

Summary
Cancer is a leading cause of mortality worldwide. Over 50% of cancers are diagnosed late, rendering many treatments ineffective. Existing liquid biopsy studies demonstrate a minimally invasive and inexpensive approach for disease detection but lack parsimonious biomarker selection, exhibit poor cancer detection performance and lack appropriate validation and testing. We established a tailored machine learning pipeline, DEcancer, for liquid biopsy analysis that addresses these limitations and improved performance. In a test set from a published cohort of 1,005 patients including 8 cancer types and 812 cancer-free individuals, DEcancer increased stage 1 cancer detection sensitivity across cancer types from 48 to 90%. In addition, with a test set cohort of patients from a high dimensional proteomics dataset of 61 lung cancer patients and 80 cancer-free individuals, DEcancer’s performance using a 14-43 protein panel was comparable to 1,000 original proteins. DEcancer is a promising tool which may facilitate improved cancer detection and management.
Subject areas: Diagnostics, Cancer, Machine learning
Graphical abstract
Highlights
-
•
DEcancer multi-cancer detection over double sensitivity of existing cited approaches
-
•
DEcancer successfully identifies distinct cancer types
-
•
DEcancer selects a succinct panel of biomarkers indicative of cancer type, including lung
-
•
DEcancer provides a foundation for clinical blood-based tests for cancer detection
Diagnostics; Cancer; Machine learning
Introduction
Cancer is among the leading cause of mortality worldwide, accounting for 10 million deaths per annum.1 Early detection through screening and monitoring of the most common cancers such as breast, colorectal, and prostate cancers have significantly helped to reduce mortality and enabled treatments with curative intent.2 However, many cancers such as lung and liver cancer that traditionally lacked effective early detection approaches remain among malignancies that account for the highest proportion of cancer related deaths globally.1 For example, lung cancer, though not the most prevalent, is the leading cause of cancer mortality worldwide in men and second in women.1,3 Unfortunately, the majority of lung cancers are diagnosed at a late stage because most patients remain asymptomatic until late disease, and only 30% are diagnosed at stage 1.3 The five-year survival rate for stage 1 lung cancer is 65% and drastically decreases to 5% by stage 4.3 Therefore, late detection is a main driving factor for lung malignancies that account for the highest cancer mortality. In contrast, breast cancer being the most common cancer in women, has an overall five-year survival rate of 90% because the majority of breast cancers are detected at an early localized stage.4 The wide adoption of mammography for screening has enabled a paradigm shift in the early detection of breast cancer, and hence improved patient survival and outcome.5 As with most cancers, there is a direct correlation between early detection and improved survival and outcome. Therefore, early detection and scalable screening approaches are critical to improving survival in cancers that continue to have a high mortality rate.
Liquid biopsy using human blood samples has recently been shown to be a promising modality for early cancer detection and screening. Cohen et al. reported a multi-analyte test approach called CancerSEEK in 2018 that can detect multiple cancers using blood samples by evaluating mutations in circulating tumor DNA (ctDNA) and protein biomarkers.6 Although CancerSEEK’s detection sensitivity and localization accuracy were promising in less prevalent cancers such as ovarian cancer, or cancers with plausible screening recommendations for early detection such as breast cancer, its performance was limited for cancers with high mortality that lack effective screening tests. For instance, CancerSEEK detects stage 1 and 2 lung cancer with only 48% and 63% sensitivity, respectively.6 In 2020, Blume et al. reported an increased depth in proteomic profiling of non-small cell lung cancer (NSCLC) patient blood samples with the integration of nanoparticles (NPs) protein corona with LC-MS/MS.7 In this study, the early lung cancer detection achieved an area under the receiver operating characteristic curve (AUC) of 0.91 with no definitive panel of biomarkers for early detection.7 However, such detection capacity is limited by the physiochemical properties of each specific nanoparticle used and its ability to recapitulate an unbiased representation of the endogenous blood proteome remains to be assessed.
Various machine learning approaches can be applied to perform robust feature selection from liquid biopsy analytes and achieve a high sensitivity and specificity of cancer detection. For example, Wong et al.8 employed machine learning approaches including deep learning, decision trees, naive Bayes and averaged one-dependence estimators and achieved a sensitivity of 77% for stage 1 cancers from the Cohen et al. dataset, compared to 48% in the original report. However, feature selection was not conducted in this study8 and the reports described previously6,7,8 did not assess performance of respective chosen algorithms on a hold-out test set separate from cross-validation. As a result, it is possible that the reported cancer detection sensitivity and specificity is over-optimistic in these studies. The objective of our study is to identify the most parsimonious set of features which enable optimal cancer detection performance and establish a pipeline that can be robustly applied to various liquid biopsy datasets.
We have developed a machine learning pipeline called DEcancer which uses a novel approach of data augmentation and feature selection methods with a variety of machine learning classifier models to achieve feature selection and high cancer detection performance. We apply our approach to the Cohen et al.6 and the Blume et al.7 datasets to demonstrate strong performance of our cancer detection algorithms for different cancer types and in the face of feature selection from high dimensional data, respectively. We demonstrate the superior performance of our algorithm using a rigorous hold-out test set approach.
Results
Multicancer detection
We developed a data analytics pipeline, DEcancer, tailored for robust analysis of liquid biopsy samples and detect multiple cancer types. We applied DEcancer to a 1,817 patient dataset from Cohen et al. Stage 1 and 2 cancers together accounted for the majority of the patient cohort in all cases apart from ovarian cancer. The calculated summary demographics for these patients is shown in Table 1.
Table 1.
Demographics of the 1,817 individuals including patients with one of 8 cancer locations and cancer-free individuals
| Cancer-free (n = 812) | Lung (n = 104) | Breast (n = 209) | Colorectum (n = 388) | Esophagus (n = 45) | Liver (n = 44) | Ovary (n = 54) | Pancreas (n = 93) | Stomach (n = 68) | |
|---|---|---|---|---|---|---|---|---|---|
| Number of males, n (%) | 434 (53) | 63 (61) | 5 (2) | 212 (55) | 40 (89) | 34 (77) | 0 (0) | 55 (59) | 53 (78) |
| Mean (standard deviation) age, years | 49 (19) | 67 (9) | 58 (13) | 64 (13) | 60 (11) | 61 (11) | 61 (10) | 66 (9) | 63 (12) |
| Number of each stage of cancer, n (%) | |||||||||
| Stage 1 | N/A | 46 (44) | 32 (15) | 77 (20) | 5 (11) | 5 (11) | 9 (17) | 4 (4) | 21 (31) |
| Stage 2 | N/A | 27 (26) | 114 (55) | 191 (49) | 29 (64) | 19 (43) | 4 (7) | 83 (89) | 30 (44) |
| Stage 3 | N/A | 31 (30) | 63 (30) | 120 (31) | 11 (24) | 20 (45) | 41 (76) | 6 (6) | 17 (25) |
| Number of each ethnicity, n (%) | |||||||||
| Asian | 22 (3) | 5 (0) | 69 (33) | 124 (32) | 32 (71) | 26 (59) | 2 (4) | 2 (2) | 41 (60) |
| Black | 154 (19) | 0 (0) | 0 (0) | 5 (1) | 1 (0) | 0 (0) | 6 (11) | 2 (2) | 0 (0) |
| Caucasian | 332 (41) | 96 (92) | 139 (67) | 257 (66) | 12 (27) | 17 (39) | 42 (78) | 86 (92) | 26 (38) |
| Hispanic | 76 (9) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 0 (0) |
| Other | 228 (28) | 3 (3) | 1 (0) | 2 (1) | 0 (0) | 1 (2) | 3 (6) | 3 (3) | 1 (1) |
| Number of each molecular subtype, n (%) | |||||||||
| Not applicable | N/A | 34 (33) | 93 (44) | 52 (13) | 9 (20) | 20 (45) | 47 (87) | 71 (76) | 21 (31) |
| AKT1 | N/A | 0 (0) | 7 (3) | 1 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| APC | N/A | 0 (0) | 0 (0) | 15 (4) | 1 (2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| BRAF | N/A | 1 (1) | 0 (0) | 12 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| CDKN2A | N/A | 3 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (2) | 0 (0) |
| CTNNB1 | N/A | 0 (0) | 0 (0) | 8 (2) | 0 (0) | 5 (11) | 2 (4) | 0 (0) | 1 (1) |
| EGFR | N/A | 3 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| FBXW7 | N/A | 0 (0) | 0 (0) | 9 (2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| GNAS | N/A | 0 (0) | 0 (0) | 2 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| HRAS | N/A | 1 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| KRA | N/A | 18 (17) | 0 (0) | 63 (16) | 0 (0) | 2 (5) | 0 (0) | 9 (10) | 2 (3) |
| NRAS | N/A | 0 (0) | 0 (0) | 3 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| PIK3CA | N/A | 2 (2) | 44 (21) | 15 (4) | 0 (0) | 1 (2) | 0 (0) | 0 (0) | 5 (7) |
| PPP2R1A | N/A | 0 (0) | 0 (0) | 3 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1) |
| PTEN | N/A | 0 (0) | 3 (1) | 3 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| TP53 | N/A | 42 (40) | 62 (30) | 202 (52) | 35 (78) | 16 (36) | 5 (9) | 11 (12) | 38 (56) |
We first trained DEcancer to distinguish between cancer-free individuals and those with cancer, irrespective of cancer type. We sought to develop both a full variable pipeline using proteins, DNA and epidemiology (DEcancerPDE), as well as a protein-only pipeline (DEcancerP). The best DEcancerPDE classifier type was random forest with an AUC of 1.00 (Figure S1). DEcancerP selected 28 proteins and the best classifier type was again random forest with an AUC of 0.99. Next, we evaluated the test performance of the ‘all cancer’ versus ‘cancer-free’ DEcancer pipeline. 364 individuals were included in the test set (randomly selected from the 1,817 individuals to reduce bias), of whom 201 patients had cancer. Characteristics of the test set individuals are similar to the characteristics of the overall 1,817-patient Cohen et al. dataset (Table 2).
Table 2.
Demographics of the 364 test set individuals (according to cancer type and stage) from the Cohen et al. dataset to assess all cancer versus cancer-free using the DEcancer pipeline
| Cancer-free (n = 163) | Lung (n = 20) | Breast (n = 42) | Colorectum (n = 78) | Esophagus (n = 9) | Liver (n = 9) | Ovary (n = 11) | Pancreas (n = 19) | Stomach (n = 13) | |
|---|---|---|---|---|---|---|---|---|---|
| Number of males, n (%) | 80 (49) | 15 (75) | 1 (2) | 38 (49) | 8 (1) | 5 (56) | 0 (0) | 14 (74) | 13 (100) |
| Mean (standard deviation) age, years | 49 (21) | 66 (9) | 60 (13) | 62 (14) | 54 (12) | 56 (15) | 67 (9) | 67 (9) | 69 (13) |
| Number of each stage of cancer, n (%) | |||||||||
| Stage 1 | N/A | 9 (45) | 6 (14) | 16 (21) | 1 (11) | 1 (11) | 2 (18) | 1 (5) | 4 (21) |
| Stage 2 | N/A | 5 (25) | 23 (55) | 38 (49) | 6 (67) | 4 (44) | 1 (9) | 17 (89) | 6 (46) |
| Stage 3 | N/A | 6 (30) | 13 (31) | 24 (31) | 2 (22) | 4 (44) | 8 (73) | 1 (5) | 3 (23) |
| Number of each ethnicity, n (%) | |||||||||
| Asian | 3 (2) | 1 (5) | 15 (36) | 29 (37) | 7 (78) | 6 (67) | 1 (9) | 1 (5) | 5 (38) |
| Black | 34 (21) | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 1 (9) | 0 (0) | 0 (0) |
| Caucasian | 70 (43) | 19 (95) | 27 (64) | 47 (60) | 2 (22) | 2 (22) | 8 (73) | 18 (95) | 7 (54) |
| Hispanic | 13 (8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Other | 43 (26) | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 1 (11) | 1 (9) | 0 (0) | 1 (8) |
| Number of each molecular subtype, n (%) | |||||||||
| Not applicable | N/A | 6 (30) | 19 (45) | 0 (0) | 1 (11) | 6 (67) | 10 (91) | 14 (74) | 5 (38) |
| AKT1 | N/A | 0 (0) | 2 (5) | 6 (8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| APC | N/A | 0 (0) | 0 (0) | 5 (6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| BRAF | N/A | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| CTNNB1 | N/A | 0 (0) | 0 (0) | 2 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| FBXW7 | N/A | 0 (0) | 0 (0) | 1 (1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| GNAS | N/A | 0 (0) | 0 (0) | 9 (12) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| KRAS | N/A | 4 (20) | 0 (0) | 7 (9) | 0 (0) | 1 (11) | 0 (0) | 2 (11) | 0 (0) |
| PIK3CA | N/A | 0 (0) | 8 (19) | 3 (4) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| PTEN | N/A | 0 (0) | 0 (0) | 2 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| TP53 | N/A | 10 (5) | 13 (31) | 42 (54) | 8 (89) | 2 (22) | 1 (9) | 3 (16) | 8 (62) |
The test includes patients with one of 8 cancer locations and cancer-free individuals.
The test set performance of the ‘all cancer’ versus ‘cancer-free’ DEcancer pipeline represented a generalizable estimate of the ‘all cancer’ versus ‘cancer-free’ DEcancer pipeline’s expected performance for unseen individuals. The ‘all cancer’ versus ‘cancer-free’ DEcancerPDE pipeline achieved a test set AUC of 1.00 (Figure 1). With a fixed specificity of 99%, the overall sensitivity for detecting cancer was 99%. The ‘all cancer’ versus ‘cancer-free’ DEcancerP pipeline also achieved a test set AUC of 1.00 (Figure 1). In this case, with a fixed specificity of 99%, the overall sensitivity for detecting cancer was 93%.
Figure 1.

Test set receiver operating characteristic (ROC) curve and area under the curve (AUC) showing the performance of the optimized classifier model for distinguishing 201 cancer patients from 163 cancer-free individuals of the Cohen et al. dataset
DEcancerP uses proteins-only and DEcancerPDE includes all 39 proteins, DNA-based and epidemiology factors. (Blue) The DEcancerPDE approach for all cancer versus cancer-free test set ROC curve showing performance of optimal model. An AUC of 1.00 is achieved. At a fixed specificity of 99%, DEcancer achieves a sensitivity of 99%. (Orange) The 28-protein model uses the DEcancerP approach for all cancer versus cancer-free test set ROC curve. An AUC of 1.00 is achieved. At a fixed specificity of 99%, DEcancer achieves a sensitivity of 93%.
An important consideration is the sensitivity of detection which can be achieved for each stage of cancer and for different cancer types. We computed the test set sensitivity achieved by the ‘all cancer’ versus ‘cancer-free’ DEcancer pipeline according to stage and cancer type for an overall specificity threshold of 99% (Tables S1 and S2).
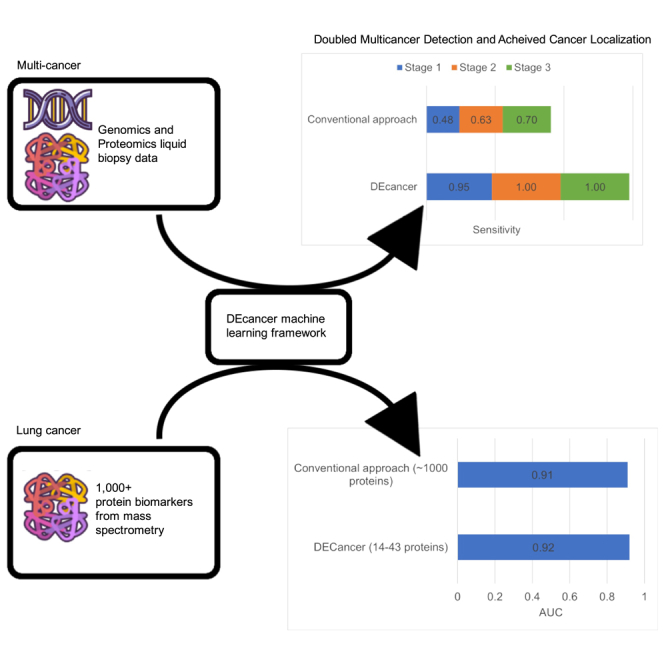
The ‘all cancer’ versus ‘cancer-free’ DEcancerPDE pipeline showed the overall sensitivity of 95%, 100% and 100% for stage 1, 2 and 3 cancers, respectively. DEcancerP model achieved slightly lower sensitivity of 90%, 94% and 95% for stage 1, 2 and 3 cancers, respectively, at a specificity fixed at 99%. DEcancer’s performance exceeded the Cohen et al. results (Table S3) where sensitivity for detecting stage 1, 2 and 3 cancers was 48%, 63% and 70%, respectively, with a minimum specificity of 99%. Notably, across all stages the ‘all cancer’ versus ‘cancer-free’ DEcancerP pipeline achieved a sensitivity of 100% for lung, 93% for breast and 97% for colorectal cancer compared to Cohen et al.’s result of 59%, 33% and 65%, respectively. DEcancer’s superior performance was especially apparent for early stage cancers, with DEcancerP achieving a sensitivity of 100%, 83%, 100% and 100% respectively for detecting stage 1 lung, breast, colorectal, and esophageal cancer compared to Cohen et al.’s result of 43%, 38%, 43% and 20%, respectively (Figure 2).
Figure 2.

Bar chart comparing the performance of DEcancerPDE using protein, DNA and epidemiological data and DEcancerP using proteins alone to Cohen et al.’s cancer detection sensitivity for stage 1, 2 and 3 of 8 cancer types
Specificity was held at 99%.
Feature selection for individual cancers
As a next step following the ‘all cancer’ analysis, we performed feature selection and assessed performance for each of lung, breast, colorectal, esphageal, liver, ovarian, pancreas and gastric cancer individually. For most cancers and classification tasks, random forest models performed best overall in the Monte Carlo cross validation (Figures S2–S9). The test set AUC for DEcancer distinguishing between a particular cancer type and cancer-free individuals ranged from 1.00 for lung cancer (both DEcancerPDE or DEcancerP using just a selected subset of 12 proteins) to 0.97 (DEcancerPDE) or 0.93 (DEcancerP using 17 proteins) for gastric cancer (Figures 3A and 3H). For distinguishing a particular cancer from other cancers or cancer-free individuals, DEcancer’s AUC ranged from 0.99 (DEcancerPDE) or 0.98 (DEcancerP using a selected subset of 12 proteins) for ovarian cancer to 0.83 (DEcancerPDE) or 0.81 (DEcancerP using a selected subset of 15 proteins) for esophageal cancer (Figures 3F and 3D).
Figure 3.

Test set receiver operating characteristic (ROC) curve and area under the curve (AUC) showing the performance of the best optimized classifier model (selected based on validation results) for detecting a target cancer from the Cohen et al. dataset
The test set results represent a generalizable estimate of performance for DEcancer’s all cancer pipeline. DEcancerP uses proteins-only and DEcancerPDE includes all 39 proteins, DNA-based and epidemiology factors. (Blue) The DEcancerPDE approach for target cancer versus cancer-free test set ROC curve showing performance of optimal model. (Orange) The DEcancerP approach for the target cancer versus cancer-free test set ROC curve. (Green) The DEcancerPDE approach for the target cancer versus other cancers test set ROC curve showing performance of optimal model. (Red) The DEcancerP approach for the target cancer versus other cancers test set ROC curve. (Purple) The DEcancerPDE approach for the target cancer versus other cancers or cancer-free test set ROC curve showing performance of optimal model. (Brown) The DEcancerP approach for the target cancer versus other cancers or cancer-free test set ROC curve.
(A) Target cancer: lung. (Blue) An AUC of 1.00 is achieved with the DEcancerPDE model. (Orange) An AUC of 1.00 is achieved with a 12-protein DEcancerP model. (Green) An AUC of 0.95 is achieved with the DEcancerPDE model. (Red) An AUC of 0.95 is achieved with a 39-protein DEcancerP model. (Purple) An AUC of 0.96 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.95 is achieved with a 22-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 95.24 and 99.39%, respectively.
(B) Target cancer: breast. (Blue) An AUC of 1.00 is achieved with the DEcancerPDE model. (Orange) An AUC of 1.00 is achieved with a 27-protein DEcancerP model. (Green) An AUC of 0.93 is achieved with a DEcancerPDE model. (Red) An AUC of 0.88 is achieved with a 29-protein DEcancerP model. (Purple) An AUC of 0.97 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.94 is achieved with a 26-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 97.62 and 100.00%, respectively.
(C) Target cancer: colorectal. (Blue) An AUC of 1.00 is achieved with the DEcancerPDE model. (Orange) An AUC of 0.99 is achieved with a 22-protein DEcancerP model. (Green) An AUC of 0.95 is achieved with the DEcancerPDE model. (Red) An AUC of 0.94 is achieved with a 22-protein DEcancerP model. (Purple) An AUC of 0.97 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.96 is achieved with a 35-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 94.87 and 99.38%, respectively.
(D) Target cancer: esophageal. (Blue) An AUC of 0.98 is achieved with the DEcancerPDE model. (Orange) An AUC of 0.99 is achieved with an 8-protein DEcancerP model. (Green) An AUC of 0.80 is achieved with the DEcancerPDE. (Red) An AUC of 0.71 achieved with a 23-protein DEcancerP model. (Purple) An AUC of 0.83 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.81 is achieved with a 15-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 88.89 and 94.48%, respectively.
(E) Target cancer: liver. (Blue) An AUC of 1.00 is achieved with the DEcancerPDE model. (Orange) An AUC of 1.00 is achieved with a 23-protein DEcancerP model. (Green) An AUC of 0.93 is achieved with the DEcancerPDE model. (Red) An AUC of 0.92 is achieved with an 8-protein DEcancerP model. (Purple) An AUC of 0.96 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.96 is achieved with a 19-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 88.89 and 98.77%, respectively.
(F) Target cancer: ovarian. (Blue) An AUC of 0.98 is achieved with the DEcancerPDE model. (Orange) An AUC of 0.98 is achieved with a 15-protein DEcancerP model. (Green) An AUC of 0.99 is achieved with the DEcancerPDE model. (Red) An AUC of 0.99 is achieved with a 13-protein DEcancerP model. (Purple) An AUC of 0.99 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.98 is achieved with a 12-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 90.91 and 95.47%, respectively.
(G) Target cancer: pancreatic. (Blue) An AUC of 0.98 is achieved with the DEcancerPDE model. (Orange) An AUC of 0.98 is achieved with a 8-protein DEcancerP model. (Green) An AUC of 0.92 is achieved with the DEcancerPDE model. (Red) An AUC of 0.91 is achieved with a 14-protein DEcancerP model. (Purple) An AUC of 0.97 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.97 is achieved with a 9-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 84.21 and 98.77%, respectively.
(H) Target cancer: gastric. (Blue) An AUC of 0.97 is achieved with the DEcancerPDE model. (Orange) An AUC of 0.93 is achieved with a 17-protein DEcancerP model. (Green) An AUC of 0.88 is achieved with the DEcancerPDE model. (Red) An AUC of 0.85 is achieved with a 19-protein DEcancerP model. (Purple) An AUC of 0.88 is achieved with the DEcancerPDE model. (Brown) An AUC of 0.88 is achieved with a 25-protein DEcancerP model. The optimal sensitivity and specificity corresponding to the top left corner of ROC curve is 85.71 and 93.21%, respectively.
Feature selection in a high dimensional low sample size context
To demonstrate the potential application of DEcancer for detecting cancers which currently lack a suitable screening test such as lung cancer as well as its ability to analyze higher dimensionality data, we sought to apply our approach to a second dataset. Furthermore, in the research and development phase of creating a sensitive and specific test for lung cancer, an important task is robust feature selection so that a succinct biomarker panel is identified enabling application of a liquid biopsy detection test in clinic at scale. Hence, we assessed the performance of DEcancer in a nanoparticle-based in-depth proteomics lung cancer dataset. We collected data published by Blume et al.7 which consisted of 61 NSCLC patients and 80 cancer-free controls (Table 3).
Table 3.
Demographics of the 141 sample high dimensional proteomics dataset from Blume et al. who analyzed depleted plasma and used nanoparticle coronas to distinguish cancer-free individuals from early non-small cell lung cancer patients
| Cancer-free (n = 80) | Non-small cell lung cancer (n = 61) | |
|---|---|---|
| Number of males, n (%) | 31 (39) | 31 (51) |
| Mean (standard deviation) age, years | 65 (8) | 67 (8) |
| Number of each stage of cancer, n (%) | ||
| Stage 1 | N/A | 23 (38) |
| Stage 2 | 7 (11) | |
| Stage 3 | 31 (51) |
Individuals in the Blume et al. dataset were age- and sex-matched. Ethnicity, pack-year smoking history and molecular subtypes of non-small cell lung cancer were not shown by Blume et al.7
Approximately half of the lung cancer patients were at stage 1 or 2 and half of the patients had cancer at stage 3. Blume et al. used two methodologies for untargeted proteomics profiling. An immunodepletion approach or a nanoparticle corona-based enrichment approach was used, with different ‘spions’ (SP-003, SP-006, SP-007, SP-333, and SP-339) each exhibiting different biophysical properties and therefore detecting particular proteins more readily. Over 400 proteins were detected using the immunodepletion approach. Of the five spions used in Blume et al.’s final analysis, the number of detected proteins ranged from over 700 to over 1,100 depending on the spion. We sought to perform feature selection and develop a lung cancer test using data from depleted plasma (DP) and each of the five spions. The characteristics of the 31 sample test set (Table 4) are comparable to those of the overall 141 sample dataset.
Table 4.
Demographics of the 31 test set individuals separated from the Blume et al. 141 individual high dimensional proteomics dataset to assess generalizability in unseen data of DEcancer machine learning framework in distinguishing cancer-free from early non-small cell lung cancer patients via proteins measured using depleted plasma and nanoparticle corona spion approaches
| Cancer-free (n = 18) | Non-small cell lung cancer (n = 13) | |
|---|---|---|
| Number of males, n (%) | 6 (33) | 6 (46) |
| Mean (standard deviation) age, years | 64 (8) | 63 (8) |
| Number of each stage of cancer, n (%) | ||
| Stage 1 | N/A | 5 (38) |
| Stage 2 | 2 (15) | |
| Stage 3 | 6 (46) | |
As a control, we first trained, validated, and assessed on the test set random forest classifiers without any feature selection. For each of the five spions, we replicated the strategy of Blume et al. in the control experiments using only proteins detected by the particular spion over and above proteins also detected in the DP. The test set AUC of our resulting classifiers (Figure 4) ranged from 0.85 (SP-333, 293 proteins used in model) to 0.95 (SP-007, 421 proteins), which are comparable to Blume et al.’s overall 0.91 AUC for distinguishing between NSCLC patients and cancer-free individuals.
Figure 4.

Control receiver operating characteristic (ROC) curve and area under the curve (AUC) showing the performance of an optimized classifier model with the full protein set used by Blume et al.’s final model to distinguish cancer-free individuals from NSCLC patients
For each spion, the full protein set detected by that spion but not detected with the depleted plasma approach is used as per the methodology in Blume et al. The final model was retrained on all 110 training and validation samples and the retrained classifier was then assessed on the 31 test set samples. An ROC curve is shown for each of the spions of Blume et al. and the number of proteins included in the model is indicated. (Blue) SP003 with 711 proteins (AUC 0.92). (Orange) SP006 with 532 proteins (AUC 0.90). (Green) SP007 with 421 proteins (AUC 0.95). (Red) SP333 with 293 proteins (AUC 0.85); (Purple) SP339 with 416 proteins (0.91).
Next, we attempted to perform training and validation of the DEcancer pipeline including feature selection to show that a clinically feasible number of proteins can be used to successfully detect NSCLC. The number of proteins identified by DEcancer’s pipeline ranged from 14 (SP-007) to 43 (SP-339). The test set AUC of DEcancer’s pipeline ranged from 0.81 (SP-006) to 0.97 (DP). Random forest performed the best overall in the validation stage (Figure S10). For different spions and DP, different classifier types performed best at validation. However, for consistency in the context of detecting one type of cancer, lung, and one classification pipeline (lung versus cancer-free), the overall best performing classifier across all spions and DP was sought out. This was the random forest, which we selected for retraining on the 110 samples before assessing on the test set (Figure 5). The best performance out of the five spions and DP was identified at the validation stage as SP-339. The 0.92 AUC test set of DEcancer using SP-339 outperforms Blume et al.’s 0.91 AUC despite the fact that the DEcancer pipeline required only 43 proteins and with a more stringent testing framework than that published by Blume et al.
Figure 5.

Receiver operating characteristic (ROC) curves and area under the curve (AUC) showing the 31-individual test set performance of the optimized randomforest classifier model for each spion or depleted plasma data of Blume et al.
For each spion or depleted plasma, the optimal subset of proteins used by the final classifier model to distinguish between cancer-free and non-lung cancer samples is indicated (Blue) Depleted plasma in which the optimal protein set included 30 proteins (AUC 0.97); (Orange) SP003 in which the optimal protein set included 32 proteins (AUC 0.93); (Green) SP006 in which the optimal protein set included 26 proteins (AUC 0.81); (Red) SP007 in which the optimal protein set included 14 proteins (AUC 0.93); (Purple) SP333 in which the optimal protein set included 36 proteins (AUC 0.92); (Brown) SP339 in which the optimal protein set included 43 proteins (AUC 0.92).
Discussion
We have provided the first machine learning framework for liquid biopsy cancer detection analysis which performs feature selection and uses a rigorous testing methodology to provide unbiased estimates of model performance (Figure 6). The use of different data augmentation procedures with several classifiers and hyperparameter optimization are key components of DEcancer which enable superior cancer detection performance. The overall slight improvement in cancer detection achieved with DEcancerPDE compared to DEcancerP is likely because of the presence of a small number of samples difficult to identify as corresponding to a cancer patient versus cancer-free individual and for such cases the additional information provided by extra proteins, DNA mutation data and epidemiology can enable a correct prediction. However, it should be noted that the difference between DEcancerP and DEcancerPDE performance was minimal. In most cases, the AUC of DEcancerP was only 0.01–0.03 lower than for DEcancerPDE. This shows that in the context of the Cohen et al.6 dataset, minimal improvement is generated by additional proteins, DNA mutations and epidemiology based information compared to the optimal protein subsets used by DEcancerP. However, it is possible that the sample size limits any improvement in cancer detection potentially achievable by adding these extra variables to our models and that using a larger sample size a significant improvement could be achieved. Nevertheless, we have demonstrated that the proteomics-only approach for analyzing blood samples is sufficient for DEcancerP to achieve more than double the sensitivity for stage 1 cancer detection compared to the Cohen et al.6 approach while maintaining specificity at 99%. This may be of clinical relevance because a protein-only approach may enable an inexpensive technology that is more cost-effective for healthcare systems, facilitating the use of early screening tests for a wider population of patients at risk of cancer.
Figure 6.

Flowchart outlining the DEcancer early cancer detection pipeline
DEcancer was applied to the Cohen et al. dataset6 of cancer-free individuals and patients with one of 8 types of cancer, as well as the Blume et al. dataset with cancer-free and lung cancer patients.7 An approximately 20% test set is first separated out. For each classification task, the remaining data not part of the test set are used to form training and validation folds as part of a 200-fold Monte Carlo cross validation scheme. Various data augmentation approaches are applied to the training data. Feature selection and hyperparameter optimization of the classifier model are carried out based on performance on the validation folds. The independent t-test is used to compare the performance of the classifier model with the best performing feature set to that of the classifier using the smallest subset of variables, such that the performance is not statistically significantly lower than that of the best feature set. The best data processing framework, classifier models and feature set are then selected. Subsequently, retraining is performed on all training and validation data combined and the models assessed on the test set samples.
DEcancer’s performance for distinguishing between cancer and cancer-free samples was high for all 8 cancer types from the Cohen et al.6 dataset. As expected, distinguishing between different types of cancer proved more challenging. Of interest, for the number of proteins selected by DEcancer to distinguish between different cancers compared to the number of proteins needed to distinguish between cancer and cancer-free individuals was similar for most cancers. The exception was lung cancer and esophageal cancer for which the number of proteins required to distinguish between cancer types was greater than to distinguish the cancer from cancer-free individuals. The performance of DEcancer in distinguishing these cancers from other cancer types was also lower than in the case of the other six cancers. This may suggest that there is more overlap in the liquid biopsy protein signature of lung cancer with other cancers and esophageal cancer with other cancers, although there is a clear distinction between the cancer and cancer-free individuals. Nevertheless, across the 8 cancer types in the Cohen et al.6 dataset, when distinguishing between different cancer types the performance of DEcancerP feature selection model was comparable to the performance of DEcancerPDE. This suggests that a protein-only approach may be sufficient for clinical uses of liquid biopsy cancer detection technology. The high dimensional nature of the Blume et al.7 dataset posed a mathematical challenge, especially in the context of low sample size (141 individuals) included in their study. However, DEcancer’s methodology is tailored to effectively perform feature selection even for high dimensional low sample size data, as demonstrated by our results. There was minimal overlap between the protein set selected by DEcancer for DP and each of the different spions. This illustrates a major limitation of the Blume et al.7 nanoparticle-based approach because a consistent set of liquid biopsy analytes would be a prerequisite for clinical application of liquid biopsy for cancer detection technology.
It must be noted that Cohen et al.6 and Blume et al.7 do not include evaluation of different classifier models for distinguishing between cancer and cancer-free individuals. Both Blume et al.7 and Wong et al.8 approaches lack feature selection in the pipeline which hinders the most parsimonious high performing cancer detection models from being developed for widespread clinical use. Cohen et al.6 performed feature selection but different classifier models were not evaluated in the study.8 Furthermore, Cohen et al.6 did not employ non-linear machine learning techniques to distinguish cancer from cancer-free individuals. This may contribute to the lower performance of the Cohen et al. cancer detection model. Unfortunately past analyses from Blume et al.,7 Cohen et al.6 and Wong et al.8 lack a hold-out test set separate from cross-validation data. This may undermine the generalizability of the reported results because the best model’s performance from cross-validation does not reflect the model’s performance for unseen individuals. Hence, DEcancer’s high performance compared to past approaches is especially promising because a rigorous testing methodology to provide generalizable estimates of model performance was employed.
A number of attempts have been made to develop liquid biopsy-based clinical products for cancer detection in addition to CancerSEEK reported by the Cohen et al.6 study. DEcancer’s results compare favorably to the performance of clinical products under development. For example, at a fixed >99% specificity, GRAIL achieved stage 1, 2, 3 and 4 cancer detection sensitivity of 17%, 40%, 77% and 90%, respectively, when evaluating 12 cancer types.9 Notably, lung cancer constituted one of GRAIL’s lowest performances for early lung cancer detection, with a stage 1 sensitivity of about 22%.9 In contrast, DEcancer achieved 100% sensitivity for all lung cancers using a protein-only model. In summary, DEcancer is a robust machine learning tool that can discover succinct subsets of biomarkers tailored to liquid biopsy-based disease detection. In the context of early cancer detection, DEcancer may help facilitate the development of novel, accessible, and effective minimally invasive clinical tests and improve cancer patient survivorship.
Limitations of the study
Some limitations of our analysis must be acknowledged. The sample size for certain cancer types in the Cohen et al.6 dataset is small, especially for esophageal, liver, ovarian, and gastric cancer. Therefore, care must be taken when interpreting hold-out test set results for cancer types with small sample sizes. For example, a(n) (in)correct test result in the case of a small number of samples may manifest as a large apparent difference in test set performance of DEcancer in terms of sensitivity and specificity. Nevertheless, we note that this limitation does not necessarily lead to inflation of apparent performance; the small sample sizes for particular cancers might just as well lead to a reduction in apparent performance. Another limitation of our analysis lies in the characteristics of the Cohen et al.6 and Blume et al.7 datasets. In Cohen et al.,6 cancer-free individuals are not matched for age, smoking history and comorbidities. It was shown that cancer-free individuals in Cohen et al. are on average younger than cancer patients, which may serve as a confounding variable. Although in Blume et al.7 NSCLC and cancer-free individuals age- and sex-matched, smoking history was not reported, and the cancer-free control group was not comorbidity matched with the NSCLC group. Thus, we envision that large-scale high-throughput proteomic studies with greater sample size and control individuals matched with cancer patients for demographic and clinical characteristics is required as a future direction. DEcancer applied to such data will help more effectively in identifying key proteins for clinical screening applications.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| CancerSEEK/Nanoparticles | https://zenodo.org/record/7544181#.Y-ZJD3bP238 | Zenodo data: https://doi.org/10.5281/zenodo.7544181 |
| Software and algorithms | ||
| DEcancer code | This paper | Github code: https://github.com/Oxford-Cancer-Analytics/DEcancer-manuscript |
| Python version 3.10.4 | Python Software Foundation | https://www.python.org |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andreas Halner (a.halner@oxcan.org).
Materials availability
This study did not generate new unique reagents.
Method details
The machine learning pipeline reported in this article can be found at https://github.com/Oxford-Cancer-Analytics/DEcancer-manuscript/.
Datasets
In order to illustrate the effectiveness of our cancer detection algorithms, we analyzed two datasets.
-
1)
Cohen et al.,6 a multicancer dataset that includes the protein- and DNA-based information in the blood of 1,005 patients diagnosed with one of eight types of cancer, including breast, colorectal, esophageal, liver, lung, ovarian, pancreatic and gastric. 812 individuals were cancer-free. The Cohen et al. dataset enabled us to assess our ability to detect and distinguish multiple different cancer types. This reflects the clinical context where both cancer detection and localization are required.
-
2)
Blume et al.,7 an in-depth proteomic dataset of 141 patients (80 cancer-free individuals) enabled us to assess our ability to perform feature selection and use a small subset of the original dataset features to detect lung cancer, which may enable more feasible clinical testing applications. This is particularly significant given that lung cancer is one of the most difficult cancers to detect early and currently lacks effective screening tests.
DEcancer pipeline
We developed a pipeline named DEcancer for processing patient data for cancer detection. DEcancer includes data pre-processing, training and optimising cancer detection classifiers and performing feature selection.
DEcancer pipeline applied to cohen et al. Dataset
DEcancer is assessed on a hold-out test set of approximately 20% of samples separated from the dataset to provide a generalisable estimate of performance. The test set is stratified according to cancer type and stage in order to reflect all cancer types and the same proportion of stage 1, 2 and 3 cancers as occurred in the overall dataset. The test set samples were selected with the constraint that samples selected for the test set were mutually exclusive from samples appearing in the remaining training and cross-validation data. In the pre-processing step, the dataset is cleaned, followed by various kernel density estimation (KDE)-based10,11data augmentation procedures. Augmentation procedures applied to the data was done through imbalancing the data in either direction, balanced, or without augmentation. Where KDE was applied, an augmentation factor of 5 was used (Table S4). The remaining patient samples constitute data for training and validation of cancer detection classifiers in a Monte Carlo cross validation12 scheme. The validation includes feature selection steps and classifier optimisation. For the Cohen et al. dataset, four classification tasks are performed for each of the eight types of cancer: 1) all cancer types versus cancer-free; 2) selected cancer type versus cancer-free; 3) selected cancer type versus other cancer types; 4) selected cancer type versus cancer-free individuals and patients with another cancer type. For each of these classification tasks, a random forest classifier is trained using 39 protein concentrations, the DNA ‘omega score’ (described in Cohen et al.) and epidemiological characteristics (age, ethnicity, sex). For DEcancerP, recursive feature elimination is performed using the random forest Gini-index based variable importance score. A random forest classifier is trained on different feature elimination subsets of proteins. Hence, two approaches were used: a selected subset of protein-only cancer detection approach (DEcancerP) and a 39 protein, DNA and epidemiological based cancer detection approach (DEcancerPDE). Using selected feature subsets, hyperparameter optimisation is performed for random forest,13support vector machine,14 logistic regression with l2 penalty15 and multilayer perceptron.16 The best performing classifier model (and in the case of DEcancerP also the optimal protein feature subset) was identified according to AUC across the Monte Carlo cross validation folds. This optimal model for DEcancerP and DEcancerPDE were retrained on all training and validation patient samples and then assessed on the hold-out test set. The same optimal model type (out of random forest, support vector machine, multilayer perceptron and logistic regression with l2 penalty) based on the best performance in the DEcancerP is used for retraining and assessment on the test set for both DEcancerP and for DEcancerDPE to enable a comparison. For the ‘all cancer’ versus ‘cancer-free’ classification, in addition to the overall test set AUC, the test set sensitivities by cancer type and stage for an overall 99% specificity were reported. This enabled a comparison with the Cohen et al. context in which minimising false positives was one of the objectives for a cancer detection algorithm.
DEcancer applied to blume et al. Dataset
Initial dataset cleaning involved imputation using Perseus software17 without considering whether a given sample is from a cancer patient or cancer-free individual. Only one classification task was performed: NSCLC versus cancer-free. However, the training and validation steps of DEcancer were applied using six different data inputs. One data input corresponded to a depleted plasma (DP) approach. The other five data inputs represented relative intensity of proteins enriched by five nanoparticle ‘spions’ each with different biophysical properties, respectively. The 141 samples were split into 110 samples for training and validation and 31 samples for the hold-out test set. The test set samples were randomly selected but with the constraint that samples selected for the test set were mutually exclusive from samples appearing in the remaining training data. A 200 Monte Carlofold cross validation strategy was used to create different training (80 samples) and validation (30 samples) data combinations. A variety of data augmentation strategies were applied to the training dataalong with feature selection and hyperparameter optimisation based on classifier performance on validation data. Augmenting the data gave us a balanced, imbalanced in favor of cancer, and an imbalanced dataset in favor of cancer-free samples. The dataset had an augmentation factor of 5 giving 200-200, 320-80, and 80–320 for balanced, imbalanced in favor of cancer, and imbalanced in favor of cancer-free, respectively, irrespective of spion or DP. Feature selection included both univariate and multivariate as initial feature filtering prior to recursive feature elimination. For DP and each of the five spions in the Blume et al. dataset, this process was used with random forest to identify optimal protein subset and best overall data processing framework. Using selected feature subsets, hyperparameter optimisation is performed for random forest,13support vector machine,14 logistic regression with l2 penalty15 and multilayer perceptron.16 The selected best model and framework are then retrained on all 110 training and validation data samples before being assessed on the 31 sample test set.
Figure 6 summarises the key steps of the DEcancer pipeline as applied to the Cohen et al. and Blume et al. datasets. All classifiers were implemented using the open-source Python scikit-learn package.18
Quantification and statistical analysis
Quantification of selected proteins can be found in the results section. All DEcancer pipelines, excluding DEcancerPDE, perform an independent t-test across the 200 Monte Carlo validation folds using the open-source Python library SciPy.19 An α of 0.05 was used to compare the AUC performance of the classifier model with the best performing feature set to that of the classifier using the smallest subset of variables, such that the performance is not statistically significantly lower than that of the best feature set.
Acknowledgments
The authors would like to thank Professor Jimmy Ba for his helpful comments for improving this paper. The work described in this paper was supported by funding from Oxford Cancer Analytics Ltd.
Author contributions
A.H. and P.J.L. co-conceived the objectives of this study. P.J.L. co-conceived the overall study approach, is the main advisor and feedback provider for data presentation and co-wrote this manuscript. A.H. co-conceived the DEcancer machine learning framework and wrote the manuscript. L.H., F.P., Z.H., A.H. and P.J.L. performed analysis. L.H. and F.P. produced figures. Z.H., A.H. and P.J.L. assisted with figure production. D.S., E.M., G.L., B.M.K. and J.S. provided feedback for data presentation and the writing of the manuscript
Declaration of interests
A.H. is a founder, an employee, and shareholder of Oxford Cancer Analytics Ltd. L.H. is an employee of Oxford Cancer Analytics Ltd. Z.L. is an employee of Oxford Cancer Analytics Ltd. F.P. was an employee and is a shareholder of Oxford Cancer Analytics Ltd. D.S. is an employee and shareholder of Oxford Cancer Analytics Ltd. E.M. is an employee of Oxford Cancer Analytics Ltd. G.L. declares no competing interests. B.K. is a shareholder and member of the Scientific Advisory Board of Oxford Cancer Analytics Ltd. J.S. is an employee of Oxford Cancer Analytics Ltd. P.J.L. is a founder, an employee and shareholder of Oxford Cancer Analytics Ltd. A.H. and P.J.L. are co-inventors of the patent “A METHOD AND SYSTEM DETECTING A HEALTH ABNORMALITY IN A LIQUID BIOPSY SAMPLE” (International Patent Application Number: PCT/EP2022/075710).
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Published: April 11, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106610.
Contributor Information
Andreas Halner, Email: a.halner@oxcan.org.
Peter Jianrui Liu, Email: p.liu@oxcan.org.
Supplemental information
Data and code availability
-
•
Data: This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
Code: Original code has been deposited at https://github.com/Oxford-Cancer-Analytics/DEcancer-manuscript/ and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Loud J.T., Murphy J. Cancer screening and early detection in the 21stCentury. Semin. Oncol. Nurs. 2017;33:121–128. doi: 10.1016/j.soncn.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller K.D., Nogueira L., Devasia T., Mariotto A.B., Yabroff K.R., Jemal A., Kramer J., Siegel R.L. Cancer treatment and survivorship statistics. CA. Cancer J. Clin. 2022;72:409–436. doi: 10.3322/caac.21731. [DOI] [PubMed] [Google Scholar]
- 4.Runowicz C.D., Leach C.R., Henry N.L., Henry K.S., Mackey H.T., Cowens-Alvarado R.L., Cannady R.S., Pratt-Chapman M.L., Edge S.B., Jacobs L.A., et al. American cancer society/American society of clinical oncology breast cancer survivorship care guideline. CA. Cancer J. Clin. 2016;66:43–73. doi: 10.3322/caac.21319. [DOI] [PubMed] [Google Scholar]
- 5.Fiorica J.V. Breast cancer screening, mammography, and other modalities. Clin. Obstet. Gynecol. 2016;59:688–709. doi: 10.1097/GRF.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 6.Cohen J.D., Li L., Wang Y., Thoburn C., Afsari B., Danilova L., Douville C., Javed A.A., Wong F., Mattox A., et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blume J.E., Manning W.C., Troiano G., Hornburg D., Figa M., Hesterberg L., Platt T.L., Zhao X., Cuaresma R.A., Everley P.A., et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020;11:3662. doi: 10.1038/s41467-020-17033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong K.C., Chen J., Zhang J., Lin J., Yan S., Zhang S., Li X., Liang C., Peng C., Lin Q., et al. Early cancer detection from multianalyte blood test results. iScience. 2019;15:332–341. doi: 10.1016/j.isci.2019.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein E.A., Richards D., Cohn A., Tummala M., Lapham R., Cosgrove D., Chung G., Clement J., Gao J., Hunkapiller N., et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021;32:1167–1177. doi: 10.1016/j.annonc.2021.05.806. [DOI] [PubMed] [Google Scholar]
- 10.Rosenblatt M. Remarks on some nonparametric estimates of a density function. Ann. Math. Statist. 1956;27:832–837. doi: 10.1214/aoms/1177728190. [DOI] [Google Scholar]
- 11.Parzen E. On estimation of a probability density function and mode. Ann. Math. Statist. 1962;33:1065–1076. [Google Scholar]
- 12.Xu Q.S., Liang Y.Z. Monte Carlo cross validation. Chemometr. Intell. Lab. Syst. 2001;56:1–11. [Google Scholar]
- 13.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 14.Cortes C., Vapnik V. Support-vector networks. Mach. Learn. 1995;20:273–297. [Google Scholar]
- 15.Hoerl A.E., Kennard R.W. Ridge regression: biased estimation for nonorthogonal problems. Technometrics. 1970;12:55–67. doi: 10.2307/1267351. [DOI] [Google Scholar]
- 16.Ripley B. Cambridge University Press; 1996. Pattern Recognition and Neural Networks. [Google Scholar]
- 17.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 18.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. Scikit-learn: machine learning in Python” J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 19.Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
Code: Original code has been deposited at https://github.com/Oxford-Cancer-Analytics/DEcancer-manuscript/ and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.