

Abstract
Background and aims:
Chronic vascular endothelial inflammation predisposes to atherosclerosis; however, the cell-autonomous roles for endothelial-expressing microRNAs (miRNAs) are poorly understood in this process. MiR-181b is expressed in several cellular constituents relevant to lesion formation. The aim of this study is to examine the role of genetic deficiency of the miR-181b locus in endothelial cells during atherogenesis.
Methods and Results:
Using a proprotein convertase subtilisin/kexin type 9 (PCSK9)-induced atherosclerosis mouse model, we demonstrated that endothelial cell (EC)-specific deletion of miR-181a2b2 significantly promoted atherosclerotic lesion formation, cell adhesion molecule expression, and the influx of lesional macrophages in the vessel wall. Yet, endothelium deletion of miR-181a2b2 did not affect body weight, lipid metabolism, anti-inflammatory Ly6Clow or the pro-inflammatory Ly6Cinterm and Ly6Chigh fractions in circulating peripheral blood mononuclear cells (PBMCs), and pro-inflammatory or anti-inflammatory mediators in both bone marrow (BM) and PBMCs. Mechanistically, bulk RNA-seq and gene set enrichment analysis of ECs enriched from the aortic arch intima, as well as single cell RNA-seq from atherosclerotic lesions, revealed that endothelial miR-181a2b2 serves as a critical regulatory hub in controlling endothelial inflammation, cell adhesion, cell cycle, and immune response during atherosclerosis.
Conclusions:
Our study establishes that deficiency of a miRNA specifically in the vascular endothelium is sufficient to profoundly impact atherogenesis. Endothelial miR-181a2b2 deficiency regulates multiple key pathways related to endothelial inflammation, cell adhesion, cell cycle, and immune response involved in the development of atherosclerosis.
Keywords: Atherosclerosis, Endothelial cells, microRNA-181b
1. Introduction
Atherosclerosis is a lipid-driven, chronic inflammatory disease of the arterial intima in which the balance of pro-inflammatory and inflammatory-resolving pathways will result in different clinical outcomes [1,2]. During this process, endothelial cells (ECs) activation represents an early hallmark of vascular inflammation and contributes importantly to the initiation and progression of atherosclerosis [3]. After activation, ECs express multiple adhesion molecules (e.g. VCAM-1, ICAM-1 and E-selectin) and release various chemotactic cytokines to recruit and foster the circulating leukocytes into the sub-endothelial space of vascular wall, where they differentiate into macrophages and eventually become foam cells, resulting in lipid plaque formation [2,4, 5]. Accumulating evidence from clinical end experimental studies highlights that targeting EC activation serves as a promising therapeutic approach for atherosclerosis prevention and treatment [6–8]. For instance, endothelium-restricted inhibition of NF- κB activity suppresses ECs activation and strongly reduces atherosclerosis in apolipoprotein E-deficient (ApoE−/−) mice [7]. Yet, the mediators controlling EC activation during the pathogenesis of atherosclerosis remain poorly understood.
MicroRNAs (miRNAs) are a class of evolutionary conserved small non-coding RNAs and regulate gene expression at the post-transcriptional level. Accumulating evidence demonstrates that miRNAs play a critical role in regulating EC activation and atherosclerosis, although their evaluation by genetic deletion specifically in the vascular endothelium remains poorly characterized in this process [6]. In our previous observations, we identified that systemic delivery of miR-181b potently suppressed EC activation and vascular inflammation in both acute and chronic diseases state such as endotoxemia and obesity [9,10]. Moreover, we found that miR-181b expression decreased in the aortic intima of ApoE−/− mice and plasma from patients with coronary artery disease and demonstrated that rescue of miR-181b expression by systemic delivery of miR-181b mimics inhibited adhesion molecules expression in the aortic intima and atherosclerosis [11]. However, a cell-autonomous role for miR-181b in the vascular endothelium has yet to be defined. Herein, we test the hypothesis that genetic deficiency of the miR-181b locus in ECs promotes atherogenesis.
2. Materials and methods
2.1. Cell culture and TNF-α treatment
HUVECs were purchased from Lonza (cc-2159) and cultured in EC growth medium EGM-2 (cc-3162). Cells were plated on 12-well plates at 60,000/well and allowed to grow to 80%–90% confluency. In some experiments, HUVECs were stimulated with 10 ng/ml of recombinant human TNF-α (210-TA/CF, R&D Systems) for 4 h. After stimulation, HUVECs were harvested for RNA isolation using TRIzol reagent (15596018, Invitrogen). Cells were passaged less than five times for all experiments. Primary lung and liver ECs from miR-181a2b2flox/flox, miR-181a2b2 knock out (miR-181a2b2−/−), or ECs-specific miR-181a2b2 deficient mice (miR-181a2b2iECKO) mice were isolated and cultured as described in our previous works [10,12]. Lung or liver ECs were stimulated with or without 20 ng/ml recombinant mouse TNF-α (410-MT/CF, R&D Systems) for 4 or 8 h and harvested for cell adhesion assay or Western blot.
2.2. Mice and high-cholesterol diet-induced atherosclerosis
MiR-181a2b2flox/flox mice (C57BL/6J) and miR-181a2b2−/− were used as previously described, and mice expressing tamoxifen-inducible endothelial-specific VE-cadherin (VECad-Cre-ERT2) (C57BL/6J) were kindly provided by R. Adams [13,14]. Inducible endothelial cells (ECs)-specific miR-181a2b2 deficient mice (miR-181a2b2flox/flox; VECad-Cre-ERT2) were generated by crossbreeding miR-181a2b2flox/flox and VECad-Cre-ERT2 mice. For induction of Cre recombination, four weeks old male miR-181a2b2flox/flox; VECad-Cre-ERT2 mice were treated with either 4-hydroxytamoxifen (H6278, Sigma) (10 mg/kg, i.p.) or the same volume of vehicle for five consecutive days to generate miR-181a2b2iECKO and control mice (Ctrl). After Cre activity induction, the miR-181a2b2iECKO and Ctrl mice at the age of eight weeks were tail vein injected a single dose of AAV-PCSK9D377Y at 5 × 10 [11] viral genomes generated by Viral Core, Boston Children’s Hospital, Boston and immediately placed on a high cholesterol diet (D12108c, Research Diets Inc.) for 12 weeks to induce atherosclerosis. Age- and cage-matched male littermates were used for experiments. Male C57BL/6J and ApoE−/− mice at eight weeks old were purchased from Charles River Laboratories and placed on a normal chow diet or high cholesterol diet (Research Diets Inc; D12108Ci) for 12 weeks to induce atherosclerosis. In some experiments (Supplemental Figs. 1F–H), C57BL/6J mice or mice on C57BL/6 genetic background were used. C57BL/6J mice at the age of 9 weeks were injected with tamoxifen (TAM) (Sigma T5648; i.p. 50 mg/kg) in corn oil on five consecutive days. Control mice were injected with corn oil in parallel. Three weeks after injection, mouse aortas were dissected for aortic intima isolation and livers and hearts were collected for endothelial cell (EC) isolation as described in our previous studies [10,11,15]. Mice on C57Bl/6 genetic background were intravenously injected with 1 × 1011 genome copies of adeno-associated viral vector (AAV) expressing mouse D377Y PCSK9 and fed with a Western Diet (TD.88137) with 18.9 g/L glucose and 23.1 g/L fructose added to the drinking water for 24 weeks. Control mice were not injected with AAV-D377Y PCSK9. Heart and liver ECs were isolated as described above. All mice were maintained under SPF conditions at an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility at the Brigham and Women’s Hospital. All animal protocols were approved by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital, Boston, MA and conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
2.3. Atherosclerotic lesions characterization and immunohistochemistry
Aortic root was embedded in optimum cutting temperature (OCT) and frozen at −80 °C. Serial cryostat sections (6 μm) were prepared using tissue processor Leica CM3050. Lesion characterizations, including Oil Red O staining of the aortic root and staining for macrophages (anti-Mac3, BD Pharmingen, 553322, 1:900), T cells (anti-CD4, BD Pharmingen, 553043, 1:90; anti-CD8, Chemicon, CBL1318, 1:100), vascular smooth muscle cells (SM-α-actin, Sigma, F-3777, 1:500) and vascular cell adhesion molecule 1 (VCAM-1) (ab134047, 1:100, abcam) were performed as previously described [11,16]. All images were captured using a Microscope VS120 Whole Slide Scanner (Olympus) and analyzed using the computer-assisted Image-Pro Plus software (Meida Cybernetics, Bethesda, MD). The quantification of atherosclerotic lesion, Mac3, VSMC, and VCAM-1 staining was performed as previously described [11,16]. The number of CD4+ and CD8+ cells was counted manually by blinded observers.
2.4. Lipid profile analysis
Lipid profile analysis was performed as previously described [11,16]. Triglyceride and total cholesterol levels were determined using the Infinity™ Triglycerides Liquid Stable Reagent and Cholesterol Reagent, respectively, from Thermo Scientific. HDL cholesterol was measured by colorimetric assay (BioAssay Systems, EnzyChrom HDL). LDL cholesterol levels were calculated using the following formula: LDL = Total Cholesterol - HDL Cholesterol - (Triglycerides)/5. Standards for cholesterol, triglyceride, and HDL were purchased from Pointe Scientific, Inc.
2.5. Intima RNA isolation from aortic arch and RNA-Seq analysis
Intima RNA from aortic arch was isolated as previously described [11,16]. Briefly, aortic arch was carefully flushed with phosphate-buffered saline (PBS), followed by intima peeling using TRIzol reagent (15596018, Invitrogen). TRIzol was flushed for 10 s, followed by 10 s pause, flushed another 10 s, collected in an Eppendorf tube, and snap-frozen in liquid nitrogen. TRIzol reagent was used for RNA isolation based on manufacturers’ protocol. Low input RNA-Seq analysis was performed after ribodepletion and standard library construction using Illumina HiSeq 2 × 150 PE HO (Genewiz, South Plainfield, NJ). All samples were processed using an RNA-seq pipeline implemented in the bcbio-nextgen project (https://bcbio-nextgen.readthedocs.org/en/latest/). Raw reads were examined for quality issues using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to ensure library generation and sequencing were suitable for further analysis. Trimmed reads were aligned to UCSC build mm10 of the Mouse genome, augmented with transcript information from Ensembl release 79 using STAR [17]. Alignments were checked for evenness of coverage, rRNA content, genomic context of alignments (for example, alignments in known transcripts and introns), complexity, and other quality checks using a combination of FastQC and Qualimap. Counts of reads aligning to known genes were generated by featureCounts [18]. Differential expression at the gene level was called with DESeq2. The total gene hit counts and counts per million (CPM) values were calculated for each gene and for downstream differential expression, analysis between specified groups was performed using DESeq2. Genes with adjusted p-value <0.1 were called as differentially expressed genes for each comparison. All samples had at least mean quality score >30 and > 70% of mapped fragments over total. MetaCore (v20.2) was used for gene set enrichment analysis.
2.6. Peripheral blood mononuclear cells (PBMCs) and bone marrow cells (BMCs) isolation
PBMCs and BMCs were isolated as previously described [11,16]. Briefly, PBMCs were isolated from blood using LSM® Lymphocyte Separation Medium (MP Biomedicals, LLC) according to the manufacturer’s instructions. For BMCs isolation, femurs were removed and cells were flushed out using a 27-gayge needle attached to a syringe containing 10 ml DMEM medium (Lonza). Cells were re-suspended by gentle pipetting, passed through a 70 μm filter, and spun down at 300×g for 10 min at 4 °C.
2.7. Flow cytometry
Isolated PBMCs were stained for surface markers (indicated in Supplemental Table SI) and subjected to Fortessa flow cytometer (BD) as described [11,16].
2.8. RNA isolation and real-time qPCR
Total RNA from intima, PBMCs, and BMCs were isolated using TRIzol Reagent according to the manufacturer’s instructions. Subsequent RT-qPCR was performed using a High-Capacity cDNA Reverse Transcription Kit (4368813, Applied Biosystems). For SyberGreen-based assay, GoTaq qPCR Master Mix (A6001, Promega) was used, and for TaqMan Universal Master Mix II, UNG (4440038, Life Technologies) was used. Expression of mRNAs and miRNA expression were normalized to β-actin or U6 (Aglient, AriaMx Real Time PCR System). Specific primers including miR-181b-5p (#001098), U6 (#001973), snoRNA234 (ID# 001234), human-Hprt (Hs05647472_cn), human-primary-miR-181b1 (Hs03302966_pri), human-primary-miR-181b2 (Hs03302963_pri), mouse-Hprt (Mm00522878_cn), mouse-primary-miR-181b1 (Mm03 307120_pri) and mouse-primary-miR-181b2 (Mm03307414_pri) were purchased from Life Technologies. VE-Cadherin forward primer CACTGCTTTGGGAGCCTTC and reverse primer GGGGCAGCGATTCATTTTTCT were used to detect VE-Cadherin mRNA expression. GAPDH primers are forward AGGTCGGTGTGAACGGATTTG and reverse TGTAGACCATGTAGTTGAGGTCA. Changes in expression were calculated using deltadelta Ct method. Primer sequences are described in (Supplemental Table SII).
2.9. Western blot
Liver tissues from C57BL/6J, Ctrl and miR-181a2b2iECKO mice were homogenized using TissueLyser II (Qiagen) according to manufacturer’ instructions. Proteins were isolated using RIPA buffer (Boston Bio-Productus, BP-115) with protease inhibitor and phosphatase inhibitor. Protein concentrations were determined using Pierce BCA kit (Thermo Scientific). 20 μg protein was loaded per lane on a 4–20% Mini-PROTEAN TGX Gel (Bio-Rad, 456–1096). Separated proteins were transferred to PVDF membranes using the Transfer Turbo Blot system (Bio-Rad) and Trans-Blot Turbo RTA Transfer Kit (Bio-Rad, 170–4272). The membrane was blocked with 5% nonfat milk in TBST for 1 h at room temperature. After blocking, the membrane was incubated overnight at 4 °C with antibodies against PCSK9 (1:1000, ab185194, abcam) or β-actin (1:4000, 4970, Cell Signaling). Quantification of protein bands was performed using a luminescent image analyzer (BioRad, Chemidoc).
2.10. Single-cell RNA sequencing
Two aortic arches were pooled and freshly digested into a single cell suspension from Ctrl (WT) and miR-181a2b2iECKO (KO) mice after a single dose of AAV-PCSK9D377Y gain-of-function transgene and 12 weeks of high cholesterol diet. The cells were processed using a 10X Genomics microfluidics chip to generate barcoded Gel Bead-In Emulsions according to manufacturer protocols. Indexed single-cell libraries were then created according to 10X Genomics specifications (Chromium Next GEM Single Cell 3ʹ v3.1-Dual Index Libraries). Samples were multiplexed and sequenced in pairs on an Illumina HiSeq 4000 device.
2.11. Single-cell RNA sequencing data analysis
The sequenced data were processed into expression matrices with the Cell Ranger Single-cell software 6.0.0 (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/6.0/what-is-cell-ranger). FASTQ files were obtained from the base-call files from HiSeq4000 sequencer and subsequently aligned to the mouse transcriptome (mm10). Cellular barcodes and unique molecular identifiers (UMIs) were filtered and corrected by Cell Ranger pipeline. The filtered counts matrices were analyzed using the Seurat package in R [19]. Filtering during this step included only genes detected in >3 cells, cells with >400 distinct genes, >600 UMI and a mitochondrial percentage <20%. A total of 3,867 sequenced cells combining all 4 samples met these quality control thresholds. Data normalization and scaling were performed using the SCTransform command under default settings. Samples were integrated following the default integration guidelines from https://satijalab.org/seurat/articles/integration_introduction.html. Principal component analysis and nonlinear dimensional reduction using Uniform Manifold Approximation and Projection (UMAP) were applied over the integrated dataset using 20 dimensions. A combination of previously known markers and newly identified markers using FindConservedMarkers function were used to assign cell types after cell clustering, ensuring a separation of known major aortic cell types. A total of 7 major aortic cell types were identified. The FindConservedMarkers function, applying Wilcoxon rank-sum test with default parameters, was used to obtain the significant markers for each cluster.
2.12. Pathway enrichment analysis
Differentially expressed genes (DEGs) were identified using the FindConservedMarkers function, applying Wilcoxon rank-sum test with default parameters, to obtain the significant markers for each cluster with adjusted p-value < 0.05 (false discovery rate). The DEGs were visualized using volcano plots. DEGs were subjected to gene set enrichment analyses using Ingenuity Pathway Analysis (IPA) software. The significant values for the canonical pathways were calculated by Fisher exact test and the top 20 pathways were visualized in dot plots.
2.13. Statistical analysis
GraphPad 7.0 software package (GraphPad Software, Inc) was used for statistical analysis. Unpaired two-tailed Student’s t-test was used to determine statistical significance between two groups for normally distributed continuous variables. For data without normal distribution, non-parametric Mann-Whitney U test was used. All data were expressed as mean ± SEM. p < 0.05 was considered significant for all test.
3. Results
The human and mouse mature miR-181b is a highly conserved miRNA encoded by two loci, including miR-181b1 and miR-181b2, located on two different chromosomes [20]. Moreover, miR-181b1 is clustered very closely with miR-181a1 and miR-181b2 is clustered together with miR-181a2 [20]. To begin interrogating which miR-181b transcript plays an important role in endothelial activation and atherosclerosis, we measured the expression of each miR-181b transcripts from cultured HUVECs exposed to vehicle alone or TNF-α and C57BL/6J mice fed a normal chow diet and ApoE−/− mice fed a western diet. The miR-181b2 transcript was decreased with inflammatory stimuli including in TNF-α-stimulated HUVECs and intima of ApoE−/− mice fed a western diet, while the expression of miR-181b1 transcript was not (Supplemental Fig. I), indicating that the reduction of miR-181b2 may play a crucial role in inflammatory responses in ECs.
To examine whether endothelial miR-181b2 contributes to atherosclerotic lesion formation, we generated mice with inducible EC-specific deficiency of the miR-181a2b2 locus (miR-181a2b2flox/lox; VECad-Cre-ERT2) by crossing the miR-181a2b2flox/flox mice with tamoxifen-inducible endothelial-specific Cre (vascular endothelial cadherin promoter, iVE-cdh5-Cre). Those mice were randomly intraperitoneal injected with 4-hydroxytamoxifen (4-OHT) to induce Cre-mediated excision of the loxP-flanked miR-181a2b2 allele in endothelial cells (miR-181a2b2iECKO) or corn oil vehicle control (Ctrl) at 4-week-old and tail vein injected with a single dose of AAV-PCSK9D377Y gain-of-function transgene, followed by 12 weeks high cholesterol diet to induced atherosclerosis (Fig. 1A). RT-qPCR analysis of RNA isolated from endothelial enriched aortic intima (Supplemental Fig. IIA) taken at the end of the experiment demonstrated that the expression of miR-181b was reduced by ~40% in miR-181a2b2iECKO mice compared to Ctrl mice and no differences were observed in the aortic media/adventitia (Fig. 1B). Genetic deletion of the miR-181a2b2 locus was also verified in ECs (Supplemental Fig. IE) and by RT-qPCR analysis of RNA isolated from endothelial enriched aortic intima (Supplemental Fig. IB and D). There were no differences in the expression of pri-miR-181a1, pri-miR-181a2, or miR-181a from the intima of KO and Ctrl mice, whereas there was a non-significant reduction in pri-miR-181b2, which was the dominantly expressed isoform (Supplemental Fig. 1F). Tamoxifen had no effect on cdh5 expression in VECad-Cre-ERT2 mice compared to corn oil vehicle control (Supplemental Fig. 1G). After 24 weeks of a modified Western diet in C57Bl/6 mice injected with AAV-D377Y-PCSK9, expression of miR-181b was moderately reduced by 31.2% and 49.7% in the liver and heart ECs, respectively (Supplemental Fig. IH), consistent with our prior report using ApoE−/− mice [11]. Interestingly, expression of miR-181b increased in the aortic intima of VECad--Cre-ERT2 mice injected with AAV-D377Y PCSK9 (and the corn oil vehicle control) after 12 weeks of Western diet compared with age-matched, 20-week old C57Bl/6 mice (Supplemental Fig. II). These data highlight that expression of miR-181b can be dynamically regulated in response to AAV-PCSK9 depending on dietary conditions or genetic background.
Fig. 1.
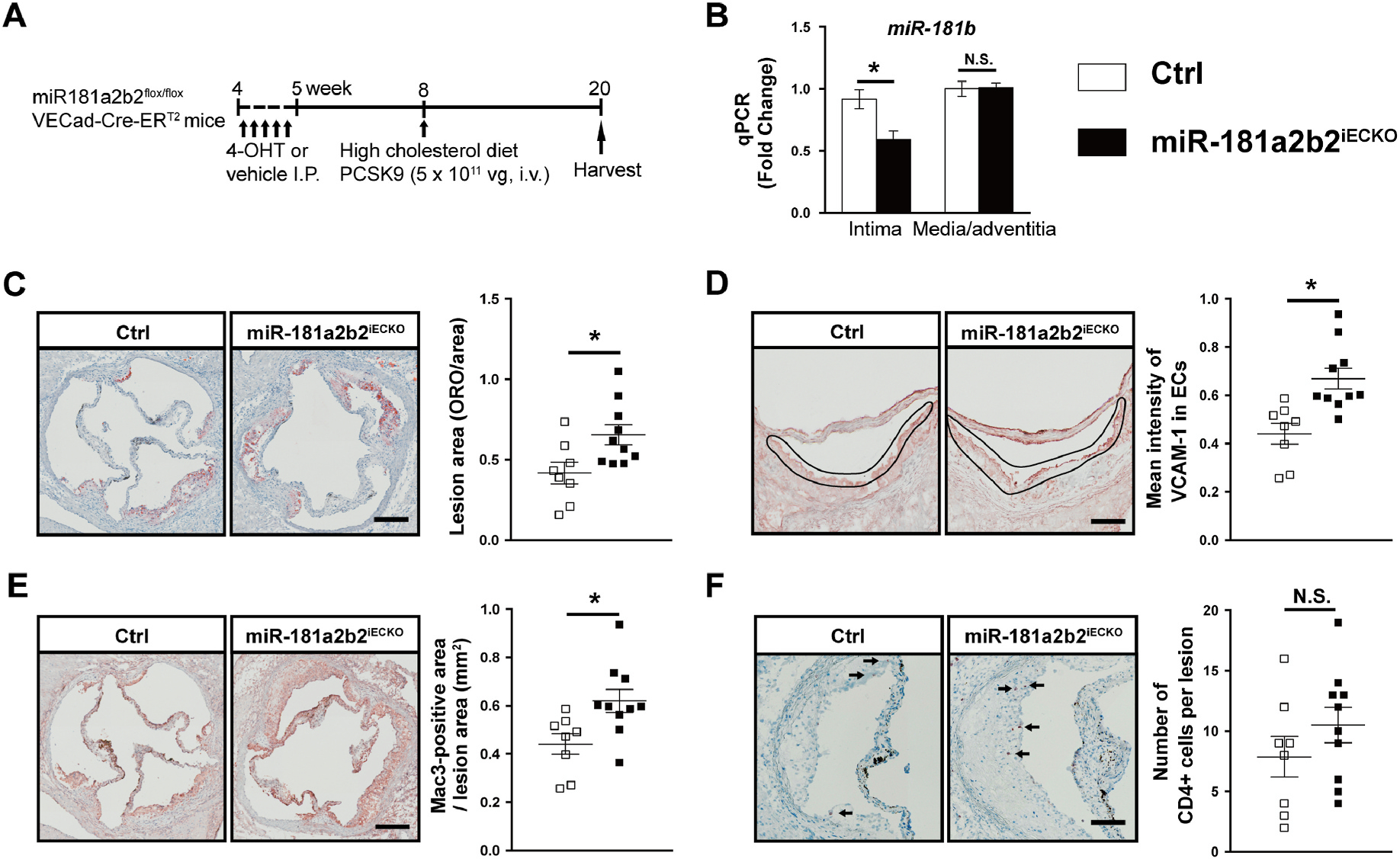
Endothelial cell-specific deficiency of miR-181a2b2 promotes atherosclerosis in a PCSK9-induced atherosclerosis mouse model.
(A) Schema of experimental procedure. miR-181a2b2 sufficient (Ctrl) or 4-hydroxytamoxifen (4-OHT)-induced endothelial cell (EC)-specific miR-181a2b2 deficient (miR-181a2b2ECKO) mice were injected with a single dose of AAV-PCSK9D377Y gain-of-function transgene (5 × 1011 vector genomes, i.v.) and fed a high cholesterol diet for 12 weeks. (B) Quantitative polymerase chain reaction (qPCR) analysis of miR-181b expression in the endothelial-enriched intima from the aortic arch and aortic media and adventitial tissues. The expression of miR-181b was normalized to U6 expression. (C) Lesion areas shown were quantified as areas between the lumen and tunica media on Oil-red O-stained aortic sinus sections. (D) Representative images and quantification of vascular cell adhesion molecule-1 (VCAM-1) staining in the intima layer in the aortic root lesions. (E) Representative images and quantification show Mac3-positive macrophages in aortic root lesions. (F) Representative images and quantification show CD4-positive T cells in aortic root lesions. Data shown are mean ± SEM. (B) n = 4 mice per group. (C to F), n = 8–10 mice per group. *p < 0.05, N.S. indicates nonsignificant.
After 12 weeks on western diet, miR-181a2b2iECKO mice showed similar body weights and levels of total cholesterol, triglyceride, low-density lipoprotein, and high-density lipoprotein compared to Ctrl mice (Supplemental Fig. IIB and C), suggesting endothelial-specific deletion of miR-181a2b2 did not affect body weights or lipid metabolism. Moreover, expression of PCSK9 in the liver was not different between Ctrl and miR-181a2b2iECKO mice and significantly higher than in C57BL/6J mice fed with normal chow diet (Supplemental Fig. IID). Analysis of atherosclerotic lesion formation by Oil Red O staining revealed markedly accelerated lesion size by 52% in miR-181a2b2iECKO mice compared to Ctrl group (Fig. 1C). The increase of atherosclerotic lesion formation in miR-181a2b2iECKO mice was associated with a 51% increase expression of the cell adhesion molecule VCAM-1 (Fig. 1D). In line with this observation, histological assessment of atherosclerotic lesions revealed a 40% accumulation of macrophages by Mac3 staining in miR-181a2b2iECKO compared to Ctrl mice and a non-significant increased trend in the numbers of CD4 and CD8 positive T cells (Fig. 1E, and Supplemental Fig. IIIA). No significant difference was observed in the accumulation of vascular smooth muscle cells in the lesion area (Supplemental Fig. IIIB). Moreover, endothelial-deficiency of miR-181a2b2 did not alter miR-181b expression and the mRNAs that encode pro-inflammatory or anti-inflammatory mediators in both bone marrow (BM) and peripheral blood mononuclear cells (PBMCs) (Supplemental Fig. IVA–D). Moreover, the anti-inflammatory Ly6Clow or the pro-inflammatory Ly6Cinterm and Ly6Chigh fractions were not significantly different in the circulating PBMCs from miR-181a2b2iECKO mice compared to those cells from Ctrl mice (Supplemental Fig. IVE). Collectively, these data indicate that endothelium-deficiency of miR-181a2b2 accelerates atherosclerotic lesion formation, at least in part, by activation of ECs and subsequent facilitation of circulating monocyte adhesion and accumulation.
It is well established that miRNAs exert their biological function by modulating a range of gene networks and pathways at the post transcriptional level [21]. To identify gene networks regulated by endothelial miR-181a2b2 in atherosclerotic lesions related to endothelial activation and inflammation, we sequenced RNA isolated from the aortic arch intima of western diet fed and PCSK9-injected Ctrl and miR-181a2b2iECKO mice (Fig. 2A). We identified 1031 genes that were differentially expressed (adjusted p-value < 0.1, log2 fold-change(1)). Top up- and downregulated genes, sorted by log2fold change, are listed in Fig. 2B. Consistent with our previous results (Fig. 1D and E, and Supplemental Fig. IIIA), gene set enrichment analysis (GSEA) of the differentially expressed genes uncovered several significantly regulated pathways associated with cell adhesion, inflammation, immune response, and cell cycle (Fig. 2C). Further analysis of the network of regulated gene interactions within the cell adhesion pathway showed several genes known to regulate monocyte adhesion (VCAM-1, ICAM-1, DOCK2) or macrophage foam cell formation (VAV-1) [22–24] (Fig. 2D). Moreover, the increased mRNA expression of VCAM-1, ICAM-1, and E-selectin was confirmed by RT-qPCR in ECs enriched from the aortic intima from miR-181a2b2iECKO mice (Fig. 2E). Expression of NF-κB p65, one of the main upstream drivers of these adhesion molecules, was also markedly increased in the vascular endothelium of miR-181a2b2iECKO mice compared to controls (Supplemental Figure VI). In line with this observation, isolated ECs from miR-181a2b2iECKO or Ctrl mice treated with TNF-α had higher leukocyte adhesion compared to ECs from Ctrl mice (Supplemental Figure VII). In addition to the cell adhesion pathway, pathways associated with the cell cycle and immune response were significantly changed by endothelium deficiency of miR-181a2b2 (Fig. 2C and Supplemental Fig. V). Finally, to explore new potential miR-181b targets in these lesions, we evaluated the common set of differentially expressed genes (DEG; p adj <0.01) from the RNA-seq and prediction algorithms (TargetScan)) to uncover 14 upregulated genes. Several of these are implicated in regulation of inflammation (e.g. Cst6, Tmem88), cell growth and differentiation (e.g. Twist1, Six2, Id4, Smad4), and cell cycle (e.g. Mycn). Taken together, these results indicate that endothelial miR-181a2b2 regulates a network of genes controlling endothelial activation, cell adhesion, cell cycle, and immune response in the accelerated lesions.
Fig. 2.
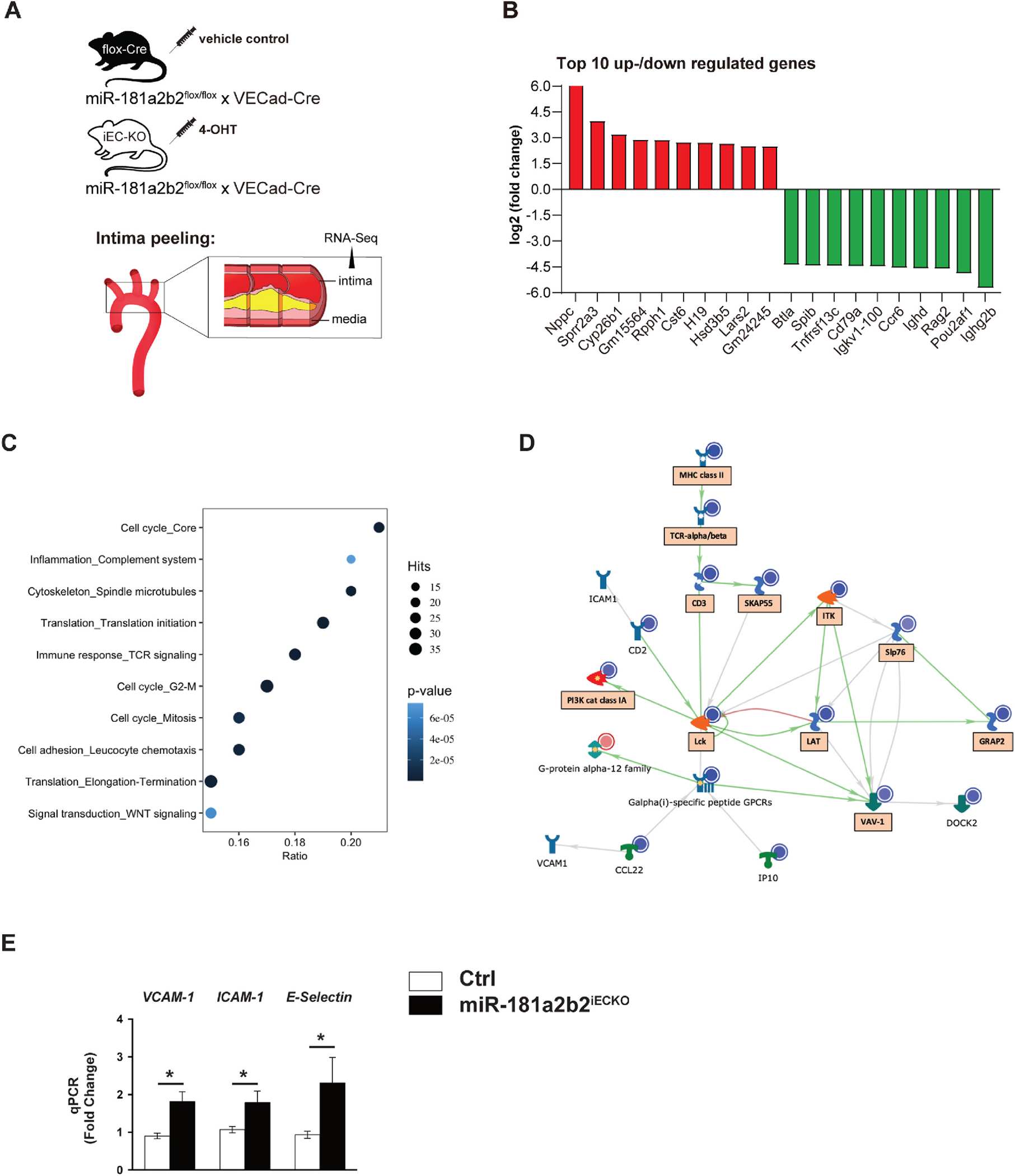
Lesional RNA-Seq studies from miR-181a2b2iECKO mice reveal activation of pathways involved in cell adhesion, cell cycle, and immune response.
(A) Scheme of experimental procedure. RNA sequence analysis of RNA isolated from aortic arch intimal ECs miR-181a2b2 sufficient (Ctrl) or 4-hydroxytamoxifen (4-OHT)-induced endothelial cell (EC)-specific miR-181a2b2 deficient (miR-181a2b2ECKO) mice after 12 weeks of high cholesterol diet and AAV-PCSK9D377Y injection as described in Fig. 1. (B) Top 10 up- or downregulated genes by fold change. (C) Gene set enrichment analysis of differentially expressed genes. Hits represent the number of differentially expressed genes per network. Color denotes p-value. Ratio represents the number of regulated genes divided by the number of expected genes from each network. (D) The network of regulated gene interactions within the ‘cell adhesion’ pathway. The objects that are up- or downregulated are marked with red or blue circles, respectively. Arrow color represents activation (green), inhibition (red), or unspecified (grey) effects. Genes in boxes are also found in ‘immune response’ and ‘inflammation’ pathways. (E) qPCR analysis of the indicated proinflammatory genes in the endothelial enriched intima from aortic arch. The expression of inflammatory genes was normalized to mouse β-actin expression. Data shown are mean ± SEM. (B to D), n = 3 mice per group. (E) n = 4 mice per group. *p < 0.05, N.S. indicates nonsignificance.
To further understand mechanistically how endothelial deficiency of miR-181a2b2 regulates the progression of atherosclerosis in mice, we performed single cell RNA-sequencing (scRNA-seq) of the aortic arch from Ctrl (WT) and miR-181a2b2iECKO (KO) after a single dose of AAV-PCSK9D377Y gain-of-function transgene and 12 weeks high cholesterol diet (Fig. 3A). Single-cell capture and library preparation were performed using a 10X Genomics Chromium Single Cell 3.1’ GEM, Library and Gel Bead Kit v3, followed by next-generation sequencing using Illumina platform. A total of 3,867 cells from 4 aortic arches (2 from Ctrl and 2 from miR-181a2b2iECKO) met quality control metrics and were then integrated and jointly subjected to downstream bioinformatics analysis (Supplemental Fig. VIII). Using the Seurat package [25], we identified 7 major aortic cell types based on enriched marker genes in each cluster (Fig. 3B and C, Supplemental Fig. IX and Supplemental Fig. XI). Interestingly, we found 2 distinct endothelial cell (EC) populations. Aortic endothelial cell subcluster 1 (EC_1) was identified based on the canonical EC markers, including Cdh5 and Egfl7 and has similar identity to the EC_2 subcluster in mouse aortas as reported previously [26]. In contrast, aortic endothelial cell subcluster 2 (EC_2) was found to highly express characteristics of immune cell-like markers, including Icam1, Vcam1, Il1b, cxcl2, S100a9, and Retnlg, and is distinct from the EC subclusters reported [26]. The percentage and the number of each aortic cell from Ctrl (WT) and miR-181a2b2iECKO (KO) aorta are shown in Fig. 3C and Supplemental Fig. X. Notably, we found that the percentage and the number of conventional EC (EC_1) decreased, whereas the percentage and the number of immune cell-like EC (EC_2), as well as macrophage and monocyte cells, increased in the KO aortas compared to WT. These observations were consistent with our increased Mac3 staining in the atherosclerotic lesions from miR-181a2b2iECKO (KO) (Fig. 1E). To further discover the regulatory role of endothelial deficiency of miR-181a2b2 in the lesion, we performed differential expression (DE) analysis between KO and WT. In the two EC subclusters, we found that Edn1, Icam1, Il-1β, Cxcl2, Lyz2, Nfkbia, and Aim2 were the top-ranking DE transcripts (FDR <0.05) in KO vs WT as shown by scatter plot and violin plot (Fig. 3E and F). Notably, endothelin-1 (Edn1), that was significantly increased in the EC_1 cluster, is known to be increased in experimental atherosclerosis in mice [27,28]. Several of these top-ranking regulated genes, i.e. Icam1, Il-1, Cxcl2, are also known to regulate inflammatory processes in atherosclerosis. To further assess the potential functional heterogeneity and signaling pathways enriched in aortic cell subsets, we performed gene ontology (GO) term analysis of the DEGs in the 2 EC subclusters, as well as on the macrophage and monocyte subclusters. GO term analysis of the EC subcluster DEGs (FDR <0.05) (Fig. 4A) uncovered several significantly regulated pathways associated with immune response, endothelial adherens and tight junction signaling (Fig. 4B). The GO term analysis in the macrophage and monocyte subclusters (Fig. 4C) showed several inflammatory associated biological processes enriched; including thrombin, IL-8 signaling, ILK signaling, and reactive oxygen species, among other inflammatory terms (Fig. 4D). These observations are consistent with our in vivo results, indicating that endothelial miR-181a2b2 regulates endothelial function and immune response in these accelerated lesions.
Fig. 3.

Single-cell RNA sequencing revealed the major cell types and key transcription signatures regulating atherosclerosis in miR-181a2b2iECKO mice.
(A) Schematic diagram of aortic arch cells isolation from Ctrl (WT) and miR-181a2b2iECKO(KO) mice for single-cell RNA sequencing. Two aortic arches were pooled for one sample for sequencing in WT and KO groups. (B) Uniform Manifold Approximation and Projection (UMAP) of different aortic cell types. EC_1: endothelial cell cluster 1; EC_2: endothelial cell cluster 2; VSMC: vascular smooth muscle cell cluster. (C) Pie chart showing the percentage of different cell types. (D) Heatmap of top 10 marker genes for each cluster. Selected cell type-specific markers labeled. Full marker list is shown in Supplemental Fig. VIII. (E) Scatter plot showing selected significantly regulated genes in endothelial cell groups (EC_1 and EC_2) (KO vs WT, FDR, <0.05). (F) The expression levels of Edn1, Icam1, Il1b, Lyz2, Hba-a1, Hba-a2, Tnfaip3, Cxcl2, Nfkia, Srgn, Wfdc17 are shown by Violin plots.
Fig. 4.

Single-cell RNA sequencing studies from miR-181a2b2iECKO mice revealactivation of inflammatory and cellular pathways in endothelial and macrophage and monocyte subclusters.
(A) Volcano plot highlighting significantly regulated genes in EC_1 and EC_2 subclusters from Ctrl (WT) and miR-181a2b2iECKO(KO) mice. (B) Ingenuity Pathway Analysis (IPA) of Canonical pathways from differentially expressed genes from EC_1 and EC_2 reveals the top 20 pathways significantly regulated. (C) Volcano plot highlighting significantly regulated genes in macrophage and monocyte subclusters from Ctrl (WT) and miR-181a2b2iECKO(KO) mice. (D) IPA Canonical pathway analysis showing the top 20 pathways from differentially expressed genes in macrophages and monocytes.
4. Discussion
EC activation is a hallmark of chronic atheroinflammation. Pharmacotherapies that inhibit EC inflammation hold promise for treating atherosclerosis [1]. We previously demonstrated that systemic delivery of miR-181b mimics overexpression of miR-181b in the aortic intima of ApoE−/− mice and significantly inhibits atherosclerotic lesion formation, pro-inflammatory genes expression and the accumulation of lesional macrophages and CD4 positive T cells in the vessel wall [11]. However, a cell-autonomous role for miR-181b in vascular endothelium, vascular inflammation, and atherosclerosis remained unknown. The impact of miRNAs in atherogenesis by endothelial-specific genetic targeting has not been characterized to date. Moreover, we previously demonstrated that endogenous miR-181b expression is reduced by six weeks of high cholesterol diet in ApoE−/− mice [11]. Deficiency of miR-181a2b2 under the control of the VE-cadherin (Cdh5) promoter allowed for assessment of immediate and sustained reduction in its expression and contribution, specifically in the vascular endothelium in this mouse model. Herein, we provide evidence that endothelium-specific deletion of miR-181a2b2 promotes ECs activation, accelerates lesional macrophage accumulation, and aggravates atherosclerosis. That these effects became manifest with only a ~40% reduction of endothelial miR-181b expression in the miR-181a2b2iECKO mice highlights that miRNAs can exert robust phenotypic effects with moderate changes in abundance. Moreover, these atherosclerotic phenotype changes were not associated with alterations in lipid metabolism or inflammatory gene expression in the bone marrow or PBMCs, suggesting miR-181a2b2-driven effects in EC activation and in turn atherosclerotic lesion formation. Although we observed that the expression of primary miR-181b2 transcript was rapidly reduced by pro-inflammatory stimuli and decreased in ECs enriched from the aortic intima of ApoE−/− mice, future studies will be of interest to explore the role of the related primary miR-181b1 transcript on EC inflammation and atherosclerosis.
Emerging evidence from experimental studies indicates that miRNAs act in a tissue and/or cell type specific manner and regulate expression of large gene networks in certain conditions, not just for an individual gene [21,29,30]. In agreement with these results, gene expression analysis and GSEA from RNA-seq data isolated from aortic arch intimal-enriched ECs showed an abundant number of changes in gene expression after endothelial-specific deletion of miR-181a2b2 during atherosclerosis. Moreover, several differentially expressed gene networks related to critical pathways controlling cell adhesion, cell cycle, and immune response emerged in the accelerated lesions of miR-181a2b2iECKO mice and are known to contribute to the initiation and progression of atherosclerosis [24,31,32]. We also uncovered a set of new putative miR-181b targets that were upregulated in the lesions of miR-181a2b2iECKO mice using the overlapping set of differentially expressed genes from the RNA-Seq and the predicted miR-181b gene targets (Fig. 2E). Given that several of these genes are implicated in regulation of inflammation (e.g. Cst6, Tmem88), cell growth and differentiation (e.g. Twist1, Six2, Id4, Smad4), and cell cycle (e.g. Mycn), future studies will be of interest to assess their role and relative dependency in atherosclerotic miR-181a2b2iECKO mice. For example, Cst6, a member of the superfamily of cysteine protease inhibitors, is a ley regulatory protein of the extracellular matrix by inhibiting cathepsin B, L, V, and legumain [33]; however, it remains unclear whether the increased expression found here is related to a direct or compensatory mechanism in the vessel wall. Similarly, TWIST1 is expressed at low shear stress in regions of adult arteries and contributes to progression of atherosclerosis by triggering inflammation, EC permeability and proliferation, an effect that may underlie the increased lesions of the miR-181a2b2iECKO mice [34]. Increased expression of Smad4, known as a common Smad that mediates signal transduction of the TGF-β family, may impact extracellular matrix, fibrosis, or arterial stiffening [35]. Finally, increased MycN, a member of the MYC family known to regulate transcriptional activation in multiple tumor types, may facilitate the growth of nascent vascular lesions [36]. Although the GSEA studies identified TCR signaling as different between groups, there was only a non-statistical trend for higher CD4+ T cell accumulation in the miR-181a2b2iECKO mice compared to controls. Given the higher sensitivity for RNA-seq than histological analysis to uncover specific signaling pathways, we interpret this data from these different approaches as closely aligned. Future studies will need to clarify TCR-specific pathways that may be impacted by EC-specific miR-181a2b2 deficiency. These data underscore the cell-autonomous impact of a miRNA on critical pathways and targets involved in the vascular endothelium and subsequent atherosclerotic lesion progression.
Advances in scRNA-sequencing provide the necessary resolution to identify the heterogeneous subpopulations in aortic cells during progression of atherosclerosis. Based on the scRNA-seq gene expression profiles of two distinct EC subpopulations, EC_1 subcluster represented a conventional type of ECs and highly expressed canonical EC markers such as Cdh5. In contrast, the EC_2 subcluster highly expressed several inflammatory or immune-related gene markers including Vcam1 and Retnlg. Endothelial-specific deletion of miR-181a2b2 decreased the percentage and number of the conventional type of ECs and increased the percentage and number of immune-like ECs, indicating an inflammatory shift in the vessel wall. Consistent with the immuno-histological observation of increased Mac3 in the lesion area, scRNA-seq also showed increased accumulation of macrophage and monocytes. Among the most expressed genes in endothelial cells, endothelin-1 (Edn1) was specifically enriched in the EC_1 subcluster and was significantly increased in miR-181a2b2 deficient mice. Interestingly, Edn1 contains a putative miR-181b binding site in its 3′-UTR (Supplemental Figure XII), is a well-established marker of inflammation, exacerbates atherosclerosis in mice, and has been linked causally to coronary artery disease in human subjects [27,28,37–39]. The increased expression of a group of immune-related genes, including Cxcl2, Icam1, Il-1β, Cxcl2, Lyz2, Aim2 and Nfkbia [40], in the EC subclusters of miR-181a2b2iECKO (KO) mice also highlights a broader level of inflammation triggered in these cells. Together with the GO term analysis of EC subclusters and macrophage and monocytes, our observations collectively indicated that endothelial-specific deletion of miR-181a2b2 triggers vascular wall inflammation and accelerated lesional macrophage activation and accumulation in the progression of atherosclerosis.
In summary, endothelial-specific deficiency of miR-181a2b2 triggers EC activation, vascular wall inflammation, and atherosclerotic lesion development by regulating key endothelial pathways related to EC inflammation, cell adhesion, immune response, and cell cycle. Therapies aimed at rescuing miR-181b expression in the vascular endothelium may provide an interesting target to limit atherosclerosis and cardiovascular disease.
Supplementary Material
Acknowledgments
The authors would like to thank Ana Lay-Hong for their assistance with immunofluorescence imaging (Harvard Digestive Disease Center, NIH P30DK034854) and Vijaya Bhaskar Baki for help with endothelial cell isolation.
Financial support
This work was supported by the National Institutes of Health (HL115141, HL134849, HL148207, HL148355, HL153356 to M.W.F.; HL150536 to X.S), and the American Heart Association (18SFR N33900144 and 20SFRN35200163 to M.W.F., 18POST34030395 to S. H.)
Footnotes
CRediT authorship contribution statement
Dafeng Yang: performed the experiments, designed and interpreted the results, wrote the manuscript. Stefan Haemmig: performed the experiments, designed and interpreted the results, wrote the manuscript. Jingshu Chen: performed the experiments, designed and interpreted the results, wrote the manuscript. Michael McCoy: performed the experiments, designed and interpreted the results. Henry S. Cheng: performed the experiments, designed and interpreted the results. Haoyang Zhou: performed the experiments, designed and interpreted the results. Daniel Pérez-Cremades: performed the experiments, designed and interpreted the results. Xiao Cheng: performed the experiments. Xinghui Sun: performed the experiments, designed and interpreted the results. Jorge Haneo-Mejia: designed and interpreted the results. Shamsudheen K. Vellarikkal: designed and interpreted the results. Rajat M. Gupta: designed and interpreted the results. Victor Barrera: designed and interpreted the results. Mark W. Feinberg: Conceived the hypothesis, designed and interpreted the results, wrote the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.atherosclerosis.2022.04.010.
References
- [1].Back M, Yurdagul A Jr., Tabas I, Oorni K, Kovanen PT, Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities, Nat. Rev. Cardiol. 16 (2019) 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Libby P, Inflammation in atherosclerosis, Arterioscler. Thromb. Vasc. Biol. 32 (2012) 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gimbrone MA Jr., Garcia-Cardena, Endothelial cell dysfunction and the pathobiology of atherosclerosis, Circ. Res. 118 (2016) 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ley K, Laudanna C, Cybulsky MI, Nourshargh S, Getting to the site of inflammation: the leukocyte adhesion cascade updated, Nat. Rev. Immunol. 7 (2007) 678–689. [DOI] [PubMed] [Google Scholar]
- [5].Hansson GK, Inflammation, atherosclerosis, and coronary artery disease, N. Engl. J. Med. 352 (2005) 1685–1695. [DOI] [PubMed] [Google Scholar]
- [6].Sun X, Belkin N, Feinberg MW, Endothelial microRNAs and atherosclerosis, Curr. Atherosclerosis Rep. 15 (2013) 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M, Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis, Cell Metabol. 8 (2008) 372–383. [DOI] [PubMed] [Google Scholar]
- [8].Moss ME, Lu Q, Iyer SL, Engelbertsen D, Marzolla V, Caprio M, Lichtman AH, Jaffe IZ, Endothelial mineralocorticoid receptors contribute to vascular inflammation in atherosclerosis in a sex-specific manner, Arterioscler. Thromb. Vasc. Biol. 39 (2019) 1588–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Registry M, Blackwell TS, Baron RM, Feinberg MW, MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation, J. Clin. Invest. 122 (2012) 1973–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun X, Lin J, Zhang Y, Kang S, Belkin N, Wara AK, Icli B, Hamburg NM, Li D, Feinberg MW, MicroRNA-181b improves glucose homeostasis and insulin sensitivity by regulating endothelial function in white adipose tissue, Circ. Res. 118 (2016) 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sun X, He S, Wara AKM, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW, Systemic delivery of microRNA-181b inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice, Circ. Res. 114 (2014) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang D, Haemmig S, Zhou H, Perez-Cremades D, Sun X, Chen L, Li J, Haneo-Mejia J, Yang T, Hollan I, Feinberg MW, Methotrexate attenuates vascular inflammation through an adenosine-microRNA-dependent pathway, Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limon P, Kaech SM, Nakayama M, Rinn JL, Flavell RA, The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis, Immunity 38 (2013) 984–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH, Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis, Nature 465 (2010) 483–486. [DOI] [PubMed] [Google Scholar]
- [15].Cheng X, Shihabudeen Haider Ali MS, Moran M, Viana MP, Schlichte SL, Zimmerman MC, Khalimonchuk O, Feinberg MW, Sun X, Long non-coding RNA Meg3 deficiency impairs glucose homeostasis and insulin signaling by inducing cellular senescence of hepatic endothelium in obesity, Redox Biol. 40 (2021), 101863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Haemmig S, Yang D, Sun X, Das D, Ghaffari S, Molinaro R, Chen L, Deng Y, Freeman D, Moullan N, Tesmenitsky Y, Wara A, Simion V, Shvartz E, Lee JF, Yang T, Sukova G, Marto JA, Stone PH, Lee WL, Auwerx J, Libby P, Feinberg MW, Long noncoding RNA SNHG12 integrates a DNA-PK-mediated DNA damage response and vascular senescence, Sci. Transl. Med. 12 (2020). [DOI] [PubMed] [Google Scholar]
- [17].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics 29 (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features, Bioinformatics 30 (2014) 923–930. [DOI] [PubMed] [Google Scholar]
- [19].Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data, Nat. Biotechnol. 33 (2015) 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sun X, Sit A, Feinberg MW, Role of miR-181 family in regulating vascular inflammation and immunity, Trends Cardiovasc. Med. 24 (2014) 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bartel DP, Chen CZ, Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs, Nat. Rev. Genet. 5 (2004) 396–400. [DOI] [PubMed] [Google Scholar]
- [22].Garcia-Bernal D, Sotillo-Mallo E, Nombela-Arrieta C, Samaniego R, Fukui Y, Stein JV, Teixido J, DOCK2 is required for chemokine-promoted human T lymphocyte adhesion under shear stress mediated by the integrin alpha4beta1, J. Immunol. 177 (2006) 5215–5225. [DOI] [PubMed] [Google Scholar]
- [23].Rahaman SO, Swat W, Febbraio M, Silverstein RL, Vav family Rho guanine nucleotide exchange factors regulate CD36-mediated macrophage foam cell formation, J. Biol. Chem. 286 (2011) 7010–7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Blankenberg S, Barbaux S, Tiret L, Adhesion molecules and atherosclerosis, Atherosclerosis 170 (2003) 191–203. [DOI] [PubMed] [Google Scholar]
- [25].Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data, Nat. Biotechnol. 33 (2015) 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM, Single-cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell populations, Circulation 140 (2019) 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li MW, Mian MOR, Barhoumi T, Rehman A, Mann K, Paradis P, Schiffrin EL, Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice, Arterioscler. Thromb. Vasc. Biol. 33 (2013) 2306–2315. [DOI] [PubMed] [Google Scholar]
- [28].Barton M, Haudenschild CC, d’Uscio LV, Shaw S, Münter K, Lüscher TF, Endothelin ETA receptor blockade restores NO-mediated endothelial function and inhibits atherosclerosis in apolipoprotein E-deficient mice, Proc. Natl. Acad. Sci. Unit. States Am. 95 (1998) 14367–14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Virtue AT, McCright SJ, Wright JM, Jimenez MT, Mowel WK, Kotzin JJ, Joannas L, Basavappa MG, Spencer SP, Clark ML, Eisennagel SH, Williams A, Levy M, Manne S, Henrickson SE, Wherry EJ, Thaiss CA, Elinav E, Henao-Mejia J, The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs, Sci. Transl. Med. 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cheng HS, Besla R, Li A, Chen Z, Shikatani EA, Nazari-Jahantigh M, Hammoutene A, Nguyen MA, Geoffrion M, Cai L, Khyzha N, Li T, MacParland SA, Husain M, Cybulsky MI, Boulanger CM, Temel RE, Schober A, Rayner KJ, Robbins CS, Fish JE, Paradoxical suppression of atherosclerosis in the absence of microRNA-146a, Circ. Res. 121 (2017) 354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Foks AC, Kuiper J, Immune checkpoint proteins: exploring their therapeutic potential to regulate atherosclerosis, Br. J. Pharmacol. 174 (2017) 3940–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zettler ME, Pierce GN, Cell cycle proteins and atherosclerosis, Herz 25 (2000) 100–107. [DOI] [PubMed] [Google Scholar]
- [33].Lalmanach G, Kasabova-Arjomand M, Lecaille F, Saidi A, Cystatin M/E (cystatin 6): a janus-faced cysteine protease inhibitor with both tumor-suppressing and tumor-promoting functions, Cancers 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mahmoud MM, Kim HR, Xing R, Hsiao S, Mammoto A, Chen J, Serbanovic-Canic J, Feng S, Bowden NP, Maguire R, Ariaans M, Francis SE, Weinberg PD, van der Heiden K, Jones EA, Chico TJ, Ridger V, Evans PC, TWIST1 integrates endothelial responses to flow in vascular dysfunction and atherosclerosis, Circ. Res. 119 (2016) 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wara AK, Foo S, Croce K, Sun X, Icli B, Tesmenitsky Y, Esen F, Lee JS, Subramaniam M, Spelsberg TC, Lev EI, Leshem-Lev D, Pande RL, Creager MA, Rosenzweig A, Feinberg MW, TGF-beta1 signaling and Kruppel-like factor 10 regulate bone marrow-derived proangiogenic cell differentiation, function, and neovascularization, Blood 118 (2011) 6450–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu Z, Chen SS, Clarke S, Veschi V, Thiele CJ, Targeting MYCN in pediatric and adult cancers, Front. Oncol. 10 (2020), 623679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gupta RM, Libby P, Barton M, Linking regulation of nitric oxide to endothelin-1: the Yin and Yang of vascular tone in the atherosclerotic plaque, Atherosclerosis 292 (2020) 201–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CR, Bianchi V, Mueller C, A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression, Cell 170 (2017) 522–533, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, The allelic landscape of human blood cell trait variation and links to common complex disease, Cell 167 (2016) 1415–1429, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba A-E, Zernecke A, Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis, Circ. Res. 122 (2018) 1661–1674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.