

Abstract
The discovery of the benefits of castration for prostate cancer treatment in 1941 led to androgen deprivation therapy, which remains a mainstay of the treatment of men with advanced prostate cancer. However, as early as this original publication, the inevitable development of castration-resistant prostate cancer was recognized. Resistance first manifests as a sustained rise in the androgen-responsive gene, PSA, consistent with reactivation of the androgen receptor axis. Evaluation of clinical specimens demonstrates that castration-resistant prostate cancer cells remain addicted to androgen signalling and adapt to chronic low-testosterone states. Paradoxically, results of several studies have suggested that treatment with supraphysiological levels of testosterone can retard prostate cancer growth. Insights from these studies have been used to investigate administration of supraphysiological testosterone to patients with prostate cancer for clinical benefits, a strategy that is termed bipolar androgen therapy (BAT). BAT involves rapid cycling from supraphysiological back to near-castration testosterone levels over a 4-week cycle. Understanding how BAT works at the molecular and cellular levels might help to rationalize combining BAT with other agents to achieve increased efficacy and tumour responses.
Introduction
Adenocarcinoma of the prostate gland is the second most common cancer in men, with ~2.2 million new instances and ~375,000 deaths estimated to occur during 2022 (ref. 1).
Androgen signalling has an important role in prostate cancer progression, Androsterone was the first androgen to be isolated from men’s urine2,3. Subsequently, a more potent androgen than androsterone was discovered in the testes, which are a rich source of androgenic hormones, and was termed testosterone from the words testes, sterol and ketone4. Testosterone is primarily produced by Leydig cells in the testes in response to luteinizing hormone secreted by the anterior pituitary, and mostly circulates bound to serum hormone-binding globulin5,6 with only the free form gaining entry into cells owing to its lipophilic nature7,8. Upon entry into prostate cells, testosterone is converted to 5 α-dihydrotestosterone (DHT), a highly potent androgen, by the enzyme 5-α reductase9. Testosterone is sufficient for the development of embryonic Wolffian ducts but insufficient for the complete development of prostate and external genitalia, which requires 5-α reductase activity and formation of DHT10,11. Results of early studies showed that radiolabelled DHT or testosterone was selectively retained by the prostate nucleus9,12. These initial observations led to the subsequent identification and cloning of the androgen receptor gene (AR)8,13,14. AR encodes a 100-kDa protein that shares structural similarities with other steroid hormone nuclear receptors, including glucocorticoid receptor, progesterone receptor, mineral corticoid receptor and the oestrogen receptor15. AR protein can be functionally divided into four domains: the N-terminal activation domain, the central DNA binding domain, the hinge domain and the C-terminal ligand-binding domain (Fig. 1a). Ligand binding results in dimerization and translocation of AR to the nucleus and subsequent activation or repression of its target genes, such as KLK3, TMPRSS2, and NKX3.1 (ref. 16) (Fig. 1b). Specificity of AR binding to androgen binding sites (ARBS) is determined by chromatin-binding proteins and co-regulators17–20. Androgen signalling is important in the development and progression of all stages of prostate cancer21,22.
Fig. 1 |. AR structure and signalling.
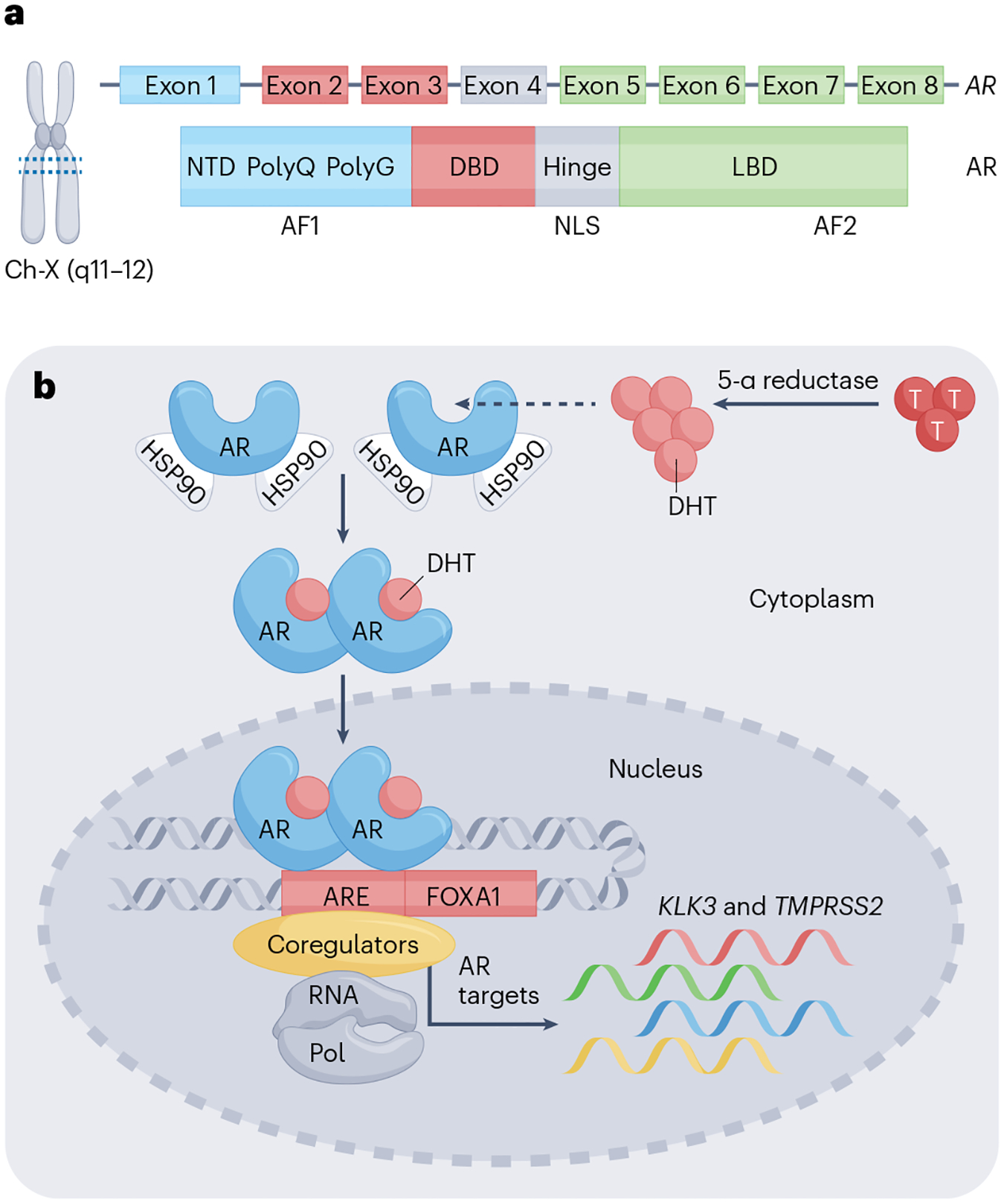
a, Structure of the androgen receptor (AR). Location of AR on the q arm of the X chromosomes (Xq12). AR contains eight exons coding for a 110-kDa protein that has 919 amino acids. The N-terminal domain (NTD) is encoded by exon 1 and has an intrinsically disordered structure. The DNA binding domain (DBD) is encoded by exons 2–3, which contain two zinc finger motifs. The DBD is linked to the ligand-binding domain by the hinge region, which is encoded by exon 4. The ligand-binding domain is encoded by exons 5–8. Both the N terminus and C terminus consist of activation functions called AF1 and AF2, respectively. b, Nuclear AR signalling. Testosterone is converted into its highly active metabolite dihydrotestosterone (DHT) by 5-α reductase, which binds to AR sequestered in the cytoplasm by chaperone proteins that include HSP90. Upon binding of DHT, AR dissociates from HSP90, dimerizes, and translocates to the nucleus to bind to androgen response elements (AREs) present in its target genes such as KLK3 and TMPRSS2. Specificity of binding is regulated by co-regulators and pioneer factors such as FOXA1.
The role of androgen signalling in prostate cancer progression forms the basis for using androgen deprivation therapy (ADT) as a standard of care for metastatic or recurrent disease23–25. Androgen deprivation is known to provide initial therapeutic benefits, but eventually all men with prostate cancer develop castration-resistant disease26,27. Intriguingly, high-dose androgens at supraphysiological levels lead to a paradoxical decrease in the growth of some models of prostate cancer through poorly understood mechanisms. Understanding how androgens promote or inhibit the growth of prostate cancer will help to develop effective clinical strategies to inhibit prostate cancer growth and progression. Bipolar androgen therapy (BAT) is an innovative therapeutic strategy in which high doses of testosterone are periodically administered to achieve supraphysiological serum testosterone levels to inhibit prostate tumour growth28.
In this Review, the role of androgens in prostate homeostasis and prostate cancer and mechanistic findings of growth inhibition by supraphysiological androgens are described, and insights from the results of prostate cancer clinical trials using supraphysiological testosterone (supraphysiological T) are provided. Finally, the future clinical development of BAT as a therapeutic option against prostate cancer is discussed.
The role of androgens and the AR in the prostate
Accumulated evidence from cellular, molecular and developmental studies indicates that androgens are necessary for the development of the prostate gland and dysregulated AR signalling aids prostate cancer growth and survival.
Androgens and the AR in prostate homeostasis
The prostate gland consists of branched epithelial ducts made up of a pseudostratified epithelium comprising luminal and basal epithelial cells29,30. The underlying stroma contains fibroblast cells, smooth muscle cells, nerve cells, endothelial cells, immune cells and rare neuroendocrine cells (Fig. 2). Results of studies conducted with seminal tissue recombination using urogenital sinus mesenchyme showed that paracrine AR signalling in the stromal compartment, but not the epithelial compartment, is essential for prostate development31,32. Results of studies using rats further indicated that the adult prostate has a profound regenerative capacity following repeated cycles of androgen withdrawal and replacement33. These pivotal studies suggested the presence of castration-resistant stem cells that survive androgen deprivation can regenerate the prostate gland. Prostate regeneration was initially attributed to stem cells in the basal cell compartment, which were largely unaffected by androgen deprivation34–37. However, lineage-tracing studies indicated that regeneration after androgen replacement might be mediated by rare luminal cells called castration-resistant Nkx3-1-expressing (CARN) luminal cells that survive androgen deprivation to have a vital role as stem cells in prostate regeneration, with rare basal cells also contributing to proliferation38,39. A number of subsequent studies indicated that the adult prostate in mice has self-sustaining basal and luminal compartments40–42. The adult prostate is mainly quiescent, but these self-sustaining epithelial cellular compartments might have a role during tissue homeostasis, injury and disease (Fig. 2). However, many of these mechanistic studies to elucidate the role of AR signalling in prostate regeneration involve AR-knockout models using cre recombinase driven by the probasin promoter, which is activated during early postnatal development43. Results from these studies leave an open question of whether the observed effects seen are developmental or homeostatic in nature. To address this question, experiments in which basal-specific and luminal-specific AR ablation using inducible cre were performed in adult mouse prostates44. The results of these studies revealed that cell-autonomous AR signalling is dispensable for basal cell maintenance and required for luminal cell morphology and the bipotentiality of rare basal stem cells. Intriguingly, AR signalling was necessary to maintain daughter cells produced by CARN cells upon androgen replacement, indicating that, unlike average luminal cells of the regressed prostate, CARNs selectively require cell-autonomous AR signalling to produce viable luminal cells during prostate regeneration. Results of a single-cell transcriptomic study suggest that prostate regeneration is driven by all persisting luminal cells that acquire stem cell transcriptional features, not just by rare stem cells45. Cumulative evidence from early tissue recombination studies and subsequent knockout and single-cell transcriptomic studies suggests that paracrine AR activity occurs in the mesenchyme rather than in the epithelial compartment, which might be responsible for androgen-driven regeneration of the normal prostate. Understanding the androgen response by the healthy and regenerating prostate could help to delineate the type of prostate cells that are likely to initiate cancer.
Fig. 2 |. Androgens in prostate homeostasis and regeneration.
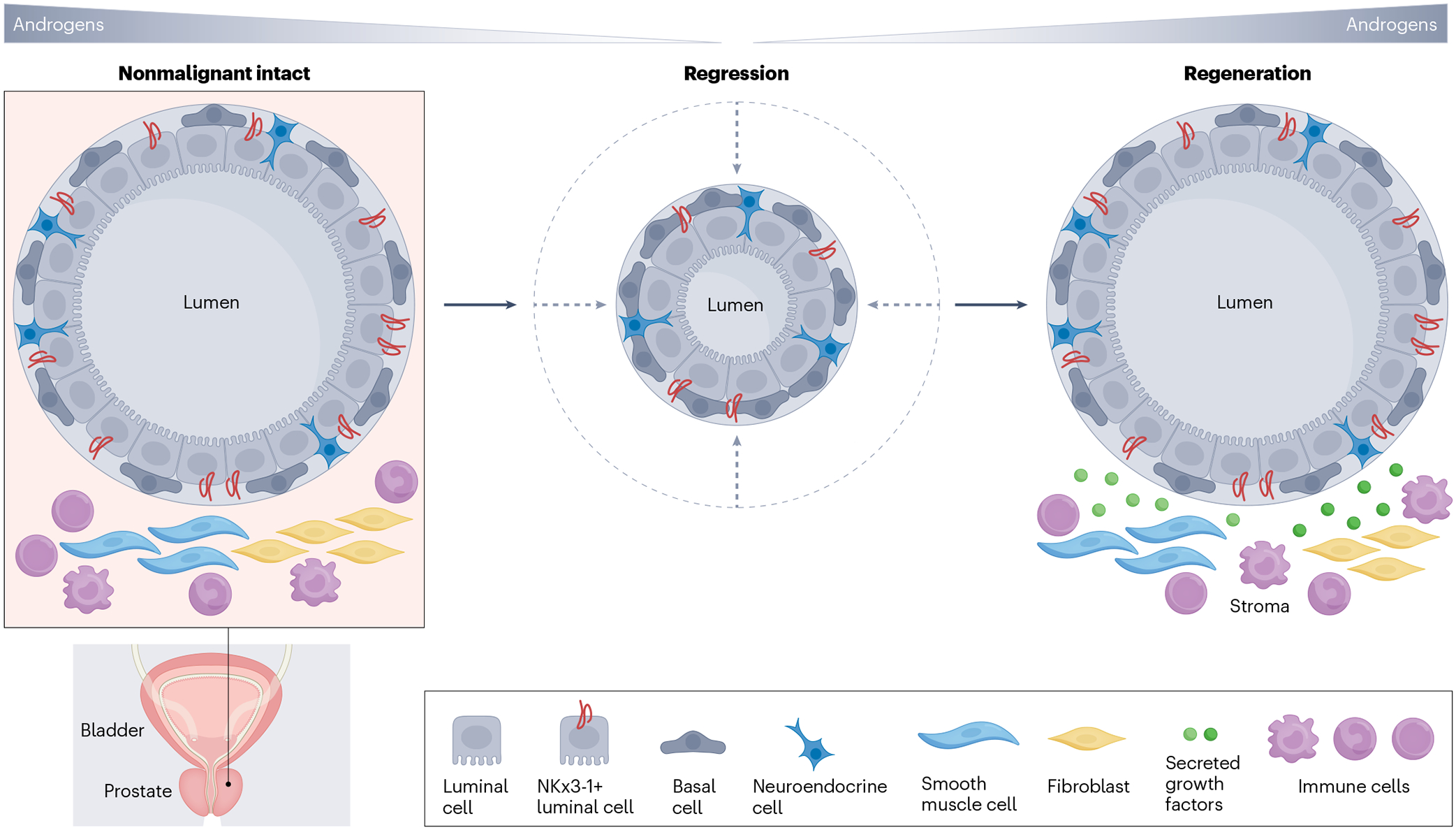
Lineage-tracing studies indicate that the regenerative capacity of the prostate gland following withdrawal and re-administration of androgens can be attributed to luminal cells that acquire stem-like transcriptional features and survive castration. Luminal cell survival and regrowth might be determined by microenvironmental niche factors such as fibroblast growth factor (FGF), insulin-like growth factor (IGF), epidermal growth factor (EGF) and hepatocyte growth factor (HGF).
Androgen signalling in prostate cancer
Unlike non-malignant prostate epithelial cells in which AR is dispensable, cell-autonomous AR signalling fuels prostate cancer growth31,32,46. The modulation of AR signalling through AR amplification21,47, splice variants48,49, AR mutation50–52, co-activator and co-repressor alteration19,53 in human prostate cancer underscores the importance of AR signalling in prostate cancer. In the absence of a ligand, the AR receptor is bound to chaperone proteins that keep it in a ligand-binding poised state. Once bound to a ligand, AR dimerizes and enters the nucleus to bind to thousands of ARBS scattered throughout the genome20,54. The majority (~90%) of AR binding sites are located hundreds of kilobases away from promoters of target genes in distal enhancer regions, which require chromatin looping to promote or repress AR-target genes55,56. In co-operation with its co-regulators and pioneering transcription factors such as FOXO1, AR can influence a number of cancer-relevant cellular processes, such as cell cycle, cell death, metabolism, chromatin remodelling, invasion and DNA repair46,57–59 (Table 1). Besides its nuclear or genomic role, evidence suggests that AR might also have a non-genomic role60 in cancer metabolism, proliferation, survival and invasion61,62 (Table 1).
Table 1 |.
AR-influenced genomic and non-genomic cellular processes
| Biological process | Biomolecules involved | Mechanism | Refs. |
|---|---|---|---|
| Cell cycle and proliferation | Cyclin-dependent kinases 2 and 4 and cyclins D1 and E | Increase in cyclin-dependent kinase activity and stimulation of the cell to enter the S phase | 170 |
| Genetic fusion | TMPRSS2 and ETS oncogene families (ERG, ETV1) and other non-random fusion events | ERG overexpression induced MMPs and plasminogen activation and cell invasion | 171–174 |
| Cistrome modification | AR cistrome reprogramming | 55,175–177 | |
| Enrichment of HOXB13 and FOXA1 motifs near AR binding sites | |||
| Growth inhibition | p21, p27 | G1 cycle arrest, inhibition of CDK2 activity | 178,179 |
| Apoptosis | G1 cell-cycle arrest | G1 cycle arrest, fragmentation of DNA | 87,119 |
| Cell survival and anti-apoptosis | HSP27, FLIP, and FOXO3a | AR-mediated upregulation of anti-apoptotic FLIP | 180 |
| Phosphatidylinositol 3-kinase and AKT and PTEN loss | MTORC2-mediated AKT activation and increased AR activity PTEN Loss causes increased FLIP expression, constitutive PI3K activity-mediated AKT phosphorylation | 181 | |
| Invasion, migration and metastasis development | MMP-2 upregulation | AR-mediated increase in pro-MMP-2 levels | 182 |
| Ezrin expression and phosphorylation | Androgen-mediated direct increase in ezrin followed by androgen-activated PKC-α-induced ezrin phosphorylation (Thr567) | 183,184 | |
| Interaction of AR with filamin A and regulation of FAK, paxillin and RAC | AR interaction with filamin A and control integrin beta 1 and FAK, paxillin and RAC | 185 | |
| Metabolism | 186,187 | ||
| Lactate dehydrogenase A and MCT4 | Pyruvate to lactate metabolism | ||
| Combined targets of AR and SREBP: ELOV6, SCD1, FASN, and A-CoA carboxylase | Increased fatty acid synthesis (monounsaturated and saturated FA) | 188,189 | |
| Amino acid transporters (LATs and ASCTs) | ASCT2-mediated glutamine uptake | 190,191 | |
| Folate cycle pathway and methionine cycle | Trans-sulfuration and polyamine synthesis | 192,193 | |
| Poly (ADP-Ribose) polymerase1 | PARylation of XRCC1 | 194,195 | |
| DNA repair | DNAPKcs | PRKDC (encoding the protein product DNAPKcs) XRCC2 and XRCC3 (RAD51) | 196 |
| Non-genomic ligand-independent crosstalk with growth factors, cytokines and non-receptor tyrosine kinase pathway | 197–201 | ||
| HER2neu | AR stabilization, increased binding of AR to AREs | ||
| MAPK and effectors SRC, ERF1 and ERF2, and PI3K and AKT signaling | Increased ERK1 and ERK2 phosphorylation RAF and ERK2 activation | 202 | |
| Calcium signaling | Increased intracellular calcium by GPCR and/or EGFR | 203,204 |
A-CoA carboxylase, acetyl-CoA carboxylase; AR, androgen receptor; ASCTs, alanine/serine/cysteine/threonine transporter; CDK2, cyclin-dependent kinase; DNAPKcs, DNA-dependent protein kinase catalytic subunit; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ERG, ETS-related gene; ERK, extracellular signal-related kinase; ETS, E-26 transformation specific; ETV1, ETS translocation variant 1; FAK, focal adhesion kinase; FASN, fatty acid synthase; FLIP, FLICE-like inhibitory protein; FOXA1, Forkhead Box A1; FOXO1, Forkhead box O1; FOXO3a, Forkhead box protein O3a; GPCR, G protein-coupled receptor; HER2neu, human epidermal growth factor receptor 2; HOXB13, Homeobox protein B13; HSP27, heat shock protein 27; IGF1, insulin-like growth factor 1; IL6, interleukin 6; IL8, interleukin 8; JAK–STAT3, Janus kinase-signal transducer and activator of transcription; LATs, linker for activation of T cells; MAPK, mitogen-activated protein kinase; MCT4, monocarboxylate transporter 4; MMP-2, matrix metalloproteinase-2; MTORC2, mechanistic target of rapamycin kinase; NADPH, nicotinamide adenine dinucleotide phosphate; PARylation, poly(ADP-ribose)-ylation; PI3K, phosphatidylinositol-4,5-bispohosphate 3-kinase; PTEN, phosphatase and tensin homologue; RAD51, RAD51 recombinase; SREBP, sterol regulatory element binding protein; TMPRSS2, transmembrane protease serine 2; XRCC, X-ray repair cross complementing.
Clinical utility of reducing AR signalling
Inhibition of AR signalling is the mainstay of the systemic treatment of prostate cancer. Inhibition of AR signalling in patients with prostate cancer can be achieved in three ways: reduction of serum testosterone; inhibition of AR; and degradation of AR. Reduction of serum testosterone can be achieved by blocking its production from the testes and/or adrenal glands63. Huggins and Scott first showed the efficiency of this therapeutic strategy by surgical removal of the testes and adrenal glands64. Currently, use of medical castration is more common than surgical castration, using luteinizing hormone-releasing hormone (LHRH) agonists (such as leuprolide and goserelin) and antagonists (such as degarelix and relugolix) to block testosterone production from the testes and the CYP17A1 inhibitor abiraterone acetate to block testosterone production by the adrenal glands. Abiraterone acetate in combination with an LHRH agonist has been shown to prolong the survival of patients with prostate cancer when used as a treatment for metastatic castration-sensitive and castration-resistant disease65–67. Direct inhibition of AR can be achieved by using antiandrogens that bind to the ligand-binding domain of AR and prevent its nuclear localization and transcriptional activity68. First-generation antiandrogens, including flutamide, bicalutamide and nilutamide, have now been replaced by second-generation antiandrogens enzalutamide, darolutamide and apalutamide, which bind AR with higher affinity69. These second-generation antiandrogens combined with an LHRH agonist can prolong the survival of patients with prostate cancer when used as a treatment for non-metastatic castration-resistant, metastatic castration-sensitive and castration-resistant disease70–74. The use of AR degraders to inhibit AR signalling is in clinical development. For example, ARV-110 is a proteolysis-targeting chimaera (PROTAC) protein degrader that creates a complex of AR with E3 ubiquitin ligase to result in ubiquitination of AR and degradation by the proteasome75. A phase II expansion study testing the efficacy of ARV-110 as a treatment for patients with metastatic castration-resistant prostate cancer (CRPC) with enrichment of T878 and H875 mutations in AR is currently underway (NCT03888612)76.
The clear clinical benefit of using agents that inhibit AR signalling with increased potency despite previous failure of alternative AR-axis inhibitors reflects the biology of prostate cancer to develop mechanisms to persistently signal through AR despite varied therapeutic approaches to obstruct this pathway. Indeed, the major mechanisms of resistance to AR signalling inhibition include AR overexpression, amplification and mutation, the production of ligand-independent variants and reprogramming of the AR cistrome21,77–81, all of which can enable ongoing AR signalling in the face of therapeutic inhibition. Reduced dependency on AR signalling, such as trans-differentiation to neuroendocrine, small-cell, or double-negative prostate cancer, presently only seems to occur in a minority of patients. This observation indicates that ongoing efforts to develop agents that target AR signalling are warranted, despite our current relatively large armamentarium of such agents.
The testosterone paradox
Huggins was the first to note that an excess of hormones can cause paradoxical regression of tumours82. His observation was based on regression of breast tumours upon treatment with a combination of supraphysiological levels of oestrogens and progesterone. Huggins called this phenomenon ‘hormone interference’ and noted it as a novel therapeutic approach to treating cancer. To understand the mechanism of this paradoxical effect, the effect of supraphysiological T on prostate cancer cells was tested. Initial studies mainly focused on the effect of supraphysiological T on cell-cycle and cell-death pathways; results of subsequent investigations showed a number of possible mechanisms using both in vitro and in vivo preclinical models; however, the supraphysiological T paradox is not clearly understood.
Initial characterization of lymph node metastasis-derived, AR-positive LNCaP prostate cancer cell line demonstrated a biphasic response to testosterone83,84, that is, LNCaP cells respond to treatment with low (0.01 nM R1881, synthetic testosterone) testosterone doses by rapidly proliferating, but proliferation is inhibited at supraphysiological T (≥1 nM R1881) concentrations83,84. When transfected with AR, AR-negative cell lines such as PC3 cells responded to the synthetic androgen R1881 with growth inhibition85, suggesting the importance of AR expression in the observed effect. Castration-resistant sublines of LNCaP cells were found to have an adaptive increase in AR expression and their growth was acutely inhibited upon R1881 (0.1 nM and above) treatment86,87. The growth repression by R1881 in these sublines was attributed to a decrease in MYC at the mRNA and protein levels. Furthermore, ectopic expression of MYC reversed the observed growth inhibition, suggesting its importance in supraphysiological T-induced growth inhibition86. Results of subsequent investigations indicated that growth inhibition was accompanied by an increase in expression of p21 and p27 and their association with CDK2, which results in G1 cell-cycle arrest86. p21 harbours an ARBS in its promoter and is a direct AR target gene, but p27 expression was found to be regulated indirectly by supraphysiological T through AR-mediated downregulation of its degrader SKP2, a subunit of SCF E3 ubiquitin ligase complex88. Results of a number of studies in which primary and immortalized normal prostate epithelial cells were used also suggest that ligand-bound AR signalling causes downregulation of MYC, leading to growth arrest and terminal differentiation89–91. Another mechanism by which AR can cause a G1 arrest was shown by investigating the role of AR as a DNA replication licensing factor92–94. Licensing factors ensure that genomic DNA is replicated once per cell cycle and they are assembled on replication origins in G1 phase, an obligatory event for activation of replication origins in the S-phase95. These factors are tightly regulated in the G1 phase either through inactivating phosphorylation or proteasomal degradation96,97. AR was found to interact with many licensing factors, namely ORC2, CDC6, CDT1 and MCM7 (ref. 98). Moreover, AR, like other licensing factors, undergoes proteasomal degradation in mitosis93,94 before the next cell cycle. Ligand-bound AR under supraphysiological T conditions was proposed to prevent AR from degradation during mitosis. This inhibition of degradation would result in origins of replication with bound AR, preventing relicensing and causing a G1 arrest.
Another mechanism for growth suppression by supraphysiological T could be through self-regulation of AR transcription. A decrease in both mRNA and protein levels of AR in castration-resistant LNCAP cell sublines treated with R1881 had been observed86. A reduction in AR transcript upon androgen stimulation was also noted in other studies99,100. In a subsequent investigation, a highly conserved ARBS site was identified in the second intron of AR101. Ligand-bound AR was shown to decrease AR expression by recruiting the lysine-specific histone demethylase LSD1 (ref. 101), a known transcriptional repressor102. Recruitment of LSD1 leads to demethylation of H3K4 and repression of AR transcription. This phenomenon is intriguing as LSD1 has been shown to primarily act as an AR co-activator, which it achieves by demethylating the K270 residue of the pioneering factor FOXA1 to enhance its chromatin binding, maintaining the AR enhancer accessibility that is needed to transcribe AR target genes18. These observations also highlight how the AR transcript increases under castration conditions to enhance prostate cancer growth and survival.
A decrease in tumour growth can also be brought about by senescence, quiescence or cell death103. All of these mechanisms have been investigated in the context of supraphysiological T treatment. Re-expression of AR in AR-negative prostate cancer cells was shown to induce apoptosis104. However, apoptosis in AR-negative DU145 cells was contingent upon co-expression of retinoblastoma (RB) protein104. AR-negative PC3 cells, when transfected with full-length AR (PC3-AR), exhibited effects ranging from a decrease in proliferation without apoptosis to a G1 arrest that culminated in apoptosis with an increase in time of treatment87. Castration-resistant LNCaP sublines have also been reported to induce BAX-mediated apoptosis upon androgen treatment105,106. Results of other studies also indicate that supraphysiological T can induce senescence in LNCaP cells107,108. Treatment of LNCaP cells with 1 nM R1881 for 72 h was sufficient to induce the formation of senescence-associated heterochromatic foci and senescence-associated β-galactosidase activity107. Supraphysiological T treatment increased p16, a known senescence marker that mediates the hypophosphorylation of RB, which resulted in downregulation of its target cyclin D1 and E2F1. These results indicated that supraphysiological T might regulate the p16–RB–E2F1 pathway to mediate cellular senescence. In line with these observations, results of another study demonstrated that supraphysiological T could be combined with a CDK4 and CDK6 inhibitor, strengthening the chromatin binding of the RB–E2F repressor complex by blocking the hyperphosphorylation of RB proteins109. Results of a previous study using PC3-AR cells had shown that androgen-mediated senescence proceeds after a G1 arrest108. Senescence was brought about by AR-dependent expression of p21 and depletion of p63. In this study, RB hypophosphorylation was mediated through AR-induced reactive oxygen species (ROS)108. Intriguingly, MTORC1 activity remained high in PC3-AR cells after supraphysiological T treatment, which was also shown to be active in LNCaP cells treated with supraphysiological T: MTOR activity promotes cellular senescence, but the mechanism is not well understood110,111. Transient exposure to androgens in AR-positive LNCaP and VCaP cells plated at low density in hypotonic growth media has been shown to induce quiescence or dormancy through redox imbalance and TGFβ–BMP signalling112. Some of the responses to supraphysiological T might seem to be varied and depend upon the cellular models, passage number, supraphysiological T treatment concentration and duration, but many of these effects might be true and not mutually exclusive (Fig. 3a).
Fig. 3 |. Mechanisms of action of supraphysiological testosterone.
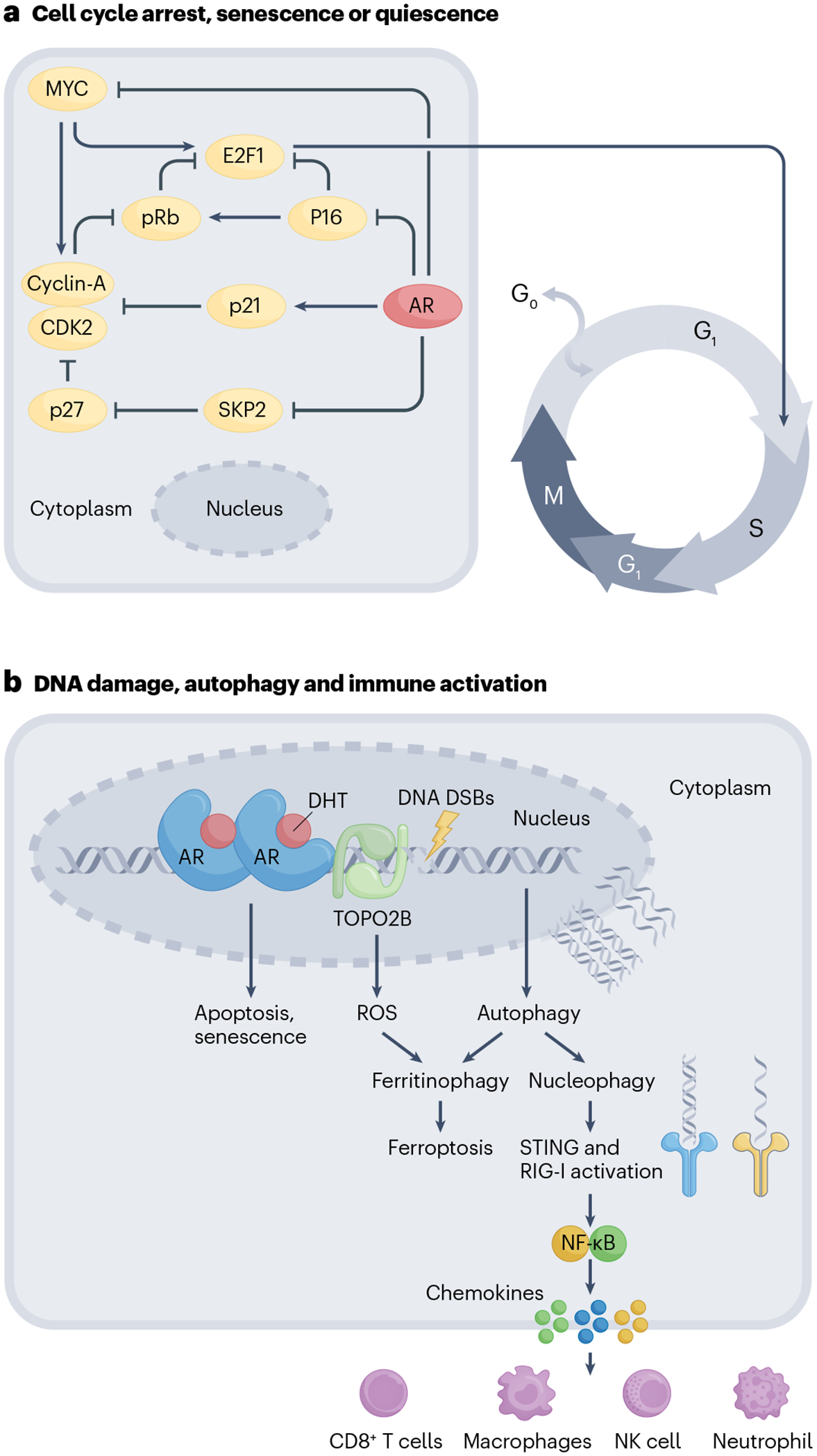
a, Cell-cycle regulation. Supraphysiological testosterone (supraphysiological T) inhibits the transcription of MYC, which is required for cyclin and cyclin-dependent kinase-mediated passage of cells from the G1 to the S phase. Downregulation of MYC suppresses CDK2 and CyclinA activity, which prevents phosphorylation-mediated degradation of RB leading to cell-cycle arrest. Supraphysiological T also increases p21 levels through transcriptional upregulation by the androgen receptor (AR) and inhibits the expression of S-phase kinase-associated protein (SKP2), a subunit of SCF-type cullin ubiquitin ligase. Downregulation of SKP2 by supraphysiological T increases p27, which, in conjunction with p21 and p16 upregulation, causes a G1 phase arrest leading to cell death and quiescence and/or senescence. b, Autophagy and immune activation. Supraphysiological T mediates DNA double-stranded breaks (DSBs) by recruiting TOPO2B to DNA binding sites. Unrepaired DNA lesions cause apoptosis, cell-cycle arrest or senescence. Supraphysiological T also causes an induction of two parallel autophagy-mediated pathways: ferritinophagy and nucleophagy. Ferritinophagy, which involves autophagy-mediated degradation of ferritin, results in increased lipid reactive oxygen species (ROS) and ferroptotic cell death. Supraphysiological T-damaged DNA can be degraded in the autophagosomes by the process of nucleophagy. Cytoplasmic autophagosomal DNA activates a nucleic acid-sensing mechanism through STING and RIG-I. Activated STING and RIG-I signal through NF-κB and cause the release of pro-inflammatory chemokines, including CXCL10, attracting natural killer (NK) cells, T cells, macrophages and neutrophils. DHT, dihydrotestosterone.
An interesting aspect of ligand-bound steroid receptors, including AR, is their ability to cause DNA damage113–115. Response of cells to DNA damage can range from apoptosis to growth arrest and senescence, an effect that is observed in supraphysiological T treatment. The exact mechanism of how androgens cause DNA damage is unknown; evidence suggests a role for ligand-bound AR in recruiting enzymes that actively induce DNA double-strand breaks (DSBs). Insights into this mechanism came from the observation that in prostate cancer, translocations of AR-driven TMPRSS2, which is located on chromosome 21, were common with ERG or ETV1 located on chromosomes 21 and 7, respectively116. Ligand-bound AR was observed to rapidly locate to these translocation sites to recruit cytidine deaminase (AID) and LINE-1 repeat-encoded ORF2 endonuclease, which induce DNA DSBs and proximity-mediated gene rearrangements leading to TMPRSS2–ERG fusions114. Recruitment of TOP2B to these sites was shown to generate DSBs, leading to TMPRSS2–ERG rearrangements115. The effects of TOPO2B are probably not restricted to rearrangement of this genomic region but are likely to occur at other AR binding sites as well. Furthermore, transcription induced by AR would be expected to lead to DNA opening, making it susceptible to ROS-induced DNA damage117,118. Cells with defects in the DNA repair pathway might be particularly susceptible to androgen-induced DNA damage under supraphysiological T conditions. In agreement with this notion, prostate cancer cell lines and patient-derived xenografts that harbour DNA repair mutations have been shown to have inhibited growth on supraphysiological T treatment119,120. Moreover, patients with prostate cancer whose disease responds well to treatment with supraphysiological T had mutations in DNA repair genes, suggesting mutations in DNA repair genes could be positively associated with response to therapy121–123. In AR-positive LNCaP cells that harbour mutations in DNA repair genes, two parallel autophagy-mediated pathways could be triggered: ferritinophagy and nucleophagy124. Ferritinophagy involves selective degradation of the iron-storage molecule ferritin, increasing the labile pool of intracellular iron, leading to non-apoptotic death by ferroptosis upon supraphysiological T treatment. Supraphysiological T-treated cells shuttled their damaged DNA to autophagosomes for degradation through nucleophagy. Activation of nucleophagy in this context might be a cytoprotective phenomenon, enabling cells to get rid of their damaged DNA; however, it can also trigger cytosolic nucleic acid sensors, and NF-κB-mediated innate immune signalling, which includes secretion of cytokines and chemokines that attract innate and adaptive immune cells124. This mechanism might occur in vivo to cause immune clearance of the tumour. Supraphysiological T considerably increased immune cell infiltration in preclinical animal xenograft models of prostate cancer and an increase in cytotoxic CD8 T cells was observed in biopsy samples from patients with prostate cancer after supraphysiological T treatment124 (Fig. 3b).
The above observations show that perturbation of transcription proteins such as AR, which affect many cellular processes, is likely to have a pleiotropic effect. One aspect of supraphysiological T biology that remains to be studied is how supraphysiological T might regulate immune cells and the tumour microenvironment. Androgens are also known to affect the development of lymphocytes in both the thymus and the bone marrow. AR expression has been found on endothelial cells, thymic epithelial cells and innate and adaptive immune systems, including T cells, B cells, innate lymphoid cells and many cell populations present in the bone marrow125–128. Neutrophils also have considerable levels of AR protein expression126. AR is universally expressed on all neutrophil lineages starting from proliferative to terminally differentiated matured phenotype. Upon activation, neutrophils give rise to pro-inflammatory cytokine expression (IL-6, IL-1β and TNF) and chemokines (CCL2, CCL3, CCL4, CXCL1, CXCL4 and CXCL7), and the expression of these were reduced upon AR knockout. Similarly, the expression of AR on monocytes and macrophages suppresses cutaneous wound healing by increased TNF production. A mouse model of myeloid-specific AR-knockout showed rescued wound healing by inhibiting the TNF-mediated inflammatory response129. Supraphysiological T is likely to directly influence the function of these cells, which might contribute to the observed tumour growth inhibition.
Testosterone as a drug
In the past decade, in spite of its reputation as a growth factor for prostate cancer, testosterone has been tested as a therapeutic agent for treatment of this disease.
Early use of testosterone for patients with prostate cancer
Testosterone was initially given to patients with prostate cancer to confirm that the beneficial effect of castration was a result of the reduction of testosterone23. Indeed, many early reports indicated that testosterone administration reversed the benefits of castration, resulting in elevation of tumour markers that were used at that time (including acid phosphatase and alkaline phosphatase) and symptomatic progression130–132, supporting the role of androgens as growth factors for prostate cancer. Given this observation, androgens were given to patients with the intent of stimulating cancer cell proliferation to sensitize them to subsequent DNA damaging agents, such as radioactive phosphorus (32 P), cyclophosphamide, 5-FU, methotrexate and doxorubicin133–135; however, the results of these studies were uniformly negative in improving patient outcomes. Yet, scattered among these initial descriptions of testosterone administration for patients with prostate cancer are anecdotal case reports of patients who paradoxically improved with testosterone monotherapy. In 1957, patient HG, a 68-year-old man with metastatic prostate cancer that had progressed following orchiectomy and hypophysectomy, was described as having a dramatic decrease in serum acid phosphatase from near 200 BU/100 cc to undetectable levels and improvement in cancer symptoms following treatment with testosterone propionate. In 1967, patient CJS, a 76-year-old man with ‘preterminal’ metastatic CRPC (mCRPC), was described to improve from an “extremely feeble” state, “unable to sit without assistance,” to “totally pain-free” and “dancing weekly” following treatment with testosterone propionate 100 mg three-times weekly136. Yet these case reports were anti-dogmatic, and further clinical investigation into whether testosterone could be used as a therapy for prostate cancer was slow. Notably, a substantial body of literature describes the use of androgen replacement in men with hypogonadism and prostate cancer. The results of these studies suggest that androgen replacement does not result in rapid prostate cancer disease progression, contrary to the previously widely held view that androgens would rapidly increase prostate cancer growth137–142. They established a precedent that testosterone could be safely administered to patients with prostate cancer, which enabled subsequent studies assessing testosterone as a prostate cancer therapy. Thus, in 2009, two groups reported on the use of transdermal testosterone as a treatment for patients with CRPC143,144. Using transdermal testosterone, physiological levels of serum testosterone of 300–850 ng/dl, which were generally well tolerated, were achieved in both studies. However, the efficacy of this approach was quite modest, with 3 of 15 patients with non-metastatic CRPC demonstrating a decrease in PSA (no patient with >50% decrease) in one study, and only 1 of 12 patients with mCRPC demonstrating a reduction in PSA of 50% in the other study143. Despite this limited efficacy, these studies supported the growing appreciation that testosterone could be administered safely to patients with advanced prostate cancer.
Bipolar androgen therapy
BAT is the administration of testosterone cypionate 400 mg intramuscularly every 28 days concurrent with an LHRH agonist to result in oscillation of serum testosterone from supraphysiological (>1,500 ng/dl) to near-castration levels145. This therapy was first tested when it was given to 16 patients involved in a pilot clinical trial in combination with etoposide as a treatment for asymptomatic mCRPC145. Remarkably, this combination therapy resulted in PSA and radiographic responses in about half of the patients involved, with 4 patients treated with BAT for >1 year145. The design of this trial was such that patients received BAT and etoposide for the first 3 months, then subsequently received BAT monotherapy if they were experiencing a PSA decline. Notably, most patients who responded to BAT and etoposide continued to respond to BAT monotherapy; thus, etoposide was thought to contribute minimal benefit but considerable toxic effects and was omitted from subsequent trials of BAT.
BAT differs in two important ways from transdermal testosterone administration: first, it achieves supraphysiological levels of serum testosterone; and second, the testosterone level is not clamped but rather is cycled between high and low levels (hence the name ‘bipolar’ androgen therapy)28. This strategy was selected given preclinical data suggesting that CRPC exhibits a biphasic response to re-exposure to androgens, whereby physiological levels of androgens induce growth and proliferation, and supraphysiological levels of androgens are required to induce growth arrest and cell death146. Moreover, this cycled approach was hypothesized to target the heterogeneity and adaptability of prostate cancer cells present in metastases, some of which might be inhibited by high testosterone and others by low testosterone.
Following the promising results of the pilot clinical trial, BAT has been tested in five subsequent clinical trials for patients with advanced prostate cancer: a single-arm trial for castration-sensitive prostate cancer (BATMAN)147; a single-arm, multicohort trial for CRPC (RESTORE)148–150; a randomized trial for mCRPC comparing BAT with enzalutamide (TRANSFORMER)151; a single-arm trial of BAT in combination with the anti-PD1 agent nivolumab for patients with mCRPC (COMBAT)152; and a single-arm, multicohort trial of BAT in combination with the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib for mCRPC153. Overall, nearly 300 patients with prostate cancer have been treated with BAT, and a great deal has been learned regarding safety, efficacy, and novel vulnerabilities and opportunities for synergistic combination therapies with BAT, although much remains to be understood.
Safety and tolerability of BAT
Given the efficacy of ADT in treating prostate cancer67, the safety and tolerability of BAT (as the opposite therapy of ADT) have been heavily scrutinized. Evidence from early reports suggested that testosterone can exacerbate pain owing to bone metastases130,134,154,155, and many have voiced concern that testosterone could induce tumour flare that might result in the dangerous spinal cord or urethral compression. Thus, all clinical trials of BAT have excluded patients with pain caused by prostate cancer requiring opiate medications and those with evidence of disease in sites that might put the patient at risk of complications should tumour flare occur. With these exclusion criteria in place, BAT has seemed to be relatively safe and very well-tolerated among treated patients. Overall, the rate and severity of adverse events seem similar to the standard-of-care agent enzalutamide151. Common adverse events tend to be low grade and include musculoskeletal pain, lower extremity oedema and breast tenderness149,151. Notably, spinal cord compression, urethral compression causing urinary obstruction or other objective evidence of tumour flare have not been observed with the use of BAT. This observation suggests that BAT does not cause tumour flare, but this possibility will be continuously assessed as increased numbers of patients are treated.
Efficacy of BAT monotherapy
The efficacy of BAT monotherapy has been tested in patients with castration-sensitive prostate cancer (BATMAN)147, CRPC that has progressed on only ADT (RESTORE cohort C)149, CRPC that has progressed on abiraterone (RESTORE cohort B and TRANSFORMER)148,151, and CRPC that has progressed on enzalutamide (RESTORE cohort A)150; however, only the TRANSFORMER trial151 was a randomized controlled trial, which means it included a control arm to assess the benefit of this therapy most accurately. On average, among patients with mCRPC, BAT results in a PSA decline ≥50% (PSA50 response) in 20–25% of patients, an objective response in 30–40% of patients, and a median progression-free survival of ~6 months. Efficacy end points studied include the PSA50 response rate (the percentage of patients with at least a 50% decline in PSA on therapy), the objective response rate (ORR) per RECIST 1.1 (ref. 156) and Prostate Cancer Working Group 3 (PCWG3) definitions157, clinical or radiographic progression-free survival PCWG3 definition157 and overall survival (OS) (Table 2).
Table 2 |.
Efficacy of BAT
| Trial | Therapy | Patient population | Number of patients | Efficacy | Clinicaltrials.gov number | Ref. |
|---|---|---|---|---|---|---|
| Pilot | Single arm: BAT plus etoposide | nmCRPC and Low-volume mCRPC | 16 | PSA50 RR: 4 of 14 ORR: 5 of 10 |
NCT01084759 | 205 |
| BATMAN | Single arm: alternating ADT plus BAT | nmCSPC | 29 | PSA <4ng/mL at 18 months: 17 of 29 | NCT01750398 | 206 |
| RESTORE | Single arm: BAT | Cohort A: mCRPC that has progressed on enzalutamide | 30 | PSA50 RR: 9 of 30 ORR: 6 of 12 Median crPFS: 8.6 months |
NCT02090114 | 159 |
| Cohort B: mCRPC that has progressed on abiraterone | 29 | PSA50 RR: 5 of 29 ORR: 2 of 7 Median crPFS: 4.3 months |
||||
| Cohort C: De novo CRPC | 29 | PSA50 RR: 4 of 29 ORR: 4 of 13 Median rPFS for mCRPC: 8.5 months |
||||
| TRANSFORMER | Randomized: BAT versus enzalutamide | mCRPC that has progressed on abiraterone | 94 (BAT) and 101 (enzalutamide) | PSA50 RR: 24 of 85 (BAT) and 24 of 94 (enzalutamide) Median rPFS: 5.7 months (BAT) and 5.7 months (enzalutamide) Median OS: 32.9 months (BAT) and 29 months (enzalutamide) |
NCT02286921 | 207 |
| COMBAT | Single arm: BAT followed by BAT plus nivolumab | mCRPC that has progressed on enzalutamide and/or abiraterone, plus or minus taxane therapy | 45 | PSA50 RR: 18 of 45 ORR: 10 of 42 Median rPFS: 5.7 months |
NCT03554317 | 208 |
| BAT plus olaparib | Single arm: BAT plus olaparib | mCRPC that has progressed on enzalutamide and/or abiraterone | 36 | PSA50 RR: 14 of 30 Median PFS: 12.6 months |
NCT03516812 | 209 |
ADT, androgen deprivation therapy; BAT, bipolar androgen therapy; crPFS, clinical or radiographic progression-free survival; mCRPC, metastatic castration-resistant prostate cancer; nmCRPC, non-metastatic castration-resistant prostate cancer; ORR, objective response rate; OS, overall survival; PSA50, PSA decline ≥50%; PFS, progression-free survival; rPFS, radiographic progression-free survival; RR, response rate.
Biomarkers for predicting response to BAT
Given that tumour regression seems to occur in a minority of patients treated with BAT, identifying biomarkers that predict sensitivity could enhance the utility of this therapy. Preclinical cell line and mouse xenograft models suggest that high AR expression induced by prolonged castration might improve sensitivity to growth inhibition by supraphysiological androgens146. The expression of full-length AR and the splice variant AR-V7 in circulating tumour cells had no correlation with response in patients included in the TRANSFORMER trial86,151. However, this approach was limited given that circulating tumour cells were not detectable in most patients, and the assay reported a binary, rather than continuous, measurement of AR expression.
High AR activity predicts growth inhibition by supraphysiological androgens and BAT in patients158. High androgen receptor activity is required for growth inhibition of prostate cancer by supraphysiological androgens by enabling downregulation of MYC158. A gene score that estimates AR activity based on a ranking of expression of 10 canonical AR target genes among the top expressed genes in tumours before BAT therapy (ARAMW score) enabled prediction of PSA response and objective response and increased progression-free survival (PFS) and OS on BAT treatment. Notably, BAT results in significant downregulation of AR (P < 0.0001), which was found to be a mechanism of resistance to growth inhibition by supraphysiological androgens. Future prospective trials are required for validation of the ARAMW score as a predictive biomarker of response to BAT.
Beyond AR, results of retrospective analyses of patients treated with BAT have suggested that patients with mutations in TP53 and/or homologous recombination in DNA repair genes might exhibit enhanced responses to BAT119,121. These observations support the idea that BAT can induce AR-mediated DNA damage that is enhanced in cancer cells with defective DNA repair mechanisms. Ongoing studies are being conducted to prospectively assess the benefit of BAT in a biomarker-selected group of patients with TP53, PTEN or RB1 pathogenic alterations (NCT02090114)159 and separately in the biomarker-selected group of patients with homologous recombination defect mutations (NCT03522064)160.
Sequencing of BAT with AR-axis inhibitory therapies
A notable finding of the pilot clinical trial of BAT was that it seemed to re-sensitize CRPC to AR-axis inhibition145. Overall, 12 of 13 patients exhibited a PSA decline to subsequent AR-directed therapy administered after progression on BAT, despite previous progression on similar agents before BAT. This idea was further explored in the RESTORE148–150 and TRANSFORMER151 trials. In RESTORE, patients who had previously progressed on enzalutamide subsequently exhibited a PSA50 response rate of 52% on enzalutamide after BAT, whereas patients who had previously progressed on abiraterone subsequently exhibited a PSA50 response rate of 16% on abiraterone after BAT148. In TRANSFORMER, the PSA50 response rate to enzalutamide without previous BAT was 25.5%, the PSA50 response to enzalutamide following BAT was 77.8%151. Moreover, the PSA PFS was 3.8 months and OS 28.6 months on enzalutamide without previous BAT, but improved to 10.9 months and 37.1 months, respectively, on enzalutamide following BAT.
Mechanistically, given that AR inhibition results in AR overexpression that can confer resistance to AR inhibition161, BAT might result in AR downregulation that can confer re-sensitization to AR inhibition. Indeed, BAT did cause downregulation of AR in all samples analysed in the COMBAT trial158. However, the results of these studies suggest that AR antagonism and AR agonists (BAT) might be repeatedly alternated to pre-empt and/or overcome resistance to either therapeutic modality. This approach is currently being tested in a prospective clinical trial of BAT alternating with enzalutamide in the STEP-UP trial (NCT04363164)162.
Opportunities for synergistic combination therapies
BAT is generally well tolerated151. Moreover, in contrast to second-generation AR-axis inhibitors, BAT is associated with minimal financial toxicity and requires no commitment of compliance on behalf of the patient, as it is administered by rapid intramuscular injection monthly in the clinic151. Thus, BAT is an ideal foundation on which to layer additional therapies that might augment responses. Treatments that have been tested in combination with BAT include the anti-PD1 agent nivolumab (COMBAT152) and the PARP inhibitor olaparib153. Outcomes of these clinical trials have been reported currently in abstract form only152,153.
The rationale for combining BAT with nivolumab comes from three anecdotal instances of patients with microsatellite-stable mCRPC exhibiting remarkable responses to anti-PD1 following progression on BAT122. These responses were notable given that microsatellite-stable mCRPC is immunologically cold and shows near-uniform resistance to anti-PD1 therapy163. The responses were hypothesized to occur through the induced vulnerability of AR-mediated activation of nucleic acid sensors and immune signalling that might recruit and activate cytotoxic immune cells to the tumour bed124. The design of the COMBAT trial152 was a 3-month lead-in of BAT monotherapy followed by combined therapy with BAT and nivolumab. The complete analysis describing the antitumour benefit attributed to nivolumab is currently in preparation; however, the overall PSA50 response rate was 40%, and the median radiographic PFS was 5.7 months152. The PSA50 response rate was slightly higher than in previous trials, but the median rPFS was identical to BAT monotherapy in the TRANSFORMER152 trial. This observation suggests that further research into the effect of BAT on prostate cancer tumour immunity is needed to understand whether BAT has a role in enhancing durable immune responses to prostate cancer.
The other combination therapy approach that has been tested is BAT in combination with olaparib153. The rationale for this approach is that supraphysiological androgens can induce AR-mediated DNA DSBs115,119 that are hypothesized to be more detrimental in the presence of PARP inhibition than not, similar to the synthetic lethality of BRCA1 and BRCA2 deficiency and PARP inhibition in prostate cancer and other cancer types164. The possible sensitivity of prostate cancer with homologous recombination deficiency mutations to BAT165 further supports the idea that efficient DNA repair is crucial to the persistence of CRPC treated with BAT. Of note, the results of the pilot clinical trial of BAT suggested minimal additional benefit from concurrent treatment with etoposide145, which exerts antitumour effects through induction of DNA DSBs. Nonetheless, olaparib has a different mechanism of action from etoposide by inhibiting PARP and impairing the repair of DNA DSBs166, which might provide enhanced synergy with BAT. Some results from this trial were presented at European Society of Medical Oncology 2021, and a PSA50 response rate of 47% and a median PFS of 12.6 months were reported153. Teasing out whether synergy between BAT and olaparib occurs in this trial will probably be challenging, given that both agents are known to be active agents as treatment for mCRPC when given as monotherapy (unlike anti-PD1).
Future directions
Many questions remain in a quest to define the optimal clinical application of the testosterone paradox in prostate cancer. The optimal schedule and dose of testosterone administration remains to be determined. Results of previous studies indicate that strategies that achieve sustained physiological serum levels of testosterone are not as effective as BAT143,144, which produces cycling of serum testosterone from supraphysiological to near-castration levels over the course of 28 days145; however, whether BAT is more effective simply owing to its ability to expose tumours to increased concentrations of testosterone or whether the cycling of testosterone is important to prevent rapid adaptation of the cancer cells to high levels of testosterone (or both) is currently unknown. One feature of testosterone cypionate is that it has variable pharmacokinetics145. Future clinical studies should consider whether other forms of AR agonists, such as novel formulations of oral testosterone including Jatenzo, an oral lipoprotein-coated testosterone undecanoate, or selective AR modulators, small-molecular non-steroidal AR agonists, might be more or less effective than testosterone cypionate.
Patient factors that predict sensitivity to BAT also need to be determined. Clinical studies of BAT have shown that only 20–40% of patients with CRPC are sensitive to BAT151. Thus, understanding mechanisms of sensitivity and primary resistance are essential to limiting the use of BAT to only patients who are likely to respond and developing novel strategies to overcome primary resistance to BAT to expand the population of patients who benefit. Promising features that might predict response to BAT include high AR activity158 and homologous recombination repair mutations119, although these biomarkers require prospective validation. A related question is the optimal timing of administration of BAT in the sequence of therapy for patients with CRPC. Current evidence suggests that progression on prolonged and potent AR-axis inhibitors might enhance sensitivity to BAT151; however, BAT priming can improve sensitivity to AR-axis inhibitors149. Thus, future studies should assess the optimal timing of BAT usage for the treatment of patients with advanced prostate cancer.
A challenge is that we have not tested BAT among patients with pain from prostate cancer. An understanding of the molecular mechanisms by which testosterone administration causes or exacerbates pain is needed to broaden the population of patients who might receive and benefit from BAT. Given the usual rapid onset of pain flares, it seems unlikely that this pain is a result of cancer cell proliferation and is more probably a neuromodulatory effect owing to production of cytokines or other pain-inducing chemical substances, but this idea is currently speculation and future research should directly address this question.
The drivers of acquired resistance to BAT also need to be determined. The majority of patients who initially respond to BAT unfortunately go on to develop resistance at around 6 months to 1 year151. BAT results in considerable downregulation of AR expression158 and this reduction is probably a substantial driver of acquired resistance to therapy. Deciphering this mechanism is important given that this adaptive resistance might be reversible. Alternative mechanisms of resistance should also be considered and studied.
The key mechanisms of tumour growth inhibition by BAT occurring in patients are important to discover. Given the diverse maladaptive effects of supraphysiological androgens in models of prostate cancer158, clinically, several mechanisms probably occur. This knowledge might lead to an understanding of novel vulnerabilities or adaptive responses induced by BAT that could be targeted concurrently with BAT to result in expanded efficacy.
Finally, the cancer cell-extrinsic effects of supraphysiological androgen and BAT that might alter prostate cancer progression need to be understood. Androgens can affect the function of diverse cell types, including immune and stromal cells within the tumour microenvironment167,168, and those of distant tissues such as bone and muscle, which might indirectly affect cancer progression169.
Conclusions
Despite the fundamental function of androgens as growth factors for prostate cancer, preclinical and clinical studies have established that supraphysiological androgens can paradoxically suppress the growth of CRPC. Accumulated preclinical evidence suggests that this growth inhibition can result from multiple mechanisms including cell cycle arrest, senescence, apoptosis, non-apoptotic cell death and immune clearance. The scientific community has made substantial progress in defining and elucidating mechanisms of the testosterone paradox of advanced prostate cancer, but considerable knowledge still needs to be gained to maximize opportunities for patient benefit. BAT is an innovative approach based on paradoxical growth inhibition of prostate cancer by supraphysiological testosterone; however, it has not been incorporated into standard-of-care practices, given the uncertainty in the optimal use of such therapy. We hope that ongoing research efforts will soon establish a role for this therapy to expand options and improve outcomes for patients with advanced prostate cancer.
Key points.
Androgens can drive prostate cancer growth providing the rationale for using deprivation of androgens as a first line of treatment for prostate cancer. Unfortunately, prostate cancer cells adapt to low androgen levels and eventually progress to a castration-resistant state.
Results of several studies have indicated a paradoxical decrease in tumour growth in prostate cancer models upon treatment with supraphysiological levels of testosterone. Evidence indicates several complementary mechanisms, including cell death and cytostasis, which might be responsible for paradoxical growth inhibition by supraphysiological testosterone.
Adaptive reliance on androgen signalling by castration-resistant prostate cancer cells becomes a therapeutic liability that can be exploited clinically through the administration of supraphysiological testosterone, an approach termed ‘bipolar androgen therapy’ (BAT). The term bipolar is used to emphasize that, with this strategy, rapid cycling occurs between two extremes: from supraphysiological back to near-castration testosterone levels over a 4-week cycle.
Understanding how BAT works at the molecular and cellular levels might help to develop biomarkers for patient stratification and to rationally combine BAT with other agents to achieve increased efficacy.
Acknowledgements
S.K. is partly supported by the W81XWH1910724, 1R01CA243184 and PCF Challenge awards. R.K. is supported by the W81XWH2210118 and PCF Young Investigator Award 21YOUN22. L.A.S. is supported by W81XWH2010079 and Johns Hopkins University Clinician-Scientist Award.
Footnotes
Competing interests
All the authors declare no competing interests.
References
- 1.Sung H et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 71, 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Butenandt A Über die chemische Untersuchung des SexualHormons. Angew. Chem 44, 905–908 (1931). [Google Scholar]
- 3.Butenandt A & Tscherning K Androsterone, a crystalline male sex hormone. I. Isolation and purification from male urine. Z. Physiol. Chem 229, 167 (1934). [Google Scholar]
- 4.David k, E. D, Freud J & Laqueur E Über krystallinisches männliches Hormon aus Hoden (Testosteron), wirksamer als aus Harn oder aus Cholesterin bereitetes Androsteron. Hoppe Seylers Z. Physiol. Chem 233, 281–283 (1935). [Google Scholar]
- 5.Pearlman WH & Crepy O Steroid-protein interaction with particular reference to testosterone binding by human serum. J. Biol. Chem 242, 182–189 (1967). [PubMed] [Google Scholar]
- 6.Rosner W & Deakins SM Testosterone-binding globulins in human plasma: studies on sex distribution and specificity. J. Clin. Invest 47, 2109–2116 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearlman WH & Pearlman MR The metabolism in vivo of Δ4-androstene-3, 17-dione-7-H3; its localization in the ventral prostate and other tissues of the rat. J. Biol. Chem 236, 1321–1327 (1961). [PubMed] [Google Scholar]
- 8.Fang S, Anderson KM & Liao S Receptor proteins for androgens. On the role of specific proteins in selective retention of 17-β-hydroxy-5-α-androstan-3-one by rat ventral prostate in vivo and in vitro. J. Biol. Chem 244, 6584–6595 (1969). [PubMed] [Google Scholar]
- 9.Bruchovsky N & Wilson JD The conversion of testosterone to 5-α-androstan-17-β-ol-3-one by rat prostate in vivo and in vitro. J. Biol. Chem 243, 2012–2021 (1968). [PubMed] [Google Scholar]
- 10.Imperato-McGinley J, Guerrero L, Gautier T & Peterson RE Steroid 5α-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science 186, 1213–1215 (1974). [DOI] [PubMed] [Google Scholar]
- 11.Siiteri PK & Wilson JD Testosterone formation and metabolism during male sexual differentiation in the human embryo. J. Clin. Endocrinol. Metab 38, 113–125 (1974). [DOI] [PubMed] [Google Scholar]
- 12.Anderson KM & Liao S Selective retention of dihydrotestosterone by prostatic nuclei. Nature 219, 277–279 (1968). [DOI] [PubMed] [Google Scholar]
- 13.Lubahn DB et al. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science 240, 327–330 (1988). [DOI] [PubMed] [Google Scholar]
- 14.Chang CS, Kokontis J & Liao ST Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science 240, 324–326 (1988). [DOI] [PubMed] [Google Scholar]
- 15.Mangelsdorf DJ et al. The nuclear receptor superfamily: the second decade. Cell 83, 835–839 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velasco AM et al. Identification and validation of novel androgen-regulated genes in prostate cancer. Endocrinology 145, 3913–3924 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Sahu B et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 73, 1570–1580 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Gao S et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat. Genet 52, 1011–1017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahu B et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 30, 3962–3976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia L et al. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS One 3, e3645 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Visakorpi T et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet 9, 401–406 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Li Y et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73, 483–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huggins C & Hodges CV Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1, 293–297 (1941). [DOI] [PubMed] [Google Scholar]
- 24.Fu AZ et al. Mortality and androgen deprivation therapy as salvage treatment for biochemical recurrence after primary therapy for clinically localized prostate cancer. J. Urol 197, 1448–1454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharifi N, Gulley JL & Dahut WL Androgen deprivation therapy for prostate cancer. JAMA 294, 238–244 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Tangen CM et al. Ten-year survival in patients with metastatic prostate cancer. Clin. Prostate Cancer 2, 41–45 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Pienta KJ & Bradley D Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res 12, 1665–1671 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Denmeade SR & Isaacs JT Bipolar androgen therapy: the rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate 70, 1600–1607 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McNeal JE Regional morphology and pathology of the prostate. Am. J. Clin. Pathol 49, 347–357 (1968). [DOI] [PubMed] [Google Scholar]
- 30.McNeal JE Normal histology of the prostate. Am. J. Surg. Pathol 12, 619–633 (1988). [DOI] [PubMed] [Google Scholar]
- 31.Cunha GR & Chung LW Stromal-epithelial interactions — I. Induction of prostatic phenotype in urothelium of testicular feminized (Tfm/y) mice. J. Steroid Biochem 14, 1317–1324 (1981). [DOI] [PubMed] [Google Scholar]
- 32.Cunha GR et al. Normal and abnormal development of the male urogenital tract. Role of androgens, mesenchymal-epithelial interactions, and growth factors. J. Androl 13, 465–475 (1992). [PubMed] [Google Scholar]
- 33.Isaacs JT & Coffey DS Etiology and disease process of benign prostatic hyperplasia. Prostate Suppl. 2, 33–50 (1989). [DOI] [PubMed] [Google Scholar]
- 34.English HF, Santen RJ & Isaacs JT Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. Prostate 11, 229–242 (1987). [DOI] [PubMed] [Google Scholar]
- 35.Collins AT, Habib FK, Maitland NJ & Neal DE Identification and isolation of human prostate epithelial stem cells based on α2β1-integrin expression. J. Cell Sci 114, 3865–3872 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Bonkhoff H & Remberger K Widespread distribution of nuclear androgen receptors in the basal cell layer of the normal and hyperplastic human prostate. Virchows Arch. A Pathol. Anat. Histopathol 422, 35–38 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Bonkhoff H, Stein U & Remberger K The proliferative function of basal cells in the normal and hyperplastic human prostate. Prostate 24, 114–118 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Germann M et al. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. Stem Cell 30, 1076–1086 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Wang X et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 461, 495–500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi N, Zhang B, Zhang L, Ittmann M & Xin L Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 21, 253–265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ousset M et al. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat. Cell Biol 14, 1131–1138 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Wang ZA et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol 15, 274–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu X et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Dev 101, 61–69 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Xie Q et al. Dissecting cell-type-specific roles of androgen receptor in prostate homeostasis and regeneration through lineage tracing. Nat. Commun 8, 14284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karthaus WR et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 368, 497–505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai C, Heemers H & Sharifi N Androgen signaling in prostate cancer. Cold Spring Harb. Perspect. Med 10.1101/cshperspect.a030452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar A et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med 22, 369–378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ware KE, Garcia-Blanco MA, Armstrong AJ & Dehm SM Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr. Relat. Cancer 21, T87–T103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen EJ et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res 21, 1273–1280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korpal M et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 3, 1030–1043 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Gottlieb B, Beitel LK, Wu JH & Trifiro M The androgen receptor gene mutations database (ARDB): 2004 update. Hum. Mutat 23, 527–533 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Robinson JL et al. Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene 33, 5666–5674 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Q et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 27, 380–392 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pomerantz MM et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet 47, 1346–1351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stelloo S, Bergman AM & Zwart W Androgen receptor enhancer usage and the chromatin regulatory landscape in human prostate cancers. Endocr. Relat. Cancer 26, R267–R285 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Westaby D et al. A new old target: androgen receptor signaling and advanced prostate cancer. Annu. Rev. Pharmacol. Toxicol 62, 131–153 (2022). [DOI] [PubMed] [Google Scholar]
- 58.Uo T, Sprenger CC & Plymate SR Androgen receptor signaling and metabolic and cellular plasticity during progression to castration resistant prostate cancer. Front. Oncol 10, 580617 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Culig Z & Santer FR Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 33, 413–427 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Deng Q et al. Non-genomic action of androgens is mediated by rapid phosphorylation and regulation of androgen receptor trafficking. Cell. Physiol. Biochem 43, 223–236 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Leung JK & Sadar MD Non-genomic actions of the androgen receptor in prostate cancer. Front. Endocrinol 8, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zarif JC & Miranti CK The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell Signal. 28, 348–356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harris WP, Mostaghel EA, Nelson PS & Montgomery B Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol 6, 76–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huggins C & Scott WW Bilateral adrenalectomy in prostatic cancer: clinical features and urinary excretion of 17-ketosteroids and estrogen. Ann. Surg 122, 1031–1041 (1945). [PMC free article] [PubMed] [Google Scholar]
- 65.de Bono JS et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med 364, 1995–2005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryan CJ, Smith MR & Bono JS Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med 368, 138–148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fizazi K, Tran N & Fein L Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med 377, 352–360 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Liao S, Howell DK & Chang TM Action of a nonsteroidal antiandrogen, flutamide, on the receptor binding and nuclear retention of 5 α-dihydrotestosterone in rat ventral prostate. Endocrinology 94, 1205–1209 (1974). [DOI] [PubMed] [Google Scholar]
- 69.Tran C et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beer TM et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med 371, 424–433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chi KN et al. Apalutamide for metastatic, castration-sensitive prostate cancer. N. Engl. J. Med 381, 13–24 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Fizazi K et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med 380, 1235–1246 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Hussain M et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med 378, 2465–2474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scher HI et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med 367, 1187–1197 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Maron SB et al. Pembrolizumab with trastuzumab and chemotherapy (PTC) in HER2-positive metastatic esophagogastric cancer (mEG): plasma and tumor-based biomarker analysis. J. Clin. Oncol 38 (Suppl. 15), 4559 (2020). [Google Scholar]
- 76.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT03888612 (2021). [DOI] [PubMed]
- 77.Linja MJ, Savinainen KJ & Saramäki OR Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 61, 3550–3555 (2001). [PubMed] [Google Scholar]
- 78.Azad AA, Volik SV & Wyatt AW Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res 21, 2315–2324 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Isaacs JT & Isaacs WB Androgen receptor outwits prostate cancer drugs. Nat. Med 10, 26–27 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Antonarakis ES, Lu C & Wang H AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scher HI & Sawyers CL Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol 23, 8253–8261 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Huggins C & Yang NC Induction and extinction of mammary cancer. A striking effect of hydrocarbons permits analysis of mechanisms of causes and cure of breast cancer. Science 137, 257–262 (1962). [DOI] [PubMed] [Google Scholar]
- 83.Horoszewicz JS et al. LNCaP model of human prostatic carcinoma. Cancer Res. 43, 1809–1818 (1983). [PubMed] [Google Scholar]
- 84.Berns EM, de Boer W & Mulder E Androgen-dependent growth regulation of and release of specific protein(s) by the androgen receptor containing human prostate tumor cell line LNCaP. Prostate 9, 247–259 (1986). [DOI] [PubMed] [Google Scholar]
- 85.Dai JL, Maiorino CA, Gkonos PJ & Burnstein KL Androgenic up-regulation of androgen receptor cDNA expression in androgen-independent prostate cancer cells. Steroids 61, 531–539 (1996). [DOI] [PubMed] [Google Scholar]
- 86.Kokontis J, Takakura K, Hay N & Liao S Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res. 54, 1566–1573 (1994). [PubMed] [Google Scholar]
- 87.Heisler LE et al. Androgen-dependent cell cycle arrest and apoptotic death in PC-3 prostatic cell cultures expressing a full-length human androgen receptor. Mol. Cell. Endocrinol 126, 59–73 (1997). [DOI] [PubMed] [Google Scholar]
- 88.Kokontis JM et al. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of Cdk2, CyclinA, and Skp2. PLoS One 9, e109170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ling MT, Chan KW & Choo CK Androgen induces differentiation of a human papillomavirus 16 E6/E7 immortalized prostate epithelial cell line. J. Endocrinol 170, 287–296 (2001). [DOI] [PubMed] [Google Scholar]
- 90.Berthon P et al. Androgens are not a direct requirement for the proliferation of human prostatic epithelium in vitro. Int. J. Cancer 73, 910–916 (1997). [DOI] [PubMed] [Google Scholar]
- 91.Antony L, van der Schoor F, Dalrymple SL & Isaacs JT Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/β-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate 74, 1118–1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.D’Antonio JM, Vander Griend DJ & Isaacs JT DNA licensing as a novel androgen receptor mediated therapeutic target for prostate cancer. Endocr. Relat. Cancer 16, 325–332 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vander Griend DJ, Litvinov IV & Isaacs JT Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle 6, 647–651 (2007). [DOI] [PubMed] [Google Scholar]
- 94.Litvinov IV et al. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc. Natl Acad. Sci. USA 103, 15085–15090 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fragkos M, Ganier O, Coulombe P & Mechali M DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol 16, 360–374 (2015). [DOI] [PubMed] [Google Scholar]
- 96.Nishitani H, Taraviras S, Lygerou Z & Nishimoto T The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J. Biol. Chem 276, 44905–44911 (2001). [DOI] [PubMed] [Google Scholar]
- 97.Nishitani H & Lygerou Z Control of DNA replication licensing in a cell cycle. Genes Cell 7, 523–534 (2002). [DOI] [PubMed] [Google Scholar]
- 98.Shi YK et al. MCM7 interacts with androgen receptor. Am. J. Pathol 173, 1758–1767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wolf DA, Herzinger T, Hermeking H, Blaschke D & Horz W Transcriptional and posttranscriptional regulation of human androgen receptor expression by androgen. Mol. Endocrinol 7, 924–936 (1993). [DOI] [PubMed] [Google Scholar]
- 100.Henttu P & Vihko P Growth factor regulation of gene expression in the human prostatic carcinoma cell line LNCaP. Cancer Res. 53, 1051–1058 (1993). [PubMed] [Google Scholar]
- 101.Cai C et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 20, 457–471 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rudolph T, Beuch S & Reuter G Lysine-specific histone demethylase LSD1 and the dynamic control of chromatin. Biol. Chem 394, 1019–1028 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Cerella C, Grandjenette C, Dicato M & Diederich M Roles of apoptosis and cellular senescence in cancer and aging. Curr. Drug. Targets 17, 405–415 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Wang X, Deng H, Basu I & Zhu L Induction of androgen receptor-dependent apoptosis in prostate cancer cells by the retinoblastoma protein. Cancer Res. 64, 1377–1385 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Lin Y et al. Androgen and its receptor promote Bax-mediated apoptosis. Mol. Cell Biol 26, 1908–1916 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Joly-Pharaboz MO et al. Inhibition of growth and induction of apoptosis by androgens of a variant of LNCaP cell line. J. Steroid Biochem. Mol. Biol 73, 237–249 (2000). [DOI] [PubMed] [Google Scholar]
- 107.Roediger J et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src-Akt pathway. Mol. Cancer 13, 214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mirochnik Y et al. Androgen receptor drives cellular senescence. PLoS One 7, e31052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Han W et al. Exploiting the tumor-suppressive activity of the androgen receptor by CDK4/6 inhibition in castration-resistant prostate cancer. Mol. Ther 10.1016/j.ymthe.2022.01.039 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Demidenko ZN et al. Rapamycin decelerates cellular senescence. Cell Cycle 8, 1888–1895 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Herranz N et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol 17, 1205–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bui AT et al. Transient exposure to androgens induces a remarkable self-sustained quiescent state in dispersed prostate cancer cells. Cell Cycle 16, 879–893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ju BG et al. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802 (2006). [DOI] [PubMed] [Google Scholar]
- 114.Lin C et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139, 1069–1083 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haffner MC et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet 42, 668–675 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tomlins SA et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648 (2005). [DOI] [PubMed] [Google Scholar]
- 117.Kim N & Jinks-Robertson S Transcription as a source of genome instability. Nat. Rev. Genet 13, 204–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cristini A, Geraud M & Sordet O Transcription-associated DNA breaks and cancer: a matter of DNA topology. Int. Rev. Cell Mol. Biol 364, 195–240 (2021). [DOI] [PubMed] [Google Scholar]
- 119.Chatterjee P et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Invest 129, 4245–4260 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lam HM et al. Durable response of enzalutamide-resistant prostate cancer to supraphysiological testosterone is associated with a multifaceted growth suppression and impaired DNA damage response transcriptomic program in patient-derived xenografts. Eur. Urol 77, 144–155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Markowski MC et al. Molecular and clinical characterization of patients with metastatic castration resistant prostate cancer achieving deep responses to bipolar androgen therapy. Clin. Genitourin. Cancer 10.1016/j.clgc.2021.08.001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Markowski MC et al. Extreme responses to immune checkpoint blockade following bipolar androgen therapy and enzalutamide in patients with metastatic castration resistant prostate cancer. Prostate 80, 407–411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Teply BA, Kachhap S, Eisenberger MA & Denmeade SR Extreme response to high-dose testosterone in BRCA2- and ATM-mutated prostate cancer. Eur. Urol 71, 499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kumar R et al. Supraphysiologic testosterone induces ferroptosis and activates immune pathways through nucleophagy in prostate cancer. Cancer Res. 81, 5948–5962 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Torres-Estay V et al. Androgen receptor in human endothelial cells. J. Endocrinol 224, R131–R137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mantalaris A et al. Localization of androgen receptor expression in human bone marrow. J. Pathol. 193, 361–366 (2001). [DOI] [PubMed] [Google Scholar]
- 127.Blanquart E, Laffont S & Guéry J-C Sex hormone regulation of innate lymphoid cells. Biomed. J 44, 144–156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guan X et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 10.1038/s41586-022-04522-6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lai J-J et al. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-α expression. J. Clin. Invest 119, 3739–3751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tagnon HJ, Schulman P, Whitmore WF & Leone LA Prostatic fibrinolysin: study of a case illustrating role in hemorrhagic diathesis of cancer of the prostate. Am. J. Med 15, 875–884 (1953). [DOI] [PubMed] [Google Scholar]
- 131.Bonner CD, Fishman WH & Homburger F Serum prostatic acid phosphatase and cancer of the prostate. N. Engl. J. Med 255, 925–933 (1956). [DOI] [PubMed] [Google Scholar]
- 132.Fowler JE Jr & Whitmore WF Jr The response of metastatic adenocarcinoma of the prostate to exogenous testosterone. J. Urol 126, 372–375 (1981). [DOI] [PubMed] [Google Scholar]
- 133.Manni A, Bartholomew M & Caplan R Androgen priming and chemotherapy in advanced prostate cancer: evaluation of determinants of clinical outcome. J. Clin. Oncol 6, 1456–1466 (1988). [DOI] [PubMed] [Google Scholar]
- 134.Suarez AJ, Lamm DL & Radwin HM Androgen priming and cytotoxic chemotherapy in advanced prostatic cancer. Cancer Chemother. Pharmacol 8, 261–265 (1982). [DOI] [PubMed] [Google Scholar]
- 135.Donati RM, Ellis H & Gallagher NI Testosterone potentiated 32P therapy in prostatic carcinoma. Cancer 19, 1088–1090 (1966). [DOI] [PubMed] [Google Scholar]
- 136.Prout GR Jr & Brewer WR Response of men with advanced prostatic carcinoma to exogenous administration of testosterone. Cancer 20, 1871–1878 (1967). [DOI] [PubMed] [Google Scholar]
- 137.Khera M et al. Testosterone replacement therapy following radical prostatectomy. J. Sex. Med 6, 1165–1170 (2009). [DOI] [PubMed] [Google Scholar]
- 138.Pastuszak AW et al. Testosterone replacement therapy in patients with prostate cancer after radical prostatectomy. J. Urol 190, 639–644 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pastuszak AW et al. Testosterone replacement therapy in the setting of prostate cancer treated with radiation. Int. J. Impot. Res 25, 24–28 (2013). [DOI] [PubMed] [Google Scholar]
- 140.Ahlering TE et al. Testosterone replacement therapy reduces biochemical recurrence after radical prostatectomy. BJU Int. 126, 91–96 (2020). [DOI] [PubMed] [Google Scholar]
- 141.Morgentaler A et al. Testosterone therapy in men with untreated prostate cancer. J. Urol 185, 1256–1260 (2011). [DOI] [PubMed] [Google Scholar]
- 142.Cui Y, Zong H, Yan H & Zhang Y The effect of testosterone replacement therapy on prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 17, 132–143 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Morris MJ, Huang D & Kelly WK Phase 1 trial of high-dose exogenous testosterone in patients with castration-resistant metastatic prostate cancer. Eur. Urol 56, 237–244 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Szmulewitz R, Mohile S & Posadas E A randomized phase 1 study of testosterone replacement for patients with low-risk castration-resistant prostate cancer. Eur. Urol 56, 97–103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schweizer MT et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci. Transl. Med 7, 269ra2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Umekita Y, Hiipakka RA, Kokontis JM & Liao S Human prostate tumor growth in athymic mice: inhibition by androgens and stimulation by finasteride. Proc. Natl Acad. Sci. USA 93, 11802–11807 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schweizer MT et al. Bipolar androgen therapy for men with androgen ablation naive prostate cancer: results from the phase II BATMAN study. Prostate 76, 1218–1226 (2016). [DOI] [PubMed] [Google Scholar]
- 148.Markowski MC et al. A multicohort open-label phase II trial of bipolar androgen therapy in men with metastatic castration-resistant prostate cancer (RESTORE): a comparison of post-abiraterone versus post-enzalutamide cohorts. Eur. Urol 79, 692–699 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sena LA et al. Bipolar androgen therapy sensitizes castration-resistant prostate cancer to subsequent androgen receptor ablative therapy. Eur. J. Cancer 144, 302–309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Teply BA et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol. 19, 76–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Denmeade SR et al. TRANSFORMER: a randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castration-resistant metastatic prostate cancer. J. Clin. Oncol 39, 1371–1382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Markowski MC et al. COMBAT-CRPC: concurrent administration of bipolar androgen therapy (BAT) and nivolumab in men with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol 10.1200/JCO.2021.39.15_suppl.5014 (2021). [DOI] [Google Scholar]
- 153.Schweizer M et al. 592P Bipolar androgen therapy (BAT) plus olaparib in men with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol 32, S639–S640 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Manni A et al. Androgen depletion and repletion as a means of potentiating the effect of cytotoxic chemotherapy in advanced prostate cancer. J. Steroid Biochem 27, 551–556 (1987). [DOI] [PubMed] [Google Scholar]
- 155.Johnson D & Haynie T Phosphorus-32 for intractable pain in carcinoma of prostate: analysis of androgen priming, parathormone rebound, and combination therapy. Urology 9, 137–139 (1977). [DOI] [PubMed] [Google Scholar]
- 156.Schwartz LH et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 62, 132–137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Scher HI et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the Prostate Cancer Clinical Trials Working Group 3. J. Clin. Oncol 34, 1402–1418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sena LA et al. Prostate cancer androgen receptor activity dictates efficacy of bipolar androgen therapy through MYC. J. Clin. Invest 10.1172/JCI162396 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT02090114 (2022). [DOI] [PubMed]
- 160.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT03522064 (2021). [DOI] [PubMed]
- 161.Abida W, Cyrta J & Heller G Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl Acad. Sci. USA 116, 11428–11436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT04363164 (2022). [DOI] [PubMed]
- 163.Sena LA, Denmeade SR & Antonarakis ES Targeting the spectrum of immune checkpoints in prostate cancer. Expert. Rev. Clin. Pharmacol 14, 1253–1266 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hussain M, Mateo J & Fizazi K Survival with olaparib in metastatic castration-resistant prostate cancer. N. Engl. J. Med 383, 2345–2357 (2020). [DOI] [PubMed] [Google Scholar]
- 165.Nyquist MD et al. Selective androgen receptor modulators activate the canonical prostate cancer androgen receptor program and repress cancer growth. J. Clin. Invest 10.1172/JCI146777 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.D’Andrea AD Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair. 71, 172–176 (2018). [DOI] [PubMed] [Google Scholar]
- 167.Bouman A, Heineman MJ & Faas MM Sex hormones and the immune response in humans. Hum. Reprod. Update 11, 411–423 (2005). [DOI] [PubMed] [Google Scholar]
- 168.Isaacs JT Resolving the Coffey Paradox: what does the androgen receptor do in normal vs. malignant prostate epithelial cells? Am. J. Clin. Exp. Urol 6, 55–61 (2018). [PMC free article] [PubMed] [Google Scholar]
- 169.Notelovitz M Androgen effects on bone and muscle. Fertil. Steril 77 (Suppl. 4), S34–S41 (2002). [DOI] [PubMed] [Google Scholar]
- 170.Lu S, Tsai SY & Tsai M-J Regulation of androgen-dependent prostatic cancer cell growth: androgen regulation of CDK2, CDK4, and CKI p16 genes. Cancer Res. 57, 4511–4516 (1997). [PubMed] [Google Scholar]
- 171.Berger MF et al. The genomic complexity of primary human prostate cancer. Nature 470, 214–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chuang K-H et al. Neutropenia with impaired host defense against microbial infection in mice lacking androgen receptor. J. Exp. Med 206, 1181–1199 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tomlins SA et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 10, 177–IN179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tomlins SA et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur. Urol 56, 275–286 (2009). [DOI] [PubMed] [Google Scholar]
- 175.Heemers HV & Tindall DJ Unraveling the complexities of androgen receptor signaling in prostate cancer cells. Cancer Cell 15, 245–247 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sharma NL et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23, 35–47 (2013). [DOI] [PubMed] [Google Scholar]
- 177.Wang Q et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245–256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chuu CP et al. Androgen suppresses proliferation of castration‐resistant LNCaP 104‐R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer Sci. 102, 2022–2028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kokontis JM, Hay N & Liao S Progression of LNCaP prostate tumor cells during androgen deprivation: hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol 12, 941–953 (1998). [DOI] [PubMed] [Google Scholar]
- 180.Cornforth A, Davis J, Khanifar E, Nastiuk K & Krolewski J FOXO3a mediates the androgen-dependent regulation of FLIP and contributes to TRAIL-induced apoptosis of LNCaP cells. Oncogene 27, 4422–4433 (2008). [DOI] [PubMed] [Google Scholar]
- 181.Wang Y et al. Regulation of androgen receptor transcriptional activity by rapamycin in prostate cancer cell proliferation and survival. Oncogene 27, 7106–7117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liao X et al. Androgen stimulates matrix metalloproteinase-2 expression in human prostate cancer. Endocrinology 144, 1656–1663 (2003). [DOI] [PubMed] [Google Scholar]
- 183.Chuan Y-C et al. Androgen induction of prostate cancer cell invasion is mediated by ezrin. J. Biol. Chem 281, 29938–29948 (2006). [DOI] [PubMed] [Google Scholar]
- 184.Hara T, Miyazaki H, Lee A, Tran CP & Reiter RE Androgen receptor and invasion in prostate cancer. Cancer Res. 68, 1128–1135 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Teh M-T et al. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinoma. PLoS One 7, e34329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tsouko E et al. Regulation of the pentose phosphate pathway by an androgen receptor–mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 3, e103–e103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Choi SYC et al. The MCT4 gene: a novel, potential target for therapy of advanced prostate cancer. Clin. Cancer Res 22, 2721–2733 (2016). [DOI] [PubMed] [Google Scholar]
- 188.Koundouros N & Poulogiannis G Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Poulose N, Mills IG & Steele RE The impact of transcription on metabolism in prostate and breast cancers. Endocr. Relat. Cancer 25, R435–R452 (2018). [DOI] [PubMed] [Google Scholar]
- 190.Ono M et al. [14C] fluciclovine (alias anti-[14C] FACBC) uptake and ASCT2 expression in castration-resistant prostate cancer cells. Nucl. Med. Biol 42, 887–892 (2015). [DOI] [PubMed] [Google Scholar]
- 191.White MA et al. Glutamine transporters are targets of multiple oncogenic signaling pathways in prostate cancer. Mol. Cancer Res 15, 1017–1028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Corbin JM & Ruiz-Echevarría MJ One-carbon metabolism in prostate cancer: the role of androgen signaling. Int. J. Mol. Sci 17, 1208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shukla-Dave A et al. Ornithine decarboxylase is sufficient for prostate tumorigenesis via androgen receptor signaling. Am. J. Pathol 186, 3131–3145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Polkinghorn WR et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 3, 1245–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sandhu S et al. Poly (ADP-ribose) polymerase (PARP) inhibitors for the treatment of advanced germline BRCA2 mutant prostate cancer. Ann. Oncol 24, 1416–1418 (2013). [DOI] [PubMed] [Google Scholar]
- 196.Goodwin JF et al. A hormone–DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 3, 1254–1271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Guo Z et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10, 309–319 (2006). [DOI] [PubMed] [Google Scholar]
- 198.Liu Y et al. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene 29, 3208–3216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mellinghoff IK et al. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 6, 517–527 (2004). [DOI] [PubMed] [Google Scholar]
- 200.Seaton A et al. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis 29, 1148–1156 (2008). [DOI] [PubMed] [Google Scholar]
- 201.Fan W et al. Insulin-like growth factor 1/insulin signaling activates androgen signaling through direct interactions of Foxo1 with androgen receptor. J. Biol. Chem 282, 7329–7338 (2007). [DOI] [PubMed] [Google Scholar]
- 202.Migliaccio A et al. Steroid-induced androgen receptor–oestradiol receptor β–Src complex triggers prostate cancer cell proliferation. EMBO J. 19, 5406–5417 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Oliver VL, Poulios K, Ventura S & Haynes JM A novel androgen signalling pathway uses dihydrotestosterone, but not testosterone, to activate the EGF receptor signalling cascade in prostate stromal cells. Br. J. Pharmacol 170, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sun YH, Gao X, Tang YJ, Xu CL & Wang LH Androgens induce increases in intracellular calcium via a G protein-coupled receptor in LNCaP prostate cancer cells. J. Androl 27, 671–678 (2006). [DOI] [PubMed] [Google Scholar]
- 205.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT01084759 (2016). [DOI] [PubMed]
- 206.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT01750398 (2016). [DOI] [PubMed]
- 207.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT02286921 (2020). [DOI] [PubMed]
- 208.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT03554317 (2022). [DOI] [PubMed]
- 209.ClinicalTrials.gov. US National Library of Medicine. https://ClinicalTrials.gov/show/NCT03516812 (2022). [DOI] [PubMed]