

Abstract
As atherosclerosis begins in childhood, early diagnosis and treatment of familial hypercholesterolemia (FH) is considered necessary. The basic diagnosis of pediatric FH (under 15 years of age) is based on hyper-low-density lipoprotein (LDL) cholesterolemia and a family history of FH; however, in this guideline, to reduce overlooked cases, “probable FH” was established. Once diagnosed with FH or probable FH, efforts should be made to promptly provide lifestyle guidance, including diet. It is also important to conduct an intrafamilial survey, to identify family members with the same condition. If the level of LDL-C remains above 180 mg/dL, drug therapy should be considered at the age of 10. The first-line drug should be statin. Evaluation of atherosclerosis should be started using non-invasive techniques, such as ultrasound. The management target level is an LDL-C level of less than 140 mg/dL. If a homozygous FH is suspected, consult a specialist and determine the response to pharmacotherapy with evaluating atherosclerosis. If the response is inadequate, initiate lipoprotein apheresis as soon as possible.
Keywords: Pediatric, Familial Hypercholesterolemia, Homozygote, Heterozygote, Diagnostic criteria, Treatment guidelines, Lifestyle, Pharmacological therapy, Low-density lipoprotein apheresis, Statins
Introduction
Atherosclerotic pathological changes can already begin to develop in childhood; this is evidenced by the autopsy findings of the Bogalusa Heart Study 1) and Pathological Determinants of Atherosclerosis in Youth (PDAY) 2) . Familial hypercholesterolemia (FH) is an inherited disorder caused by mutations in the low-density lipoprotein (LDL) receptor (LDLR) and its related genes, resulting in hyper-LDL cholesterolemia. This condition where one is life-long exposed to elevated LDL-C puts him or her at high-risk for atherosclerosis, making early diagnosis and early intervention important. For patients with FH, there has long been no consensus in Japan on its screening in childhood (<15 years of age), the type and starting age of treatment, the assessment of atherosclerosis, and the goals of treatment. In 2017, the Japan Pediatric Society and the Japan Atherosclerosis Society jointly established the Guidance for Pediatric Familial Hypercholesterolemia 2017 (hereafter, Pediatric FH Guide 2017) 3) and proposed one guideline. This time, after 5 years, the entire guideline was reviewed and revised. Although there is little evidence in pediatric patients, we set up clinical questions, conducted an evidence evaluation, and used them in this new guideline for medical practice. As in the previous edition, the aim is to prevent possible future atherosclerosis by diagnosing the disease at an early stage and applying therapeutic intervention.
1. Background Question (BQ)/ Foreground Question (FQ)
BQ1. What is the Prevalence of FH in Japan?
In general, it is found in approximately 1 in 300 people in the general population, 1 in 30 people with coronary artery disease, and 1 in 15 people with premature coronary artery disease (CAD) or severe hyper-LDL cholesterolemia. (Level of evidence: E-2)
The prevalence of FH has been reported to be 1 in 500 people, but in recent years, a series of cross-sectional and cohort studies in the United States and Europe have shown a clearly higher prevalence than that. Moreover, in Japan, Mabuchi et al. examined the frequency of molecular epidemiology in the Hokuriku region and reported a prevalence rate of 1 in 208 people 4) .
A systematic review/meta-analysis published in 2017 (not including Japanese) reported a frequency of 1 in 250 in the general population 5) . In addition, a subsequent systematic review/meta-analysis (including Japanese) reported a frequency of 1 in 313 6) and 311 7) people, respectively, in the general population. The meta-analysis, which reported 1 in 250 people had FH, included literature reporting extremely high frequencies due to the so-called founder effect, whereas the latter two reports excluded such studies. As the effect of increased frequency due to founder effect is assumed to exist in some regions, it is reasonable to assume that the number of cases is about 1 in 300 people, contrary to the report by Mabuchi et al. The number of Japanese in the above meta-analysis is small, and it is assumed that differences in frequency may be attributed to factors such as consanguineous marriages in some regions and demographic bottleneck effects, but there is no evidence of significant differences among races, so the results of the above meta-analysis may be applicable to Japanese as well.
BQ2. What are the Prognosis and Main Complications of Patients with FH ?
• CAD: odds ratio 10–20 times higher than non-FH (Level of evidence: E-1a*)
• Peripheral arterial disease: odds ratio 5–10 times higher than non-FH (Level of evidence: E-1a)
• Stroke: no clear impact (Level of evidence: E-1a)
• Aortic valve stenosis: no epidemiological association has been shown, but there have been case reports of FH complicating the disease. (Level of evidence: E-3)
• Abdominal aortic aneurysm: no epidemiological association has been shown, but there have been case reports of FH complicating the disease. (Level of evidence: E-3)
*Although there was no meta-analysis of cohort studies, we chose E-1a because of the existence of multiple cohort studies and identical results.
The major complication of systemic atherosclerosis in patients with FH is CAD 8) . A significantly higher prevalence of peripheral arterial disease and carotid atherosclerosis has been reported in addition to CAD compared to non-FH (systematic review/meta-analysis) 9 , 10) . On the other hand, with regard to stroke, many reports state that its effects are not clear. Regarding aortic disease and valvular disease (aortic aneurysm, aortic stenosis, supravalvular stenosis, etc.), there are case reports showing an association with the disease, although no epidemiological reports have shown such an association.
Although there are no randomized controlled trials (RCTs) or systematic reviews on the prognosis of FH, Mabuchi et al. presented a study comparing the prognosis of FH heterozygotes (HeFH) and FH homozygotes (HoFH) in the pre- and post-statin era in Japan 11) . This report shows that before the advent of statins, 73% of men and 64% of women who suffered from HeFH died of cardiac death; the age of death for HeFH increased from an average of 63 years before statins to 76 years after statins; meanwhile, for HoFH, the average age of death increased from 28 years to 59 years after statins.
FQ1. Can Statins be Recommended as the First Choice in Drug Therapy for HeFH?
Strict lipid management with statins as first-line drug is recommended for the treatment of HeFH. (Level of evidence: 3; Recommendation level: A)
In total, there are 13 randomized, double-blind trials examining the LDL-C lowering effect of statin in HeFH 12 - 24) , including 9 placebo-controlled trials (2 in adult HeFH 12 , 13) , 6 in children to adolescent HeFH 15 - 20) , and 1 in adult FH subjects without mention of whether they are HeFH or HoFH 14) ). Its efficacy and safety have been established in both adult and pediatric cases. Furthermore, a randomized, double-blind, crossover study in HoFH has shown its efficacy 21) . In Japan, reports have been published showing that statin therapy reduces LDL-C in adults and pediatric patients 23 , 24) , which is consistent with the results of clinical trials in other countries.
A randomized, double-blind, comparative study has been published in adults 25) and pediatric patients 20) , respectively, to investigate the efficacy of high-intensity statin therapy (80 mg atorvastatin, twice the Japanese approved dose in Japan) compared to standard-intensity statin therapy (40 mg simvastatin, twice the approved dose in Japan) in adults and standard-intensity statin therapy (20–40 mg pravastatin, the approved dose is 20 mg in Japan and no pediatric indication) compared to placebo in children. Both studies showed inhibition of the development of carotid artery intima Media Thickness (IMT). A sub-analysis of pediatric cases showed that statin initiation at an earlier age was associated with less IMT thickening 26) . In addition, a report of an observational study examining 2447 cases from the Netherlands has shown that statin use is associated with a lower incidence of CAD and lower all-cause mortality 27) . Although scientific evidence for prevention of atherosclerotic cardiovascular disease by direct comparison is deemed insufficient, statins appear to be the most recommended drug therapy at this time, given the abundant evidence in non-FH.
Four clinical studies that compare the LDL-C lowering effects of statins and other lipid-lowering drugs in FH have been published (all in adults, all with pravastatin, 2 in HeFH, and 2 in FH subjects). In a report comparing pravastatin 40 mg with cholestyramine 4 g (or cholestipol 5 g), both groups had significantly lower LDL-C from baseline, but the group of pravastatin had significantly lower LDL-C than the cholestyramine group 13) . On the other hand, reports comparing 40 mg of pravastatin with 16–24 g of cholestyramine (1.5–2 times the Japanese approved dose) did not show significant differences between the two groups, although both groups had significantly lower LDL-C from baseline 12 , 14 , 28) . These studies were conducted with pravastatin in the early 1990s, and since the newer generation of statins shows stronger lipid-lowering effects, statins are expected to be more effective than cholestyramine. Although there exist no direct comparative studies between statins and probucol, cholesterol transporter inhibitor at small intestine (ezetimibe), or proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, there have been reports of trials of ezetimibe and PCSK9 inhibitors on top of statins 29 - 32) .
FQ2. Is Lipoprotein Apheresis Therapy Recommended for HoFH and Severe HeFH with Drug Resistance ?
For HoFH and severe HeFH with drug resistance, strict control of LDL-C is recommended with lipoprotein apheresis therapy. (Level of evidence: 3; Recommendation level: A)
In the 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk 33) , although there is no mention of evidence levels, etc., the lipoprotein apheresis is recommended as a treatment for HoFH.
A systematic review reported in 2016 analyzed a total of 38 articles (8 open-label clinical trials, 11 observational studies, 17 reviews/guidelines, and 2 medical technology evaluations) 34) . Although RCTs were not included, they have been shown to have clinical benefits in terms of lowering LDL-C, lowering lipoprotein (a) [Lp (a)], and preventing cardiovascular events. As each country has different rates of diagnosis of FH, availability and access to apheresis treatment, indications, methods, and costs, it will be necessary to evaluate the situation in Japan in the future. However, although there is a physical and social burden on the patient, we believe that lipoprotein apheresis is recommended for patients with FH who do not respond adequately to drug therapy in Japan, where access is relatively easy in terms of transportation and cost.
In 2019, a systematic review that examined 76 case reports (209 patients) of children with HoFH was reported 35) . Although it has not been shown whether lipoprotein apheresis is more or less beneficial than drug therapy alone for cardiovascular outcomes, it has been reported to lower LDL-C and reduce xanthomas with fewer adverse events, making it safe in general.
FQ3. Is it Recommended to Start Treatment Early in Pediatric Patients with FH?
FH is a high-risk condition for atherosclerotic diseases; thus, early initiation of treatment is recommended, depending on LDL-C levels*.
(Level of evidence: Consensus, Level of recommendation: A)
*See Flowchart of pediatric HeFH treatment ( Fig.3 in the text)
Fig.3.
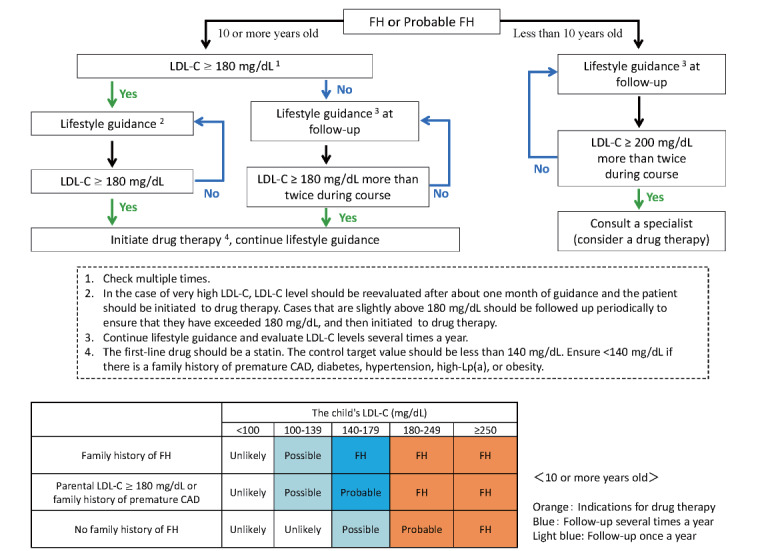
Flowchart of treatment for pediatric FH heterozygotes
The US Food and Drug Administration guidelines “approve” pravastatin for pediatric FH starting at age 8 and other statins starting at age 10 36) . Our policy of “approving” pitavastatin for ages 10 and older, as we stated in Pediatric FH Guide 2017 3) , is in line with the global trend. The 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk 33) also has a level of evidence/recommendation of Class IIa, Level C, but it states that pediatric FH treatment should begin with statins at 8–10 years of age, with a goal of <135 mg/dL at ages 10 years and older. “Statins for children with familial hypercholesterolemia,” which summarized nine studies using statins, found that statin therapy in children with FH successfully and safely reduced LDL-C without affecting liver function, muscle symptoms, muscle damage, and growth. They have also reported that carotid atherosclerosis can be reduced and some endothelial function improved, although the level of evidence is not high 37) . Furthermore, to date, RCTs and meta-analyses of RCTs and systematic reviews have shown that the use of statins, resins, and ezetimibe in pediatric patients of 10 years and older is safe and effective in lowering LDL-C levels.
Due to the limited number of RCTs that used statins in children due to the nature of their subjects and the short duration of statin treatment, no studies have been able to evaluate the incidence of atherosclerotic cardiovascular disease (ASCVD), cardiovascular death, and long-term safety. However, a 20-year follow-up study showed that starting statins at 13.0±2.9 years (mean LDL-C 237.3 mg/dL) did not cause any mortality related to ASCVD by at least the age of 39 years, although the mean LDL-C level reached 160.7 mg/dL 38) . Since LDL-C accumulation levels, over time, are believed to be associated with the development of ASCVD, and since FH is a high-risk condition for atherosclerotic disease, early initiation of treatment in children is thus recommended.
2. Outline of FH
Key Points
• FH is an inherited disease associated with mutations in the LDL receptor (LDLR) and related genes and is inherited mainly in an autosomal dominant form.
• The frequency of FH in the general population is approximately 1 in 300 people and is even more frequent in patients with CAD, especially those with premature CAD.
• FH is classified into two categories, namely, heterozygotes and homozygotes, according to the number of pathogenic gene mutations.
What is FH: Its Etiology, Frequency, and Major Signs
FH is defined as an autosomal dominant hereditary disease in which mutations occur in the LDLR and its related genes. A recent meta-analysis of molecular epidemiological studies 6 , 7) suggests that the frequency of HeFH in the general population is approximately 1 in 300 people worldwide. The meta-analysis of these molecular epidemiological studies includes data from Japan, with a total sample size of more than several million people, and is extremely important to determine the frequency of this disease. Furthermore, the frequency of FH in patients with CAD is thought to be approximately 1 in 30 people, while the frequency of FH in patients with premature CAD is approximately 1 in 15 people 6) . Currently, the diagnosis rate of FH, especially in children, is estimated to be extremely low, despite the fact that FH is considered the most common hereditary metabolic disease and is commonly encountered in daily medical practice.
The main clinical features of FH are hyper-LDL cholesterolemia, tendon and skin xanthomas, and premature CAD (onset of CAD before age 55 in men and 65 in women). However, it should be noted in pediatric HeFH that LDL-C levels decrease during puberty, as discussed below, and tendon xanthomas and premature CAD are rarely seen.
FH Causative Genes
Three genes are known to cause FH, namely, LDLR, which encode the LDL receptor; APOB, which encode the apolipoprotein B-100; and PCSK9, which encode the PCSK9 protein. If there is one pathogenic mutation, it corresponds to HeFH, and if there are two, it refers to HoFH (including compound heterozygotes and double heterozygotes). Most FH is caused by LDLR mutations; however, in Japan, about 5% of HeFH is determined to be caused by PCSK9 gain-of-function mutations 11) . APOB mutations are relatively frequent in Europe due to the founder effect, but extremely rare in Japan 39) .
On the other hand, autosomal recessive hypercholesterolemia (ARH) is an extremely rare disorder caused by the biallelic pathogenic mutations in low-density lipoprotein receptor adaptor protein 1 (LDLRAP1). ARH should be treated as HoFH. Carrier parents may present with mild hyper-LDL cholesterolemia, but usually do not develop the FH heterozygote phenotype 40) . ARH is very rare, with only a few cases reported in Japan 40) .
Recently, in addition to FH associated with pathogenic genetic mutations of the LDLR and related genes as described above, it is suggested that there is so-called polygenic FH associated with the superposition of high-frequency genetic polymorphisms at loci involved in LDL metabolism 41) . However, there is no evidence that the superposition of high-frequency gene polymorphisms alone, in any combination known at this time, causes the so-called FH, which is believed to influence the phenotype of FH (and other hyper-LDL cholesterolemia) rather than being involved in the pathogenesis of FH.
Genotype Designations
Basically, HeFH has one pathogenic mutation and HoFH has two. HoFH may be further differentiated according to genotype as follows ( Fig.1 ) : Autosomal mutated genes are inherited from both the paternal and maternal sides of the family. Two identical mutations of the same gene are identified as true homozygotes, a combination of the same gene but different mutations is a compound heterozygote, and a combination of different genes is a double heterozygote.
Fig.1.
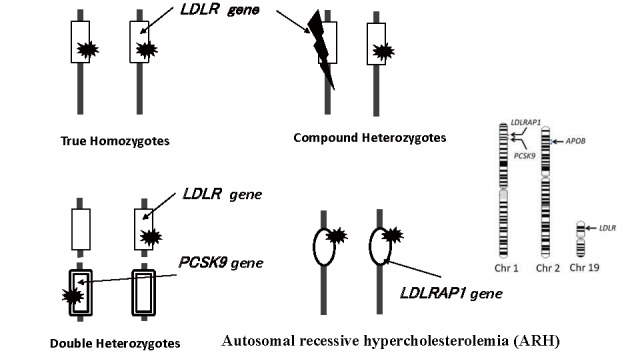
Combination of genetic mutation showing HoFH clinically
Signs of FH (Arteriosclerosis, Xanthomas, etc.)
Atherosclerotic disease is believed to progress in proportion to cumulative LDL-C levels 42) . In cases of FH, their cumulative LDL-C levels reach the threshold for developing CAD earlier due to prolonged exposure to marked hyper-LDL cholesterolemia since birth. Therefore, early intervention is necessary to prevent the development of atherosclerosis. Although premature CAD is one of the common phenotypes of FH in general, the degree of atherosclerotic lesions throughout the body can vary with age and the degree of concomitant risk factors. In particular, complications of aortic valve disease should be given the proper attention in FH practice, as it can be a cause of death at a young age. Xanthomas are the result of exposure to marked hyper-LDL cholesterolemia during childhood when the tissues are flexible. Tendon xanthomas are a highly specific physical finding in FH, except in rare conditions such as sitosterolemia and cerebral tendon xanthomatosis. However, it is difficult to use the presence of tendon xanthomas to diagnose a pediatric FH, because these often manifest in adulthood or later. On the other hand, if a child presents with xanthomas of the skin or tendons, homozygosity is strongly suspected, and consultation with a specialist is mandatory.
3. Diagnosis of Pediatric FH ( Table 1 )
Table 1. Diagnostic criteria for pediatric FH (under the age of 15).
| 1. Hyper-LDL cholesterolemia (untreated LDL-C level ≥ 140 mg/dL, confirmed multiple times) |
| 2. Family history of FH (Parents or siblings) |
| 3. Parental LDL-C ≥ 180 mg/dL or family history of premature coronary artery disease (Grand parent or parent) |
| After ruling out other primary and secondary Hyper-LDL cholesterolemia, |
|
|
|
|
|
|
|
Key Points
• Diagnose “FH” when there is hyper-LDL cholesterolemia (≥ 140 mg/dL) and a family history of FH (in parents and siblings).
• Diagnose “probable FH” when there is hyper-LDL cholesterolemia with a high parental LDL-C level (≥ 180 mg/dL) or with a family history of premature CAD in grandparents or parents (item 3).
• If the LDL-C is ≥ 180 mg/dL, diagnose “probable FH” on that basis alone. If item 3 is present in addition to that, diagnose “FH.”
• Cases with LDL-C ≥ 250 mg/dL and those who are positive for pathogenic gene mutations of FH are diagnosed as “FH.”
• Whenever a patient with “FH” or “probable FH” is diagnosed, a family survey should be conducted, in order to identify family members with FH.
Need for Proactive Diagnosis of FH from Childhood
In pediatric HeFH, hypercholesterolemia is present from birth, as described above, but few symptoms related to atherosclerosis have been observed. Therefore, in most cases, hyper-LDL cholesterolemia is detected by chance after a blood test at a pediatric checkup of lifestyle-related diseases or for other diseases, and a diagnosis rate of FH is low in Japan 42) . In Europe and the United States, pediatric FH diagnosis and treatment guidelines have been established, and active medical care is being provided 43) . In Japan, the Japan Pediatric Society and the Japan Arteriosclerosis Society jointly developed the first Pediatric FH Guide 2017 3) 5 years ago. Based on subsequent clinical evidence, the diagnostic criteria have been revised in 2022.
The diagnosis of FH can be made clinically or genetically, but the former is the basis for the diagnosis of pediatric FH. However, since it cannot be ruled out that there may be some cases that fall outside the clinical diagnosis, additional genetic testing should be considered.
HeFH typically does not show cutaneous xanthomas or thickening of the Achilles tendon in childhood. Therefore, the diagnosis is based on hyper-LDL cholesterolemia and family history after excluding other dyslipidemias. In the Pediatric FH Guide 2017 3) , the diagnosis was based on two items: hyper-LDL cholesterolemia and family history of FH or premature CAD. In reality, however, it remains difficult to obtain a detailed family history, and there are many cases in which FH is not diagnosed, especially in lifestyle-related disease checkups 44) . Therefore, in the diagnostic criteria of the current guideline, a new category of “probable FH” was established. There are some differences from the simultaneously revised guidelines for adult FH (15 years and older), not only in terms of the value of high LDL-C but also items of family history. This is because we believe that it is important to increase sensitivity while maintaining specificity and to actively follow-up probable cases from the early stage to reduce the number of missed cases of FH in childhood.
Diagnostic Criteria for Pediatric FH
Table 1 shows the diagnostic criteria of this newly developed Guidelines for the Diagnosis and Treatment of Pediatric FH 2022.
The first step is to differentiate primary or secondary hyper-LDL cholesterolemia. It is possible that the patient originally has FH and is concurrently suffering from other diseases. Obese children do not always have elevated LDL-C. It is thus necessary to conduct a thorough family history interview and always keep FH in mind. When hyper-LDL cholesterolemia is found, FH and other diseases should actually be differentiated simultaneously.
Item 1 is the criterion for hyper-LDL cholesterolemia; a value of ≥ 140 mg/dL (95th percentile) is considered hyper-LDL cholesterolemia 45) . If total cholesterol (TC) is tested and is ≥ 220 mg/dL (95th percentile value), an LDL-C value should be obtained. This reference value can be used throughout childhood, but some infants can exhibit hyper-LDL cholesterolemia due to breastfeeding, in which case, the test should be repeated after weaning. Serum LDL-C measurement is based on the Friedewald method, although a fasting blood sample is required. If fasting blood sampling is difficult, the direct method is then used, and non-HDL-C (≥ 150 mg/dL is considered high) is also used as a reference 46) . Furthermore, because LDL-C levels fluctuate (decrease) physiologically during puberty, LDL-C should be measured and evaluated multiple times 47) .
Serum LDL-C levels in childhood decrease continuously from the age of puberty onset 48) and begin to increase after puberty is completed 47) . In other words, by considering the stage of puberty, it may be possible to predict the next variation in the LDL-C measurements 47) . The Tanner classification is a useful and convenient way to assess the stage of puberty 49) . The average decrease in LDL-C levels during puberty is approximately 10 mg/dL 48) , but naturally there are individual differences in the age at which puberty begins and its duration. The range of variability in LDL-C is also likely to vary between individuals. Although multiple reference values for LDL-C appear in this guideline, multiple tests should be performed to ensure that the reference values are met.
If the LDL-C is ≥ 250 mg/dL, this alone is sufficient to diagnose “FH.” It is difficult to imagine any other dyslipidemia in children with such high levels of LDL-C other than FH, and FH is strongly suspected even in adults 50) . Furthermore, in studies in which children were genetically tested, children with ≥ LDL-C 250 mg/dL were generally found positive for pathogenic gene mutations in the LDLR or PCSK9 44 , 51) ; negative cases are extremely rare. In such cases of markedly elevated LDL-C, the presence of xanthomas should also be investigated, and the possibility of HoFH should be considered.
Furthermore, since 180 mg/dL of LDL-C is approximately the 99.7th percentile value in children 44) , LDL-C ≥ 180 mg/dL alone was considered “probable” FH. FH with pathogenic gene mutation-positive cases have significantly higher levels of LDL-C than mutation-negative cases. Many FH cases with the pathogenic gene mutation would be included if ≥ 180 mg/dL. The cutoff value of 180 mg/dL is considered to be fair for receiver operating characteristic analysis 44 , 51) .
Item 2 is the family history of FH. In the previous Guidance for Pediatric FH 2017, the family history of FH and that of premature CAD were treated equally, but in this guideline, we separated the family history of FH from that of premature CAD. This is because premature CAD is sometimes due to causes other than FH. In addition, instead of using the term “parentage” as previously employed, FH’s family history is to be examined by blood parents and siblings (first-degree relatives). Diagnose “FH” on items 1 and 2. If the grandfather or grandmother was diagnosed with FH, the father or mother is likely to have FH and thus requires close examination.
Item 3 is another family history set as an item followed by the item 2. When diagnosing pediatric FH, it is not always clear whether the parents have FH or not because information on grandparents is not often available. So, if the LDL-C of the father or mother is ≥ 180 mg/dL, it is highly likely associated with FH due to age in the absence of other diseases; in the 20s, the mean +2 SD is approximately 160 mg/dL, and in the 30s, it is approximately 180 mg/dL for men and 170 mg/dL for women 47 , 52) . This content was noted within the previous diagnostic flowchart 3) .
Premature CAD is defined as CAD that occurs at <55 years of age in men or <65 years of age in women. For premature CAD, it was decided to interview both parents and grandparents, since it is impossible to determine this in children without including grandparents. The combination of item 1 and item 3 results in “probable FH.” However, if the child’s LDL-C is very high (≥ 180 mg/dL) and item 3 is present, the diagnosis of “FH” is made. If there is a family history of premature CAD, a close examination of the family for FH is also recommended to improve the diagnostic yield.
“Probable FH” cases are those that are considered to require further scrutiny and follow-up. Continue detailed family research, including blood tests. Since LDL-C is elevated, lifestyle guidance, including diet, should be provided even in “probable FH” cases. If necessary, refer to a specialist, and consider genetic testing of the child and family members. Consider pharmacotherapy in cases of persistently high LDL-C (≥ 180 mg/dL, see Treatment section). The reason for setting “probable FH” cases in this guideline is that although they do not currently meet the diagnostic criteria for FH, they are suspected to have FH and are meant to provide guidance to prevent complications such as obesity, type 2 diabetes, and hypertension to prevent future atherosclerotic diseases.
Genetic testing is not essential for diagnosis, but cases with FH pathogenic gene mutations are diagnosed as “FH” (see section on FH Causative Genes). Approximately half of the children clinically diagnosed with FH in Japan have a pathogenic gene mutation that causes FH 51) . In difficult to diagnose cases or in severe cases, such as homozygotes, genetic testing should be performed with consent, if necessary. If a parent or sibling is found to have a pathogenic gene mutation for FH, this constitutes a family history of FH and is considered as item 2 (see About Genetic Testing and Genetic Counseling in FH).
Although diagnostic criteria differ from country to country even in Europe 53) , where FH research is active, the European Atherosclerosis Society (EAS) has recommended to screen at 5 years old and established the following criteria for pediatric FH: (1) LDL-C ≥ 190 mg/dL, (2) LDL-C ≥ 160 mg/dL and family history of premature CAD or hyper-LDL cholesterolemia in one parent, or (3) LDL-C ≥ 130 mg/dL and a pathogenic gene mutation in the parent (or a pathogenic gene mutation in the patient), while LDL-C 110 mg/dL or higher should be followed 43) .
Flowchart of Diagnosis ( Fig.2 )
Fig.2.
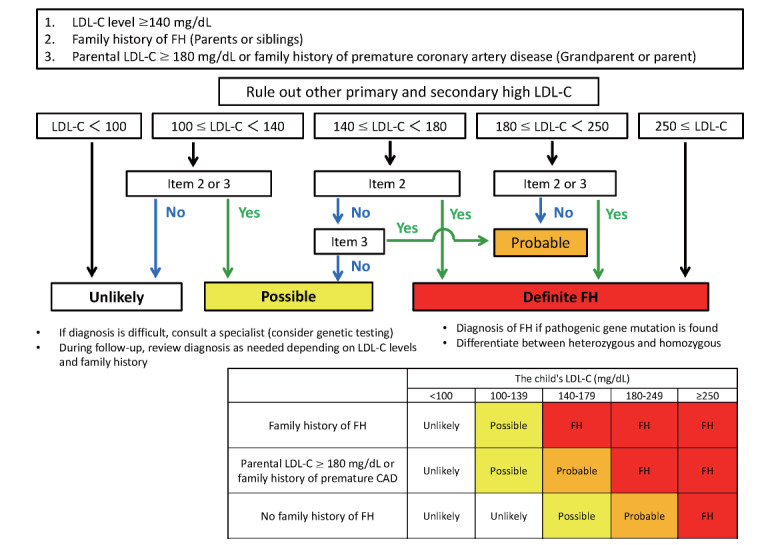
Flowchart of pediatric FH diagnosis
FH is diagnosed when (1) both item 1 (≥ LDL-C 140 mg/dL) and item 2 are met, (2) both the individual’s LDL-C is ≥ 180 mg/dL and item 3 is present, or (3) the LDL-C is ≥ 250 mg/dL. Once the diagnosis of FH is made, differentiate between heterozygotes and homozygotes as needed. In addition, the diagnosis of “FH” is made if the genetic test shows pathogenic mutations. (1) If items 1 and 3 are present, or (2) if the individual’s LDL-C is only ≥ 180 mg/dL, the patient is “probable FH.” If item 2 or item 3 is present but LDL-C is 100–139 mg/dL, or if LDL-C is 140–179 mg/dL but family history is not clear, FH cannot be ruled out, and annual follow-up should be done (yellow on chart). Otherwise, i.e., if there is a family history of FH, including item 3, but LDL-C is <100 mg/dL, or if there is no family history at all and LDL-C is <140 mg/dL, it is considered “unlikely” (not inherited). In this case, no follow-up is required (white on the chart).
In cases that are difficult to diagnose, consult with a specialist and consider genetic testing. If LDL-C increases during the course of the disease or if a family history becomes apparent, the diagnosis should be re-assessed.
Screening for Pediatric FH
With the enactment of the Basic Law for Child and Maternal Health and Child Development in 2019, it is required to provide the seamless provision of necessary child-rearing medical care, etc., to children in the process of growing up and their guardians and others. It is important to establish a screening system that enables early diagnosis, early treatment, and follow-up of pediatric FH.
In the 1990s, universal screening was conducted in Kumamoto City with the cooperation of the local government using 1 year and 6-month health checkups, wherein many pediatric HeFH were identified 54) . Currently, several municipalities and medical associations are taking the lead in implementing pediatric lifestyle-related disease checkups, including LDL-C testing. However, the follow-up system is not sufficient in many areas. In Kagawa Prefecture, universal screening for FH has been conducted since 2018 using the Kagawa Lifestyle-related Disease Prevention Health Examination for 4th graders in elementary schools throughout the prefecture. Many pediatric HeFHs have been identified and followed through the combined efforts of the government, medical associations, and core hospitals 44) . With children as the probands, CAD has become evident in their parents, and the establishment of a pediatric FH screening system (reverse cascade screening), in addition to traditional cascade screening, is expected to dramatically improve the diagnosis rate of FH in both children and adults.
When conducting cascade screening, it is important to remember that FH is primarily an autosomal dominant inherited disease. If one parent is HeFH, there is a 50% chance that the child will also be HeFH, and if both parents are HeFH, the child has a 25% chance of being HoFH and a 50% chance of being HeFH. Additionally, one of the parents of an FH-affected child found by reverse cascade screening is FH.
Since early diagnosis of FH can prevent atherosclerotic heart disease, it is recommended that at least TC be tested once there is an opportunity to draw blood. High-risk cases should be diagnosed by testing for LDL-C at 10 years at the latest (earlier for HoFH). Pediatric HeFHs are eligible for chronic pediatric diseases and are subsidized for medical expenses.
Differential Diseases
Secondary hyper-LDL cholesterolemia, as shown in Table 2 , should be excluded. Many of these are conditions that can be diagnosed and treated. Other diseases that are particularly important to differentiate from HoFH are listed in Table 3 . All these present an autosomal recessive form of inheritance.
Table 2. Differential Diagnosis of Secondary Hyper-LDL cholesterolemia.
| disease name | Points of differentiation |
|---|---|
| Nephrotic syndrome | Increased cholesterol and apoprotein synthesis in the liver. Edema is observed. It is important to check proteinuria and serum albumin levels. |
| Obesity | Measure height, weight, percentage of body fat, waist circumference, etc. and correctly evaluate the degree of obesity. High TG and low HDL-C occurs mainly by visceral fat accumulation. LDL-C also tend to be high. |
| Hypothyroidism | Systemic metabolic function decreases. Hyper-LDL cholesterolemia is caused by decreased expression and activity of the LDL receptor. Because Hashimoto’s disease is common, autoantibodies should be measured. |
| Anorexia nervosa | The patient presents with a high degree of emaciation. Low T3 syndrome leading to hyper-LDL cholesterolemia. Sex hormones and other factors are also suppressed. |
| Diabetes mellitus | Diagnosis is based on blood glucose and HbA1c levels. Hyper-LDL cholesterolemia, hypertriglyceridemia, and hypo-HDL cholesterolemia are often seen. Diabetes is a major risk for atherosclerotic disease, and glycemic control alone has little preventive effect. |
| Cushing’s syndrome | Excessive cortisol secretion promotes VLDL synthesis in the liver, resulting in hyper-LDL cholesterolemia. In children, the disease is characterized by weight gain without height growth and early onset of puberty due to androgen excess. |
| Pheochromocytoma | Excessive secretion of catecholamines promotes VLDL synthesis in the liver, resulting in hyper-LDL cholesterolemia. Suspected when symptoms such as headache, palpitations, sweating, and hypertension (paroxysmal or persistent) are present. |
|
Diet related (excessive intake of cholesterol) |
Excessive cholesterol intake may cause hyper-LDL cholesterolemia. In particular, if breastfeeding results in marked hyper-LDL cholesterolemia, the child should be retested after weaning to differentiate from sitosterolemia. Examine for the presence of xanthomas. |
| Cholestatic liver disease | FH-like hyper-LDL cholesterolemia may occur with biliary atresia and cholestatic liver disease. |
|
Drug-induced (steroids, cyclosporin, etc.) |
The diagnosis and treatment can be made by with drawal or reduction of the suspected drug. |
Table 3. Differential Diagnosis of Primary hyper LDL cholesterolemia.
| Disease name | Cause | Identifying points |
|---|---|---|
| Sitosterolemia |
ATP-binding cassette sub- family G member 5/8 (ABCG5, ABCG8) abnormality |
Autosomal recessive inheritance Raised serum sitosterol Hyper-LDL cholesterolemia may not be significant even in the presence of xanthomas. Transiently in infancy, patients may present with hyper-LDL cholesterolemia at levels suspicious for HoFH. |
| Cerebrotendinous xanthomatosis (27-hydroxylase deficiency) | Sterol 27-hydroxylase (CYP27A1) abnormality |
Autosomal recessive inheritance Progressive neurological disorders Raised serum cholestanol Serum cholesterol levels may not be high, but xanthomas are prominent. It also accumulates in the brain. |
|
Wolman disease Cholesteryl ester storage disease (lysosomal acid lipase deficiency) |
Lysosomal acid lipase (LIPA) abnormality |
Autosomal recessive inheritance Hepatomegaly Typical cases present with marked hepatosplenomegaly and liver damage leading to fatty liver and cirrhosis, but the severity of the disease varies and some cases are not diagnosed until adulthood. Adults often present with hyper-LDL cholesterolemia . |
About Genetic Testing
Ethical considerations must be made regarding genetic testing (germline human genetic testing) for FH, whether in adults or children. The Japanese Association of Medical Science’s “Guidelines for Genetic Testing and Diagnosis in Medical Care” is a guideline for conducting genetic testing 55) . According to this, when testing a child, it is necessary to obtain the consent of a person who is in a position to consent to the test on behalf of the child, and the best interests of the examinee should be fully considered in doing so. In addition, it is desirable to provide explanations that are appropriate to the level of understanding of the examinee and to obtain his/her consent (informed assent). Also provide genetic counseling as needed. For a definitive diagnosis in cases where the disease is already present (e.g., high LDL-C levels), genetic testing can be considered in children as needed, since there are treatment options for FH. On the other hand, for diagnosis of asymptomatic disease (no symptoms at all, including cholesterol), genetic diagnosis should be postponed until the patient is an adult and able to make autonomous decisions. The handling of incidental/secondary findings in genetic testing as part of the research framework is the policy of each principal investigator and should be well explained and understood when obtaining informed consent from the participant 56) .
Genetic testing for HoFH has been covered by insurance from April 2022 in Japan and is expected to be used for the definitive diagnosis and selection of treatment options.
Genetic Counseling in FH 57)
Genetic counseling should be provided at the appropriate time, if necessary, at the time of genetic testing and diagnosis. Genetic counseling should provide not only information but also psychological and social support to allow the patient/subject to make autonomous choices. Therefore, genetic counseling should be performed as a team medicine, with cooperation between physicians with extensive experience treating FH and persons skilled in genetic counseling (such as clinical genetic specialists and certified genetic counselors). As the basic form of FH is an autosomal dominant inherited disease, the case of autosomal dominant inheritance due to abnormalities in three etiologic genes (LDLR, APOB, PCSK9) is described here. If the child is HeFH, either parent is HeFH. If one parent is HeFH, there is a 50% chance that the child will also be HeFH, and if both parents are HeFH, the child has a 75% chance of developing this disease (25%: HoFH, 50%: HeFH). When both parents of an HeFH child are clinically unremarkable, it is necessary to consider the possibility of a de novo mutation in the affected child, or both parents have mild symptoms, or the child is not truly related to the parents by adoption. Prenatal diagnosis and preimplantation diagnosis are attempted in some cases; however, these procedures should be considered very carefully, given that there is no intellectual abnormality and treatments exist even if the child is homozygous for FH.
Notes on Genetic Test Results for FH
Like other genetic diseases, not just FH, current genetic testing also has its limitations. The detection rate of pathogenic mutations in clinically diagnosed cases of FH is reported to be approximately 60%–80% in adults 58) . Furthermore, approximately 10% of the causative genetic mutations in FH are structural mutations in the LDLR 58) . Therefore, care should be taken because some analytical methods do not identify such structural variations.
4. Diagnosis of Pediatric HoFH
Key Points
• The child meets the diagnostic criteria for pediatric FH; in many cases, the parents are heterozygous for FH.
• Skin or tendon xanthomas are present, and LDL-C levels are approximately twice those of the parent.
• Two pathogenic mutations are identified in the FH causative gene.
Clinical Presentation of Pediatric HoFH
As a rule, pediatric HoFH inherit the FH-causing gene mutation from both parents and are clinically diagnosed when serum LDL-C levels are approximately twice those of parents and other family members who are HeFH. Untreated LDL-C is usually ≥ 500 mg/dL; if ≥ 400 mg/dL, suspect HoFH. It is important to confirm serum levels of LDL-C in family members (parents and siblings), clinical characteristics such as tendon xanthomas, premature CAD, and the presence of consanguineous marriages as much as possible. HoFH are exposed to very high LDL-C from fetal life, and the appearance of cutaneous xanthomas at skin flexures such as wrists and ankles during infancy is often a trigger for consultation. Nodular xanthomas, interdigital xanthomas, and buttock xanthomas are also findings suggestive of HoFH. These manifestations improve with early LDL-C lowering therapy. Tendon xanthomas are usually unremarkable in infancy, but appear earlier than HeFH and are highly thickened.
Genetic Testing for HoFH
As mentioned above, the diagnosis of HoFH, in the broad sense, is confirmed when two pathogenic mutations in the FH causative gene (LDLR, PCSK9, APOB) are identified at two loci. When two pathogenic mutations in LDLRAP1 are identified across two loci, it leads to the diagnosis of ARH.
In Japan, double heterozygotes with LDLR and PCSK9 gain-of-function mutations have been reported. Their phenotypes were milder than that of true homozygotes or compound heterozygotes with LDLR, and the response to pharmacotherapy such as statins was preserved 59 , 60) . Even when two LDLR mutations are present, the receptor-defective type is considered less severe than the receptor-negative type. As HoFH patients with different mutations respond differently to treatment, it is advisable to confirm the genetic mutation as much as possible when FH homozygosity is suspected. Note that in the case of double heterozygotes, phenotypic inheritance may not follow a simple Mendelian inheritance form, as the HoFH phenotype may be inherited from one parent, or, conversely, the child may have the normal form. This kind of definitive diagnosis, including comprehensive genetic tests, such as panel analysis, may lead to a differential diagnosis of sitosterolemia and other disorders 61) .
Notes on Diagnosis
The clinical presentation of HoFH can vary to some extent, and even patients whose LDL-C below 500 mg/dL may be diagnosed as HoFH based on genetic testing.
HoFH are known to have a very poor prognosis, as atherosclerotic diseases such as coronary atherosclerosis and aortic valve disease ( Appendix Fig.1 ) rapidly progress from childhood, and they may die at their childhood. In addition, cutaneous xanthomas are often seen in HoFH ( Appendix Fig. 2 ) .
Appendix Fig.1.
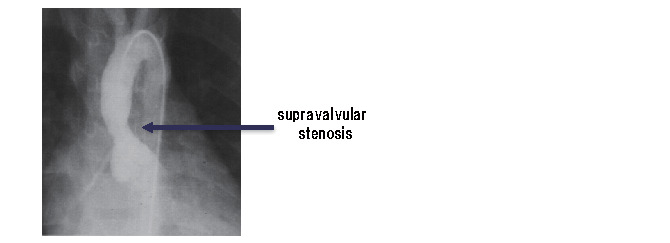
Supravalvular aortic stenosis
Appendix Fig.2.
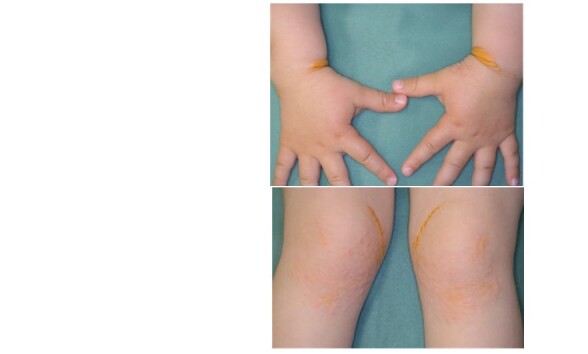
3-year-old boy, HoFH, xanthoma cutis
If HoFH is suspected, such as the presence of cutaneous xanthomas, marked tendon xanthomas, or higher levels of LDL-C levels compared with HeFH, a diagnosis and treatment plan must be determined by a specialist such as a Japan Atherosclerosis Society Board Certified Specialist. For a list of facilities where FH patients can be referred and the physicians in charge, please refer to the website of the Japan Arteriosclerosis Society (https://www.j-athero.org/jp/wp-content/uploads/specialist/pdf/fh_institution.pdf).
HoFH are also designated as intractable diseases in addition to chronic childhood diseases and are eligible for subsidies for medical expenses.
Differential Diagnosis
If there is no clinical evidence of HeFH in both parents, ARH or other conditions are suspected. Other diseases in which xanthomas are prominent include sitosterolemia, in which serum plant sterols are elevated, and cerebrotendinous xanthomatosis, in which serum cholestanol is elevated ( Table 3 ) . Sitosterolemia, in particular, can present with highly variable cholesterol levels and skin and tendon xanthomas with LDL-C levels similar to those of HoFH in infancy 62) .
5. Evaluation of Atherosclerotic Disease in Pediatric FH
Key Points
• If Achilles tendon thickening and carotid atherosclerosis are observed in patients with HeFH, regular examination considering the possibility of CAD is advisable.
• Due to the early development of atherosclerotic lesions in patients with HoFH, regular evaluation of systemic atherosclerotic disease by a specialist is necessary. In particular, evaluation for the presence of CAD, aortic valve and supravalvular stenosis, and thoracic and abdominal aortic aneurysms is recommended.
• In children, the examination should start with ultrasound methods (carotid ultrasound, transthoracic echocardiography, and abdominal ultrasound) to avoid the effects of radiation exposure as much as possible.
• If deemed necessary, CAD is screened by coronary computed tomography (CT), and if stenosis is suspected, coronary angiography is performed after hospitalization.
FH Heterozygote
HeFH develop CAD less frequently in childhood, but atherosclerotic lesions develop earlier than in healthy children, so non-invasive testing should be performed primarily when necessary. The presence of thickening or plaque on carotid ultrasound, or thickening of the Achilles tendon, should prompt suspicion of atherosclerotic disease and periodic examination.
HoFH
1) Interview
Ischemic symptoms of CAD can occur even in childhood, including chest pain and anterior chest tightness during exertion and radiating pain to the neck and left hand, which should be assessed for improvement with rest. For peripheral artery disease (PAD), the patient is asked if he or she has muscle pain (intermittent claudication) when walking and if it disappears with rest. In aortic valve and supravalvular stenosis, the patient should be asked about shortness of breath during exertion.
2) Physical Findings
Although the physical findings of CAD remain scarce, confirm the presence or absence of arterial pulsatility and attenuation, especially in the lower extremities by palpation of the femoral, popliteal and dorsal ankle arteries, and measurement of the ankle-brachial index when PAD is suspected. A systolic murmur of the aortic valve in auscultation suggests aortic valve/supravalvular stenosis, and a vascular murmur in the limb arteries suggests arterial stenosis.
3) Biochemical Examination
CK-MB and cardiac troponin are elevated during acute myocardial infarction.
4) Morphological Examination
Non-invasive examination
Exercise stress EKG: Exercise stress EKG using a treadmill is useful, but it should be performed after evaluating the presence of aortic and supravalvular stenosis by transthoracic echocardiography. Be aware of the risk of ventricular fibrillation, etc., due to induced myocardial ischemia.
Ultrasound: It is useful for assessing the degree of peripheral arteries, especially the carotid arteries, and is evaluated by the presence or absence of plaque and changes in IMT, and the rate of change over time is also considered. Transthoracic echocardiography is used to diagnose aortic valve and supravalvular stenosis and to evaluate cardiac function.
CT: Cerebrovascular lesions are rare in pediatric HoFH, and the evaluation of the coronary artery is the main focus. Due to the high radiation dose, coronary CT should be performed only when necessary in children.
Magnetic resonance imaging (MRI), MRA (MR angiography): It is now possible to delineate stenotic lesions of the aorta, peripheral arteries, and coronary arteries. CT and MRI scans require sedation depending on the age of the patient, and care must be taken to avoid accidents.
Invasive Examination
If progression of atherosclerosis is suspected, angiography is used to evaluate the narrowing of the luminal diameter of the vessel, but radiation exposure should be reduced as much as possible. In particular, if CAD is suspected, the patient should be admitted to the hospital for coronary angiography and left ventricular and aortic angiography as well.
6. Treatment of Pediatric FH
Once FH is diagnosed, lifestyle advice should be provided as early as possible in order to reduce the risk of atherosclerosis, including lowering LDL-C levels. If lifestyle modification is insufficient, consider starting drug therapy at around 10 years of age. Pediatric HoFH should consult with a specialist to determine response to pharmacotherapy, and if the response is inadequate, initiate lipoprotein apheresis immediately.
1) Lifestyle Improvement/Guidance
Key Points
• Provide guidance on lifestyle, including diet, as early as possible. The guidance should continue after the initiation of drug therapy.
• Diet: Total energy intake should be calorie requirements by age and height. The energy ratio should be 20%–25% fat and 50%–60% carbohydrates. Saturated fatty acids should be less than 7% of the energy ratio and cholesterol less than 200 mg/day. Reduce trans-fatty acid intake. Focus on traditional Japanese food and eat enough vegetables.
• Obesity control: Maintain an appropriate weight. Severe obesity also requires limiting energy intake. Develop a proper rhythm of life, eating habits, and exercise routines.
• Exercise therapy: Attempt to develop and maintain an exercise routine. Encourage patients to lead active lifestyles, which include avoiding long periods of sedentary time.
• Prevention of smoking and passive smoking: Emphasize the point that the patient should never smoke during their lifetime. In addition, the cooperation of family and those around the patient should be obtained in preventing passive smoking.
Lifestyle Guidance
Once FH is diagnosed, lifestyle guidance should begin as early as possible for HoFH and heterozygotes alike 45) . Investigate lifestyle habits, including diet, exercise habits and rhythm of life, and guide improvements 45) . However, it is often difficult to reduce and manage LDL-C levels by lifestyle modification alone. In such cases, pharmacotherapy should be added, and lipoprotein apheresis should be considered for patients homozygous for FH 63) . After the start of these treatments, the guidance regarding lifestyle habits should continue ( Figs.3 and 4 ) .
Fig.4.
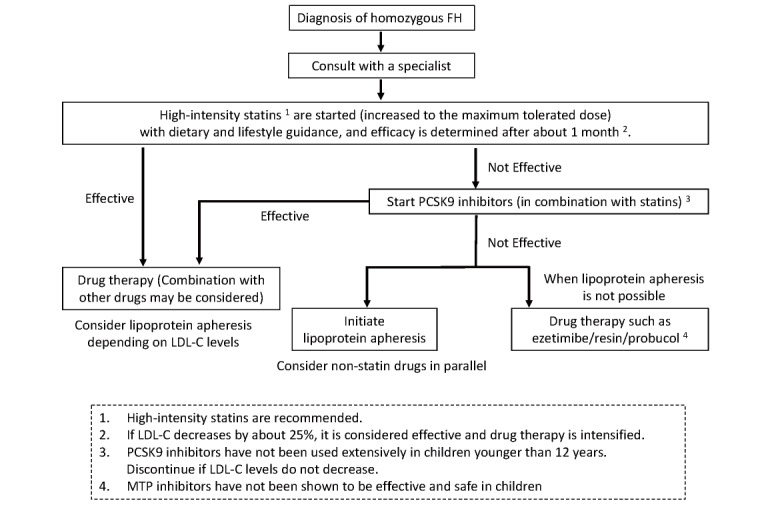
Flowchart of treatment for pediatric FH homozygotes
Dietary Therapy
Even in the absence of obesity, total daily energy intake should be the required calorie intake by age and height to maintain an appropriate body weight 64) . Evaluate the progress of height and weight over time, and adjust diet and exercise accordingly.
For energy distribution, the 2017 edition of the Guidelines for the Prevention of Atherosclerotic Disease recommends a fat energy ratio of 20%–25% and a carbohydrate energy ratio of 50–60% for dyslipidemia in adults 45) . The Dietary Reference Intakes for Japanese (2020) recommend a fat energy ratio of 20%–30% and a carbohydrate energy ratio of 50%–65% 64) , which is the same from age 1 to 75 years and older, so children should also follow the same standards as adults.
The Dietary Reference Intakes for Japanese (2020) do not set an upper limit for cholesterol intake because of the large individual differences in cholesterol absorption from the diet and the inconsistent effects of diet therapy 64) . However, in hypercholesterolemic patients, cholesterol intake restriction has also been suggested to be effective in lowering LDL-C 65) . In the 2017 edition of the Guidelines for the Prevention of Atherosclerotic Diseases, the Japan Atherosclerosis Society recommends less than 7% saturated fatty acids, reduced trans-fatty acids, and less than 200 mg/day of cholesterol for hypercholesterolemia 45 , 63 , 66) .
In recent years, due to westernization of diets, fat intake has tended to be high even in children. As in adults, a slight moderation in fat and carbohydrates is needed. In other words, children should be instructed to eat a diet based on traditional Japanese food patterns, with low intake of animal fat and a good balance of fish, vegetables, soybeans (products), mushrooms, and fruits. Also, beware of excessive salt intake 45 , 63) .
Anti-Obesity Therapy
1) Prevention of Obesity
Use the percentage of overweight (POW) to determine obesity in children 67 , 68) . POW is a physique index obtained by [(actual weight/standard weight)/standard weight]×100(%). Use the standard weight by sex, age, and height according to school health statistics as the standard weight. The appropriate POW is within ±20% for schoolchildren and ±15% for young children (<6 years old) 67) .
Visceral fat evaluation is also important in children, as excessive accumulation of visceral fat can lead to abnormal secretion of various adipocytokines 68 , 69) . Waist circumference is a useful alternative to CT for a simple marker of visceral fat accumulation. The standard for school-age children is 80 cm, above which visceral fat over-accumulation is suspected 68) .
In dietary guidance, reference should be made to the guidelines for intake by four food groups based on the Dietary Reference Intakes for Japanese (2020) 64) . Use the “Dietary Balance Guide 70) ” for meal distribution.
The 2017 Guidelines for the Management of Obesity Disease in Children and Adolescents recommend at least a total of 60 minutes per day of moderate- to high-intensity exercise as physical activity to prevent obesity 67) . It is also important to regulate the rhythm in life. For young children, it is recommended that they sleep at least 10.5 hours and watch no more than 2 hours of TV; for school-age children, it is recommended that they avoid sleeping less than 8 hours and long periods of screen time 67) .
2) In Case of Obesity
If there is an excess of dietary intake, it should be restored to an appropriate level. However, due to the growth period, energy intake should not be extremely restricted, but rather nutritional balance should be maintained, and energy expenditure through exercise should be increased 67) . Energy intake should be set according to the level of physical activity for each sex and age group 64) . In the case of severe obesity, energy intake can be limited to 90%.
Exercise Therapy
In HoFH and severe cases of HeFH, exercise instruction should be given after a cardiac ultrasound and a close examination for CAD (see the Examination section). Exercise therapy is as important as dietary guidance because it improves glucose and lipid metabolism and optimizes adipocytokines in children 70) . In particular, obese children and children who do not have an exercise routine should be thoroughly instructed.
In adults, it is recommended that aerobic exercise, mainly at moderate intensity or higher, be performed regularly (at least 30 minutes total daily, with a goal of at least 3 days per week) 45 , 63) , and the same should be done in children. In the early elementary grades, the main focus will be on increasing the amount of physical activity, such as encouraging outdoor play.
Prevention of Smoking and Passive Smoking
Smoking is not only a risk factor for atherosclerotic disease, but it also increases the risk of developing diabetes, dyslipidemia, and metabolic syndrome and is associated with a general increased risk of atherosclerotic disease 45 , 63) . Teach children from childhood to avoid smoking throughout their lives. Moreover, passive smoking is a risk factor for CAD and stroke. It has also been reported to increase the risk of diabetes 45 , 63) . Promptly encourage all those around children, including family members, to quit smoking.
2) Pharmacotherapy of Pediatric HeFH
Key Points
• If LDL-C 180 mg/dL or higher persists after adequate lifestyle guidance, drug therapy should be considered at age 10, regardless of sex.
• The target of management is an LDL-C level of less than 140 mg/dL.
• First-line drug therapy is statins, starting with the lowest dose. In addition to lipid levels, blood tests such as liver function and CK, and other parameters, as well as symptoms such as muscle pain should be followed every month at first and then every 3–4 months once the dosage has stabilized, to monitor for the onset of side effects. Simultaneously monitor growth and also secondary sexual characteristics.
• The cases of “probable FH” are considered in the same way as FH. Adequate lifestyle guidance should be provided, and drug therapy should be considered, taking into account LDL-C levels and background factors.
Criteria for Consideration of Drug Treatment Initiation
Hyper-LDL cholesterolemia from childhood is an independent risk of atherosclerosis, and IMT thickening is already known to develop in many pediatric HeFH from late school-age 71) . In recent years, many overseas guidelines have highlighted the importance of treatment from childhood for the prevention of future cardiovascular events 43 , 72 , 73) . In administering drug therapy to pediatric HeFH, it is thus important to fully explain the need for treatment to parents and, whenever possible, to the affected child, to gain their understanding, given the long-term nature of the medication.
Fig.3 shows the treatment flowchart for pediatric HeFH. If LDL-C 180 mg/dL or higher persists despite lifestyle modifications such as diet and exercise, consider starting drug therapy at 10 years of age or older, regardless of gender 43 , 72 , 73) . In cases of markedly elevated LDL-C, drug therapy should be administered as early as possible, as improved diet and lifestyle alone are not sufficient to lower LDL-C. Cases around 180 mg/dL should be monitored, taking into account pubertal fluctuations, and if they continue to exceed 180 mg/dL (multiple times), drug therapy should be administered. The “probable FH” cases are also likely to be FH, and persistent LDL-C 180 mg/dL or higher is an indication for pharmacotherapy. Cases requiring pharmacotherapy should be referred or consulted with a specialist to determine a treatment plan. Even in patients younger than 10 years, consult a specialist in cases of persistently markedly high levels of 200 mg/dL or higher even after dietary guidance. Early initiation of drug therapy should also be considered, taking into account LDL-C, age, risk factors, and family history.
Drug Selection
The first-line drug is statin, starting at the lowest dose. In Japan, pitavastatin has been indicated for children 10 years of age and older since 2015. Looking at the status of pediatric indications for statins in each country, simvastatin, atorvastatin, pravastatin, fluvastatin, and rosuvastatin have been approved for pediatric use in the United States and Europe. Most are approved for use in children as young as 10 years, but pravastatin has been approved for use at ages 8 and up in the United States, rosuvastatin at ages 6 and up in Europe, and atorvastatin at ages 6 and up in Australia. If statin alone is found to be not effective enough, consider (1) increasing the dose, (2) changing to a more intensive one and increasing the dose, or (3) combining a lipid-lowering drug with another mechanism in addition to the statin.
Adjunctive agents that have been reported to be effective in childhood are small intestinal cholesterol transporter inhibitors (ezetimibe) and resins (anion exchange resins: cholestyramine and cholestimide). Ezetimibe is indicated in the United States and Europe for children 10 years of age and older. Resin had also been the first-line drug for children in Japan due to its history as the first-line drug for hypercholesterolemia in the United States in the past. However, it has many side effects such as abdominal pain, bloating, and constipation and thus is not indicated for pediatric use in Europe, and there is little evidence of its effectiveness in preventing atherosclerosis. It inhibits the absorption of folic acid and fat-soluble vitamins, so regular monitoring and occasional supplementation are required. In a recent survey of eight European countries, about half of the statin-treated children were on combination therapy, although there were differences from country to country, and ezetimibe was used very commonly as a concomitant drug (96% of all concomitant cases) 53) . Even in children, the combination of high-intensity statins and ezetimibe can reduce LDL-C by approximately 50% of the pretreatment values. Combined agents of statin and ezetimibe are currently available in Japan.
LDL-C Control Target Values
The target value for the management of pediatric FH is the same as in Pediatric FH Guide 2017 3) , with an LDL-C level of 140 mg/dL. Ensure to maintain below 140 mg/dL in cases with a family history of premature CAD or risk factors such as diabetes (described later). Although it is difficult to achieve the goal in severe cases, try to get as close to the goal with the use of drug combination therapy. Even after the start of drug therapy, lifestyle guidance, including diet, should be provided. The LDL-C control goal for adults is less than 100 mg/dL. For reference, the ESC/EAS guideline is <135 mg/dL for children 43) .
Risk Factors
In addition to common risk factors such as smoking (passive smoking), hypertension, diabetes, other dyslipidemias, and obesity, Lp(a) has been emphasized abroad.
Lp(a) is a lipoprotein consisting of apoprotein(a) S-S bound to B-100, the apoprotein of LDL, and its molecular weight is noted to vary from individual to individual. Apoprotein (a) is highly homologous to plasminogen and is considered highly atherosclerosis-promoting in terms of both lipid deposition and thrombus formation 74) . Hyper-Lp(a)emia is associated with a 1.5-fold increased risk of premature CAD 43) . There are also reports of Lp(a) deposition in the aorta (mainly near the internal elastic plate) from the age of 10 75) . Serum Lp(a) concentrations can vary widely among individuals, are independent of LDL-C, and are strongly influenced by genetic factors. In Japan, a level above 30 mg/dL is generally considered high for adults 63) , and the same is true for children. Sometimes, there are cases of very high Lp(a) even in pediatric FH. In patients with hyper-Lp(a)emia, it is recommended that LDL-C should be maintained at <140 mg/dL after starting therapy, considering the same risk factors.
Follow-Up
The safety and tolerability of statins in children are similar to those in adults. Statin use in children should be started at the lowest dose, and symptoms such as liver function, including AST and ALT, CK, serum lipid levels, and muscle pain should be evaluated 1 month after initiation. Compared to values prior to drug initiation, the development of side effects such as hepatic dysfunction, myopathy, and, although extremely rare, rhabdomyolysis, should be noted. The patient continues to be examined and tested in the following month, if necessary, depending on the state of LDL-C decline and side effects. If there are no side effects and the LDL-C level and other parameters are stable, follow-up visits should be conducted three to four times a year.
In children, growth and pubertal state (secondary sexual characteristics) should also be routinely assessed. In children, CK elevation is often seen with strenuous exercise, so it is important to distinguish CK elevation from drug-induced elevation. It is advisable to follow-up on blood glucose levels and HbA1c, as statins have been reported to increase new onset of diabetes in adults 76) . Special attention should be paid to side effects when taking high doses of statins.
For Cases where Statins are Difficult to Continue
There are rare cases in which statins are difficult to continue due to strong side effects. Currently, six statins are available in Japan, with slightly different strengths of LDL-C lowering and metabolic pathways. If side effects occur with one statin, it is advisable to start with a lower dose of a statin with a different metabolic pathway. For the cases of so-called statin intolerance, in which multiple statins cannot be used, the Japan Atherosclerosis Society has published the “Statin Intolerance Clinical Guide 2018 77) ”. The “Muscle Flowchart” and “Liver Flowchart” are defined for two major categories of reasons for the difficulty in continuing statins: muscle damage and liver dysfunction.
For Patients under 10 Years of Age
As mentioned above, in cases of persistently high LDL-C (≥ 200 mg/dL) in children younger than 10 years, drug therapy should also be considered, taking into account the background of the affected child. Although there is no evidence, the theory of cumulative LDL-C (Chol-year) suggests that the earlier it is lowered, the better the prognosis. It needs to be lowered immediately, especially if it is markedly high, as in the case of HoFH.
The problem is that younger children cannot take the tablets. Crushed statin tablets are not suitable for administration. Pitavastatin and rosuvastatin are available in tablets that disintegrate orally, making them relatively easy to take for those under 10 years of age, but only pravastatin is available in a powder formulation. Ezetimibe is a tablet, but it is the size of a grain of rice (8.1 mm×4.1 mm), making it relatively easy to take internally. Currently in Japan, the indication for pitavastatin is strictly for ages 10 and older, and ezetimibe also does not have a pediatric indication. In general, many drugs in the pediatric field have “no established safety profile for children,” but in reality, they are used empirically for many diseases. For younger children, there is also an option of waiting until the age of 10 years to intensify the treatment, since it is not necessary to lower the level to the control target (140 mg/dL), as it can be considered a popular HeFH level if it is lowered to 200 mg/dL or less.
Pregnancy
Since there have been reports of malformations in newborns of pregnant women who accidentally took statins in early pregnancy 78) , statins are contraindicated in women who wish to become pregnant, pregnant women, and nursing mothers 45) . When starting statins in girls, explain the risks of teratogenicity and the importance of a planned pregnancy to the patient and her parents prior to starting.
(3) Pharmacotherapy of Pediatric HoFH
Key points
• Once HoFH is diagnosed, they should consult with a medically experienced specialist. Determine the effect of statins as early as possible, and if the reduction in LDL-C levels is not sufficient, PCSK9 inhibitors and further lipoprotein apheresis therapy should be initiated. In parallel, additional administration of other drugs should be considered.
• Probucol is effective in promoting the reduction and disappearance of skin and tendon xanthomas, but the side effect of QT prolongation should be noted.
Genetic Mutations and the Effects of Drug Therapy
The efficacy of lipid-lowering drugs such as statins, ezetimibe, and resins is primarily attributed to increased LDLR activity, and HoFH are often less responsive to drugs than HeFH. For HoFH of receptor-negative type that have no LDLR activity, the above drugs are not effective in lowering LDL-C levels, but improved prognosis has been reported with statin administration 79) . On the other hand, for the receptor-defective type, which has a small amount of LDLR activity, there are reports of significant efficacy of combination therapy with resins, statins, nicotinic acid, etc. 80) . In cases called double heterozygotes, in which the patient has an LDLR loss-of-function mutation and a PCSK9 gain-of-function mutation, a certain level of efficacy can be expected.
Pharmacotherapy Practice
The treatment of HoFH should be performed by a specialist with medical experience. Treatment and drug use should only be started after the effects and possible side effects have been fully explained to parents or guardians and their consent obtained. If diagnosed as HoFH, any of the above types should first be started on high-intensity statins and increased to the maximum tolerated dose. After approximately 1 month, the effect of statins will be determined, and if the effect is insufficient, a PCSK9 inhibitor will be started to check the response of LDL-C levels. If the target values are not reached after three doses, lipoprotein apheresis is then initiated. Parallelly, examine the effects of drugs other than statins (e.g., ezetimibe, resin, probucol) ( Fig.4 ) . A report examining the effect of atorvastatin on HoFH under treatment with lipoprotein apheresis showed an average decrease in LDL-C levels of approximately 20%, with little response in the receptor-negative type 81) . Probucol has been reported to have a certain lowering effect on TC levels (lowering of LDL-C and HDL-C) and reduction or disappearance of skin and tendon xanthomas in HoFH 82) . In a retrospective study examining adult HeFH in Japan, significantly fewer recurrent cardiovascular events were reported in patients treated with probucol 83) . However, probucol has the side effect of QT prolongation 84) and thus requires periodic ECG testing 85) . Ezetimibe has been reported to be effective in delaying post-treatment re-elevation of LDL-C levels in HoFH undergoing lipoprotein apheresis treatment 86) .
PCSK9 inhibitors, which inhibit LDLR degradation, have been shown to be effective in lowering LDL-C even in adult HoFH 31) , but there is not much experience with their use in younger than 12 years of age, even overseas. Although there are benefits in lowering LDL-C early in children, it should be noted that safety has not yet been fully tested. If LDL-C levels do not decrease with receptor-negative type, discontinue early. In addition, PCSK9 inhibitors are supposed to be used in combination with the maximum tolerated dose of statins in Japan. Microsomal triglyceride transfer protein inhibitor (lomitapide) is an oral drug indicated only for HoFH, but has been reported to have side effects of gastrointestinal symptoms, liver dysfunction, and fatty liver; thus, caution should be exercised, especially in pediatric patients. Currently, pediatric indications are not approved in any country.
Liver transplantation is an option for patients who are resistant or intolerant to all of the above treatments, but there are currently very few cases of liver transplantation in Japan 87 , 88) .
7. Lipoprotein Apheresis
Key Points
• If drug therapy is not adequate, initiate lipoprotein apheresis therapy promptly.
• Lipoprotein apheresis therapy should be performed once every 1–2 weeks while monitoring LDL-C levels before and after treatment.
• Three different methods are used: simple plasma exchange therapy, double membrane filtration, and selective LDL adsorption.
Start of Lipoprotein Apheresis Therapy
HoFH generally respond poorly to drugs, and lipoprotein apheresis therapy is indicated in many cases. Realistically, treatment should begin at 4–6 years of age, when the patient is bedridden and extracorporeal circulation can be performed, although there have been reports of treatment beginning at 3.5 years of age 89) . As there are cases of coronary artery stenosis, complete occlusion, aortic stenosis, and supravalvular stenosis noted in childhood, and the later the time of initiation, the worse the prognosis, one should not hesitate to start lipoprotein apheresis therapy if the response to the drug remains poor 90) . Patients who are too young to be treated with lipoprotein apheresis should have regular checkups and examinations. If possible, pharmacotherapy should be used until a time when apheresis is possible.
Methods of Lipoprotein Apheresis
Lipoprotein apheresis is performed once every 1–2 weeks while monitoring LDL-C levels before and after treatment. Blood access is usually to the elbow veins of both upper extremities. In cases of poor access, shunt surgery may be performed, but care should be taken due to the high-risk of obstruction due to high levels of LDL-C and other factors. Currently, there are three major types of lipoprotein apheresis therapy in Japan, namely, simple plasma exchange, double membrane filtration, and LDL adsorption therapy. Double membrane filtration and LDL adsorption therapy are widely used because they can selectively remove LDL. Plasma exchange therapy may be used for these treatments due to the large extracorporeal circulating volume when the patient weighs less than 30 kg.
Long-Term Therapeutic Effect of Lipoprotein Apheresis for HoFH
There are many reports of favorable long-term effects of lipoprotein apheresis treatment for HoFH, including regression of skin and tendon xanthomas, lessening of symptoms of angina pectoris, and inhibition of the development of atherosclerotic lesions in the coronary arteries, as well as the effect of regression 90 - 93) . A systematic review from reports of 209 pediatric FH homozygotes also stated that lipoprotein apheresis has few adverse events and is generally safe in lowering LDL-C levels and further reduce xanthomas 35) . On the other hand, if lipoprotein apheresis is delayed in HoFH, there have been reports of deaths from myocardial infarction 93) ; thus, lipoprotein apheresis should be introduced in cases where it is necessary.
8. Supplementary Provisions
Public Subsidies
Pediatric HeFH are eligible for the chronic childhood diseases 94) . HoFH, in addition to being designated as the chronic childhood diseases, are also designated as an intractable disease based on the “Law Concerning Medical Care for Patients with Intractable Diseases 95) ” and are eligible for public subsidies for medical expenses.
Systematic Review Committee Member
Hirofumi Okada (Department of Cardiovascular Medicine, Kanazawa University Hospital), Masatsune Ogura (Department of Metabolism and Endocrinology, Eastern Chiba Medical Center/Department of General Medical Science, Chiba University Graduate School of Medicine), Yu Kataoka (Department of Cardiovascular Medicine, National Cerebral and Cardiovascular Center), Yoshihiro Tanaka (Department of Preventive Medicine, Northwestern University), Takahito Doi (Department of Clinical Biochemistry, Copenhagen University Hospital, Herlev and Gentofte/Department of Cardiovascular Medicine, National Cerebral and Cardiovascular Center), Tetsuo Nishikawa (Department of Transplant Medicine, National Cerebral and Cardiovascular Center), Akihiro Nomura (College of Transdisciplinary Sciences for Innovation, Department of Cardiovascular Medicine, Kanazawa University), and Masashi Yamamoto (Clinical Cell Biology and Medicine, Chiba University)
Classification of Level of Evidence for Treatment and Diagnosis
| 1+ | High-quality RCTs* and their MA/SR |
| 1 | Other RCTs and their MA/SR |
| 2 | Prospective cohort studies, their MA/SR, (predefined) RCT sub-analysis |
| 3 | Non-randomized controlled trials, before and after studies, retrospective cohort studies, case-control studies, and their MA/SR and RCT post hoc sub-analysis |
| 4 | Cross-sectional studies, case series |
| Consensus | By consensus of the supervisory committee members and the members who prepared the report |
RCT: randomized controlled trial, MA: meta-analysis, SR: systematic review
*A high-quality RCT is defined as (1) large number of subjects; (2) double-blind, independent assessment; (3) high follow-up rate (low dropout rate); (3) low protocol deviation, (4) clear random allocation method, etc.
Classification of Level of Evidence for Epidemiological Studies
| E-1a | Meta-analysis of cohort studies |
| E-1b | Cohort studies |
| E-2 | Case-control studies, cross-sectional studies |
| E-3 | Descriptive studies (case series) |
Recommended level
| A | Strongly Recommended |
| B | Weakly Recommended |
Conflicts of Interest
The following discloses the conflicts of interest of the members of the Joint Working Group of the Japan Pediatric Society and the Japan Atherosclerosis Society for the Development of Guidelines for the Treatment of Pediatric Familial Hypercholesterolemia for the years 2017 - 2021.
| Names | Disclosure Item 1 | Disclosure Item 2 | Disclosure Item 3 | Disclosure Item 4 | Disclosure Item 5 | Disclosure Item 6 |
| Disclosure Item 7 | Disclosure Item 8 | Disclosure Item 9 | Disclosure Item 10 | Disclosure Item 11 | Disclosure Item 12 | |
| Ohtake A. | None | None | None | Sanofi K.K. | None |
SBI Pharmaceuticals Co., Ltd. |
| None | None | None | None | None | None | |
| Okada H. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Ogura M. | None | None | None |
Astellas Pharma Inc., Amgen Astellas BioPharma K.K., Sanofi K.K., Kowa Company, Ltd., Amgen K.K. |
None | None |
| None | None | None | None | None | None | |
| Kataoka Y. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Harada- Shiba M. | None | Liid Pharmaceuticals, Inc. | None | Astellas Pharma Inc., Amgen K.K., Amgen Astellas BioPharma K.K., Kaneka Medix Corporation, Sanofi K.K. | None | Aegerion Pharmaceuticals, Inc. , Parexel International Inc. |
| Aegerion Pharmaceuticals, Inc. , MSD K.K., Kaneka Medix Corporation, Kowa Pharmaceutical Co. Ltd., Sanofi K.K., Takeda Pharmaceutical Company Limited, Recordati Rare Diseases | None | None | None | None | None | |
| Sugiyama D. | Ajinomoto Frozen Foods Co., Inc. | None | None | None | None | None |
| None | None | None | None | None | None | |
| Tada H. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Tanaka Y. | None | None | None | None | None | None |
| American Heart Association (AHA), JAPAN LIFELINE Co., Ltd. | None | None | None | None | None | |
| Doi T. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Dobashi K. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Nishikawa T. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Nomura A. | CureApp, Inc. | None | None | None | None | None |
| None | None | None | None | None | None | |
| Matsuki K. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Minamino T. | None | None | None | Daiichi Sankyo Company, Limited, Nippon Boehringer Ingelheim Co., Ltd. | None | A&D Company, Limited, OMRON Corporation, Matsutani Chemical Industry Co.,Ltd., Melody International Ltd. |
| Astellas Pharma Inc., Otsuka Pharmaceutical Co., Ltd.,Kyowa Hakko Kirin Co., Ltd., Kyowa Kirin Co., Ltd., Sanofi K.K.,Takeda Pharmaceutical Company Limited, MitsubishiTanabe Pharma Corporation | None | None | None | None | None | |
| Yamashita S. | None | None | None | Amgen K.K., MSD K.K., Otsuka Pharmaceutical Co., Ltd., Kowa Company, Ltd., Skylight Biotech,Inc., Novartis Pharma K.K., Bayer Yakuhin,Ltd,, Hayashibara Co., Ltd., East Japan Institute ofTechnology Co., Ltd. | None | Medical Photonics Co.,Ltd. |
|
ONO PHARMACEUTICAL CO., LTD., Nippon Boehringer Ingelheim Co., Ltd., MSD K.K. |
None | None | None | None | None | |
| Yamamoto Y. | None | None | None | None | None | None |
| None | None | None | None | None | None | |
| Yamamoto M. | None | None | None | None | None | None |
| None | None | None | None | None | None |
【Disclosure Item】
1.Assuming a position of a board member or advisor in a profit-making business,/ Advisory role (1,000,000 yen or more annual compensation from a single business entity, or group)
2.Stock holdings or options (Annual profit of 1,000,000 yen or more/ownership of 5% or more of total shares)
3.Patent royalties/licensing fees (1,000,000 yen or more annual income per patent)
4.Honoraria (e.g. lecture fees) (500,000 yen or more total annual income from a single company or organization)
5.Fees for promotional materials (e.g. manuscript fee) (500,000 yen or more total annual income from a single company or organization)
6.Total clinical research funding (1,000,000 yen or more total annual research grants paid from a single company or organization to you or your department)
7.Total scholarship grants (1,000,000 yen or more total annual scholarship contributed by a single company or organization to you or your department)
8.Courses endowed by companies (Fill in if you belong to any course endowed by a company, etc.)
9.Others (e.g. trips, travel, or gifts, which are not related to research) (50,000 yen or more annually from a single company or organization)
Matters declared by the author's spouse, relatives within the first degree of consanguinity, or persons who share income or property
10.Assuming a position of a board member or advisor in a profit-making business,/ Advisory role (1,000,000 yen or more annual compensation from a single business entity, or group)
11.Stock holdings or options (Annual profit of 1,000,000 yen or more/ownership of 5% or more of total shares)
12.Patent royalties/licensing fees (1,000,000 yen or more annual income per patent)
*The company name is the name of the company as it was at the time of filing.
References
- 1).Li S, Chen W, Srinivasan SR, Bond MG, Tang R, Urbina EM, Berenson GS: Childhood cardiovascular risk factors and carotid vascular changes in adulthood: the Bogalusa Heart Study. Jama, 2003; 290: 2271-2276 [DOI] [PubMed] [Google Scholar]
- 2).Natural history of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY Study. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb, 1993; 13: 1291-1298 [DOI] [PubMed] [Google Scholar]
- 3).Harada-Shiba M, Ohta T, Ohtake A, Ogura M, Dobashi K, Nohara A, Yamashita S, Yokote K: Guidance for Pediatric Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 539-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Tada H, Nakanishi C, Mori M, Yamagishi M, Inazu A, Koizumi J: Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis, 2011; 214: 404-407 [DOI] [PubMed] [Google Scholar]
- 5).Akioyamen LE, Genest J, Shan SD, Reel RL, Albaum JM, Chu A, Tu JV: Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open, 2017; 7: e016461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG: Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol, 2020; 75: 2553-2566 [DOI] [PubMed] [Google Scholar]
- 7).Hu P, Dharmayat KI, Stevens CAT, Sharabiani MTA, Jones RS, Watts GF, Genest J, Ray KK, Vallejo-Vaz AJ: Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation, 2020; 141: 1742-1759 [DOI] [PubMed] [Google Scholar]
- 8).Austin MA, Hutter CM, Zimmern RL, Humphries SE: Familial hypercholesterolemia and coronary heart disease: a HuGE association review. Am J Epidemiol, 2004; 160: 421-429 [DOI] [PubMed] [Google Scholar]
- 9).Hutter CM, Austin MA, Humphries SE: Familial hypercholesterolemia, peripheral arterial disease, and stroke: a HuGE minireview. Am J Epidemiol, 2004; 160: 430-435 [DOI] [PubMed] [Google Scholar]
- 10).Akioyamen LE, Tu JV, Genest J, Ko DT, Coutin AJS, Shan SD, Chu A: Risk of Ischemic Stroke and Peripheral Arterial Disease in Heterozygous Familial Hypercholesterolemia: A Meta-Analysis. Angiology, 2019; 70: 726-736 [DOI] [PubMed] [Google Scholar]
- 11).Mabuchi H: Half a Century Tales of Familial Hypercholesterolemia (FH) in Japan. J Atheroscler Thromb, 2017; 24: 189-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Betteridge DJ, Bhatnager D, Bing RF, Durrington PN, Evans GR, Flax H, Jay RH, Lewis-Barned N, Mann J, Matthews DR: Treatment of familial hypercholesterolaemia. United Kingdom lipid clinics study of pravastatin and cholestyramine. Bmj, 1992; 304: 1335-1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Hoogerbrugge N, Mol MJ, Van Dormaal JJ, Rustemeijer C, Muls E, Stalenhoef AF, Birkenhäger JC: The efficacy and safety of pravastatin, compared to and in combination with bile acid binding resins, in familial hypercholesterolaemia. J Intern Med, 1990; 228: 261-266 [DOI] [PubMed] [Google Scholar]
- 14).Wiklund O, Angelin B, Fager G, Eriksson M, Olofsson SO, Berglund L, Lindén T, Sjöberg A, Bondjers G: Treatment of familial hypercholesterolaemia: a controlled trial of the effects of pravastatin or cholestyramine therapy on lipoprotein and apolipoprotein levels. J Intern Med, 1990; 228: 241-247 [DOI] [PubMed] [Google Scholar]
- 15).Avis HJ, Hutten BA, Gagné C, Langslet G, McCrindle BW, Wiegman A, Hsia J, Kastelein JJ, Stein EA: Efficacy and safety of rosuvastatin therapy for children with familial hypercholesterolemia. J Am Coll Cardiol, 2010; 55: 1121-1126 [DOI] [PubMed] [Google Scholar]
- 16).Clauss SB, Holmes KW, Hopkins P, Stein E, Cho M, Tate A, Johnson-Levonas AO, Kwiterovich PO: Efficacy and safety of lovastatin therapy in adolescent girls with heterozygous familial hypercholesterolemia. Pediatrics, 2005; 116: 682-688 [DOI] [PubMed] [Google Scholar]
- 17).de Jongh S, Ose L, Szamosi T, Gagné C, Lambert M, Scott R, Perron P, Dobbelaere D, Saborio M, Tuohy MB, Stepanavage M, Sapre A, Gumbiner B, Mercuri M, van Trotsenburg AS, Bakker HD, Kastelein JJ: Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized, double-blind, placebo-controlled trial with simvastatin. Circulation, 2002; 106: 2231-2237 [DOI] [PubMed] [Google Scholar]
- 18).Knipscheer HC, Boelen CC, Kastelein JJ, van Diermen DE, Groenemeijer BE, van den Ende A, Büller HR, Bakker HD: Short-term efficacy and safety of pravastatin in 72 children with familial hypercholesterolemia. Pediatr Res, 1996; 39: 867-871 [DOI] [PubMed] [Google Scholar]
- 19).Stein EA, Illingworth DR, Kwiterovich PO, Jr., Liacouras CA, Siimes MA, Jacobson MS, Brewster TG, Hopkins P, Davidson M, Graham K, Arensman F, Knopp RH, DuJovne C, Williams CL, Isaacsohn JL, Jacobsen CA, Laskarzewski PM, Ames S, Gormley GJ: Efficacy and safety of lovastatin in adolescent males with heterozygous familial hypercholesterolemia: a randomized controlled trial. Jama, 1999; 281: 137-144 [DOI] [PubMed] [Google Scholar]
- 20).Wiegman A, Hutten BA, de Groot E, Rodenburg J, Bakker HD, Büller HR, Sijbrands EJ, Kastelein JJ: Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. Jama, 2004; 292: 331-337 [DOI] [PubMed] [Google Scholar]
- 21).Stein EA, Dann EJ, Wiegman A, Skovby F, Gaudet D, Sokal E, Charng MJ, Mohamed M, Luirink I, Raichlen JS, Sundén M, Carlsson SC, Raal FJ, Kastelein JJP: Efficacy of Rosuvastatin in Children With Homozygous Familial Hypercholesterolemia and Association With Underlying Genetic Mutations. J Am Coll Cardiol, 2017; 70: 1162-1170 [DOI] [PubMed] [Google Scholar]
- 22).Lambert M, Lupien PJ, Gagné C, Lévy E, Blaichman S, Langlois S, Hayden M, Rose V, Clarke JT, Wolfe BM, Clarson C, Parsons H, Stephure DK, Potvin D, Lambert J: Treatment of familial hypercholesterolemia in children and adolescents: effect of lovastatin. Canadian Lovastatin in Children Study Group. Pediatrics, 1996; 97: 619-628 [PubMed] [Google Scholar]
- 23).Harada-Shiba M, Arisaka O, Ohtake A, Okada T, Suganami H: Efficacy and Safety of Pitavastatin in Japanese Male Children with Familial Hypercholesterolemia. J Atheroscler Thromb, 2016; 23: 48-55 [DOI] [PubMed] [Google Scholar]
- 24).Nozue T, Michishita I, Ito Y, Hirano T: Effects of statin on small dense low-density lipoprotein cholesterol and remnant-like particle cholesterol in heterozygous familial hypercholesterolemia. J Atheroscler Thromb, 2008; 15: 146-153 [DOI] [PubMed] [Google Scholar]
- 25).Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF: Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet, 2001; 357: 577-581 [DOI] [PubMed] [Google Scholar]
- 26).Rodenburg J, Vissers MN, Wiegman A, van Trotsenburg AS, van der Graaf A, de Groot E, Wijburg FA, Kastelein JJ, Hutten BA: Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation, 2007; 116: 664-668 [DOI] [PubMed] [Google Scholar]
- 27).Besseling J, Hovingh GK, Huijgen R, Kastelein JJP, Hutten BA: Statins in Familial Hypercholesterolemia: Consequences for Coronary Artery Disease and All-Cause Mortality. J Am Coll Cardiol, 2016; 68: 252-260 [DOI] [PubMed] [Google Scholar]
- 28).Jay RH, Sturley RH, Stirling C, McGarrigle HH, Katz M, Reckless JP, Betteridge DJ: Effects of pravastatin and cholestyramine on gonadal and adrenal steroid production in familial hypercholesterolaemia. Br J Clin Pharmacol, 1991; 32: 417-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, Hovingh GK, Cariou B, Gouni-Berthold I, Somaratne R, Bridges I, Scott R, Wasserman SM, Gaudet D: PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet, 2015; 385: 331-340 [DOI] [PubMed] [Google Scholar]
- 30).Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, Blom D, Civeira F, Krempf M, Lorenzato C, Zhao J, Pordy R, Baccara-Dinet MT, Gipe DA, Geiger MJ, Farnier M: ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J, 2015; 36: 2996-3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA: Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet, 2015; 385: 341-350 [DOI] [PubMed] [Google Scholar]
- 32).Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, Visseren FL, Sijbrands EJ, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E: Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med, 2008; 358: 1431-1443 [DOI] [PubMed] [Google Scholar]
- 33).Authors/Task Force Members; ESC Committee for Practice Guidelines (CPG); ESC National Cardiac Societies ; 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis, 2019; 290: 140-205 [DOI] [PubMed] [Google Scholar]
- 34).Wang A, Richhariya A, Gandra SR, Calimlim B, Kim L, Quek RG, Nordyke RJ, Toth PP: Systematic Review of Low-Density Lipoprotein Cholesterol Apheresis for the Treatment of Familial Hypercholesterolemia. J Am Heart Assoc, 2016; 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Luirink IK, Determeijer J, Hutten BA, Wiegman A, Bruckert E, Schmitt CP, Groothoff JW: Efficacy and safety of lipoprotein apheresis in children with homozygous familial hypercholesterolemia: A systematic review. J Clin Lipidol, 2019; 13: 31-39 [DOI] [PubMed] [Google Scholar]
- 36).Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics, 2011; 128 Suppl 5: S213-256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Vuorio A, Kuoppala J, Kovanen PT, Humphries SE, Tonstad S, Wiegman A, Drogari E, Ramaswami U: Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev, 2019; 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Luirink IK, Wiegman A, Kusters DM, Hof MH, Groothoff JW, de Groot E, Kastelein JJP, Hutten BA: 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N Engl J Med, 2019; 381: 1547-1556 [DOI] [PubMed] [Google Scholar]
- 39).Hori M, Takahashi A, Son C, Ogura M, Harada-Shiba M: The first Japanese cases of familial hypercholesterolemia due to a known pathogenic APOB gene variant, c.10580 G>A: p.(Arg3527Gln). J Clin Lipidol, 2020; 14: 482-486 [DOI] [PubMed] [Google Scholar]
- 40).Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S, Yamamoto A: Clinical features and genetic analysis of autosomal recessive hypercholesterolemia. J Clin Endocrinol Metab, 2003; 88: 2541-2547 [DOI] [PubMed] [Google Scholar]
- 41).Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, Neil HA, Descamps OS, Langenberg C, Lench N, Kivimaki M, Whittaker J, Hingorani AD, Kumari M, Humphries SE: Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet, 2013; 381: 1293-1301 [DOI] [PubMed] [Google Scholar]
- 42).Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A: Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-3490a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, Ose L, Averna M, Boileau C, Borén J, Bruckert E, Catapano AL, Defesche JC, Descamps OS, Hegele RA, Hovingh GK, Humphries SE, Kovanen PT, Kuivenhoven JA, Masana L, Nordestgaard BG, Pajukanta P, Parhofer KG, Raal FJ, Ray KK, Santos RD, Stalenhoef AF, Steinhagen-Thiessen E, Stroes ES, Taskinen MR, Tybjærg-Hansen A, Wiklund O: Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J, 2015; 36: 2425-2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Matsunaga K, Mizobuchi A, Fu HY, Ishikawa S, Tada H, Kawashiri MA, Yokota I, Sasaki T, Ito S, Kunikata J, Iwase T, Hirao T, Yokoyama K, Hoshikawa Y, Fujisawa T, Dobashi K, Kusaka T, Minamino T: Universal Screening for Familial Hypercholesterolemia in Children in Kagawa, Japan. J Atheroscler Thromb, 2022; 29: 839-849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Kinoshita M, Yokote K, Arai H, Iida M, Ishigaki Y, Ishibashi S, Umemoto S, Egusa G, Ohmura H, Okamura T, Kihara S, Koba S, Saito I, Shoji T, Daida H, Tsukamoto K, Deguchi J, Dohi S, Dobashi K, Hamaguchi H, Hara M, Hiro T, Biro S, Fujioka Y, Maruyama C, Miyamoto Y, Murakami Y, Yokode M, Yoshida H, Rakugi H, Wakatsuki A, Yamashita S: Japan Atherosclerosis Society (JAS) Guidelines for Prevention of Atherosclerotic Cardiovascular Diseases 2017. J Atheroscler Thromb, 2018; 25: 846-984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Abe Y, Okada T, Sugiura R, Yamauchi K, Murata M: Reference Ranges for the Non-High-Density Lipoprotein Cholesterol Levels in Japanese Children and Adolescents. J Atheroscler Thromb, 2015; 22: 669-675 [DOI] [PubMed] [Google Scholar]
- 47).Dobashi K: Changes in Serum Cholesterol in Childhood and its Tracking to Adulthood. J Atheroscler Thromb, 2022; 29: 5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Eissa MA, Mihalopoulos NL, Holubkov R, Dai S, Labarthe DR: Changes in Fasting Lipids during Puberty. J Pediatr, 2016; 170: 199-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Tanner JM: Growth at adolescence, Blackwell Scientific Publications, Oxford, 1962 [Google Scholar]
- 50).Harada-Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, Dobashi K, Nohara A, Bujo H, Miyauchi K, Yamashita S, Yokote K: Guidelines for Diagnosis and Treatment of Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 751-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51).Nagahara K, Nishibukuro T, Ogiwara Y, Ikegawa K, Tada H, Yamagishi M, Kawashiri MA, Ochi A, Toyoda J, Nakano Y, Adachi M, Mizuno K, Hasegawa Y, Dobashi K: Genetic Analysis of Japanese Children Clinically Diagnosed with Familial Hypercholesterolemia. J Atheroscler Thromb, 2022; 29: 667-677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Ministry of Health, Labour and Welfare: The National Health and Nutrition Survey in Japan, 2018, https://www.mhlw.go.jp/content/000681200.pdf, 2018 in Japanese [Google Scholar]
- 53).Ramaswami U, Futema M, Bogsrud MP, Holven KB, Roeters van Lennep J, Wiegman A, Descamps OS, Vrablik M, Freiberger T, Dieplinger H, Greber-Platzer S, Hanauer-Mader G, Bourbon M, Drogari E, Humphries SE: Comparison of the characteristics at diagnosis and treatment of children with heterozygous familial hypercholesterolaemia (FH) from eight European countries. Atherosclerosis, 2020; 292: 178-187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).Ohta T, Kiwaki K, Endo F, Umehashi H, Matsuda I: Dyslipidemia in young Japanese children: its relation to familial hypercholesterolemia and familial combined hyperlipidemia. Pediatr Int, 2002; 44: 602-607 [DOI] [PubMed] [Google Scholar]
- 55).The Japanese Association of Medical Science: Guidelines for Genetic Testing and Diagnosis in Medical Care, https://jams.med.or.jp/guideline/genetics-diagnosis.pdf, 2011 [Google Scholar]
- 56).Ministry of Education C, Sports, Science and Technology: Ethical Guidelines for Life Sciences and Medical Research Involving Human Subjects, https://www.mhlw.go.jp/content/000769923.pdf, 2021 in Japanese [Google Scholar]
- 57).Youngblom E, Pariani M, Knowles JW: Familial Hypercholesterolemia.In: Familial Hypercholesterolemia, ed by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, University of Washington, Seattle Copyright © 1993-2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., Seattle (WA), 1993 [Google Scholar]
- 58).Tada H, Hori M, Nomura A, Hosomichi K, Nohara A, Kawashiri MA, Harada-Shiba M: A catalog of the pathogenic mutations of LDL receptor gene in Japanese familial hypercholesterolemia. J Clin Lipidol, 2020; 14: 346-351.e349 [DOI] [PubMed] [Google Scholar]
- 59).Noguchi T, Katsuda S, Kawashiri MA, Tada H, Nohara A, Inazu A, Yamagishi M, Kobayashi J, Mabuchi H: The E32K variant of PCSK9 exacerbates the phenotype of familial hypercholesterolaemia by increasing PCSK9 function and concentration in the circulation. Atherosclerosis, 2010; 210: 166-172 [DOI] [PubMed] [Google Scholar]
- 60).Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Inoue T, Mori M, Tada H, Nakanishi C, Yagi K, Yamagishi M, Ueda K, Takegoshi T, Miyamoto S, Inazu A, Koizumi J: Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis, 2014; 236: 54-61 [DOI] [PubMed] [Google Scholar]
- 61).Tada H, Okada H, Nomura A, Yashiro S, Nohara A, Ishigaki Y, Takamura M, Kawashiri MA: Rare and Deleterious Mutations in ABCG5/ABCG8 Genes Contribute to Mimicking and Worsening of Familial Hypercholesterolemia Phenotype. Circ J, 2019; 83: 1917-1924 [DOI] [PubMed] [Google Scholar]
- 62).Tada H, Kawashiri MA, Takata M, Matsunami K, Imamura A, Matsuyama M, Sawada H, Nunoi H, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, Mabuchi H, Yamagishi M: Infantile Cases of Sitosterolaemia with Novel Mutations in the ABCG5 Gene: Extreme Hypercholesterolaemia is Exacerbated by Breastfeeding. JIMD Rep, 2015; 21: 115-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Japan Atherosclerosis Society: Dyslipidemia Practice Guide 2018, Japan Atherosclerosis Society, 2018 in Japanese [Google Scholar]
- 64).Ministry of Health, Labour and Welfare: Dietary Reference Intakes for Japanese (2020) https://www.mhlw.go.jp/content/10904750/000586553.pdf 2019 in Japanese [Google Scholar]
- 65).Berger S, Raman G, Vishwanathan R, Jacques PF, Johnson EJ: Dietary cholesterol and cardiovascular disease: a systematic review and meta-analysis. Am J Clin Nutr, 2015; 102: 276-294 [DOI] [PubMed] [Google Scholar]
- 66).Japan Atherosclerosis Society: Statement on Cholesterol Intake, https://www.j-athero.org/jp/outline/cholesterol_150501/, 2015 [Google Scholar]
- 67).Japan Society for the Study of Obesity: Guidelines for the management of obesity disease in children and adolescents 2017, Life Science Publishing 2017 in Japanese [Google Scholar]
- 68).Dobashi K: Evaluation of Obesity in School-Age Children. J Atheroscler Thromb, 2016; 23: 32-38 [DOI] [PubMed] [Google Scholar]
- 69).Amemiya S, Dobashi K, Urakami T, Sugihara S, Ohzeki T, Tajima N: Metabolic syndrome in youths. Pediatr Diabetes, 2007; 8 Suppl 9: 48-54 [DOI] [PubMed] [Google Scholar]
- 70).Dietary Balance Guide Food Guide (tentative name) Study Group Report: Ministry of Health, Labor and Welfare and Ministry of Agriculture, Forestry and Fisheries Decision. Diichi-shuppan 2005 in Japanese [Google Scholar]
- 71).Wiegman A, de Groot E, Hutten BA, Rodenburg J, Gort J, Bakker HD, Sijbrands EJ, Kastelein JJ: Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia. Lancet, 2004; 363: 369-370 [DOI] [PubMed] [Google Scholar]
- 72).Wierzbicki AS, Humphries SE, Minhas R: Familial hypercholesterolaemia: summary of NICE guidance. Bmj, 2008; 337: a1095 [DOI] [PubMed] [Google Scholar]
- 73).Kwiterovich PO, Jr.: Recognition and management of dyslipidemia in children and adolescents. J Clin Endocrinol Metab, 2008; 93: 4200-4209 [DOI] [PubMed] [Google Scholar]
- 74).Tada H, Takamura M, Kawashiri MA: Lipoprotein(a) as an Old and New Causal Risk Factor of Atherosclerotic Cardiovascular Disease. J Atheroscler Thromb, 2019; 26: 583-591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75).Kusumi Y, Sakurai I: Significance of Lp(a) deposition in the aortic intima of children and young adults. Doumyakukouka, 2000; 27: 223-227 [Google Scholar]
- 76).Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, Sofat R, Stender S, Johnson PC, Scott RA, Leusink M, Verweij N, Sharp SJ, Guo Y, Giambartolomei C, Chung C, Peasey A, Amuzu A, Li K, Palmen J, Howard P, Cooper JA, Drenos F, Li YR, Lowe G, Gallacher J, Stewart MC, Tzoulaki I, Buxbaum SG, van der AD, Forouhi NG, Onland-Moret NC, van der Schouw YT, Schnabel RB, Hubacek JA, Kubinova R, Baceviciene M, Tamosiunas A, Pajak A, Topor-Madry R, Stepaniak U, Malyutina S, Baldassarre D, Sennblad B, Tremoli E, de Faire U, Veglia F, Ford I, Jukema JW, Westendorp RG, de Borst GJ, de Jong PA, Algra A, Spiering W, Maitland-van der Zee AH, Klungel OH, de Boer A, Doevendans PA, Eaton CB, Robinson JG, Duggan D, Kjekshus J, Downs JR, Gotto AM, Keech AC, Marchioli R, Tognoni G, Sever PS, Poulter NR, Waters DD, Pedersen TR, Amarenco P, Nakamura H, McMurray JJ, Lewsey JD, Chasman DI, Ridker PM, Maggioni AP, Tavazzi L, Ray KK, Seshasai SR, Manson JE, Price JF, Whincup PH, Morris RW, Lawlor DA, Smith GD, Ben-Shlomo Y, Schreiner PJ, Fornage M, Siscovick DS, Cushman M, Kumari M, Wareham NJ, Verschuren WM, Redline S, Patel SR, Whittaker JC, Hamsten A, Delaney JA, Dale C, Gaunt TR, Wong A, Kuh D, Hardy R, Kathiresan S, Castillo BA, van der Harst P, Brunner EJ, Tybjaerg-Hansen A, Marmot MG, Krauss RM, Tsai M, Coresh J, Hoogeveen RC, Psaty BM, Lange LA, Hakonarson H, Dudbridge F, Humphries SE, Talmud PJ, Kivimäki M, Timpson NJ, Langenberg C, Asselbergs FW, Voevoda M, Bobak M, Pikhart H, Wilson JG, Reiner AP, Keating BJ, Hingorani AD, Sattar N: HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet, 2015; 385: 351-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77).Kajinami K, Tsukamoto K, Koba S, Inoue I, Yamakawa M, Suzuki S, Hamano T, Saito H, Saito Y, Masuda S, Nakayama T, Okamura T, Yamashita S, Kagawa T, Kaneyama J, Kuriyama A, Tanaka R, Hirata A: Statin Intolerance Clinical Guide 2018. J Atheroscler Thromb, 2020; 27: 375-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78).Edison RJ, Muenke M: Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med, 2004; 350: 1579-1582 [DOI] [PubMed] [Google Scholar]
- 79).Uauy R, Vega GL, Grundy SM, Bilheimer DM: Lovastatin therapy in receptor-negative homozygous familial hypercholesterolemia: lack of effect on low-density lipoprotein concentrations or turnover. J Pediatr, 1988; 113: 387-392 [DOI] [PubMed] [Google Scholar]
- 80).Malloy MJ, Kane JP, Kunitake ST, Tun P: Complementarity of colestipol, niacin, and lovastatin in treatment of severe familial hypercholesterolemia. Ann Intern Med, 1987; 107: 616-623 [DOI] [PubMed] [Google Scholar]
- 81).Yamamoto A, Harada-Shiba M, Kawaguchi A, Oi K, Kubo H, Sakai S, Mikami Y, Imai T, Ito T, Kato H, Endo M, Sato I, Suzuki Y, Hori H: The effect of atorvastatin on serum lipids and lipoproteins in patients with homozyous familial hypercholesterolemia undergoing LDL-apheresis therapy. Atherosclerosis, 2000; 153: 89-98 [DOI] [PubMed] [Google Scholar]
- 82).Yamamoto A, Matsuzawa Y, Kishino B, Hayashi R, Hirobe K, Kikkawa T: Effects of probucol on homozygous cases of familial hypercholesterolemia. Atherosclerosis, 1983; 48: 157-166 [DOI] [PubMed] [Google Scholar]
- 83).Yamashita S, Bujo H, Arai H, Harada-Shiba M, Matsui S, Fukushima M, Saito Y, Kita T, Matsuzawa Y: Long-term probucol treatment prevents secondary cardiovascular events: a cohort study of patients with heterozygous familial hypercholesterolemia in Japan. J Atheroscler Thromb, 2008; 15: 292-303 [DOI] [PubMed] [Google Scholar]
- 84).Jones DB, Simpson HC, Slaughter P, Lousley S, Carter RD, Cobbe SM, Mann JI: A comparison of cholestyramine and probucol in the treatment of familial hypercholesterolaemia. Atherosclerosis, 1984; 53: 1-7 [DOI] [PubMed] [Google Scholar]
- 85).Buckley MM, Goa KL, Price AH, Brogden RN: Probucol. A reappraisal of its pharmacological properties and therapeutic use in hypercholesterolaemia. Drugs, 1989; 37: 761-800 [DOI] [PubMed] [Google Scholar]
- 86).Yamamoto A, Harada-Shiba M, Endo M, Kusakabe N, Tanioka T, Kato H, Shoji T: The effect of ezetimibe on serum lipids and lipoproteins in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis therapy. Atherosclerosis, 2006; 186: 126-131 [DOI] [PubMed] [Google Scholar]
- 87).Shirahata Y, Ohkohchi N, Kawagishi N, Syouji M, Tsukamoto S, Sekiguchi S, Koyamada N, Oikawa S, Satomi S: Living-donor liver transplantation for homozygous familial hypercholesterolemia from a donor with heterozygous hypercholesterolemia. Transpl Int, 2003; 16: 276-279 [DOI] [PubMed] [Google Scholar]
- 88).Kawagishi N, Satoh K, Akamatsu Y, Sekiguchi S, Ishigaki Y, Oikawa S, Satomi S: Long-term outcome after living donor liver transplantation for two cases of homozygous familial hypercholesterolemia from a heterozygous donor. J Atheroscler Thromb, 2007; 14: 94-98 [DOI] [PubMed] [Google Scholar]
- 89).Stefanutti C, Di Giacomo S, Vivenzio A, Colloridi V, Bosco G, Berni A, Rabbone I, Cerutti F, Bertolini S: Low-density lipoprotein apheresis in a patient aged 3.5 years. Acta Paediatr, 2001; 90: 694-701 [DOI] [PubMed] [Google Scholar]
- 90).Makino H, Harada-Shiba M: Long-term effect of low-density lipoprotein apheresis in patients with homozygous familial hypercholesterolemia. Ther Apher Dial, 2003; 7: 397-401 [DOI] [PubMed] [Google Scholar]
- 91).Mol MJ, Stalenhoef AF: Homozygous familial hypercholesterolaemia: metabolic studies and treatment with LDL apheresis. Neth J Med, 1990; 36: 279-287 [PubMed] [Google Scholar]
- 92).Naito C, Yamamoto A, Saito Y, Muto E, Nishide T, Shinomiya M, Mukai M, Tomono S, Sato T, Yasuda K, Yamauchi T, Mabuchi H, Kobayashi J, Ueda H, Nonaka K, Wada N, Yamasak S, Ono T, Yoshimura, A, Harada M, Nakamura H, Kondo K, Ageta M, Matsui T, Mayuyama H, Fukuo Y, Matsuzawa Y, Irie Y, Kon T, Endo M, Yuasa Y, Takahashi Y, Koda Y, Hara Y, Kikkawa T, Oikawa S, Ito H, Homma Y, Iino Y, Takahashi Y, Ito H, Teramoto T, Takahashi K, Mimori A, Takada H, Suzuki M, Kawanishi H, Yashiro A, Tazaki H, Segawa J, Sugano K, Kawashima T, Matsushima T, Nakajima Y: Long term effect of LDL apheresis in Japan. LDL Apheresis Study Group. Biomater Artif Cells Immobilization Biotechnol, 1991; 19: 19-26 [DOI] [PubMed] [Google Scholar]
- 93).Yamamoto A, Harada-Shiba M, Kawaguchi A, Tsushima M: Apheresis technology for prevention and regression of atherosclerosis. Ther Apher, 2001; 5: 221-225 [DOI] [PubMed] [Google Scholar]
- 94).Information Center for Specific Pediatric Chronic Diseases, Japan, https://www.shouman.jp/disease/details/08_12_130/ [Google Scholar]
- 95).Ministry of Health, Labour and Welfare: Japan Intractable Diseases Information Center, https://www.nanbyou.or.jp/entry/65 [Google Scholar]