

Abstract
Cryo-electron tomography can uniquely probe the native cellular environment for macromolecular structures. Tomograms feature complex data with densities of diverse, densely crowded macromolecular complexes, low signal-to-noise, and artifacts such as the missing wedge effect. Post-processing of this data generally involves isolating regions or particles of interest from tomograms, organizing them into related groups, and rendering final structures through subtomogram averaging. Template-matching and reference-based structure determination are popular analysis methods but are vulnerable to biases and can often require significant user input. Most importantly, these approaches cannot identify novel complexes that reside within the imaged cellular environment. To reliably extract and resolve structures of interest, efficient and unbiased approaches are therefore of great value. This review highlights notable computational software and discusses how they contribute to making automated structural pattern discovery a possibility. Perspectives emphasizing the importance of features for user-friendliness and accessibility are also presented.
Keywords: Cryo-ET, image pattern recognition, data mining, machine learning, cellular structural biology
Graphical Abstract
Introduction
Cryo-electron tomography (cryo-ET) enables in situ visualization of subcellular structures in their native states inside a cell, which is not possible by any other imaging technology [1]. X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single particle cryo-electron microscopy (cryo-EM) can resolve atomic details of macromolecular structures but are in vitro methods that lack cell context. Conversely, fluorescence imaging methods can image an entire cell and the organelles within but cannot resolve macromolecular structures. Cryo-ET uniquely bridges these imaging methods by providing molecular resolution information of structures in a cellular context [2]. The absence of disruptive sample preparation, such as chemical fixation [3], further enables the observation of spatial- and temporal-regulated proteinprotein interactions, permitting the study of a cell’s native molecular sociology [4]. Thus, cryo-ET can reveal complex and novel macromolecular structures inside the cells which represent active function-conducting cellular machines. In preferred scenarios, the resolution can achieve the sub-nanometer range, thereby directly revealing structural mechanisms in situ [5–7].
In cryo-ET, specimens are immobilized via cryo-fixation and subsequently imaged as the sample is progressively tilted about an axis perpendicular to the electron beam. This generates a series of 2D transmission electron micrographs that can then be used to reconstruct a 3D image, or tomogram [8]. The tomograms contain readily visible organelles and macromolecular structures, represented as recurring density patterns that appear throughout the volume. By recognizing and then averaging multiple instances of a pattern, the underlying structure can be determined. A complete molecular structural landscape visualization of a cell can be pursued by determining multiple structures and localizing them throughout the tomogram (Figure 1a–e). Yet, this is challenging given that raw tomograms contain large, complex data that is further complicated by artifacts such as the missing wedge effect and low signal-to-noise ratio (SNR) [9]. Manual analysis of such data is not only cumbersome but at risk of bias. Hence, automated methods to extract and analyze unique cellular structures captured by cryo-ET are of great importance.
Figure 1. Exploring the diverse, crowded environment within cellular tomograms.

A tomogram slice (a) and corresponding isosurface rendering (b) of an endoplasmic reticulum (ER)-derived vesicle decorated in ribosome-transolocon complexes. (c) The cryo-EM structure of the translocon complex bound to the translocon associated protein complex (TRAP) and oligosaccharyl-transferase (OST), determined from subtomogram averaging of the complex on vesicles. A tomogram slice (d) and the isosurface rendering of the determined complexes (e) in intact cells demonstrates the increased complexity of studying structures in the native cellular context and demonstrates the generation of a limited visual atlas [78]. A tomogram slice (f) of a PC12 cell and (g) the training and annotation of regions of interest in the EMAN2 analysis software. (h) The automated segmentation of four independently trained neural networks for each labeled structure of interest. (i) The final merged annotation of the four features identified by EMAN2 segmentation neural network [14].
Tomogram processing suites such as IMOD [10–12], EMAN2 [13–16], PyTom [17–20], Protomo [21–25], and others provide comprehensive tools for general tomogram analysis (see Table 1). In addition, the development of new methods that target specific tasks to advance cryo-ET analysis is also being pursued from many facets (see Table 2). Below we follow the general sequence for a structural determination that follows tomographic reconstruction, discussing the advantages, shortcomings, and accessibility of currently available methods to contextualize the current state of quantitative cryo-ET analysis. We primarily focus on approaches that reduce the manual intervention required by researchers and highlight algorithmic developments that we envision will advance the field towards mining structures de novo from cryo-ET tomograms.
Table 1:
Cryo-ET image software for pattern-mining tasks.
| Software tool | GUI | Segmentation | Template matching | Template- free |
Subtomogram Averaging | Classification |
|---|---|---|---|---|---|---|
| Amira + EMPackage [26] |
Yes | Yes | Yes (for fiber tracing) | No | No | No |
| Bsoft (w/ Jsubtomo) [27,28] |
No | Yes | Yes | No | Yes | No |
| Dynamo [29–33] |
Yes | No | Manual | No | Yes | Yes |
| EMAN2 [13–16] |
Yes | Yes | Yes | Yes | Yes | Yes |
| emClarity [34,35] |
No | No | Yes | No | Yes | No |
| IMOD (w/ PEET) [10–12] |
Yes | Yes | Manual | No | Yes | No |
| Protomo [21–25] |
No | No | Yes | No | Yes | Yes |
| PySeg [36–38] |
No | Yes | No | Yes | Yes | Yes |
| PyTom [17–20] |
Partial | No | Yes | No | Yes | Yes |
| RELION [39–43] |
Yes | No | No | No | Yes | Yes |
| SuRVoS [44] |
Yes | Yes | No | No | No | No |
| Tomominer,TomoMinerCloud [45] |
No | No | Yes | Yes | Yes | Yes |
Table 2:
Recent notable technical developments for specific cryo-ET pattern-mining tasks.
| Software tool | Task | Notes |
|---|---|---|
| SuRVoS [44] |
Segmentation | Allows users to varythe degree of manual segmentation required. |
| DSM-Net [46] |
Segmentation Classification (supervised) Structure determination |
Multi-task deep learning model to train three networks simultaneously on (1) semantic segmentation, (2) classification, and (3) coarse structure determination. Outperformed single-task models and was able to identify structures absent in training data. |
| VP-Detector [47] |
Segmentation Particle picking Classification (supervised) | Uses a CNN to take user-annotated data and automatically segment, localize, and classify particles of interest. |
| DeepFinder [48] |
Particle picking | Efficient multiclass particle picking of structures with various sizes and shapes. |
| 3D-UCaps [49] |
Particle picking | Efficient multiclass particle picking was given limited training data. |
| PySeg [36] |
Particle picking Classification (unsupervised) | Particle picking of small membrane-bound complexes. |
| DSRF3D-v2, RB3D, CB3D [50] |
Classification (supervised) | Classifies complexes spanning a range of molecular weights. |
| DISCA [51] |
Classification (unsupervised) | High-throughput, fully unsupervised approach to cluster particles using deep learning. |
| MPP [52]7 |
Classification (unsupervised) Structure determination |
Iterative alignment to sort a heterogenous set of particles into structure patterns, capable of identifying patterns de novo. |
| Jim-Net [53] |
Classification (unsupervised) Structure determination |
Uses pair-matching alignment and hierarchical clustering to achieve comparable accuracy with supervised method to determine structures de novo. |
| RELION-4.0 Bayesian single-particle tomography structure determination [43] |
Structure determination | Structure determination via optimization of a regularized likelihood function. In contrast to previous RELION renditions, features a new weighting system and refinement methods. |
| FAML [54] |
Structure determination | Combines features of FRM and maximum likelihood structure determination methods to achieve higher robustness to noise and artifacts compared to FRM methods and requires fewer subtomograms. |
| Harmony [126] |
Structure determination | Isolates shape information of a particle, allowing for further clustering or alignment. |
| HEMNMA-3D [55] |
Structure determination | Creates a conformational space of a complex that maps how a structure may transition between different configurations in situ. |
| - [56] |
Data augmentation | High density packing by modeling structures with multiple spheres. |
| - [57] |
Data augmentation | Computationally efficient packing by modeling structures as single spheres with gradient descent algorithm. |
| CryoETGAN [58] |
Data augmentation | Deep learning model designed to train on tomograms to generate imitations that can be used to increased training data without the need to simulate artificial tomograms, which may not fully reflect experimental data. |
| 3D-ADA [59] |
Domain adaptation | Utilizes deep learning to map particles from images with different parameters to an intermediate latent space. |
| Cryo-Shift [60] |
Domain adaptation | Transforms training data to improve the generalization ability of classifiers to predict on different tomography datasets. |
1. Segmentation
Segmentation is frequently the first step of examining the tomogram. Voxels that correspond to biological features of interest are annotated in the tomogram for 3D illustration. Segmentations can be performed manually in software such as IMOD [10], where a user traces the region of interest slice-by-slice, but this strategy is tedious and sometimes inconsistent [61]. Low SNR, low contrast, missing wedge effects, and crowded densities exacerbate the difficulty of this task [26,44,62]. Developing strategies for fast, reproducible, and high-quality segmentations is crucial for reliable data annotation and structural analyses.
Different methods have been developed to reduce the level of human intervention required for segmentation. Early approaches largely focused on boundary detection [63–65], watershed algorithms [66], and comparisons of regions to pre-defined models of structures [62,67]. Amira [26] is a popular graphics software by Thermo Fisher Scientific that implements watershed thresholding algorithms to semi-automatically segment biological structures such as membranes, nuclei, and filaments. Methods that unite watershed and boundary approaches have also been investigated [68]. Each approach has its own set of weaknesses, causing its performance to be highly dependent on the dataset. Moreover, they require significant input from the user for reasonable segmentations.
SuRVoS [44] is a more recent tool that employs machine learning on manually segmented slices to predict regions on subsequent slices or automatically segment boundaries by analyzing local density changes within the tomogram, effectively reducing the amount of annotation required. Deep learning methods have also been developed toward fully automated segmentation. These are capable of robustly segmenting multiple subcellular structures at once. For instance, the EMAN2 software suite [14,15] includes a deep learning-based automated segmentation method that requires only 10 manually annotated images to locate similar particles throughout the tomogram. This vastly reduces the need for manual annotation for feature extraction (Figure 1f–i). The software can also utilize a neural network trained on one tomogram to segment other tomograms, provided they have a similar imaging quality and voxel size. This semi-automated segmentation approach has supported the investigations of Trypanosoma brucei as well as thylakoid membranes [69,70]. In addition to annotation, segmentation is commonly used to facilitate other structural pattern-mining tasks. Many software introduced in later sections of the review can also perform automated segmentation to facilitate particle picking by restricting the search space (e.g., using membrane segmentations to locate membrane-bound complexes) and informing on particle orientation [2,36,61].
2. Particle picking
Particle picking can be considered a variation of segmentation. This process picks subvolumes, also known as subtomograms, of equal size in the tomogram and each contains a structure of interest (the “particle”) [71]. The subtomograms can then be computationally cropped out and the collection of particles is overlaid to generate a subtomogram average with enhanced contrast and resolution of the structure of interest. While software packages often allow users to manually select positions to generate subtomograms [29,72], this can be time-consuming, subjective, and incomplete [73]. Template matching and template-free approaches for particle picking have been developed to facilitate this task.
2.1. Template matching
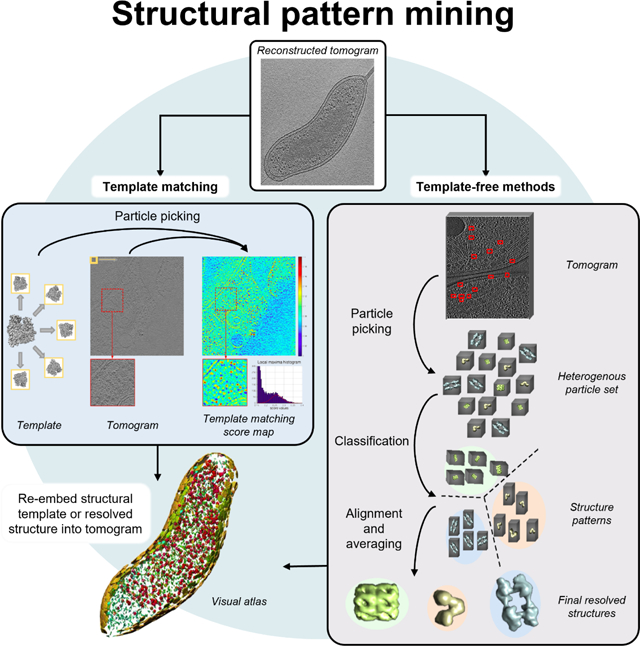
Templates are used to find corresponding density matches in the tomogram. They are generally macromolecular structures resolved with X-ray crystallography, NMR spectroscopy, or single-particle cryo-electron microscopy (cryo-EM). These reference structures must be properly low-pass-filtered and adjusted to reflect the tomogram’s resolution and distortions from the cryo-ET imaging process [74,75]. Templates may also be generated de novo from subtomogram averages if densities are highly abundant and regular [76]. All possible orientations of the templates are scanned through the tomogram to calculate correlation maps. Statistical analyses, such as peak extraction, are applied to the map to discern macromolecule positions throughout the tomogram. The reference structure can even be mapped back to these positions to create a visual atlas of the macromolecule populating the tomogram [18] (Figure 2a).
Figure 2. Particle picking via template matching or template-free approaches.

. (a) Template mapping to achieve a visual protein atlas. Template matching structures from a template library identifies the cross-correlation values of each reference against regions of a tomogram, ultimately identifying positions that most likely represent particles from the library. The rendered structure models at the calculated positions and orientations forms a protein atlas, providing information on the distribution of these structures throughout the cell and their relation to other structures [91]. (b) manually segmented membranes (gray and yellow) and particle picking positions Rubisco holoenzymes (orange) in native Chlamydomonas pyrenoid determined by DeepFinder [48]. (c) A tomogram slice (top) with the segmentations as performed by DeepFinder (middle; orange) and template matching (cleaned with CPCA classification). Scale bar, 50 nm. Subtomogram averages of holoenzymes generated from each particle picking method with Rubisco molecular structure (d) and FSC curves showing final resolution of 15 Å (FSC > 0.143; e). Slice of tomogram (f) and zoom-in comparing DeepFinder to template matching score map (g). (h) Membranogram (topological view of membrane surface by projecting tomogram densities onto segmented membrane surface) and corresponding comparisons between DeepFinder-, template matching-, and expert-derived particle picking positions.
Many software packages offer template matching, including Dynamo [29–33], emClarity [34,35], PyTom [17–20], EMAN2 [13–16], and TomoMiner [45] (see Table 1). Template matching has enabled the determination of highly resolved structures of large macromolecular complexes, such as ribosomes, the 26S proteosome, and viral Gag assembles and is widely used to automatically identify particles of interest from cryo-ET tomograms [75,77–79]. Aside from discrete particles, it can be used in concert with tracing algorithms to segment filamentous structures, such as those present in the cytoskeleton [80–82].
Despite its popularity, traditional template matching suffers from several drawbacks. It is low throughput and can be extremely time-consuming [52]. Performance depends on both the image quality of the tomogram and the reference structure. Molecular crowding in the tomogram is another major concern [83]. Dense packing of particles in tomograms may increase the presence of off-target densities, making it more challenging to locate densities of interest. While a tight mask enables algorithms to ignore extraneous densities and focus on the most relevant regions of the candidate object when calculating template matching scores, this solution can only be used in limited situations where molecules are rigid and well-resolved [74]. False positives are another issue, resulting in references being matched to not only the true location of structures but also regions of noise [84,85].
The most inherent disadvantage of template matching is template bias, where detectable structures are limited to those with an existing template obtained from other structural biology methods [2,61,86]. Likewise, template-matching algorithms may suffer sample-specific biases that can occur if templates do not reflect unexpected binding partners or conformations. On the other hand, averaging manually selected subtomograms potentially introduces user-dependent bias. Consequently, template matching cannot be utilized effectively in studies involving flexible macromolecules or complex interactions, and alternative methods must be considered.
2.2. Template-free particle picking
To forgo the need for preexisting structural references to recover cellular structures de novo for pattern recognition, template-free approaches are of growing interest for cryo-ET data analysis. To achieve the nonspecific extraction of subtomogram volumes, some approaches have turned to deep learning. Convolutional neural networks (CNNs) are deep learning architectures that are powerful tools especially adept at image processing, which is useful given that subtomograms are essentially 3D images representing particles [87]. Neural networks such as DeepPicker [73] and SPHIRE-crYOLO [88] exist for automated particle picking in single-particle cryo-EM micrographs. Performing the same task in cellular tomograms is a greater challenge because the native cellular environment is more crowded and contains a more diverse set of structures compared to cryo-EM micrographs [15].
Neural networks for feature extraction have also been successfully implemented for cryo-ET. As previously noted, the EMAN2 package [14,15] implements deep learning for automated segmentation, but can also perform automatic particle picking. Although this approach can identify particles from more than just a single class, the computation time scales linearly for each additional class. Because particles of different structures could be used to inform the recognition of each other, it is possible to develop a more efficient system to leverage this information. DeepFinder [48] is a CNN that can simultaneously segment multiple classes of varying shapes and sizes (Figure 2b–h). While the EMAN2 classifier has a shorter training time to identify a single class, DeepFinder outperforms EMAN2 in multi-class automatic particle picking. Moreover, it reduces the need for extensive postprocessing for reliable results. Improving on these attempts, 3D-UCaps [49] is a deep learning framework that can also perform binary and multi-class particle identification. Replacing the data-hungry CNN architecture with the data-efficient CapsNet, this method demonstrates comparable picking performance even in situations with limited training data (defined as less than 10,000 annotated particles).
Specialized segmentation methods exist for membrane-bound complexes. Pyto [89] is specifically designed to detect small, flexible complexes linking larger structures such as membranes, organelles, or filaments. Multiple connectivity-based segmentation at different thresholds is organized into a hierarchy to establish a final segmentation. This is in turn used to identify particles. Pyto has a high detection rate and offers statistical analyses to characterize the segmentations. Another software for particle picking is PySeg [36], a software package that also focuses on the automatic localization of small, membrane-bound complexes. It incorporates TomoSegMemTV [37], a package that utilizes tensor voting to achieve robust segmentation of membranes, as well as PyOrg [38], which offers statistical analyses to characterize the segmentations.
3. Structure determination
Once particles have been identified from a tomogram, the underlying structures must be recovered by further processes. The particles in subtomograms are typically positioned in various orientations but should contain common features with other particles of the same structural class. Thus, subtomograms are aligned and averaged to recover the high-resolution structure shared by the individual particles within each class. The better-resolved structures can then be re-embedded into the original tomogram space to obtain the visual landscape. This is similar to template matching but replaces the reference with the obtained structure. Several well-established analysis packages provide subtomogram averaging and manipulation protocols (see Table 1). Note that these strategies are designed to analyze a relatively homogenous particle set. The following section (3. Structure determination) discusses strategies to handle heterogeneous particles, which is especially relevant for de novo structure mining.
There are two primary approaches to subtomogram averaging. The first is alignment-based determination, where an angular search is performed to align the subtomogram to a reference structure. This is the subtomogram averaging approach utilized by software such as Dynamo [29]. The reference structure is first masked and rotated in real space. To account for the missing wedge, the reference is then masked in the Fourier space. The 3D cross correlation values are calculated between the reference and subtomogram. The algorithm identifies the transformations that produce highest cross correlation score. These transformations are applied to the subtomograms, a weighted average is performed to account for the previous masking to yield a final structure. However, the search risks converging onto local minima, resulting in a suboptimal structure. Without a priori information on the search parameters, the algorithm may also be computationally intensive due to the Fourier transforms and rotations, which require the costly step of linear interpolation [71]. Fast rotational matching (FRM) is a form of alignment-based determination that forgo explicit rotation of volumes by decoupling alignment into a rotational and translational search to achieve faster global alignment [9,90], and has been implemented into tomogram analysis packages [12,91]. However, this comes at the cost of reaching suboptimal solutions and is prone to be biased by image noise and the missing wedge effect [54].
The second approach to subtomogram averaging, maximum likelihood-based determination, is more robust to the abovementioned effects, and which is used by software such as RELION [43,92]. Expectation-maximization algorithms [93] optimize a probability function of observing the experimental data given the initial reference structure [71,92,94]. The probability-weighted average of each subtomogram in each orientation becomes the new reference and the process is repeated. This is repeated in an iterative process as the reference and probability function are updated with a more refined sampling of possible orientations. The process converges into a well-refined high-resolution and identifiable structural map. This approach also overcomes the need to extensively tune parameters, as they are instead inferred during estimation [40]. Thus, maximum likelihood-based determination is more amenable for non-experts. However, the convergence is relatively slow and requires significant computational resources. For recovering structures from a very large data set, this may not be practical [54,71].
Efforts have been made to improve subtomogram averaging methods. One technique that has been developed to combine these methods to overcome the weaknesses of FRM and maximum likelihood-based determination is the Fast Alignment Maximum Likelihood (FAML) [54] method. This uses fast alignment to sample transformations; the sampled transformations then approximate integrals to update the maximum likelihoods of subtomogram averages using the expectation-maximization algorithm. Compared to FRM alone, FAML exhibits higher robustness to noise and missing wedge effects, and it performs better with fewer subtomograms.
If the underlying structure of subtomograms is flexible, they will exhibit densities with a degree of variation. HEMNMA-3D [55] is an analysis method that maps the low-dimensional representation of heterogeneity within a set of subtomograms. This technique relies on an initial atomic or pseudo-atomic reference structure of the complex to perform elastic deformations and achieve the mapping. The resulting conformational space allows for grouping and averaging, identification of unanticipated conformations, and animations of the reference structure deforming along the densest regions of the space (i.e., conformations shared by the most subtomograms) or following alternative, rarer trajectories.
4. Particle classification
As previously noted, if the total set of subtomograms contains particles with different structures, it must then be sorted into homogenous groups with similar features, known as structure patterns. Each structure pattern should correspond to a distinct macromolecule or alternate conformation of the same macromolecule. For de novo structure mining, the classification of particles is an inevitable hurdle to overcome. It can also serve to identify false positives from particle picking.
A common approach to classification is multireference alignment [71,95]. This slightly modifies the structure determination strategies discussed in the previous section (3. Structure determination). Rather than being compared to a single reference, the particle is aligned against a set of different references. The particle is ultimately assigned to the reference that yields the closest similarity. This roughly separates the initial particles into separate classes for each reference, which are then averaged to generate a new reference for subsequent iterations. This approach is implemented in popular software packages such as EMAN2 and Dynamo [29,96]. These strategies require knowledge of particles a priori, in the form of initial references, so are not conducive to identifying novel structures that populate the tomogram. Classification approaches that forgo the use of references are therefore especially desirable for end-to-end pattern mining.
Reference-free classification can either be supervised or unsupervised. Supervised classification aims to group subtomograms into homogeneous structure patterns based on knowledge provided by the user. Instead of providing a reference structure, a user manually annotates a set of training subtomograms, assigning a label to the subtomogram based on the structure present. With the labeled training data, the classification algorithm aims to imitate the labeling scheme for new data. Deep learning models are powerful tools for this task, especially the CNN architecture, which as noted before, is especially adept at image processing. Many of these frameworks localize particles prior to classification, and so are also capable of automated particle picking.
To this end, Che et al. investigated three CNN architectures (DSRF3D-v2, RB3D, and CB3D) [50]. Compared to previous, non-deep learning approaches, the CNNs demonstrate improved discrimination and scalability for structure determination. They found CB3D displays the best performance with a high level of accuracy, even with low SNR (0.01) and the missing wedge effect present. However, the classifier’s performance was dependent on the size of the macromolecules, as larger complexes were associated with higher accuracies.
Additional supervised deep learning networks have been employed to locate and classify heterogeneous particles, many of which are introduced in the Shape Retrieval Challenge (SHREC) for the classification of cryo-ET data [97–99]. Such methods include 3D MS-D, 3D ResNet [100,101], DN3DUnet [102–104], YOPO, and UMC [103,105,106] (introduced in SHREC 2020, see more details in [98]), as well as URFinder [100,101,107,108], U-CLSTM [102,103,109–112], MC DS Net [103,113], and CFN [100,114–116] (introduced in SHREC 2020, see more details in [99]). The performance of these techniques is evaluated on a fully annotated simulated tomogram dataset containing particles of various molecular weights (Figure 3a) and compared to the baseline of template matching methods performed in PyTom [19]. This assessment measured training time, prediction time, performance compared to network architecture size, and observed general trends for these approaches. The SHREC evaluations found that neural networks are more robust to noise than template matching while also significantly reducing computation time, depending on the architecture [97–99]. Of these methods, YOPO does not require semantic segmentation, and instead only relies on the coordinate and particle class for training making it more accessible for users compared to the other learning-based methods introduced in SHREC 2020 [98].
Figure 3. Recent supervised deep learning methods to locate and classify particles from tomograms and early work on unsupervised classification.

(a) Isosurface rendering of protein structures across a range of molecular weights in the SHREC data [47]. Neural network performance was evaluated for each particle and PyTom template matching (TM/TM-F) acted as a baseline. These approaches were evaluated based on their F1 score (i.e., the harmonic average of precision, the proportion of true positives to all positives, and recall, the proportion of true positives to all possible relevant elements). (b,c) F1 score versus the molecular weight of the particle for deep learning localization and classification frameworks assessed in SHREC 2020 [98] and SHREC 2021 [99], respectively. Classification by CPCA [117] using the reference structure shown in (d), generated from the GroEL14GroES7 complex on tomograms containing GroEL and GroEL/GroES. (e) The average of all subtomograms gathered. (f) Class averages produced from CPCA method. Isosurface rendering of subtomogram averages of two classes determined by CPCA corresponding to (g) GroEL and (h) GroEL/ES. (i) A schematic demonstrating the classification of mammalian ribosomes bound to native endoplasmic reticulum translocons by AC3D [85]. Four classes were identified: 80S ribosomes bound to translocons with TRAP only (class #1), 80S ribosomes bound to translocons with TRAP and OST (class #2), 60S ribosomes with TRAP only (class #3), and finally 60S ribosomes with both TRAP and OST.
Similar to the case of CB3D, the performance of neural networks featured in SHREC tends to increase with size of the particle, a trend that is especially drastic for smaller particles [97–99] (Figure 3b,c). VP-detector [47] is a deep learning framework designed to better predict on complexes across a wide range of molecular weights and features high accuracy, fewer training parameters, and good performance despite small training sets or class imbalances.
In contrast to fully or partially supervised classification, unsupervised approaches do not rely on any manual annotation inform on structure pattern and therefore recover novel or unlabeled structures de novo. k-means is a popular machine learning approach for clustering that is the basis for several approaches to unsupervised subtomogram classification. This strategy was often employed in early unsupervised classification strategies. For instance, Förster et al. developed the CPCA method [117] to perform binary classification while also reducing the impact of the missing wedge effect. A principal component analysis is performed on pair-wise constrained correlation values and used in conjunction with k-means clustering. This procedure discriminated between GroEL and GroEL/GroES complexes (Figure 3d–h) but was sensitive to SNR and the computational cost of calculating the correlation matrix increased quadratically with the number of subtomograms. AC3D [85] is an algorithm based upon a modified k-means clustering algorithm. This classification scheme is designed to focus on subtomogram regions with large structural discrepancies between class averages and successfully discriminates between structures with and without cofactors (Figure 3i). Both the CPCA and AC3D methods are implemented in PyTOM [19,85].
One drawback of k-means is that the method requires the user to estimate the initial number of structural patterns. It also assumes the classes are evenly populated and well-distributed. A machine learning method capable of forming structure patterns without an initial cluster number is affinity propagation, which evaluates similarities between subtomograms by transferring information between data points. PySeg [36] implements this technique to achieve the automated detection and classification of heterogeneous membrane-bound complexes.
Deep learning approaches are another alternative for unsupervised classification. PySeg [36], previously noted to be capable of locating membrane-bound complexes in a template-free manner, is also able to perform unsupervised classification on the acquired subtomograms. This approach was validated in microsomal membranes and subsequently tested on tomograms of P19 embryonic carcinoma cells (Figure 4a–b). Of the determined structure patterns, many were consistent with known structures (Figure 4c), such as the ribosome-bound translocon, while other averages were not immediately recognizable (Figure 4d). However, the particles contained in these classes nonetheless appeared to be homogenous, suggesting that new structures may have been identified de novo.
Figure 4. Template-free pattern mining approach examples.

. (a) A P19 cell tomogram slice superimposed with membranes (yellow) and particles (blue) identified by PySeg [36]. Tomogram slices (b) where circles encompass the x-y position of particles identified for classes shown in (c). Scale bar, 50 nm. (c) Average densities of identified particle classes. From left to right: membrane-associated ribosome (red), ribosome-associated translocon (light red), ribosome-free translocon (yellow), two putative PLC complexes (magenta and purple), putative IP3 receptor (blue). Scale bar, 10 nm. (d) Examples of additional class averages identified by PySeg. (e) A tomogram slice of intact Synechocystis cell with circles depicting particle picking results. (f) Unsupervised, iterative clustering of extracted features, plotted in 2D, performed by YOPO neural network [51]. (g) Isosurface rendering of discovered patterns. A tomogram slice (h) of intact Acetonema longum and (i) rendered structure models of patterns identified via multi-pattern pursuit (MPP). (j) Magnified region from (i) showing a close-up of the embedded structures. (k) A comparison of an acquired GroEL-like pattern compared to the known atomic model. The average of all subtomograms of this structure pattern aligned to the known structure of GroEL; template is shown [52].
DISCA [51] is a fully unsupervised method to identify structure patterns. It first uses template-free particle picking to collect a large set of heterogenous particles (~105 subtomograms from 10 tomograms; Figure 4e–g). The approach makes use of the YOPO CNN [98,99], which achieved the third highest accuracy of the tested frameworks and outperformed template matching for classification on the SHREC 2020 dataset. In DISCA, YOPO is used to extract second-order statistics on translation and rotation invariant features. These are then modeled as multivariate Gaussian distributions, effectively clustering the particles according to their underlying structure. DISCA is a time-efficient analysis, where recovering structures of multiple classes only takes a day or two of computation for what would take about a month using template matching.
Classification and structure determination can also be implemented concurrently, as subtomograms that fail to align well with each other can be indicative of particles from distinct classes. A template-free framework known as multi-pattern pursuit (MPP) [52] structure patterns from a large, highly heterogeneous collection of particles from a crowded environment (Figure 4h–k). This strategy uses a series of alignments based on constrained correlation and FRM to generate structures aligned to common frames. It discards redundant patterns until high-quality patterns remain. The results of MPP depend on the shape and size of the complex, image resolution, and levels of crowding. Although the final structures are coarsely defined, they may serve as references for multireference alignment to yield higher resolution structures.
Simultaneous alignment and classification are also possible through multi-task learning, where a single deep learning model is trained to perform multiple tasks at once to optimize the entire pipeline as opposed to each step. DSM-Net [46] is a multi-task model for simultaneously performing semantic segmentation, classification, and coarse structure determination. Three networks each perform one of these tasks individually, and to achieve multi-task learning, the losses of these networks are combined into a global loss function through linear combination. Each task reinforces the others, and DSM-Net demonstrates improved feature extraction, reaches greater accuracy, and outperforms single-task models. The model even identified new structures that were not present in the training data. Jim-Net [53] is an end-to-end model for simultaneous, unsupervised alignment and clustering. After hierarchical clustering of subtomograms, a pair-matching alignment algorithm couples and aligns image pairs from a given branch. This architecture performed better than when each task was completed alone, and its accuracy was on par with state-of-the-art supervised methods. Harmony [126] is a model to disentangle the semantic structure from confounded transformations in the subtomograms, which can be further used for joint clustering and alignment.
5. Challenges & future directions
Although there have been significant advancements in cryo-ET data mining methods, there remain some outstanding challenges that need to be overcome to establish the approach’s routine use.
5.1. Limitation of annotated data
Models perform better when trained on large quantities of unbiased data, but obtaining tomograms is labor and time intensive, and more so if manual annotations are required. Although developments in cryo-ET technology expedite the imaging process [78], issues remain in objectively and efficiently labeling tomograms. Data augmentation addresses the issue by simulating realistic data, while domain adaptation extrapolates information in previously labeled data sources to predict new tomograms.
5.1.1. Data augmentation via simulated tomograms and image synthesis
Via simulated tomograms, data augmentation generates substantial amounts of objectively labeled training data. In contrast, annotating real tomograms suffers from subjectivity, as users may locate and label particles differently. Because simulations offer a ground truth, they can benchmark the performance of data analysis methods. Simulations should closely resemble real tomograms, considering factors such as crowding conditions, image distortions, and noise.
To replicate the heterogeneous, crowded environment of tomograms, Liu et al. [56] simulated macromolecular complexes and ultra-structures as multiple spheres with fixed relative positions that could deform under a force field. This achieves a high density of packing with considerations of structural diversity and deformation but is time-consuming and unsuitable for large data generation. A more efficient approach [57] packs macromolecules, represented as a single sphere, in a noise-free density map that is then used to simulate a tomographic image.
Simulating realistic data must incorporate a variety of factors, and neural networks trained on such data tend to exhibit reduced performance once applied to real data. Rather than creating tomogram simulations from scratch, CryoETGAN [58] is a generative adversarial network designed to use real density maps of particles to synthesize imitations of subtomograms and efficiently infer undiscovered elements present in a tomogram of interest.
5.1.2. Domain adaptation to facilitate model performance between distinct data sets
Domain adaptation is another strategy to produce more training data by permitting a model to train on a set of annotated data and apply it to a target set. The primary challenge is overcoming the domain shift between the annotated data and targets – where the cross-data prediction is biased due to a difference in the imaging quality of the two image sets, or domains. 3D-ADA [59] is an adversarial domain adaptation framework that overcomes this by mapping subtomograms into a latent space existing between the domains. This setup improves cross-data source prediction and even the determination of novel structures absent from the training data. Cryo-Shift [60] is a framework to address domain shifts using multi-adversarial domain adaptation. Simulated tomograms are used as training data and are transformed by Cryo-Shift through domain randomization. This randomization improves the generalization ability of the classifier to better predict in the case of the experimental data. Despite being fully unsupervised and not training on any labeled experimental samples, Cryo-Shift still outperforms other cross-domain subtomogram classification approaches.
5.2. Accessibility
For pattern mining methods to be used extensively, they must be convenient for a wide variety of users, including those without substantial computational backgrounds or resources.
5.2.1. User convenience
While publicly releasing software code is now routine for reproducibility and to foster research developments, there are additional considerations that can increase their ease of use for researchers. For instance, including a graphic user interface attracts a broader audience to utilize the protocols, as in the case of IMOD and Dynamo [10,29]. Consolidating scripts into a single suite is convenient for a user to select analyses of interest. Similarly, ScipionTomo [118] is a framework that enables a user to select protocols from multiple suites into a single analysis pipeline. Computational speed-ups such as GPU acceleration and parallel processing should also be considered, especially for machine learning processes, which typically require GPUs to achieve a reasonable timeframe [61]. Finally, cloud support or web-based portals enable research labs without in-house hardware resources to utilize analysis software [29,34,45].
5.2.2. Model size and efficiency must be considered during tool design
Deep learning models such as CNNs are advantageous for many cryo-ET pattern mining tasks but also can require considerable amounts of state-of-the-art hardware to train and store, diminishing their accessibility [119]. Thus, it is ideal to reduce the computational load of software when possible. To this end, Matuszewski et al. [119] aimed to reduce the size of CNN models, specifically the popular U-Net architecture [103] (utilized by the UMC, U-CLSTM, and MC DS Net supervised classifiers [98,99]). The study characterized the impact of a deep learning model’s size on its accuracy. The resulting lighter U-Net featured a parameterized architecture with fewer trainable weights and achieved comparable accuracy in a particle recognition task with four times fewer weights but at the cost of more sensitive hyperparameters. Deeper architectures tended to feature better performance at the cost of longer training, more required training data, and susceptibility to overfitting. Thus, researchers must make practical design considerations to avoid making overly complex systems while also retaining some level of robustness.
5.3. Performance benchmarks
Evaluating proposed frameworks is commonly achieved by comparing their performance against current standard approaches. However, researchers may apply different evaluation methods or standards, and the performance of methods against each other may be ambiguous. Thus, implementing benchmark studies that examine several models at once using the same data set and assessment serves to properly determine the performance of cutting-edge methods.
The previously noted SHREC track [97–99] is an annual study to evaluate learning-based localization and classification methods developed by various groups using the same set of validation data. Studies such as these not only introduce new quantitative cryo-ET analysis strategies, but also clarify their strengths, weaknesses, and promising directions compared to current state-of-the-art technologies. Benchmark studies should aim to assess new methods with ground-truth simulated data that closely reflects experimental data, well-validated experimental data, or both. Furthermore, factors such as runtime and user accessibility should also be important considerations in evaluating the strengths and weaknesses of each approach.
6. Perspectives
In terms of studying protein structure and interactions, cryo-ET offers a large-scale perspective, presenting a unique opportunity to investigate the molecular sociology within a cell. To this end, computational pattern mining methods can achieve visual proteomics, mapping positions of complexes in tomograms to elucidate the spatial and population distributions of identified macromolecules [78]. Given that a tomogram is both information dense and costly to image and annotate, effective pattern mining is critical for high-throughput analysis. Ideal methods should perform segmentation, classification, and structure determination with great speed, spatial resolution, and structure identification accuracy.
Template-free, unsupervised approaches are especially promising, as they overcome the substantial annotation requirements that render supervised methods laborious and introduce a pathway towards de novo structure discovery as they forego a priori knowledge for complex identification. Aside from generating structure patterns, unsupervised classification can facilitate supervised methods. Given sets of patterns, a user only needs to label groups of subtomograms rather than each subtomogram individually. This could be exploited to generate training data for supervised classification with much less effort than fully manual approaches. The advancement of these methods will address a major bottleneck in the cryo-ET pipeline. Moreover, it will open new opportunities to revisit existing cryo-ET data and search for previously overlooked biomolecular interactions. Such a development would enable exciting discoveries of proteinprotein interactions and structural conformations at a rapid rate, significantly accelerating research in numerous biological contexts.
However, to achieve this goal, it is not sufficient to simply develop highly effective algorithms. To ensure that these developments are widely disseminated, software packages should encourage implementation of cutting-edge cryo-ET analysis techniques in the broader research community. Cross-comparative studies of the latest methods should regularly assess the state of the field. As methods become more advanced, integrative modeling becomes essential, as pattern mining results should be validated or informed with other techniques such as quantitative or cross-linking mass spectrometry and computational structural predictions [2,78,120–123]. Advancements in tomogram processing for pattern mining will translate into a deeper and more complete understanding of countless cellular processes.
Cryo-ET is a unique imaging technology capable of investigating protein and complex interactions in situ, where the development of visual atlases can map distribution of complexes throughout the cell enabling an understanding of the molecular sociology of a cell.
Although popular Cryo-ET image analysis software enable researchers to generate visual atlases via template matching and manual annotation, these methods are insufficient to efficiently identify numerous structures de novo from a tomogram.
Template-free particle picking and structure determination methods must be developed in order to achieve de novo structure pattern mining, and approaches aiming to accomplish this are in development.
Thoughtful design choices and complementary computational strategies should be implemented in order to maximize the accessibility and performance of computational methods.
Standardized benchmarks are critical to objectively evaluate the performance of new computational methods and assess the current state of these technologies.
Acknowledgements
YWC is supported by the David and Lucile Packard Fellowship for Science and Engineering (201969645), Mark Foundation for Cancer Research ASPIRE Award (19–044-ASP), Pennsylvania Department of Health FY19 Health Research Formula Fund, NIH R01GM134020, RF1AG065341, R01AI127798 and RM1GM126511. HK is supported by the Biomedical Graduate Studies at University of Pennsylvania Perelman School of Medicine. MX and MRU are supported by NIH R01GM134020 and P41GM103712, NSF DBI-1949629, IIS-2007595, IIS-2211597 and MCB2205148, Mark Foundation for Cancer Research 19–044-ASP, and AMD COVID-19 HPC Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Oikonomou CM, Jensen GJ, Cellular electron cryotomography: Toward structural biology in situ, Annu Rev Biochem. 86 (2017). 10.1146/annurev-biochem-061516-044741. [DOI] [PubMed] [Google Scholar]
- [2].Turk M, Baumeister W, The promise and the challenges of cryo‐electron tomography, FEBS Lett. 594 (2020) 3243–3261. 10.1002/1873-3468.13948. [DOI] [PubMed] [Google Scholar]
- [3].Adrian M, Dubochet J, Lepault J, McDowall AW, Cryo-electron microscopy of viruses, Nature. 308 (1984). 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- [4].Robinson C. v., Sali A, Baumeister W, The molecular sociology of the cell, Nature. 450 (2007). 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- [5].Schur FK, Toward high-resolution in situ structural biology with cryo-electron tomography and subtomogram averaging, Curr Opin Struct Biol. 58 (2019) 1–9. 10.1016/j.sbi.2019.03.018. [DOI] [PubMed] [Google Scholar]
- [6].Koning RI, Koster AJ, Sharp TH, Advances in cryo-electron tomography for biology and medicine, Annals of Anatomy - Anatomischer Anzeiger. 217 (2018) 82–96. 10.1016/j.aanat.2018.02.004. [DOI] [PubMed] [Google Scholar]
- [7].Frangakis AS, It’s noisy out there! A review of denoising techniques in cryo-electron tomography, J Struct Biol. 213 (2021) 107804. 10.1016/j.jsb.2021.107804. [DOI] [PubMed] [Google Scholar]
- [8].Lučić V, Rigort A, Baumeister W, Cryo-electron tomography: The challenge of doing structural biology in situ, Journal of Cell Biology. 202 (2013). 10.1083/jcb.201304193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bartesaghi A, Sprechmann P, Liu J, Randall G, Sapiro G, Subramaniam S, Classification and 3D averaging with missing wedge correction in biological electron tomography, J Struct Biol. 162 (2008) 436–450. 10.1016/J.JSB.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kremer JR, Mastronarde DN, McIntosh JR, Computer Visualization of Three-Dimensional Image Data Using IMOD, J Struct Biol. 116 (1996) 71–76. 10.1006/JSBI.1996.0013. [DOI] [PubMed] [Google Scholar]
- [11].Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR, The molecular architecture of axonemes revealed by cryoelectron tomography, Science (1979). 313 (2006). 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- [12].Heumann JM, Hoenger A, Mastronarde DN, Clustering and variance maps for cryo-electron tomography using wedge-masked differences, J Struct Biol. 175 (2011). 10.1016/j.jsb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ, EMAN2: An extensible image processing suite for electron microscopy, J Struct Biol. 157 (2007) 38–46. 10.1016/J.JSB.2006.05.009. [DOI] [PubMed] [Google Scholar]
- [14].Chen M, Dai W, Sun SY, Jonasch D, He CY, Schmid MF, Chiu W, Ludtke SJ, Convolutional neural networks for automated annotation of cellular cryo-electron tomograms, Nat Methods. 14 (2017) 983–985. 10.1038/nmeth.4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bell JM, Chen M, Durmaz T, Fluty AC, Ludtke SJ, New software tools in EMAN2 inspired by EMDatabank map challenge, J Struct Biol. 204 (2018). 10.1016/j.jsb.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen M, Bell JM, Shi X, Sun SY, Wang Z, Ludtke SJ, A complete data processing workflow for cryo-ET and subtomogram averaging, Nat Methods. 16 (2019). 10.1038/s41592-019-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nickell S, Förster F, Linaroudis A, del Net W, Beck F, Hegerl R, Baumeister W, Plitzko JM, TOM software toolbox: acquisition and analysis for electron tomography, J Struct Biol. 149 (2005) 227–234. 10.1016/J.JSB.2004.10.006. [DOI] [PubMed] [Google Scholar]
- [18].Förster F, Han BG, Beck M, Visual Proteomics, Methods Enzymol. 483 (2010) 215–243. 10.1016/S0076-6879(10)83011-3. [DOI] [PubMed] [Google Scholar]
- [19].Hrabe T, Chen Y, Pfeffer S, Kuhn Cuellar L, Mangold A-V, Förster F, PyTom: A python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis, J Struct Biol. 178 (2012) 177–188. 10.1016/j.jsb.2011.12.003. [DOI] [PubMed] [Google Scholar]
- [20].Chen Y, Pfeffer S, Hrabe T, Schuller JM, Förster F, Fast and accurate reference-free alignment of subtomograms, J Struct Biol. 182 (2013) 235–245. 10.1016/J.JSB.2013.03.002. [DOI] [PubMed] [Google Scholar]
- [21].Taylor KA, Liu J, Winkler H, Localization and classification of repetitive structures in electron tomograms of paracrystalline assemblies, in: Electron Tomography: Methods for Three-Dimensional Visualization of Structures in the Cell, 2006. 10.1007/978-0-387-69008-7_16. [DOI] [Google Scholar]
- [22].Winkler H, 3D reconstruction and processing of volumetric data in cryo-electron tomography, J Struct Biol. 157 (2007). 10.1016/j.jsb.2006.07.014. [DOI] [PubMed] [Google Scholar]
- [23].Winkler H, Taylor KA, Multivariate statistical analysis of three-dimensional cross-bridge motifs in insect flight muscle, Ultramicroscopy. 77 (1999). 10.1016/S0304-3991(99)00035-2. [DOI] [Google Scholar]
- [24].Winkler H, Zhu P, Liu J, Ye F, Roux KH, Taylor KA, Tomographic subvolume alignment and subvolume classification applied to myosin V and SIV envelope spikes, J Struct Biol. 165 (2009). 10.1016/j.jsb.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wu S, Liu J, Reedy MC, Winkler H, Reedy MK, Taylor KA, Methods for identifying and averaging variable molecular conformations in tomograms of actively contracting insect flight muscle, J Struct Biol. 168 (2009). 10.1016/j.jsb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pruggnaller S, Mayr M, Frangakis AS, A visualization and segmentation toolbox for electron microscopy, JStruct Biol. 164 (2008) 161–165. 10.1016/j.jsb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- [27].Heymann JB, Belnap DM, Bsoft: Image processing and molecular modeling for electron microscopy, J Struct Biol. 157 (2007). 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- [28].Heymann JB, High resolution electron tomography and segmentation-by-modeling interpretation in Bsoft, Protein Science. 30 (2021). 10.1002/pro.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Castaño-Díez D, Kudryashev M, Arheit M, Stahlberg H, Dynamo: A flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments, J Struct Biol. 178 (2012) 139–151. 10.1016/J.JSB.2011.12.017. [DOI] [PubMed] [Google Scholar]
- [30].Castaño-Díez D, Kudryashev M, Stahlberg H, Dynamo Catalogue: Geometrical tools and data management for particle picking in subtomogram averaging of cryo-electron tomograms, J Struct Biol. 197 (2017) 135–144. 10.1016/J.JSB.2016.06.005. [DOI] [PubMed] [Google Scholar]
- [31].Castaño-Díez D, The Dynamo package for tomography and subtomogram averaging: Components for MATLAB, GPU computing and EC2 Amazon Web Services, in: Acta Crystallogr D Struct Biol, 2017. 10.1107/S2059798317003369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Navarro PP, Stahlberg H, Castaño-Díez D, Protocols for subtomogram averaging of membrane proteins in the Dynamo software package, Front Mol Biosci. 5 (2018). 10.3389/fmolb.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Scaramuzza S, Castaño-Díez D, Step-by-step guide to efficient subtomogram averaging of virus-like particles with Dynamo, PLoS Biol. 19 (2021). 10.1371/journal.pbio.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Himes BA, Zhang P, emClarity: software for high-resolution cryo-electron tomography and subtomogram averaging, Nat Methods. 15 (2018). 10.1038/s41592-018-0167-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ni T, Frosio T, Mendonça L, Sheng Y, Clare D, Himes BA, Zhang P, High-resolution in situ structure determination by cryo-electron tomography and subtomogram averaging using emClarity, Nat Protoc. 17 (2022). 10.1038/s41596-021-00648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Martinez-Sanchez A, Kochovski Z, Laugks U, Meyer J zum Alten Borgloh, S. Chakraborty, S. Pfeffer, W. Baumeister, V. Lučić, Template-free detection and classification of membrane-bound complexes in cryo-electron tomograms, Nature Methods 2020 17:2. 17 (2020) 209–216. 10.1038/s41592-019-0675-5. [DOI] [PubMed] [Google Scholar]
- [37].Martinez-Sanchez A, Garcia I, Asano S, Lucic V, Fernandez JJ, Robust membrane detection based on tensor voting for electron tomography, J Struct Biol. 186 (2014) 49–61. 10.1016/J.JSB.2014.02.015. [DOI] [PubMed] [Google Scholar]
- [38].Martinez-Sanchez A, Baumeister W, Lučić V, Statistical spatial analysis for cryo-electron tomography, Comput Methods Programs Biomed. 218 (2022) 106693. 10.1016/J.CMPB.2022.106693. [DOI] [PubMed] [Google Scholar]
- [39].Scheres SHW, RELION: Implementation of a Bayesian approach to cryo-EM structure determination, J Struct Biol. 180 (2012). 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Scheres SHW, A bayesian view on cryo-EM structure determination, J Mol Biol. 415 (2012). 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bharat TAM, Russo CJ, Löwe J, Passmore LA, Scheres SHW, Advances in Single-Particle Electron Cryomicroscopy Structure Determination applied to Sub-tomogram Averaging, Structure. 23 (2015). 10.1016/j.str.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bharat TAM, Scheres SHW, Resolving macromolecular structures from electron cryo-tomography data using subtomogram averaging in RELION, Nature Protocols 2016 11:11. 11 (2016) 2054–2065. 10.1038/nprot.2016.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zivanov J, Otón J, Ke Z, von Kügelgen A, Pyle E, Qu K, Morado D, Castaño-Díez D, Zanetti G, Bharat TA, Briggs JA, Scheres SH, A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0, Elife. 11 (2022). 10.7554/elife.83724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Luengo I, Darrow MC, Spink MC, Sun Y, Dai W, He CY, Chiu W, Pridmore T, Ashton AW, Duke EMH, Basham M, French AP, SuRVoS: Super-Region Volume Segmentation workbench, J Struct Biol. 198 (2017) 43–53. 10.1016/J.JSB.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Frazier Z, Xu M, Alber F, TomoMiner and TomoMinerCloud: A Software Platform for Large-Scale Subtomogram Structural Analysis, Structure. 25 (2017). 10.1016/j.str.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu C, Zeng X, Wang K, Guo Q, Xu M, Multi-task Learning for Macromolecule Classification, Segmentation and Coarse Structural Recovery in Cryo-Tomography, (2018). http://arxiv.org/abs/1805.06332 (accessed May 30, 2022). [PMC free article] [PubMed] [Google Scholar]
- [47].Hao Y, Wan X, Yan R, Liu Z, Li J, Zhang S, Cui X, Zhang F, VP-Detector: A 3D multi-scale dense convolutional neural network for macromolecule localization and classification in cryo-electron tomograms, Comput Methods Programs Biomed. 221 (2022) 106871. 10.1016/J.CMPB.2022.106871. [DOI] [PubMed] [Google Scholar]
- [48].Moebel E, Martinez-Sanchez A, Lamm L, Righetto RD, Wietrzynski W, Albert S, Lariviamp D, Fourmentin E, Pfeffer S, Ortiz J, Baumeister W, Peng T, Engel BD, Kervrann C, Deep learning improves macromolecule identification in 3D cellular cryo-electron tomograms, Nat Methods. 18 (2021) 1386–1394. 10.1038/s41592-021-01275-4. [DOI] [PubMed] [Google Scholar]
- [49].Hajarolasvadi N, Sunkara V, Khavnekar S, Beck F, Brandt R, Baum D, Volumetric macromolecule identification in cryo-electron tomograms using capsule networks, BMC Bioinformatics. 23 (2022) 360. 10.1186/s12859-022-04901-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Che C, Lin R, Zeng X, Elmaaroufi K, Galeotti J, Xu M, Improved deep learning-based macromolecules structure classification from electron cryo-tomograms, Mach Vis Appl. 29 (2018) 1227–1236. 10.1007/s00138-018-0949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zeng X, Kahng A, Xue L, Mahamid J, Chang Y-W, Xu M, DISCA: high-throughput cryo-ET structural pattern mining by deep unsupervised clustering, BioRxiv. (2021) 2021.05.16.444381. 10.1101/2021.05.16.444381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xu M, Singla J, Tocheva EI, Chang YW, Stevens RC, Jensen GJ, Alber F, De Novo Structural Pattern Mining in Cellular Electron Cryotomograms, Structure. 27 (2019) 679–691.e14. 10.1016/J.STR.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zeng X, Howe G, Xu M, End-to-end robust joint unsupervised image alignment and clustering, in: 2021 IEEE/CVF International Conference on Computer Vision (ICCV), IEEE, 2021: pp. 3834–3846. 10.1109/ICCV48922.2021.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhao Y, Zeng X, Guo Q, Xu M, An integration of fast alignment and maximum-likelihood methods for electron subtomogram averaging and classification, Bioinformatics. 34 (2018) i227–i236. 10.1093/bioinformatics/bty267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Harastani M, Eltsov M, Leforestier A, Jonic S, HEMNMA-3D: Cryo Electron Tomography Method Based on Normal Mode Analysis to Study Continuous Conformational Variability of Macromolecular Complexes, Front Mol Biosci. 8 (2021). 10.3389/fmolb.2021.663121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu S, Ban X, Zeng X, Zhao F, Gao Y, Wu W, Zhang H, Chen F, Hall T, Gao X, Xu M, A unified framework for packing deformable and non-deformable subcellular structures in crowded cryo-electron tomogram simulation, BMC Bioinformatics. 21 (2020). 10.1186/s12859-020-03660-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu S, Ma Y, Ban X, Zeng X, Nallapareddy V, Chaudhari A, Xu M, Efficient Cryo-Electron Tomogram Simulation of Macromolecular Crowding with Application to SARS-CoV-2, in: Proceedings - 2020 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2020, Institute of Electrical and Electronics Engineers Inc., 2020: pp. 80–87. 10.1109/BIBM49941.2020.9313185. [DOI] [Google Scholar]
- [58].Wu X, Li C, Zeng X, Wei H, Deng H-W, Zhang J, Xu M, CryoETGAN: Cryo-Electron Tomography Image Synthesis via Unpaired Image Translation, Frontiers in Physiology | Www.Frontiersin.Org. 1 (2022) 760404. 10.3389/fphys.2022.760404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lin R, Zeng X, Kitani K, Xu M, Adversarial domain adaptation for cross data source macromolecule in situ structural classification in cellular electron cryo-tomograms, Bioinformatics. 35 (2019) i260–i268. 10.1093/bioinformatics/btz364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bandyopadhyay H, Deng Z, Ding L, Liu S, Uddin MR, Zeng X, Behpour S, Xu M, Cryo-shift: reducing domain shift in cryo-electron subtomograms with unsupervised domain adaptation and randomization, Bioinformatics. 38 (2022) 977–984. 10.1093/bioinformatics/btab794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Navarro PP, Quantitative Cryo-Electron Tomography, Front Mol Biosci.9 (2022). 10.3389/fmolb.2022.934465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Martinez-Sanchez A, Garcia I, Fernandez J-J, A differential structure approach to membrane segmentation in electron tomography, J Struct Biol. 175 (2011) 372–383. 10.1016/j.jsb.2011.05.010. [DOI] [PubMed] [Google Scholar]
- [63].Whitaker RT, Elangovan V, A direct approach to estimating surfaces in tomographic data, Med Image Anal. 6 (2002) 235–249. 10.1016/S1361-8415(02)00082-8. [DOI] [PubMed] [Google Scholar]
- [64].Bartesaghi A, Sapiro G, Subramaniam S, An energy-based three-dimensional segmentation approach for the quantitative interpretation of electron tomograms, IEEE Transactions on Image Processing. 14 (2005) 1314–1323. 10.1109/TIP.2005.852467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ress DB, Harlow ML, Marshall RM, McMahan UJ, Methods for Generating High-Resolution Structural Models from Electron Microscope Tomography Data, Structure. 12 (2004) 1763–1774. 10.1016/j.str.2004.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Volkmann N, A novel three-dimensional variant of the watershed transform for segmentation of electron density maps, J Struct Biol. 138 (2002) 123–129. 10.1016/S1047-8477(02)00009-6. [DOI] [PubMed] [Google Scholar]
- [67].Moussavi F, Heitz G, Amat F, Comolli LR, Koller D, Horowitz M, 3D segmentation of cell boundaries from whole cell cryogenic electron tomography volumes, J Struct Biol. 170 (2010) 134–145. 10.1016/J.JSB.2009.12.015. [DOI] [PubMed] [Google Scholar]
- [68].Nguyen Hieu, Ji Qiang, Shape-Driven Three-Dimensional Watersnake Segmentation of Biological Membranes in Electron Tomography, IEEE Trans Med Imaging. 27 (2008) 616–628. 10.1109/TMI.2007.912390. [DOI] [PubMed] [Google Scholar]
- [69].Sun SY, Kaelber JT, Chen M, Dong X, Nematbakhsh Y, Shi J, Dougherty M, Lim CT, Schmid MF, Chiu W, He CY, Flagellum couples cell shape to motility in Trypanosoma brucei, Proc Natl Acad Sci U S A. 115 (2018). 10.1073/pnas.1722618115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Huokko T, Ni T, Dykes GF, Simpson DM, Brownridge P, Conradi FD, Beynon RJ, Nixon PJ, Mullineaux CW, Zhang P, Liu LN, Probing the biogenesis pathway and dynamics of thylakoid membranes, Nat Commun. 12 (2021). 10.1038/s41467-021-23680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wan W, Briggs JAG, Cryo-Electron Tomography and Subtomogram Averaging, in: Methods Enzymol, Academic Press, 2016: pp. 329–367. 10.1016/bs.mie.2016.04.014. [DOI] [PubMed] [Google Scholar]
- [72].Galaz-Montoya JG, Flanagan J, Schmid MF, Ludtke SJ, Single particle tomography in EMAN2, J Struct Biol. 190 (2015) 279–290. 10.1016/J.JSB.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wang F, Gong H, Liu G, Li M, Yan C, Xia T, Li X, Zeng J, DeepPicker: A deep learning approach for fully automated particle picking in cryo-EM, J Struct Biol. 195 (2016) 325–336. 10.1016/J.JSB.2016.07.006. [DOI] [PubMed] [Google Scholar]
- [74].Frangakis AS, Förster F, Computational exploration of structural information from cryo-electron tomograms, Curr Opin Struct Biol. 14 (2004) 325–331. 10.1016/j.sbi.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [75].Böhm J, Frangakis AS, Hegerl R, Nickell S, Typke D, Baumeister W, Toward detecting and identifying macromolecules in a cellular context: Template matching applied to electron tomograms, Proc Natl Acad Sci U S A. 97 (2000). 10.1073/pnas.230282097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Asano S, Engel BD, Baumeister W, In Situ Cryo-Electron Tomography: A Post-Reductionist Approach to Structural Biology, J Mol Biol. 428 (2016) 332–343. 10.1016/j.jmb.2015.09.030. [DOI] [PubMed] [Google Scholar]
- [77].Frangakis AS, Böhm J, Förster F, Nickell S, Nicastro D, Typke D, Hegerl R, Baumeister W, Identification of macromolecular complexes in cryoelectron tomograms of phantom cells, Proc Natl Acad Sci U S A. 99 (2002). 10.1073/pnas.172520299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Beck M, Baumeister W, Cryo-Electron Tomography: Can it Reveal the Molecular Sociology of Cells in Atomic Detail?, Trends Cell Biol. 26 (2016) 825–837. 10.1016/J.TCB.2016.08.006. [DOI] [PubMed] [Google Scholar]
- [79].Lucas BA, Himes BA, Xue L, Grant T, Mahamid J, Grigorieff N, Locating macromolecular assemblies in cells by 2D template matching with cisTEM, Elife. 10 (2021). 10.7554/eLife.68946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rigort A, Günther D, Hegerl R, Baum D, Weber B, Prohaska S, Medalia O, Baumeister W, Hege HC, Automated segmentation of electron tomograms for a quantitative description of actin filament networks, J Struct Biol. 177 (2012) 135–144. 10.1016/J.JSB.2011.08.012. [DOI] [PubMed] [Google Scholar]
- [81].Rusu M, Starosolski Z, Wahle M, Rigort A, Wriggers W, Automated Tracing of Filaments in 3D Electron Tomography Reconstructions using Sculptor and Situs, (2012). 10.1016/j.jsb.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Weber B, Greenan G, Prohaska S, Baum D, Hege HC, Müller-Reichert T, Hyman AA, Verbavatz JM, Automated tracing of microtubules in electron tomograms of plastic embedded samples of Caenorhabditis elegans embryos, J Struct Biol. 178 (2012) 129–138. 10.1016/J.JSB.2011.12.004. [DOI] [PubMed] [Google Scholar]
- [83].Grünewald K, Medalia O, Gross A, Steven AC, Baumeister W, Prospects of electron cryotomography to visualize macromolecular complexes inside cellular compartments: Implications of crowding, Biophys Chem. 100 (2002). 10.1016/S0301-4622(02)00307-1. [DOI] [PubMed] [Google Scholar]
- [84].Shatsky M, Hall RJ, Brenner SE, Glaeser RM, A method for the alignment of heterogeneous macromolecules from electron microscopy, J Struct Biol. 166 (2009) 67–78. 10.1016/j.jsb.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chen Y, Pfeffer S, Fernández JJ, Sorzano COS, Förster F, Autofocused 3D Classification of Cryoelectron Subtomograms, Structure. 22 (2014) 1528–1537. 10.1016/J.STR.2014.08.007. [DOI] [PubMed] [Google Scholar]
- [86].Henderson R, Avoiding the pitfalls of single particle cryo-electron microscopy: Einstein from noise, Proc Natl Acad Sci U S A. 110 (2013). 10.1073/pnas.1314449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chen Y, Hrabe T, Pfeffer S, Pauly O, Mateus D, Navab N, Förster F, Detection and identification of macromolecular complexes in cryo-electron tomograms using support vector machines, 2012. 10.1109/ISBI.2012.6235823. [DOI] [Google Scholar]
- [88].Wagner T, Merino F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D, Quentin D, Roderer D, Tacke S, Siebolds B, Schubert E, Shaikh TR, Lill P, Gatsogiannis C, Raunser S, SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM, Commun Biol. 2 (2019). 10.1038/s42003-019-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lučić V, Fernández-Busnadiego R, Laugks U, Baumeister W, Hierarchical detection and analysis of macromolecular complexes in cryo-electron tomograms using Pyto software, J Struct Biol. 196 (2016) 503–514. 10.1016/J.JSB.2016.10.004. [DOI] [PubMed] [Google Scholar]
- [90].Kovacs JA, Wriggers W, Fast rotational matching, Acta Crystallogr D Biol Crystallogr. 58 (2002). 10.1107/S0907444902009794. [DOI] [PubMed] [Google Scholar]
- [91].Nickell S, Kofler C, Leis AP, Baumeister W, A visual approach to proteomics, Nat Rev Mol Cell Biol. 7 (2006) 225–230. 10.1038/nrm1861. [DOI] [PubMed] [Google Scholar]
- [92].Scheres SHW, Melero R, Valle M, Carazo JM, Averaging of Electron Subtomograms and Random Conical Tilt Reconstructions through Likelihood Optimization, Structure. 17 (2009) 1563–1572. 10.1016/J.STR.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Dempster AP, Laird NM, Rubin DB, Maximum Likelihood from Incomplete Data Via the EM Algorithm , Journal of the Royal Statistical Society: Series B (Methodological). 39 (1977). 10.1111/j.2517-6161.1977.tb01600.x. [DOI] [Google Scholar]
- [94].Scheres SHW, Gao H, Valle M, Herman GT, Eggermont PPB, Frank J, Carazo JM, Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization, Nat Methods. 4 (2007). 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- [95].Castaño-Díez D, Zanetti G, In situ structure determination by subtomogram averaging, Curr Opin Struct Biol. 58 (2019) 68–75. 10.1016/J.SBI.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Galaz-Montoya JG, Flanagan J, Schmid MF, Ludtke SJ, Single particle tomography in EMAN2, J Struct Biol. 190 (2015) 279–290. 10.1016/J.JSB.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Gubins I, van der Schot G, Veltkamp RC, Förster F, Du X, Zeng X, Zhu Z, Chang L, Xu M, Moebel E, Martinez-Sanchez A, Kervrann C, Lai TM, Han X, Terashi G, Kihara D, Himes BA, Wan X, Zhang J, Gao S, Hao Y, Lv Z, Yang Z, Ding Z, Cui X, Zhang F, SHREC’19 Track: Classification in Cryo-Electron Tomograms, Eurographics Workshop on 3D Object Retrieval. (2019). 10.2312/3dor.20191061. [DOI] [Google Scholar]
- [98].Gubins I, Chaillet ML, van der Schot G, Veltkamp RC, Förster F, Hao Y, Wan X, Cui X, Zhang F, Moebel E, Wang X, Kihara D, Zeng X, Xu M, Nguyen NP, White T, Bunyak F, SHREC 2020: Classification in cryo-electron tomograms, Comput Graph. 91 (2020) 279–289. 10.1016/J.CAG.2020.07.010. [DOI] [Google Scholar]
- [99].Gubins I, Chaillet ML, van der Schot G, Cristina Trueba M, Veltkamp RC, Förster F, Wang X, Kihara D, Moebel E, Nguyen NP, White T, Bunyak F, Papoulias G, Gerolymatos S, Zacharaki EI, Moustakas K, Zeng X, Liu S, Xu M, Wang Y, Chen C, Cui X, Zhang F, SHREC 2021: Classification in cryo-electron tomograms, Eurographics Workshop on 3D Object Retrieval. (2021). 10.2312/3dor.20211307. [DOI] [Google Scholar]
- [100].He K, Zhang X, Ren S, Sun J, Deep Residual Learning for Image Recognition, in: 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), IEEE, 2016: pp. 770–778. 10.1109/CVPR.2016.90. [DOI] [Google Scholar]
- [101].Hara K, Kataoka H, Satoh Y, Learning Spatio-Temporal Features with 3D Residual Networks for Action Recognition, in: 2017 IEEE International Conference on Computer Vision Workshops (ICCVW), IEEE, 2017: pp. 3154–3160. 10.1109/ICCVW.2017.373. [DOI] [Google Scholar]
- [102].Zhang K, Zuo W, Chen Y, Meng D, Zhang L, Beyond a Gaussian Denoiser: Residual Learning of Deep CNN for Image Denoising, IEEE Transactions on Image Processing. 26 (2017) 3142–3155. 10.1109/TIP.2017.2662206. [DOI] [PubMed] [Google Scholar]
- [103].Ronneberger O, Fischer P, Brox T, U-net: Convolutional networks for biomedical image segmentation, in: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2015. 10.1007/978-3-319-24574-4_28. [DOI] [Google Scholar]
- [104].Sudre CH, Li W, Vercauteren T, Ourselin S, Jorge Cardoso M, Generalised Dice Overlap as a Deep Learning Loss Function for Highly Unbalanced Segmentations, in: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2017: pp. 240–248. 10.1007/978-3-319-67558-9_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dai J, He K, Sun J, Instance-Aware Semantic Segmentation via Multi-task Network Cascades, in: 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), IEEE, 2016: pp. 3150–3158. 10.1109/CVPR.2016.343. [DOI] [Google Scholar]
- [106].Salehi SSM, Erdogmus D, Gholipour A, Tversky Loss Function for Image Segmentation Using 3D Fully Convolutional Deep Networks, in: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2017: pp. 379–387. 10.1007/978-3-319-67389-9_44. [DOI] [Google Scholar]
- [107].Huang H, Lin L, Tong R, Hu H, Zhang Q, Iwamoto Y, Han X, Chen Y-W, Wu J, UNet 3+: A Full-Scale Connected UNet for Medical Image Segmentation, in: ICASSP 2020 – 2020 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), IEEE, 2020: pp. 1055–1059. 10.1109/ICASSP40776.2020.9053405. [DOI] [Google Scholar]
- [108].Zhou Z, Rahman Siddiquee MM, Tajbakhsh N, Liang J, UNet++: A Nested U-Net Architecture for Medical Image Segmentation, in: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2018: pp. 3–11. 10.1007/978-3-030-00889-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Milletari F, Rieke N, Baust M, Esposito M, Navab N, CFCM: Segmentation via Coarse to Fine Context Memory, in: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2018: pp. 667–674. 10.1007/978-3-030-00937-3_76. [DOI] [Google Scholar]
- [110].Shi X, Chen Z, Wang H, Yeung D-Y, Wong W, Woo W, Convolutional LSTM Network: A Machine Learning Approach for Precipitation Nowcasting, Adv Neural Inf Process Syst. 2015-January (2015). 10.48550/arxiv.1506.04214. [DOI] [Google Scholar]
- [111].Chen L-C, Papandreou G, Kokkinos I, Murphy K, Yuille AL, DeepLab: Semantic Image Segmentation with Deep Convolutional Nets, Atrous Convolution, and Fully Connected CRFs, IEEE Trans Pattern Anal Mach Intell. 40 (2018) 834–848. 10.1109/TPAMI.2017.2699184. [DOI] [PubMed] [Google Scholar]
- [112].Nguyen NP, Ersoy I, Gotberg J, Bunyak F, White TA, DRPnet: automated particle picking in cryo-electron micrographs using deep regression, BMC Bioinformatics. 22 (2021) 55. 10.1186/s12859-020-03948-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gubins I, Veltkamp RC, Deeply Cascaded U-Net for Multi-Task Image Processing, (2020). http://arxiv.org/abs/2005.00225 (accessed February 14, 2023). [Google Scholar]
- [114].He K, Zhang X, Ren S, Sun J, Identity Mappings in Deep Residual Networks, in: Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 2016: pp. 630–645. 10.1007/978-3-319-46493-0_38. [DOI] [Google Scholar]
- [115].Yu F, Koltun V, Multi-Scale Context Aggregation by Dilated Convolutions, 4th International Conference on Learning Representations, ICLR 2016 - Conference Track Proceedings. (2015). http://arxiv.org/abs/1511.07122. [Google Scholar]
- [116].Lin T-Y, Goyal P, Girshick R, He K, Dollar P, Focal Loss for Dense Object Detection, IEEE Trans Pattern Anal Mach Intell. 42 (2020) 318–327. 10.1109/TPAMI.2018.2858826. [DOI] [PubMed] [Google Scholar]
- [117].Förster F, Pruggnaller S, Seybert A, Frangakis AS, Classification of cryo-electron sub-tomograms using constrained correlation, J Struct Biol. 161 (2008) 276–286. 10.1016/J.JSB.2007.07.006. [DOI] [PubMed] [Google Scholar]
- [118].de la Morena J. Jiménez, Conesa P, Fonseca YC, de Isidro-Gómez FP, Herreros D, Fernández-Giménez E, Strelak D, Moebel E, Buchholz TO, Jug F, Martinez-Sanchez A, Harastani M, Jonic S, Conesa JJ, Cuervo A, Losana P, Sánchez I, Iceta M, del Cano L, Gragera M, Melero R, Sharov G, Castaño-Díez D, Koster A, Piccirillo JG, Vilas JL, Otón J, Marabini R, Sorzano COS, Carazo JM, Tomo Scipion: Towards cryo-electron tomography software integration, reproducibility, and validation, J Struct Biol. 214 (2022) 107872. 10.1016/J.JSB.2022.107872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Matuszewski DJ, Sintorn IM, Reducing the U-Net size for practical scenarios: Virus recognition in electron microscopy images, Comput Methods Programs Biomed. 178 (2019) 31–39. 10.1016/J.CMPB.2019.05.026. [DOI] [PubMed] [Google Scholar]
- [120].Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Dustin Schaeffer R, Millán C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues A. v., van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Christopher Garcia K, Grishin N. v., Adams PD, Read RJ, Baker D, Accurate prediction of protein structures and interactions using a three-track neural network, Science (1979). 373 (2021). 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Hassabis D✉, Highly accurate protein structure prediction with AlphaFold, Nature. 596 (2021) 583. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M, ColabFold: making protein folding accessible to all, Nat Methods. 19 (2022). 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Rantos V, Karius K, Kosinski J, Integrative structural modeling of macromolecular complexes using Assembline, Nat Protoc. 17 (2022). 10.1038/s41596-021-00640-z. [DOI] [PubMed] [Google Scholar]