


Abstract
The ontogeny and dynamics of mtDNA heteroplasmy remain unclear due to limitations of current mtDNA sequencing methods. We developed individual Mitochondrial Genome sequencing (iMiGseq) of full-length mtDNA for ultra-sensitive variant detection, complete haplotyping, and unbiased evaluation of heteroplasmy levels, all at the individual mtDNA molecule level. iMiGseq uncovered unappreciated levels of heteroplasmic variants in single cells well below the conventional NGS detection limit and provided accurate quantitation of heteroplasmy level. iMiGseq resolved the complete haplotype of individual mtDNA in single oocytes and revealed genetic linkage of de novo mutations. iMiGseq detected sequential acquisition of detrimental mutations, including large deletions, in defective mtDNA in NARP/Leigh syndrome patient-derived induced pluripotent stem cells. iMiGseq identified unintended heteroplasmy shifts in mitoTALEN editing, while showing no appreciable level of unintended mutations in DdCBE-mediated mtDNA base editing. Therefore, iMiGseq could not only help elucidate the mitochondrial etiology of diseases, but also evaluate the safety of various mtDNA editing strategies.
INTRODUCTION
Mitochondria play vital roles in cellular metabolic and signaling processes. Each human mitochondrion contains on average 1.4 copies of a 16.5 kb circular genome (1)—mtDNA—that is densely packed with 13 genes encoding core subunits of the oxidative phosphorylation complexes, 24 RNA genes, and a non-coding control region (D-loop region). Mitochondria and hence mtDNA undergo constant turnover even in non-dividing cells (2,3). The demand for frequent DNA replication and the lack of histonized chromatin in mtDNA contribute to a mutation rate that is at least one order of magnitude higher than that of the human nuclear genome (3–5). Because mammalian cells typically contain 1000–10 000 copies of mtDNA, mutations arisen in individual mtDNA produce an admixture of mutant and wild-type mtDNA (heteroplasmy) within a cell (6). Heteroplasmic mutations inherited from the oocyte can cause inborn disorders and are associated with late-onset complex diseases (4).
Our understanding of mtDNA genetics has been dramatically shaped by available technologies. In recent years, deep short-read next-generation sequencing (NGS) based methods have been instrumental in revealing the rich diversity of mtDNA. They were used to show that heteroplasmic mutations are present in the general population and are among the most common causes of inherited metabolic diseases when present above a heteroplasmy threshold (7,8). However, conventional NGS-based studies of mtDNA are unable to detect rare heteroplasmic variants that exist below 1% allele frequency due to the background NGS error rate (0.1–1%) and contamination of nuclear mitochondrial DNA-like sequences (NUMTs) in short-amplicon PCR (3,9,10). Even though single-mitochondrion sequencing (11) and duplex mtDNA sequencing (12) have been developed with improved sensitivity, these methods are limited in throughput or variant detection. Single-mitochondrion sequencing can only survey around one mtDNA per cell and is thus impractical for studying heteroplasmy. Duplex mtDNA sequencing can detect single nucleotide variants (SNVs) from a mixed mtDNA population but provides no haplotype.
Current methods of mtDNA sequencing suffer from two seemingly paradoxical issues–amalgamation and fragmentation. Most studies of mtDNA genetics are based on short-read shotgun or amplicon NGS (2,9,10,13–15). These techniques average out the heterogeneity of mtDNA in two ways. Firstly, mtDNA genotypes of thousands of cells are averaged in bulk sequencing, thus masking variants in rare cells and underestimating cell-to-cell heterogeneity (16). The higher mutational load, relaxed replication, and vegetative segregation of mtDNA could lead to extreme high- or low-frequency mutations in different cells. Indeed, recent published data revealed that high-frequency mtDNA mutations are found in vivo only at the single-cell level in mid-aged humans and their heteroplasmy levels can vary greatly in different cells (16,17). It has been shown that the variance of heteroplasmy level increases linearly during mouse aging (18). Similarly, in humans some new somatic mtDNA mutations reach frequencies high enough to have functional consequences only after decades of aging (10,19). Therefore, so-called ‘low-frequency’ mutations observed in bulk mtDNA sequencing should not be disregarded as functionally unimportant, and there is unmet need for analyzing heteroplasmy dynamics at the single-cell level throughout human lifespan. Secondly, even in single-cell studies (17,20), what was obtained was a composite genotype of all mtDNA rather than the true genotypes of individual mtDNA. Additionally, the phenotypic significance of an mtDNA variant is often strongly modified by other co-inherited variants (4). However, conventional methods cannot provide full haplotypes due to fragmentation of mtDNA molecules, which largely prevents the genetic study of linkage between heteroplasmy variants.
High-throughput analysis of the whole mutational landscape of single mtDNA molecules in single cells is still beyond the reach of current methodologies (16). To overcome these hurdles, we developed a series of single-cell and long-amplicon manipulation procedures to adapt our long-read individual molecule sequencing strategy (IDMseq (21), capable of detecting allele frequency as low as 0.004%) to sequence individual complete mtDNA in single cells. This new technology–individual Mitochondrial Genome sequencing (iMiGseq)–labels each mtDNA in single cells with a unique molecular identifier (UMI). UMI-labeled mtDNA are further amplified by high-fidelity long-range PCR and sequenced on long-read sequencing platforms to obtain full-length mtDNA, generate variant details and resolve the complete haplotype of individual mtDNA (Figure 1A).
Figure 1.

Validation of iMiGseq using 293T cells. (A) Schematic representation of iMiGseq. Single oocytes (5 cells for cultured cells) are manually picked and lysed in RIPA buffer. iMiGseq applies the IDMseq strategy to specifically label individual mtDNA with UMIs by a single round of primer extension. The UMI oligo contains a 3′ gene-specific sequence (red), a UMI sequence (yellow), and a 5′ universal primer sequence (green). The UMI-labeled mtDNA is further amplified by long-range PCR using the universal primer (green) and gene-specific reverse primer (purple) as a single amplicon. The sequencing of long amplicons is performed on long-read Nanopore and PacBio sequencing platforms. The sequencing data are analyzed by a bioinformatic toolkit—VAULT—to identify UMI sequences, bin reads based on UMI and call variants. (B) Distribution of effectively covered length of UMI groups detected by Nanopore and PacBio sequencing. Color-coded triangles indicate N50 values. (C) VAF distribution of SNVs detected in iMiGseq of 293T cells. The majority of unique SNVs are below the current 1% detection limit (the vertical dotted line). (D) Circular plots showing the distribution of SNVs in the mitochondrial genome of 293T cells determined by Nanopore and PacBio sequencing. The innermost circle (grey) shows the depth of reads of all detected UMI groups in linear scale as indicated by the scale bar in the center. The red triangle indicates the position of the primers. The middle circle (light blue) represents common SNPs from the human dbSNP-151 database. Individual SNVs are plotted in the outer barplot circle, in which the height of bar represents the VAF. Red color indicates VAF >0.6. The outermost circle is a color-coded diagram of the human mtDNA. Blue: protein-coding genes. Yellow: rRNA genes. Dark green: tRNA genes. Light green: D-loop. (E) ddPCR results showing the detected positive events (droplets) for variants with different VAFs. The VAFs calculated by Nanopore iMiGseq are shown under the variants. Around 80 cells are used in ddPCR to ensure the generation of enough events for analysis, as compared to 5 cells in iMiGseq. The data shown here and in Supplementary Table S2 are from two independent biological replicates.
As compared with low-throughput single-mitochondrion sequencing (11) that requires manual isolation of single mitochondrion, iMiGseq uses a simple and fast procedure to directly label individual mtDNA in lysed single cells in 30 min, and thus reduces the loss of mtDNA or cells in multi-step mitochondria isolation and eliminates the need for complicated and labor-intensive manual dissection of mitochondria (11,22). This results in a dramatic increase in throughput from sequencing a few mtDNA per cell to up to thousands of mtDNA per cell. Thus, iMiGseq allowed us to address several key open questions in the field. It provided true heteroplasmy level by counting molecules directly. It uncovered whole spectrum of pathogenic mtDNA mutations, including SNVs and large structural variants (SVs) that lie below the current 1% detection limit in single cells. It showed the first haplotype-resolved mitochondrial genomes from single oocytes and illustrated sequential acquisition of mutations in defective mtDNA in patient-derived cells. It revealed the linkage of heteroplasmic mutations, allowed the study of the genetic linkage between pathogenic mtDNA mutations. iMiGseq identified unintended heteroplasmy shifts in mitoTALEN mtDNA editing, while showing no appreciable level of unintended mutations in DdCBE-mediated mtDNA base editing.
MATERIALS AND METHODS
Cell lines
The 293T cell line was purchased from ATCC and cultured in Gibco™ DMEM medium (high glucose) containing 10% Gibco™ Fetal Bovine Serum (heat inactivated) and 1× Gibco™ Penicillin–Streptomycin (5000 U/ml). Cells were maintained at 37 °C in a humidified incubator with sea-level air enriched with 5% CO2.
Oocyte isolation
The animal experiments in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of KAUST. The NSG and C57BL/6 mice were purchased from the Jackson Laboratory and Charles River Laboratories and kept in KAUST animal resources core lab. The NSG oocytes were collected from naturally ovulating female mice. For B6 oocytes, superovulation was induced in C57BL/6 (6 weeks) female mice by sequential intraperitoneal injection of five international units (IU) of pregnant mare's serum gonadotrophin (PMSG) (USBiological Life Sciences G8575A) and 5 IU of human chorionic gonadotrophin (hCG) (Sigma-Aldrich C1063) 46–48 h later. C57BL/6 mice were sacrificed 14 h after hCG injection. Oviducts were dissected from NSG mice (16–20 weeks) or from the super-ovulated C57BL/6 mice, and the oocytes were isolated by mouth pipette and washed in cold PBS. Then, the oocytes were dissociated from cumulus cells using Accutase (5–10 min at RT), washed in cold PBS by pipetting up and down, transferred to PCR tubes in a small volume of PBS, and frozen at –80°C.
Derivation and culture of hEPS cells
To generate mitochondria diseases iPSCs, endometrium tissues fibroblast cells were transfected with a Sendai virus reprogramming kit (Life Technologies, A16517). The transfected cells were then plated onto Matrigel-coated culture dishes according to the manufacturer's instructions. The iPSCs were cultured on Matrigel-coated tissue culture dishes (ES-qualified, BD Biosciences) with mTeSR1 (STEMCELL Technologies) at 37°C and 5% CO2 in a humidified atmosphere incubator. The iPSCs culture medium was changed daily. The cells were passaged every 3–4 days using Accutase (Stemcell Technologies). The iPSCs conversion to EPS was preformed as previously reported (23).
Cell manipulation, lysis and DNA purification
All cells were washed twice by cold PBS and counted by Invitrogen Countess 3 Automated Cell Counter to determine the concentration. A serial dilution with PBS was performed to achieve the desired cell concentration. The diluted cells were further checked under microscope to confirm the concentration and the desired number of cells were obtained by adding different volumes of diluted cell suspension. Single oocytes were obtained by mouth pipette and transferred into cold PBS under a stereo microscope. One microliter of diluted 293T cells (∼5 cells), single oocytes, previously frozen hEPSm.8993T>C cells (5-30 cells), or gene edited cells were lysed in 5 μl RIPA buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) on ice for 15 mins. The cell lysate was diluted by adding 10 μl H2O, and further purified to extract total DNA using 15 μl Beckman Coulter AMPure XP beads (A63882). Two-round of 70% ethanol washes were performed to remove detergents. The DNA was eluted in 10 μl H2O to be used for the UMI labeling of mtDNA.
UMI labeling and amplification of mtDNA
The targeted UMI labeling of individual mtDNA was achieved by mtDNA specific UMI oligos. The oligos were selected to enable efficient amplification of full-length human or mouse mitochondrial genomes. A 5′ universal primer sequence and middle UMI sequence were added to the five-prime end of mtDNA specific oligos to form UMI oligos. The full list of oligos used in this study is shown in Supplementary Table S3. The BLAST of human primers to the human reference genome showed that no primers will amplify NUMTs. Since iMiGseq amplified the whole circular mtDNA genome using inverse primers, linear NUMTs were less likely to be amplified by inverse primers. The data analysis can efficiently detect and exclude NUMTs since they share a different structure than the full-length mtDNA. A screen of DNA polymerases was performed to ensure high efficiency in UMI labeling and PCR amplification.
The UMI labeling reaction was set up as follows: 10 μl purified DNA, 2.5 μl UMI oligos (10 μM), 12.5 μl 2× Platinum™ SuperFi™ PCR Master Mix (Invitrogen, 12358010). The reaction was incubated on a thermocycler with a ramp rate of 1°C per second using the following program: 98°C 1 min, 70°C 5 s, 69°C 5 s, 68°C 5 s, 67°C 5 s, 66°C 5 s, 65°C 5 s, 72°C 10 min, 4°C hold. After UMI labeling, DNA was purified by 0.8X AMPure XP beads, washed twice by 70% ethanol and eluted 10 μl H2O. The universal primer and mtDNA specific reverse primer were used to amplify only the UMI labeled mtDNA. The 50 μl PCR reaction contains 1.25 U PrimeSTAR GXL DNA Polymerase (Takara, R050), 1× PrimeSTAR GXL Buffer, 200 μM dNTP mixture, 0.2 μM each primer, and 10 μl purified UMI-labeled DNA. The thermocycler program was set as follows: 95°C 1 min, (98°C 10 s, 68°C 14 min, 30 cycles), 68°C 5 min, 4°C hold. The amplicon was validated by agarose gel electrophoresis. If a specific DNA band was observed, the rest DNA would be purified by 0.8X AMPure XP beads. A second round of PCR amplification for 15–20 cycles with PrimeSTAR GXL DNA Polymerase could be performed to obtain enough DNA for sequencing. A typical UMI labeling and mtDNA amplification experiment can be finished in 10 hours with reagents cost less than eight USD.
We considered if the variant calling results could be affected by PCR artifacts, which is the main source of errors in UMI consensus sequencing originating from polymerase replication error in the barcoding step (24). The Platinum SuperFi DNA polymerase we used has the highest reported fidelity (>300× that of Taq polymerase), and meanwhile captures twice more molecules in the library than Taq (21). Theoretically, this polymerase introduces one error in ∼1600 unique 16 556-bp molecules in the UMI labeling step. Accordingly, this type of inescapable error is expected to be around 1 in 1600 UMI groups, thus representing a minor fraction of the observed SNVs.
ddPCR
ddPCR was performed on a Bio-Rad QX200 Droplet Digital PCR System using ddPCR Supermix for Probes (No dUTP) kit (Bio-Rad, 1863024) according to manufacturer's protocols. The probes were synthesized by Integrated DNA Technologies Inc. as PrimeTime qPCR Probes. The wildtype probes were labeled by 5′HEX/ZEN/3′IBFQ, while the mutant probes were labeled by 5′FAM/ZEN/3′IBFQ. The primer and probe sequences were shown in Supplementary Table S3. The primer/probe ratio was set as 3.6:1. For each reaction, 0.5 ng of purified 293T cell genome were used. All experiments were performed in three independent replicates, and the positive events were combined for the analysis.
mtDNA editing
The cells for mitoTALEN mtDNA editing were previously described (25). We followed a previously described protocol of CRISPR-free mitochondrial base editing (26). Briefly, 50 000 293T cells were seeded on one well of collagen-coated 48-well plate (Corning). After 24 h, 500 ng ND4-DdCBE-right TALE (Addgene # 157843) and 500 ng ND4-DdCBE-left TALE (Addgene # 157844) were premixed with 1.5 μl lipofectamine 2000 (Thermo Fisher Scientific) and transfected to the cells. As a control, only 500 ng ND4-DdCBE-right side TALE was transfected into the cells. The cells were harvested 6 days post-transfection for genomic DNA extraction (Qiagen DNeasy® Blood & Tissue kit), Sanger sequencing, and iMiGseq. The mitoTALEN sequencing was performed with ∼20 cells. The ND4-DdCBE sequencing was done with ∼50 ng purified DNA.
Library preparation and sequencing
For Nanopore sequencing, library preparations were done using the ligation sequencing kit (Oxford Nanopore Technologies, SQK-LSK109). Most sequencing runs were performed on Oxford Nanopore MinION sequencers monitored by MinKNOW (v22.05.5). The mtDNA base editing samples were sequenced using one R9.4.1 flow cell and two R10.3 flow cells per sample. All other samples were sequenced using one R9.4.1 flow cell per sample. Base calling of Nanopore reads was done using the official basecaller termed Guppy (v3.2.1 and v5.0.7 (only for mtDNA editing and blastoid generation experiments)). For PacBio sequencing, library preparations were done using Sequel Sequencing Kit 3.0. The sequencing runs were performed by the BIOPIC core facility at Peking University (Beijing, China) on the PacBio Sequel with SMRT Cell 1M v3 LR Tray. HiFi Reads were generated by the official tool termed ccs (v3.4.1). All procedures were preformed according to manufacturer's protocols. The requirement of sequencing throughput was determined on the expected UMI group number after sequencing, and relevant to the original molecule number in the UMI labeling. In general, to identify a desired number of UMI groups, a higher sequencing throughput will be needed for more abundant initial mtDNA molecules and 4000× coverage could be used as a starting point. Multiple library preparation and sequencing of the same sample may be needed to identify a sufficient number of molecules.
Bioinformatic analysis
All iMiGseq data were analyzed by VAULT with –unmapped_reads and –group_filter options, to remove unmapped reads before UMI analysis and filter out low-confidence UMI groups after variants calling. The percentage of reads with UMI depends on base calling errors in the UMI region and/or DNA fragmentation during library preparation and sequencing. For Nanopore data, the error tolerance threshold –error in UMI identification was set to 0.11, while for PacBio CCS data, it was set to 0.05. Only perfect UMIs with correct length and structure (NNNNNTGNNNNN) were subjected to downstream analysis. The SNV calling and filter were performed using default parameters of VAULT, and involved in Samtools v1.9 (27). SNVs in primer regions were filtered out before downstream analysis.
The reference sequence –refer used in VAULT analysis was the designed amplicon sequence, which is based on CM004185.1 for NOD oocytes, NC_005089 for B6 oocytes, and NC_012920 for 293T cells and human cells. This gave rise to a different coordinate to the canonical mitochondria reference genome. The vault position command was used to revise the DNA coordinate and reference chromosome name in VCF files, to enable further functional analysis by SnpEff v4.3 (28). In the SNV annotation, the positions of SNVs in NOD oocytes were converted to the corresponding coordinates of B6 mouse strain. GRCm38.86 database of SnpEff was used in mouse SNV annotation, while hg38kg database was used for human SNVs.
The SV calling of VAULT utilized minimap2.1 (29) and sniffles v1.0.11 (30). The detected SVs were first filtered by a variant allele frequency of 0.6 and then manually checked. The consensus sequence of extensive coverage UMI groups was called using vault consensus command. It utilized canu v2.0 (31) to do de novo assembly, and the Nanopore official tool medata v0.12.1 to polish the assembled sequence. Most UMI groups failed to generate contigs in canu assembly, thus lead to a reduction of assembled mtDNA. Further assemblers with improved performance could potentially solve this problem and lead to more assembled mtDNA.
SNV allele frequency (VAF, also mentioned as NHL) is calculated as
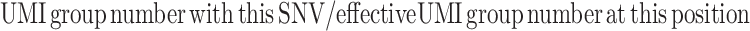 |
Effective UMI group number is defined as the number of UMI groups with depth ≥3 at the position of this SNV.
SNV number per genome is calculated as
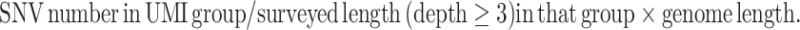 |
RESULTS
Development and validation of iMiGseq
The ideal sequencing technology for iMiGseq should offer accurate ultra-long reads (∼16.5 kb). We have previously shown that IDMseq and VAULT, the foundations of iMiGseq, provide sensitive and accurate quantitative characterization of various types of variants through error correction by the molecular consensus strategy (21). The design of iMiGseq further eliminates the influence of NUMT contaminations that perplexed the current NGS-based analysis of mtDNA variants (see Materials and Methods). Conventional mtDNA isolation methods require a large number of input cells and lose a significant number of mitochondria, we therefore first developed a method for polymerase-directed UMI labeling of mtDNA in lysed cells without complicated fractionation steps. After testing different combinations of lysis conditions (e.g. the type and concentration of detergents, temperature, lysis time, etc.), polymerases, and DNA purification methods, we devised a quick and simple protocol capable of barcoding mtDNA in single cells (Figure 1A).
We tested two state-of-the-art long-read platforms–Oxford Nanopore MinION and PacBio Sequel. Five HEK 293T cells (∼1500 copies of mtDNA/cell) were subjected to a single round of primer extension to label individual full-length mtDNA with UMI followed by high-fidelity long-range PCR (Figure 1A, Supplementary Figure S1A & B, see Materials and Methods). The trial Nanopore sequencing generated 81.8k reads mapped to the human reference mtDNA (rCRS NC_012920 was used unless indicated otherwise) with an average alignment identity of 89.9% (Table 1, Supplementary Figure S2A). Using the VAULT analysis pipeline (21), the reads were assigned to 542 UMI groups (a set of reads sharing the same UMI), of which 92.6% covered ≥95% of the full-length mtDNA (position with depth ≥ 3) and the N50 was 16 547 bp (Figure 1B). After further filtering based on in-group read consistency (see method and ref (21)), we obtained 424 high-confidence UMI groups (only high-confidence UMI groups were used hereafter), 92% of which covered ≥95% of the mtDNA, which represented 390 individual complete mitochondrial genomes from single long reads. Variant analysis showed that the 424 UMI groups contained a total of 7766 high-confidence SNVs. No SVs were detected.
Table 1.
Summary of individual sequencing runs
| Sample | Mapped read count | Reads with UMI | Detected mtDNA | mtDNA with SNV | SNV count | Median covered region of UMI groups | Extensive coverage mtDNA* | Unique SNV number |
|---|---|---|---|---|---|---|---|---|
| 293T Nanopore | 81 798 | 21 635 | 424 | 424 | 7766 | 16 546 | 390 | 58 |
| 293T PacBio | 167 103 | 52 475 | 666 | 666 | 9223 | 7510 | 0 | 33 |
| NSG mouse oocyte 1 | 1 086 527 | 234 256 | 3911 | 168 | 173 | 10 355 | 1243 | 79 |
| NSG mouse oocyte 2 | 959 446 | 182 770 | 1137 | 123 | 124 | 15 860 | 596 | 42 |
| NSG mouse oocyte 3 | 1 992 516 | 559 310 | 17 942 | 208 | 218 | 5424 | 3953 | 103 |
| B6 mouse oocyte 1 | 988 865 | 200 174 | 2493 | 1173 | 2145 | 7782 | 436 | 44 |
| B6 mouse oocyte 2 | 549 736 | 116 430 | 1849 | 945 | 1818 | 8573 | 363 | 40 |
| B6 mouse oocyte 3 | 325 899 | 63 039 | 615 | 329 | 557 | 9926 | 129 | 14 |
| hEPSm.8993T>C | 72 496 | 34 959 | 536 | 536 | 12 173 | 16 573 | 515 | 117 |
| mitoTALEN unedited | 211 901 | 111 811 | 1011 | 1011 | 21 004 | 16 193 | 619 | 66 |
| mitoTALEN edited | 284 546 | 197 783 | 2783 | 2658 | 42 890 | 3239 | 933 | 292 |
| ND4-DdCBE unedited | 614 099 | 148 096 | 274 | 251 | 2382 | 12 519 | 114 | 36 |
| ND4-DdCBE edited | 782 572 | 159 588 | 286 | 261 | 2099 | 8345 | 95 | 46 |
*UMI groups with reads covering >95% of the full-length mtDNA with depth ≥3×.
The variant allele frequency (VAF) of SNVs (calculated by counting molecules, see methods) ranged between 0.23% and 96.47%, and, surprisingly, the majority of unique SNVs had a VAF below the 1% detection limit of conventional NGS (Figure 1C and D). The mutational spectrum strongly biased toward transitions (i.e. A > G (T > C) and C > T (G > A)) (Supplementary Figure S1C) as previously reported (10,17,32). The extensive-coverage UMI groups (covering > 95% of the full-length mtDNA with depth ≥ 3) supported de novo assembly of 11 full-length mtDNA. All 11 mtDNA were placed into the U5a1 mitochondrial haplogroup by MITOMASTER (33), which fit the Dutch origin of 293 cells (34), thus proving the accuracy of the assemblies.
Compared to Nanopore sequencing, PacBio circular consensus sequencing (CCS) generated 1.5× number of UMI groups from 2× number of reads, but the N50 of read length was reduced by more than 2× and no UMI group covered > 95% of the full-length of mtDNA (Table 1, Figure 1B). The pattern of VAF of the 9223 SNVs in the 666 high-confidence UMI groups was similar to that of Nanopore iMiGseq (Figure 1C). Unlike the Nanopore data that supported de novo assembly of full-length mitochondrial genomes, the PacBio data resulted in none. All the PacBio heteroplasmic SNVs (VAF ≥ 1%) existed in the Nanopore data, thus cross validating these called variants (Figure 1D).
To ensure the reliability of rare variants detected in iMiGseq, we evaluated the extent of false-positive variants due to polymerase replication error in the barcoding step (the main source of errors in UMI consensus sequencing (24)) and determined that this type of error introduced roughly 1 mutation in 1600 labeled full-length mtDNA (see methods and shown previously (21)), thus representing a miniscule fraction of the SNVs (0.03 SNVs in the Nanopore iMiGseq data). We further asked if rare SNVs, especially those identified in single UMI groups, could be sequencing artifacts (e.g. DNA damage during library preparation (35)). The single-UMI-group SNVs showed the same mitochondrial somatic mutation signature (10,15) as high-confidence SNVs (SNVs detected in multiple UMI groups), with predominantly C > T and T > C substitutions that are thought to be caused by different mutagenesis and DNA repair processes between the nucleus and mitochondria (36,37) (Figure 2A, Supplementary Figure S3).
Figure 2.

Single-cell iMiGseq of mouse oocytes. (A) Mutational spectrum of individual samples sequenced by iMiGseq. The analysis is performed in three categories: SNVs found in only one UMI group (shown here), SNVs found in multiple UMI groups and with < 1% VAF (shown in Supplementary Figure S3), and SNVs with 1–60% VAF (shown in Supplementary Figure S3). Samples without SNVs falling in any of the three categories are excluded in the comparison. Single-UMI-group SNVs show the same mitochondrial somatic mutation signature as high-confidence SNVs, suggesting that they are true positives. (B) Strand biases of mtDNA SNVs detected in individual samples. Heteroplasmic SNVs are separated to single-UMI-group SNVs (shown here), multiple-UMI-group and <1% VAF SNVs (shown in Supplementary Figure S4), and 1–60% VAF SNVs (shown in Supplementary Figure S4) to show strand biases of detected mutations. The results are normalized by the number of available nucleotides in the strand. Significant strand biases are observed for SNVs in all three categories, suggesting that rare variants detected by iMiGseq are not sequencing artifacts. (C) Circular plots showing the distribution of SNVs in the mitochondrial genome of the NSG and B6 mouse oocytes. The arrangement of the circular plots is similar to Figure 1D, except that the middle circle (light blue) represents the common SNPs from the mouse dbSNP-142 database. (D) Comparison between in silico VAFs and VAFs as determined by iMiGseq in different subsampling experiments. The box plots show the distribution of VAFs determined by iMiGseq for the respective in silico VAFs. The percentages are those of the NSG allele. The numbers of sub-sampling iterations are shown as red numbers. The red dots inside the boxes indicate the mean values, and the thick black lines represent the median values. Potential outlier values are marked by bold black dots, while individual values of sub-samplings are shown as small grey dots. The lower and upper boundaries of the box represent the 25th and 75th percentiles, respectively. (E) Comparison of common SNVs in the three NSG oocytes. * indicates detrimental variant. The detection limit of current NSG mtDNA sequencing is indicated by the gray horizontal line. (F) Phylogenetic trees of individual detected mtDNA (shown in blue) constructed using ultra-rare SNVs (shown in red) in the NSG_3 oocyte. The DNA sequences indicate the UMIs.
The native mtDNA mutational spectrum is known to be highly asymmetric between the light and heavy strands due to the differences in the replication of the two strands (10,36). In contrast, artificial mutations introduced by DNA damage and/or polymerase error during in vitro manipulation are expected to affect the two mtDNA strands equally. Thus, we checked the strand bias of single-UMI-group and high-confidence SNVs. Significant strand biases were observed in detected SNVs (Figure 2B, Supplementary Figure S4, Supplementary Table S1), suggesting that rare variants detected by iMiGseq were not sequencing artifacts. Consistent with previous studies of mtDNA mutations in over 4000 human tumors of various types using bulk NGS sequencing (10,36), we observed more abundant C > T substitutions in the heavy strand (H) of mtDNA than in the light strand (L) (Supplementary Table S1). It is worth noting that previous studies were performed by bulk sequencing of large numbers (e.g. millions) of human cells, while our study was done in five cells (293T) or in single cells (mouse oocytes). The mtDNA heterogeneity among selected cells and the limited number of SNVs detected in small numbers of cells could result in mutational spectra distinct from previous bulk sequencing results. Together, these results strongly supported the authentic biological origin of the rare variants detected by iMiGseq.
To further validate the rare variants detected by iMiGseq, we performed digital droplet PCR (ddPCR) assays for five SNVs representing a wide range of VAFs (0.24–41.18%) in the Nanopore iMiGseq data. The three randomly selected single-UMI-group SNVs were confirmed by ddPCR (Figure 1E, Supplementary Table S2). The VAFs obtained by ddPCR were comparable to iMiGseq results. The two high-frequency SNVs showed consistent VAFs in iMiGseq by Nanopore and PacBio (m.4429G > A, 16.71% in Nanopore and 15.08% in PacBio; m.15259C > T, 41.18% in Nanopore and 39.12% in PacBio), and were further validated by ddPCR (m.4429G > A, 38.29%; m.15259C > T, 44.46%, Figure 1E). It is worth noting that the difference in the VAF of m.4429G > A between iMiGseq and ddPCR is potentially due to the different population of cells in the experiments and heterogeneity of mtDNA in 293T cells.
Because our validation results showed that Nanopore sequencing-based iMiGseq can reliably detect rare variants and quantify variant frequencies, and that PacBio failed to generate sufficiently long reads to cover the whole mtDNA, we concluded that Nanopore-based iMiGseq could provide accurate and complete mtDNA sequencing.
iMiGseq enables the analysis of mtDNA heterogeneity in single oocytes
The design of iMiGseq enabled robust capture of mtDNA in single cells. As a proof of concept, we applied iMiGseq to single oocytes of the NOD-scid IL2Rgammanull (NSG) and C57BL/6 (B6) mouse strains. These strains were chosen because of the availability of reference mtDNA sequence and of naturally occurring SNVs, which served as the ground truth for benchmarking false discovery rate of variants and accuracy of iMiGseq. iMiGseq identified 3911, 1137 and 17942 high-confidence UMI groups from three NSG oocytes (same female), and 2493, 1849 and 615 from three B6 oocytes (same female), respectively (Table 1, Supplementary Figure S2B and c). Variant analysis by VAULT identified significant number of SNVs in all six oocytes (Table 1). No large SV was detected. Most SNVs had a nominal heteroplasmy level (NHL, similar to VAF, calculated as [no. of UMI groups with SNV]/[no. of UMI groups covering the SNV position]) < 1%, which is the detection limit of most published studies (3,9,10,13) (Figure 2C, Supplementary Figure S5A and B). The mutation spectrum of rare SNVs suggested that they were not sequencing artifacts (Figure 2A and B, Supplementary Figure S3 and S4).
The NSG and B6 reference mtDNA differ in one position–m.9348 (B6/NSG: G/A), which served as the ground truth for determining false SNVs in iMiGseq. Despite the 10876 deep coverage of UMI groups, this position showed zero non-reference SNV in either mouse strain, supporting the excellent accuracy of iMiGseq. We further took advantage of these variants to construct in silico ground-truth heteroplasmies by mixing various numbers of reads randomly subsampled from NSG_2 with all reads of B6_2. iMiGseq accurately determined the heteroplasmy levels in iterated sub-samplings (71–105 times) even when the number of reads was limited (Figure 2D). It proved that iMiGseq can accurately quantitate heteroplasmy levels.
iMiGseq allowed us to compare the frequencies of variants shared by oocytes of the same mouse. We surveyed relatively high frequency SNVs (> 1% NHL) in NSG oocytes and observed a significant difference in the frequency of the same variant (Figure 2E). Two SNVs, m.606T > C and m.9941T > C (detrimental, p.Phe22Ser), showed ultra-low frequencies (<0.1% NHL) in two oocytes but a drastically higher frequency (up to 6.1%) in the third oocyte (Figure 2E). These results suggested that ultra-rare detrimental variants could under some circumstances increase their frequencies by nearly a hundredfold during oogenesis, which supports the need for unbiased studies on these variants using ultra-sensitive methods such as iMiGseq.
Several phylogenetic trees were constructed based on haplotypes of individual mtDNA and showed evidence of sequential acquisition of de novo mutations in individual mtDNA (Figure 2F). Because long reads allowed phasing of novel variants with cell type specific SNVs, NUMT could be confidently avoided. Together, the above findings demonstrated the ability of iMiGseq in accurate characterization of mtDNA heterogeneity in single cells, which enables the quantitative understanding of the genetics of mtDNA in single oocytes.
iMiGseq identifies and quantitates the disease-causing SNV and a novel large deletion in mtDNA in NARP/leigh syndrome iPSCs
Mitochondrial mutations are a common cause of inherited disease with a prevalence of ∼1 in 5000 adults (38). The diagnosis of mitochondrial disease is challenging due to the clinical complexity and genetic complexity associated with mitochondrial disorders (39). Recent advances in NGS have enabled whole-genome sequencing (WGS) as a first-line testing to unravel the molecular defect in mitochondrial disease (40). However, the diagnostic yield by WGS is still considerably low, leaving ∼1/3 of suspected mitochondrial disease cases unresolved (41). This is potentially due in part to the limitation of conventional short-read NGS in detecting large and complex mutations. Since iMiGseq enabled accurate quantitation of mtDNA mutations in a haplotype-resolved manner in single cells, we hypothesized that it could be adapted for clinical samples to facilitate genetic diagnosis and understanding of mitochondrial diseases.
Human extended pluripotent stem (hEPS) cells can generate blastocyst-like structures (hEPS-blastoids) through lineage segregation and self-organization in vitro (42,43). hEPS-blastoids could provide a unique model of mitochondrial disease during early embryogenesis. As a proof-of-concept, we performed iMiGseq in hEPS cells derived from mitochondrial disease-specific induced pluripotent stem cells (iPSCs) described previously (25). The patient was diagnosed by WGS with the m.8993T > C mutation causing the neuropathy, ataxia and retinitis pigmentosa (NARP) and maternally inherited Leigh syndrome. She gave birth to two daughters who died of Leigh syndrome and one son who was age three and asymptomatic at the time of sample collection, all confirmed carriers of m.8993T > C. The patient-derived hEPS cells (referred to as hEPSm.8993T>C hereafter) were collected for UMI labeling of whole mtDNA. iMiGseq showed an extensive coverage of full-length mtDNA in hEPSm.8993T>C for detailed analyses of variants (Figure 3A, Table 1). It detected the m.8993T > C mutation in 74.2% of mtDNA, which matched the heteroplasmy levels observed in the patient's oocytes (70–90%, Figure 3A). Furthermore, five pathogenic SNVs were identified, two of which were nonsense mutations (m.4974G > A in the ND2 gene with a VAF of 0.19% and m.13031G > A in the ND5 gene with a VAF of 0.57%), while the rest (m.7426C > T, m.9140C > T and m.8111C > T, all with a VAF of 0.19%) were missense mutations and were annotated as ‘possibly pathogenic’ by MITOMAP (33). All five SNVs were below the detection limit of WGS and missed by the current diagnosis procedure. Interestingly, we found that all five pathogenic SNVs were linked to the m.8993T > C mutation, suggesting sequential acquisitions of detrimental mutations in defective mtDNA. The m.13031G > A mutation was identified in three UMI groups potentially due to clonal expansion by mtDNA replication in the patient's somatic cells (44) or during in vitro reprogramming and cell culture (Figure 3B). The genetic linkage of diverse pathogenic mutations in patient cells revealed by iMiGseq could help to understand the complexity of clinical symptoms in mitochondrial disease.
Figure 3.

iMiGseq of patient derived hEPSm.8993T>C cells. (A) Circular plots showing the distribution of SNVs in the mitochondrial genome of hEPSm.8993T>C. The arrangement of the circular plot is similar to Figure 1D and the m.8993T > C mutation is indicated by the red frame. (B) Sequential acquisition of detrimental mutations illustrated by Integrative Genomics Viewer (IGV) plots. Three UMI groups (mtDNA) with the same pathogenic m.13031G > A mutation (indicated in black under the plot) are shown. The putative disease-causing mutation of m.8993T > C is indicated in red under the plot. A de novo m.7797T > C mutation is found in one UMI group and indicated in black above the plot. (C) A schematic map of the mutant mtDNA harboring the 251-bp deletion and m.8993T > C mutation detected in hEPSm.8993T>C and the accompanying IGV tracks showing the alignment of Nanopore reads. The read triangle shows the position 8993 in the reads.
Single-molecule long-read sequencing approaches have unique advantages in resolving SVs previously inaccessible to short-read technologies (21,45). We expected that iMiGseq could overcome the challenges for SV analysis in mtDNA (39). We found that one of the m.8993T > C mutant mtDNA also harbored a 251-bp deletion in the D-loop region (Figure 3C), which is involved in mtDNA replication and transcriptional regulation (46). As this deletion was only detected in one mtDNA among the five sequenced cells, it is possible that the gain of the D-loop large deletion was spontaneous and detrimental to mtDNA replication. These results demonstrated that iMiGseq could not only detect and quantitate disease-causing mutations but also provide comprehensive characterizations of all types of mutations, including SNVs and SVs, and their genetic linkages with high sensitivity and specificity.
iMiGseq characterizes mitochondrial genome editing by mitoTALEN and mitochondrial base editor DdCBE at single-mtDNA level
Mitochondrial genome editing is a promising tool in the study and treatment of inherent mitochondrial diseases (47). Several studies have shown that the elimination or correction of mutant mtDNA by different genome editing tools, such as mitoTALEN and mitochondrial base editing, can rescue mitochondrial functions in disease models (25,26,48,49). Despite the advances in mtDNA editing tools, the consequent editing outcomes remain poorly characterized due to the limitations of existing analysis methods. Deep NGS sequencing of mtDNA is the conventional way of evaluating mtDNA editing, but NGS is unable to detect complex SVs and VAF changes of low-frequency variants (what are often the case for on-target or off-target edits) or to study the impact of editing on linked variants. Our method offers the opportunity to reappraise the genetic changes introduced by current mtDNA editing tools at the single mitochondrial genome level.
We first applied iMiGseq to our previously published mitoTALEN-edited mitochondrial encephalomyopathy and stroke-like episodes (MELAS) patient iPSCs (MiPSCs) (Table 1, Supplementary Figure S2D) (25). To compare the performance of iMiGseq and deep NGS, we also conducted Illumina 150-bp paired-end amplicon sequencing of unedited and edited MiPSCs with >100k coverage. Consistent with the original study (25), iMiGseq showed that 56.54% of unedited extensive-coverage UMI groups harbored the m.3243A > G mutation, and this mutation was completely eliminated after mitoTALEN editing as revealed by iMiGseq and confirmed by deep Illumina sequencing. In contrast to the common outcomes observed after repair of CRISPR-Cas9 induced double strand breaks (DSBs) in the nuclear genome (50), no increase of SNV, indel, or large SV was detected around the cut site following mitoTALEN editing. It suggested the lack of error-prone mtDNA repair mechanisms upon DSBs in mitochondria, which was also demonstrated in a HEK 293 cell line model (51).
Furthermore, iMiGseq enabled the haplotype-resolved analysis of rare variants in single edited mtDNA. It identified a dramatic VAF increase of the m.16289A > G SNV from 3.07% to 96.36% after mitoTALEN editing. The haplotype analysis of SNVs revealed the predominant linkage (99.14%) between m.3243G (the mitoTALEN target) and m.16289A in mutant mtDNA in non-editing MiPSCs (Figure 4A). It thus provided definitive evidence that mitoTALEN eliminated the mutant mtDNA that contained the linked m.16289A, leading to a near homoplasmic shift of m.16289G after mitoTALEN editing. In addition, iMiGseq detected a pathogenic SNV (m.3538G > T, p.Ala78Ser in ND1) with 1.07% VAF only in mitoTALEN-edited cells, while deep Illumina sequencing failed to report it. Other ultra-rare variants with <1% VAF, such as m.625G > A (VAF of 0.55%) detected in five UMI groups in non-editing cells, were totally masked by the background errors of NGS and not detected by deep Illumina sequencing (Figure 4A). The neglect of such rare pathogenic variants by conventional methods could constitute uncertainties of the safety of mtDNA editing.
Figure 4.

Characterization of mitochondrial genome editing by mitoTALEN and DdCBE. (A) The read alignment of unedited (the top track) and mitoTALEN edited (the second track) cells by deep Illumina sequencing, and two haplotypes (the bottom two tracks) of unedited cells by iMiGseq illustrated by IGV plots. The mitoTALEN target position (m.3243) is labeled by red frame. Bulk Illumina sequencing confirms the complete elimination of the m.3243A > G mutation but fails to detect the rare m.625G > A SNV (VAF of 0.55%). The two haplotypes by iMiGseq show the predominant linkage between m.16289A and m.3243G, and the infrequent linkage of m.16289G and m.3243G (only one UMI group is shown for each haplotype). Bulk Illumina sequencing is unable to conduct haplotype analysis. (B) Schematic representation of mitoTALEN and DdCBE editing for iMiGseq. 293T cells were seeded in collagen-coated 48-well plate, cultured for 24 hours for transfection of TALEN/DdCBE left half and right half plasmids. The transfected cells were cultured for another 6 days before cell harvest, followed by DNA extraction and iMiGseq. (C) The m.11922G > A SNV (on the top H-strand, i.e. C4> T on the bottom L-strand) (red triangle) installed by ND4-DdCBE is detected by iMiGseq (left, one UMI group is shown for each genotype) and confirmed by Sanger sequencing (right). A diagram of the ND4-DdCBE left and right TALEs and the target spacing region is shown below the sequencing results. The cytosines that are within the ND4-DdCBE editing window are highlighted. Cells received only the right side ND4-DdCBE TALE are used as the unedited control. (D) The VAF changes of primary SNVs (VAF > 1%) before and after ND4-DdCBE editing. No significant VAF shift is observed.
Mitochondrial base editing is a recently developed method that can install targeted mtDNA SNVs without inducing DSBs (26). Though it holds promise in the treatment of mitochondrial disorders, several recent papers purported that mtDNA base editors could induce substantial nuclear off-target mutations (52). The effects of mitochondrial base editing on full-length mtDNA genome remain unclear due to the limitations of current evaluation methods. Thus, we performed DddA-derived cytosine base editor (DdCBE)-directed mtDNA editing in HEK 293T cells as previously described (26), and applied iMiGseq to evaluate the editing outcomes (Figure 4B, Table 1, Supplementary Figure S2E).
The previously reported ND4-DdCBE (left side + right side TALE, Figure 4B) (26) successfully installed the m.11922G > A (C4 > T on the complimentary strand) variant in 11.58% extensive-coverage UMI groups as determined by iMiGseq and Sanger sequencing, whereas the right side ND4-DdCBE TALE alone showed no evidence of editing and served as the unedited control. iMiGseq confirmed ND4-DdCBE’s preference for editing C4 over C7 (26) (Figure 4C). Variant analysis showed that ND4-DdCBE did not lead to any SV in mtDNA, and there was no evidence of SNV increase around the targeted region. No editing related variants (C > T and other mutations around the targeted region) were detected in the base-edited mtDNA. We tracked SNV VAF change before and after base editing (Figure 4D). In contrast to mitoTALEN editing, iMiGseq showed that the heteroplasmic levels of SNVs remained stable after ND4-DdCBE editing. Notwithstanding the caveats of relatively low frequency of base editing and sequencing depth, our data suggested that DdCBE did not introduce any appreciable level of unintended mutations in the mitochondrial genome above the background.
Taken together, iMiGseq revealed the potential risk of unintended heteroplasmy shifts in elimination-based mitoTALEN editing, showed the strength of haplotype-resolved analysis of full-length mtDNA in understanding the heteroplasmy changes of mitochondrial DNA, and demonstrated the need for an ultra-sensitive method in the evaluation of mtDNA editing outcomes.
DISCUSSION
To the best of our knowledge, the development of iMiGseq represents the first demonstration of an unbiased high-throughput base-resolution analysis of individual full-length mtDNA in single cells. Taking advantages of molecular consensus sequencing and a purpose-built bioinformatics pipeline, iMiGseq greatly improves the sensitivity of mtDNA heteroplasmy detection, and enables the quantitative analysis of all types of mtDNA mutations and their genetic linkage in single cells.
iMiGseq allowed us to track the frequencies of variants among different cells. The analysis of oocytes from the same NSG female showed that the frequency of rare variants can fluctuate greatly (up to a hundredfold) among different cells. Additionally, phylogenetic trees of individual mtDNA suggest a sequential accumulation of de novo mutations in the same mtDNA molecule. iMiGseq could offer new opportunities to follow the dynamics of such SNVs during development and aging in hope of deciphering the emergence of mutations and understanding their clinical significance.
The current diagnosis of mitochondrial disease relied on prior knowledge of pathogenicity of mtDNA mutations. In the sequencing of patient cells, iMiGseq identified 23 amino acid substitution SNVs, of which 8 have not been linked to mitochondrial disease. The ability to unbiasedly discover mtDNA mutations in mitochondrial disease patients using iMiGseq could promote the identification of novel disease-causing mutations and benefit the diagnosis of mitochondrial disease.
Because NGS of mtDNA necessitates fragmentation of mtDNA molecules, it is impossible to ascertain the true haplotype of individual mtDNA molecules. It is thus extremely challenging to use short-read data to study genetic interactions between mutations and their haplotype backgrounds. Short reads also could be erroneously mapped to NUMTs, causing false variants (53). Taking advantages of long reads of Nanopore sequencing, iMiGseq could provide thousands of full-length mtDNA and their variants in a cell, which avoids NUMTs (see methods) and enables studies of interactions between different heteroplasmies. Additionally, nanopore-based IDMseq has been shown to offer superior characterization of SVs induced by CRISPR-Cas9 editing in human embryonic stem cells (21). mtDNAs with large SVs are expected to have severe functional defects and are thought to be rarely transmitted in the germline (4). Large mtDNA deletions have been described in multiple mitochondrial disorders (54) and in aging postmitotic tissues (55,56), and could, in some cases, even be preferentially replicated (57). Here, we showed that iMiGseq is capable to detect de novo large SVs in NAPR/Leigh syndrome patient cells. Analysis of such SVs in mtDNA has hitherto been limited to long-range PCR followed by Sanger sequencing (55,56), which is low throughput and non-quantitative. iMiGseq offers unbiased and sensitive detection and quantification of SVs in mtDNA.
Mitochondria genome editing tools promise to treat and/or model mitochondrial diseases, but the consequent editing outcomes remain poorly characterized due to the limitations of existing analysis methods. iMiGseq data revealed the potential risk of unintended heteroplasmy shifts in elimination-based mitoTALEN editing, showed the strength of haplotype-resolved analysis of full-length mtDNA in understanding the heteroplasmy changes of mitochondrial DNA, and demonstrated the need for an ultra-sensitive method in the evaluation of mtDNA editing outcomes.
Besides mitochondria genome editing, it is logical to extend iMiGseq technology to germline and somatic tissues to unravel the direction of causality between mtDNA mutations and aging and complex diseases in the future. Similarly, because iMiGseq works for different species and cell types, we expect it to be widely applicable to many fields for the study of this ancient organelle that energizes most life forms on earth.
DATA AVAILABILITY
VAULT and sample data in this study are accessible at GitHub (https://github.com/milesjor/vault) and Zenodo (https://doi.org/10.5281/zenodo.7450392).Raw sequencing data are available in the SRA database (accession ID PRJNA657296), which is accessible with the following link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA657296.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Li laboratory, Khaled Alsayegh for helpful discussions; Jinna Xu, Marie Krenz Y. Sicat and Doreena Chen for administrative support. We thank Chenyang Geng at the BIOPIC core facility at Peking university for technical assistance in PacBio sequencing. We thank Professor Jasmeen Merzaban's lab and KAUST Animal Research Core Laboratory for sharing mouse strains.
Author contributions: C.B. and L.W. performed majority of the experiments related to sequencing. M.L., G.R.M. and C.B. performed experiments related to mouse oocyte isolation. Y.F. and P.Z. performed the human oocyte collection and experiments related to hEPS cells. C.B. performed the bioinformatics analysis. B.Y., X.Z. and Y.S. performed cell culture and mtDNA labeling experiments. C.B. and J.W. performed experiments related to PacBio sequencing. C.B. performed mitoTALEN-related experiments. B.Y. performed DdCBE base editing with technical assistance and advice from Y.S. and P.R. B.Y., Y.Z. and S.A. performed iMiGseq of base editing experiments. C.B., J.C.I.B., Y.Y., Y.H. and M.L. analyzed the data. C.B. and M.L. wrote the manuscript. Y.H., Y.Y. and J.C.I.B. contributed to the writing of the manuscript. C.B. and M.L. conceived the study. Y.Y., J.C.I.B. and M.L. supervised the study.
Notes
Present address: Lin Wang. Key Laboratory of Zoonosis Research, Ministry of Education, Institute of Zoonosis, College of Veterinary Medicine, Jilin University, Changchun, China.
Contributor Information
Chongwei Bi, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Lin Wang, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Yong Fan, Department of Obstetrics and Gynecology, Key Laboratory for Major Obstetric Diseases of Guangdong Province, The Third Affiliated Hospital of Guangzhou Medical University, 510150 Guangzhou, China.
Baolei Yuan, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Gerardo Ramos-Mandujano, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Yingzi Zhang, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Samhan Alsolami, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Xuan Zhou, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Jincheng Wang, Beijing Advanced Innovation Center for Genomics (ICG), Biomedical Pioneering Innovation Center (BIOPIC), School of Life Sciences, College of Chemistry, College of Engineering, Peking-Tsinghua Center for Life Sciences, Peking University, Beijing, China.
Yanjiao Shao, Altos Labs, San Diego, CA, 92121, USA.
Pradeep Reddy, Altos Labs, San Diego, CA, 92121, USA.
Pu-Yao Zhang, Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing, 100191, China.
Yanyi Huang, Beijing Advanced Innovation Center for Genomics (ICG), Biomedical Pioneering Innovation Center (BIOPIC), School of Life Sciences, College of Chemistry, College of Engineering, Peking-Tsinghua Center for Life Sciences, Peking University, Beijing, China; Institute for Cell Analysis, Shenzhen Bay Laboratory, Shenzhen, China.
Yang Yu, Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing, 100191, China; Stem Cell Research Center, Peking University Third Hospital, Beijing, 100191, China.
Juan Carlos Izpisua Belmonte, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Altos Labs, San Diego, CA, 92121, USA.
Mo Li, Bioscience program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Bioengineering program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Li laboratory was supported by KAUST Office of Sponsored Research (OSR) [BAS/1/1080-01] and KAUST Competitive Research Grant [URF/1/3412-01-01 to M.L., Y.H., J.C.I.B.]. National Key R&D Program of China [2021YFC2700303]; National Natural Science Funds [82225019, 82192873, and 81971381]; MMAAP foundation (to Y.Y.). National Key Research and Development Program of China [2019YFA0110804, 2021YFC2700904]; National Natural Science Foundation of China [82071723]; Guangdong Basic and Applied Basic Research Foundation [2021B1515020069] (to Y.F.). Funding for open access charge: funding grants.
Conflict of interest statement. A patent application based on methods described in this paper has been filed by King Abdullah University of Science and Technology, in which C.B., L.W. and M.L. are listed as inventors. The authors declare no other competing interest.
This paper is linked to: doi:10.1093/nar/gkad209.
REFERENCES
- 1. Kukat C., Wurm C.A., Spahr H., Falkenberg M., Larsson N.G., Jakobs S.. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Nat. Acad. Sci. U.S.A. 2011; 108:13534–13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zaidi A.A., Wilton P.R., Su M.S., Paul I.M., Arbeithuber B., Anthony K., Nekrutenko A., Nielsen R., Makova K.D.. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc. Nat. Acad. Sci. U.S.A. 2019; 116:25172–25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rebolledo-Jaramillo B., Su M.S., Stoler N., McElhoe J.A., Dickins B., Blankenberg D., Korneliussen T.S., Chiaromonte F., Nielsen R., Holland M.M.et al.. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Nat. Acad. Sci. U.S.A. 2014; 111:15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallace D.C., Chalkia D.. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013; 5:a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A.et al.. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012; 488:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stewart J.B., Chinnery P.F.. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 2015; 16:530–542. [DOI] [PubMed] [Google Scholar]
- 7. Schon E.A., DiMauro S., Hirano M.. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012; 13:878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F.. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008; 83:254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Payne B.A., Gardner K., Coxhead J., Chinnery P.F.. Deep resequencing of mitochondrial DNA. Methods Mol. Biol. 2015; 1264:59–66. [DOI] [PubMed] [Google Scholar]
- 10. Yuan Y., Ju Y.S., Kim Y., Li J., Wang Y., Yoon C.J., Yang Y., Martincorena I., Creighton C.J., Weinstein J.N.et al.. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020; 52:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morris J., Na Y.J., Zhu H., Lee J.H., Giang H., Ulyanova A.V., Baltuch G.H., Brem S., Chen H.I., Kung D.K.et al.. Pervasive within-mitochondrion single-nucleotide variant heteroplasmy as revealed by single-mitochondrion sequencing. Cell Rep. 2017; 21:2706–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arbeithuber B., Hester J., Cremona M.A., Stoler N., Zaidi A., Higgins B., Anthony K., Chiaromonte F., Diaz F.J., Makova K.D.. Age-related accumulation of de novo mitochondrial mutations in mammalian oocytes and somatic tissues. PLoS Biol. 2020; 18:e3000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duan M., Tu J., Lu Z.. Recent advances in detecting mitochondrial DNA heteroplasmic variations. Molecules. 2018; 23:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li M., Schroder R., Ni S., Madea B., Stoneking M.. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Nat. Acad. Sci. U.S.A. 2015; 112:2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kennedy S.R., Salk J.J., Schmitt M.W., Loeb L.A.. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013; 9:e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kong M., Guo L., Xu W., He C., Jia X., Zhao Z., Gu Z.. Aging-associated accumulation of mitochondrial DNA mutations in tumor origin. Life Med. 2022; 1:149–167. [Google Scholar]
- 17. Ludwig L.S., Lareau C.A., Ulirsch J.C., Christian E., Muus C., Li L.H., Pelka K., Ge W., Oren Y., Brack A.et al.. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell. 2019; 176:1325–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burgstaller J.P., Kolbe T., Havlicek V., Hembach S., Poulton J., Pialek J., Steinborn R., Rulicke T., Brem G., Jones N.S.et al.. Large-scale genetic analysis reveals mammalian mtDNA heteroplasmy dynamics and variance increase through lifetimes and generations. Nat. Commun. 2018; 9:2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H., Slone J., Fei L., Huang T.. Mitochondrial DNA variants and common diseases: a mathematical model for the diversity of age-related mtDNA mutations. Cells. 2019; 8:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ancora M., Orsini M., Colosimo A., Marcacci M., Russo V., De Santo M., D’Aurora M., Stuppia L., Barboni B., Camma C.et al.. Complete sequence of human mitochondrial DNA obtained by combining multiple displacement amplification and next-generation sequencing on a single oocyte. Mitochondrial DNA. 2017; 28:180–181. [DOI] [PubMed] [Google Scholar]
- 21. Bi C., Wang L., Yuan B., Zhou X., Li Y., Wang S., Pang Y., Gao X., Huang Y., Li M.. Long-read individual-molecule sequencing reveals CRISPR-induced genetic heterogeneity in human escs. Genome Biol. 2020; 21:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reiner J.E., Kishore R.B., Levin B.C., Albanetti T., Boire N., Knipe A., Helmerson K., Deckman K.H.. Detection of heteroplasmic mitochondrial DNA in single mitochondria. PLoS One. 2010; 5:e14359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Y., Liu B., Xu J., Wang J., Wu J., Shi C., Xu Y., Dong J., Wang C., Lai W.et al.. Derivation of pluripotent stem cells with In vivo embryonic and extraembryonic potency. Cell. 2017; 169:243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Filges S., Yamada E., Stahlberg A., Godfrey T.E.. Impact of polymerase fidelity on background error rates in next- generation sequencing with unique molecular identifiers/barcodes. Sci Rep. 2019; 9:3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Y., Wu H., Kang X., Liang Y., Lan T., Li T., Tan T., Peng J., Zhang Q., An G.et al.. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell. 2018; 9:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mok B.Y., de Moraes M.H., Zeng J., Bosch D.E., Kotrys A.V., Raguram A., Hsu F., Radey M.C., Peterson S.B., Mootha V.K.et al.. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020; 583:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Proc G.P.D.. The sequence alignment/map format and samtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X.Y., Ruden D.M.. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly. 2012; 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sedlazeck F.J., Rescheneder P., Smolka M., Fang H., Nattestad M., von Haeseler A., Schatz M.C.. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods. 2018; 15:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ni T., Wei G., Shen T., Han M., Lian Y., Fu H., Luo Y., Yang Y., Liu J., Wakabayashi Y.et al.. MitoRCA-seq reveals unbalanced cytocine to thymine transition in Polg mutant mice. Sci. Rep. 2015; 5:12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lott M.T., Leipzig J.N., Derbeneva O., Xie H.M., Chalkia D., Sarmady M., Procaccio V., Wallace D.C.. mtDNA variation and analysis using Mitomap and Mitomaster. Curr. Protoc. Bioinformatics. 2013; 44:1.23–1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin Y.C., Boone M., Meuris L., Lemmens I., Van Roy N., Soete A., Reumers J., Moisse M., Plaisance S., Drmanac R.et al.. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014; 5:4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Costello M., Pugh T.J., Fennell T.J., Stewart C., Lichtenstein L., Meldrim J.C., Fostel J.L., Friedrich D.C., Perrin D., Dionne D.et al.. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013; 41:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ju Y.S., Futreal C.A., Gerstung M., Martincorena I., Nik-Zainal S., Ramakrishna M., Davies H.R., Papaemmanuil E., Gundem G., Shlien A.et al.. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife. 2014; 3:e02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z.et al.. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016; 164:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gorman G.S., Schaefer A.M., Ng Y., Gomez N., Blakely E.L., Alston C.L., Feeney C., Horvath R., Yu-Wai-Man P., Chinnery P.F.et al.. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015; 77:753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schon K.R., Ratnaike T., van den Ameele J., Horvath R., Chinnery P.F.. Mitochondrial diseases: a diagnostic revolution. Trends Genet. 2020; 36:702–717. [DOI] [PubMed] [Google Scholar]
- 40. Parikh S., Goldstein A., Koenig M.K., Scaglia F., Enns G.M., Saneto R., Anselm I., Cohen B.H., Falk M.J., Greene C.et al.. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015; 17:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riley L.G., Cowley M.J., Gayevskiy V., Minoche A.E., Puttick C., Thorburn D.R., Rius R., Compton A.G., Menezes M.J., Bhattacharya K.et al.. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020; 22:1254–1261. [DOI] [PubMed] [Google Scholar]
- 42. Fan Y., Min Z., Alsolami S., Ma Z., Zhang E., Chen W., Zhong K., Pei W., Kang X., Zhang P.et al.. Generation of human blastocyst-like structures from pluripotent stem cells. Cell Discov. 2021; 7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sozen B., Jorgensen V., Weatherbee B.A.T., Chen S., Zhu M., Zernicka-Goetz M.. Reconstructing aspects of human embryogenesis with pluripotent stem cells. Nat. Commun. 2021; 12:5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greaves L.C., Nooteboom M., Elson J.L., Tuppen H.A., Taylor G.A., Commane D.M., Arasaradnam R.P., Khrapko K., Taylor R.W., Kirkwood T.B.et al.. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLos Genet. 2014; 10:e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Logsdon G.A., Vollger M.R., Eichler E.E.. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020; 21:597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barshad G., Marom S., Cohen T., Mishmar D. Mitochondrial DNA transcription and its regulation: an evolutionary perspective. Trends Genet. 2018; 34:682–692. [DOI] [PubMed] [Google Scholar]
- 47. Silva-Pinheiro P., Minczuk M.. The potential of mitochondrial genome engineering. Nat. Rev. Genet. 2022; 23:199–214. [DOI] [PubMed] [Google Scholar]
- 48. Mok B.Y., Kotrys A.V., Raguram A., Huang T.P., Mootha V.K., Liu D.R.. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 2022; 40:1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reddy P., Ocampo A., Suzuki K., Luo J., Bacman S.R., Williams S.L., Sugawara A., Okamura D., Tsunekawa Y., Wu J.et al.. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015; 161:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bi C., Wang L., Yuan B., Zhou X., Pang Y., Li Y., Gao Y.X., Huang Y., Li M.. Long-read individual-molecule sequencing reveals CRISPR-induced genetic heterogeneity in Human escs. Genome Biol. 2020; 21:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moretton A., Morel F., Macao B., Lachaume P., Ishak L., Lefebvre M., Garreau-Balandier I., Vernet P., Falkenberg M., Farge G.. Selective mitochondrial DNA degradation following double-strand breaks. PLoS One. 2017; 12:e0176795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lei Z., Meng H., Liu L., Zhao H., Rao X., Yan Y., Wu H., Liu M., He A., Yi C.. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature. 2022; 606:804–811. [DOI] [PubMed] [Google Scholar]
- 53. Santibanez-Koref M., Griffin H., Turnbull D.M., Chinnery P.F., Herbert M., Hudson G.. Assessing mitochondrial heteroplasmy using next generation sequencing: a note of caution. Mitochondrion. 2019; 46:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nissanka N., Minczuk M., Moraes C.T.. Mechanisms of mitochondrial DNA deletion formation. Trends Genet. 2019; 35:235–244. [DOI] [PubMed] [Google Scholar]
- 55. Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T.et al.. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006; 38:515–517. [DOI] [PubMed] [Google Scholar]
- 56. Kraytsberg Y., Kudryavtseva E., McKee A.C., Geula C., Kowall N.W., Khrapko K.. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006; 38:518–520. [DOI] [PubMed] [Google Scholar]
- 57. Diaz F., Bayona-Bafaluy M.P., Rana M., Mora M., Hao H., Moraes C.T.. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002; 30:4626–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
VAULT and sample data in this study are accessible at GitHub (https://github.com/milesjor/vault) and Zenodo (https://doi.org/10.5281/zenodo.7450392).Raw sequencing data are available in the SRA database (accession ID PRJNA657296), which is accessible with the following link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA657296.