

Abstract
Insulin-degrading enzyme (IDE) is an atypical zinc-metallopeptidase that degrades insulin and the amyloid ß-protein and is strongly implicated in the pathogenesis of diabetes and Alzheimer’s disease. We recently developed the first effective inhibitors of IDE, peptide hydroxamates that, while highly potent and selective, are relatively large (MW >740) and difficult to synthesize. We present here a facile synthetic route that yields enantiomerically pure derivatives comparable in potency to the parent compounds. Through the generation of truncated variants, we identified a compound with significantly reduced size (MW = 455.5) that nonetheless retains good potency (ki = 78 ± 11 nM) and selectivity for IDE. Notably, the potency of these inhibitors was found to vary as much as 60-fold in a substrate-specific manner, an unexpected finding for active-site directed inhibitors. Collectively, our findings demonstrate that potent, small-molecule IDE inhibitors can be developed that, in certain instances, can be highly substrate selective.
Keywords: insulin-degrading enzyme, diabetes melittus, Alzheimer disease, protease inhibitors, hydroxamic acids
Introduction
Insulin-degrading enzyme (IDE) is a structurally distinctive zinc-metalloprotease responsible for catabolizing insulin, the amyloid ß-protein (Aß) and other intermediate-sized peptide substrates1. IDE is strongly implicated in the pathogenesis of multiple highly prevalent diseases, including type 2 diabetes melittus (T2DM), Alzheimer’s disease, and varicella zoster virus infection2–4. Despite its clear biological significance, elucidation of IDE’s role in these disorders, as well as its fundamental role in regulating insulin signaling5, has been hindered by the lack of suitable pharmacological inhibitors. Moreover, considerable evidence suggests that IDE inhibitors may hold therapeutic value, particularly for T2DM and other disorders involving impaired insulin signaling2, 6.
IDE possesses a highly distinctive “clamshell”-like structure, consisting of two bowl-shaped halves (IDE-N and IDE-C), which are connected by a flexible linker, allowing the protease to transition between “closed” and “open” conformations1. In the closed conformation the two halves encapsulate a large (~13,000 Å3) internal chamber1. The active site of IDE is bipartite, being made up of residues in both the IDE-N and IDE-C domains, and is only fully formed when the protease is in the closed conformation7.
Using a substrate-based, rational design approach, we recently developed conventional and retro-inverso peptide hydroxamate inhibitors of IDE8. These compounds are >105-fold more potent than previously described inhibitors and, despite their relatively primitive design, show remarkable selectivity for IDE vis-a-vis other zinc-metalloproteases (~104-fold). Despite these merits, as relatively large (MW >740), peptidic compounds that are difficult to synthesize, these inhibitors are less than ideal as chemical probes or pharmacophores for future drug development.
To address these limitations, we describe here the synthesis and characterization of multiple variants of our original, 4-residue peptide hydroxamate IDE inhibitors, including variants developed via a facile alternative synthetic route. Using an IDE-inhibitor co-crystal structure as a guide, we focused on the effects of truncation, modification of the P2’ residue, and variation of the terminal group. Among other advances, we describe a novel variant that is considerably smaller than the parent compound (MW = 455.5) that nonetheless exhibits good potency (ki = 87 ± 11 nM). Through subsequent characterization of a diverse set of conventional and retro-inverso peptide hydroxamates, we discovered that the potency of certain compounds varies dramatically in a substrate-dependent manner, a highly unexpected finding for active site-directed inhibitors. To account for this unusual property, we propose a novel model wherein interactions among the enzyme, substrate, and inhibitor act in concert to determine the effective potency of inhibition. The insights gained from this study may facilitate the eventual development of more drug-like IDE inhibitors, including substrate-selective compounds.
Results
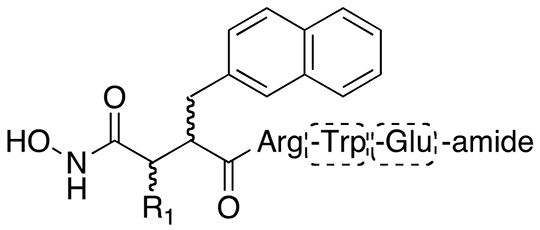
Previously, we used a substrate-based, rational-design approach9 to develop peptide hydroxamate inhibitors of IDE (Figure 1A)8. Optimization by incorporation of 2-naphthylalanine (2Nap) at the P1’ position yielded the highly potent (ki = 2.96 ± 0.20 nM), 4-residue conventional peptide hydroxamate, IDE inhibitor 1, (Ii1, 1a, Figure 1B). We also generated the corresponding retro-inverso peptide hydroxamate 2 (Figure 1C), that, though less potent (ki = 78 ± 4.2 nM), had the advantage of being readily synthesized by solid-phase peptide synthesis (SPPS) from commercially available components8.
Figure 1.
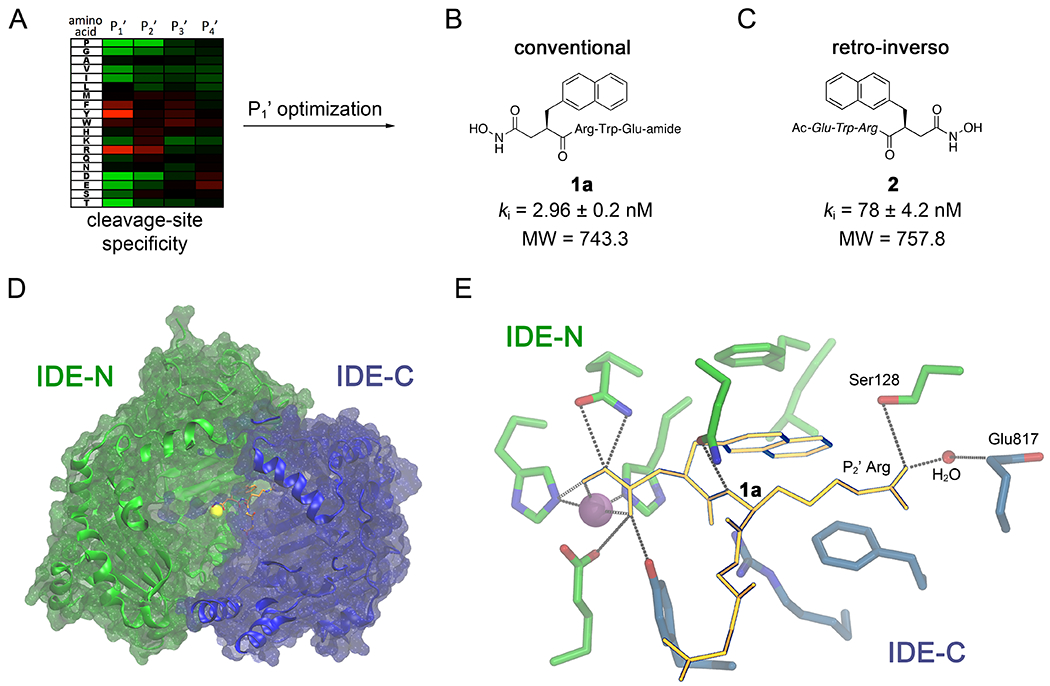
Development of the first peptide hydroxamate IDE inhibitors and mechanistic insights derived from the inhibitor 1a:IDE co-crystal structure. A, Overview of the rational design approach used to develop peptide hydroxamate inhibitor 1a (B), which involved determination of the cleavage-site specificity (heat map) and subsequent optimization of the P1’ position. Note that IDE shows comparatively little preference for specific amino acids at the P3’ and P4’ positions. B,C, Conventional (B) and retro-inverso (C) peptide hydroxamate inhibitors derived from the approach outlined in (A). Note that inhibitor 1a (B) is highly potent but relatively large in size. D-amino acids are depicted in italics. D, The co-crystal structure of inhibitor 1a (orange sticks) bound to a single monomer of human IDE (blue and green ribbons). Note that inhibitor 1a is positioned between IDE-N (green), which contains the active-site zinc (yellow sphere), and IDE-C (blue). E, Close-up depiction of inhibitor 1a (orange sticks) bound to the active-site of IDE, highlighting the interaction with the zinc atom (orange sphere) and residues within IDE-N (green sticks) and IDE-C (blue sticks). Note that the guanidinium moiety within the P2’ Arg residue makes indirect contact with Glu817 via an intervening water molecule.
The co-crystal structure of inhibitor 1a bound to human IDE was subsequently determined at 2.6-Å resolution8 (Figure 1D), revealing several salient features informing the present study. First, the positioning of the P3’ and P4’ residues was not resolvable from the co-crystal structure, and these residues were instead predicted to protrude into the internal chamber of IDE. Analysis of the cleavage-site specificity of IDE also showed no preference for specific residues at these positions. Together, these results suggest that the P3’ and P4’ residues might be dispensable for effective IDE inhibition. Second, inhibitor 1a makes extensive contacts not only with IDE-N, which contains the residues involved in zinc-binding and catalysis, but also with IDE-C, portions of which make up the second half of IDE’s bipartite active site7. This configuration suggests that inhibitor 1a exerts its inhibitory effect by maintaining IDE in the “closed” conformation. Third, the latter feature appeared to be mediated in part by the guanidinium functionality of the P2’ Arg residue, which appears well positioned to interact electrostatically with Glu817 within IDE-C. Nevertheless, this interaction is not direct but is instead mediated via an intervening water molecule, suggesting it might be strengthened by extending the linker joining the guanidinium functionality to the peptide backbone.
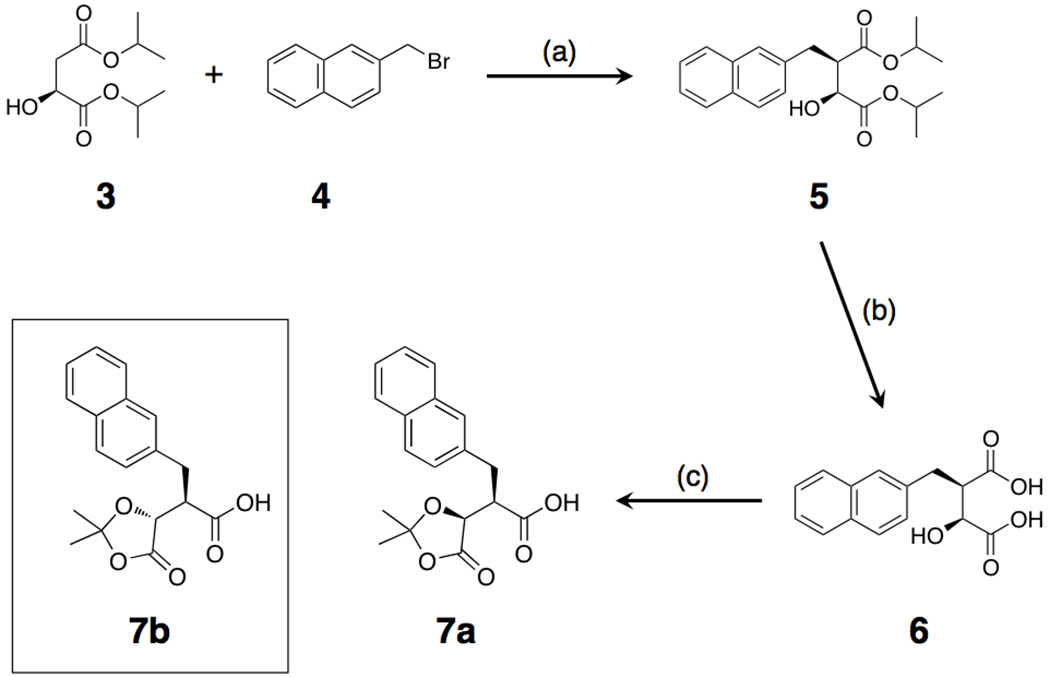
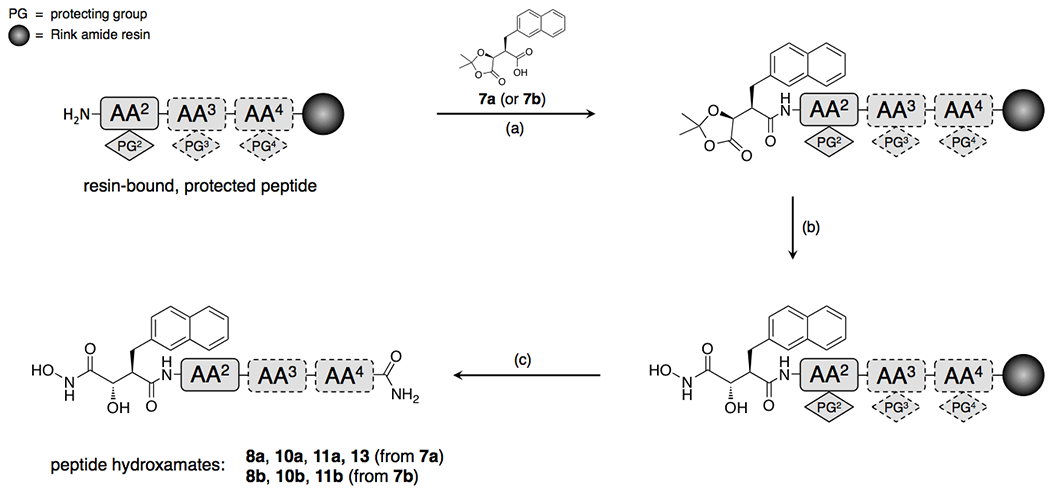
Although the bulk of inhibitor 1a is simply a conventional peptide, synthesis of the 2Nap-containing P1’ precursor in enantiomerically pure form is cumbersome, requiring a complex, multi-step synthesis with a relatively poor yield8. We therefore tested an alternative synthetic route, following Cravatt and colleagues10, wherein the P1’ residue and hydroxamate functionality are generated from ketal-based precursors 7a or 7b (Scheme 1), which can in turn be readily attached to peptides generated by SPPS (Scheme 2). This approach results in enantiomerically pure products in comparatively high yield, albeit containing an −OH group attached to the α-carbon adjacent to the hydroxamate in one of two possible orientations (Scheme 2).
Scheme 1.
Scheme 2.
Variants of the 4-residue inhibitor 1a with the α-carbon −OH group in the S and R orientation (inhibitors 8a and 8b, respectively) exhibited ki values of 25.9 ± 1.2 and 103 ± 6.9 nM, respectively (Table 1), using a well-characterized assay involving the fluorogenic peptide substrate, FRET111. Although inhibitors 8a and 8b were less potent than the parent inhibitor 1a, their ease of synthesis represents a distinct advantage so derivatives of these compounds were included in subsequent SAR studies.
Table 1.
Relative potency of 4-residue conventional peptide hydroxamate IDE inhibitors generated by alternate synthetic routes.
| |||
|---|---|---|---|
| Cmpd. | R1 | R1 (R/S) | ki (nM) (FRET1) |
| 1a | H | - | 2.96 ± 0.20 (n = 3) |
| 8a | OH | S | 25.9 ± 1.2 (n = 3) |
| 8b | OH | R | 103 ± 6.9 (n = 3) |
Based on the observation that IDE lacks preferences for specific amino acids at the P3’ and P4’ positions (Figure 1A), together with the findings that these residues were poorly resolved in the inhibitor 1a:IDE co-crystal structure, with the peptide backbone projecting into the internal chamber (Figure 1D), we investigated whether these residues were not essential for potent inhibition of IDE. To that end, we generated truncated forms of inhibitors 1a, 8a and 8b lacking one or both of these residues. The synthetic route used to generate variants of inhibitor 1a (Scheme 3) yields products with the 2Nap residue in both the S and R orientations, so diastereomers of both orientations were generated and characterized. The truncated, 2-residue variant of inhibitor 1a—inhibitor 9a—exhibited dramatically poorer potency (ki = 1.15 ± 0.38 μM). Consistent with previous results8, the variant of inhibitor 1a containing 2Nap in the S orientation, inhibitor 1b, was significantly less potent than the parent molecule (ki = 73.3 ± 4.2 nM). This pattern held for the corresponding 2-residue variant, inhibitor 9b, which was found to be inactive. Truncation of inhibitors 8a and 8b, containing the α-carbon −OH functionality in the S and R orientation, respectively, resulted in a similar diminution of potency (Table 2). Of note, the 2-residue variant of inhibitor 8a, inhibitor 11a, was unexpectedly found to be more potent (ki = 7.75 ± 1.6 μM) than the 3-residue version, inhibitor 10a (ki = 18 ± 1.1 μM). By contrast, 3- and 2-residue versions of inhibitor 7b (inhibitors 10b and 11b, respectively) exhibited potencies that decreased in proportion to the degree of truncation (ki = 498 ± 10 nM and 7.02 ± 0.33 μM, respectively).
Scheme 3.
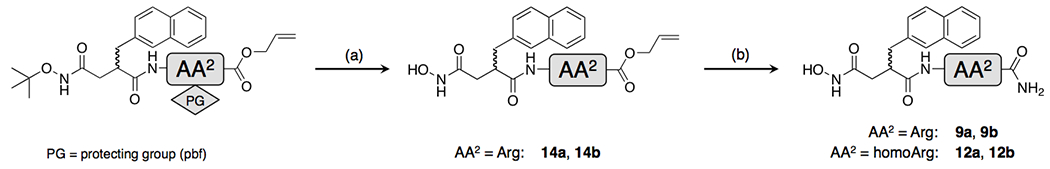
Table 2.
Effects of truncation on potency of conventional peptide hydroxamate IDE inhibitors.
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd. | R1 | R1 (R/S) | 2Nap (R/S) | AA2 | AA3 | AA4 | ki, nM (FRET1) |
| 1a | H | - | R | Arg | Trp | Glu | 2.96 ± 0.20 (n = 3) |
| 9a | H | - | R | Arg | - | - | 1150 ± 380 (n = 3) |
| 1b | H | - | S | Arg | Trp | Glu | 73.3 ± 40 (n = 3) |
| 9b | H | - | S | Arg | - | - | >33000 (n = 3) |
| 8a | OH | S | R | Arg | Trp | Glu | 26.0 ± 1.2 (n = 3) |
| 10a | OH | S | R | Arg | Trp | - | 18000 ± 1100 (n = 3) |
| 11a | OH | S | R | Arg | - | - | 7750 ± 1600 (n = 3) |
| 8b | OH | R | R | Arg | Trp | Glu | 103 ± 7.0 (n = 3) |
| 10b | OH | R | R | Arg | Trp | - | 498 ± 10 (n = 3) |
| 11b | OH | R | R | Arg | - | - | 7020 ± 330 (n = 3) |
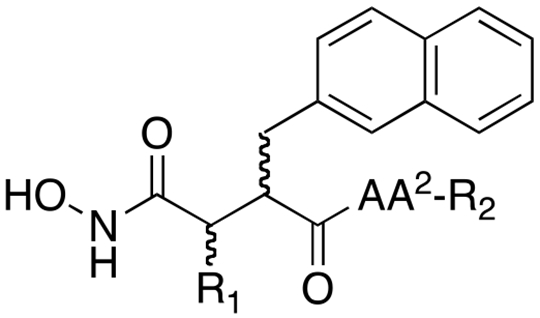
The inhibitor 1a:IDE co-crystal structure (Figure 1E) suggests that the P2’ Arg residue makes a potentially key interaction with Glu817, which we hypothesized might be further optimized by extending the linker between the guanidinium group and the peptide backbone. To test this, we generated variants of inhibitors 9a, 9b and 11a containing homoArg in the place of Arg (inhibitors 12a, 12b and 13, respectively). Rather than improving potency, this modification was found instead to abolish activity (Table 3). To investigate possible reasons for this, we conducted computational docking of inhibitor 9a with the crystal structure of IDE (see Experimental Section). In the top-scoring poses, the longer linker in homoArg unexpectedly permitted the guanidinium functionality to interact with Ser138, resulting, in turn, in the displacement of the P1’ 2Nap residue from the S1’ pocket (Figure 2). Thus, rather than promoting electrostatic interactions between the guanidinium and Glu817, as hypothesized, the longer linker in homoArg appears to promote alternative, non-optimal binding modes.
Table 3.
Effects of modification of the P2’ moiety and terminal group on potency of conventional peptide hydroxamate IDE inhibitors.
| ||||||
|---|---|---|---|---|---|---|
| Cmpd. | R1 | R1 (R/S) | 2Nap (R/S) | AA2 | R2 | ki, nM (FRET1) |
| 9a | H | - | R | Arg | NH2 | 11,500 ± 380 (n = 3) |
| 12a | H | - | R | homoArg | NH2 | >33,000 (n = 3) |
| 9b | H | - | S | Arg | NH2 | >33,000 (n = 3) |
| 12b | H | - | S | homoArg | NH2 | >33,000 (n = 3) |
| 11a | OH | S | R | Arg | NH2 | 7,750 ± 1600 (n = 3) |
| 13 | OH | S | R | homoArg | NH2 | >33,000 (n = 3) |
| 14a | H | - | R | Arg | O-allyl | 87 ± 11 (n = 3) |
| 14b | H | - | S | Arg | O-allyl | 5,280 ± 100 (n = 3) |
Figure 2.
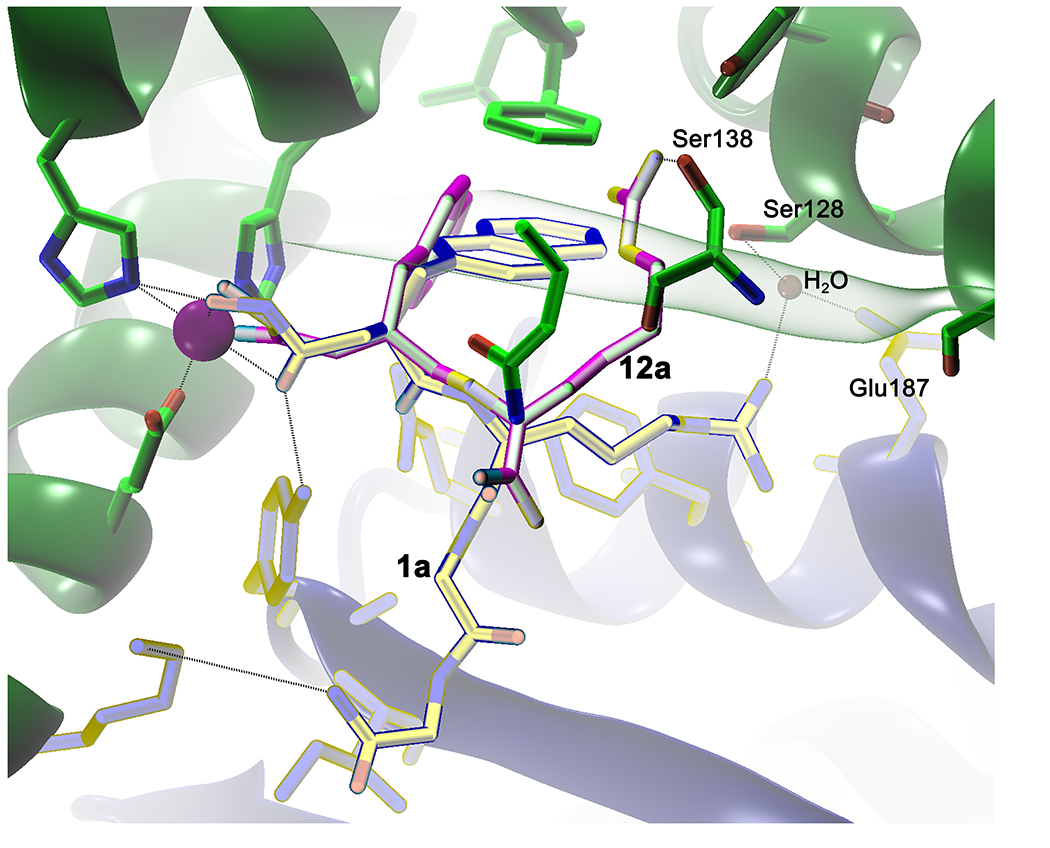
Computational docking of hydroxamate 12a suggests why substitution of homoArg for Arg at the P2’ position fails to improve potency. Comparison of the lowest energy poses for inhibitor 1a (yellow sticks) versus inhibitor 12a (magenta sticks) bound to the catalytic zinc (purple sphere) and residues within IDE-N (green) and IDE-C (blue). Note that the guanidinium moiety within the homoArg residue of compound 12a fails to form electrostatic interactions with Glu817, as hypothesized, but instead makes strong contact with Ser138. This alternate interaction appears to force the P1’ 2Nap residue into a less stable conformation, while simultaneously reducing the interactions between IDE-N and IDE-C, which are hypothesized to be essential for effective inhibition.
Synthesis of inhibitors 9a and 9b involved the generation of 2-residue intermediates containing a C-terminal O-allyl moiety as an orthogonal protecting group (inhibitors 14a and 14b, Scheme 3). When these were tested for activity, inhibitor 14a was found to unexpectedly potent (ki = 87 ± 11 nM), being ~13-fold more potent than the parent amide, 9a. Overall, inhibitor 14a proved to be the most potent of all the 2-residue IDE inhibitors investigated in this study. Given its relatively small size (MW = 455.5) and other properties, inhibitor 14a comprises the most drug-like inhibitor yet described for IDE.
As mentioned, inhibitor 1a was shown previously to be highly selective (>104-fold) for IDE vis-à-vis diverse set of zinc-metalloproteases8. To assess whether this property was adversely affected by the modifications described above, we tested the most potent variants—inhibitors 8a, 8b and 14a—against a diverse set of zinc-metalloproteases (Figure 3), including neprilysin (NEP), endothelin-converting enzyme-1 (ECE1), ACE, and MMP-2 and -7. We note that all these experiments were conducted using a common substrate (OmniMMP, see Experimental Section). Compared to the parent compound, inhibitor 1a (Figure 3A), the next most potent derivative, inhibitor 8a, exhibited considerably poorer selectivity for IDE (Figure 3B), for example, being a relatively effective inhibitor of MMP7 (IC50 = 1.15 ± 0.32 μM). By contrast, the less potent diastereomer, inhibitor 8b, exhibited excellent selectivity for IDE (Figure 3C), exhibiting no or only partial inhibition of any of the other proteases tested at concentrations up to 10 μM. Inhibitor 14a also showed reasonably good selectivity (Figure 3D), being >100-fold more potent against IDE vis-à-vis the other proteases tested.
Figure 3.
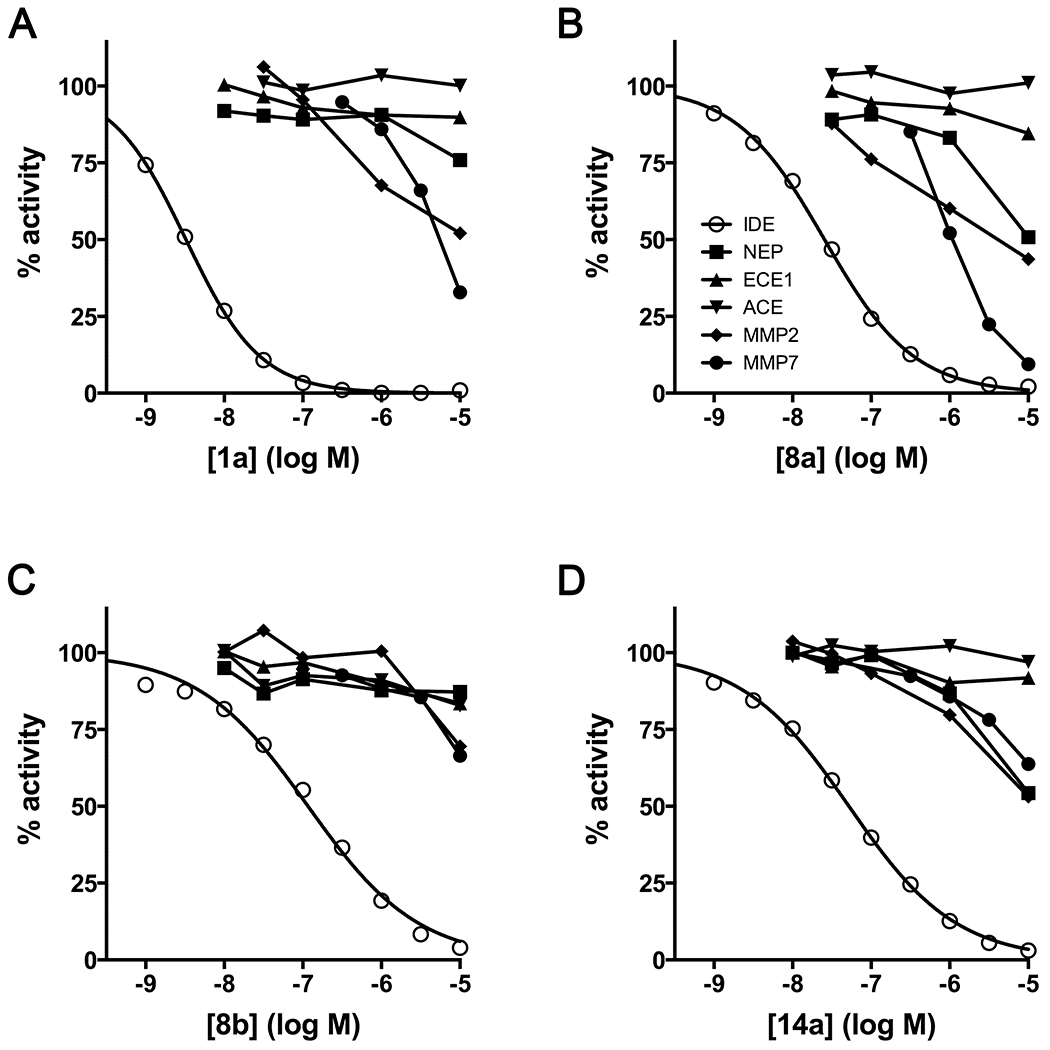
Conventional peptide hydroxamates show varying degrees of selectivity for IDE. Dose-response curves for inhibitors 1a (A), 8a (B), 8b (C) and 14a (D) against IDE, NEP, ECE1, ACE, MMP2 and MMP7. The legend in panel B (inset) applies to all graphs. Note that MMP7 is rather potently inhibited by inhibitors 1a and 8a, but not by inhibitors 8b or 14a.
In previous work8, we developed retro-inverso peptide hydroxamate inhibitors of IDE. Although such compounds are, generally speaking, considerably less potent than the corresponding conventional peptide hydroxamates (c.f., inhibitors 1a and 2, Figure 1B,C), they can be synthesized with facility by SPPS, with some variants showing excellent potency8. Through the testing of a number of retro-inverso inhibitors, we identified a diverse set of compounds exhibiting good potency in our FRET1 activity assay (Table 4), including: a retro-inverso version of inhibitor 1a with a free amino terminus (inhibitor 15; ki = 49.8 ± 3.7 nM); a variant of the latter with Tyr substituted for Trp at the P3’ position (inhibitor 16; ki = 85.1 ± 5.0 nM); a previously described inhibitor (ML1-XF)8 containing Bpa at the P2’ position, Tyr at the P3’ position, and Glu(EDANS) at the P4’ position (inhibitor 17; ki = 43.7 ± 8.0 nM); and a cyclized retro-inverso peptide hydroxamate (inhibitor 18; ki = 23.1 ± 1.2 nM). With the exception of inhibitor 18, these retro-inverso peptide hydroxamates were developed and characterized previously8.
Table 4.
Potency of retro-inverso peptide hydroxamates used for downstream characterization.
| Cmpd. | Sequence | ki, nM (FRET1) |
|---|---|---|
| 15 | Glu-Trp-Arg-2Nap-Hx | 49.8 ± 3.7 (n = 3) |
| 16 | Glu-Tyr-Arg-2Nap-Hx | 85.1 ± 5.0 (n = 3) |
| 17 | Ac-Glu(EDANS)-Tyr-Bpa-2Nap-Hx | 43.7 ± 8.0 (n = 3) |
| 18 |
NH-Asp-His-Phe-Ile-Arg-Glu-Trp-Arg-2Nap-Hx 
|
23.1 ± 1.2 (n = 3) |
2Nap = 2-naphthylalanine; Hx = hydroxamate; Ac = acetyl; EDANS = 5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid; Bpa = benzoylphenylalanine. D-amino acids are shown in italics. ß-amino acids are shown in bold text. In compound 18, the amino terminus is linked to the carboxylic acid residue in the Glu via a peptide bond.
The latter compounds, together with the most potent conventional peptide hydroxamates (inhibitors 1a, 8a, 8b and 14a), were subsequently tested using well-characterized assays for Aß12 and insulin7 degradation. Consistent with previous findings8, inhibitor 1a exhibited apparent ki (kiapp) values for Aß and insulin degradation that were ~10-fold higher than for FRET1, a trend that was maintained for most of the 8 compounds tested (Table 5, Figure 4). However, retro-inverso peptide hydroxamates 15 and 17 were found to deviate significantly from this pattern. Inhibitor 15 exhibited a kiapp value for insulin degradation that was >60-fold higher than that obtained with the FRET1 degradation assay, while showing no appreciable difference with the Aß degradation assay (Table 5, Figure 4). Conversely, inhibitor 17 exhibited a kiapp value >30-fold lower for Aß degradation relative to the ki for FRET1 degradation and ~300-fold lower relative to that for insulin degradation (Table 5, Figure 4).
Table 5.
The potency of active-site-directed IDE inhibitors varies in a substrate-selective manner.
| kiapp, nM | |||
|---|---|---|---|
| Cmpd. | FRET1 | FAßB | Insulin |
| 1a | 2.96 ± 0.20 (n = 3) | 21.7 ± 3.5 (n = 3) | 37.9 ± 4.0 (n = 3) |
| 8a | 25.9 ± 1.2 (n = 3) | 70.3 ± 12 (n = 3) | 275 ± 25 (n = 3) |
| 8b | 103 ± 7.0 (n = 3) | 927 ± 120 (n = 3) | 1890 ± 410 (n = 3) |
| 14a | 86.9 ± 11 (n = 3) | 107 ± 25 (n = 3) | 1280 ± 300 (n = 3) |
| 15 | 49.8 ± 3.7 (n = 3) | 228 ± 67 (n = 3) | 3260 ± 760 (n = 3) |
| 16 | 85.1 ± 5.0 (n = 3) | 365 ± 68 (n = 3) | 833 ± 160 (n = 3) |
| 17 | 43.7 ± 8.0 (n = 3) | 1.40 ± 0.31 (n = 3) | 410 ± 120 (n = 3) |
| 18 | 23.1 ± 1.2 (n = 3) | 246 ± 33 (n = 3) | 171 ± 26 (n = 3) |
Figure 4.
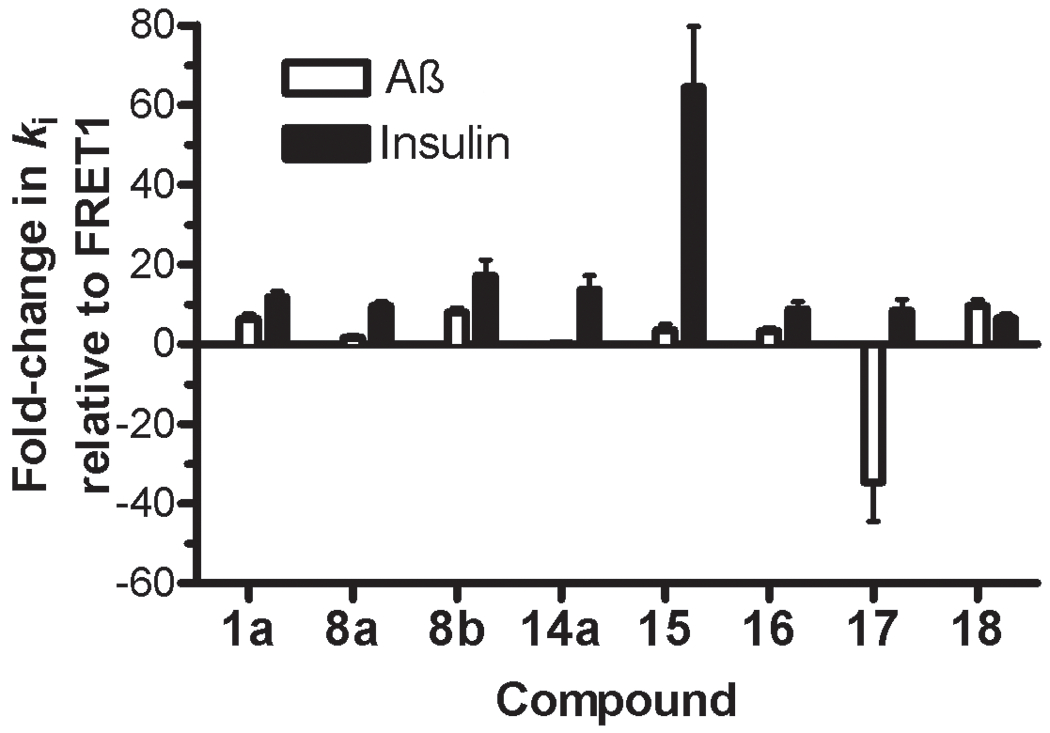
The potency of individual IDE inhibitors is markedly substrate selective. Fold-change in apparent ki (kiapp) values for Aß degradation (open bars) and insulin degradation (solid bars) relative to those obtained with the FRET1 activity assay (derived from Table 5) obtained using inhibitor 1a and the most potent inhibitors developed in this study. Note that most inhibitors show at least 10-fold higher kiapp values for both Aß and insulin degradation relative to FRET1 degradation, an effect that nonetheless varies between different inhibitors. In particular, relative to FRET1 degradation, inhibitor 15 inhibits insulin degradation >60-fold, while inhibitor 17 inhibits the Aß degradation >30-fold more effectively.
Discussion and conclusions
Since its discovery in 194913, IDE has been considered to be an attractive therapeutic target for the treatment of T2DM and other disorders involving impaired insulin signaling6. Supporting the merits of this approach, a purified endogenous inhibitor of IDE (non-proteinaceous, but of undetermined identity), was shown to potentiate and prolong the hypoglycemic effect of insulin in vivo as early as 195514. Nevertheless, despite widespread interest in their development throughout the 1950s, effective inhibitors of IDE did not emerge for more than a half-century8.
Our group has pursued the development of effective IDE inhibitors for more than a decade15. After failing to identify effective inhibitors through high-throughput screening of small-molecule compound libraries8, we employed a classical rational design approach, wherein the preferred cleavage-site specificity of IDE was determined de novo9 and used to design peptide hydroxamic acids8. Following optimization of the P1’ position, these efforts yielded inhibitor 1a (Ii1), a compound with excellent potency (ki = 2.96 ± 0.20 nM) and unexpectedly good selectivity (~104-fold) vis-à-vis other zinc-metalloproteases and representative member of other protease classes8. In the present study, we sought to further optimize inhibitor 1a, focusing in particular on its principal limitations, including its cumbersome route of synthesis and its relatively large size, with the ultimate goal of developing derivatives amenable to conversion to non-peptidic inhibitors with drug-like properties.
To circumvent the complex synthesis of inhibitor 1a, variants derived from ketal-based P1’ precursors 7a and 7b were developed and characterized. The resulting 4-residue compounds (inhibitors 8a and 78), which differ from the parent compound only by the inclusion of an −OH group on the α-carbon between the hydroxamate and the peptide backbone, were much more tractable to synthesize, and yielded enantiomerically pure products in significantly higher yield, but were ~9- and ~35-fold less potent than inhibitor 1a, depending on the orientation of the α-carbon −OH group (S and R, respectively). Nevertheless, given that inhibitor 8a retains low-nanomolar potency (ki = 25.9 ± 1.2 nM) and, in particular, is readily synthesized in high yield, we consider inhibitor 8a to be a significant improvement over inhibitor 1a. Despite its relatively poorer potency, our analysis suggests that inhibitor 8b may in fact be superior to inhibitor 8a in terms of selectivity for IDE.
The relatively large size of inhibitor 1a (MW = 743.8) was the second limitation we sought to optimize. Based on the inhibitor 1a:IDE co-crystal structure and other considerations, we hypothesized that the P4’ and/or P3’ redidues in inhibitor 1a might be dispensable. N truncated versions of inhibitors 1a, 8a and 8b were, in general, found to be far less potent than the parent molecules. On the other hand, inhibitor 14a, a 2-residue variant of inhibitor 1a containing a C-terminal O-allyl group, was found to exhibit good potency (ki = 87 ± 11 nM). These results suggest that the P3’ and P4’ residues of latter inhibitors do in fact interact substantially with the active site of IDE, although—as is evident from the lack of resolution of these moieties in the inhibitor 1a:IDE co-crystal structure, together with the lack of cleavage-site specificity at these positions8—not merely via a single mode of binding. Molecular docking of inhibitor 14a was performed (see Experimental Section), but this approach failed to identify interactions between the O-allyl moiety and specific residues within IDE of sufficient strength to account for the increased potency of this compound, suggesting that multiple binding modes are likely operative in this instance, as well. Whatever the precise mechanism(s), our findings suggest that the P3’ and P4’ residues are not strictly required to achieve potent inhibition, a result that augurs well for the eventual development of effective small-molecule (MW = <500) IDE inhibitors. Given its small size, its potency, and its selectivity for IDE, inhibitor 14a constitutes an excellent pharmacophore for future drug development.
A key insight emerging from the inhibitor 1a:IDE co-crystal structure was the finding that the inhibitor makes extensive contacts with both halves of IDE’s active site, evidently acting like a “latch” to maintain the protease in the closed conformation. In an attempt to optimize these interactions, we hypothesized that the P2’ Arg residue could be modified to interact more strongly with Glu817 within IDE-C by extending the linker between the guanidinium moiety and the peptide backbone. We tested this by incorporating homoArg into 2-residue conventional peptide hydroxamates (inhibitors 12a, 12b and 13). Contrary to expectations, this modification actually resulted in substantial decreases in potency relative to the parent compounds. Subsequent analysis by computational docking suggests that longer alkane linker in homoArg permits multiple sub-optimal modes of binding, in part by increasing the flexibility of the linker. It remains possible that incorporation of homoArg into 3- or 4-residue hydroxamates might help to overcome these alternate binding modes, and favor interaction with Glu817, by limiting the range of motion of the P2’ residue. However, in light of the substantially diminished potency of most 3- and 2-residue inhibitors, de novo optimization of the P2’ residue in 2-residue inhibitors (e.g., inhibitor 14a) might be a more fruitful route to improving potency. In this connection, we note that Bpa was well tolerated at the P2’ position (e.g., inhibitor 17) in retro-inverso peptide hydroxamates, suggesting that relatively large residues can be tolerated at this position, a feature that might permit more substantial interactions with the C-terminal half of IDE’s active site.
One of the more notable aspects of the current study was the discovery that the potency of individual inhibitors, though all presumably interacting with the active-site zinc, varied substantially in a substrate-dependent manner. We observed both increases and decreases in potency, depending on the particular inhibitor and the particular substrate tested. Substrate-dependent pharmacologal modulation has been observed in multiple contexts previously. For example, ATP inhibits the degradation of insulin16 and Aß11, yet profoundly activates the degradation of FRET111 or other short fluorogenic substrates17. Unrelated drug-like compounds have also been identified that exhibit comparable effects11. Conversely, Çakir and colleagues used computational methods to identify compounds interacting with IDE’s exosite that are reported to activate insulin degradation18. Although these unusual properties are undoubtedly attributable to the many novel structural features of IDE—including its bipartite active site and the fact that the substrate and inhibitors can both be contained within the internal chamber—it is nevertheless difficult to postulate a definitive model that cleanly accounts for all of the reported effects.
In sum, this study made considerable progress in optimizing the first effective inhibitor of IDE, inhibitor 1a. We developed variants that are more tractable to synthesize, and also identified truncated versions that retain good potency while being considerably smaller in size and retaining good selectivity for IDE. In addition, we demonstrate that active site-directed compounds can inhibit IDE in a highly substrate-selective manner. Collectively, these findings are expected to facilitate the development of drug-like IDE inhibitors suitable for a variety of experimental and medicinal applications.
Experimental Section
Synthesis of 1a and 1b
Inhibitors 1a and 1b were synthesized, purified and characterized in a previous publication8.
Synthesis of ketal-based P1’ precursors 7a and 7b.
(2S,3R)-Diisopropyl 2-hydroxy-3-(naphthalen-2-ylmethyl)succinate (5):
Diisopropyl-2S-hydroxybutanedioate (3) (5.00 g, 22.91 mmol) was added dropwise to a solution of 1M LDA (2.5 eq) in THF maintained at the temperature at −70 °C. After the addition the reaction mixture was slowly warmed to −15° C and stirred for 8 h (CCl4-dry ice). The reaction mixture was then cooled to −70 °C and 2-naphthylmethyl bromide (4) (5.57 g, 25.2 mmol) dissolved in THF (5 mL) was added slowly, ensuring that the temperature did not exceeded −65° C. The mixture was warmed to −40 °C (ACN-dry ice bath) and stirred for 18 h before quenching at −15 °C with citric acid. The organic layer was separated, washed with 10% NaHCO3 (50 mL) and brine (30 mL), then dried over MgSO4, filtered and concentrated. The crude product was purified over flash chromatography using 5-15% ethyl acetate/hexane as eluent, affording the product as bright yellow oil (3.50 g, 43%). 1H NMR (300 MHz, CDCl3): δ 1.13-1.48 (m, 12H), 3.05-3.23 (m, 1H), 3.27-3.37 (m, 2H), 4.09 (dd, 1H, J = 2.9, 5.3 Hz), 4.94-5.13 (m, 2H), 7.39-7.48 (m, 3H), 7.73-7.82 (m, 4H). MS m/z (ESI) 380.90 (M+Na)+.
(2S,3R)-2-hydroxy-3-(naphthalen-2-ylmethyl)succinic acid (6):
(2S,3R)-Diisopropyl 2-hydroxy-3-(naphthalen-2-ylmethyl)succinate (5) (3.25 g, 9.07 mmol) was dissolved in dioxane (7 mL) and water (7 mL), and a solution of KOH (2.035 g, 36.30 mmol) in water (11 mL) was added and the mixture was heated at 90 °C overnight. The solution was then allowed to cool at room temperature and then treated with an ion exchange resin (Dowex 50X4-400, 100 mL), filtered and washed with methanol. The filtrate was evaporated under reduced pressure to yield the title compound as yellow gummy sold (2.462 g, 99%). 1H NMR (300 MHz, CDCl3): δ 3.17-3.47 (m, 3H), 4.13 (d, 1H, J = 2.3 Hz), 7.39-7.50 (m, 3H), 7.75-7.84 (m, 4H). MS m/z (ESI) 272.76 (M-1)+.
(R)-2-((S)-2,2-Dimethyl-5-oxo-1,3-dioxolan-4-yl)-3-(naphthalen-2-yl)propanoic acid (7a):
((2S,3R)-2-Hydroxy-3-(naphthalen-2-ylmethyl)succinic acid (1.88 g, 6.85 mmol) was dissolved in dimethoxypropane (70 mL) and DMF (10 mL) and p-toulenesulfonic acid (0.354 g, 2.056 mmol) was added. The reaction mixture was stirred overnight at 40° C overnight. The solvents were then removed under reduced pressure. The residue was dissolved in ether and the organic solution was washed with water and brine, and dried over MgSO4. After filtration and evaporation, the product was obtained as a brownish solid (1.46 g, 68%). 1H NMR (300 MHz, CDCl3): δ 1.50 (s, 3H), 1.62 (s, 3H), 3.15 (dd, 1H, J = 9.3, 13.2 Hz), 3.36-3.40 (m, 1H), 3.48 (dd, 1H, J = 5.9, 13.2 Hz), 4.32 (d, 1H, J = 3.4 Hz), 7.36 (dd, 1H, J = 1.5, 8.4 Hz), 7.42-7.50 (m, 2H), 7.70 (s, 1H), 7.78-7.83 (m, 3H). 13C NMR (125 MHz, DMSO) 25.8, 26.3, 33.7, 47.8, 73.2, 110.6, 125.6, 126.1, 127.3, 127.4, 127.5, 127.5, 128.0, 131.9, 133.1, 136.3, 171.7, 171.8. HR ESI-MS [M+H]+ m/z = 315.1239 (calc. for C18H19O5: 315.1232)
(R)-2-((R)-2,2-Dimethyl-5-oxo-1,3-dioxolan-4-yl)-3-(naphthalen-2-yl)propanoic acid (7b):
This compound was synthesized following the procedure described for compound 7a, starting with diisopropyl-2R-hydroxybutanedioate and proceding via purification of the appropriate diasteromer of 5 ((2R,3R)-Diisopropyl 2-hydroxy-3-(naphthalen-2-ylmethyl)succinate). 1H NMR (500 MHz, DMSO): δ 1.50 (s, 3H), 1.57 (s, 3H), 3.03 (m, 1H), 3.30-3.36 (m, 2H), 4.53 (d, 1H, J = 3.1 Hz), 7.42-7.52 (m, 3H), 7.77 (s, 1H), 7.84-7.90 (m, 3H), 12.82 (brs, 1H). 13C NMR (125 MHz, DMSO) 25.7, 26.3, 32.7, 48.5, 73.4, 110.5, 125.6, 126.1, 127.1, 127.3, 127.4, 127.5, 127.9, 131.9, 133.0, 136.1, 171.8, 173.1. HR ESI-MS [M+H]+ m/z = 315.1227 (calc. for C18H19O5: 315.1232).
Synthesis of peptide hydroxamates by SPPS from ketal-based P1’ precursors 7a and 7b.
General procedure.
Rink amide resin (Chem-impex international) (403 mg, 0.65 mmol/g of Fmoc-blocked amino groups) was placed in a clamped glass filter funnel with a glass frit. A slow, steady flow of dry nitrogen was allowed to pass through the resin from the lower end via a piece of Tygon tubing. The resin was swollen with DCM for 30 min, then drained. Resin swelling procedure was repeated for 10 min. After draining, the resin was washed with DMF (10 mL x 4). Fmoc on the Rink amide resin was removed by shaking with 10 ml of 20% piperidine (PIP) in dimethylformamide (DMF) under dry nitrogen for 15 min and the liquid phase drained. This process was repeated once. After washing the deprotected resin with DMF (10 mL x 5), the first Fmoc-amino acid was coupled to the resin by addition of a clear preformed reagent cocktail prepared by vortexing a mixture of 2.2 ml of O-(7-azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU), DIPEA (1.0 mmol each) in DMF, and solid Fmoc (tBu)-protected amino acid (1.00 mmol). The resulting suspension was shaken under nitrogen for 1 h at room temperature. Completion of the coupling reaction was monitored by Kaiser test. The coupling was repeated as needed. After ensuring the coupling reaction, the liquid residue was drained off with nitrogen pressure and the resin was washed with dry DMF (10 ml x 4). The Fmoc amine protecting group on the newly added amino acid was removed with 20% PIP as described above. This process of Fmoc removal and coupling was repeated with the remaining Fmoc (Boc) or (Pbf)-protected amino acids and in the peptide sequence. After final coupling with (R)-2-((S)-2,2-dimethyl-5-oxo-1,3-dioxolan-4-yl)-3-(naphthalen-2-yl)propanoic acid or its enantiomer and thorough washing with DMF (10 ml x 3), the resin was treated with NH2OH.HCl (40 mmol) and of DIPEA (80 mmol) in DMF (2 mL) and heated at 50° C under nitrogen over night. The solvent was removed by filtration and the residue was washed sequentially with ethanol and with dichloromethane (DCM) (10 ml x 4). Finally, the resin-bound (tBu)-, (Boc)- and Pbf-protected peptide was cleaved off the resin by wrist action shaking for 2h with 10 ml of 2.5% triisopropyl silane, 2.5% H2O in trifluoroacetic acid (TFA) under nitrogen. After final draining of the cleaved peptide solution, the resin was thoroughly washed with DCM (10 ml x 4). The extracts and the DCM washes were combined. The volatiles were removed under rotary evaporation to afford the crude peptide. Dry diethyl ether was added and the resulting precipitate was removed by centrifugation and dried under high vacuum. The peptide in the crude residue was purified by reverse-phase HPLC using a linear of acetonitrile:water gradient containing 0.1%TFA. Compounds 8a, 8b, 10a, 10b, 11a, 11b, and 13 were synthesized by following this general procedure.
(S)-5-Amino-4-((S)-2-((S)-5-guanidino-2-((2R,3S)-3-hydroxy-4-(hydroxyamino)-2-(naphthalen-2-ylmethyl)-4-oxobutanamido)pentanamido)-3-(1H-indol-2-yl)propanamido)-5-oxopentanoic acid (8a):
(Yield: 89 mg, 80%) 1H NMR (300 MHz, CD3OD): δ 1.19-1.31 (m, 2H), 1.33-1.44 (m, 1H), 1.56-1.79 (m, 2H), 1.91-2.02 (m, 1H), 2.15 (t, 2H, J = 7.6 Hz), 2.86-2.97 (m, 4H), 3.02-3.09 (m, 2H), 4.02 (d, 1H, J = 4.8 Hz), 4.13-4.20 (m, 2H), 4.35-4.39 (m, 1H), 6.90-7.01 (m, 3H), 7.19-7.34 (m, 4H), 7.46 (d, 1H, J = 7.4 Hz), 7.58-7.67 (m, 4H), 7.83 (d, 1H, J = 7.1 Hz). HR ESI-MS [M+H]+ m/z = 760.3441 (calc. for C37H46N9O9: 760.3418).
(S)-5-Amino-4-((S)-2-((S)-5-guanidino-2-((2S,3R)-3-hydroxy-4-(hydroxyamino)-2-(naphthalen-2-ylmethyl)-4-oxobutanamido)pentanamido)-3-(1H-indol-2-yl)propanamido)-5-oxopentanoic acid (8b):
(Yield: 68 mg, 87%) 1H NMR (300 MHz, CD3OD): δ 1.18-1.29 (m, 2H), 1.31-1.41 (m, 1H), 1.52-1.77 (m, 2H), 1.91-2.00 (m, 1H), 2.17 (t, 2H, J = 7.7 Hz), 2.81-2.93 (m, 4H), 3.00-3.10 (m, 2H), 4.05 (d, 1H, J = 4.7 Hz), 4.15-4.19 (m, 2H), 4.36-4.40 (m, 1H), 6.91-7.02 (m, 3H), 7.18-7.33 (m, 4H), 7.456 (d, 1H, J = 7.3 Hz), 7.57-7.66 (m, 4H), 7.82 (d, 1H, J = 7.2 Hz). HR ESI-MS [M+H]+ m/z = 760.3442 (calc. for C37H46N9O9: 760.3418).
(2R,3S)-N1-((S)-1-((S)-1-Amino-3-(1H-indol-2-yl)-1-oxopropan-2-ylamino)-5-guanidino-1-oxopentan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (10a):
(Yield: 51 mg, 77%) 1H NMR (300 MHz, CD3OD): δ 0.59-0.64 (m, 2H), 1.04-1.11 (m, 1H), 1.31 (s, 2H), 1.40-1.44 (m, 1H), 2.44 (t, 2H, J = 6.9 Hz), 2.98-3.16 (m, 4H), 4.11-4.14 (m, 1H), 4.20 (d, 1H, J = 6.8 Hz). 4.59-4.67 (m, 1H), 6.98-7.11 (m, 3H), 7.30-7.48 (m, 4H), 7.62 (d, 1H, J = 7.7 Hz), 7.66 (s, 1H), 7.76-7.82 (m, 3H), 7.88 (d, 1H, J = 8.7 Hz), 8.07 (d, 1H, J = 7.8 Hz). 13C NMR (125 MHz, DMSO) 24.4, 27.6, 28.1, 34.0, 50.9, 52.0, 53.6, 69.8, 71.2 110.3, 111.2, 118.2, 118.4, 120.8, 123.8, 125.3, 126.0, 126.7, 127.2, 127.3, 127.4, 127.5, 127.7, 131.7, 133.0, 136.0, 136.7, 156.7, 168.4, 170.7, 172.5, 173.5. HR ESI-MS [M+H]+ m/z = 631.3008 (calc. for C32H39N8O6: 631.2992).
(2S,3R)-N1-((S)-1-((S)-1-amino-3-(1H-indol-2-yl)-1-oxopropan-2-ylamino)-5-guanidino-1-oxopentan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (10b):
(Yield: 90 mg, 85%) 1H NMR (300 MHz, CD3OD): δ 0.61-0.66 (m, 2H), 1.06-1.13 (m, 1H), 1.33 (s, 2H), 1.41-1.46 (m, 1H), 2.43 (t, 2H, J = 6.8 Hz), 2.99-3.15 (m, 4H), 4.13-4.17 (m, 1H), 4.22 (d, 1H, J = 6.8 Hz). 4.60-4.68 (m, 1H), 6.99-7.12 (m, 3H), 7.33-7.49 (m, 4H), 7.63 (d, 1H, J = 7.6 Hz), 7.65 (s, 1H), 7.75-7.81 (m, 3H), 7.88 (d, 1H, J = 8.6 Hz), 8.05 (d, 1H, J = 7.7 Hz). HR ESI-MS [M+H]+ m/z = 631.3007 (calc. for C32H39N8O6: 631.2992).
(2R,3S)-N1-((S)-1-Amino-5-guanidino-1-oxopentan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (11a):
(Yield: 69 mg, 88%) 1H NMR (300 MHz, CD3OD): δ 1.56 (br s, 3H), 1.84 (br s, 1H), 3.11 (br s, 5H), 4.29 (br s, 1H), 7.24-7.83 (m, 3H), 7.72 (s, 1H), 7.78-7.83 (m, 3H). 13C NMR (125 MHz, DMSO) 24.8, 28.7, 34.1, 40.3, 50.3, 52.1, 69.7, 125.2, 125.9, 126.7, 127.3, 127.4, 127.5, 127.6, 131.6, 132.9, 136.7, 156.6, 172.0, 173.0. HR ESI-MS [M+H]+ m/z = 445.2208 (calc. for C21H29N6O5: 445.2199).
(2S,3R)-N1-((S)-1-amino-5-guanidino-1-oxopentan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (11b)
(Yield: 50 mg, 88%) 1H NMR (300 MHz, CD3OD): δ 1.54 (br s, 3H), 1.88 (br s, 1H), 3.13 (br s, 5H), 4.30 (br s, 1H), 7.22-7.84 (m, 3H), 7.71 (s, 1H), 7.79-7.85 (m, 3H). HR ESI-MS [M+H]+ m/z = 445.2210 (calc. for C21H29N6O5: 445.2199).
(2R,3S)-N1-((S)-1-amino-6-guanidino-1-oxohexan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (13):
(Yield: 93 mg, 81%) 1H NMR (300 MHz, CD3OD): δ 1.30-1.36 (m, 2H), 1.44-1.57 (m, 3H), 1.78-1.83 (m, 1H), 3.06-3.24 (m, 5H), 3.99 (s, 1H), 4.16 (d, 1H, J = 4.5 Hz). 4.26-4.30 (m, 1H), 7.42-7.49 (m, 3H), 7.74 (s, 1H), 7.79-7.84 (m, 3H), 8.07 (d, 1H, J = 8.2 Hz). 13C NMR (125 MHz, DMSO) 22.2, 27.9, 31.2, 34.2, 40.6, 50.3 52.2, 70.8, 125.3, 126.0, 126.8, 127.3, 127.4, 127.5, 127.6, 131.7, 133.0, 136.8, 156.8, 168.8, 172.1, 173.4. HR ESI-MS [M+H]+ m/z = 459.2365 (calc. for C22H31N6O5: 459.2355).
Synthesis of truncated variants of inhibitors 1a and 1b (9a, 9b, 12a, 12b, 14a and 14b).
General procedure.
Compounds 9a, 9b, 14a and 14b were synthesized by solution phase peptide synthesis using methods described previously8. Compounds 12a, 12b were synthesized by the same methods used to generate 9a and 9b, albeit utilizing homoArg in the place of Arg.
(S)-Allyl5-guanidino-2-((S)-4-(hydroxyamino)-2-(naphthalen-2-ylmethyl)-4-oxobutanamido)pentanoate (14a)
A solution of (6S,9S)-allyl 1-imino-14,14-dimethyl-9-(naphthalen-2-ylmethyl)-8,11-dioxo-1-(2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-sulfonamido)-13-oxa-2,7,12-triazapentadecane-6-carboxylate8 (47.0 mg, 0.064 mmol) in 5% anisole in TFA (6 ml) was stirred at 40° C for 4.5 h. The reaction mixture was cooled and the volatiles were removed in vacuo. The resultant oil was washed with ether (10 mL), dissolved in water (5ml) and filtered. The filtrate was lyophilized to give a crude solid that was further purified by reverse-phase HPLC using a linear 0.1%TFA in acetonitrile:water gradient to give the title compound as a white powder (20.1 mg, 72%). 1H NMR (300 MHz, CD3OD): δ 0.76 (br s, 2H), 1.15-1.42 (m, 2H), 2.17 (dd, 1H, J = 7.4, 14.5 Hz), 2.34 (dd, 1H, J = 7.0, 14.5 Hz), 2.49 (br s, 2H), 2.81 (d, 2H, J = 7.3 Hz) 3.05 - 3.10 (m, 1H), 3.05 (m, 1H), 4.05-4.06 (m, 1H), 4.43 (d, 2H, J = 4.7 Hz), 5.05 (d, 1H, J = 10.4 Hz), 5.15 (d, 1H, J = 17.2 Hz), 5.71-5.80 (m, 1H), 7.22-7.31 (m, 3H), 7.50 (s, 1H), 7.64-7.66 (m, 2H). HR ESI-MS [M+H]+ m/z = 470.2423 (calc. for C24H32N5O5: 470.2403).
(S)-Allyl5-guanidino-2-((R)-4-(hydroxyamino)-2-(naphthalen-2-ylmethyl)-4-oxobutanamido)pentanoate (14b):
This compound was synthesized following the procedure mentioned above (112 mg, 92%. 1H NMR (300 MHz, CD3OD): δ 1.49-1.64 (m, 3H), 1.73-1.75 (m, 1H), 2.13 (dd, 1H, J = 4.2, 14.8 Hz), 2.41 (dd, 1H, J = 10.1, 14.8 Hz), 2.77 (dd, 1H, J = 6.7, 13.1 Hz), 2.88 - 3.06 (m, 3H), 4.14-4.29 (m, 2H), 4.34-4.36 (m, 1H), 4.43 (d, 2H, J = 4.7 Hz), 5.03 (d, 1H, J = 10.5 Hz), 5.08 (d, 1H, J = 18.6 Hz), 5.55-5.68 (m, 1H), 7.26-7.37 (m, 3H), 7.55 (s, 1H), 7.67-7.72 (m, 2H). 13C NMR (125 MHz, DMSO) δ 24.9, 27.9, 33.6, 37.7, 40.3, 42.6, 51.5, 64.7, 117.7, 125.9, 127.0, 127.3, 124.4, 127.6, 131.7, 132.3, 133.0, 136.8, 156.6, 167.6, 171.4, 173.7. HR ESI-MS [M+H]+ m/z = 470.2420 (calc. for C24H32N5O5: 470.2403).
(S)-N1-((S)-1-amino-5-guanidino-1-oxopentan-2-yl)-N4-hydroxy-2-(naphthalen-2-ylmethyl)succinamide (9a):
(S)-Allyl5-guanidino-2-((S)-4-(hydroxyamino)-2-(naphthalen-2-ylmethyl)-4-oxobutanamido)pentanoate (50.0 mg, 0.064 mmol) was dissolved in 6 M solution anhydrous ammonia in methanol (1.6 mL) at −10° C and stirred under nitrogen at room temperature overnight. The solvent was removed under reduced pressure to afford a white solid (47.0 mg, 99%). 1H NMR (300 MHz, CD3OD): δ 0.56-0.65 (m, 2H), 1.12-1.24 (m 1H), 1.53-1.59 (m, 1H), 2.32 (dd, 1H, J = 3.7, 15.2 Hz), 2.49 (br s, 2H), 2.60 (dd, 1H, J = 10.2, 15.2 Hz), 2.88-2.94 (m, 2H), 3.04-3.07 (m, 1H), 3.96-4.00 (m, 1H), 6.89 (br s, 1H), 7.32-7.43 (m, 3H), 7.59 (s, 1H), 7.73-7.78 (m, 3H), 8.08 (d, 1H, J = 8.0 Hz). 13C NMR (125 MHz, DMSO) 24.7, 28.1, 34.2, 38.3, 40.2, 43.3, 51.9, 125.3, 125.9. 127.0, 127.3, 127.4, 127.5, 127.6, 131.7, 133.0, 136.7, 156.6, 167.9, 173.6, 173.7. HR ESI-MS [M+H]+ m/z = 429.2263 (calc. for C21H29N6O4: 429.2250).
(R)-N1-((S)-1-amino-5-guanidino-1-oxopentan-2-yl)-N4-hydroxy-2-(naphthalen-2-ylmethyl)succinamide (9b).
This compound was synthesized following the procedure mentioned above (60.2 mg, 97%). 1H NMR (300 MHz, CD3OD): δ 1.53 (m, 3H), 1.79 (br s, 1H), 2.23 (br d, 1H, J = 15.0 Hz), 2.43-.246 (m, 1H), 2.82-2.88 (m, 1H), 2.99-3.07 (m, 4H), 4.25 (br s, 1H), 7.32-7.40 (m, 3H), 7.61 (s, 1H), 7.74-7.76 (m, 3H), 8.08 (d, 1H, J = 7.8 Hz). 13C NMR (125 MHz, DMSO) 24.8, 28.8, 33.7, 37.9, 40.2, 43.0, 51.9, 125.3, 126.0, 127.0, 127.3, 127.4, 127.5, 127.6, 131.7, 133.0, 136.8, 156.6, 167.9, 173.2, 173.4. HR ESI-MS [M+H]+ m/z = 429.2264 (calc. for C21H29N6O4: 429.2250).
(2R,3S)-N1-((S)-1-Amino-5-guanidino-1-oxopentan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (12a):
This compound was synthesized following the procedure for 9a, albeit substituting homoArg in the place of Arg (69.5 mg, 95%). 1H NMR (300 MHz, CD3OD): δ 1.56 (br s, 3H), 1.84 (br s, 1H), 3.11 (br s, 5H), 4.29 (br s, 1H), 7.24-7.83 (m, 3H), 7.72 (s, 1H), 7.78-7.83 (m, 3H). 13C NMR (125 MHz, DMSO) 24.8, 28.7, 34.1, 40.3, 50.3, 52.1, 69.7, 125.2, 125.9, 126.7, 127.3, 127.4, 127.5, 127.6, 131.6, 132.9, 136.7, 156.6, 172.0, 173.0. HR ESI-MS [M+H]+ m/z = 443.2314 (calc. for C22H31N6O4: 443.2329).
(2S,3R)-N1-((S)-1-amino-5-guanidino-1-oxopentan-2-yl)-N4,3-dihydroxy-2-(naphthalen-2-ylmethyl)succinamide (12b):
This compound was synthesized following the procedure for compound 12a (66.7 mg, 92%). 1H NMR (300 MHz, CD3OD): δ 1.54 (br s, 3H), 1.88 (br s, 1H), 3.13 (br s, 5H), 4.30 (br s, 1H), 7.22-7.84 (m, 3H), 7.71 (s, 1H), 7.79-7.85 (m, 3H). HR ESI-MS [M+H]+ m/z = 443.2318 (calc. for C22H31N6O4: 443.2329).
Synthesis of retro-inverso peptide hydroxamates (2, 15, 16, 17, 18)
All retro-inverso inhibitors were synthesized by conventional SPPS from commercially available Fmoc-protected amino acids using 2-chlorotrityl hydroxylamine resin, as described8. The synthesis, purification and characterization of inhibitors 2, 15, 16 and 17 is described in a previous publication8. Inhibitor 18 was synthesized by a commercial vendor (Anaspec Corp.).
Purification of compounds.
All compounds were purified by reverse-phase HPLC (purity >95%) as described8.
IDE activity assays
Dose-response determinations for IDE inhibitors were obtained using previously described assays for the degradation of FRET111 (5 μM), OmniMMP (5 μM)8, Aß12 (200 nM), and insulin7 (2 nM). All assays were conducted at 22 °C in Assay Buffer (100 mM NaCl, 10 mM MgCl2, 50 mM HEPES, pH 7.4, supplemented with 0.05% BSA) using recombinant human IDE19. IC50 values were calculated in Prism 5.0 (Graphpad Software, Corp.) then converted to ki values via the Cheng-Prussoff equation20 using published kM values for the different substrates (10.1 μM, 0.82 μM and 71 nM for FRET121, Aß12 and insulin8, respectively).
Molecular Docking
The starting conformation of ligands was obtained by the method of Polak-Ribière Conjugate Gradient (PRCG) energy minimization with the Optimized Potentials for Liquid Simulations (OPLS) 2005 force field for 5000 steps, or until the energy difference between subsequent structures was less than 0.001 kJ/mol-Ǻ22. Our docking methodology has been previously described23. Briefly, in order to generate the grids for docking, the ions, molecular refracting molecules 1,4-diethylene dioxide (DIO) were removed from the IDE crystal structure (PDB Code 3E4A)8. Inhibitor 1a was used as the reference ligand to generate the grids. The binding site was generated via multiple overlapping grids with a default rectangular box centered on inhibitor 1a. Ligands were docked into the catalytic binding site of IDE using Glide extra precision (XP) (Glide, version 5.6, Schrödinger, LLC, New York, NY). Molecular conformations were sampled using previous methods in the literature24.
We generated XP descriptors to obtain atom-level energy terms such as hydrogen bond interactions, electrostatic interactions, hydrophobic enclosure and pi-pi stacking interactions that resulted during the docking run. The final conformation of the docked ligand was obtained from the optimized, best scoring poses with descriptors of the Glide XP score using a recently developed approach23,25, 26. Briefly, we used Phase to automatically generate the pharmacophoric sites (Phase, v3.2, Schrödinger, LLC, New York, NY) using the default set of six chemical features: namely, hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic (H), negative ionizable (N), positive ionizable (P), and aromatic ring (R) site. The energetic value assigned to each pharmacophore feature was arrived at as the sum of the Glide XP contributions of the atoms comprising the site. Overall dockings at the active site were quantified and ranked on the basis of these energetic terms25, 26. From these features, a common pharmacophore model was developed to evaluate the ability to reproduce known inhibitors using Phase. The distance matching tolerance was set to 2.0 Å. To account for protein flexibility and lessen the effects of minor steric clashes, excluded volumes spheres were created for all receptor atoms within 5 Å around every ligand. Each sphere has a radius corresponding to 50% of the van der Waals radius of the receptor atom. Receptor atoms within 1.5 Å from the ligand were ignored. Figures were generated either using Linux command-line Tachyon, Version 0.99b2, or Tachyon within the Visual Molecular Dynamics (VMD) Version 1.9.1 program27.
Acknowledgments.
We thank the University of South Florida Mass Spectrometry Facility for acquisition of HR-MS data. Supported by grants from the Ellison Medical Foundation and the American Diabetes Association to M.A.L.
Abbreviations Used:
- Aß
amyloid ß-protein
- ECE1
endothelin-converting enzyme-1
- IDE
insulin-degrading enzyme
- IDE-C
IDE C-terminal domain
- IDE-N
IDE N-terminal domain
- Ii1
IDE inhibitor 1
- NEP
neprilysin
- SPPS
solid-phase peptide synthesis
- T2DM
type 2 diabetes mellitus
References
- 1.Shen Y; Joachimiak A; Rosner MR; Tang W-J Structures of human insulin-degrading enzyme reveal a new substrate mechanism. Nature 2006, 443, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdul-Hay SO; Kang D; McBride M; Li L; Zhao J; Leissring MA Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. PLoS ONE 2011, 6, e20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiu WQ; Folstein MF Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging 2006, 27, 190–198. [DOI] [PubMed] [Google Scholar]
- 4.Li Q; Ali MA; Cohen JI Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 2006, 127, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Authier F; Posner BI; Bergeron JJ Insulin-degrading enzyme. Clin Invest Med 1996, 19, 149–160. [PubMed] [Google Scholar]
- 6.Mirsky IA Insulinase, insulinase-inhibitors, and diabetes mellitus. Recent Prog Horm Res 1957, 13, 429–465; discussion 465-471. [PubMed] [Google Scholar]
- 7.Neant-Fery M; Garcia-Ordonez RD; Logan TP; Selkoe DJ; Li L; Reinstatler L; Leissring MA Molecular basis for the thiol sensitivity of insulin-degrading enzyme. Proc Natl Acad Sci U S A 2008, 105, 9582–9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leissring MA; Malito E; Hedouin S; Reinstatler L; Abdul-Hay SO; Choudhry S; Fauq AH; Huzarska M; May PS; Choi S; Logan TP; Turk BE; Cantley LC; Manolopoulou M; Tang WJ; Stein RL; Cuny GD; Selkoe DJ Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One 2010, 5, e10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turk BE; Huang LL; Piro ET; Cantley LC Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol 2001, 19, 661–667. [DOI] [PubMed] [Google Scholar]
- 10.Saghatelian A; Jessani N; Joseph A; Humphrey M; Cravatt BF Activity-based probes for the proteomic profiling of metalloproteases. Proc Natl Acad Sci U S A 2004, 101, 10000–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabrol C; Huzarska MA; Dinolfo C; Rodriguez MC; Reinstatler L; Ni J; Yeh L-A; Cuny GD; Stein RL; Selkoe DJ; Leissring MA Small-molecule activators of insulin-degrading enzyme discovered through high-throughput compound screening. PLoS ONE 2009, 4, e5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leissring MA; Lu A; Condron MM; Teplow DB; Stein RL; Farris W; Selkoe DJ Kinetics of amyloid beta-protein degradation determined by novel fluorescence- and fluorescence polarization-based assays. J Biol Chem 2003, 278, 37314–37320. [DOI] [PubMed] [Google Scholar]
- 13.Mirsky IA; Broth-Kahn RH The inactivation of insulin by tissue extracts. I. The distribution and properties of insulin inactivating extracts (insulinase). Arch Biochem 1949, 20, 1–9. [PubMed] [Google Scholar]
- 14.Mirsky IA; Perisutti G Effect of insulinase-inhibitor on hypoglycemic action of insulin. Science 1955, 122, 559–560. [PubMed] [Google Scholar]
- 15.Leissring MA; Farris W; Turk BE; Cantley LC; Yeh L; Caclin C; Xing X; Cuny GD; Stein RL; Selkoe DJ Small-molecule inhibitors of insulin-degrading enzyme. Society for Neuroscience Abstract Viewer/Itinerary Planner 2004, Program No. 488.11. [Google Scholar]
- 16.Camberos MC; Perez AA; Udrisar DP; Wanderley MI; Cresto JC ATP inhibits insulin-degrading enzyme activity. Exp Biol Med (Maywood) 2001, 226, 334–341. [DOI] [PubMed] [Google Scholar]
- 17.Song ES; Juliano MA; Juliano L; Fried MG; Wagner SL; Hersh LB ATP effects on insulin-degrading enzyme are mediated primarily through its triphosphate moiety. J Biol Chem 2004, 279, 54216–54220. [DOI] [PubMed] [Google Scholar]
- 18.Cakir B; Dagliyan O; Dagyildiz E; Baris I; Kavakli IH; Kizilel S; Turkay M Structure based discovery of small molecules to regulate the activity of human insulin degrading enzyme. PLoS ONE 2012, 7, e31787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farris W; Leissring MA; Hemming ML; Chang AY; Selkoe DJ Alternative splicing of human insulin-degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid beta-protein. Biochemistry 2005, 44, 6513–6525. [DOI] [PubMed] [Google Scholar]
- 20.Cheng Y; Prusoff WH Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 21.Song ES; Mukherjee A; Juliano MA; Pyrek JS; Goodman JP Jr.; Juliano L; Hersh LB Analysis of the subsite specificity of rat insulysin using fluorogenic peptide substrates. J Biol Chem 2001, 276, 1152–1155. [DOI] [PubMed] [Google Scholar]
- 22.Still WC; Tempczyk A; Hawley RC; Hendrickson T Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. Journal of the American Chemical Society 1990, 112, 6127–6129. [Google Scholar]
- 23.Caulfield T; Medina-Franco JL Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J Struct Biol 2011, 176, 185–191. [DOI] [PubMed] [Google Scholar]
- 24.Caulfield TR; Devkota B Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 2012. [DOI] [PubMed] [Google Scholar]
- 25.Loving K; Salam NK; Sherman W Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J Comput Aided Mol Des 2009, 23, 541–554. [DOI] [PubMed] [Google Scholar]
- 26.Salam NK; Nuti R; Sherman W Novel method for generating structure-based pharmacophores using energetic analysis. J Chem Inf Model 2009, 49, 2356–2368. [DOI] [PubMed] [Google Scholar]
- 27.Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. Journal of molecular graphics 1996, 14, 33–38, 27-38. [DOI] [PubMed] [Google Scholar]