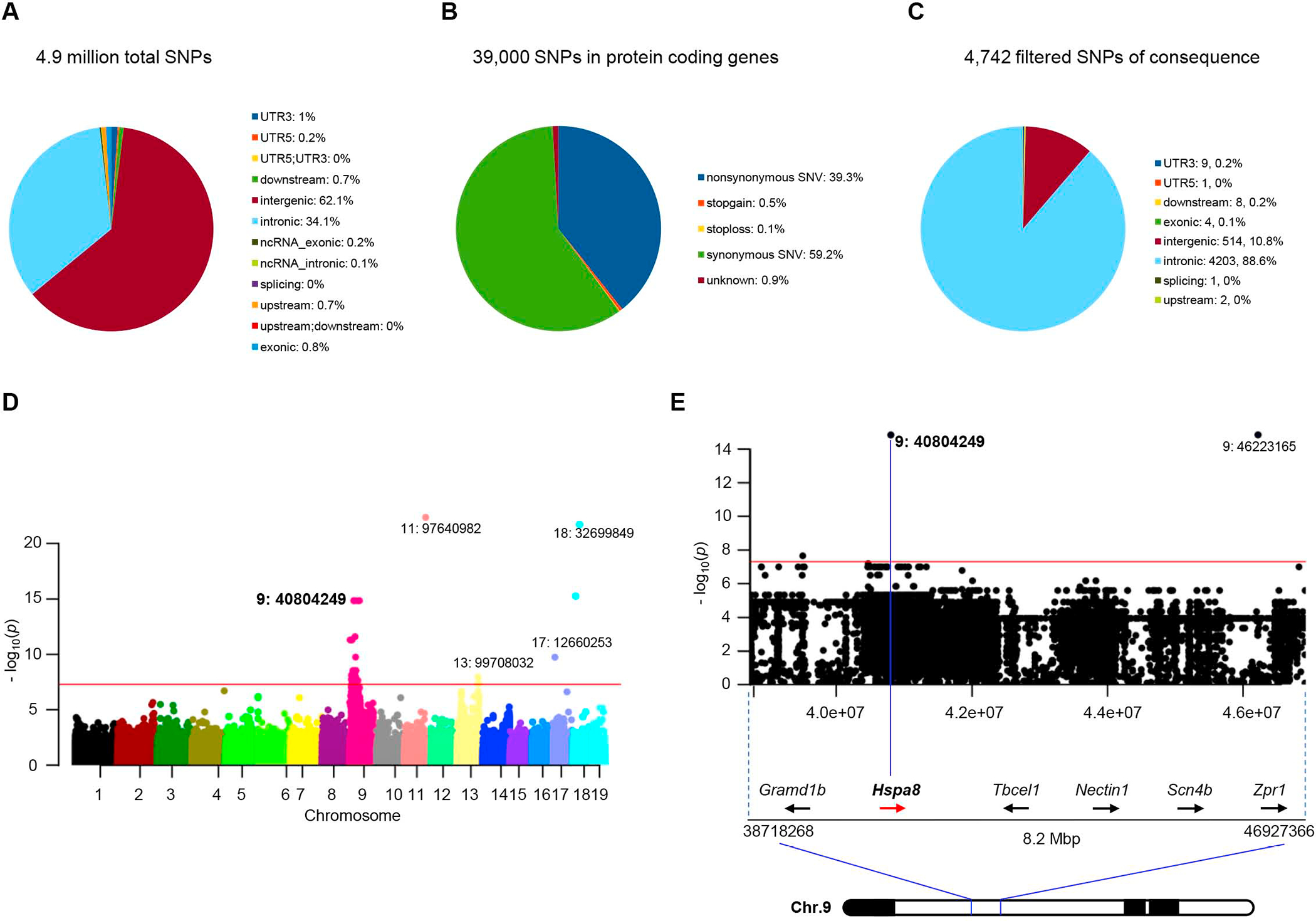
Graphical representations of (A) the total number of variant SNPs in the modified SMA mice, and (B) those in protein-coding genes. (C) Pie chart of SNPs filtered for P values < 5×10−8 and predicted for mutational consequences by ANNOVAR. (D) Manhattan plot of highly significant variant SNPs linked to the modified SMA phenotype, emphasizing those clustered in the Chr. 9 ROI; magenta line signifies P = 5×10−8. (E) Restricted view of the Manhattan Plot depicting the Chr. 9 Hspa8 c.G1408C SNP (id: 9:40804249, exon 7) underlying the Hspa8G470R variant (P = 1.5×10−15). See also Tables S2, S3.
To demonstrate unequivocally that Hspa8G470R underlies the modified SMA phenotype, we edited the wild-type GGG (Gly470) codon to CGG – for Arginine – in FVB/N-derived Δ7 carriers (Figure 3A). Two independent lines termed C3 and C5 were thus obtained. Each was used to generate SMA mutants. Consistent with Hspa8G470R being the SMA-modifying factor, we found that median survival of mutants from the two lines was increased vis-à-vis typically affected SMA mice, although lifespans of the modified mutants did not differ substantially from each other (Figure S4A). The latter observation suggested that the two lines were equivalent, and this was reflected in similar body weights, motor performance and relative levels of SMN and Hspa8 in the mutants (Figure S4B – S4E). Importantly, we also showed equivalent amounts of Hspa8 in animals with or without G470R suggesting that the variant does not alter protein stability (Figure S4F and S4G). Considering the equivalence between the C3 and C5 lines, all subsequent analyses were conducted on mutants from the C5 line.
Figure 3. Editing of WT Hspa8 to the G470R variant is sufficient to protect against severe SMA.
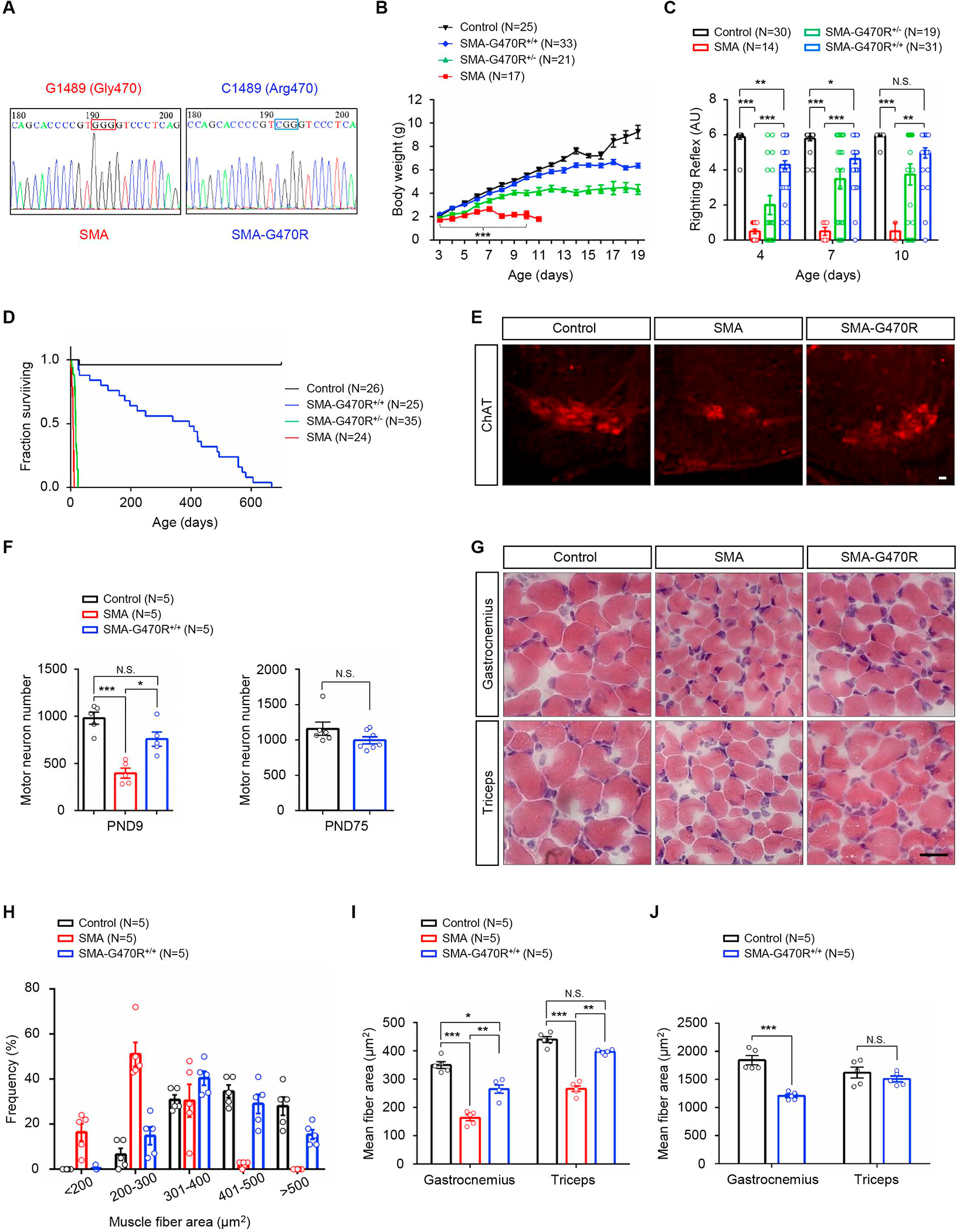
(A) Sequence chromatograms highlighting the ancestral (GGG) and edited (CGG) codons in Hspa8 from severe and modified SMA mutants. Hspa8G470R-expressing mutants are (B) larger, (C) perform significantly better in a motor performance assay and (D) have markedly longer lifespans than do mutants expressing the WT protein. Note: *, **, ***, P < 0.05, P < 0.01, P < 0.001 respectively; t tests (SMA and SMA-G470R+/+), one-way ANOVA and log-rank test respectively for panels B, C and D. (E) Representative immunostains of lumbar spinal cord sections depicting normal numbers of motor neurons in the SMA-G470R mutant at PND9. (F) Morphometric counts of lumbar motor neurons in SMA and littermate controls. (G) H&E-stained muscle sections from PND9 SMA mutants with or without the G470R variant and a littermate control; myofibers in the SMA-G470R+/+ mouse are larger than those of the SMA mutant. (H) Frequency distributions and (I) average sizes of myofiber in PND9 SMA and littermate controls. (J) Graph of mean myofiber areas in young adult (PND75) SMA-G470R+/+ mutants and littermate controls. Note: *, **, ***, P < 0.05, P < 0.01, P < 0.001 respectively; one-way ANOVA for comparisons at PND9 and t tests for comparisons at PND75 in panels F, I and J. N.S. – not significant. Scale bars: 20μm (panel E), 25μm (panel G). Data: mean ± SEM. See also Fig. S4.
Hspa8G470R mitigates neuromuscular pathology in SMA model mice
We next investigated how Hspa8G470R might mitigate neuromuscular dysfunction. Expectedly, we found that mutants harboring the variant were larger, more agile and considerably longer-lived than mutants expressing WT Hspa8 (Figure 3B – 3D). The disease-modifying effects of the G470R variant were furthermore confirmed in a more severe model of SMA (Figure S5A and S5B), suggesting that the modification is not line-specific. Moreover, on the Δ7 background, Hspa8G470R expression restored spinal motor neuron numbers in PND9 and PND75 mutants and resulted in muscle fibers that were larger than those of age-matched severe SMA mice expressing WT Hspa8 (Figure 3E – 3J). An examination of the NMJs, which in severe SMA mice feature small, immature, denervated endplates juxtaposed against nerve terminals engorged with neurofilament (NF) protein, revealed significantly fewer of these defects in the presence of Hspa8G470R (Figure 4A – 4D). This was accompanied by restoration of function, as assessed electro-physiologically, in hindlimb muscles of adult, modified mutants (Figure 4E). In fact, consistent with a role for Hspa8 in neurotransmission, end-plate currents in Hspa8G470R-expressing mutants were statistically assessed to be even greater than those observed in controls heterozygous for murine Smn and WT for Hspa8. This interesting phenomenon was also seen when controls expressing the variant (SMN2;Δ7;Hspa8G470R;Smn+/−) were compared to littermates absent the modifier (SMN2;Δ7;Smn+/−), and extended to most parameters analyzed, indicating that the modifier potentiates neurotransmission (Figure 4F). NMJ function in PND8-10 SMA mice, at a time point when all three mouse cohorts were viable, also demonstrated restored neurotransmission in the presence of the modifier (Figure S5C – F). Collectively, this aspect of the study unambiguously assigned a potent SMA-modifying property to the Hspa8G470R protein.
Figure 4. NMJ defects in SMA mice are suppressed by the Hspa8G470R variant.
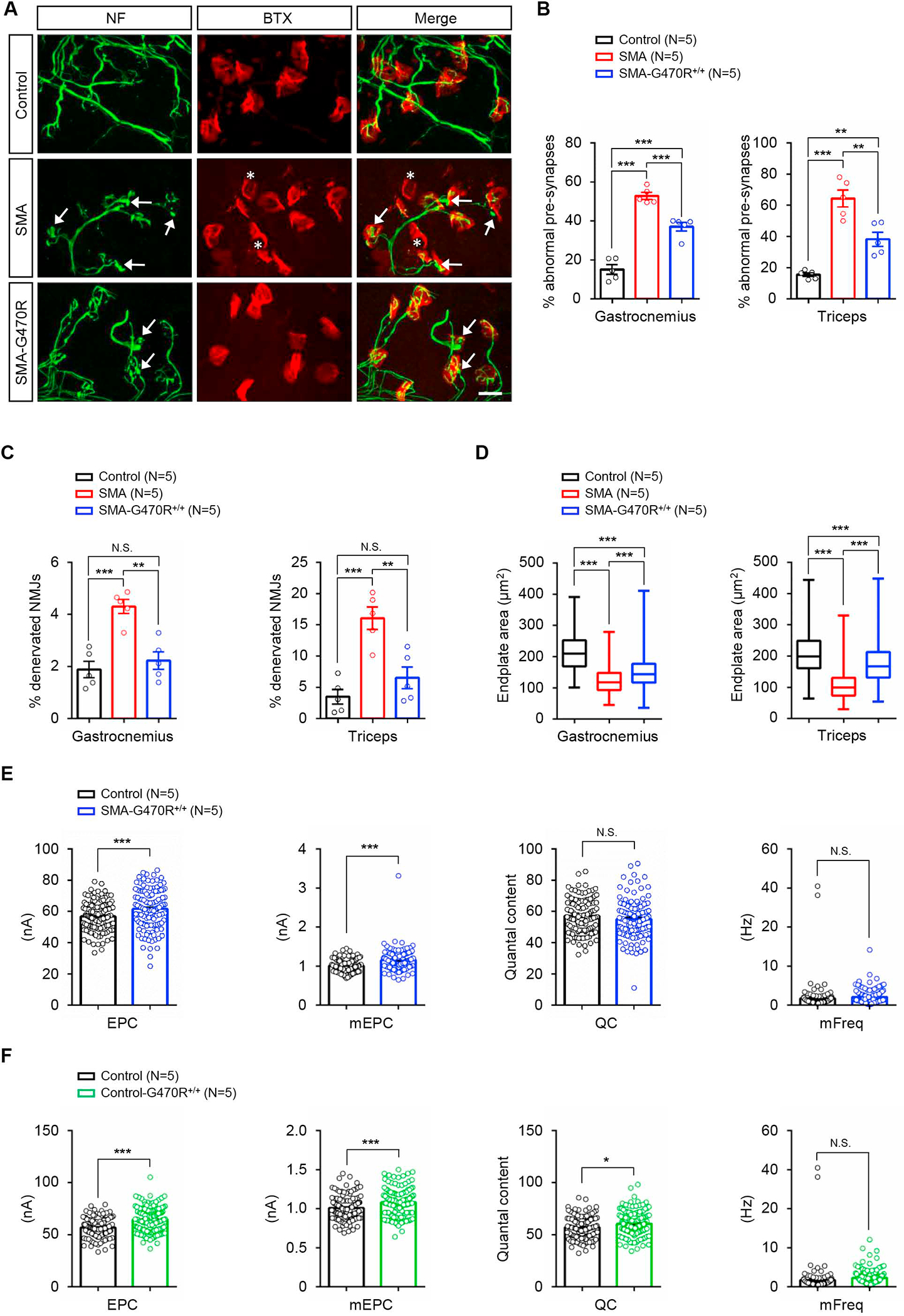
(A) Immunostains of NMJs in the triceps of PND9 controls and SMA mutants with or without Hspa8G470R; the modifier reduces denervation (asterisks) and the incidence of nerve terminals with abnormal NF varicosities (arrows). Scale bar: 20μm. Enumeration of NMJs in the three cohorts of mice displaying (B) nerve terminals abnormally swollen with NF protein and (C) denervated endplates. (D) Graphs depict relative enlargement of endplates in SMA-G470R+/+ versus SMA mutants. Note: **, ***, P < 0.01, P < 0.001, one-way ANOVA (panels B – D). Electrophysiological measures from EDL muscles of (E) PND75 SMA-G470R+/+ mutants and controls and (F) similarly aged controls with or without the variant illustrate the potentiating effect of the modifier on neurotransmission. Note: *, ***, P < 0.05, P < 0.001, t tests, N.S. – not significant. Data: mean ± SEM. See also Fig. S5.
Hspa8G470R modestly raises SMN levels by inducing SMN2 exon 7 inclusion
Considering the disease-mitigating effects of Hspa8G470R, we sought to explain its mechanism(s) of action. We began, notwithstanding evidence of acutely low protein in F2 mutants, by again assessing SMN levels. This analysis was undertaken as it had proved challenging, prior to identifying Hspa8G470R, to reliably assort severe and modified mutants into distinct groups for comparison. To our surprise, we found that although modified mutants continue to express only residual protein (≤20%) relative to SMN2+/+;SMNΔ7+/+;Smn+/− controls, they nevertheless expressed more SMN than mutants devoid of the modifier (Figure 5A, B). This suggested that one means by which Hspa8G470R acts is by increasing SMN. Considering the only source of SMN in Δ7 SMA mice is the human SMN2 transgene and that inducing exon 7 inclusion into SMN2 transcripts is one commonly reported means by which SMN may be raised, we examined levels of the full-length (FL) and truncated (SMNΔ7) transcripts from SMN2 in our various cohorts of mice by Q-PCR. We found that levels of FL-SMN were indeed modestly enhanced (~2-fold) in modified versus severe mutants (Figure 5C and 5D). This result is, moreover, consistent with reports of a feedback loop wherein low SMN protein exacerbates SMN2 exon 7 exclusion, and small increases in protein can stimulate production of FL-SMN transcript.27,28 Congruent with an increase in FL-SMN in the modified mutants, levels of SMNΔ7 transcript dropped relative to those in severely affected SMA mice (Figure 5E). Since measuring these transcripts in Δ7 mice may be confounded by the presence of the SMNΔ7 cDNA transgene16 in them, we repeated the analysis with mice harboring SMN2 but devoid of SMNΔ7.29 The outcome of the experiments with the latter set of mice reflected results obtained in the Δ7 line of mice (Figure S5G and S5H); FL-SMN increased while levels of the truncated isoform correspondingly dropped in the presence of Hspa8G470R. This suggested that murine Hspa8G470R raises SMN levels by altering SMN2 splicing.
Figure 5. Hspa8G470R alters SMN2 splicing and modestly raises SMN levels.
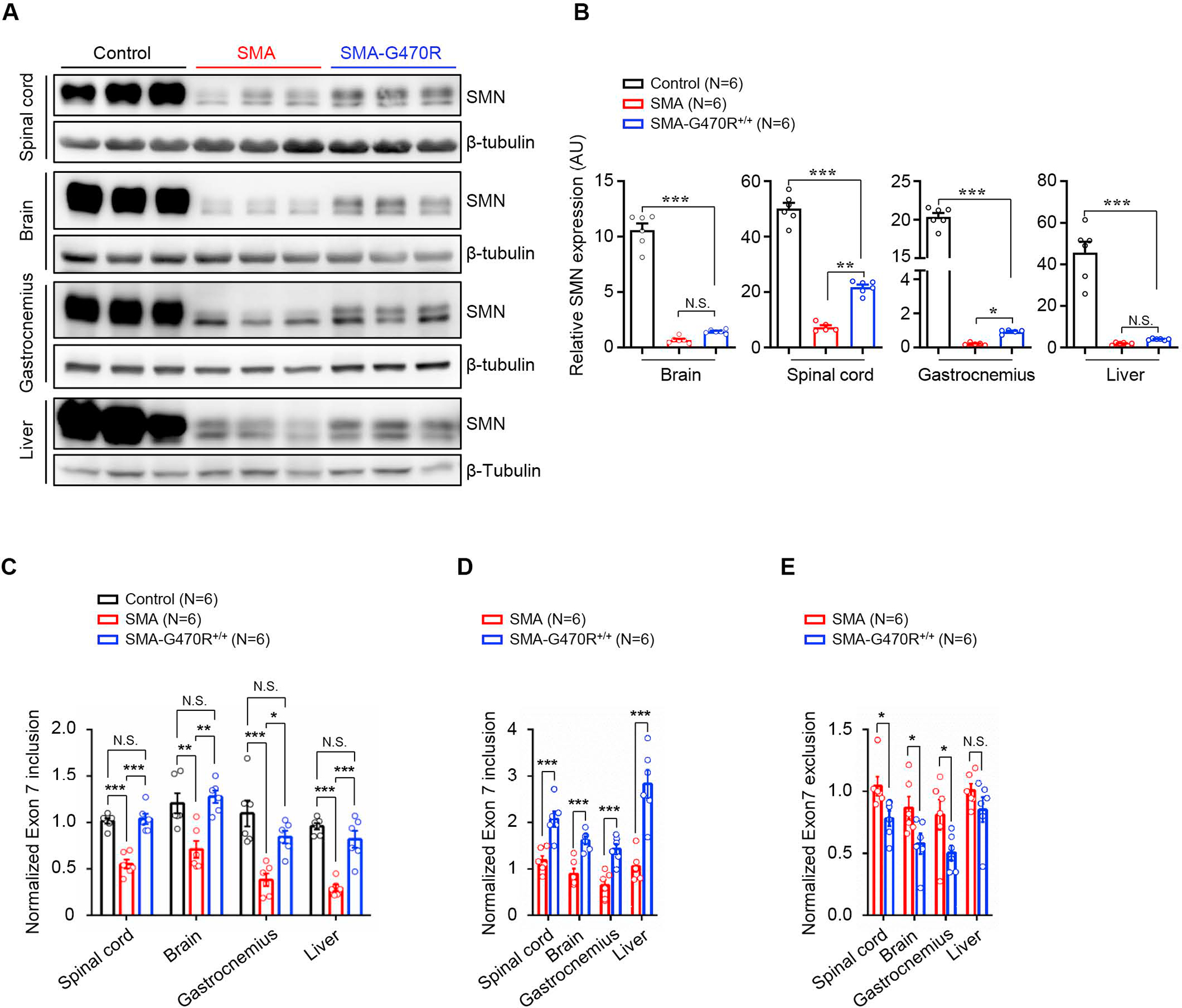
(A) Western blots of SMN protein in PND9 controls and SMA mutants with or without the G470R modifier and (B) quantified results of the blots in the three cohorts of mice. (C) Analysis of FL-SMN transcript levels by Q-PCR at PND9 in the three cohorts of mice. Two-way comparisons of (D) FL-SMN transcript levels and (E) the SMNΔ7 isoform in the two sets of SMA mutants illustrate an increase in the intact transcript and a corresponding drop in the truncated form. Note: *, **, ***, P < 0.05, P < 0.01, P < 0.001 respectively, one-way ANOVA (panels B and C), t tests, panels (D and E). Data: mean ± SEM. See also Figs. S5, S6.
In a final set of experiments and as measure of the consequences of the Hspa8G470R –dependent increase in SMN expression, we examined the levels of a number of downstream targets of SMN.30,31 We found that the reductions in the U12 and U4atac snRNAs seen in severe SMA were reversed and defects of Mdm2 and Clcn7 splicing rescued in modified mutants (Figure S6A – S6D). However, aberrant splicing of Tmem41b, defective 3’-UTR processing of histone H1c and abnormal Cdkn1a expression persisted in the mildly affected mutants (Figure S6E – S6G). Unexpectedly, U11 levels were found to increase in severe SMA, and perturbed concentrations of this snRNA, U2 and mis-spliced Tspan31 abnormally expressed or actually exacerbated in modified mutants (Figure S6H – S6J). These results suggest that although Hspa8G470R does raise SMN, the increase is insufficient to fully rescue splicing and/or expression defects universally in genes downstream of SMN.
Hspa8G470R exhibits an enhanced affinity for its synaptic co-chaperone partners
The discordance between the very modest increase in SMN and the striking rescue of the SMA phenotypes in our mutants led us to speculate that Hspa8G470R modifies disease directly and in addition to its effects on SMN. This idea was especially appealing considering the many synaptic functions of Hspa8 32,33 and our own observations suggesting a potentiation of neurotransmission by Hspa8G470R. Indeed, Hspa8 is an indispensable member of a tripartite chaperone complex critical for proteostasis and recycling of a number of synaptic proteins including those of the SNARE complex involved in neurotransmission.34,35 We therefore inquired if the other two members, SGTA and CSPα, of the tripartite chaperone of which Hspa8 is a component were affected under conditions of low SMN or by the Hspa8G470R variant. We found that levels of SGTA and CSPα were unaffected by low SMN (Figure 6A and 6B). Yet, interestingly, the G470R variant enhanced the affinity of Hspa8 for its binding partners (Figure 6C and 6D). That the strengthened interaction was not just SMN-mediated, given the incremental increase in SMN in modified versus severe mutants, was confirmed by extending the analysis to control samples. A large increase in SMN in these control samples, WT for Hspa8, did not augment the interaction of the latter for SGTA to the extent discerned in mutants harboring the variant (Figure S7A and S7B). Congruently, Hspa8WT-SGTA binding remained weaker than the binding of the G470R variant to SGTA in Smn heterozygotes (Figure S7C and S7D). We also discovered that Hspa8 is an SMN-interacting partner, and this association was significantly weakened by the variant (Figure 6E and 6F). We suggest that binding of Hspa8 to the two other members of the tripartite chaperone complex occurs while potentially being competed for by other client protein interactors such as SMN. These interactions, which likely influence one another and have downstream functional consequences, are altered by Hspa8G470R.
Figure 6. An enhanced affinity of Hspa8G470R for synaptic co-chaperone proteins.
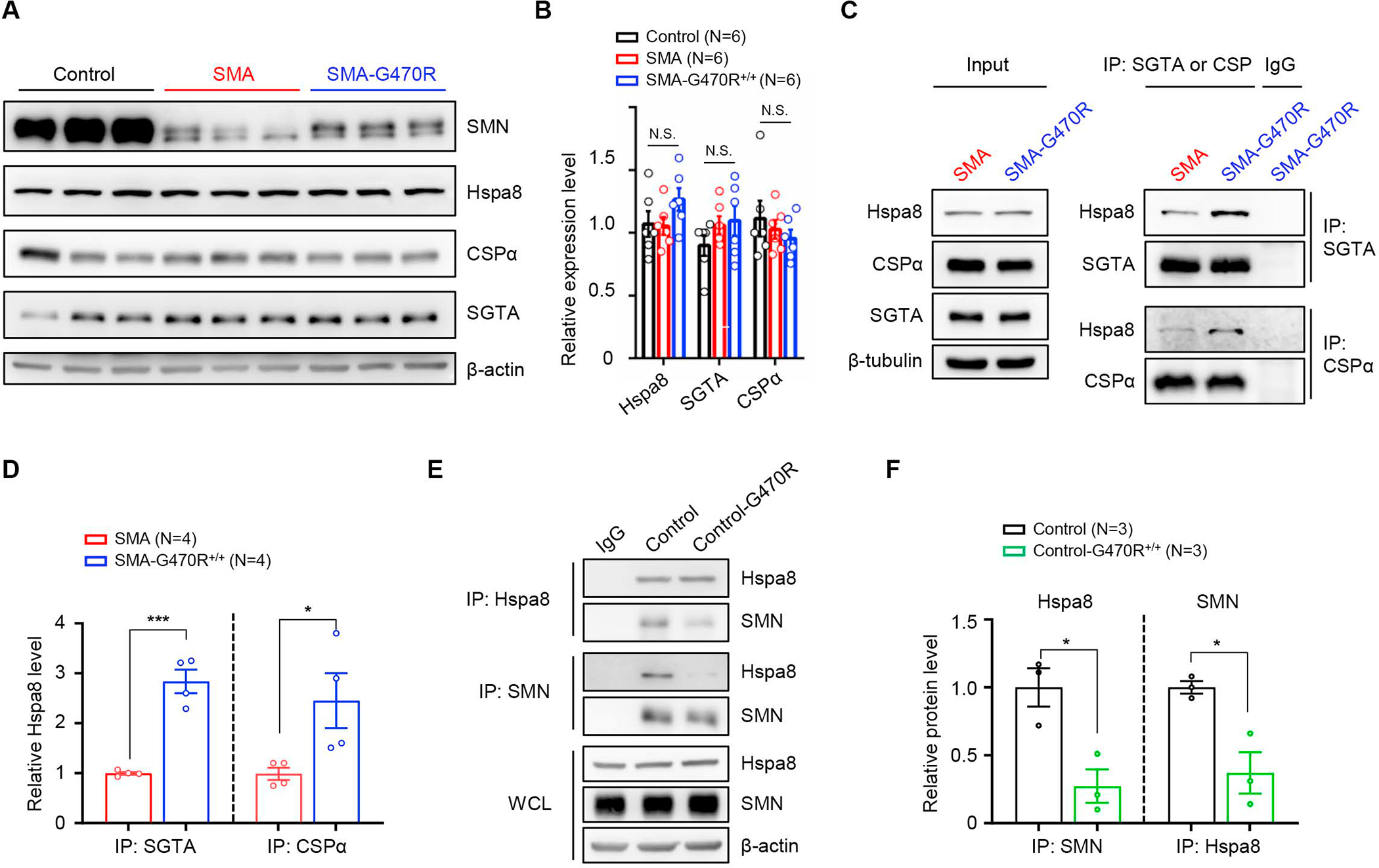
(A) Western blot analysis depicting equivalent levels of Hspa8 and other constituent members of a synaptic chaperone complex in PND9 brain tissue of controls and SMA mutants expressing WT or the G470R Hspa8 variant. (B) Quantified results of blot; N.S. – not significant, one-way ANOVA. (C) Co-immunoprecipitation (co-IP) analysis of relative affinities of WT Hspa8 or the G470R variant for its co-chaperones, SGTA and CSPα; the variant binds better to SGTA and CSPα. (D) Graph depicting the affinities of Hspa8WT and Hspa8G470R for their interacting partners. (E) Reciprocal co-IP analysis of brain-derived Hspa8 and SMN illustrates that the two interact and that there is weakened affinity of the G470R variant for SMN. (F) Quantification of relative affinities of WT Hspa8 or the G470R variant for SMN. Note: *, ***, P < 0.05, P < 0.001 respectively, t tests for analyses in panels D and F; brain lysates from PND9 mice were used for co-IP experiments. Data: mean ± SEM. See also Fig. S7.
SNARE complex assembly is disrupted by low SMN and recovers in SMA;Hspa8G470R mice
Considering the implications of proper chaperone complex formation and function for SNARE complex assembly, and the upshot – poor neurotransmission – of inefficient SNARE complex formation, we quantified assembled SNARE complexes in modified and severe mutants. We began this investigation in the traditional manner – by using brain tissue, a source of CNS synapses. However, we failed to detect changes (Figure S8A and S8B). We therefore turned to muscle (triceps) tissue, a source of neuromuscular synapses; these synapses are particularly vulnerable in SMA. Interestingly, this analysis revealed a clear and reproducible difference between severe SMA mice and healthy littermate controls heterozygous for murine Smn; assembled SNARE complexes as detected with an antibody against SNAP25, a core component of the complex, were significantly less abundant under conditions of low SMN, but absent any attendant drop in levels of SNAP25, Stx1 or Syb2 monomers. Importantly, their levels were restored in modified mutants, and this pattern observed, albeit to a lesser extent, in gastrocnemius muscle (Figure 7A, 7B and Figure S8C – S8E).
Figure 7. SNARE complex assembly is disrupted in SMA NMJs and restored by the Hspa8G470R variant.
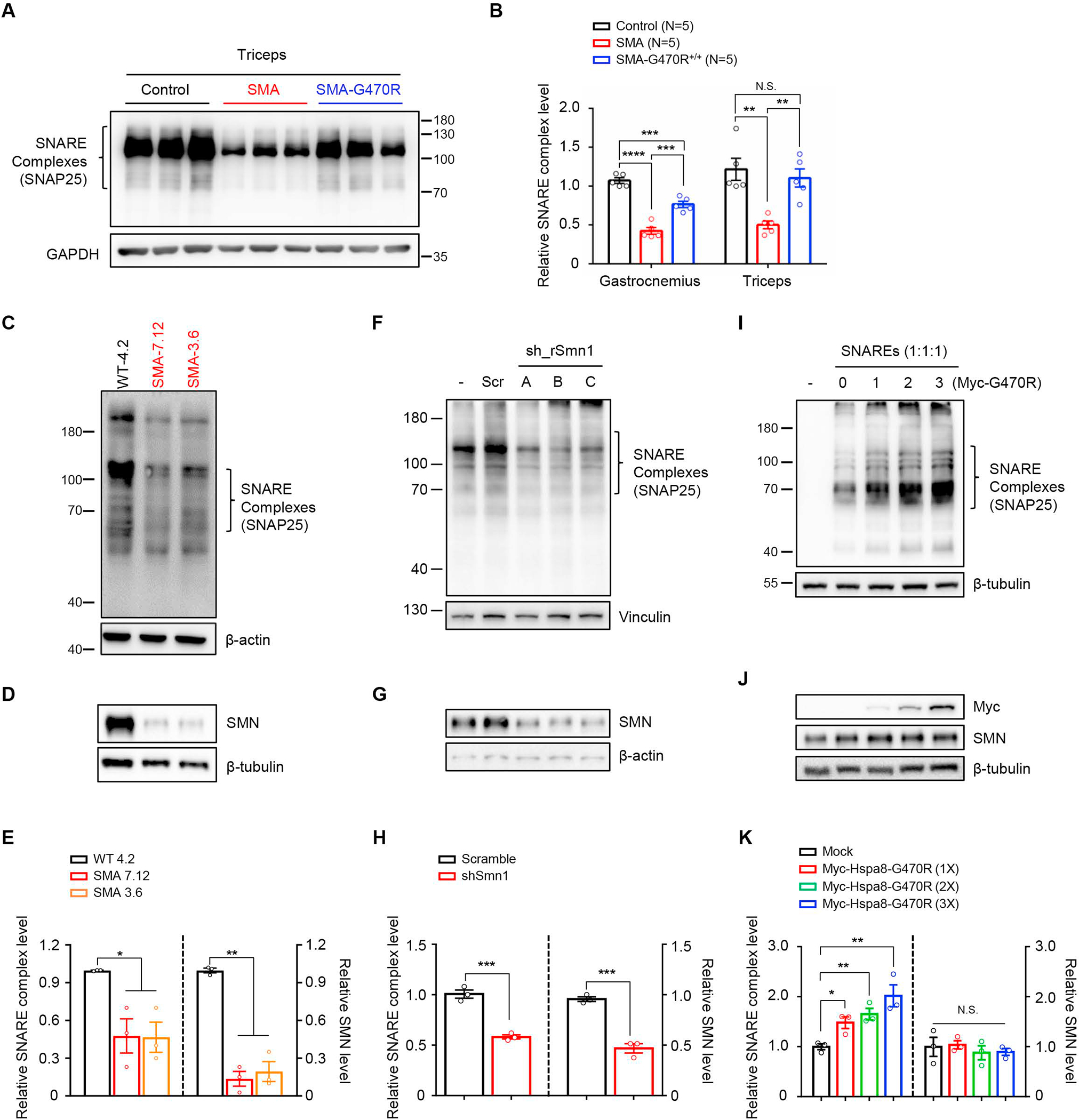
(A) Representative immunoblot, probed for SNAP25, illustrating reduced high molecular weight SDS-resistant SNARE complexes in PND9 SMA NMJs derived from triceps; complex levels are restored in SMA-G470R mutants. Note: Samples were not boiled. (B) Quantified SNARE complex levels in triceps and gastrocnemius muscles of PND9 controls and SMA mutants with or without the G470R variant. Note: **, ***, P < 0.01, P < 0.001 respectively, one-way ANOVA. N.S. – not significant. (C) Representative immunoblot depicting reduced SNARE complexes in two iPSC-derived motor neuron lines from severe SMA patients; samples were not boiled. (D) Western blot analysis of boiled samples from panel c confirm low levels of SMN in the SMA lines. (E) Graph showing relative SNARE complex concentrations and SMN levels in panels C and D respectively. (F) Reduced SNARE complex assembly is observed in the immunoblot of samples from PC-12 cells expressing shRNAs against SMN; samples were not boiled. (G) Western blot analysis of boiled samples from panel F confirm low levels of SMN in shRNA-mediated knockdown lines. (H) Quantified SNARE complex and SMN levels in samples analyzed for study depicted in panels F and G. Note: ***, P < 0.001, respectively, t tests for analysis of data in panel (H). (I) Immunoblot depicting effect of raising Hspa8G470R levels (from a plasmid) on SNARE complex formation in HEK293 cells transfected with the core SNARE components; samples were not boiled. (J) Western blot analysis of boiled samples from panel (I) showing relatively stable levels of SMN notwithstanding increasing concentrations of Myc-Hspa8G470R, as detected with anti-myc antibody. (K)Quantified levels of SNARE complexes and SMN respectively in panels I and J. Note: *, **, P < 0.05, P < 0.01, respectively, one-way ANOVA. N.S. – not significant. Data: mean ± SEM. See also Figs. S8, S9.
Given the novelty of SNARE complex assembly deficits in SMA, we sought to confirm our findings and investigate underlying mechanisms. Accordingly, we first repeated assessments of assembled SNARE complexes, probing our blots with antibodies against the two remaining components, Stx1 and Syb2, of the complex. These experiments also revealed inefficient SNARE complex assembly in severe SMA and rescue of the assembly in Hspa8G470R –expressing mutants (Figure S8F – S8H). Next, we examined SNARE complex assembly in iPSC-derived motor neurons from severe SMA patients. This too revealed fewer assembled complexes under low SMN conditions (Figure 7C – 7E). The lower abundance of SNARE complexes detected in the iPSC-derived motor neurons with the SNAP25 antibody were confirmed with antibodies against Stx1 and Syb2 (Figure S8I – S8L). Similarly, knockdown of SMN in neuron-like PC-12 cells resulted in inefficient SNARE complex assembly (Figure 7F – 7H). Third, we ascertained the likelihood that rescue of SNARE complex assembly in modified mutants was mediated exclusively through induction of SMN by Hspa8G470R or, alternatively, at least in part through a direct effect of the variant chaperone on the SNARE complex. For this, we examined complex assembly in two sets of Smn+/− controls expected to assemble sufficient complexes for normal neurotransmission. Yet, controls expressing the Hspa8G470R variant were found to assemble SNARE complexes more efficiently than cohorts harboring WT Hspa8 (Figure S8M and S8N). This suggested that Hspa8G470R promoted SNARE complex assembly even under non disease-relevant SMN conditions and without a requirement for large increases in SMN. Indeed, we found that over-expressing Hspa8G470R in HEK293 cells transfected to express equimolar quantities of Stx1, Syb2 and SNAP25 resulted in correspondingly greater levels of SNARE complexes (Figure 7I – 7K). Interestingly, in these experiments, an appreciable increase in SMN was not detected (Figure 7J and 7K) indicative of a direct effect of Hspa8G470R on SNARE complex formation. Moreover, forced expression of SMN in HEK293 cells beyond WT levels failed to stimulate SNARE complex assembly the way Hspa8G470R did (Figure S8O and S8P). This suggested that Hspa8G470R has a direct and potent effect of potentiating SNARE complex assembly. In the context of fewer such complexes forming in SMA NMJs, Hspa8G470R likely restores their levels, rescuing neurotransmission at these synapses. In aggregate, we posit that Hspa8G470R has a significantly more robust and direct effect on restoring SNARE complex assembly in our mutants instead of merely acting to promote the formation of these complexes by raising SMN. Notwithstanding such an inference, and given the clear dependency of SNARE complex assembly on SMN levels, we performed a fourth set of experiments to inquire if poor complex formation was a peculiarity of just the Δ7 line or more generally observed in SMA. To do so, we examined complex formation in a second SMA mouse model – the Smn2B/− mouse.36 Poor SNARE complex assembly was determined to be a feature of this model too (Figure S9A, and S9B) suggesting that this phenomenon is a likely bona fide synaptic defect in SMA. In a final set of experiments, we inquired if inefficient SNARE complex formation in SMA mutants is a mere by-product of denervation by examining how well the complexes formed in a mouse model of ALS.37 We found that complex assembly in symptomatic (PND110 – 127) ALS mutants was no different from that in controls (Figure S9C and S9D). Indeed, in this particular mutant SOD-1 line, expressing Hspa8G470R failed to modify the ALS phenotype (Figure S9E). In aggregate, the data reveals a new aspect of SMN function – a capacity to modulate, likely indirectly, SNARE complex assembly. The data also suggests that Hspa8G470R functions in the same pathway to independently promote complex formation and, finally, that inefficient formation of this synaptic complex is not merely a general consequence of a denervating disease.
Discussion
The durability of current SMA treatments remains unclear, and the precise function(s) of the SMN protein in maintaining the vigor of the neuromuscular system continues to be debated.38 Here we report findings that center primarily on SMN functions but could also impact future therapy development and, more broadly, inform how nerve-muscle function is preserved by proteins best recognized for their housekeeping properties. Two notable outcomes of our work are worth summarizing. The first is the discovery of Hspa8G470R, a novel and potent SMA suppressor. Morphological and functional defects in model mice expressing Hspa8G470R were rescued, motor neuron numbers restored and lifespan enhanced from approximately 10 days to roughly 300 days. Such a profound rescue has not previously been reported for severe SMA. Our second noteworthy finding emerges from functions attributed to Hspa8, implicating SMN in a novel role consistent with the classic, neuromuscular SMA phenotype – influencing SNARE complex assembly at nerve-muscle synapses. Building on the discovery that the G470R variant is better able than WT Hspa8 to bind other constituents of a tripartite synaptic chaperone critically important for SNARE complex formation, we found that assembly of this complex is disrupted at NMJs and spinal motor neurons respectively of severely affected SMA mice and humans. In contrast, SNARE complex formation was restored in the presence of the modifier; NMJ function was concomitantly normalized, indeed potentiated, in Hspa8G470R-expressing SMA mutants. These findings cast new light on how low SMN triggers motor neuron dysfunction.
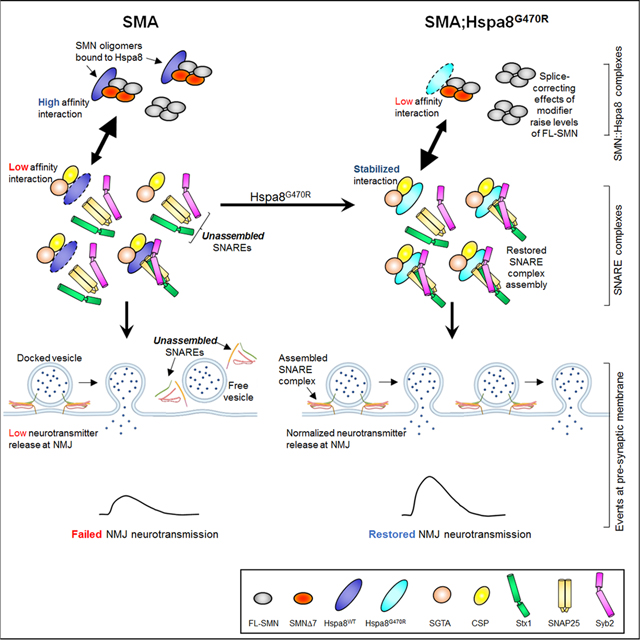
Aside from a handful of human studies that identified perturbations of endocytosis in SMA12–14,15, positional cloning strategies and unbiased genetic screens to probe disease mechanisms have not been particularly informative for this condition. Moreover, none of the investigations employed humanized SMA model mice. Here, for the first time, we describe the outcome of one such exploration for SMA modifiers using the rodent models. Initially somewhat puzzling as a candidate modifier, Hspa8G470R as a modulator of the SMA phenotype is perhaps not altogether a surprise. Wild-type Hspa8 is an especially abundant component of synapses39 and best known for ensuring cellular homeostasis through the sequestration or degradation of nascent, misfolded or aggregated proteins.40 However, it is in two lesser-known capacities – as a chaperone of neuronal SNARE complexes and as a modulator of splicing that the G470R variant most likely alters the SMA phenotype. Hspa8 is not widely reported to be involved in splicing. Still, it has been detected by mass spectrometry analysis in mammalian spliceosome complexes and, additionally, emerged in an unbiased screen for splice modulators as a weak activator of PSD-95 splicing.41–43 It is therefore reasonable to assume that WT Hspa8 is a constituent of the factors that regulate SMN2 splicing, either directly as a member of the spliceosome or, indirectly, by altering an SMN2 splicing protein in its capacity as a chaperone. Discerning these possibilities is an exciting avenue for future studies. Substituting Gly470 in Hspa8 with Arg likely alters the levels and/or activities of these factors resulting in exon 7 inclusion in the SMN2 transcript. The substitution concomitantly weakens Hspa8-SMN interaction but, importantly, enhances the affinity of Hspa8 for its binding partners in the tripartite chaperone complex, likely increasing the efficiency with which it assembles into a functional unit to promote SNARE complex formation. The precise dynamics of these altered interactions leading to SMA suppression remain to be fully elucidated but are nevertheless captured in a hypothetical model premised, in part, on competing interactions for Hspa8 by various partners (Figure 8).
Figure 8. Modulation of SNARE complex assembly by SMN and the G470R variant of Hspa8.
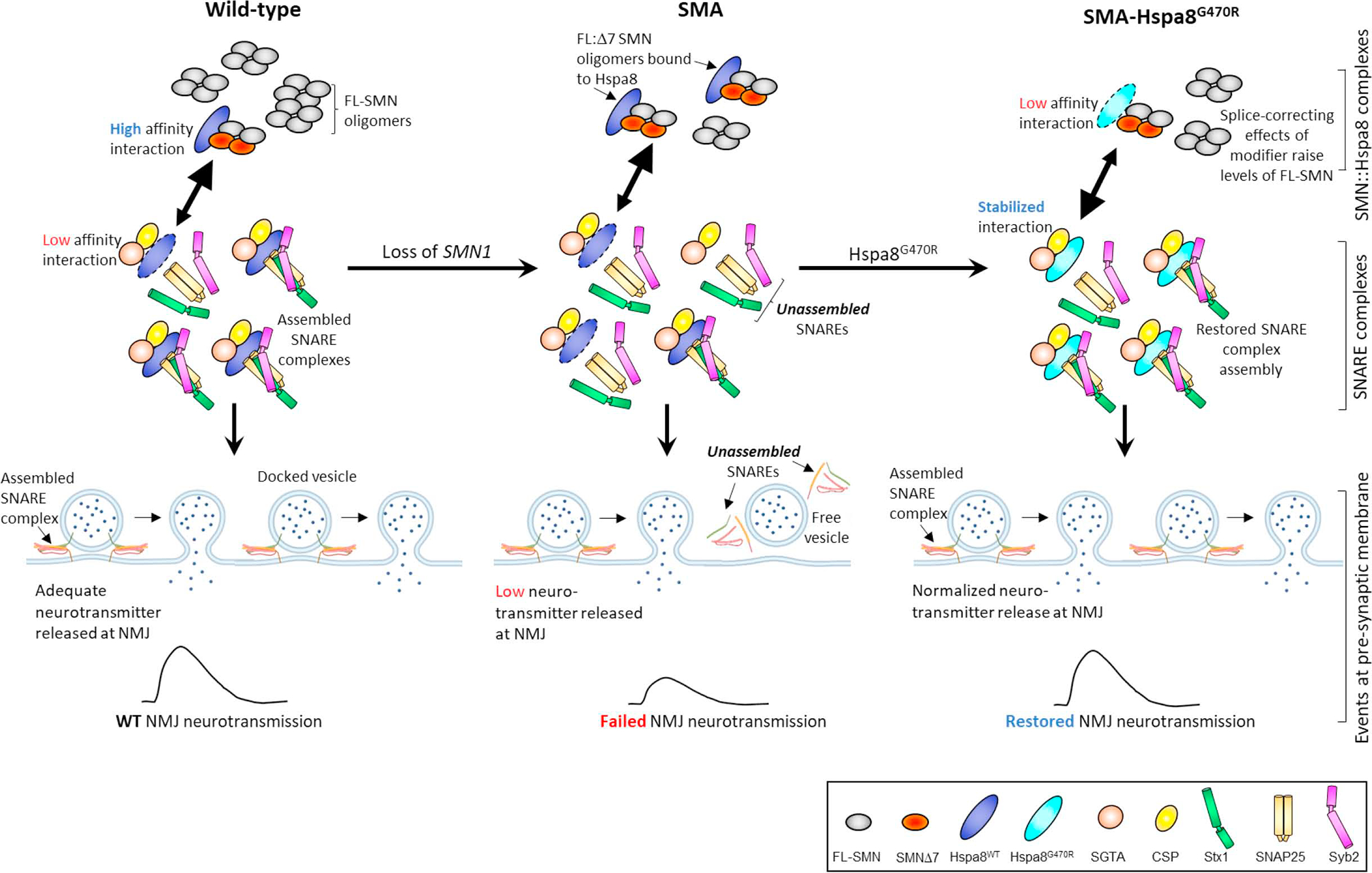
Depicted is a hypothetical model that encapsulates the relationship between SNARE complex assembly, SMN and the Hspa8G470R SMA modifier at NMJs. In the WT state, the preponderance of SMN exists as intact (FL), stable oligomeric complexes, SMNΔ7 isoforms being excluded from the complexes. Consequently, relatively little Hspa8 is required for SMN turnover in synapses. Hspa8 is therefore free to engage with its synaptic co-chaperones, CSPα and SGTA, notwithstanding a potentially weak affinity for these proteins. Still, this ensures proper SNARE complex assembly and efficient neurotransmission. In the absence of SMN1 (SMA), total FL-SMN levels fall and relative concentrations of oligomers of the protein containing the unstable SMNΔ7 species and proteins such as NF rise. Hspa8 is diverted away from the tripartite chaperone complex to effect turnover of dysregulated proteins such as the less stable FL-SMN:SMNΔ7 hybrid oligomeric complexes. Consequently, repeated cycles of SNARE complex assembly are disrupted. The G470R variant reverses this effect by stabilizing the interaction of Hspa8 with its co-chaperones and concomitantly weakening its association with SMN and perhaps other clients too. SNARE complex assembly is thus restored. A weak, SMN2 splice-switching property inherent in Hspa8G470R modestly raises levels of the intact SMN and combines with the effect of the variant on assembling SNAREs to potently suppress the SMA phenotype. Note: Larger arrowheads in double-headed arrows signify direction of preferred interaction.
What are the structural consequences of the G470R substitution? Glycine-470 lies in the substrate-binding domain (SBD) of Hspa8. That this position is occupied, in all species examined, by glycine is not surprising; this section of Hspa8 constitutes a bend/loop between two β-sheets. Loops or bends are secondary structures in proteins, generally composed of small amino acids with low steric restrictions.44 Still, our finding that Hspa8 accommodates Arg at this position is not entirely unexpected. Arginine is charged. Such amino acids are not normally tolerated in tight bends owing to their charged nature and large size. Moreover, in a number of studies Gly → Arg substitutions were found to be amongst the most common disease-causing missense mutations45–47, with arginine a particularly frequent mutant residue in bends and turns.45 Yet paradoxically, arginine is also the most statistically over-represented amino acid in turns/bends of WT proteins.45 This is consistent with the G470R variant we report and suggests that the bend/loop between the β5 and β6 sheets harboring this residue is not just tolerant of the substitution but having acquired it, in fact turns Hspa8 into a disease suppressor. Still, precisely how the G470R variant alters the 3-dimensional structure of Hspa8 and the implications of the variant for the chaperone’s multifarious clients including Tau – a protein associated with Alzheimer’s disease48 – remain to be investigated.
Arguably, the most noteworthy finding to have emerged from this work is that of disrupted SNARE complex assembly in SMA. We suggest that this results, at least in part, via unintended competition between client proteins such as SMN and SGTA/CSPα for Hspa8 (see Figure 8), entrapment of Hspa8 into SMN-containing units and, consequently, inadequate chaperone complex to maintain synaptic SNAREs in proper conformation to sustain repeated cycles of neurotransmission. Precisely why SMN binds Hspa8 is not clear. However, the interaction has been observed in prior studies49–51, and it is quite possible that SMN is subject to Hspa8-mediated proteostasis via autophagy, a process reportedly elevated in SMA52 when SMN oligomers are likely to consist of unstable SMNΔ7 units instead of mainly intact, full-length SMN1-derived protein. A second, equally intriguing outcome of our study is the selective disruption of SNARE complex assembly in neuromuscular synapses; we were unable to detect similar perturbations in brain synapses. While these findings are consistent with especially profound dysfunction of neuromuscular synapses in SMA, an explanation for the differential vulnerability of CNS and peripheral synapses to disruptions of SNARE complex assembly in the disease is not readily apparent. The cellular machinery that is responsible for neurotransmission is conserved in central synapses and NMJs. Yet, there are important distinctions and these can produce markedly different effects not only at these two synapses but also between excitatory and inhibitory CNS synapses.53 Thus, for instance, combined knockout of Munc13-1 and Munc13-2, proteins involved in neurotransmitter release, evoked quite distinct effects in brain and peripheral synapses, abolishing miniature end-plate potentials (MEPPs) in hippocampal synapses while augmenting them two-fold at NMJs.54,55 It is possible that bMunc13-2, a brain-specific isoform of Munc13-2 underlies this difference. Similarly, while double knockout of ELKS1 and ELKS2, proteins responsible for vesicle priming, failed to affect EPPs in the calyx of Held, ablation of ELKS1 at fly NMJs was sufficient to reduce evoked currents significantly.56,57 Expression of both ELKS proteins in brain but only ELKS1 at NMJs is one plausible explanation of this finding. Whether differential expression in brain and peripheral synapses of these or other neurotransmission-related proteins explain the selective vulnerability of NMJs in SMA to SNARE complex assembly remains to be determined and could, more generally, elucidate nuanced differences in the way brain and neuromuscular synapses operate.
The Hspa8G470R SMA suppressor we report here is the first to have emerged from observations of discordant phenotypes in humanized model mice and arguably, the most potent reported so far. This likely stems from multiple mechanisms of action. Our study provides evidence for involvement of at least two pathways, one based on increased SMN from the human SMN2 gene, the other most likely acting directly on neurotransmission via effects on SNARE complex assembly. Interestingly, the fly homolog of Hspa8 (Hsc70-4) is also reported to mitigate neurodegenerative disease via effects at the synapse.58 Still, the relative contributions of the two pathways identified here to overall disease mitigation remain to be determined, as small changes in SMN can significantly alter disease.59–61 Future studies to dissociate the effects of Hspa8G470R on promoting SNARE complex assembly on the one hand and raising SMN levels on the other will enable one to discern the extent to which each pathway modifies SMA. Despite this question, the net effect observed here was a complete rescue of neuromuscular dysfunction – as assessed in young adult mutant mice – but failure to resolve defects of splicing fully. This bolsters the notion that the neuromuscular SMA phenotype is not immutably linked to defects of splicing. Indeed, we suggest that directly targeting other disease-relevant pathways be actively considered if existing SMN-augmenting therapies fail to provide optimal benefit. Improving function at the synapse via Hspa8-associated or other related pathways is one appealing prospect. In this vein, it is pertinent to consider the low (~6%) frequency with which we obtained mild SMA mutants in our mapping studies – one that predicts a second FVB/N-derived modifier acting in concert with Hspa8G470R to produce the rescue we report. Revealing the identity of this second modifier will cast additional light on basic SMA biology and be an important driver of future investigations.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Umrao R. Monani (um2105@columbia.edu).
Materials availability
Unique reagents generated in this study will be made available on request, but we may require a payment and/or MTA if there is potential for commercial application.
Data & code availability
Mouse whole genome sequencing data that support the findings of this study have been deposited in the BioProjects database with ID number PRJNA855239.
METHOD DETAILS
Animals
Δ7 SMA model mice (Jax #005025), and their progenitors harboring only human SMN2 (Jax #005024), were created by us16,29 and are available from the Jackson Labs. Transfer of the various transgenic and knockout alleles constituting the Δ7 model onto the FVB/N and C57BL/6 strain backgrounds was carried out at Columbia University. Smn2B/− mice were a kind gift from Rashmi Kothary, Ottawa University. SOD1G86R mutants were acquired from the Jackson Labs (#05110). Hspa8G470R mice were created at the Columbia University mouse core facility using CRISPR technology. To generate Hspa8G470R mice, a donor template (5’ –TATGAAGGTGAAAGGGCCATGACCAAGGACAACAAC CTGCTTGGAAAGTTCGAGCTCACAGGCATCCCTCCAGCACCCCGTCGGGTCCCTCAGATTGAGGTTACTTTTGACATCGATGCCAATGGCATCCTCAATGTTTCTGCTGTAGATAAGAGCACA – 3’) and Hspa8-G470R guide RNA (sgRNA: 5’-GCAUCCCUCCAGC ACCCCGGUUUUAGAGCUAUGCU −3’), were synthesized (Integrated DNA Technologies). S. pyogenes CAS9 nuclease was purchased (New England Biolabs). Equimolar concentrations of CAS9 enzyme and sgRNA were mixed in injection buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA, pH 8.0) to form 0.15 μM RNP complex. The RNP complex (0.15 μM) and the donor template (0.5 μM) were injected into the pro-nuclei of fertilized FVB/NJ eggs and transferred after a 24h incubation period into the oviducts of pseudo-pregnant surrogate females. Analyses of the various mice employed for the study were carried out in a blinded fashion except for behavioral studies when severe phenotypes displayed by mutants precluded blinding. All animal procedures adhered to protocols described in the Guide for the Care and Use of Lab Animals (National Academies Press, 2011) and were approved by Columbia University’s IACUC. The subjects of this study were randomly selected, mixed or pure background male and female mice housed in a controlled environment on a 12-hour light/12-hour dark cycle with food and water. Δ7 SMA carrier mice heterozygous for murine Smn constituted healthy controls unless otherwise specified. Genotyping was performed by PCR on tail DNA using primers listed in Table S4. Righting ability was assessed as previously described.20 Briefly, mice were placed on their backs and latency of the animals to place all four limbs on the tabletop recorded. Time to right was converted into a score as described 20 and reported as such in the manuscript. For the behavioral studies, GraphPad Prism was used to determine sample sizes to detect differences of at least 2 SDs with a power of 80% (P < 0.05). Mice were not randomized, but in any instance where mutants did not exhibit an overt disease phenotype, it was possible to blind the investigator to the particular cohort being assessed.
Sequencing and GWAS
Genomic DNA from 25 F2 SMA mutants exhibiting either severe or mild disease was isolated and subjected to whole genome sequencing using the NovaSeq 6000 platform (Novogene Inc., Table S3A). An average of 52 billion bases per sample were sequenced and roughly 333 million reads per subject mapped by aligning the reads to the C57BL/6 reference sequence (GRCm38.p6/mm10) using the Burrows-Wheeler Aligner algorithm.62 Removal of duplication reads from the BAM files thus generated (Table S3B) resulted in sequence coverage of 11X – 14X (> 95% of genomic regions covered at a depth of 4X and 98% of the genome covered at a depth of 1X). Samtools63 and ANNOVAR64 were used to call SNPs from BAM files and to predict the consequences of the variants, respectively (Table S3C and S3D). Genome-wide association tests were performed with the following phenotypic groups stratified based on increasing degree of disease severity (LL1-11, I1-8, and S9-14; LL: long-lived, I: Intermediate; S: Severe) and assigned numeric values; LL1-11 = 1, I1-8 = 2, S9-14 = 3. Tests were run under linear models by using the --glm function in PLINK2 software (https://www.cog-genomics.org/plink/2.0/) (Table S2B). ANNOVAR64 was used to predict consequences of the 4742 significant SNPs, P values < 5×10−8 (Table S3D).
Motor neuron, NMJ and muscle histology
Motor neuron, NMJ and muscle histology was essentially carried out as previously described20,65 and detailed below.
Motor neurons:
Spinal cord tissue was dissected following transcardial perfusion (4% PFA in 1X PBS) of mice. The tissue was post-fixed in the same fixative, cryo-protected in 20% and then 30% sucrose before embedding the material in Tissue-Tek OCT medium for cryostat sections. 20 μm sections from lumbar spinal cords were overlaid for 10 minutes with 4% PFA, washed in 1X PBS and the tissue then permeabilized with 1% Triton X-100, 5 min. To stain the motor neurons, sections were placed in blocking buffer (2% normal goat serum, 2% BSA, 0.5% Triton X-100 in PBS) for 1 hr at room temperature. The sections were then incubated (4°C, overnight) with primary antibodies against ChAT (1:100, Millipore) or vGlut1 (1:10,000, Millipore) diluted in blocking buffer, and washed (15 min × 4) with 1X PBS. Sections were subsequently incubated with appropriate secondary antibodies (Alexa Fluor-594 conjugated donkey anti-goat IgG or Alexa Fluor-488 conjugated goat anti-guinea pig IgG, Invitrogen) at dilutions of 1:1000, 2 hr at room temperature. After washing (15 min ×4) with 1X PBS, the sections were mounted in anti-fade mounting media (Vector Labs) and motor neurons visualized on either a Nikon 80i fluorescent microscope (Nikon) or a Leica TCS SP5 II laser scanning confocal microscope (Leica). Motor neuron numbers were assessed in the L1-L5 region of the spinal cord and the raw numbers extrapolated to the entire region based on the length and thickness of tissue segment.
Muscle histology:
The proximal triceps muscle and distal gastrocnemius muscle were flash-frozen in isopentane cooled with liquid nitrogen. 12 μm thick sections were stained with hematoxylin and eosin (H&E; Sigma), and morphology, size and numbers of fibers were determined using ImageJ software following image acquisition with a SPOT 4.5 camera and associated software (Diagnostic Instruments). Fiber sizes were determined by assessing ≥ 100 fibers from individual samples. Cardiac structure was assessed using transverse sections cut at the level of the ventricles. The thickness of the left ventricles in mutants was normalized to heart size.
NMJ assessment:
NMJ analysis was performed on whole muscle. Tissues were fixed and permeabilized with 100% methanol for 10 min at −20°C and incubated with blocking buffer (2% normal goat serum, 3% BSA, 1% Triton X-100 in PBS) for 1 hr. at room temperature. The tissue was incubated overnight at 4°C with a primary antibody against neurofilament (NF, 1:1000, Millipore) and washed (20 min × 4) by 1X PBS. The following secondary antibodies were subsequently applied: Alexa Fluor-488 conjugated goat anti-rabbit IgG secondary antibody (1:1000, Invitrogen), and rhodamine-α-bungarotoxin (BTX, 1:1000, Invitrogen) for 3 hr at room temperature. After washing (20 min × 4) by 1X PBS, the tissue was mounted in anti-fade medium (Vector Labs) and images of NMJs were acquired with Leica TCS SP5 II laser scanning confocal microscope (Leica). Morphological analyses were conducted using LAS-X software (Leica). NMJ abnormalities were quantified as previously described.20
NMJ electrophysiology
NMJ function was assessed by electrophysiological means as described by us in prior reports.21,66 Detailed protocols follow. Methods described in a previous report66 were used to evaluate NMJ function in modified F2 SMA mutants. In short, EDL muscle with the sciatic nerve attached was extracted from 4 – 5 month-old mice and placed in oxygenated mammalian Ringer’s solution. Muscle contraction was suppressed by incubating the tissue (45min) in 2–3μm μ-Conotoxin. Recordings were subsequently carried out in toxin-free Ringer’s solution. Between 40 and 70 MEPPs and 20 EPPs were gathered from each NMJ. EPPs were triggered with a 1Hz train, normalized to −75mV and corrected for non-linear summation. Mean quantal content was calculated from the ratio of EPPs to MEPPs. The protocol for assessing NMJs in SMA-G470R+/+ mutants and controls differed somewhat and is detailed in a previous report.21 Briefly, TA (tibialis anterior) muscle with the intact nerve was dissected from mice at ~PND75 and placed in oxygenated mammalian Ringer’s solution. NMJs were visualized with 10 μm 4-(4-diethylaminostyryl)-N-methylpyridinium iodide (4-Di-2ASP), then impaled at a distance no greater than 100μm from the endplate. Myofibers were crushed away from the endplate band and voltage clamped to −45 mV to prevent movement after nerve stimulation. Two-electrode voltage clamp was employed to determine miniature endplate current (MEPC) amplitudes and endplate currents (EPCs) evoked after nerve stimulation. Voltage clamp precludes issues stemming from differences in muscle fiber size, e.g., differences in capacitance and input resistance. Quantal content was calculated by dividing EPC amplitude by the mean MEPC amplitude for a given NMJ. NMJ function in PND8-10 mice was assessed in the TVA muscle as described previously.67
Cardiovascular function
Cardiac function was evaluated according to protocols previously employed by us.68,69 Briefly, mice were lightly anesthetized (1–2% isoflurane) before performing transthoracic echocardiography using a Visualsonics Vevo 770 ultrasound system (Visualsonics) with a 30 MHz transducer applied to the chest wall. M-mode images and two-dimensional (2D) parasternal short-axis images at the mid-papillary muscle level were recorded. Measurements were made offline by a single individual in a group-blinded fashion. Assessments of left ventricular (LV) end-diastolic and end-systolic internal diameters were made enabling calculation of LV fractional shortening (FS), LV end-diastolic volume, LV end-systolic volume, and ejection fraction (EF). End-diastolic and end-systolic cavity areas were quantified at the mid-papillary level by tracing the endocardial border. The % LV fractional area change was estimated using the formula: LV end-diastolic cavity areas – LV end-systolic cavity areas)/LV end-diastolic cavity areas] × 100. M-mode images were used to determine heart rates. All parameters represent the mean of three beats.
Transcript and protein measurements
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and then treated with RNase-free DNase I (Thermofisher). cDNA was synthesized using the RevertAid RT kit (Thermo) or the iScript Reverse Transcription Supermix (Bio-Rad). Quantitative PCR was performed in triplicate on a CFX96 Real-Time System (Bio-Rad) using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), and normalized to endogenous Gapdh or β-Actin mRNA levels; for primer sequences, see Table S4. For protein estimation, western blot analysis was performed. Protein was extracted from cells or tissues using lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1mM EDTA, 1% NP-40, protease Inhibitor Cocktail; Roche - #4693159001). In the case of tissue-derived protein, tissue was homogenized and incubated in ice for 30 min. Lysates were cleared by centrifugation (15,000g for 30 min at 4°C) and then protein levels quantified using the BCA protein assay kit (Thermo). Prior to loading protein samples on SDS gels for immunoblotting, lysates were denatured by boiling for 5 min with 4X Laemmli sample buffer (Bio-Rad), or were mixed with 2X Laemmli sample buffer (Bio-Rad) – without boiling – to measure SDS-resistant SNARE complexes. To detect the lower abundance of SNARE complexes in the hiPSC lines, the protocol was slightly modified such that the lysis buffer indicated above was replaced with 1X PBS. For co-IP studies, 0.5–1 mg of protein lysate was incubated (4°C, overnight) with primary antibodies, then 20 μl of protein A/G PLUS-Agarose beads (SantaCruz) added to the lysate and the mix further incubated (4 °C for 4–6 hr). The beads were washed 5 times with lysis buffer, and bound proteins eluted in 2X Laemmli sample buffer (Bio-Rad) before performing SDS-PAGE and immunoblotting. Separated proteins were transferred to PVDF membrane (Millipore), membranes blocked in TBS containing 5% non-fat milk and 0.1% Tween 20, and primary antibodies used to probe (2 hr at room temperature or overnight at 4°C) for the presence of relevant proteins. Following incubation with primary antibodies, membranes were washed (1X TBST) and then incubated (1hr, room temperature) with appropriate secondary antibodies. Protein bands were visualized using the ECL kit (Bio-Rad) and images captured with a ChemiDoc Imaging System (Bio-Rad). Band intensities were determined with the Image Lab (Bio-Rad) or ImageJ software.
Cell cultures and transfections
PC-12 cells (ATCC CRL-1721) for our experiments were maintained in RPMI-1640 supplemented with 10% horse serum and 5% fetal bovine serum (FBS); HEK293T cells were maintained in DMEM supplemented with 10% FBS at 37°C with 5% CO2. Knockdown of SMN in the PC-12 cells was accomplished using lentivirus expressing a rat Smn shRNA. Lentivirus was produced by co-transfecting the following three plasmids into HEK293 cells: pGFP-C-shSmn1lenti (Origene, #TL710710), the pCMV-Δ8.2 packaging vector (Addgene, #12263) and pMD2.G (Addgene #12259), which codes for the virus envelope. Culture media from the transfected cells was collected 48h and 72h post-transfection, combined and viral particles in the liquid concentrated using the Lenti-X™ Concentrator (Clontech) according to the manufacturer’s instruction. To assess SNARE-complex assembly in HEK293 cells, a plasmid expressing rat Syntaxin 1A, VAMP2 and SNAP25 (1:1:1) under the control of the CMV promoter (generous gift from T. Sudhof, Stanford University and M. Sharma, Weill Cornell Medical College) was transiently co-transfected into the cells (Lipofectamine 3000, Invitrogen) along with a construct containing Myc-tagged mouse Hspa8-G470R or human SMN1. Forty-eight hours later, the cells were harvested, lysed and protein extracted for immunoblotting. SMA iPS cells were generated and maintained on irradiated mouse embryonic fibroblasts. Motor neurons from the iPS cells (provided by the Ebert laboratory at the Medical College of Wisconsin) were derived from human induced pluripotent stem cell (hiPSC) lines from 1x healthy individual (line 4.2), 1x SMA Type 1 patient (line 7.12) and 1x SMA Type 2 patient (line 3.6; previously classified as Type 1). hiPSCs were differentiated into spinal cord-like motor neurons using a previously established protocol.70,71 iPSCs were maintained on Corning® Matrigel® Growth Factor Reduced Basement Membrane Matrix coated 6-well plates in Essential 8 medium. Neurospheres were generated from iPSCs via dual SMAD inhibition (SB 431542 and LDN 1931899) and Wnt activation (Chir 99021) and subsequently patterned with retinoic acid and smoothened agonist to induce a ventral-caudal cell fate. Spinal motor neuron progenitors were dissociated from neurospheres and plated down on to Matrigel® coated plates with maturation media supplemented with DAPT, BDNF and GDNF for terminal differentiation. Cells were cultured to Day 50, collected and snap frozen for protein extraction.
Quantification and statistical analysis
Kaplan-Meier survival curves were assessed for differences using the log-rank test equivalent to the Mantel-Haenszel test. The unpaired, 2-tailed Student’s t test, one-way ANOVA followed by Tukey’s post-hoc comparison or Kruskal-Wallis test followed by Tukey’s or Dunn’s post-hoc comparison, where indicated, were used to compare means for statistical differences. In instances where categorical binomial variables had to be analyzed, the Fisher’s exact test was employed. Data are represented as mean ± SEM unless otherwise indicated. P < 0.05 was considered significant. Statistical analyses were performed with GraphPad Prism v6.0 (GraphPad Software).
Supplementary Material
Table S1. SNP analysis to localize the SMA modifier to chromosome 9. (A) Analysis with 180 polymorphic markers. (B) Analysis with 727 informative markers. Related to Figure 1.
Table S2. Results of GWAS of F2 SMA mutants on a mixed strain background to identify the disease modifier. (A) Compilation of all coding-region SNP variants identified. (B) List of the 4742 statistically significant disease-associated SNPs. Related to Figure 2.
Table S3. Sequence analysis of F2 SMA mutants. (A) Summary of the analysis. (B) Results of Burrows-Wheeler alignment of sequence reads with the C57BL/6 reference sequence. (C) Annotation of SNPs in the F2 SMA cohorts. (D) Summary of SNPs significantly associated with disease modification. Related to Figure 2.
Table S4. Oligonucleotide primers employed in the study. Related to Methods.
Video S1. Overt phenotype of representative modified F2 SMA mice at 1.5 months, 4 months and 6 months of age. Related to Figure 1
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SMN | BD biosciences | Cat#610647; RRID: AB_397973 |
| β-tubulin | Santa Cruz | Cat#SC53140; RRID: AB_793543 |
| β-actin | Santa Cruz | Cat#SC47778; RRID: AB_626632 |
| GAPDH | Santa Cruz | Cat#sc32233; RRID: AB_627679 |
| Hspa8 | Invitrogen | Cat#MA1-26078; RRID: AB_794523 |
| SGTA | Proteintech | Cat#11019-2-AP; RRID: AB_2188830 |
| CSPα | Invitrogen | Cat#PA5-87937; RRID: AB_2804520 |
| SNAP25 | BioLegend | Cat#836303; RRID: AB_2715864 |
| Syntaxin 1 | SYSY | Cat#110 011; RRID: AB_887842 |
| Synaptobrevin 2 | SYSY | Cat#104 211; RRID: AB_2619757 |
| Myc-Tag | Santa Cruz | Cat#SC40; RRID: AB_627268 |
| Goat anti-Rabbit IgG-HRP | Jackson ImmunoResearch | Cat#111-035-003; RRID: AB_2313567 |
| Goat anti-Mouse IgG-HRP | Jackson ImmunoResearch | Cat# 115-035-003; RRID: AB_10015289 |
| Goat anti-Rat IgG-HRP | Jackson ImmunoResearch | Cat# 112-035-003; RRID: AB_2338128 |
| Choline acetyltransferase (ChAT) | Millipore | Cat#AB144P; RRID: AB_2079751 |
| vGlut1 | Millipore | Cat#AB5905; RRID: AB_2301751 |
| Neurofilament | Millipore | Cat#AB1987; RRID: AB_91201 |
| Rhodamine-α-bungarotoxin | Invitrogen | Cat#B13424; RRID: AB_2313931 |
| Synaptophysin | Santa Cruz | Cat#SC9116; RRID: AB_2199007 |
| Alexa Fluor-488 conjugated donkey anti-mouse | Invitrogen | Cat#A-21202; RRID: AB_141607 |
| Alexa Fluor-488 conjugated goat anti-rabbit | Invitrogen | Cat#A-11034; RRID: AB_2576217 |
| Alexa Fluor-594 conjugated donkey anti-goat | Invitrogen | Cat#A-11058; RRID: AB_2534105 |
| Alexa Fluor-594 conjugated goat anti-mouse | Invitrogen | Cat#A-11032; RRID: AB_2534091 |
| Oligonucleotide | ||
| Primers for genotyping, see Table S4 | This paper | N/A |
| Primers for qRT-PCR, see Table S4 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Δ7 mouse | Jackson Laboratory | Stock #005025; RRID: IMSR_JAX:005025 |
| SMN2 mouse | Jackson Laboratory | Stock #005024; RRID: IMSR_JAX:005024 |
| Smn2B/- mice | Bowerman et al. 200936 | N/A |
| SOD1G86R mouse | Jackson Laboratory | Stock #005110; RRID: IMSR_JAX:005110 |
| Software and Algorithms | ||
| GraphPad Prism | Graph Pad Software | RRID: SCR_002798 |
| ImageJ | NIH | RRID: SCR_003070 |
| ImageQuantTL | GE Healthcare | RRID: SCR_018374 |
| SPOT v4.5 | Diagnostic Instruments | RRID: SCR_014313 |
| Leica LAS X | Leica | RRID: SCR_013673 |
Highlights.
An Hspa8G470R synaptic chaperone variant genetically suppresses SMA severity.
SNARE complex formation is disrupted in SMA neuromuscular junctions (NMJs).
Disrupted SNARE complexes in SMA NMJs link SMN to motor neuron disease.
Hspa8G470R stimulates SNARE complex formation and functions in splice-switching.
Acknowledgements
We thank members of the Monani lab for comments and suggestions. We are also very appreciative of T.C. Südhof, K.H. Fischbeck, C.J. Sumner and D.C. De Vivo for critically reading this manuscript and for their feedback. Miriam H. Meisler is gratefully acknowledged for her advice and comments on our mapping strategy and for reviewing our manuscript. This article is dedicated to the memory of Christine E. Beattie who inspired, championed and never stopped believing in the work described here.
Funding:
N.N.J and T.A. were recipients respectively of Muscular Dystrophy Association Development Grants, MDA-DG-217087 and MDA-DG-871922. Additional support provided by Cure SMA, AFM-France and NIH (R01 NS104218, R01 NS123292) to U.R.M., R21 NS102911 to A.D.E. and R01 AR074985 to M.M.R.
Footnotes
Competing interests: J-K.K. and U.R.M. are inventors on a provisional patent application filed by Columbia University on the use of Hspa8 as a means of treating neurodegenerative disease.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, and Burghes AH (1997). The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6, 1205–1214. 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, and et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, and Melki J (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16, 265–269. 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 4.Lorson CL, Hahnen E, Androphy EJ, and Wirth B (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 96, 6307–6311. 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, and McPherson JD (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8, 1177–1183. 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 6.Chaytow H, Faller KME, Huang YT, and Gillingwater TH (2021). Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine. Cell Rep Med 2, 100346. 10.1016/j.xcrm.2021.100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaytow H, Huang YT, Gillingwater TH, and Faller KME (2018). The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol Life Sci 75, 3877–3894. 10.1007/s00018-018-2849-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchner DA, Trudeau M, and Meisler MH (2003). SCNM1, a putative RNA splicing factor that modifies disease severity in mice. Science 301, 967–969. 10.1126/science.1086187. [DOI] [PubMed] [Google Scholar]
- 9.Nadeau JH (2003). Modifier genes and protective alleles in humans and mice. Curr Opin Genet Dev 13, 290–295. 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 10.Dimitriadi M, Sleigh JN, Walker A, Chang HC, Sen A, Kalloo G, Harris J, Barsby T, Walsh MB, Satterlee JS, et al. (2010). Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet 6, e1001172. 10.1371/journal.pgen.1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen A, Dimlich DN, Guruharsha KG, Kankel MW, Hori K, Yokokura T, Brachat S, Richardson D, Loureiro J, Sivasankaran R, et al. (2013). Genetic circuitry of Survival motor neuron, the gene underlying spinal muscular atrophy. Proc Natl Acad Sci U S A 110, E2371–2380. 10.1073/pnas.1301738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosseinibarkooie S, Peters M, Torres-Benito L, Rastetter RH, Hupperich K, Hoffmann A, Mendoza-Ferreira N, Kaczmarek A, Janzen E, Milbradt J, et al. (2016). The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am J Hum Genet 99, 647–665. 10.1016/j.ajhg.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janzen E, Mendoza-Ferreira N, Hosseinibarkooie S, Schneider S, Hupperich K, Tschanz T, Grysko V, Riessland M, Hammerschmidt M, Rigo F, et al. (2018). CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 141, 2343–2361. 10.1093/brain/awy167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riessland M, Kaczmarek A, Schneider S, Swoboda KJ, Lohr H, Bradler C, Grysko V, Dimitriadi M, Hosseinibarkooie S, Torres-Benito L, et al. (2017). Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am J Hum Genet 100, 297–315. 10.1016/j.ajhg.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, and Wirth B (2008). Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524–527. 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, and Burghes AH (2005). SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet 14, 845–857. 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 17.Crawford TO, and Pardo CA (1996). The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3, 97–110. 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 18.Harding BN, Kariya S, Monani UR, Chung WK, Benton M, Yum SW, Tennekoon G, and Finkel RS (2015). Spectrum of neuropathophysiology in spinal muscular atrophy type I. J Neuropathol Exp Neurol 74, 15–24. 10.1097/NEN.0000000000000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramos DM, d’Ydewalle C, Gabbeta V, Dakka A, Klein SK, Norris DA, Matson J, Taylor SJ, Zaworski PG, Prior TW, et al. (2019). Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J Clin Invest 129, 4817–4831. 10.1172/JCI124120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, and Monani UR (2008). Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17, 2552–2569. 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marce M, Griffin JW, Rich MM, and Sumner CJ (2009). Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29, 842–851. 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, and Gillingwater TH (2008). Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet 17, 949–962. 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 23.Heier CR, Satta R, Lutz C, and DiDonato CJ (2010). Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet 19, 3906–3918. 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo J, Kwon SK, Nam J, Choi S, Takahashi H, Krueger D, Park J, Lee Y, Bae JY, Lee D, et al. (2013). The adhesion protein IgSF9b is coupled to neuroligin 2 via S-SCAM to promote inhibitory synapse development. J Cell Biol 201, 929–944. 10.1083/jcb.201209132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, and Stahl B (2001). A trimeric protein complex functions as a synaptic chaperone machine. Neuron 31, 987–999. 10.1016/s0896-6273(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 26.Zinsmaier KE, and Bronk P (2001). Molecular chaperones and the regulation of neurotransmitter exocytosis. Biochem Pharmacol 62, 1–11. 10.1016/s0006-2952(01)00648-7. [DOI] [PubMed] [Google Scholar]
- 27.Jodelka FM, Ebert AD, Duelli DM, and Hastings ML (2010). A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum Mol Genet 19, 4906–4917. 10.1093/hmg/ddq425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruggiu M, McGovern VL, Lotti F, Saieva L, Li DK, Kariya S, Monani UR, Burghes AH, and Pellizzoni L (2012). A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 32, 126–138. 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossoll W, Prior TW, et al. (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9, 333–339. 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 30.Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, and Pellizzoni L (2012). An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151, 440–454. 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tisdale S, Lotti F, Saieva L, Van Meerbeke JP, Crawford TO, Sumner CJ, Mentis GZ, and Pellizzoni L (2013). SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3’-end formation of histone mRNAs. Cell Rep 5, 1187–1195. 10.1016/j.celrep.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudhof TC, and Rizo J (2011). Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol 3. 10.1101/cshperspect.a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, Kuenen S, Wittocx R, Corthout N, Marrink SJ, et al. (2015). Hsc70–4 Deforms Membranes to Promote Synaptic Protein Turnover by Endosomal Microautophagy. Neuron 88, 735–748. 10.1016/j.neuron.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, and Sudhof TC (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 35.Sharma M, Burre J, and Sudhof TC (2011). CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol 13, 30–39. 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- 36.Bowerman M, Anderson CL, Beauvais A, Boyl PP, Witke W, and Kothary R (2009). SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci 42, 66–74. 10.1016/j.mcn.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Ripps ME, Huntley GW, Hof PR, Morrison JH, and Gordon JW (1995). Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 92, 689–693. 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JK, and Monani UR (2018). Augmenting the SMN Protein to Treat Infantile Spinal Muscular Atrophy. Neuron 97, 1001–1003. 10.1016/j.neuron.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, et al. (2014). Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–1028. 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Daniels CK, and Cao S (2012). Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Ther 136, 354–374. 10.1016/j.pharmthera.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 41.Barbosa-Morais NL, Carmo-Fonseca M, and Aparicio S (2006). Systematic genome-wide annotation of spliceosomal proteins reveals differential gene family expansion. Genome Res 16, 66–77. 10.1101/gr.3936206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rappsilber J, Ryder U, Lamond AI, and Mann M (2002). Large-scale proteomic analysis of the human spliceosome. Genome Res 12, 1231–1245. 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng S, Damoiseaux R, Chen L, and Black DL (2013). A broadly applicable high-throughput screening strategy identifies new regulators of Dlg4 (Psd-95) alternative splicing. Genome Res 23, 998–1007. 10.1101/gr.147546.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, and Hendrickson WA (1996). Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614. 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan S, and Vihinen M (2007). Spectrum of disease-causing mutations in protein secondary structures. BMC Struct Biol 7, 56. 10.1186/1472-6807-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molnar J, Szakacs G, and Tusnady GE (2016). Characterization of Disease-Associated Mutations in Human Transmembrane Proteins. PLoS One 11, e0151760. 10.1371/journal.pone.0151760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen L, and Ji HF (2015). Mutational Spectrum Analysis of Neurodegenerative Diseases and Its Pathogenic Implication. Int J Mol Sci 16, 24295–24301. 10.3390/ijms161024295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor IR, Ahmad A, Wu T, Nordhues BA, Bhullar A, Gestwicki JE, and Zuiderweg ERP (2018). The disorderly conduct of Hsc70 and its interaction with the Alzheimer’s-related Tau protein. J Biol Chem 293, 10796–10809. 10.1074/jbc.RA118.002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meister G, Buhler D, Pillai R, Lottspeich F, and Fischer U (2001). A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol 3, 945–949. 10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- 50.Ryu SW, Stewart R, Pectol DC, Ender NA, Wimalarathne O, Lee JH, Zanini CP, Harvey A, Huibregtse JM, Mueller P, and Paull TT (2020). Proteome-wide identification of HSP70/HSC70 chaperone clients in human cells. PLoS Biol 18, e3000606. 10.1371/journal.pbio.3000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shafey D, Boyer JG, Bhanot K, and Kothary R (2010). Identification of novel interacting protein partners of SMN using tandem affinity purification. J Proteome Res 9, 1659–1669. 10.1021/pr9006987. [DOI] [PubMed] [Google Scholar]
- 52.Garcera A, Bahi N, Periyakaruppiah A, Arumugam S, and Soler RM (2013). Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell Death Dis 4, e686. 10.1038/cddis.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takikawa K, and Nishimune H (2022). Similarity and Diversity of Presynaptic Molecules at Neuromuscular Junctions and Central Synapses. Biomolecules 12. 10.3390/biom12020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, and Rosenmund C (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A 99, 9037–9042. 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Varoqueaux F, Sons MS, Plomp JJ, and Brose N (2005). Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol Cell Biol 25, 5973–5984. 10.1128/MCB.25.14.5973-5984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong W, Radulovic T, Goral RO, Thomas C, Suarez Montesinos M, Guerrero-Given D, Hagiwara A, Putzke T, Hida Y, Abe M, et al. (2018). CAST/ELKS Proteins Control Voltage-Gated Ca(2+) Channel Density and Synaptic Release Probability at a Mammalian Central Synapse. Cell Rep 24, 284–293 e286. 10.1016/j.celrep.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tokoro T, Higa S, Deguchi-Tawarada M, Inoue E, Kitajima I, and Ohtsuka T (2007). Localization of the active zone proteins CAST, ELKS, and Piccolo at neuromuscular junctions. Neuroreport 18, 313–316. 10.1097/WNR.0b013e3280287abe. [DOI] [PubMed] [Google Scholar]
- 58.Coyne AN, Lorenzini I, Chou CC, Torvund M, Rogers RS, Starr A, Zaepfel BL, Levy J, Johannesmeyer J, Schwartz JC, et al. (2017). Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell Rep 21, 110–125. 10.1016/j.celrep.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michaud M, Arnoux T, Bielli S, Durand E, Rotrou Y, Jablonka S, Robert F, Giraudon-Paoli M, Riessland M, Mattei MG, et al. (2010). Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol Dis 38, 125–135. 10.1016/j.nbd.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 60.Feng Z, Ling KK, Zhao X, Zhou C, Karp G, Welch EM, Naryshkin N, Ratni H, Chen KS, Metzger F, et al. (2016). Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum Mol Genet 25, 964–975. 10.1093/hmg/ddv629. [DOI] [PubMed] [Google Scholar]
- 61.Iyer CC, Corlett KM, Massoni-Laporte A, Duque SI, Madabusi N, Tisdale S, McGovern VL, Le TT, Zaworski PG, Arnold WD, et al. (2018). Mild SMN missense alleles are only functional in the presence of SMN2 in mammals. Hum Mol Genet 27, 3404–3416. 10.1093/hmg/ddy251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park GH, Maeno-Hikichi Y, Awano T, Landmesser LT, and Monani UR (2010). Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci 30, 12005–12019. 10.1523/JNEUROSCI.2208-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ling KK, Lin MY, Zingg B, Feng Z, and Ko CP (2010). Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One 5, e15457. 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruiz R, Casanas JJ, Torres-Benito L, Cano R, and Tabares L (2010). Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30, 849–857. 10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caine C, Shohat M, Kim JK, Nakanishi K, Homma S, Mosharov EV, and Monani UR (2017). A pathogenic S250F missense mutation results in a mouse model of mild aromatic l-amino acid decarboxylase (AADC) deficiency. Hum Mol Genet 26, 4406–4415. 10.1093/hmg/ddx326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kariya S, Obis T, Garone C, Akay T, Sera F, Iwata S, Homma S, and Monani UR (2014). Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 124, 785–800. 10.1172/JCI72017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maury Y, Come J, Piskorowski RA, Salah-Mohellibi N, Chevaleyre V, Peschanski M, Martinat C, and Nedelec S (2015). Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol 33, 89–96. 10.1038/nbt.3049. [DOI] [PubMed] [Google Scholar]
- 71.Sison SL, Patitucci TN, Seminary ER, Villalon E, Lorson CL, and Ebert AD (2017). Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum Mol Genet 26, 3409–3420. 10.1093/hmg/ddx230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. SNP analysis to localize the SMA modifier to chromosome 9. (A) Analysis with 180 polymorphic markers. (B) Analysis with 727 informative markers. Related to Figure 1.
Table S2. Results of GWAS of F2 SMA mutants on a mixed strain background to identify the disease modifier. (A) Compilation of all coding-region SNP variants identified. (B) List of the 4742 statistically significant disease-associated SNPs. Related to Figure 2.
Table S3. Sequence analysis of F2 SMA mutants. (A) Summary of the analysis. (B) Results of Burrows-Wheeler alignment of sequence reads with the C57BL/6 reference sequence. (C) Annotation of SNPs in the F2 SMA cohorts. (D) Summary of SNPs significantly associated with disease modification. Related to Figure 2.
Table S4. Oligonucleotide primers employed in the study. Related to Methods.
Video S1. Overt phenotype of representative modified F2 SMA mice at 1.5 months, 4 months and 6 months of age. Related to Figure 1