

Abstract
Nuclear epidermal growth factor receptor (EGFR) has been shown to be correlated with drug resistance and a poor prognosis in patients with cancer. Previously, we have identified a tripartite nuclear localization signal (NLS) within EGFR. To comprehensively determine the functions and underlying mechanism of nuclear EGFR and its clinical implications, we aimed to explore the nuclear export signal (NES) sequence of EGFR that is responsible for interacting with the exportins. We combined in silico prediction with site-directed mutagenesis approaches and identified a putative NES motif of EGFR, which is located in amino acid residues 736-749. Mutation at leucine 747 (L747) in the EGFR NES led to increased nuclear accumulation of the protein via a less efficient release of the exportin CRM1. Interestingly, L747 with serine (L747S) and with proline (L747P) mutations were found in both tyrosine kinase inhibitor (TKI)-treated and -naïve patients with lung cancer who had acquired or de novo TKI resistance and a poor outcome. Reconstituted expression of the single NES mutant EGFRL747P or EGFRL747S, but not the dual mutant along with the internalization-defective or NLS mutation, in lung cancer cells promoted malignant phenotypes, including cell migration, invasiveness, TKI resistance, and tumor initiation, supporting an oncogenic role of nuclear EGFR. Intriguingly, cells with germline expression of the NES L747 mutant developed into B cell lymphoma. Mechanistically, nuclear EGFR signaling is required for sustaining nuclear activated STAT3, but not for Erk. These findings suggest that EGFR functions are compartmentalized and that nuclear EGFR signaling plays a crucial role in tumor malignant phenotypes, leading to tumorigenesis in human cancer.
Keywords: EGFR, nuclear translocation, activating mutations, NES, NLS, CRM1, exportin, internalization, non-small cell lung cancer
Introduction
A large number of receptor tyrosine kinases, including EGFR/ErbB family members, FGFR, IGFR, TrkA, c-Met, and VEGFR, in the form of holoreceptors or C-terminal fragments, have now been reported to be trafficked from the cell surface to the nucleus [1-7]. Nuclear localization of EGFR has been observed for more than three decades since nuclear EGFR was first detected in regenerating hepatocytes and primary adrenocortical carcinomas [8,9]. Nuclear EGFR was further detected in developing mouse embryos, as well as the uterus, placenta, thyroid, and some immortalized epithelial cells derived from ovaries and kidneys [10]. A growing body of evidence has emerged that nuclear EGFR plays an important role in tissue regeneration, cancer progression, survival, and radiotherapy and chemotherapy response [11-14]. Higher nuclear levels of EGFR were found in a variety of cancers (e.g. breast, oral cavity, ovary, skin, bladder, cervix, adrenocortex, lung, brain, and thyroid), and nuclear EGFR can serve as an indicator of invasiveness and poor clinical outcomes in cancer patients [15-22]. In addition, nuclear EGFR signaling is involved in resistance to cetuxiumab in lung and triple-negative breast cancers [23,24]. Mechanistically, accumulating evidence supports the model in which nuclear EGFR functions as a transcriptional cofactor to regulate target genes such as cyclin D1, iNOS, B-Myb, Aurora A, c-Myc, cyclooxygenase-2, and breast cancer-resistant protein [25-31]. In addition, nuclear EGFR interacts with RNA helicase A, proliferating cell nuclear antigen, and DNA-PK to affect cell proliferation, DNA repair, cisplatin resistance, radioprotection, and radioresistance [32-36]. Interestingly, the results of our and other studies uncovered the connection between nuclear EGFR and the STAT3 signaling pathway that contributes to tumor malignancy [29,30,37,38].
Transport of macromolecules in and out of the nucleus through nuclear pore complexes (NPCs) is mediated by importins and exportins via the nuclear localization signal (NLS) and nuclear export signal (NES), respectively [39]. Importins bind to their substrates in the cytoplasm and release them in the nucleus upon RanGTP binding; by contrast, exportins-for example, CRM1 (chromosomal maintenance 1, also known as exportin 1)-bind cooperatively to RanGTPs and their cargos, assembling a trimeric export complex in the nucleus and dissociating in the cytoplasm through the NPC [40,41]. CRM1 is one of the well-characterized exportins that has the broadest-known substrate range and all CRM1 cargos share a short, hydrophobic, leucine-rich NES [42,43]. Unlike other exportin-cargo complexes, the affinity of such NES for binding to CRM1 is usually relatively low [41]. Notably, Engelsma et al. demonstrated that the increased affinity of NES for binding to CRM1 impaired the efficient release of the NES-CRM1 complex from the NPC, leading to sequestration of CRM1 at the nuclear rim and in the cytoplasm; as a result, the recycling of CRM1 was reduced and cargo accumulated in the nucleus [44]. Our previous findings showed that addition of leptomycin B, a specific CRM1 inhibitor, enhances the nuclear accumulation of EGFR, suggesting that EGFR is exported out of the nucleus via a CRM1-dependent mechanism [39].
Non-small cell lung cancer is the leading cause of cancer death in the USA [45]. Targeted therapy of EGFR with tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, or the ATP-competitive anilinoquinazoline inhibitors shows promise in the treatment of metastatic non-small cell lung cancer [46]. Clinical data revealed that responsiveness to these drugs is dependent on somatic EGFR gene mutations; in most patients who are highly responsive to EGFR TKIs, tumors harbor the two most prevalent EGFR mutations, i.e. short-in-frame deletion of exon 19 (Δ746-750) and a point mutation in exon 21 (L858R) [47-49]. However, prolonged TKI treatment induces secondary mutations such as T790M on EGFR, resulting in acquired resistance to TKI therapy [50,51]. Recently, other minor secondary mutations-L747P, L747S, and C797S-were also found in EGFR TKI-treated and -naïve lung cancers [52-59]. Clinical data showed that EGFR L747P and L747S mutations were not sensitive to first- and third-generation EGFR-TKIs and were associated with poor prognosis [60-69]. In particular, a case report showed that EGFR L747P mutation was resistant to the third-generation TKI osimertinib [62]. However, other reports demonstrated that the irreversible inhibitor afatinib was effective and resulted in improvements in the patient progression-free with EGFR L747P or L747S mutation [63,64]. Therefore, the identification of EGFR L747P or L747S mutation would provide helpful guidance for the effective treatment of these patients. Interestingly, compared to wild-type (WT) EGFR, the L858R mutant protein is exclusively localized in the cytosol after exposure to cisplatin or ionizing radiation [70]. It remains unclear whether these minor activating mutations on EGFR change its subcellular localization and function.
A tripartite NLS within EGFR has been identified [71], whereas the NES sequence of EGFR that is responsible for interacting with the exportins has not been well clarified. In this report, using an in silico analysis combined with site-directed mutagenesis and confocal imaging, we found that the amino acid 736-749 region of EGFR is a putative NES sequence that is highly conserved among ErbB family members and all vertebrates. The replacement of L747 with alanine (L747A), proline (L747P), or serine (L747S) led to the increased accumulation of EGFR in the nuclei. The L747 mutation also greatly enhanced cell mobility, invasive potential, TKI resistance, and tumorsphere formation in vitro and promoted tumorigenesis in vivo. Importantly, the increased malignant properties of these cancer cells were mainly determined by EGFR nuclear signaling but not canonical membrane-bound signaling, and cells with germline EGFR NES mutant expression by the CRISPR/Cas9-mediated approach developed into hematopoietic tumors. These findings suggest that EGFR NES mutation that leads to enhanced nuclear signaling plays a crucial role in tumor initiation, invasiveness, and TKI resistance and might be a critical factor in tumorigenesis, disease progression, and poor prognosis in patients with lung cancer.
Materials and methods
Cell culture, transfection conditions, and stable cell line establishment
All cell lines were maintained in Dulbecco’s modified Eagle’s medium/F-12 with 10% fetal calf serum and 100 μg/mL penicillin and streptomycin. To observe the effect of EGF, the cells were serum-starved for 20-24 h and then stimulated with EGF in various concentrations for different times. Transient transfection was performed with the homemade cationic liposome. To establish stable cell lines expressing WT and mutant EGFR, the H1299 and MD-MB-231 lung and breast cancer cell lines with EGFR knockdown by lentiviral infection were transfected and selected in the presence of both G418 and puromycin. The resultant colonies were further sorted on the basis of GFP.
Plasmid constructs
All of the EGFR expression plasmids were cloned in pcDNA6-EGFR or pCMV-EGFR-GFP as previously described [71]. To generate NES EGFR mutants, specific alanine mutations were constructed by site-directed mutagenesis using either pcDNA6A-EGFR or pCMV-EGFR-GFP plasmids with a site-directed mutagenesis kit (Stratagene, Santa Clara, CA) according to the manufacturer’s instructions. The EGFR mutants were constructed by PCR with the following primers: (1) L747A: Forward 5’-cc gtc gct atc aag gaa gca aga gaa gca aca tct ccg, Reverse 5’-cgg aga tgt tgc ttc tct tgc ttc ctt gat agc gac gg; (2) L747S: Forward 5’-cc gtc gct atc aag gaa tca aga gaa gca aca tct ccg, Reverse 5’-cgg aga tgt tgc ttc tct tga ttc ctt gat agc gac gg; (3) L747P: Forward 5’-cc gtc gct atc aag gaa cca aga gaa gca aca tct ccg, Reverse 5’-cgg aga tgt tgc ttc tct tgg ttc ctt gat agc gac gg; and (4) LL1010-1011AA: Forward 5’-tcc acg tca cgg act ccc gcc gcg agc tct ctg agt gca acc, Reverse 5’-ggt tgc act cag aga gct cgc ggc ggg agt ccg tga cgt gga. All mutant constructs were confirmed via nucleotide sequencing by the DNA Core Facility at The University of Texas MD Anderson Cancer Center (Houston, TX).
Cellular fractionation, co-immunoprecipitation, and Western blot analysis
Cellular fractionation was performed as described previously [72]. In brief, cells were washed with ice-cold phosphate-buffered saline, harvested by scraping with a rubber policeman. Nuclear extracts were prepared from these cells using an NP-40 lysis method. In brief, 1 × 107 cells were resuspended in 200 μl buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, and 1 mM DTT) and incubated on ice for 25 min before adding 5 μl of 10% NP-40. The cytosolic fraction and nuclei were collected by centrifugation at 1,500 g for 5 min, and the resultant nuclear pellets were extracted by vortexing vigorously in 50 μl of buffer C (10 mM HEPES [pH 7.9], 400 mM NaCl, 1 mM EDTA, and 1 mM DTT). The nuclear extracts were obtained by collecting the supernatants after centrifugation at 16,100 g for 30 min. The protein concentration of each fraction was measured using the BCA reagents from Pierce (Rockford, IL). Samples were subjected to SDS-PAGE on 8% polyacrylamide gels, and the proteins were transferred to nitrocellulose membranes. Prestained molecular mass standards for electrophoresis were obtained from Bio-Rad (Hercules, CA). Membranes were probed with monoclonal or polyclonal antibodies, followed by horseradish peroxidase-labeled secondary antibodies. The proteins in the samples were detected with an enhanced chemiluminescence reagent (Pierce or Amersham Biosciences, Piscataway, NJ). For the co-immunoprecipitation assays, cells were lysed in a lysis buffer (150 mM NaCl, 0.1 mM EDTA, 10% glycerol, 0.5% NP-40, 1 mM dithiothreitol, 20 mM NaF, 1 mM Na3VO4, and a cocktail of protease inhibitors). The supernatants obtained after centrifugation were pre-cleared with protein-A+G agarose beads. Three micrograms of antibodies were incubated with the supernatants for 4 h or overnight at 4°C. The precipitates were collected by being incubated with protein-A+G agarose beads, followed by washing three times with the lysis buffer. The precipitates bound to the beads were analyzed by Western blot analysis with appropriate antibodies.
Antibodies
We used the following antibodies in this study: anti-EGFR (Santa Cruz Biotechnology [Santa Cruz, CA] and Thermo Fisher Scientific Lab Vision [Kalamazoo, MI]), anti-EGFR-pY1068 (Cell Signaling Technology, Danvers, MA), anti-lamin B (Calbiochem, Billerica, MA), anti-α-tubulin (Sigma-Aldrich, St. Louis, MO), anti-actin (Sigma-Aldrich), anti-c-Myc (Roche Applied Science, Madison, WI), and anti-GFP (Santa Cruz). Anti-pStat3 and anti-Stat3 were purchased from Epitomics (Burlingame, CA) and Cell Signaling Technology (Danvers, MA), respectively. All secondary antibodies were obtained from Vector Laboratories (Burlingame, CA) and Jackson ImmunoResearch Laboratories (West Grove, PA).
Confocal imaging assay and IHC staining
GFP-EGFR-expressing cells grown on chamber slides (Nalge Nunc International, Rochester, NY) were fixed in 3.5% paraformaldehyde for 30 min at ambient temperature, permeabilized with 4% NP-40, and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, Eugene, OR). Serial confocal images 1 µm apart along the Z axis were obtained using a LSM710 fluorescence confocal microscope (Carl Zeiss, Germany) under a 63× oil immersion lens. To quantify the fraction of EGFR inside the nucleus, we chose the confocal slice at the center of the nucleus. Fractions of EGFR that were co-localized with DAPI (inside the nucleus) pixels vs. total EGFR pixels at the confocal plane were calculated using LSM710 software. Data were compiled from three independent experiments. Images were processed using Adobe Photoshop software (Adobe Systems, San Jose, CA). IHC staining of paraformaldehyde-fixed tumor tissues was performed using specific Brilliant Violet 510™ anti-mouse CD19 (Biolegend) and NucRed Dead 647 (ThermoFisher).
Three dimension (3D) modeling
The 3D model of the kinase domain of human EGFR L747 mutants was constructed using 1M17 as the template for homology modeling using the program (PS)2 [73]. The figures were produced by PyMOL (Schrödinger, Cambridge, MA) [74] and the matchmaker tool of UCSF Chimera [75].
FACS analysis
The monolayer-cultured stable cell lines expressing EGFR/WT and mutants were trypsinized into single cells and put on petri dishes for 2 h. The cell suspension was blocked with 5% fetal bovine solution in Hank’s Balanced Salt Solution (Life Technologies, Grand Island, NY) for 30 min. The cells were stained with a monoclonal cocktail solution of anti-CD44-APC and anti-EpCAM-Pacific Blue stem cell markers for 20 min. The cells were washed and fixed with 1% paraformaldehyde before FACS.
Tumorsphere formation assay
All stable cells expressing EGFR/WT, L747 and L1010LL/AA, and L858R mutants were derived from the H1299 and MD-MB-231 cells with endogenous EGFR knocked-down. We cultured 2.5 × 104 or 1 × 105 cells/well in serum-free stem cell culture medium with supplements (Stemcell Technologies, Vancouver, BC, Canada) for the indicated times. The tumor spheroids larger than 100 μM in diameter were counted. To observe the inhibitory effect of TKIs on tumor spheroid formation, 1 × 105 cells were re-suspended in the stem cell culture medium containing either 2.5 μM erlotinib or vehicle DMSO; the media was refreshed every other day.
3D cell culture and time-lapse imaging
Matrigel was used to perform 3D cultures of the stable cell lines reconstituted with EGFR/WT and L747 mutants according to the assay instructions of the product. In brief, 2 × 104 cells were mixed with 0.4 ml of the chilled collagen solution; the mixtures were immediately transferred into a 12-well plate. After incubating these cell mixtures at 37°C in the cell culture incubator for 1 h, the wells were covered with regular culture medium. The cells and spheroids were visualized using ZESS Cell Observer Microscopy. The random migration of cells in Matrigel 3D culture was recorded using automated time-lapse video microscopy. The time-lapse imaging interval was 30 min.
Limiting-dilution transplantation assay
The endogenous EGFR knocked-down H1299 cells were reconstituted with EGFR WT, WT/LL1010-1011AA; L747P(S), and L747P(S)/LL1010-1011AA. The stable cell lines were transplanted into mice at different dilutions mixed with Matrigel by being subcutaneously injected into flanks. Tumor formation was examined every 3 days. The mice were euthanized 2 months after injection.
Microarray analysis of transcriptome
Transcriptome analysis by cDNA array. Total RNAs were isolated from the stable cell lines using the RNeasy® Mini Kit (QIAGEN). The microassay analyses were then performed (Arraystar Inc.).
Generation of CRISPR-Cas9-mediated L749 and LL1012-1013 mutant knock-in mice
To generate knock-in mice of EGFR L749P or L749S (equivalent to human L747) and LL1012-1013AA (equivalent to human LL1010-1011AA), we used a CRISPR/Cas9-mediated genome editing approach by performing pronuclear injection with the sgRNAs, donor L749P and L749S donor oligos, and Cas9 mRNAs (PNA Bio, Inc.) in the MD Anderson genetically engineered mouse core facility. The genotypings were carried out by genomic high-fidelity PCR, followed by DNA sequencing. sgRNA-L749 sequence: 5’-TGAGAAAGTAAAAATCCCGGTGG-3; and sgRNA-L1010-1011s sequence: 5’-GGCATACCAGAGAACTCAAGAGG-3’. L749 mutant PCR was performed under the following condition: 94°C 3’, 94°C 30”, 58°C 1’, and 72°C 1’, 35 cycles; PCR primers: Forward 5’-ctctgatatgggatggtatttgt; Reverse 5’-cctcctcccacgtccctataa; the PCR products were purified and sequenced with sequencing primer: gtaatgtaga gcccttgagg actccggtcc. LL1012-1013 mutant PCR was performed under the following condition: 94°C 3’, 94°C 30”, 55°C 1’ and 72°C 1’, 35 cycles; genotyping PCR primers: Forward 5’-agatggaaaagggacata; Reverse 5’-cttggagagaagaatttttgtct and sequencing primer: ACCGA GCCCTGATGG ATGAAGAGGA CATGG. The donor oligo sequences are as follows: (1) L749P: gccctcttaatctcagGGTCTCTGGATCCCAGAAGGTGAGAAAGTAAAAATCCCGGTcGCCATCAAGGAGcctcGAGAAGCCACATCTCCAAAAGCCAACAAAGAAATCCTTGACgtga. (2) L749S: gccctcttaatctcagGGTCTCTGGATCCCAGAAGGTGAGAAAGTAAAAATCCCGGTcGCCATCAAGGAGTctAGAGAAGCCACATCTCCAAAAGCCAACAAAGAAATCCTTGACgtga. (3) LL1012/1013AA: CTGATGAGTATCTTATCCCACAGCAAGGCTTCTTCAACAGCCCGTCCACGTCGAGGACTCCCgcggcGAGTTCTCTGgtatgccccccctcttgatttaaattgatttactttcc.
Results
Mapping putative NES of EGFR
The results of our previous studies demonstrated that nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin β1, and CRM1 [39], and we have identified tripartite NLS on EGFR [71]; however, the NES that controls EGFR export from the nucleus to the cytosol remains elusive. To identify the putative NES on EGFR, an in silico approach was used to analyze the human EGFR protein sequence using the NESbase 1.0 database. On the basis of prediction and sequence conservation comparisons, we identified four putative NES motifs throughout EGFR (Figure 1A; M1-M4). To evaluate the functional NES, we generated five EGFR mutants with leucine or isoleucine replaced by alanine using site-directed mutagenesis (Figure 1A). To determine whether the disruption of EGFR NES affects its nuclear localization, HEK293T cells were transiently transfected with myc-tagged WT or mutant (M1, M2, M3a, M3b, and M4) EGFR. The transfected cells were then serum-starved for 20 h, followed by stimulation with 50 ng/mL of EGF for 30 min. The cell fractionations were created and analyzed using Western blot analysis. As shown in Figure 1B, among the five mutants, only EGFRL747A in the M4 region had increased nuclear localization compared with EGFRWT, suggesting that the M4 mutant (hereafter referred to as EGFRL747A) is a functional NES sequence of EGFR. In addition to HEK293T cells, the increased nuclear localization of EGFRL747A was further confirmed in HeLa cervical cancer and MDA-MB-231 breast cancer cells (Figure S1A and S1B). In addition, we used confocal microscopy to verify nuclear localization of EGFRWT- and EGFRL747A-green fluorescent protein (GFP) fusion proteins in response to EGF in HEK293T cells (Figure S1C). Upon EGF stimulation for 60 min, EGFRL747A likely exhibited greater nuclear accumulation than did EGFRWT. Interestingly, even in the absence of EGF, some EGFRL747A mutant proteins were found in the nucleus (Figure S1C, right upper panel).
Figure 1.

Characterization of the putative NES on EGFR. (A) Diagram of the putative NES on EGFR in an in silico analysis. The human EGFR amino acid sequence was analyzed using the NESbase 1.0 database. The predicted NES sequences are referred to as M1, M2, M3, and M4. The red highlighted amino acid residue is replaced by alanine to generate NES mutants. (B) The putative NES regions were point-mutated with site-directed mutagenesis. The cytosolic and nuclear fractions were immunoblotted with anti-myc (EGFR). Lamin B and α-Tubulin were used as nuclear and cytosolic protein markers. (C) Myc-tagged deletion mutants of EGFR plasmids were transiently transfected into HEK293T cells. Whole cell lysates of the transfected cells were immunoprecipitated with anti-CRM1 antibodies. The precipitates were separated on SDS-polyacrylamide gel and immunoblotted with anti-myc and anti-CRM1. (D and E) The putative NES region sequence (M4) alignment of EGFR among four members of the ErbB family (D) and vertebrates (E). (F) Amino acid sequence similarity of EGFR and protein kinase inhibitor (PKI)-type NES. WT, wild-type; ECD, extracellular domain; ICD, intracellular domain; TM, transmembrane domain; JM, juxtamembrane domain; KD, kinase-dead.
Because functional NES is usually associated with CRM1, we tested the physical interaction between EGFR and CRM1 using a co-immunoprecipitation assay (Figure 1C). The results showed that the full-length, kinase-dead (KD), and intracellular domain (ICD) of EGFR had strong interactions with CRM1, and there was weak binding of the transmembrane-juxtamembrane (TMJ) mutant. No interaction between CRM1 and the extracellular domain (ECD) was observed in the immunoprecipitates. Consistent with our in silico prediction, the ICD of EGFR which contains one putative NES sequence within the M4 region is responsible for the receptor interacting with CRM1 (Figure 1C).
A sequence alignment showed that the M4 region (amino acids 736-749) was highly conserved in the ErbB family (Figure 1D). The conserved sequence also broadly existed among vertebrates (Figure 1E). Amino acid sequence alignments of the identified leucine/isoleucine-rich NES from protein kinase inhibitor (PKI) and EGFR indicated that the critical hydrophobic residues (highlighted in red) in the M4 motif of EGFR had a highly conserved consensus NES sequence (Figure 1F). Taken together, these findings suggest that the M4 motif is a putative NES sequence on EGFR.
NES mutation of EGFR leads to constitutive activation of tyrosine kinase
Because the M4 NES motif is located in the EGFR kinase domain (Figure 1A), we determined whether disruption of the EGFR NES affected EGFR kinase activity. To test this possibility, we examined the kinase activity of the NES mutant, as indicated by autophosphorylation or transphosphorylation of EGFR proteins after EGF stimulation. HEK293T cells were transiently transfected with myc-tagged EGFRWT, EGFRL747A mutant, and the EGFR KD-expressing plasmids. The transfected HEK293T cells were serum-starved overnight and stimulated with or without EGF for 30 min. The phosphorylated EGFR and its mutants in the immunoprecipitates were probed with anti-phosphotyrosine antibodies (4G10). As indicated in Figure 2A, in the absence of EGF, the L747A mutant protein in the precipitates was strongly phosphorylated (lane 4); EGF stimulation only slightly increased the phosphorylated form of this protein (lane 8). Moreover, the L747A mutant activated the EGFR canonical downstream target STAT3 in the absence of EGF, as demonstrated by the occurrence of phospho-STAT3 (Figure 2B, lane 3). These results suggest that the EGFRL747A mutant is a constitutively activating mutation.
Figure 2.
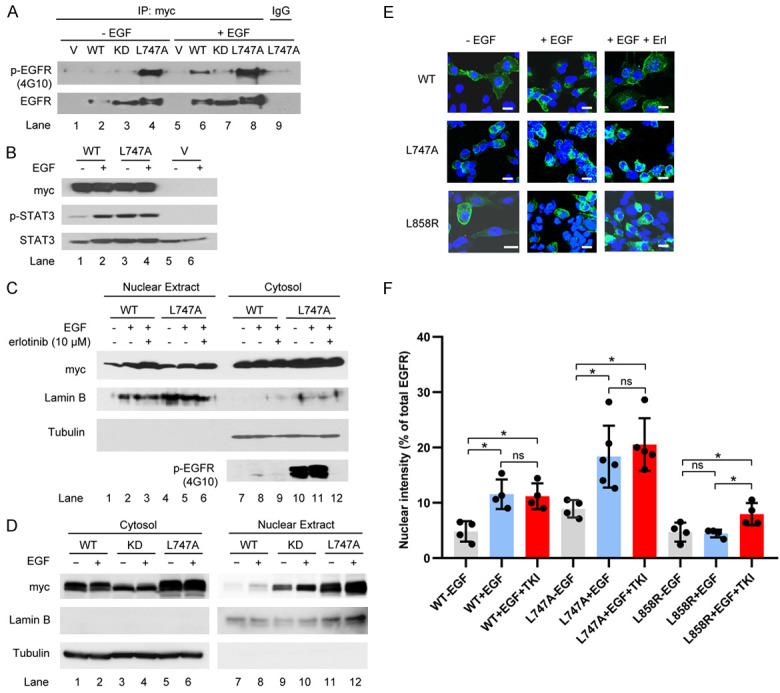
Influence of NES mutation on EGFR kinase activity and nuclear translocalization. (A) Transfection of EGFR WT and mutants was performed and the transfected cells were serum-starved for 20 h and stimulated with 50 ng/ml EGF for 30 min. The precipitates with anti-myc antibodies were immunoblotted with anti-phospho-tyrosine antibodies (4G10) and anti-EGFR. (B) HEK293T cells were transfected with EGFRWT and EGFRL747A mutant, starved for 20 h, and then treated with or without EGF for 30 min. A Western blot analysis was performed with specific antibodies against myc (EGFR), phospho-STAT3 (p-STAT3), and STAT3. (C) HEK293T cells were transfected with the indicated plasmids. The transfected cells were starved for 20 h and pretreated with the EGFR inhibitor erlotinib at 10 µM for 30 min, followed by EGF stimulation for 30 min. The cell fractions were separated and immunoblotted with the indicated antibodies. (D) HEK293T cells were transiently transfected with the indicated plasmids. EGF treatment and analysis were performed as described in (C). (E) H1299 cells with endogenous knockdown EGFR were reconstituted with EGFR WT, L747, and L858R mutants. The stable cell lines were starved for 16 h and stimulated with 100 ng/ml EGF, with or without pretreatment with erlotinib (10 µM) for 45 min. Images were obtained using confocal microscopy. Bar, 10 µm. (F) Semi-quantitative analysis of confocal images. Fractions of EGFR co-localized with DAPI (inside the nucleus) pixels vs. total EGFR pixels at the confocal plane were calculated using LSM710 software. Data were compiled from three independent experiments. Nuclear EGFR is represented as the ratio of nuclear vs. total EGFR intensities. ns, not significant, *P < 0.05.
Enhanced kinase activity of EGFR does not contribute to its nuclear localization
The point mutation in the NES region of EGFR resulted in the constitutive activation of kinase activity, leading us to determine whether the enhanced nuclear accumulation of mutated EGFR is due to its higher kinase activity. To this end, serum-starved EGFRWT and EGFRL747A mutant-expressing stable cell lines were pretreated with EGFR TKI erlotinib (10 μM) for 30 min, followed by EGF stimulation for an additional 30 min. The results of the biochemical analysis showed that the inhibition of EGFR and NES mutant kinase activity did not block EGF-induced nuclear localization (Figure 2C, lane 2 vs. lane 3, lane 5 vs. lane 6). Moreover, increased nuclear localization of the well-defined EGFR KD was also observed in the presence of EGF, suggesting that kinase activity is not necessary for the ligand-triggered nuclear translocation of EGFR (Figure 2D, lane 9 vs. lane 10).
In addition, in confocal imaging of EGRFWT and EGFRL747A mutant-expressing H1299 lung cancer stable cell lines in which the endogenous EGFR had been depleted, we found that the TKI pretreatment did not change the nuclear localization following EGF stimulation (Figure 2E and 2F). It is worth noting that the EGFRL858R mutant, a well-identified TKI-sensitizing mutant, was predominantly localized in the cytoplasm, even after EGF stimulation; pretreatment of TKI resulted in a mild increase in nuclear accumulation in the EGFRL858R-expressing cells upon EGF stimulation (Figure 2E and 2F). It is possible that TKI binding to the kinase domain of EGFRL858R mutant leads to conformational change, which in turn affects the NLS or NES interfaces to interact with their partners, importin β1 and CRM1.
NES mutation of EGFR affects its interaction with CRM1
In our previous study, we demonstrated that there is a dynamic balance between EGFR binding to importin β1 and CRM1 during cytoplasmic-nuclear transport and that EGFR binding to CRM1 is time-dependent following EGF stimulation, reaching a maximal peak at 45 min [39]. To understand how the L747A mutant of EGFR increased EGFR nuclear accumulation, we examined the interactions among EGFRWT, mutated EGFR protein, importin β1, and CRM1. First, we determined whether the EGFR L747 mutant protein binds to importin β1 with higher affinity and in turn increases EGFR mutant translocation into the nucleus. Interestingly, no significant increase was found in EGFRL747A mutant protein and importin β1 compared with the EGFRWT-importin β1 interaction in the co-immunoprecipitation assay (Figure 3A). These results suggest that the nuclear import efficiency of the mutant protein does not contribute to increased nuclear localization. Next, we determined whether the EGFRL747A mutant protein has lower export efficiency because of its decreased affinity to bind to CRM1. As shown in Figure 3B, CRM1 was co-immunoprecipitated with both EGFRWT and EGFRL747A following stimulation with EGF for 45 min. Unexpectedly, compared with EGFRWT, EGFRL747A mutant protein pulled down more CRM1 proteins, suggesting that it has a higher affinity to bind CRM1; as a result, the recycling of CRM1 was reduced [44]. These findings indicate that increased nuclear localization of EGFR NES mutant is due to the impaired release of CRM1 from the export complexes, leading to less effective export of nuclear EGFR.
Figure 3.
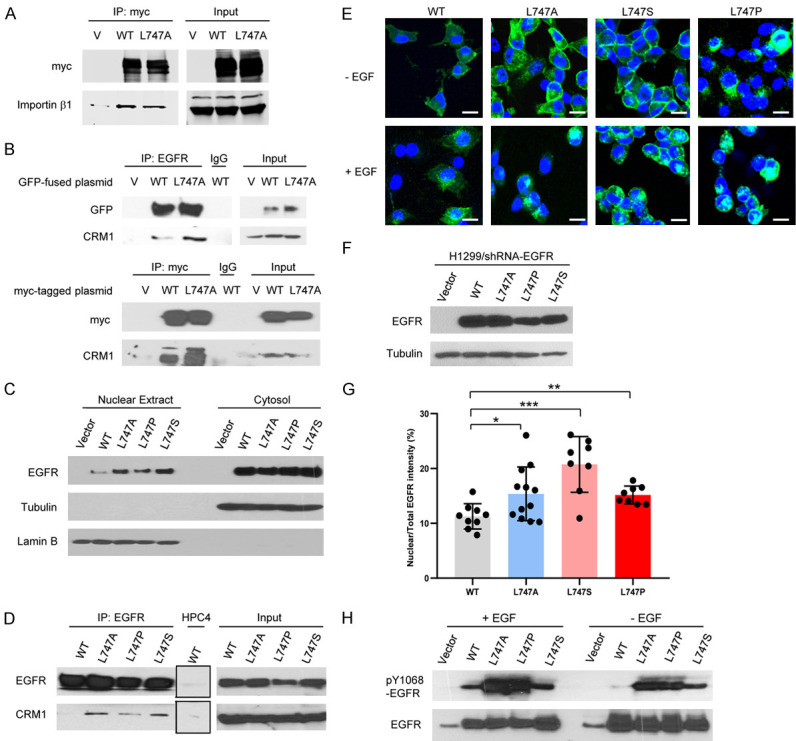
Expression of EGFR NES mutants increases the interaction with CRM1. (A) HEK293T cells were transfected with myc-tagged EGFR plasmids. The immunoprecipitates of anti-myc antibodies from the transfected HEK293T cells were analyzed with anti-myc (EGFR) and anti-importin β1. (B) GFP-fused or myc-tagged EGFR WT and L747A mutant expressed in HEK293T cells were immunoprecipitated with anti-EGFR or anti-myc antibodies. The precipitates were immunoblotted with anti-GFP or anti-myc and anti-CRM1 antibodies. (C) The nuclear extract and cytosolic fractions of H1299 stable cell lines expressing EGFR WT and L747 mutants were immunoblotted with the indicated antibodies. (D) The immunoprecipitates of anti-EGFR or anti-HPC4 (negative control) from H1299 stable cell lines expressing EGFR WT and L747 mutants were immunoblotted with the indicated antibodies. (E) Representative confocal images of H1299 cancer cells stably expressing GFP-fused EGFR WT and L747 mutants. The stable cell lines were serum-starved overnight and treated with 60 ng/ml EGF for 50 min. The treated cells were fixed and permeabilized with 4% NP-40 and then mounted with ProLong Gold antifade reagent with DAPI. The images were obtained using a LSM710 fluorescence confocal microscope. Bar, 10 µm. (F) Expression levels of EGFR WT and NES L747 mutants were immunoblotted with anti-EGFR antibodies. Tubulin was used as a loading control. (G) Quantitative analyses of the nuclear EGFR WT and NES L747 mutants in the stable cell lines in (C). *P < 0.05, **P < 0.01, ***P < 0.001. (H) Whole cell lysates of HEK293T cells transfected with EGFR WT and NES mutants were immunoblotted with antibodies against phospho-EGFR (pY1068) and total EGFR.
L747 mutation of EGFR presents in patients with lung cancer
We searched the Catalogue of Somatic Mutations in Cancer EGFR mutation database (COSMIC); L747 mutations were reported in lung cancer patients with little or no response to TKI therapy and poor outcomes [61-69]. L747 was replaced with serine or proline in both TKI-naïve and -treated patients with lung cancer, suggesting that these mutants are primary or secondary mutations in lung cancer. To determine whether L747S and L747P mutants change their nuclear localization, we first knocked down the endogenous EGFR in the H1299 lung cancer cell line with lentiviral shRNAs targeting either the coding region or the 3’-UTR region; the EGFR knock-down (EGFRkd) stable clone pool was selected by puromycin. The endogenous EGFR of the lung cancer cells was effectively depleted by both shRNAs (Figure S2A). The EGFRkd stable cell line with shRNA targeting 3’-UTR regions was chosen to re-express EGFRWT, EGFRL747A, EGFRL747P, or EGFRL747S-GFP-fused proteins, and the cytosolic and nuclear extracts from these stable cell lines were immunoblotted with specific antibodies. As indicated in Figure 3C, the Western blot analysis results showed that the EGFR NES mutants, including L747A, L747S, and L747P, had a higher protein level in the nuclear extracts than did EGFRWT. Moreover, like EGFRL747A, both EGFRL747S and EGFRL747P mutants also exhibited a higher affinity to bind CRM1, leading to reduced exporting efficacy from the nucleus (Figure 3D).
In addition, a confocal imaging analysis of the EGFR L747 mutant-expressing stable cells displayed a subcellular distribution similar to that of EGFRL747A mutant, namely much higher accumulation in the nuclei than EGFRWT (Figure 3E), although their expression levels were similar to those of EGFRWT (Figure 3F). To verify whether EGFR WT and L747 mutants localize within the nucleus, confocal Z-stack scanning of H1299 cells expressing GFP fused EGFRWT and EGFR L747 mutants was performed using a LSM710 fluorescence confocal microscope. We obtained serial confocal images 1 µm apart along the Z axis. The image data demonstrated that EGFRWT and NES mutant EGFRL747A, EGFRL747P, and EGFRL747S were indeed localized in the nuclei (Figure S2B). A quantitative analysis showed that these L747 mutations in the NES region significantly enhanced the nuclear translocation of EGFR following EGF stimulation (Figure 3G). We further demonstrated that, like EGFRL747A, both EGFRL747P and EGFRL747S mutants were also constitutively kinase active, as indicated by highly tyrosine-phosphorylated EGFR, with or without EGF stimulation (Figure 3H). These results indicate that L747 mutations of EGFR found in patients with lung cancer, i.e. EGFRL747P and EGFRL747S, increase EGFR accumulation in the nucleus.
NES mutation of EGFR enhances cell migration, invasion, and TKI resistance
It has been shown that nuclear EGFR contributes to drug resistance and poor outcomes in lung cancer patients [19,23]. Therefore, we asked whether the cells expressing EGFR L747 mutants that nuclear EGFR is increased promote malignant phenotypes, such as cell migration, invasion, and EGFR TKI resistance. To test this hypothesis, we first determined whether EGFR NES mutant expression alters cancer cell migration and invasion. A wound-healing assay was used to determine the effect of EGFR NES mutant on cancer cell migration. We found that reconstituted expression of EGFRWT in the H1299 cells with endogenous EGFRkd accelerated wound healing ability, as expected, and re-expression of the NES mutant (L747A, L747P, and L747S) increased cell migration even more (Figure 4A). A cell invasion in vitro assay showed that stable cells expressing EGFR L747 mutants possessed much higher invasive potential than did vector control and EGFRWT cells (Figure 4B and 4C). These data suggest that NES mutation of EGFR confers higher migration and invasive potential on the tumor cells.
Figure 4.
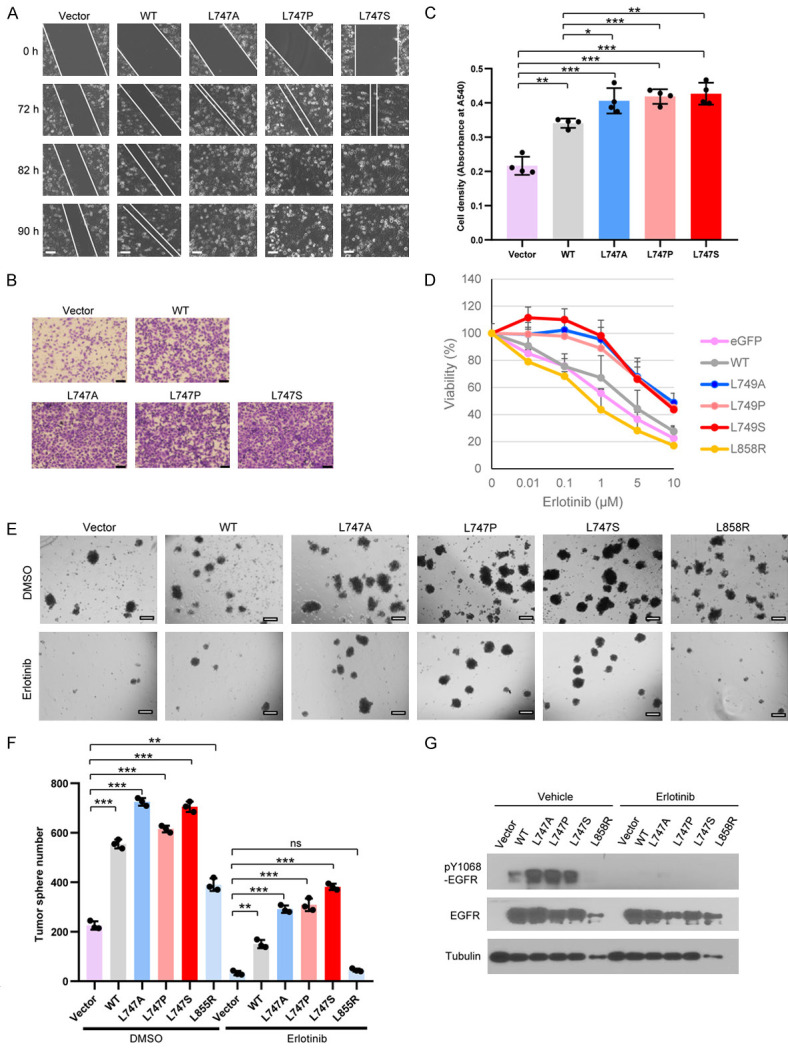
Expression of EGFR NES mutants enhances migration, invasion, and TKI resistance. (A) Cells expressing the indicated plasmids were seeded in a 6-well plate. Live microimaging of wound healing was performed using Cell Observer Microscopy (ZEISS). Bar, 100 µm. (B) H1299 cells (1 × 104) expressing the indicated plasmids in culture medium without serum were seeded into the invasion chambers coated with Matrigel. The chambers were inserted into 24-well plates filled with 10% fetal calf serum-containing medium. The cells were cultured for 48 h before fixation and staining with crystal violet. Bar, 200 µm. (C) Quantitative analysis of the invasive potential of the lung cancer cells expressing EGFR WT and NES mutants. Bar graphs were presented as the mean value ± SD of four independent experiments. (D) Cells expressing the indicated plasmids were seeded in 96-well plates at 3,000 cells/well and allowed to grow overnight. Erlotinib at the indicated concentrations was added and was replaced with new medium containing erlotinib at 36 h. The viable cells were determined by Trypan Blue staining. Each point was triplicated. The curve data represented were the mean values from at least two independent experiments. (E) A tumor spheroid formation assay was performed in H1299 cells expressing the indicated plasmids in the presence of erlotinib (2.5 μM) or vehicle DMSO for 12 days. Bar, 100 μM. (F) Bar graphs of tumorsphere numbers are presented as the mean value ± SE of three independent experiments. (G) Whole lysates of the tumorspheres in the presence and absence of 2.5 μM erlotinib, isolated from the Matrigel 3D sphere cultures (E), were immunoblotted with the indicated antibodies. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
Next, to determine whether EGFR NES mutation confers more resistance to TKI, the EGFRkd H1299 cells were stably reconstituted with EGFRWT or EGFR NES L747 mutants, treated with erlotinib at various concentrations for 72 h. The cell viability assays showed that the cell lines expressing EGFR L747 mutants had higher resistance to erlotinib, in particular within the concentration range of 0.1-1 μM, than did those expressing EGFRWT and EGFRL858R mutant, known as an TKI-sensitizing mutation (Figure 4D). We further validate the results using a 3D Matrigel culture assay, which better recapitulates the extracellular matrix of cancer cells in vivo [76]. Compared to vector, EGFRWT, and EGFRL858R-expressing cells, EGFR NES mutant-expressing cells formed more tumorspheres in the absence of erlotinib (Figure 4E and 4F), likely because of the increased phospho-EGFR Y1068 status, which is indicative of kinase activation (Figure 4G). Interestingly, in the presence of erlotinib, the difference in the number and size of tumorspheres among EGFR WT and NES mutants was more obvious (Figure 4E and 4F), yet phospho-EGFR Y1068 signals in different cells were barely detectable (Figure 4G), indicating that EGFR kinase activity was suppressed with erlotinib treatment. Thus, the significant difference in tumorsphere formation among EGFR WT, L858R, and L747 mutants cannot be due to differential kinase activity; it might be as a result of the difference in nuclear accumulation, as EGFR L747 mutation gave rise to intensified nuclear signaling. Taken together, these data support that cancer cells expressing EGFR NES mutants are predisposed to TKI resistance. This phenotype is not attributed to increased kinase activity; rather, it is highly related to escalated nuclear accumulation of the mutated EGFR protein.
L747 mutation of EGFR does not significantly affect the overall structure
Because the putative NES sequence is located on the surface of N-Lobe and is close to the αC-helix, which regulates the kinase active and inactive switch [77,78], we determined whether L747 mutation changes the conformation of the kinase domain by constructing a 3D model of the kinase domain of human EGFR L747 mutants using 1M17 as the template for homology modeling and the program (PS)2 [73] and the figures were produced by PyMOL [74]. Surprisingly, we found no significant conformational changes between the WT and L747 mutants (Figure S3A). Notably, WT L747 forms a hydrophobic interaction with A755, while L747A and L747P weaken this hydrophobic interaction because the distances between L747A and A755 and between L747P and A755 are greater than those between WT L747 and A755. L747S does not have such a hydrophobic interaction. However, the hydrophobic interaction is quite weak compared with the hydrogen bond and charge-charge interaction. Moreover, L747 is located in the loop, which is a more flexible structure; thus, the L747 mutants should not significantly affect the overall structure of the molecules (Figure S3A). In contrast, the well-identified mutant L858R, which is sensitive to TKI, had significant conformational change (Figure S3B). The major change in EGFRL858R was the interaction between L858R and D837. In EGFRWT protein, there was no interaction between L858 and D837, but when L858 mutated to R858, two hydrogen bonds formed between R858 nitrogen and D837 oxygen (Figure S3B). Therefore, L858R mutation may increase its kinase activity or facilitate TKI inhibition.
To determine whether L747 mutation of EGFR affects the interaction between erlotinib and the kinase domain, we performed a superimposition of the inhibitor binding sites of WT and mutated EGFR; the L747 residue of EGFR is located at the more flexible loop region, far from the inhibitor-binding site, and is thus not directly involved in the formation of the TKI/ATP-binding pocket. Therefore, replacement of L747 with alanine, proline, or serine did not affect erlotinib binding to the ATP pocket (Figure S3C). These data suggest that the mechanism of EGFR L747 mutants’ resistance to TKI might differ from that of the EGFR T790M mutant in which threonine is replaced by methionine, resulting in alterations of the topology of the ATP-binding pocket and ATP affinity [77-79].
NES mutation of EGFR increases CSC-like properties in vitro and in vivo
It is well-documented that cancer stem cells (CSCs), also known as tumor-initiating cells (TICs), contribute to TKI resistance in chronic myeloid leukemia and lung cancer cells [80-82]. Therefore, we tested whether EGFR mutation in the NES region, which enhances TKI resistance, alters the CSC population. We performed a fluorescent-activation cell sorting (FACS) analysis of H1299 cells that stably expressed EGFRWT and EGFR L747 mutants and used CD44hi and EpCAM- as promising biomarkers for lung CSCs because CD44 population is enriched for CSC-like properties in vitro and in vivo [83] and epithelial cell adhesion molecule (EpCAM), one of the well-identified CSC markers, is also used for epithelial cell-derived lung CSCs [84,85]. Results from the FACS analyses showed that, compared with EGFRWT-expressing cells, EGFR L747 mutant-expressing cells had higher populations of CD44hiEpCAM-, suggesting that the CSC population was enriched in the EGFR L747 mutants (Figure 5A, upper panel). In addition, the median values of cell fluorescence intensity for total surface CD44 on the cells expressing EGFR L747 mutants were greater than those on EGFRWT and vector control cells (Figure 5A, bottom panel). Interestingly, compared with EGFRWT, the expression of EGFRL858R, which is nuclear translocation-deficient but has higher kinase activity, resulted in a less population of CD44hiEpCAM- with a lower density of surface CD44 (Figure 5A), suggesting that the increased kinase activity is not sufficient to increase the CSC population.
Figure 5.

Expression of EGFR NES mutants increases CSC-like properties. (A) FACS analysis of the EGFR WT- and L747 mutant-expressing cells. The monolayer-cultured cells were trypsinized into single cells and recovered in petri dishes for 2 h before staining. Cells (0.5 × 106) were stained in an aliquot of the cocktail solution of conjugated monoclonal antibody CD44-APC and EpCAM-Pacific Blue (PacBl). An FACS analysis was carried out using a BD FACS Aria II sorter. Bottom, the median values of cell fluorescence intensity for total surface CD44 on the cells expressing EGFR WT and L747 mutants. (B) H1299 cells (1 × 105) with EGFR knocked down and stably re-expressing EGFR WT and L747 mutants were plated onto low-attachment plates and cultured in serum-free stem cell culture medium plus supplements. Half of the culture medium volume was refreshed every 3 days. The tumor spheroid number (> 100 μM in diameter) was counted at 12 to 14 days. Bar, 100 µm. (C) Bar graphs of primary tumorsphere numbers in (B) are presented as the mean value ± SE of three independent experiments. (D) Increased numbers of secondary tumor spheroids in lung cancer cells expressing EGFR L747 mutants. The cells were harvested from primary spheres and trypsinized into single cells. Cells (4 × 104 per plate) were seeded and cultured as described in (B). Bar, 200 µm. (E) Bar graphs of secondary tumorsphere numbers (D) are presented as the mean value ± SE of three independent experiments. (F) In vivo limiting-dilution assay of the lung cancer cells harboring EGFR NES mutation. H1299 cells expressing EGFR WT and NES mutants were subcutaneously transplanted into NOD/SCID mice and tumor formation of the transplants was observed for 2 months. n = 6. Bar, 100 mm. *P < 0.05, **P < 0.01, ***P < 0.001.
An in vitro tumorsphere formation-based functional assay for CSCs was used to further determine whether the EGFR L747 mutants enhance the CSC population and promote self-renewal capacity. We found that stable expression of EGFR NES mutants significantly increased the primary tumorsphere numbers to a greater degree than vector or EGFRWT cells, suggesting that the L747 mutants expended the CSC population, which may lead to drug resistance and other malignant phenotypes (Figure 5B and 5C). Similar results were also observed when the EGFR L747 mutants were expressed in the MDA-MB-231 breast cancer cell line (Figure S4A and S4B). Next, we tested whether the expression of EGFR L747-mutated proteins in lung cancer cells enhanced the self-renewal capacity of lung CSCs. We collected and trypsinized the primary tumorspheres derived from the stable cell lines expressing EGFRWT, EGFRL747P or L747S, and EGFRL858R, as well as vector control, and then cultured these cells in the serum-free stem cell culture medium to observe secondary tumorsphere formation. The results revealed that the expression of EGFR L747-mutated protein significantly increased the number and diameter of secondary tumorspheres, compared with that in cells expressing EGFRWT, suggesting that EGFR NES mutants promote the self-renewal capacity of lung cancer cells (Figure 5D and 5E). It is well-known that patients with lung cancer with the EGFRL858R mutant are TKI-sensitive and have a good prognosis. Consistently, compared with the L747 mutation, the EGFRL858R-activating mutation had fewer CSCs, as indicated by a smaller number of tumorspheres in the primary and secondary tumorsphere formation assays (Figure 5C and 5E).
To further evaluate the above in vitro findings, we performed a limiting-dilution transplantation assay subcutaneously injected H1299 cells that stably express EGFR WT and EGFR L747 mutants at serial dilutions into NOD/SCID mice. The palpable tumors were examined and measured every 3 days and the mice were euthanized 2 months after transplantation. As shown in Figure 5F, the tumor formation frequency of EGFR L747 mutant-expressing cells was much higher than that of EGFRWT cells (n = 4 or 6) (Table 1 and Figure 5F), suggesting that the CSC population was enriched in transplanted lung cancer cells expressing EGFRL747P and EGFRL747S mutants but not in the cells expressing EGFRWT.
Table 1.
Limiting dilution transplantation assay of the cancer cells expressing EGFR WT or NES mutants
| Cell line | Cell dilution | Mouse number | Tumor formation rate (%) |
|---|---|---|---|
| WT | 1 × 105 | 6 | 3/6 (50) |
| 5 × 104 | 6 | 0/6 (0) | |
| 1 × 104 | 6 | 0/6 (0) | |
| L747P | 1 × 105 | 6 | 5/6 (83.3) |
| 5 × 104 | 6 | 4/6 (66.7) | |
| 1 × 104 | 6 | 4/6 (66.7) | |
| L747S | 1 × 105 | 4 | 4/4 (100) |
| 5 × 104 | 6 | 5/6 (83.3) | |
| 1 × 104 | 6 | 0/6 (0) |
Transcriptome analysis of cells expressing EGFRWT, EGFRL747S, and EGFRL858R reveals different expression profiles
To further determine the distinct effects of increased nuclear localization but not kinase activity of EGFR mutants on the malignant phenotypes, we analyzed the transcriptomes of the lung cancer cell line H1299 reconstitutively expressing EGFRWT, pro-nuclear NES mutant EGFRL747S, and nuclear-deficient but TKI-sensitizing mutant EGFRL858R. Indeed, phospho-EGFR levels in whole cell lysates from the EGFRL747S and EGFRL858R-expressing cell lines were much higher than those from the EGFRWT-expressing cells in the presence or absence of EGF stimulation, demonstrating that both EGFRL747S and EGFRL858R are activating mutants with the same kinase activity levels (Figure S5A, left panel). In line with the above results, EGFRL747S-expressing cells acquired stronger invasive potential than did those expressing EGFRWT and EGFRL858R-expressing cells (Figure S5B).
Compared with EGFRWT, heat maps showed that the protein coding and non-coding gene expression profiles in EGFRL747S and EGFRL858R-expressing cell lines were very different, but they indeed shared some target genes, which may have resulted from the enhanced kinase activities (Figure S5C-E and Table S1). Both EGFRL747S and EGFRL858R are activating mutants with intact membrane-bound and cytosolic signaling pathways; thus, the distinct phenotypes may be at least partially attributed to the different magnitude of nuclear translocations. Interestingly, compared with EGFRL858R, EGFRL747S expression in lung cancer cells had significantly up-regulated oncogenes such as IKBKB (IKK2) and FOXM1 but down-regulated tumor suppressor genes such as PTPN12 and 14 (Figure S5D). Notably, ATP-binding cassette transport protein A3 (ABCA3) gene was specifically up-regulated in EGFRL747S-expressing cells; this gene is associated with multi-drug resistance [86-89]. Unlike EGFRL858R, L747 mutation in the kinase domain does not affect ATP pocket conformation (Figure S3B and S3C). Therefore, the altered sensitivity of EGFR L747 mutants to TKI is not related to the conformational change in the kinase domain but may be due to altered subcellular localization, which in turn regulates the downstream pathways. In addition, microassay data revealed that non-coding RNA profiles of EGFRL747S-expressing cells were significantly different from those of EGFRL858R cells; for instance, Lnc-LIF-AS was specifically up-regulated in EGFRL747S but not EGFRL858R-expressing cells (Figure S5E). Lnc-LIT-AS, an LIF natural antisense lncRNA, stabilizes LIF mRNA by overlapping a 3’-UTR of LIF mRNA and thus inhibits LIF mRNA degradation that is mediated by other RNAs [90,91]; LIF is the cytokine leukemia inhibitory factor that activates the JAK-STAT signaling pathway and is a key factor in stem cell self-renewal and maintenance [92-94]. Consequently, the observed gene expression profiles are highly consistent with the functional roles of the cancer cells harboring EGFR NES mutations.
Internalization-deficient and NES dual mutations of EGFR results in less malignant phenotypes than the NES singal mutant
To gain further mechanistic insights into the role of EGFR NES mutants, we generated EGFR double mutations at NES L747 along with LL1010-1011 that is deficient in internalization of the receptor induced by ligand binding [95,96]. Our previous findings demonstrated that nuclear translocation of full-length EGFR is an internalization-dependent process [39]; as a result, the EGFR internalization-deficient mutant is expected to reduce the nuclear translocation but not change the membrane-bound signaling. We found that the autophosphorylation levels of membrane-bound forms of double mutant EGFRL747A/LL1010-1011AA, EGFRL747P/LL1010-1011AA, or EGFRL747S/LL1010-1011AA did not significantly change in the presence of EGF, indicating that adding LL1010-1011AA mutation to EGFR NES mutants did not result in a significant reduction in their kinase activity (Figure 6A). However, a subcellular fraction analysis showed that the LL1010-1011AA mutation together with EGFR WT or L747 mutants significantly reduced the nuclear EGFR levels (Figure 6B). A confocal image analysis also showed that these membrane-bound EGFR mutants had reduced nuclear localization compared with their corresponding parental proteins (Figure 6C).
Figure 6.

Internalization-deficient and NES dual mutations of EGFR results in less malignant phenotypes than the NES singal mutant. (A) Whole cell lysates from HEK293T cells expressing EGFR/WT and L747 mutants, minus or plus LL1010-1011AA mutation, were used for the immunoblotting assay. The autophosphorylation of EGFR proteins was probed with anti-EGFR-pY1068 antibodies. (B) Nuclear extracts and cytosolic fractions of cells expressing the indicated plasmids were immunoblotted with indicated antibodies. (C) H1299 cells knocked down with shRNAs against endogenous EGFR were reconstituted with EGFR WT, NES mutant, or NES combined with internalization-defective double mutants and used for the confocal imaging analysis. Bar, 10 µm. (D) H1299 cells (2 × 104) expressing EGFR and their mutants were mixed with collagen gel and 3D cultures were visualized using ZESS Cell Observer Microscopy. The photos were obtained at day 7. Bar, 10 µm. (E) Bar graphs are presented as tumorsphere in (D) mean values ± SE of at least 10 fields. (F) The cell migration ability of H1299 cells expressing the indicated plasmids, along with their parental molecules, was determined using a wound-healing assay. The photos are representatives of time-lapse microscopy recording at 0 and 24 h. Bar, 200 µm. (G) H1299 cells expressing EGFR WT and L747 single mutants, along with their double mutants, were treated with various concentrations of erlotinib for 3 days. Viability curves were generated from two independent experiments in triplicate. (H) Tumorsphere numbers of H1299 cells expressing EGFR WT and LL1010-1011AA mutants were counted 7 days after treatment with 2.5 µM of erlotinib or vehicle DMSO. Bar graphs are presented as the mean value ± SE. (I) H1299 cells (1 × 106) expressing EGFR WT and L747P and their internalization-deficient mutants (LL1010-1011AA) were mixed with Matrigel and subcutaneously injected into NOD/SCID mice (n = 6). The palpable tumors were examined every 3 days and mice were euthanized 2 months after injection. *P < 0.05, **P < 0.01, ***P < 0.001.
We performed a 3D Matrigel culture assay and found that while EGFRWT cells displayed smooth and round solid spheres, the cells that stably expressed EGFR NES mutant alone (L747A, L747P, and L747S) gave rise to more tumorspheres and the morphologies of these tumorspheres were of a more aggressive phenotype to be mass and stellate in shape [97,98] (Figure 6D, upper panel, and Figure 6E). In contrast, the cells expressing EGFRWT/LL1010-1011AA or EGFRL747A(P,S)/LL1010-1011AA compound mutants formed fewer spheres and their morphologies were round with less aggressiveness, supporting the notion that decreased nuclear EGFR attenuated their malignant potential (Figure 6D, bottom panel, and Figure 6E). Moreover, the cells expressing membrane-bound EGFR L747 mutants displayed significantly slower migration in a wound-healing assay (Figure 6F). To determine whether a reduction in nuclear EGFR signaling in collaboration with the EGFRLL1010-1011AA membrane-bound form sensitizes cancer cells to TKI, we performed a cell viability assay in the presence of erlotinib at different concentrations for 72 h. All cells expressing the membrane-bound EGFR L747 mutants had a greater decrease in resistance to TKI compared to the corresponding L747 single mutants, suggesting that increased nuclear EGFR signaling contributes at least partially to TKI resistance in these cells (Figure 6G). Furthermore, a tumorsphere formation assay in serum-free stem cell suspension cultures showed that the expression of the membrane-bound EGFR WT or L747 mutants produced significantly fewer tumorspheres, both in the presence and absence of erlotinib, suggesting that nuclear EGFR signaling is indeed required for the CSC expansion (Figure 6H). We then performed an in vivo transplantation assay to clarify whether nuclear EGFR signaling but not canonical membrane-bound EGFR signaling leads to the enrichment of CSC population. H1299 lung cancer cells (5 × 104 and 1 × 105) with endogenous EGFRkd and stably reconstituted with EGFRWT, EGFRL747P(S), or their compound mutants EGFRWT/LL1010-1011AA and EGFRL747P(S)/LL1010-1011AA were subcutaneously injected into the right and left flanks of NOD/SCID mice, respectively. The palpable tumors were examined and measured every 3 days. The mice were euthanized 2 months after injection. As shown in Table S2, in comparison with EGFRWT, EGFRL747P(S)-expressing cells were more likely to form tumors, suggesting that there were more CSCs within the cell line. Adding LL1010-1011AA mutation to the L747P(S) mutant resulted in a significant decrease in tumor formation frequency and a concomitant reduction in tumor size (Figure 6I and Table S2), suggesting that nuclear EGFR is a key up-regulator CSCs in tumorigenesis. It should be noted that membrane-bound EGFRL747P(S) still has a higher tendency to grow tumors than does the WT counterpart (Table S2), suggesting that constitutive activation of the membrane-bound EGFR signaling pathway also plays a minor role in tumor initiation and growth.
NLS and NES dual mutations of EGFR compromised the malignant phenotypes mediated by the NES singal mutant
To exclude the possibility that the higher malignant potential of the cancer cells harboring EGFR NES mutants may be due to their cytoplasmic function, the key cofactor for EGFR translocating from the cytoplasm into the nucleus, Sec61β was knocked down using siRNAs (Figure S6A). Since deletion of Sec61β in the EGFRWT and L747 mutant-expressing cells supposedly led to sequestration of EGFR in the cytoplasm [99], we found that the cytoplasmic sequestration of EGFR NES-mutated proteins by knocked-down Sec61β significantly sensitized the cells to TKI compared with the control cells (Figure S6B). These results further support the notion that nuclear EGFR signaling contributes to TKI resistance.
To further assess the nuclear function of EGFR in malignant phenotypes, we generated the NLS deletion mutant of EGFR that lack nuclear translocation ability but possess normal membrane-bound and cytoplasmic signaling activities. The results of previous reports and our current data revealed that deletion of the EGFR NLS motif abolishes EGFR kinase activity [36] (Figure S7). Such NLS motif-deleted EGFR mutant is difficult to exclude the possibility of a putative kinase-dead effect on the malignant phenotype. To clarify the cause-effect of nuclear EGFR, we further generated point mutations at the NLS domain instead of deletion and determined which had nuclear translocation deficiency with maximal remaining kinase activity. As shown in Figure 7A, we generated four types of mutation combination at the NLS domain. A Western blot analysis indicated that compared to EGFRWT, all NLS mutants displayed a much lower level of nuclear EGFR protein accumulation; of four types of NLS mutation, the NLSm mutant had barely accumulated in the nuclei (Figure 7B). To determine whether cells expressing NLSm affected EGFR kinase activity, HEK293T cells were transiently transfected with NLSm combined with EGFR L747 mutants. Interestingly, the kinase activities of the NLS-NES compound EGFR mutants were not significantly reduced compared to those of the L747 single mutant (Figure 7C), indicating that NES mutation-increased kinase activity is dominant in the kinase activity of the compound mutations. However, the accumulation of NLS-NES compound EGFR mutants was significantly reduced in the nucleus after EGF treatment (Figure 7D), suggesting that NLS mutation is predominant in directing cytoplasmic location. These NES-NLS compound EGFR mutants are optimal candidates to clarify the nuclear roles of EGFR in malignant phenotypes.
Figure 7.
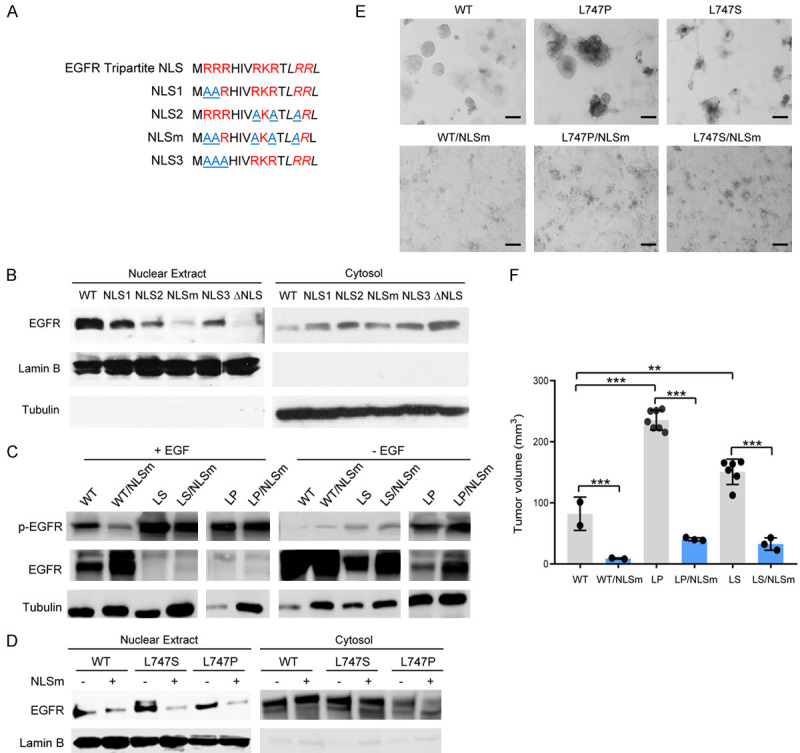
NLS and NES dual mutations of EGFR compromised the malignant phenotypes mediated by the NES singal mutant. A. Schematic diagram of the NLS mutation of EGFR. B. The cytosolic and nuclear fractions of the H1299 cells expressing EGFR WT, NLS point mutations, and NLS deletion (ΔNLS) were immunoblotted with the indicated antibodies. C. The phosphorylated EGFR at Y1068 in whole cell lysates, with or without EGF treatment, was immunoblotted with specific anti-EGFR pY1068 antibodies. D. The cytosolic and nuclear fractions of the H1299 cells expressing EGFR WT, NES mutants, and NES and NLS double mutants were immunoblotted with the indicated antibodies. E. Tumorsphere formation of cells expressing the indicated plasmids in 3D Matrigel culture was performed. Bar, 10 µm. F. H1299 cells (1 × 106) expressing the indicated plasmids were subcutaneously injected into Nude/Nude mice (n = 6). Bar graphs are presented as the tumor volume mean values ± SE. **P < 0.01, ***P < 0.001.
To determine whether the NES-NLS compound mutations of EGFR affect their biological functions, we examined the tumorsphere formation and motility of the cancer cells expressing EGFR L747 mutants, with or without NLS mutation, in 3D Matrigel cultures. As shown in Figure 7E, the lung cancer cells expressing EGFR NLS-NES dual mutants grew as a monolayer and displayed different morphologies from those of L747 single mutants, which formed spheres in 3D cultures. As the p-EGFR levels of NLS-NES compound mutants and NES single mutants were similar (Figure 7C), the reduction in the nuclear accumulation of EGFR predominantly contributed to a loss of potency to generate tumorspheres, suggesting that a desirable level of nuclear EGFR is required for the maintenance of stemness of tumor cells (Figure 7E). Furthermore, using long-term time-lapse imaging of living cells, we monitored the motility of the lung cancer cells that expressed EGFRWT and EGFRL747 mutant, with or without NLS mutation. The time-lapse imaging unveiled that in contrast to EGFRWT, EGFRL747P-expressing cancer cells were prone to form spheres and barely built monolayers at the interface of 3D Matrigel (Movie S1A and S1B). The addition of NLS mutations to EGFRWT and EGFRL747P in 3D gel biasedly exhibited horizontal migration on the gel interface and rapidly formed monolayers with a few spheres (Movie S1C and S1D). These observations that the NLS mutant of EGFRL747P-expressing cancer cells compromised the observed unique motility of EGFRL747P-expressing cells clearly indicate that nuclear EGFR plays a crucial role in controlling cell motion and sphere morphology.
Next, we determined whether NLS mutation of EGFR affects the NES mutant-mediated tumorigenesis and tumor growth in vivo. To this end, H1299 cells with endogenous EGFRkd were stably reconstituted with EGFRWT, EGFRL747P(S), and their NLS-NES compound mutants. The stable cell lines were subcutaneously injected in parallel into the left and right flanks of Nude/Nude mice and the palpable tumors were regularly examined and measured every 3 days for 2 months. The NLS mutation in the EGFR NES mutants led to a significantly lower tumor formation incidence and smaller tumor size (Table 2 and Figure 7F). The in vivo findings provided additional evidence that nuclear EGFR plays a more crucial role than cytosolic EGFR in tumorigenic potential and tumor growth.
Table 2.
Tumor incidence of WT or EGFR NES mutants, with or without NLS mutation
| Genotype | Tumor incidence (%) | Genotype | Tumor incidence (%) |
|---|---|---|---|
| WT | 2/7 (28.6) | WT/NLSm | 2/7 (28.6) |
| L747S | 6/8 (75.0) | L747S/NLSm | 3/8 (37.5) |
| L747P | 7/8 (87.5) | L747P/NLSm | 3/8 (37.5) |
Taken together, compared with EGFRWT, EGFR NES mutants have two unique characteristics: higher kinase activity and enhanced nuclear accumulation. Our in vitro and in vivo findings greatly support the theory that the increased nuclear accumulation but not kinase activity of the EGFR NES mutants plays a predominant role in promoting tumorigenesis and the malignant phenotypes of cancer cells (Table 3).
Table 3.
Summary of the characterizations of EGFR
| Mutation | Nuclearlocalization | Kinase activity | Migration | TKI resistance | Stemness | Tumorigenic potential | Signaling | ||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Ma | Ca | Na | |||||||
| WT | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| L747A | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | +++ | +++ | +++ |
| L747P | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | +++ | +++ | +++ |
| L747S | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | +++ | +++ | +++ |
| L858R | - | +++ | n/a | - | + | + | +++ | +++ | - |
| LL1010-1011AA | +/- | ++ | + | + | + | + | ++ | - | - |
| WT-LL1010-1011AA | +/- | ++ | + | + | +/- | + | ++ | - | - |
| L747A-LL1010-1011AA | + | ++++ | + | + | +/- | + | +++ | - | - |
| L747P-LL1010-1011AA | + | ++++ | + | + | - | + | +++ | - | - |
| L747S-LL1010-1011AA | + | ++++ | + | + | - | + | +++ | - | - |
| WT-NLSm | - | + | n/a | n/a | - | +/- | ++ | ++ | - |
| L747P-NLSm | + | ++++ | n/a | n/a | - | + | +++ | +++ | - |
| L747S-NLSm | + | ++++ | n/a | n/a | - | + | +++ | +++ | - |
M, membrane-bound; C, cytosolic; N, nuclear.
Germline expression of pro-nuclear EGFR NES mutant drives tumorigenesis
Previous studies’ findings and our in vitro, in vivo, and ex vivo findings demonstrated that EGFR function is cellularly compartmentalized [1,100,101]. It is imperative to assess these phenotypes using genetically engineered mouse models. To this end, the CRISPR-Cas9-mediated genome editing approach was used to generate three types of EGFR mutant knock-in mice: internalization-deficient mutation (membrane-bound), pro-nuclear (NES) mutation, and dual mutation (Figure 8A). The oligos of LL1012-1013AA, L749P(S), and their sgRNAs were co-injected with Cas9 mRNA into early-stage embryos using microinjection. Genotyping was performed using DNA sequencing with specific primers and a Western blot analysis. To verify the germline expression of L749P(S) and LL1012-1013AA mutants in F1 offspring, nuclear fraction from primary cultures of hepatocytes from the knock-in and its littermate control mice were used for the Western blot analysis. We found that upon EGF stimulation, nuclear EGFR accumulation increased in EGFRL749P heterozygous mouse hepatocytes; conversely, a significant reduction in nuclear EGFR was observed in EGFTRLL1012-1013AA homozygous mouse hepatocytes (Figure 8B).
Figure 8.
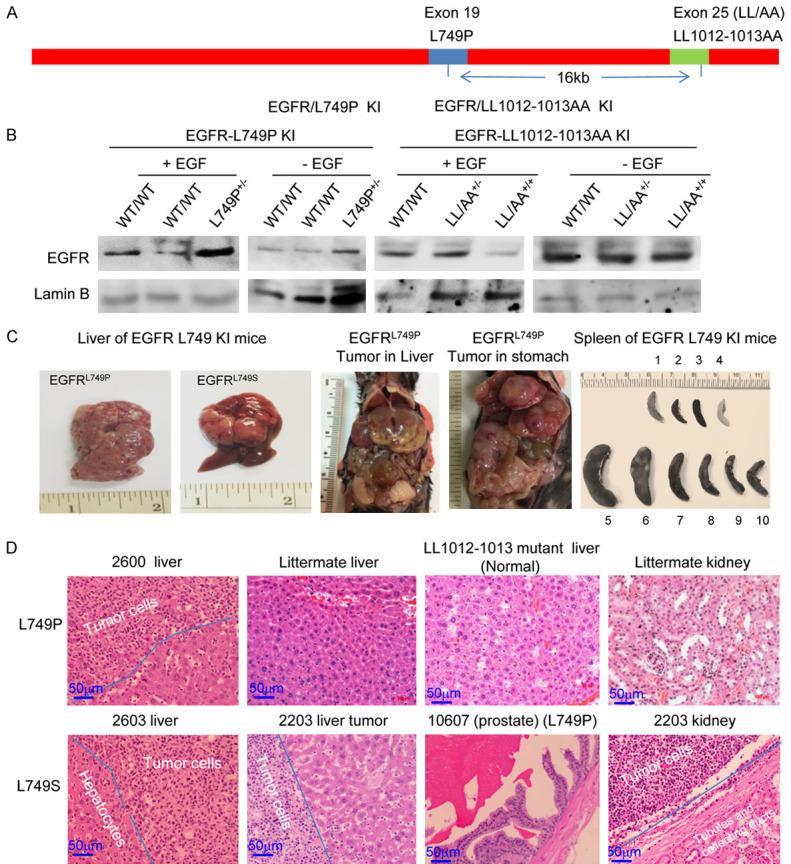
Cells with germline expression of EGFR NES mutant develope B cell lymphoma. A. Diagram of EGFR allele-specific genome editing using a CRISPR/Cas9 editing approach. B. Western blot analysis of primary hepatocytes from EGFR WT and NES knock-in mice. The primary cultures of the hepatocytes were serum-starved and treated with 60 ng/ml EGF for 30 min. Subcellular fractions were immunoblotted with the indicated antibodies. C. Gross phenotypes of EGFR L749P(S) knock-in mice. Representative photos of major abdominal organs. D. H & E staining of major tumor tissues from EGFR NES knock-in mice.
The EGFR knock-in founders were back-crossed with WT C57BL/6 mice to obtain F1 offspring. As shown in Table S3, genotyping results of F1 EGFRL749P(S) knock-in mice indicated that 5.7% of the offspring were heterozygous (EGFRL749/P749 ) and thus did not significantly follow predictable inheritance rules (< 25%) according to Mendel’s principle of segregation, suggesting that germline expression of EGFRL749P(S) gave rise to a deficiency in breeding. In contrast, EGFRLL1012-1013AA mutant mice displayed no gross abnormalities, even in homozygous offspring (Table S4), suggesting that the depletion of nuclear EGFR has no significant phenotype. To generate homozygotes of EGFR NES mutant mice, F1 heterozygous females were crossed with heterozygous male or vice versa. All offspring of these mating pairs were WT. F1 heterozygous EGFRL749 mutant mice crossed with WT C57BL/6 also failed to produce offspring with the genotype of EGFRL749P(S) mutation. Alternatively, the oligos of L749P and their sgRNAs were co-injected with Cas9 mRNA into early-stage embryos of homozygous EGFRLL1012-1013AA mice. The resultant founders also exclusively failed to produce F1 offspring with the L749P and LL1012-1013AA double mutation genotype. These findings suggest that EGFR NES mutant is embryonic lethal or infertile. The causal effect of EGFR L749 mutant on the impaired breeding phenotype in knock-in mice remains unknown.
Of note, the female and male founders and F1 EGFRL749P mice developed tumors in major organs, including the stomach, pancreas, liver, and prostate gland, with no gender difference (Figure 8C and 8D). Compared with littermates, EGFRL749(S) mice had enlarged spleens with numerous white clusters of the tumor colonies, indicating that clonal expansions of lymphoma cells occurred (Figure 8C). H & E staining revealed that along with the normal histologic structures of the examined organ tissues, many infiltrating lymphocytes were present that always formed clusters in the homing organ tissues (Figure 8D). To further identify the subpopulation of lymphocytes, the tumor tissues were stained with specific antibodies against B cell marker CD19. The IHC staining results indicated that the infiltrating lymphocytes were B cells; we did not find tumor cells in blood smears or bone marrow (data not shown), suggesting that the tumor type was B cell lymphoma (Figure S8). Lymphoma cells were found in the stomach, liver, pancreas, and kidneys, but not in the lungs (Figure 8D). The latency of tumor development was 17 to 23 months, and the tumor incidence in F1 offspring of EGFRL749P(S) knock-in mice was 10 of 15 (67%). These findings indicate that a single allele mutation in EGFR NES domain is capable of driving hematopoietic tumorigenesis. In other words, the EGFR NES mutant possesses strong oncogenic activity.
Nuclear EGFR biasedly regulates downstream signaling pathways
It has been reported that STAT3 interacts with nuclear EGFR [29,30,37,38], and therefore we examined the activation of the representative downstream targets, e.g. STAT3 and Erk, in cells expressing EGFRWT and L747 mutants. As shown in Figure 9A, phospho-STAT3 (p-STAT3) in the nuclear extracts from EGFRL747 mutant-expressing cells was much higher than that from EGFRWT-expressing cells. However, the addition of a secondary mutation at LL1010-1011 to L747 mutants resulted in a significant reduction in nuclear p-STAT3 levels compared with a L747 single mutation. By contrast, there were no significantly differences in Erk activation by EGF stimulation, suggesting that different subcellular localizations of EGFR can be attributed to differential regulation of the downstream targeted signaling pathways (Figure 9A). As indicated in Figure 6A, the addition of LL1010-1011AA mutation to EGFRWT and L747 mutants did not significantly change the protein kinase activities. Presumably, the expression of a single or compound EGFR mutation has equal potential to activate STAT3 in the cytoplasm; thus, enhanced nuclear p-STAT3 may specifically result from an increased nuclear interaction between EGFR and STAT3, which stabilizes p-STAT3 (Figure 9B). Taken together, cells expressing the NES mutation of EGFR resulted in subcellular specific regulation of the downstream target signaling pathways; as a result, these cells displayed distinct gene expression profiles (Figure S5 and Table S1).
Figure 9.
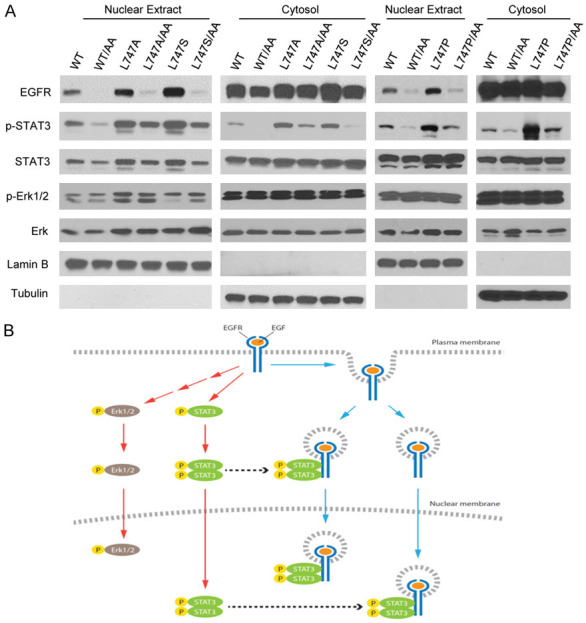
Nuclear EGFR is required to sustain the activated STAT3 in the nucleus. A. Cell fractions from transfected cells with the indicated EGFR and their mutant plasmids were immunoblotted with the indicated antibodies. B. Working model for canonical and non-canonical EGFR signaling. Ligand-triggered EGFR membrane-bound signaling activates Erk and STAT3, and nuclear EGFR is required for sustaining activated STAT3 via EGFR/STAT3 interaction.
Discussion
In this report, we identified a putative NES in the kinase domain (amino acid residue 736-749) of EGFR. The putative NES sequence is highly conserved among ErbB family members and vertebrates. A mutation analysis of the NES sequence of EGFR demonstrated that replacement of L747A resulted in significantly increased nuclear translocation compared with that in EGFRWT. Moreover, the L747A mutant of EGFR is constitutively active, as indicated by the autophosphorylation and activation of the downstream target STAT3 in the absence of EGF. However, the enhanced kinase activity did not contribute to the increased nuclear translocation of EGFR because TKI treatment and KD mutation were not able to significantly to reduce nuclear EGFR. In particular, ectopically expressed EGFR KD was also capable of translocating into the nucleus in response to EGF stimulation. These findings suggest that nuclear accumulation of EGFR is not kinase activity-dependent. We also observed that the EGFRL858R mutant, which is a well-identified TKI-sensitive EGFR mutation, was predominantly localized in the cytoplasm. Taken together, these data suggest that the nuclear translocation of EGFR is not dependent on its tyrosine kinase activity but on ligand-induced dimerization and internalization.
It is known that cell surface EGFR is trafficked to the inner nuclear membrane through the NPC mediated by importin β1 and the membrane-bound EGFR is then released from the inner nuclear membrane to the nucleus [99,102]. Our previous studies characterized tripartite NLS in the EGFR family and provided evidence that the dynamic interaction between EGFR/importin β1 in the cytoplasm and EGFR/CRM1 in the nucleus is ligand- and time-dependent [39,71]. Therefore, the nuclear protein level of EGFR is dependent on the balance between import and export efficiency. Our current data indicate that L747 mutation in the putative NES region did not affect the interaction between the proteins and importin β1 but increased the binding affinity of the mutant proteins to CRM1. Previous reports revealed that most leucine-rich NES sequences bind to CRM1 with relatively low affinity and that high-affinity NES binding to CRM1 impairs the efficient release of CRM1 from the NES export complex at the NPC and cytosol [41,44]. Therefore, L747 mutation of EGFR that leads to increased nuclear retention may result from reduced CRM1 recycling for dynamic EGFR/CRM1 complex assembly.
The identified NES region is found on the surface of the EGFR N-Lobe, located between αC-Helix and the nucleotide phosphate binding loop (P-Loop) [77]. Therefore, replacement of L747 by the amino acid with a different side chain will affect the interaction between the kinase and CRM1. The electron density of residues 721-722 and 747-749 cannot be observed from the crystal structure of the WT EGFR/AMPPNP complex (3VJO) [78], demonstrating its flexible properties in WT EGFR. The flexible loop usually has important functions, such as protein-protein interaction. However, the conformation of these two regions was obtained at the EGFR L858R/AMPPNP complex structure (2ITV) [103]; the crystal structure shows that the side chain carbon atoms of L747 provide an important hydrophobic interaction with F723, K745, and A755 to stabilize the conformations of α-helix C and the P-Loop. K745 is a conserved residue that forms a salt bridge to E762 that interact with the α- and β-phosphates when ATP or a close homologue is present [104]. Therefore, the observed mutations at L747 may affect the side chain orientation of K745 and the conformation of the P-Loop (residues 721-724) and may play a key role in the interaction between EGFR and CRM1 by affecting the local environment of the CRM1 binding site on EGFR, leading to increased binding affinity between EGFR and CRM1.
It is worth mentioning that Gururaj et al. reported another putative NES motif for EGFR, and identified its location in the transmembrane domain at I640 residue [105]. Since EGFR I640 is required for the transmembrane domain dimerization motif, mutation of this amino acid may affect clustering- and ligand-induced endocytosis/internalization of the receptors, leading to altered receptor trafficking [106]. In addition, the hydrophobic motif of the putative NES in the transmembrane region is involved in transmembrane domain dimerization so that the interface for interacting with CRM1 supposedly is occupied after the receptor is activated [39,106]. Further study of the protein-protein interaction between EGFR and CRM1 is required to clarify the detailed interaction.
Interestingly, somatic L747 mutations were found in patients with lung cancer with poor outcomes. L747S mutation was first detected in a patient with EGFRL858R mutation who experienced a prolonged response to single-agent gefitinib before detection of the L747S secondary mutation [59,60]. The L747S-L858R double mutation is more resistant to TKIs, although it is not as strongly resistant as the L790M-L858R double mutation [59]. More recently, a L747S or a L747P single mutation was also detected in patients with TKI-naïve lung cancer [107]. van der Wekken et al. observed stable disease upon EGFR-TKI treatment in a patient with a lung adenocarcinoma that harbored a L747P point mutation in exon 19 [108]. Taken together, the tumors harboring L747S and L747P mutations did not show a response to gefitinib and therefore had a poor prognosis. Our experimental data revealed that all L747 mutants have increased nuclear retention and consequently possessed higher TKI drug-resistant potential, mobility, and invasiveness than did EGFRWT-expressing cells. Furthermore, we provided evidence that reconstituted expression of EGFR L747 mutants, but not L858R and WT, in EGFR-depleted lung and breast cancer cells promoted primary and secondary tumor spheroid formation, suggesting that these cell lines have expanded CSC populations and that EGFR L747 mutant expression is capable of maintaining CSC self-renewal. In contrast, EGFRL858R mutant-expressing cells, which lack of nuclear localization, were very sensitive to TKI; thus, few tumorspheres were observed in the presence of erlotinib. This finding is consistent with clinical observations of patients with lung cancer [59,60]. EGFRL858R mutant affects its kinase confirmation, leading to TKI sensitivity. However, EGFRL747P and EGFRL747S mutation in patients with lung cancer did not change their kinase domain conformation; therefore, L858R mutant is not the best control.
To precisely determine the role of EGFR signaling in the nucleus, we generated membrane-bound EGFR double mutants at L747P(S) and LL1010-1011AA. C-terminal LL1010-1011 (LL1012-1013 in mice) is required for EGFR internalization triggered by ligand. Replacement of LL1010-1011 with AA abolished this internalization; as a result, the mutated protein became a membrane-bound receptor [95,96]. The in vitro experimental data showed that the cells expressing membrane-bound EGFR mutants significantly reduced their nuclear localization and concomitantly attenuated the malignant potential of cancer cells. Importantly, a limiting-dilution transplantation assay revealed that the cancer cells with reconstituted expression of EGFRL747P(S) mutant gave rise to a much higher frequency of tumors than did those with EGFRWT, suggesting that there are more TICs in EGFRL747P(S)-expressing cells. Moreover, the cancer cells expressing the membrane-bound forms of EGFRWT/LL1010-1011AA and EGFRL747P(S)/LL1010-1011AA mutants significantly lost their tumor initiation ability and slowed down tumor growth in NOD/SCID mice in comparison with their parental molecule-expressing cells. These results strongly support the hypothesis that nuclear EGFR signaling is necessary for the maintenance or expansion of the CSC population. Of note, cells expressing EGFRLL1010-1011AA-NESm compound mutants still possessed a higher potential to develop into palpable tumors than did WT cells. This result implies that constitutive activation of canonical membrane-bound EGFR signaling also plays a partial role in driving tumorigenesis.
Previous studies demonstrated that transgenic mouse models with targeted overexpression of human EGFR mutants, including L858R, T790M, DelE746-A750, or double-point mutants of EGFR, developed lung cancers [109-112]. Notably, EGFR was overexpressed in all conventional and inducible transgenic mouse models reported; mutated EGFRs are activating forms that drive the development of tumors with oncogene addition [109-112]. However, hotspot mutations of EGFR gene in cancer patients are frequently heterozygous (Cosmic database, https://cancer.sanger.ac.uk/cosmic) [47]. The results of a previous study revealed that familial lung cancers have concurrent somatic EGFR mutations, along with EGFR T790M or germline mutation [113]; a statistical analysis showed that germline EGFR V843I mutation predispose patients to lung cancer [114]. Here, we demonstrated that one allele mutation at the NES domain confers EGFR oncogenic activity in a genetically engineered mouse model. The phenotypes of the genetically engineered mouse model demonstrated that the EGFR NES mutant is a tumor driver that has a profound effect on reproductive function. Interestingly, by searching the database of EGFR mutations in human tumors (Cosmic database, https://cancer.sanger.ac.uk/cosmic), we found that among the somatic mutations in EGFR gene, gene amplification represents only a small part of the gene alternations in human cancers and that the majority of human B lymphocyte-originated tumors harboring EGFR NES mutation are heterozygotes (Table S5). Therefore, the phenotype of EGFR NES knock-in mice reported here recapitulated human plasma cell myeloma (Table S5). Noticeably, somatic mutations of EGFR at L747 were found in patients with lung cancer; mice with germline expression of this type of EGFR mutant could develop lung cancer. However, no pathological features of lung cancer were observed in EGFR NES knock-in mice until the mice were euthanized because of tumor progression. It is possible that lung cancer development driven by EGFR NES mutant has a longer latency than does lymphoma.
Notably, activation of the downstream signaling pathways by EGFR was subcellular location-specific. We found no significant difference between nuclear translocation-deficient or membrane-bound EGFR and pro-nuclear EGFR mutants in activating canonical EGFR membrane-bound signaling, such as Erk. However, pro-nuclear EGFR L747 single mutants have strong potential to activate and sustain high levels of nuclear STAT3 signaling via EGFR/STAT3 interaction (Figure 9). It has been well-documented that activation of STAT3 signaling is necessary for the proliferation and survival of tumor-initiating cells and drug resistance and becomes a novel molecular target for cancer prevention [115-117]. Our findings demonstrated that pro-nuclear EGFR NES mutants led to the constitutive activation and sustaining of the STAT3 signaling pathway, thereby increasing the tumor-initiating cell population, and conferring more malignant properties to tumor cells. In accord with these phenotypes, the gene expression profiles of EGFRWT-, EGFRL747S-, and EGFRL858R-expressing cell lines in microarray analyses were significantly different and some identified STAT3 target genes were specifically up-regulated in EGFRL747S-expressing cells (Figure S5 and Table S1).
It must be pointed out that the L747S mutated protein had the highest nuclear level among EGFR WT and other L747 mutants, but autophosphorylation of L747S proteins is similar to that of WT proteins, followed by EGF stimulation, suggesting again that the nuclear translocation of EGFR is not correlated with its kinase activity. These data indicate that the altered phenotypes driven by EGFR-mutated proteins were predominantly associated with the nuclear protein levels and that an increased CSC population contributes, at least partially, to TKI resistance and poor prognosis in lung cancer patients harboring EGFRL747P and EGFRL747S mutation [60-69]. In addition, a homologous analysis revealed that mutations within the highly conserved putative NES motif also occur in other kinases, such as ErbB2, ErbB4, ABL1, and BRAF [118-121]. An in silico analysis showed that these mutation regions met the required NES score and that mutations in the NES-like sequences of the above-mentioned well-known kinases are highly associated with tumor progression and drug resistance, indicating that NES mutation of kinases might be critical contributors to tumorigenesis, invasion, de novo or acquired drug resistance development, and cancer recurrence. Whether NES-like mutations in these tyrosine kinases enhance their nuclear localization is unknown and requires further experimentation. Thus, a better understanding of the nuclear roles of tyrosine kinases and kinase receptors will provide new insights into tumorigenesis and tumor-targeted therapies.
Acknowledgements
We acknowledge the editing service of Sutton, Ann M, Research Medical Library, at The University of Texas MD Anderson Cancer Center. This study was supported in part by the following: MD Anderson Startup Fund, NIH/NCI Cancer Center Support Grant (P30 CA016672), The University of Texas MD Anderson Cancer Center-China Medical University Sister Institution Fund, and National Science and Technology Council, Taiwan (NSTC 111-2639-B-039-001-ASP).
Disclosure of conflict of interest
None.
Figures S1-S8 and Tables S2-S5
Table S1
Movie S1A
Movie S1B
Movie S1C
Movie S1D
References
- 1.Chen MK, Hsu JL, Hung MC. Nuclear receptor tyrosine kinase transport and functions in cancer. Adv Cancer Res. 2020;147:59–107. doi: 10.1016/bs.acr.2020.04.010. [DOI] [PubMed] [Google Scholar]
- 2.Hsu JL, Hung MC. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016;35:575–588. doi: 10.1007/s10555-016-9649-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpenter G, Liao HJ. Receptor tyrosine kinases in the nucleus. Cold Spring Harb Perspect Biol. 2013;5:a008979. doi: 10.1101/cshperspect.a008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song S, Rosen KM, Corfas G. Biological function of nuclear receptor tyrosine kinase action. Cold Spring Harb Perspect Biol. 2013;5:a009001. doi: 10.1101/cshperspect.a009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang YN, Hung MC. Nuclear functions and subcellular trafficking mechanisms of the epidermal growth factor receptor family. Cell Biosci. 2012;2:13. doi: 10.1186/2045-3701-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene. 2010;29:3997–4006. doi: 10.1038/onc.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun. 1994;203:1589–1598. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- 8.Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, Jones AL. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology. 1991;13:15–20. [PubMed] [Google Scholar]
- 9.Kamio T, Shigematsu K, Sou H, Kawai K, Tsuchiyama H. Immunohistochemical expression of epidermal growth factor receptors in human adrenocortical carcinoma. Hum Pathol. 1990;21:277–282. doi: 10.1016/0046-8177(90)90227-v. [DOI] [PubMed] [Google Scholar]
- 10.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen MK, Hung MC. Regulation of therapeutic resistance in cancers by receptor tyrosine kinases. Am J Cancer Res. 2016;6:827–842. [PMC free article] [PubMed] [Google Scholar]
- 12.Lee HH, Wang YN, Hung MC. Non-canonical signaling mode of the epidermal growth factor receptor family. Am J Cancer Res. 2015;5:2944–2958. [PMC free article] [PubMed] [Google Scholar]
- 13.Chen MK, Hung MC. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015;282:3693–3721. doi: 10.1111/febs.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brand TM, Iida M, Li C, Wheeler DL. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med. 2011;12:419–432. [PMC free article] [PubMed] [Google Scholar]
- 15.Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005;65:338–348. [PubMed] [Google Scholar]
- 16.Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R, Rimm D, Burtness BA. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res. 2005;11:5856–5862. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- 17.Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, Huo LF, Miller S, Hung MC. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog. 2009;48:610–617. doi: 10.1002/mc.20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burel-Vandenbos F, Turchi L, Benchetrit M, Fontas E, Pedeutour Z, Rigau V, Almairac F, Ambrosetti D, Michiels JF, Virolle T. Cells with intense EGFR staining and a high nuclear to cytoplasmic ratio are specific for infiltrative glioma: a useful marker in neuropathological practice. Neuro Oncol. 2013;15:1278–1288. doi: 10.1093/neuonc/not094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traynor AM, Weigel TL, Oettel KR, Yang DT, Zhang C, Kim K, Salgia R, Iida M, Brand TM, Hoang T, Campbell TC, Hernan HR, Wheeler DL. Nuclear EGFR protein expression predicts poor survival in early stage non-small cell lung cancer. Lung Cancer. 2013;81:138–141. doi: 10.1016/j.lungcan.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saloura V, Vougiouklakis T, Zewde M, Deng X, Kiyotani K, Park JH, Matsuo Y, Lingen M, Suzuki T, Dohmae N, Hamamoto R, Nakamura Y. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci Rep. 2017;7:40664. doi: 10.1038/srep40664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, You L, Xie J, Pan H, Han W. The roles of subcellularly located EGFR in autophagy. Cell Signal. 2017;35:223–230. doi: 10.1016/j.cellsig.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Liu N, Lai W, Yan B, Chen L, Liu S, Liu S, Wang X, Xiao D, Liu X, Mao C, Jiang Y, Jia J, Liu Y, Yang R, Cao Y, Tao Y. Nuclear EGFR-PKM2 axis induces cancer stem cell-like characteristics in irradiation-resistant cells. Cancer Lett. 2018;422:81–93. doi: 10.1016/j.canlet.2018.02.028. [DOI] [PubMed] [Google Scholar]
- 23.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28:3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brand TM, Iida M, Dunn EF, Luthar N, Kostopoulos KT, Corrigan KL, Wleklinski MJ, Yang D, Wisinski KB, Salgia R, Wheeler DL. Nuclear epidermal growth factor receptor is a functional molecular target in triple-negative breast cancer. Mol Cancer Ther. 2014;13:1356–1368. doi: 10.1158/1535-7163.MCT-13-1021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 25.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 26.Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45:10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- 28.Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, Chuang YH, Lai CH, Chang WC. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–4351. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS One. 2011;6:e19605. doi: 10.1371/journal.pone.0019605. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Lo HW, Cao X, Zhu H, Ali-Osman F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res. 2010;8:232–245. doi: 10.1158/1541-7786.MCR-09-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, Chiu PC, Huang WP, Wang YN, Chen CH, Chang WC, Chen AJ, Tsai CH, Hung MC. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem. 2011;286:20558–20568. doi: 10.1074/jbc.M111.240796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huo L, Wang YN, Xia W, Hsu SC, Lai CC, Li LY, Chang WC, Wang Y, Hsu MC, Yu YL, Huang TH, Ding Q, Chen CH, Tsai CH, Hung MC. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc Natl Acad Sci U S A. 2010;107:16125–16130. doi: 10.1073/pnas.1000743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 34.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 35.Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 2005;76:157–161. doi: 10.1016/j.radonc.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 36.Hsu SC, Miller SA, Wang Y, Hung MC. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am J Transl Res. 2009;1:249–258. [PMC free article] [PubMed] [Google Scholar]
- 37.Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, Chen J, Lau J, Knobbe-Thomsen C, Weller M, Jura N, Reifenberger G, Shokat KM, Weiss WA. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell. 2013;24:438–449. doi: 10.1016/j.ccr.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han W, Carpenter RL, Cao X, Lo HW. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation with STAT3. Mol Carcinog. 2013;52:959–969. doi: 10.1002/mc.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem. 2006;98:1570–1583. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 40.Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 2007;17:193–201. doi: 10.1016/j.tcb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Kutay U, Guttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15:121–124. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23:3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu D, Farmer A, Collett G, Grishin NV, Chook YM. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell. 2012;23:3677–3693. doi: 10.1091/mbc.E12-01-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelsma D, Bernad R, Calafat J, Fornerod M. Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J. 2004;23:3643–3652. doi: 10.1038/sj.emboj.7600370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 46.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003;21:2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 47.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 48.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 49.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 51.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee PC, Fang YF, Yamaguchi H, Wang WJ, Chen TC, Hong X, Ke B, Xia W, Wei Y, Zha Z, Wang Y, Kuo HP, Wang CW, Tu CY, Chen CH, Huang WC, Chiang SF, Nie L, Hou J, Chen CT, Huo L, Yang WH, Deng R, Nakai K, Hsu YH, Chang SS, Chiu TJ, Tang J, Zhang R, Wang L, Fang B, Chen T, Wong KK, Hsu JL, Hung MC. Targeting PKCdelta as a therapeutic strategy against heterogeneous mechanisms of EGFR inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell. 2018;34:954–969. e954. doi: 10.1016/j.ccell.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia XL, Chen G. EGFR and KRAS mutations in Chinese patients with adenosquamous carcinoma of the lung. Lung Cancer. 2011;74:396–400. doi: 10.1016/j.lungcan.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 54.Santis G, Angell R, Nickless G, Quinn A, Herbert A, Cane P, Spicer J, Breen R, McLean E, Tobal K. Screening for EGFR and KRAS mutations in endobronchial ultrasound derived transbronchial needle aspirates in non-small cell lung cancer using COLD-PCR. PLoS One. 2011;6:e25191. doi: 10.1371/journal.pone.0025191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tochigi N, Dacic S, Nikiforova M, Cieply KM, Yousem SA. Adenosquamous carcinoma of the lung: a microdissection study of KRAS and EGFR mutational and amplification status in a western patient population. Am J Clin Pathol. 2011;135:783–789. doi: 10.1309/AJCP08IQZAOGYLFL. [DOI] [PubMed] [Google Scholar]
- 56.Wu JY, Yu CJ, Shih JY, Yang CH, Yang PC. Influence of first-line chemotherapy and EGFR mutations on second-line gefitinib in advanced non-small cell lung cancer. Lung Cancer. 2010;67:348–354. doi: 10.1016/j.lungcan.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 57.Pallis AG, Voutsina A, Kalikaki A, Souglakos J, Briasoulis E, Murray S, Koutsopoulos A, Tripaki M, Stathopoulos E, Mavroudis D, Georgoulias V. ‘Classical’ but not ‘other’ mutations of EGFR kinase domain are associated with clinical outcome in gefitinib-treated patients with non-small cell lung cancer. Br J Cancer. 2007;97:1560–1566. doi: 10.1038/sj.bjc.6604068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sunaga N, Tomizawa Y, Yanagitani N, Iijima H, Kaira K, Shimizu K, Tanaka S, Suga T, Hisada T, Ishizuka T, Saito R, Dobashi K, Mori M. Phase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non-small cell lung cancer with EGFR mutations, irrespective of previous chemotherapy. Lung Cancer. 2007;56:383–389. doi: 10.1016/j.lungcan.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 59.Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–1679. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costa DB, Nguyen KS, Cho BC, Sequist LV, Jackman DM, Riely GJ, Yeap BY, Halmos B, Kim JH, Janne PA, Huberman MS, Pao W, Tenen DG, Kobayashi S. Effects of erlotinib in EGFR mutated non-small cell lung cancers with resistance to gefitinib. Clin Cancer Res. 2008;14:7060–7067. doi: 10.1158/1078-0432.CCR-08-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu G, Xie X, Sun D, Geng J, Fu F, Zhang L, Wang H. EGFR mutation L747P led to gefitinib resistance and accelerated liver metastases in a Chinese patient with lung adenocarcinoma. Int J Clin Exp Pathol. 2015;8:8603–8606. [PMC free article] [PubMed] [Google Scholar]
- 62.Huang J, Wang Y, Zhai Y, Wang J. Non-small cell lung cancer harboring a rare EGFR L747P mutation showing intrinsic resistance to both gefitinib and osimertinib (AZD9291): a case report. Thorac Cancer. 2018;9:745–749. doi: 10.1111/1759-7714.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang SK, Ko JC, Yang JC, Shih JY. Afatinib is effective in the treatment of lung adenocarcinoma with uncommon EGFR p.L747P and p.L747S mutations. Lung Cancer. 2019;133:103–109. doi: 10.1016/j.lungcan.2019.05.019. [DOI] [PubMed] [Google Scholar]
- 64.Zhou T, Zhou X, Li P, Qi C, Ling Y. EGFR L747P mutation in one lung adenocarcinoma patient responded to afatinib treatment: a case report. J Thorac Dis. 2018;10:E802–E805. doi: 10.21037/jtd.2018.12.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seki Y, Fujiwara Y, Kohno T, Takai E, Sunami K, Goto Y, Horinouchi H, Kanda S, Nokihara H, Watanabe S, Ichikawa H, Yamamoto N, Kuwano K, Ohe Y. Picoliter-droplet digital polymerase chain reaction-based analysis of cell-free plasma DNA to assess EGFR mutations in lung adenocarcinoma that confer resistance to tyrosine-kinase inhibitors. Oncologist. 2016;21:156–164. doi: 10.1634/theoncologist.2015-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He Q, Shi X, Zhu H, Yan J, Yu XM. A case treated with crizotinib after secondary MET amplification of A double rare L747S and G719S EGFR mutation pulmonary sarcomatoid carcinoma. Ann Oncol. 2020;31:544–546. doi: 10.1016/j.annonc.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 67.Grolleau E, Haddad V, Boissiere L, Falchero L, Arpin D. Clinical efficacy of osimertinib in a patient presenting a double EGFR L747S and G719C mutation. J Thorac Oncol. 2019;14:e151–e153. doi: 10.1016/j.jtho.2019.02.034. [DOI] [PubMed] [Google Scholar]
- 68.Swami U, Ma D, Zhang J. Response to Erlotinib in a patient with compound EGFR L747S and Exon 19 deletion. J Thorac Oncol. 2018;13:e129–e130. doi: 10.1016/j.jtho.2018.03.034. [DOI] [PubMed] [Google Scholar]
- 69.Wang YT, Ning WW, Li J, Huang JA. Exon 19 L747P mutation presented as a primary resistance to EGFR-TKI: a case report. J Thorac Dis. 2016;8:E542–546. doi: 10.21037/jtd.2016.05.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011;71:1103–1114. doi: 10.1158/0008-5472.CAN-10-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007;282:10432–10440. doi: 10.1074/jbc.M610014200. [DOI] [PubMed] [Google Scholar]
- 72.Wang YN, Huo L, Hsu JL, Hung MC. Biochemical fractionation of membrane receptors in the nucleus. Methods Mol Biol. 2015;1234:99–112. doi: 10.1007/978-1-4939-1755-6_9. [DOI] [PubMed] [Google Scholar]
- 73.Chen CC, Hwang JK, Yang JM. (PS)2: protein structure prediction server. Nucleic Acids Res. 2006;34:W152–157. doi: 10.1093/nar/gkl187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeLano WL. The PyMOL molecular graphics system. Online Database. 2002 [Google Scholar]
- 75.Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics. 2006;7:339. doi: 10.1186/1471-2105-7-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langhans SA. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front Pharmacol. 2018;9:6. doi: 10.3389/fphar.2018.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008;26:1742–1751. doi: 10.1200/JCO.2007.12.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshikawa S, Kukimoto-Niino M, Parker L, Handa N, Terada T, Fujimoto T, Terazawa Y, Wakiyama M, Sato M, Sano S, Kobayashi T, Tanaka T, Chen L, Liu ZJ, Wang BC, Shirouzu M, Kawa S, Semba K, Yamamoto T, Yokoyama S. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene. 2013;32:27–38. doi: 10.1038/onc.2012.21. [DOI] [PubMed] [Google Scholar]
- 79.Liu B, Bernard B, Wu JH. Impact of EGFR point mutations on the sensitivity to gefitinib: insights from comparative structural analyses and molecular dynamics simulations. Proteins. 2006;65:331–346. doi: 10.1002/prot.21111. [DOI] [PubMed] [Google Scholar]
- 80.Ghosh G, Lian X, Kron SJ, Palecek SP. Properties of resistant cells generated from lung cancer cell lines treated with EGFR inhibitors. BMC Cancer. 2012;12:95. doi: 10.1186/1471-2407-12-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang CP, Tsai MF, Chang TH, Tang WC, Chen SY, Lai HH, Lin TY, Yang JC, Yang PC, Shih JY, Lin SB. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013;328:144–151. doi: 10.1016/j.canlet.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 82.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–935. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 83.Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, Fink LM, Ma Y, Wong MP. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5:e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Karimi-Busheri F, Zadorozhny V, Li T, Lin H, Shawler DL, Fakhrai H. Pivotal role of CD38 biomarker in combination with CD24, EpCAM, and ALDH for identification of H460 derived lung cancer stem cells. J Stem Cells. 2011;6:9–20. [PubMed] [Google Scholar]
- 85.van der Gun BT, Melchers LJ, Ruiters MH, de Leij LF, McLaughlin PM, Rots MG. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis. 2010;31:1913–1921. doi: 10.1093/carcin/bgq187. [DOI] [PubMed] [Google Scholar]
- 86.Steinbach D, Gillet JP, Sauerbrey A, Gruhn B, Dawczynski K, Bertholet V, de Longueville F, Zintl F, Remacle J, Efferth T. ABCA3 as a possible cause of drug resistance in childhood acute myeloid leukemia. Clin Cancer Res. 2006;12:4357–4363. doi: 10.1158/1078-0432.CCR-05-2587. [DOI] [PubMed] [Google Scholar]
- 87.Efferth T, Gillet JP, Sauerbrey A, Zintl F, Bertholet V, de Longueville F, Remacle J, Steinbach D. Expression profiling of ATP-binding cassette transporters in childhood T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2006;5:1986–1994. doi: 10.1158/1535-7163.MCT-06-0086. [DOI] [PubMed] [Google Scholar]
- 88.Chapuy B, Koch R, Radunski U, Corsham S, Cheong N, Inagaki N, Ban N, Wenzel D, Reinhardt D, Zapf A, Schweyer S, Kosari F, Klapper W, Truemper L, Wulf GG. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia. 2008;22:1576–1586. doi: 10.1038/leu.2008.103. [DOI] [PubMed] [Google Scholar]
- 89.Overbeck TR, Arnemann J, Waldmann-Beushausen R, Trumper L, Schondube FA, Reuter-Jessen K, Danner BC. ABCA3 phenotype in non-small cell lung cancer indicates poor outcome. Oncology. 2017;93:270–278. doi: 10.1159/000477619. [DOI] [PubMed] [Google Scholar]
- 90.Song W, Wang J, Liu H, Zhu C, Xu F, Qian L, Shen Z, Zhu J, Yin S, Qin J, Chen L, Wu D, Nashan B, Shan G, Xiao W, Zhou Y. Effects of LncRNA Lnc-LIF-AS on cell proliferation, migration and invasion in a human cervical cancer cell line. Cytokine. 2019;120:165–175. doi: 10.1016/j.cyto.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 91.Qian L, Xu F, Wang X, Jiang M, Wang J, Song W, Wu D, Shen Z, Feng D, Ling B, Cheng Y, Xiao W, Shan G, Zhou Y. LncRNA expression profile of DeltaNp63alpha in cervical squamous cancers and its suppressive effects on LIF expression. Cytokine. 2017;96:114–122. doi: 10.1016/j.cyto.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 92.Niwa H, Ogawa K, Shimosato D, Adachi K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- 93.Onishi K, Zandstra PW. LIF signaling in stem cells and development. Development. 2015;142:2230–2236. doi: 10.1242/dev.117598. [DOI] [PubMed] [Google Scholar]
- 94.Nicola NA, Babon JJ. Leukemia inhibitory factor (LIF) Cytokine Growth Factor Rev. 2015;26:533–544. doi: 10.1016/j.cytogfr.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Q, Zhu F, Wang Z. Identification of EGF receptor C-terminal sequences 1005-1017 and di-leucine motif 1010LL1011 as essential in EGF receptor endocytosis. Exp Cell Res. 2007;313:3349–3363. doi: 10.1016/j.yexcr.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 96.Wu P, Wee P, Jiang J, Chen X, Wang Z. Differential regulation of transcription factors by location-specific EGF receptor signaling via a spatio-temporal interplay of ERK activation. PLoS One. 2012;7:e41354. doi: 10.1371/journal.pone.0041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, Gray JW, Bissell MJ. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Harma V, Virtanen J, Makela R, Happonen A, Mpindi JP, Knuuttila M, Kohonen P, Lotjonen J, Kallioniemi O, Nees M. A comprehensive panel of three-dimensional models for studies of prostate cancer growth, invasion and drug responses. PLoS One. 2010;5:e10431. doi: 10.1371/journal.pone.0010431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, Wang H, Hsu JM, Hung MC. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem. 2010;285:38720–38729. doi: 10.1074/jbc.M110.158659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 102.Wang YN, Lee HH, Lee HJ, Du Y, Yamaguchi H, Hung MC. Membrane-bound trafficking regulates nuclear transport of integral epidermal growth factor receptor (EGFR) and ErbB-2. J Biol Chem. 2012;287:16869–16879. doi: 10.1074/jbc.M111.314799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 105.Gururaj AE, Gibson L, Panchabhai S, Bai M, Manyam G, Lu Y, Latha K, Rojas ML, Hwang Y, Liang S, Bogler O. Access to the nucleus and functional association with c-Myc is required for the full oncogenic potential of DeltaEGFR/EGFRvIII. J Biol Chem. 2013;288:3428–3438. doi: 10.1074/jbc.M112.399352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heukers R, Vermeulen JF, Fereidouni F, Bader AN, Voortman J, Roovers RC, Gerritsen HC, van Bergen En Henegouwen PM. Endocytosis of EGFR requires its kinase activity and N-terminal transmembrane dimerization motif. J Cell Sci. 2013;126:4900–4912. doi: 10.1242/jcs.128611. [DOI] [PubMed] [Google Scholar]
- 107.Yamaguchi F, Kugawa S, Tateno H, Kokubu F, Fukuchi K. Analysis of EGFR, KRAS and P53 mutations in lung cancer using cells in the curette lavage fluid obtained by bronchoscopy. Lung Cancer. 2012;78:201–206. doi: 10.1016/j.lungcan.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 108.van der Wekken AJ, Stigt JA, A’T Hart N. A novel EGFR mutation in exon 19 showed stable disease after TKI treatment. J Thorac Oncol. 2012;7:e8. doi: 10.1097/JTO.0b013e31825ccae8. [DOI] [PubMed] [Google Scholar]
- 109.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, Koutcher JA, Solit DB, Rosen N, Zakowski MF, Pao W. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One. 2007;2:e810. doi: 10.1371/journal.pone.0000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ohashi K, Rai K, Fujiwara Y, Osawa M, Hirano S, Takata K, Kondo E, Yoshino T, Takata M, Tanimoto M, Kiura K. Induction of lung adenocarcinoma in transgenic mice expressing activated EGFR driven by the SP-C promoter. Cancer Sci. 2008;99:1747–1753. doi: 10.1111/j.1349-7006.2008.00875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, Mahmood U, Mitchell A, Sun Y, Al-Hashem R, Chirieac LR, Padera R, Bronson RT, Kim W, Janne PA, Shapiro GI, Tenen D, Johnson BE, Weissleder R, Sharpless NE, Wong KK. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 113.Yamamoto H, Yatabe Y, Toyooka S. Inherited lung cancer syndromes targeting never smokers. Transl Lung Cancer Res. 2018;7:498–504. doi: 10.21037/tlcr.2018.06.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ikeda K, Nomori H, Mori T, Sasaki J, Kobayashi T. Novel germline mutation: EGFR V843I in patient with multiple lung adenocarcinomas and family members with lung cancer. Ann Thorac Surg. 2008;85:1430–1432. doi: 10.1016/j.athoracsur.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 115.Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, Lin J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011;71:7226–7237. doi: 10.1158/0008-5472.CAN-10-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA, Polyak K. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers (Basel) 2014;6:926–957. doi: 10.3390/cancers6020926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, Barthorpe A, Blow M, Brackenbury L, Butler A, Clarke O, Cole J, Dicks E, Dike A, Drozd A, Edwards K, Forbes S, Foster R, Gray K, Greenman C, Halliday K, Hills K, Kosmidou V, Lugg R, Menzies A, Perry J, Petty R, Raine K, Ratford L, Shepherd R, Small A, Stephens Y, Tofts C, Varian J, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Knowles M, Leung SY, Louis DN, Looijenga LH, Malkowicz B, Pierotti MA, Teh B, Chenevix-Trench G, Weber BL, Yuen ST, Harris G, Goldstraw P, Nicholson AG, Futreal PA, Wooster R, Stratton MR. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 119.Soung YH, Lee JW, Kim SY, Wang YP, Jo KH, Moon SW, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH. Somatic mutations of the ERBB4 kinase domain in human cancers. Int J Cancer. 2006;118:1426–1429. doi: 10.1002/ijc.21507. [DOI] [PubMed] [Google Scholar]
- 120.Testoni E, Stephenson NL, Torres-Ayuso P, Marusiak AA, Trotter EW, Hudson A, Hodgkinson CL, Morrow CJ, Dive C, Brognard J. Somatically mutated ABL1 is an actionable and essential NSCLC survival gene. EMBO Mol Med. 2016;8:105–116. doi: 10.15252/emmm.201505456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.