

Abstract
Centromeres attach chromosomes to spindle microtubules during cell division and, despite this conserved role, show paradoxically rapid evolution and are typified by complex repeats. We used long-read sequencing to generate the Col-CEN Arabidopsis thaliana genome assembly that resolves all five centromeres. The centromeres consist of megabase-scale tandemly repeated satellite arrays, which support CENTROMERE SPECIFIC HISTONE H3 (CENH3) occupancy and are densely DNA methylated, with satellite variants private to each chromosome. CENH3 preferentially occupies satellites that show the least amount of divergence and occur in higher-order repeats. The centromeres are invaded by ATHILA retrotransposons, which disrupt genetic and epigenetic organization. Centromeric crossover recombination is suppressed, yet low levels of meiotic DNA double-strand breaks occur that are regulated by DNA methylation. We propose that Arabidopsis centromeres are evolving through cycles of satellite homogenization and retrotransposon-driven diversification.
INTRODUCTION:
The centromeres of eukaryotic chromosomes assemble the multiprotein kinetochore complex and thereby position attachment to the spindle microtubules, allowing chromosome segregation during cell division. The key function of the centromere is to load nucleosomes containing the CENTROMERE SPECIFIC HISTONE H3 (CENH3) histone variant [also known as centromere protein A (CENPA)], which directs kinetochore formation. Despite their conserved function during chromosome segregation, centromeres show radically diverse organization between species at the sequence level, ranging from single nucleosomes to megabase-scale satellite repeat arrays, which is termed the centromere paradox. Centromeric satellite repeats are variable in sequence composition and length when compared between species and show a high capacity for evolutionary change, both at the levels of primary sequence and array position along the chromosome. However, the genetic and epigenetic features that contribute to centromere function and evolution are incompletely understood, in part because of the challenges of centromere sequence assembly and functional genomics of highly repetitive sequences. New long-read DNA sequencing technologies can now resolve these complex repeat arrays, revealing insights into centromere architecture and chromatin organization.
RATIONALE:
Arabidopsis thaliana is a model plant species; its genome was first sequenced in 2000, yet the centromeres, telomeres, and ribosomal DNA repeats have remained unassembled, owing to their high repetition and similarity. Genomic repeats are difficult to assemble from fragmented sequencing reads, with longer, high-identity repeats being the most challenging to correctly assemble. As sequencing reads have become longer and more accurate, eukaryotic de novo genome assemblies have captured an increasingly complete picture of the repetitive component of the genome, including the centromeres. For example, Oxford Nanopore Technologies (ONT) reads have become longer and more accurate, now reaching >100 kilo–base pairs (kbp) in length with 95 to 99% modal accuracy. PacBio high-fidelity (HiFi) reads, although shorter (~15 kbp), are >99% accurate. Using ONT and HiFi reads, it is possible to bridge across interspersed unique marker sequences and accurately assemble centromere sequences. In this study, we used long-read DNA sequencing to generate a genome assembly of the A. thaliana accession Columbia (Col-CEN) that resolves all five centromeres. We use the Col-CEN assembly to derive insights into the chromatin and recombination landscapes within the Arabidopsis centromeres and how these regions evolve.
RESULTS:
The Col-CEN assembly reveals that the Arabidopsi centromeres consist of mega-base-scale tandemly repeated satellite arrays, which support high CENH3 (the centromere-specific histone variant that recruits kinetochores) occupancy and are densely DNA methylated. We show patterns of higher-order repetition within centromeres and that many satellite variants are private to each chromosome, which has implications for the recombination pathways acting in the centromeres. CENH3 preferentially occupies the satellites with the least amount of divergence and that show higher-order repetition. The Arabidopsis centromeres are mainly composed of satellite repeats that are ~178 bp in length, termed the CEN180 satellites. Arabidopsis centromeres have also been invaded by ATHILA long terminal repeat–class retrotransposons, which disrupt the genetic and epigenetic organization of the centromeres. Using chromatin immunoprecipitation sequencing (ChIP-seq) and immunofluorescence, we demonstrate that the centromeres show a hybrid chromatin state that is distinct from euchromatin and heterochromatin. We show that crossover recombination is suppressed within the centromeres, yet low levels of meiotic double-strand breaks occur, which are regulated by DNA methylation. Together, our Col-CEN assembly reveals the genetic and epigenetic landscapes within the Arabidopsis centromeres.
CONCLUSION:
Our Col-CEN assembly and functional genomics analysis have implications for understanding centromere sequence evolution in eukaryotes. We propose that a recombination-based homogenization process, occurring between allelic or nonallelic locations on the same chromosome, maintains the CEN180 library close to the consensus optimal for CENH3 recruitment. The advantage conferred to ATHILA retrotransposons by integration within the centromeres is presently unclear. They may be engaged in centromere drive, supporting the hypothesis that centromere satellite homogenization acts as a mechanism to purge driving elements. Each Arabidopsis centromere appears to represent different stages in cycles of satellite homogenization and ATHILA-driven diversification. These opposing forces provide both a capacity for homeostasis and a capacity for change during centromere evolution. In the future, assembly of centromeres from multiple Arabidopsis accessions and closely related species may further clarify how centromeres form and the evolutionary dynamics of CEN180 and ATHILA repeats.
Graphical Abstract
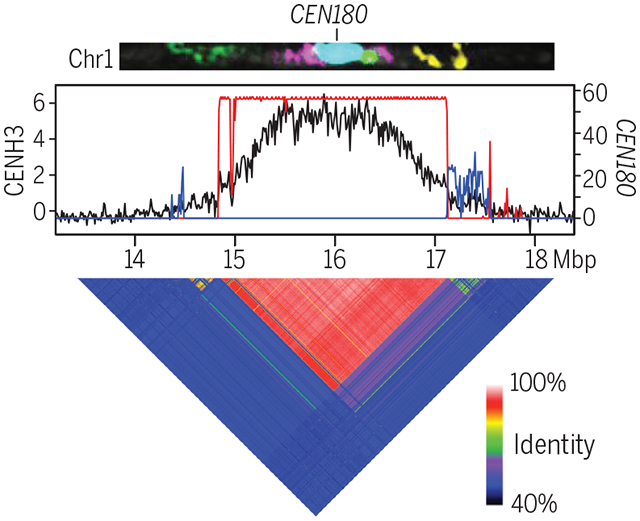
Assembly of the Arabidopsis centromeres. The structure of Arabidopsis centromere 1 is shown by fluorescence in situ hybridization (top) [upper-arm bacterial artificial chromosomes (BACs) (green), ATHILA (purple), CEN180 (blue), the telomeric repeat (green), and bottom-arm BACs (yellow)] and a long-read genome assembly (middle). The density of centromeric histone CENH3 binding measured by ChIP-seq is shown (black), alongside the frequency of CEN180 centromere satellite repeats. Red and blue represent forward- and reverse-strand satellites, respectively. The heatmap (bottom) shows patterns of sequence identity across the centromere between nonoverlapping 5-kbp windows. Chr, chromosome 1.
Despite their conserved function during chromosome segregation, centromeres show diverse organization between species, ranging from single nucleosomes to megabase-scale tandem repeat arrays (1). Centromere “satellite” repeat monomers are commonly ~100 to 200 base pairs (bp) long, with each repeat capable of hosting a CENTROMERE SPECIFIC HISTONE H3 (CENH3) [also known as centromere protein A (CENPA)] variant nucleosome (1, 2). CENH3 nucleosomes ultimately assemble the kinetochore and position spindle attachment on the chromosome, allowing segregation during cell division (3). Satellites are highly variable in sequence composition and length when compared between species (2). The library of centromere repeats present within a genome often shows concerted evolution, yet they have the capacity to change rapidly in structure and sequence within and between species (1, 2, 4). However, the genetic and epigenetic features that contribute to centromere evolution are incompletely understood, in large part because of the challenges of centromere sequence assembly and functional genomics of highly repetitive sequences.
Genomic repeats, especially long or high-similarity repeats, are notoriously difficult to assemble from fragmented sequencing reads (5). As sequencing reads have become longer and more accurate, eukaryotic de novo genome assemblies have captured an increasingly complete picture of repetitive elements. Oxford Nanopore Technologies (ONT) long reads have become substantially longer and more accurate (>100 kbp with 95 to 99% modal accuracy), owing to improved DNA extraction and library preparation, together with advanced machine learning–based base calling. Additionally, PacBio high-fidelity (HiFi) reads, although shorter (~15 kbp), are highly accurate (>99%). Using these technologies with new computational methods, researchers have assembled a complete telomere-to-telomere representation of a human genome, including the centromere satellite arrays (6–8). This work revealed that ONT and HiFi reads are sufficient to span interspersed unique marker sequences in human centromeres and other complex repeats, suggesting that truly complete genome assemblies for diverse eukaryotes are on the horizon.
Arabidopsis thaliana is a major model plant species; its genome was sequenced in 2000, yet the centromeres, telomeres, and ribosomal DNA repeats have remained unassembled, owing to their high repetition and similarity (9). The Arabidopsis centromeres contain millions of base pairs of the CEN180 satellite, which support CENH3 loading (10–14). We used long-read ONT sequencing, followed by polishing with high-accuracy PacBio HiFi reads, to establish the Col-CEN reference assembly, which wholly resolves all five Arabidopsis centromeres from the Columbia (Col-0) accession. The assembly contains a library of 66,131 CEN180 satellites, with each chromosome possessing mostly private satellite variants. Chromosome-specific higher-order CEN180 repetition is prevalent within the centromeres. We identified ATHILA retrotransposons that have invaded the satellite arrays and interrupt the genetic and epigenetic organization of the centromeres. By analyzing SPO11-1-oligonucleotide data from mutant lines, we demonstrate that DNA methylation epigenetically silences initiation of meiotic DNA double-strand breaks (DSBs) within the centromeres. Our data suggest that satellite homogenization and retrotransposon invasion are driving cycles of centromere evolution in Arabidopsis.
Complete assembly of the Arabidopsis centromeres
We collected Col-0 genomic ONT and HiFi sequencing data comprising a total of 73.6 Gbp (~56×, >50 kbp) and 14.6 Gbp (111.3×, 15.6 kbp mean read length), respectively. These data yielded an improved assembly of the Col-0 genome (Col-CEN v1.2), where chromosomes 1, 3, and 5 are wholly resolved from telomere to telomere, and chromosomes 2 and 4 are complete apart from the short-arm 45S ribosomal DNA (rDNA) clusters and adjacent telomeres (Fig. 1). After telomere patching and repeat-aware polishing with ONT, HiFi, and Illumina reads (15), the Col-CEN assembly has a quality value of 45.99 and 51.71 inside and outside of the centromeres, equivalent to approximately one error per 40,000 and 148,000 bases, respectively (figs. S1 and S2A and table S1). Additionally, Hi-C and Bionano optical maps validate the large-scale structural accuracy of the assembly (fig. S2). The Col-CEN assembly is highly concordant with TAIR10, showing no large structural differences within the chromosome arms (Fig. 1B). Of the Col-0 bacterial artificial chromosome (BAC) contigs, 97.5% align to both TAIR10 and Col-CEN with high coverage and identity (>95%), and 99.9% of TAIR10 gene annotations are represented in Col-CEN.
Fig. 1. Complete assembly of the Arabidopsis centromeres.
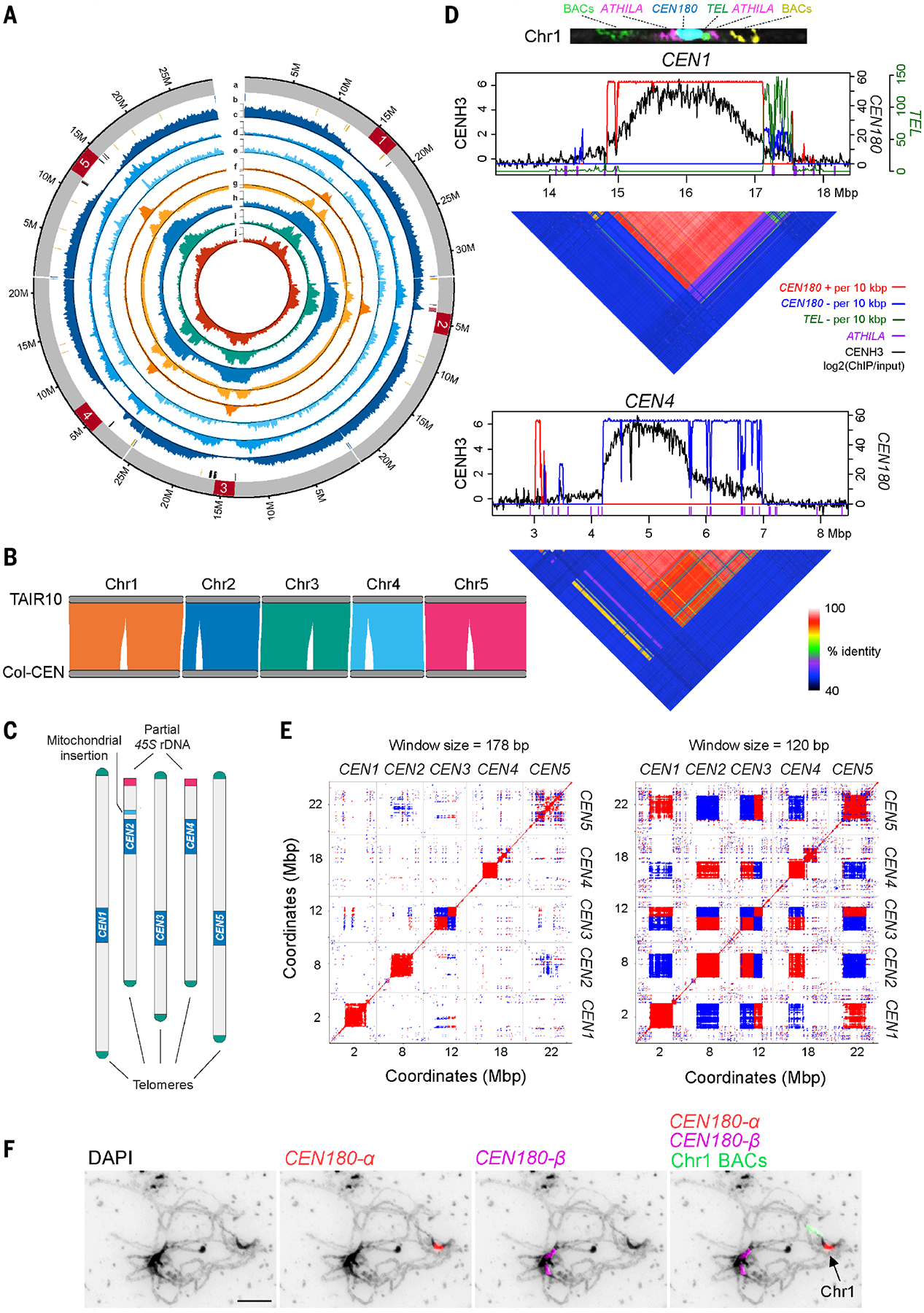
(A) Circos plot of the Col-CEN assembly. Quantitative tracks (labeled c to j) are aggregated in 100-kbp bins, and independent y-axis labels are given as (low value, mid value, high value, measurement unit) as follows: (a) chromosome with centromeres shown in red; (b) telomeres (blue), 45S rDNA (yellow), 5S rDNA (black), and the mitochondrial insertion (pink); (c) genes (0, 25, 51, gene number); (d) transposable elements (0, 84, 167, transposable element number); (e) Col×Ler F2 crossovers (0, 7, 14, crossover number); (f) CENH3 [−0.5, 0, 3, log2(ChIP/input)]; (g) H3K9me2 [−0.6, 0, 2, log2(ChIP/input)]; (h) CG methylation (0, 47, 95, %); (i) CHG methylation (0, 28, 56, %); and (j) CHH methylation (0, 7, 13, %). (B) Syntenic alignments between the TAIR10 and Col-CEN assemblies. (C) Col-CEN ideogram with annotated chromosome landmarks (not drawn to scale). (D) CENH3 log2(ChIP/input) (black) plotted over centromeres 1 and 4 (10). CEN180 per 10-kbp plotted for forward (red) or reverse (blue) strand orientations. ATHILA are indicated by purple x-axis ticks. Heatmaps show pairwise sequence identity between all nonoverlapping 5-kbp regions. A FISH-stained chromosome 1 at pachytene is shown at the top, probed with upper-arm BACs (green), ATHILA (purple), CEN180 (blue), the telomeric repeat (green), and bottom-arm BACs (yellow). (E) Dot plots comparing the five centromeres using a search window of 120 or 178 bp. Red and blue indicate forward- and reverse-strand similarity, respectively. (F) Pachytene-stage chromosomes stained with 4′,6-diamidino-2-phenylindole (DAPI) (black) and CEN180-α (red), CEN180-β (purple), and chromosome 1 BAC (green) FISH probes. The scale bar represents 10 μM.
Col-CEN reconstructs all five centromeres spanning 12.6 Mbp of new sequence, 120.0 and 97.6 kbp of 45S rDNA in the chromosome 2 and 4 nucleolar organizer regions (NORs), and the complete telomeres of the eight chromosome arms without subtelomeric NORs (Fig. 1, A to C; and figs. S1 to S3). We found several instances of apparently genuine variation between the Col-0 strains used to generate TAIR10 and Col-CEN (fig. S4 and tables S2 and S3). For example, a thionin gene cluster shows a deletion in Col-CEN relative to TAIR10 (fig. S4). In total, 27 TAIR10 genes are missing from Col-CEN owing to presence or absence variation, and 13 are present in multiple copies (tables S2 and S3). To comprehensively account for variation between Col-0 strains, we aligned ONT, HiFi, and Illumina reads to the Col-CEN assembly and called variants, providing a database of potential allelic differences, including heterozygous variants (https://github.com/schatzlab/Col-CEN). This revealed only 41 and 37 structural variant calls from ONT and HiFi data genome-wide, respectively, consistent with very low heterozygosity.
We confirmed chromosome landmarks flanking centromere 1 using fluorescence in situ hybridization (FISH), which included labeling a telomeric-repeat cluster located adjacent to the centromere (Fig. 1D and fig. S5). To validate centromere structure, we performed in silico digestion with AscI and NotI and compared the predicted fragments with published physical maps, which validated Col-CEN (fig. S6) (16). We also examined our Bionano optical data across the centromeres (fig. S7). The optical contigs are consistent with the structure of Col-CEN CEN180 arrays, although the low density of centromeric labeling sites prevents full resolution by optical fragments alone (fig. S7).
The centromeres are characterized by a 178-bp satellite repeat (CEN180), arranged head to tail and organized into higher-order repeats (Figs. 1D and 2 and fig. S8). We validated the structural and base-level accuracy of the centromeres using techniques from the human Telomere-to-Telomere (T2T) Consortium (6, 8) and observed even long-read coverage across the centromeres with few loci showing plausible alternate base signals (fig. S1B). We observed relatively few missing k-mers that are found in the assembly but not in Illumina short reads, which are diagnostic of residual consensus errors that remain after polishing (fig. S1B) (17). We observed that unique marker sequences are frequent, with a maximum distance between consecutive markers of 41,765 bp within the centromeres, suggesting that our reads can confidently span these markers and assemble reliably (fig. S1C). The five centromeres are relatively distinct at the sequence level, with each exhibiting chromosome-specific repeats (Figs. 1E and 2 and tables S4 and S5). Using the Col-CEN sequence, we designed CEN180 variant FISH probes to label specific centromere arrays (Fig. 1F and fig. S5). For example, the CEN180-α, CEN180-γ, and CEN180-δ probes specifically label arrays within centromere 1 (Fig. 1F and fig. S5), providing cytogenetic validation for chromosome-specific satellites.
Fig. 2. The Arabidopsis CEN180 satellite repeat library.
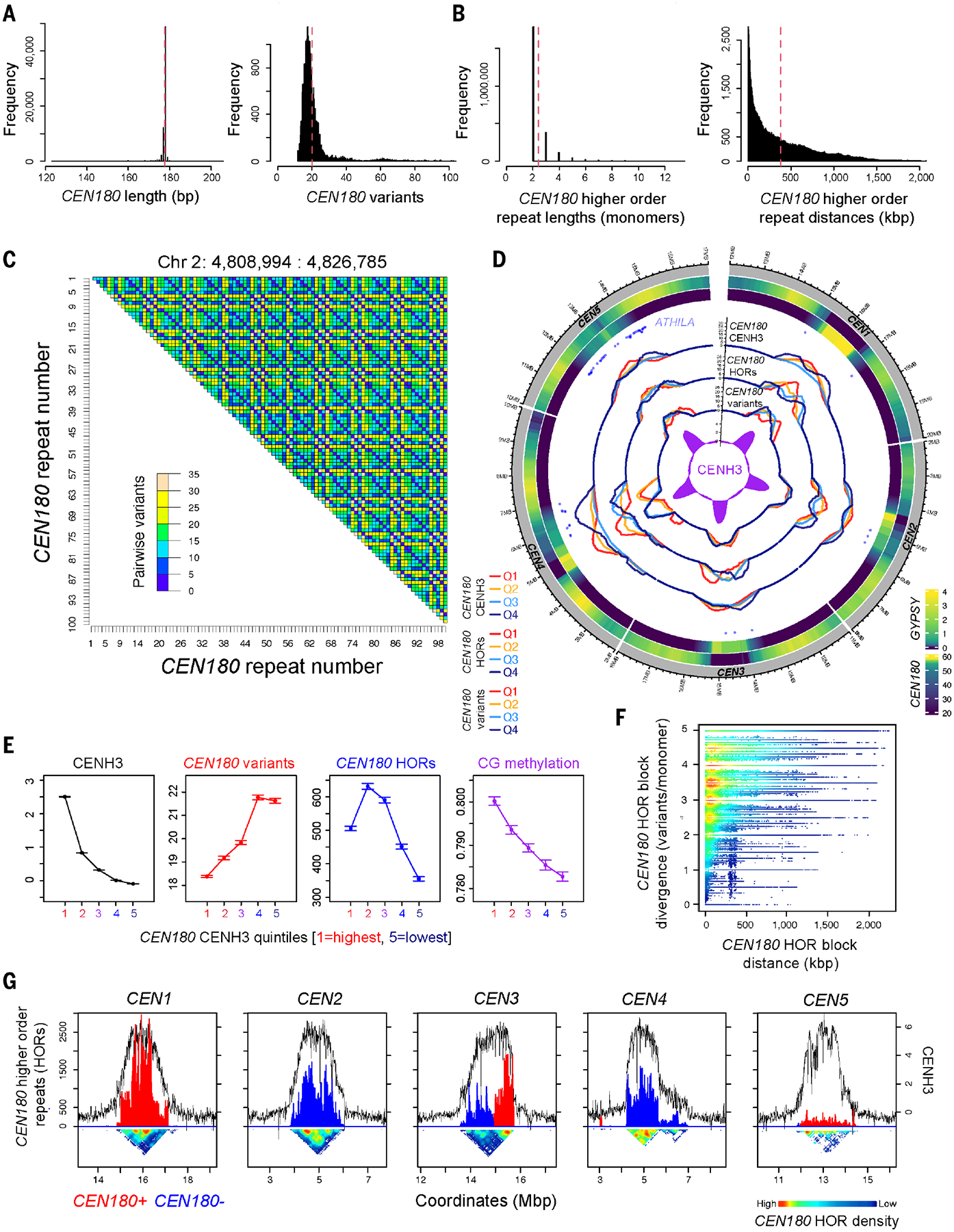
(A) Histograms of CEN180 monomer lengths (bp), and variant distances relative to the genome-wide consensus. Red dashed lines indicate mean values. (B) Same as for (A) but showing widths of CEN180 higher-order repeat blocks (monomers) and the distance between higher-order repeats (kbp). (C) Heatmap of a representative region within centromere 2, shaded according to pairwise variants between CEN180. (D) Circos plot showing (i) GYPSY density; (ii) CEN180 density; (iii) centromeric ATHILA “rainfall”; (iv) CEN180 density grouped by decreasing CENH3 log2(ChIP/input) (red, high; navy, low); (v) CEN180 density grouped by decreasing higher-order repetition (red, high; navy, low); (vi) CEN180 grouped by decreasing variant distance (red, high; navy, low); and (vii) CENH3 log2(ChIP/input) (purple) across the centromeres. (E) CEN180 were divided into quintiles according to CENH3 log2(ChIP/input) and mean values with 95% confidence intervals plotted. The same groups were analyzed for CEN180 variant distance (red), higher-order repetition (blue), and CG-context DNA methylation (purple). (F) Plot of the distance between pairs of higher-order repeats (kbp) and divergence (variants per monomer) between the higher-order repeats. (G) Plots of CENH3 log2(ChIP/input) (black) across the centromeres compared with CEN180 higher-order repetition on forward (red) or reverse (blue) strands. The heatmap beneath is shaded according to higher-order repeat density.
The Arabidopsis CEN180 satellite repeat library
We performed de novo searches for tandem repeats to define the centromere satellite library (table S4). We identified 66,131 CEN180 satellites in total, with between 11,848 and 15,613 copies per chromosome (Fig. 2, fig. S9, and table S4). The CEN180 repeats form large tandem arrays, with the satellites within each centromere found predominantly on the same strand, except for centromere 3, which is formed of two blocks on opposite strands (Fig. 1D and fig. S8). The distribution of repeat monomer length is constrained around 178 bp (Fig. 2A and fig. S9). We aligned all CEN180 sequences to derive a genome-wide consensus and calculated nucleotide frequencies at each alignment position to generate a position probability matrix (PPM). Each satellite was compared with the PPM to calculate a “variant distance” by summation of disagreeing nucleotide probabilities. Substantial sequence variation was observed between satellites and the PPM, with a mean variant distance of 20.2 (Fig. 2A). Each centromere contains essentially private libraries of CEN180 monomers, with only 0.3% sharing an identical copy on a different chromosome (Fig. 1E and table S4). By contrast, there is a high degree of CEN180 repetition within chromosomes, with 57.1 to 69.0% showing one or more duplicates (table S4). We also observed a minor class of CEN160 repeats found on chromosome 1 (1289 repeats, mean length of 158.2 bp) (14).
We aligned CENH3 chromatin immunoprecipitation sequencing (ChIP-seq) data to the Col-CEN assembly and observed, on average, 12.9-fold log2(ChIP/input) enrichment within the CEN180 arrays, compared with the chromosome arms (Fig. 1D and fig. S8) (10). CENH3 ChIP-seq enrichment is generally highest within the interior of the main CEN180 arrays (Fig. 1D and fig. S8). We observed a negative relationship between CENH3 ChIP-seq enrichment and CEN180 variant distance (Fig. 2, D and E), consistent with the idea that CENH3 nucleosomes prefer to occupy satellites that are closer to the genome-wide consensus. In this respect, centromere 4 is noteworthy because it consists of two distinct CEN180 arrays, with the right array showing higher variant distances and lower CENH3 enrichment (Figs. 1D and 2D and fig. S8). Together, these data are consistent with the possibility that satellite divergence leads to loss of CENH3 binding, or vice versa.
To define CEN180 higher-order repeats, monomers were considered the same if they shared five or fewer pairwise variants. Consecutive repeats of at least two monomers below this variant threshold were identified, yielding 2,408,653 higher-order repeats (Fig. 2D and table S5). Like the CEN180 monomer sequences, higher-order repeats are largely chromosome specific (table S5). The mean number of CEN180 monomers per higher-order repeat was 2.41 (equivalent to 429 bp) (Fig. 2B and table S5), and 95.4% of CEN180 were monomers of at least one larger repeat unit. Higher-order repeat block sizes show a negative exponential distribution, and the largest block was formed of 60 monomers (equivalent to 10,689 bp) (Fig. 2B). Many higher-order repeats are in close proximity (26% are <100 kbp apart), although they are dispersed throughout the length of the centromeres. For example, the average distance between higher-order repeats was 380 kbp and the maximum was 2365 kbp (Fig. 2B and table S5). We also observed that higher-order repeats further apart showed a higher level of variants between the blocks (variants per monomer) (Fig. 2F), consistent with the idea that satellite homogenization is more effective over repeats that are physically closer. Genome-wide, the CEN180 quantile with highest CENH3 occupancy correlates with higher-order repetition and increased CG DNA methylation (Fig. 2, D, E, and G). However, an exception to these trends is centromere 5, which has 6.8 to 13.4% of higher-order repeats compared with the other centromeres yet recruits comparable CENH3 (Fig. 2G and table S5).
Invasion of the Arabidopsis centromeres by ATHILA retrotransposons
In addition to reduced CEN180 higher-order repetition, centromere 5 is also disrupted by breaks in the satellite array (Fig. 2G and fig. S8). Most of the main satellite arrays are CEN180 (92.8%), with only 111 interspersed sequences >1 kbp. Within these breaks, we identified 53 intact and 20 fragmented ATHILA long terminal repeat (LTR) retrotransposons of the GYPSY superfamily (Fig. 3, A to C, and table S6) (18). The intact ATHILA have a mean length of 11.05 kbp, and most have similar and paired LTRs, target site duplications, primer binding sites, polypurine tracts, and GYPSY open reading frames (Fig. 3C and table S6). LTR comparisons indicate that the centromeric ATHILA are young, with, on average, 98.7% LTR sequence identity, which was significantly higher than that for ATHILA located outside the centromeres (96.9%, n = 58, Wilcox test, P = 4.89 × 10−8) (Fig. 3D and fig. S10). We also identified 12 ATHILA solo LTRs, consistent with postintegration intra-element homologous recombination (table S6). We observed six instances where centromeric ATHILA loci were duplicated on the same chromosome and located between 8.9 and 538.5 kbp apart, consistent with the idea that transposons are copied postintegration, potentially by the same mechanism that generates CEN180 higher-order repeats. For example, a pair of adjacent ATHILA5 and ATHILA6A elements within centromere 5 has been duplicated within a higher-order repeat (fig. S11). The duplicated elements share target site duplications and flanking sequences and show high identity between copies (99.5 and 99.6%) (fig. S11 and table S6). By contrast, the surrounding CEN180 show higher divergence and copy number variation between the higher-order repeats (94.3 to 97.3% identity) (fig. S11). This indicates an increased rate of CEN180 sequence change compared with that of the ATHILA, after duplication.
Fig. 3. Invasion of the Arabidopsis centromeres by ATHILA retrotransposons.
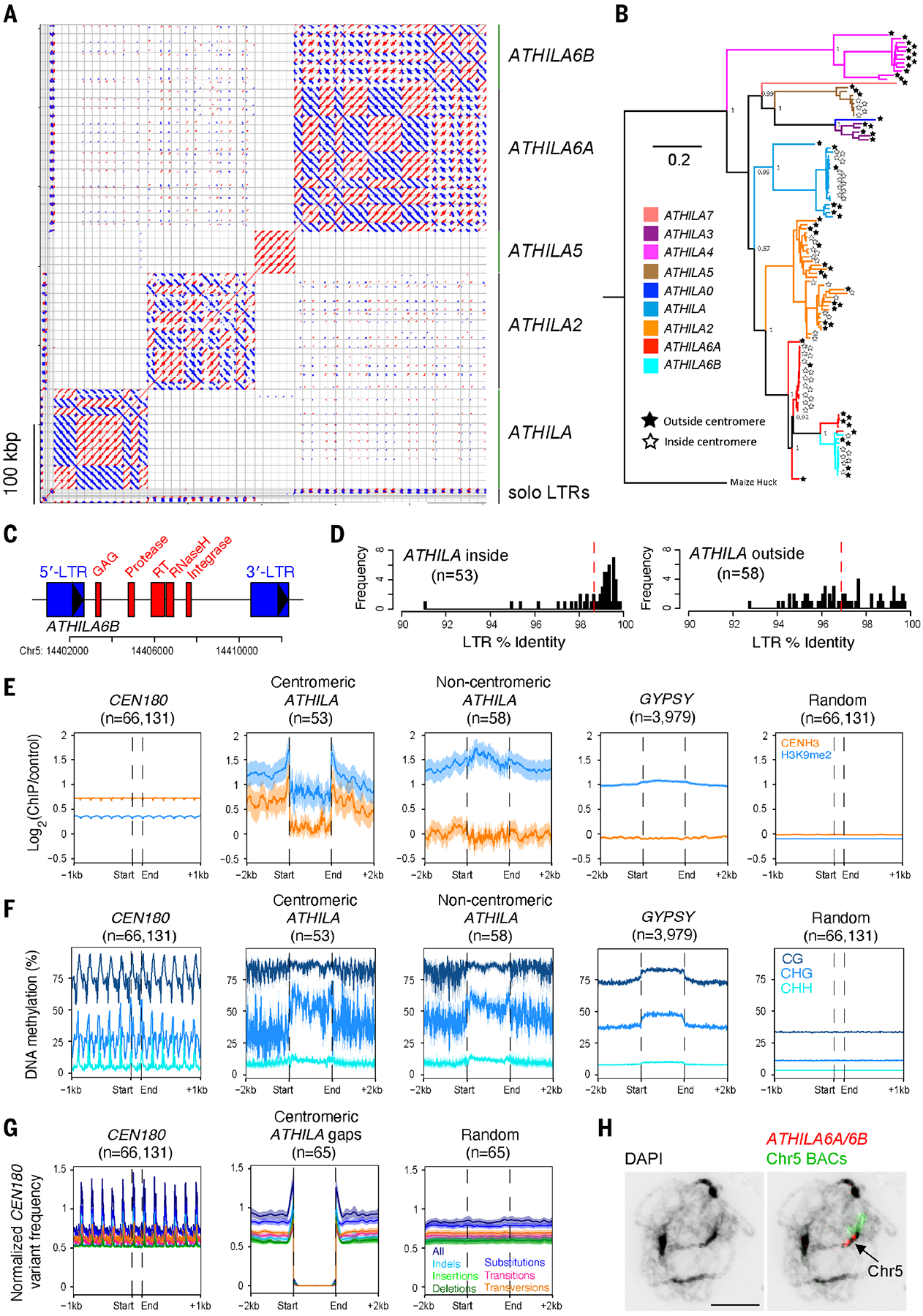
(A) Dot plot of centromeric ATHILA using a 50-bp search window. Red and blue indicate forward- and reverse-strand similarity, respectively. ATHILA subfamilies and solo LTRs are indicated. (B) Maximum likelihood phylogenetic tree of 111 intact ATHILA elements, color coded according to subfamily. Stars at the branch tips indicate ATHILA inside (white) or outside (black) the centromeres. (C) An annotated map of an ATHILA6B with LTRs (blue) and core protein domains (red) highlighted. (D) Histograms of LTR sequence identity for centromeric ATHILA elements (n = 53) compared with ATHILA outside of the centromeres (n = 58). Red dashed lines indicate mean values. (E) Metaprofiles of CENH3 (orange) and H3K9me2 (blue) ChIP-seq signals around CEN180 (n = 66,131), centromeric intact ATHILA (n = 53), ATHILA located outside the centromeres (n = 58), GYPSY retrotransposons (n = 3979), and random positions (n = 66,131). Shaded ribbons represent 95% confidence intervals for windowed mean values. (F) Same as for (E) but analyzing ONT-derived percentage of DNA methylation in CG (dark blue), CHG (blue), and CHH (light blue) contexts. (G) Meta-profiles of CEN180 sequence edits (insertions, deletions, and substitutions relative to the CEN180 consensus), normalized by CEN180 presence, in positions surrounding CEN180 gaps containing ATHILA (n = 65) or random positions (n = 65). All edits (dark blue), substitutions (blue), indels (light blue), insertions (light green), deletions (dark green), transitions (pink), and transversions (orange) are shown. Shaded ribbons represent 95% confidence intervals for windowed mean values. (H) Pachytene-stage chromosome spread stained with DAPI (black), an ATHILA6A/6B GAG FISH probe (red), and chromosome 5–specific BACs (green). The scale bar represents 10 μM.
We analyzed centromeric ATHILA for CENH3 ChIP-seq enrichment and observed a decrease relative to the surrounding CEN180, yet higher levels than in ATHILA located outside of the centromere (Fig. 3E). The ATHILA show greater histone H3 lysine 9 dimethylation (H3K9me2) enrichment compared with all CEN180 (Fig. 3E). We used our ONT reads to profile DNA methylation over the ATHILA and observed dense methylation, with higher CHG-context methylation (where H is A, T, or C) than the surrounding CEN180 (Fig. 3F). Hence, ATHILA elements are distinct from the CEN180 satellites at the chromatin level. We profiled CEN180 variants around centromeric ATHILA loci (n = 65) and observed increased satellite divergence in the flanking regions (Fig. 3G), reminiscent of Nasonia PSR tandem repeat divergence at the junction with a NATE retrotransposon (19). This indicates that ATHILA insertion was mutagenic on the surrounding satellites or that transposon insertion influenced the subsequent divergence or homogenization of the adjacent CEN180. We also used FISH to cytogenetically validate the presence of ATHILA6A/6B and ATHILA2 subfamilies within the centromeres (Fig. 3H and fig. S5). Together, these data show that ATHILA insertions interrupt the genetic and epigenetic organization of the Arabidopsis CEN180 arrays.
Epigenetic organization and meiotic recombination within the centromeres
To assess genetic and epigenetic features of the centromeres, we analyzed all of the chromosome arms along their telomere-centromere axes using a proportional scale (Fig. 4A). Centromere midpoints were defined as the point of maximum CENH3 ChIP-seq enrichment (fig. S12). As expected, CEN180 satellites are highly enriched in proximity to centromeres, and these regions are relatively GC-rich compared with the AT-rich chromosome arms (Fig. 4A). Gene density drops as the centromeres are approached, whereas transposon density increases, until they are replaced by CEN180 (Fig. 4A). Gene and transposon densities are tracked closely by H3K4me3 and H3K9me2 ChIP-seq enrichment, respectively (Fig. 4A). H3K9me2 enrichment is observed within the centromere, although there is a reduction in the center coincident with CENH3 enrichment (Fig. 4A), consistent with reduced H3 occupancy caused by CENH3 replacement. A slight increase in H3K4me3 enrichment is observed within the centromeres, relative to the flanking pericentromeres (Fig. 4A).
Fig. 4. Epigenetic organization and meiotic recombination within the centromeres.
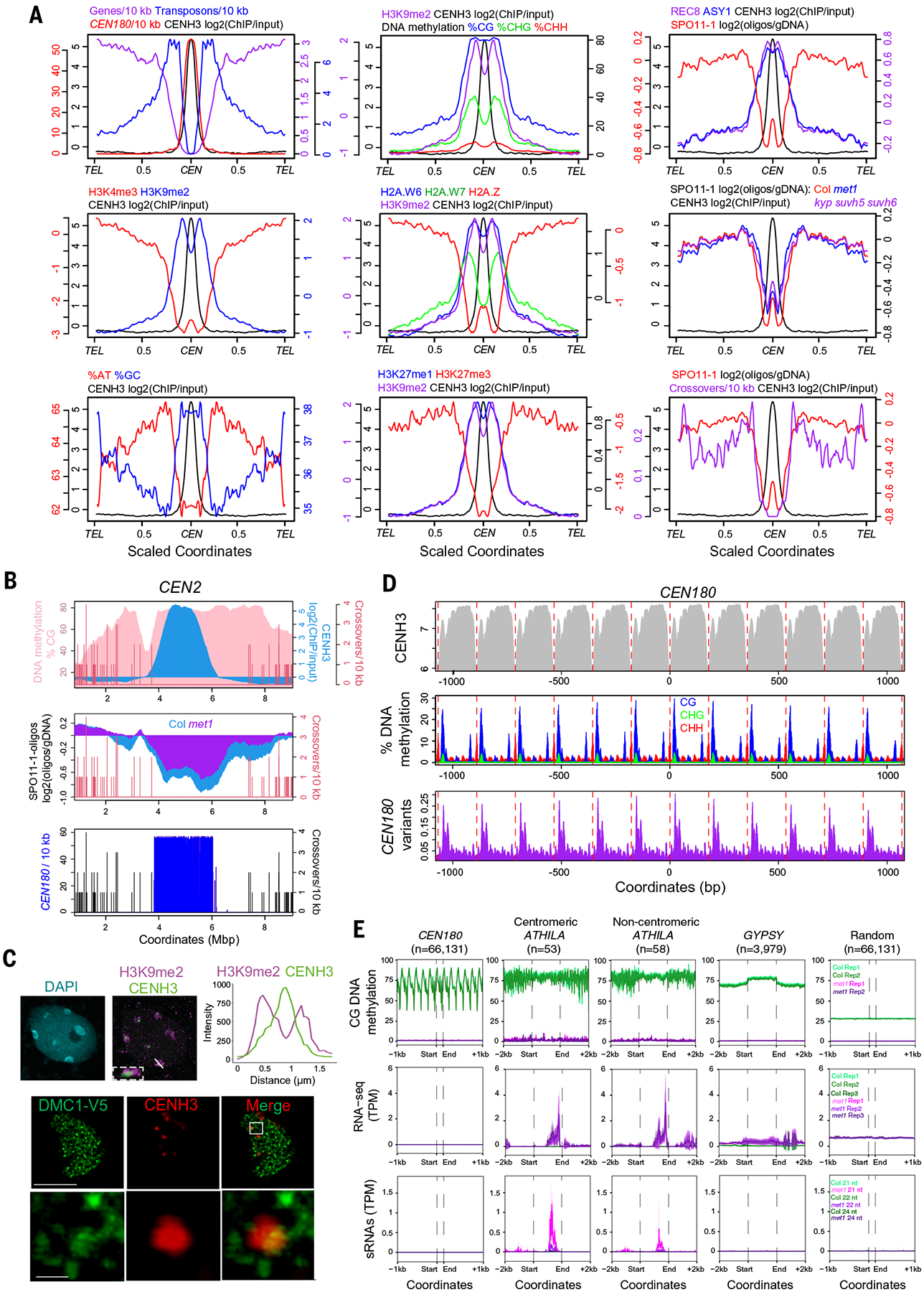
(A) Quantification of genomic features plotted along chromosome arms that were proportionally scaled between telomeres (TEL) and centromere midpoints (CEN) [defined by maximum CENH3 ChIP-seq log2(ChIP/input) enrichment]. Data analyzed were gene, transposon, and CEN180 density; CENH3, H3K4me3, H3K9me2, H2A.W6, H2A.W7, H2A.Z, H3K27me1, H3K27me3, REC8, and ASY1 log2 (ChIP/input); and percentage of AT/GC base composition, DNA methylation, SPO11-1-oligonucleotides (in wild type and met1), and crossovers (table S7). (B) Plot quantifying crossovers (red), percentage of CG DNA methylation (pink), CENH3 (blue), SPO11-1-oligonucleotides in wild type and met1, and CEN180 density along centromere 2. (C) An interphase nucleus immunostained for H3K9me2 (magenta) and CENH3-GFP (green) is shown at the top. The white line indicates the confocal section used for the intensity plot shown on the right; the region outlined by the white dashed line shows a magnified image of a centromere. The scale bar represents 5 μM. At the bottom is a male meiocyte (early prophase I) immunostained for CENH3 (red) and V5-DMC1 (green). The region outlined by the white line indicates the magnified region shown in the lower row of images. Scale bars are 10 μM (upper) and 1 μM (lower). (D) Plots of CENH3 ChIP enrichment (gray), DNA methylation in CG (blue), CHG (green) and CHH (red) contexts, and CEN180 variants (purple), averaged over windows centered on CEN180 starts. The red dashed lines show 178-bp increments. (E) Metaprofiles of CG-context DNA methylation, RNA-seq, and siRNA-seq in wild type (green) or met1 (pink and purple) (29) around CEN180 (n = 66,131), centromeric intact ATHILA (n = 53), ATHILA located outside the centromeres (n = 58), GYPSY (n = 3979), and random positions (n = 66,131). Shaded ribbons represent 95% confidence intervals for windowed mean values.
Using our ONT reads with the DeepSignal-plant algorithm (20), we observed dense DNA methylation across the centromeres in CG, CHG, and CHH contexts (Fig. 4, A and B). However, CHG DNA methylation shows relatively reduced centromeric frequency compared with CG methylation (Fig. 4A). This may reflect centromeric depletion of H3K9me2 (Fig. 4A), a histone modification that maintains DNA methylation in non-CG contexts (21). To further investigate the DNA methylation environment associated with CENH3 deposition, we performed ChIP using either H3K9me2 or CENH3 antibodies and sequenced the immunopurified DNA with ONT. We analyzed methylation frequency in reads that aligned to the centromeres and observed dense CG methylation in both read sets but depletion of CHG and CHH methylation in the CENH3 reads relative to H3K9me2 (fig. S13). This further supports that H3 replacement by CENH3 causes a decrease in non-CG methylation maintenance within the Arabidopsis centromeres.
To investigate genetic control of centromeric DNA methylation, we analyzed bisulfite sequencing (BS-seq) data from wild type and eight mutants defective in CG and non-CG DNA methylation maintenance (fig. S14) (21, 22). Centromeric non-CG methylation is eliminated in drm1 drm2 cmt2 cmt3 mutants and reduced in kyp suvh5 suvh6 mutants, whereas CG methylation is intact in these backgrounds (fig. S14) (21, 22). By contrast, both CG and non-CG methylation in the centromeres are reduced in ddm1 and met1 mutants (fig. S14) (22). Hence, centromeric CG-context methylation is relatively high compared with non-CG, and non-CG methylation shows an unexpected dependence on CG maintenance pathways.
We observed pericentromeric ChIP-seq enrichment of the heterochromatic marks H2A.W6, H2A.W7, and H3K27me1, which are relatively depleted within the centromeres (Fig. 4A) (23, 24). The polycomb-group modification H3K27me3 is low in the centromeres and found largely in the chromosome arms (Fig. 4A). Enrichment of the euchromatic histone variant H2A.Z is low in the centromeres, but, like H3K4me3, shows a slight increase in the centromeres relative to the pericentromeres (Fig. 4A), suggesting that the centromeres have a distinct chromatin state relative to neighboring heterochromatin. We performed immunofluorescent staining of Arabidopsis nuclei for CENH3-GFP (GFP, green fluorescent protein) and euchromatic and heterochromatic histone modifications (Fig. 4C and figs. S15 and S16). Quantification of fluorescence intensity confirmed that heterochromatic marks are relatively depleted where CENH3-GFP is enriched (Fig. 4C and fig. S16). Hence, the Arabidopsis centromeres show depletion of heterochromatic and enrichment of euchromatic marks relative to the pericentromeres, consistent with a hybrid chromatin state.
Meiotic recombination, including unequal crossover and gene conversion, has been proposed to mediate centromere evolution (4, 25). We mapped 2080 meiotic crossovers from Col×Ler F2 sequencing data against the Col-CEN assembly (resolved, on average, to 1047 kbp) (fig. S17). As expected, crossovers were suppressed in proximity to the centromeres (Fig. 4, A and B, and fig. S17). We observed high centromeric ChIP-seq enrichment of REC8-cohesin and ASY1, which are components of the meiotic chromosome axis (Fig. 4A) (26, 27). To investigate the potential for meiotic DSB formation within the centromeres, we aligned SPO11-1-oligonucleotides from wild type (28). Overall, SPO11-1-oligonucleotides are low within the centromeres, although we observed an increase relative to the pericentromeres, reminiscent of H3K4me3 and H2A.Z ChIP-seq enrichment (Fig. 4A). To investigate the role of DNA methylation, we mapped SPO11-1-oligonucleotides from the CG DNA methylation mutant met1-3 (28), which showed a gain of DSBs within the centromeres (Fig. 4, A and B). We immunostained meiocytes in early prophase I for CENH3 and V5-DMC1, which is a marker of meiotic interhomolog recombination (Fig. 4C and figs. S18 and S19). DMC1-V5 foci were observed along the chromosomes and adjacent to the surface of CENH3 foci, but not within them (Fig. 4C). Hence, despite suppression of crossovers, we observe evidence for low levels of meiotic recombination initiation within the centromeres, which is influenced by DNA methylation.
CENH3 nucleosomes show a phased pattern of enrichment with the CEN180, with relative depletion in spacer regions at the satellite edges (Fig. 4D). CENH3 spacer regions also associate with increased DNA methylation and CEN180 variants (Fig. 4D), consistent with the possibility that CENH3-nucleosomes influence epigenetic modification and satellite divergence. We analyzed chromatin and transcription around CEN180 and ATHILA at the fine scale and compared wild type with the DNA methylation mutant met1-3. In met1-3, CG-context DNA methylation is lost in both ATHILA and CEN180 repeats (Fig. 4E and fig. S20) (29). However, met1 RNA sequencing (RNA-seq) and small interfering RNA sequencing (siRNA-seq) signals show increased expression of ATHILA transcripts, but not CEN180 (Fig. 4E and fig. S20) (29). The greatest RNA and siRNA expression increases in met1-3 are observed in the ATHILA internal 3′ regions (Fig. 4E and fig. S20), which correspond to transcriptionally silent information (TSI) transcripts and epigenetically activated siRNA (easiRNA) populations (30, 31). This further indicates that epigenetic regulation of the CEN180 repeats is distinct from that of the ATHILA elements.
Discussion
Leveraging advances in sequencing technology and genome assembly, we have generated the Col-CEN reference genome, which resolves the centromere satellite arrays. By profiling chromatin and recombination within the centromeres, we demonstrate that Col-CEN enables biological insights from existing functional genomics data. Using ONT long reads, we have also resolved patterns of DNA methylation within the centromeres, highlighting the potential of complete reference assemblies for understanding epigenetic regulation of repeats. The Col-0 centromeres contain interspersed unique sequences that facilitate assembly with modern sequencing reads. However, similar to the human T2T Consortium, the Col-CEN assembly required extensive manual processes to polish and curate repetitive loci (8, 15, 32). We anticipate that as complete genome assembly becomes more automated, researchers will be able to compare centromere sequences across populations and species, ultimately revealing how centromere diversity and evolution affect genome function.
In the centromeres, extensive variation is observed among the CEN180, and most monomer sequences are private to each centromere. This is consistent with the model that satellite homogenization occurs primarily within chromosomes. The negative correlation between CEN180 divergence and CENH3 occupancy suggests that centromeric chromatin may promote recombination pathways that lead to homogenization, including DSB formation and repair through homologous recombination. For example, interhomolog strand invasion and noncrossover repair during meiosis, using allelic or nonallelic templates, have the potential to cause CEN180 gene conversion and structural change (fig. S21). Similarly, repair and recombination using a sister chromatid may also contribute to CEN180 change, which could occur during mitosis or meiosis (fig. S21). We note that CEN180 higher-order repeats are, on average, 432 bp long, which is within the size range of Arabidopsis gene conversions (33), although we also observe large (10 to 100 kbp) intra-centromere duplications, for which the origin is less clear. We observe a proximity effect on divergence between CEN180 higher-order repeats, with repeat blocks that are further apart showing greater differences. These patterns are reminiscent of human centromeric higher-order repeats, although duplicated blocks of α-satellites are longer and occur over greater physical distances (6, 34, 35). Because meiotic crossover repair is suppressed within the centromeres, consistent with patterns across eukaryotes (25, 36), we do not consider unequal crossover to be a major pathway driving Arabidopsis centromere evolution. However, we propose that a recombination-based homogenization process, occurring between allelic or nonallelic locations on the same chromosome, maintains the CEN180 library close to the consensus that is optimal for CENH3 recruitment (fig. S21).
Aside from homogenizing recombination within the CEN180, the centromeres have experienced invasion by ATHILA retrotransposons. The ability of ATHILA to insert within the centromeres is likely determined by their integrase protein. The Tal1 COPIA element from Arabidopsis lyrata also shows an insertion bias into CEN180 when expressed in A. thaliana (37), despite satellite sequences varying between these species (38), indicating that epigenetic information may be important for targeting. Most of the centromeric ATHILA elements appear young, based on high LTR identity, and possess many features required for transposition, although the centromeres show differences in the frequency of ATHILA insertions, with centromeres 4 and 5 being the most invaded. Compared with CEN180, centromeric ATHILA have distinct chromatin profiles and are associated with increased satellite divergence in adjacent regions. Therefore, ATHILA elements represent a potentially disruptive influence on the genetic and epigenetic organization of the centromeres. However, transposons are widespread in the centromeres of diverse eukaryotes and can directly contribute to repeat evolution (e.g., mammalian CENP-B is derived from a Pogo DNA transposase) (39). Therefore, ATHILA elements may also beneficially contribute to centromere integrity and stability in Arabidopsis.
The advantage conferred to ATHILA by integration within the centromeres is presently unclear, although we speculate that they may be engaged in centromere drive (40). Haig-Grafen scrambling through recombination has been proposed as a defense against drive elements within the centromeres (41). For example, maize meiotic gene conversion can eliminate centromeric CRM2 retrotransposons (25). Therefore, centromere satellite homogenization may serve as a mechanism to purge ATHILA, although in some cases this results in transposon duplication (fig. S22). The presence of ATHILA solo LTRs is also consistent with homologous recombination acting on the retrotransposons after integration (fig. S22). Centromere 5 and the diverged CEN180 array in centromere 4 show both high ATHILA density and reduced CEN180 higher-order repetition. This indicates that ATHILA may inhibit CEN180 homogenization or that loss of homogenization facilitates ATHILA insertion. We propose that each Arabidopsis centromere represents a different stage in cycles of satellite homogenization and ATHILA-driven diversification. These opposing forces provide a dual capacity for homeostasis and change during centromere evolution. Assembly of centromeres from multiple Arabidopsis accessions, and closely related species, has the potential to reveal new insights into centromere formation and the evolutionary dynamics of CEN180 and ATHILA repeats.
Methods summary
Genomic DNA was extracted from A. thaliana Col-0 plants and used for ONT and PacBio HiFi long-read sequencing and Bionano optical mapping. ONT reads were used to establish a draft assembly, which was then scaffolded and polished with HiFi reads to generate the Col-CEN v1.2 assembly. ONT reads were used to analyze DNA methylation with the Deep-Signal-plant algorithm (20). CEN180 monomers, higher-order repeats, and ATHILA retrotransposons were identified de novo using custom pipelines. Short-read datasets (table S7) were aligned to Col-CEN to map chromatin and recombination distributions, using standard methods. Cytogenetic analysis of the centromeres was performed using FISH and immunofluorescence staining. A full description of all experimental and computational methods can be found in the supplementary materials.
Supplementary Material
ACKNOWLEDGMENTS
This paper is dedicated to Simon Chan. We thank I. Thompson for ATHILA analysis, S. Henikoff for the generous gift of CENH3 antibodies, A. Shumate for help with gene Liftoff interpretation, B. Fischer for advice on high–molecular weight DNA isolation, and M. Pouch for assistance designing FISH probes.
Funding:
This work was supported by BBSRC grants BB/S006842/1, BB/S020012/1, and BB/V003984/1 to I.R.H.; European Research Council Consolidator Award ERC-2015-CoG-681987 “SynthHotSpot” to I.R.H.; Marie Curie International Training Network “MEICOM” to I.R.H.; Human Frontier Science Program award RGP0025/2021 to T.K., M.C.S., and I.R.H.; US National Institutes of Health grant S10OD028632-01; US National Science Foundation grants DBI-1350041 and IOS-1732253 to M.C.S.; Royal Society awards UF160222 and RGF/R1/180006 to A.B.; the Czech Science Foundation grant no. 21-03909S to M.A.L.; the Gregor Mendel Institute to F.B.; grants Fonds zur Förderung der wissenschaftlichen Forschung (FWF) P26887, P28320, P30802, P32054, and TAI304 to F.B. and chromatin dynamics W1238 to A.S. and B.J.; Leverhulme Trust Research Leadership grant RL-2012-042 to J.T.; and grants from the Howard Hughes Medical Institute and US National Institutes of Health (RO1GM067014) to R.A.M.
Footnotes
Competing interests: The authors have no competing interests.
Data and materials availability:
The ONT sequencing reads used for assembly are available for download at ArrayExpress accession E-MTAB-10272 (www.ebi.ac.uk/arrayexpress/). The PacBio HiFi reads are available for download at European Nucleotide Archive accession number PRJEB46164 (www.ebi.ac.uk/ena/browser/view/PRJEB46164). All data, code and materials are available in the manuscript or the supplementary materials and at https://github.com/schatzlab/Col-CEN.
REFERENCES AND NOTES
- 1.Malik HS, Henikoff S, Major evolutionary transitions in centromere complexity. Cell 138, 1067–1082 (2009). doi: 10.1016/j.cell.2009.08.036; [DOI] [PubMed] [Google Scholar]
- 2.Melters DP et al. , Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14, R10 (2013).doi: 10.1186/gb-2013-14-1-r10; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKinley KL, Cheeseman IM, The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Biol 17, 16–29 (2016). doi: 10.1038/nrm.2015.5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rudd MK, Wray GA, Willard HF, The evolutionary dynamics of α-satellite. Genome Res. 16, 88–96 (2006). doi: 10.1101/gr.3810906; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain M et al. , Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol 36, 338–345 (2018). doi: 10.1038/nbt.4060; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miga KH et al. , Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). doi: 10.1038/s41586-020-2547-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logsdon GA et al. , The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021). doi: 10.1038/s41586-021-03420-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nurk S et al. , The complete sequence of a human genome. bioRxiv 2021.05.26.445798 [Preprint] (2021). doi: 10.1101/2021.05.26.445798 [DOI] [Google Scholar]
- 9.Arabidopsis Genome Initiative, Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000). doi: 10.1038/35048692; [DOI] [PubMed] [Google Scholar]
- 10.Maheshwari S, Ishii T, Brown CT, Houben A, Comai L, Centromere location in Arabidopsis is unaltered by extreme divergence in CENH3 protein sequence. Genome Res. 27, 471–478 (2017). doi: 10.1101/gr.214619.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copenhaver GP et al. , Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286, 2468–2474 (1999). doi: 10.1126/science.286.5449.2468; [DOI] [PubMed] [Google Scholar]
- 12.Talbert PB, Masuelli R, Tyagi AP, Comai L, Henikoff S, Centromeric localization and adaptive evolution of an Arabidopsis histone H3 variant. Plant Cell 14, 1053–1066 (2002). doi: 10.1105/tpc.010425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Zapater JM, Estelle MA, Somerville CR, A highly repeated DNA sequence in Arabidopsis thaliana. Mol. Gen. Genet 204, 417–423 (1986). doi: 10.1007/BF00331018 [DOI] [Google Scholar]
- 14.Round EK, Flowers SK, Richards EJ, Arabidopsis thaliana centromere regions: Genetic map positions and repetitive DNA structure. Genome Res. 7, 1045–1053 (1997). doi: 10.1101/gr.7.11.1045; [DOI] [PubMed] [Google Scholar]
- 15.McCartney AM et al. , Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. bioRxiv 2021.07.02.450803 [Preprint] (2021). doi: 10.1101/2021.07.02.450803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosouchi T, Kumekawa N, Tsuruoka H, Kotani H, Physical map-based sizes of the centromeric regions of Arabidopsis thaliana chromosomes 1, 2, and 3. DNA Res. 9, 117–121 (2002). doi: 10.1093/dnares/9.4.117; [DOI] [PubMed] [Google Scholar]
- 17.Rhie A, Walenz BP, Koren S, Phillippy AM, Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020). doi: 10.1186/s13059-020-02134-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright DA, Voytas DF, Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome Res. 12, 122–131 (2002). doi: 10.1101/gr.196001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAllister BF, Werren JH, Evolution of tandemly repeated sequences: What happens at the end of an array? J. Mol. Evol 48, 469–481 (1999). doi: 10.1007/PL00006491; [DOI] [PubMed] [Google Scholar]
- 20.Ni P et al. , Genome-wide detection of cytosine methylations in plant from nanopore sequencing data using deep learning. bioRxiv 2021.02.07.430077 [Preprint] (2021). doi: 10.1101/2021.02.07.430077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stroud H et al. , Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol 21, 64–72 (2014). doi: 10.1038/nsmb.2735; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE, Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364 (2013). doi: 10.1016/j.cell.2012.10.054; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacob Y et al. , ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol 16, 763–768 (2009). doi: 10.1038/nsmb.1611; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yelagandula R et al. , The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 158, 98–109 (2014). doi: 10.1016/j.cell.2014.06.006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J et al. , Widespread gene conversion in centromere cores. PLOS Biol. 8, e1000327 (2010). doi: 10.1371/journal.pbio.1000327; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambing C et al. , Interacting genomic landscapes of REC8-cohesin, chromatin, and meiotic recombination in Arabidopsis. Plant Cell 32, 1218–1239 (2020). doi: 10.1105/tpc.19.00866; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambing C, Kuo PC, Tock AJ, Topp SD, Henderson IR, ASY1 acts as a dosage-dependent antagonist of telomere-led recombination and mediates crossover interference in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A 117, 13647–13658 (2020). doi: 10.1073/pnas.1921055117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi K et al. , Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions. Genome Res. 28, 532–546 (2018). doi: 10.1101/gr.225599.117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigal M et al. , Epigenome confrontation triggers immediate reprogramming of DNA methylation and transposon silencing in Arabidopsis thaliana F1 epihybrids. Proc. Natl. Acad. Sci. U.S.A 113, E2083–E2092 (2016). doi: 10.1073/pnas.1600672113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steimer A et al. , Endogenous targets of transcriptional gene silencing in Arabidopsis. Plant Cell 12, 1165–1178 (2000). doi: 10.1105/tpc.12.7.1165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SC et al. , Arabidopsis retrotransposon virus-like particles and their regulation by epigenetically activated small RNA. Genome Res. 30, 576–588 (2020). doi: 10.1101/gr.259044.119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhie A et al. , Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021). doi: 10.1038/s41586-021-03451-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wijnker E et al. , The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. eLife 2, e01426 (2013). doi: 10.7554/eLife.01426; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durfy SJ, Willard HF, Patterns of intra- and interarray sequence variation in alpha satellite from the human X chromosome: Evidence for short-range homogenization of tandemly repeated DNA sequences. Genomics 5, 810–821 (1989). doi: 10.1016/0888-7543(89)90123-7; [DOI] [PubMed] [Google Scholar]
- 35.Altemose N et al. , Complete genomic and epigenetic maps of human centromeres. bioRxiv 2021.07.12.452052 [Preprint] (2021). doi: 10.1101/2021.07.12.452052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahtani MM, Willard HF, Physical and genetic mapping of the human X chromosome centromere: Repression of recombination. Genome Res. 8, 100–110 (1998). doi: 10.1101/gr.8.2.100; [DOI] [PubMed] [Google Scholar]
- 37.Tsukahara S et al. , Centromere-targeted de novo integrations of an LTR retrotransposon of Arabidopsis lyrata. Genes Dev. 26, 705–713 (2012). doi: 10.1101/gad.183871.111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawabe A, Nasuda S, Structure and genomic organization of centromeric repeats in Arabidopsis species. Mol. Genet. Genomics 272, 593–602 (2005). doi: 10.1007/s00438-004-1081-x; [DOI] [PubMed] [Google Scholar]
- 39.Klein SJ, O’Neill RJ, Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 26, 5–23 (2018). doi: 10.1007/s10577-017-9569-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malik HS, The centromere-drive hypothesis: A simple basis for centromere complexity. Prog. Mol. Subcell. Biol 48, 33–52 (2009). doi: 10.1007/978-3-642-00182-6_2; [DOI] [PubMed] [Google Scholar]
- 41.Haig D, Grafen A, Genetic scrambling as a defence against meiotic drive. J. Theor. Biol 153, 531–558 (1991). doi: 10.1016/S0022-5193(05)80155-9; [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ONT sequencing reads used for assembly are available for download at ArrayExpress accession E-MTAB-10272 (www.ebi.ac.uk/arrayexpress/). The PacBio HiFi reads are available for download at European Nucleotide Archive accession number PRJEB46164 (www.ebi.ac.uk/ena/browser/view/PRJEB46164). All data, code and materials are available in the manuscript or the supplementary materials and at https://github.com/schatzlab/Col-CEN.