

Abstract
The intermolecular interactions of noble gases in biological systems are associated with numerous biochemical responses, including apoptosis, inflammation, anesthesia, analgesia, and neuroprotection. The molecular modes of action underlying these responses are largely unknown. This is in large part due to the limited experimental techniques to study protein-gas interactions. The few techniques that are amenable to such studies are relatively low throughput and require large amounts of purified proteins. Thus, they do not enable the large-scale analyses that are useful for protein-target discovery. Here we report the application of Stability of Proteins from Rates of Oxidation (SPROX) and limited proteolysis (LiP) methodologies to detect protein-xenon interactions on the proteomic scale using protein folding stability measurements. Over 5,000 methionine-containing peptides and over 5,000 semi-tryptic peptides, mapping to ~1,500 and ~950 proteins, respectively, in the yeast proteome, were assayed for Xe-interacting activity using the SPROX and LiP techniques. The SPROX and LiP analyses identified 31 and 60 Xe-interacting proteins, respectively, none of which were previously known to bind Xe. A bioinformatics analysis of the proteomic results revealed that these Xe-interacting proteins were enriched in those involved in ATP-driven processes. A fraction of the protein targets that were identified is tied to previously established modes of action related to xenon’s anesthetic and organoprotective properties. These results enrich our knowledge and understanding of biologically relevant xenon interactions. The sample preparation protocols and analytical methodologies developed here for xenon are also generally applicable to the discovery of a wide range of other protein-gas interactions in complex biological mixtures, such as cell lysates.
Keywords: Noble Gases, Mass Spectrometry, Proteomics, SPROX, Limited Proteolysis, Molecular Dynamics
Graphical Abstract
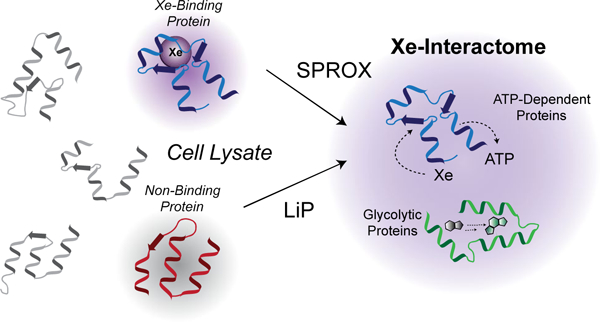
INTRODUCTION
The intermolecular interactions of noble gases (NGs) in biological systems are associated with numerous biochemical responses, including apoptosis, inflammation, anesthesia, analgesia, and neuroprotection. Little is known, however, about either the full scope of NG-biomolecular interactions or the molecular modes of action by which they exert a biological response.1 The lack of information about protein-NG interactions is tied to two intrinsic features of NGs: (1) they cannot be covalently tethered to a solid support or fluorescent (or other) tag, and (2) their isotropic shape, and the absence of charge and polarity, produce relatively weak interactions. The few analytical techniques commonly applied to protein-gas binding interactions, namely NMR spectroscopy and X-ray crystallography, provide rich structural insights but are less effective in quantifying binding affinities.2–7 They also require relatively large amounts of purified protein and are not inherently high throughput. We were therefore motivated to develop methods suitable to the high-throughput discovery and quantification of protein-noble gas interactions in physiologically relevant conditions.
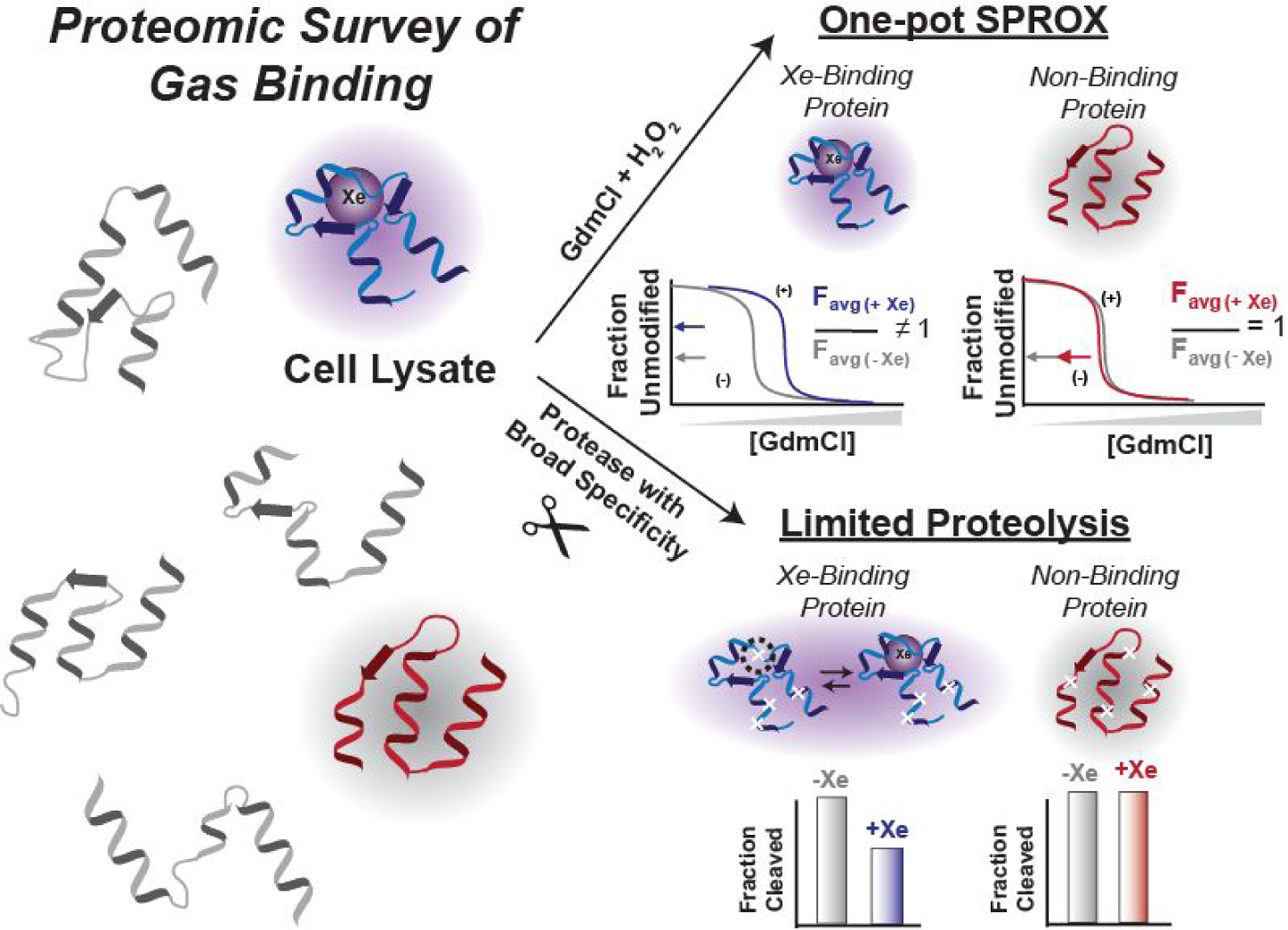
We took inspiration from the recent development of a series of mass spectrometry-based methods for the detection and quantitation of protein-ligand binding interactions in solution and on the proteomic scale. These methods include: the thermal shift assay8, the Stability of Proteins from Rates of Oxidation (SPROX) technique9–11, and proteolysis-based methods such as pulse proteolysis (PP), drug affinity responsive target stability (DARTS), and limited proteolysis (LiP)12–17. While the above mass spectrometry-based techniques have been applied to a number of different small molecule ligands to date (e.g., small molecule drugs, enzyme co-factors, and metals), they have not been applied to dissolved gaseous ligands. The thermal shift assay is not easily adapted to the analysis of such ligands, as gas solubilities are temperature dependent. The proteolysis- and SPROX-based methods, however, have the potential to detect protein-gas binding interactions in solution and obtain information about the relative binding affinities of proteins. Moreover, the proteolysis- and SPROX-based methods have an advantage over more conventional methods that rely on spectroscopic techniques, because they do not require large amounts of purified protein samples. This makes possible the large-scale analysis of ligand-binding interactions using complex biological samples such as cell lysates. We therefore set out to (i) develop a mass spectrometry-based method for the detection of noble gas binding to proteins in solution using the proteolysis- and SPROX-based methodologies (Figure 1), and (ii) apply that method to the discovery of the xenon-interactome.
Figure 1.
Overview of the proteome-wide survey of xenon-protein interactions proposed in this work.
Extension of the SPROX and LiP methodologies to the analysis of protein-gas binding interactions initially required the development of a sample preparation protocol that is conducive to protein-gas binding measurements. Such a protocol was developed as part of this work and validated using the interaction of metmyoglobin and two gases: a control gas, nitrogen, and the inert noble gas, xenon, which is known to bind metmyoglobin.18–20 Ultimately, the sample preparation protocol validated using the metmyoglobin model system was employed in large-scale, proteome-wide SPROX and limited proteolysis experiments to identify xenon-interacting proteins in yeast lysates. A total of 89 unique xenon-interacting proteins with xenon-induced stability changes were discovered, greatly enhancing both the number and the functional diversity of the known interactome. The protein hits identified in these target discovery experiments help to elucidate the mechanisms behind xenon’s biological activities, and reveal a subset of ATP-binding and ATPase proteins as molecular targets of xenon.
RESULTS
Assessment of Sample Preparation.
Extension of the SPROX and LiP techniques to the study of protein-gas interactions initially involved the development of a sample preparation protocol that maximized the concentration of solubilized gas and minimized the amount of gas required for analysis. Maximizing the gas concentration maximizes the ligand-induced stability changes expected in the SPROX and LiP experiments, and minimizing the amount of gas required for analysis is important, given the relatively high cost of Xe. Shown in Figure 2 is the freeze-pump-thaw (FPT) degassing procedure developed here to remove endogenous gases present in the SPROX and LiP samples and maximize the solution concentration of the target gas. The concentration of Xe in our samples after the FPT procedure was determined using a gas chromatography-mass spectrometry headspace analysis. Our analysis revealed that the concentration of Xe in each buffer was close to 5 mM (Supplementary Figure 1), which is the expected concentration of xenon in water exposed to 1 atm xenon based on Xe’s previously established solubility.21
Figure 2.
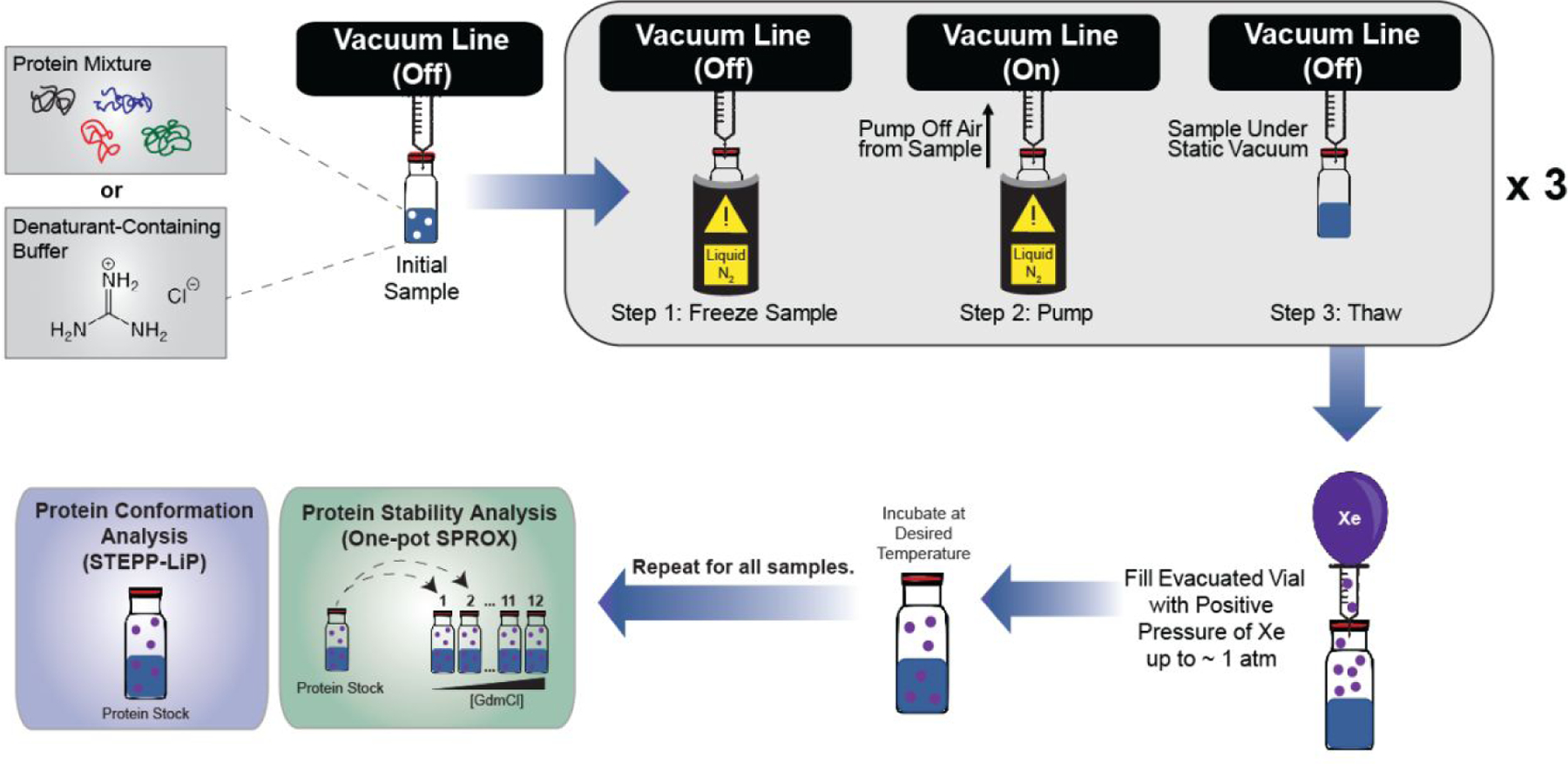
Schematic of the freeze-pump-thaw degassing technique followed by xenon saturation of samples used in this work.
The sample preparation protocol above was assessed in the context of protein binding using metmyoglobin, a known xenon-binding protein, and a UV-vis spectroscopy-based readout to probe the chemical denaturant-induced equilibrium unfolding properties of the protein and the protein-Xe complex. The method exploited the absorbance of metmyoglobin’s heme cofactor, which decreases as the protein unfolds and the heme group is exposed to solvent (Figure 3a). Chemical denaturation curves were collected for metmyoglobin in the absence and in the presence of xenon and nitrogen. Xenon binding to metmyoglobin was successfully detected by a shift in the transition midpoint of its denaturation curve (i.e., value) that averaged 0.24 ± 0.07 M in 3 replicates (Figure 3b, Supplementary Figure 2), whereas the shift was essentially zero for the metmyoglobin-nitrogen control sample (i.e., averaged 0.03 +/− 0.03 M in 3 replicates) (Figure 3c, Supplementary Figure 2). The values measured between the metmyoglobin and metmyoglobin-Xe samples were used to calculate a value of 1.9 ± 1.3 mM for the Xe-metmyoglobin interaction, which is within a factor of three of the reported literature value of 5 mM for the highest affinity xenon binding pocket in metmyoglobin at room temperature.20
Figure 3.
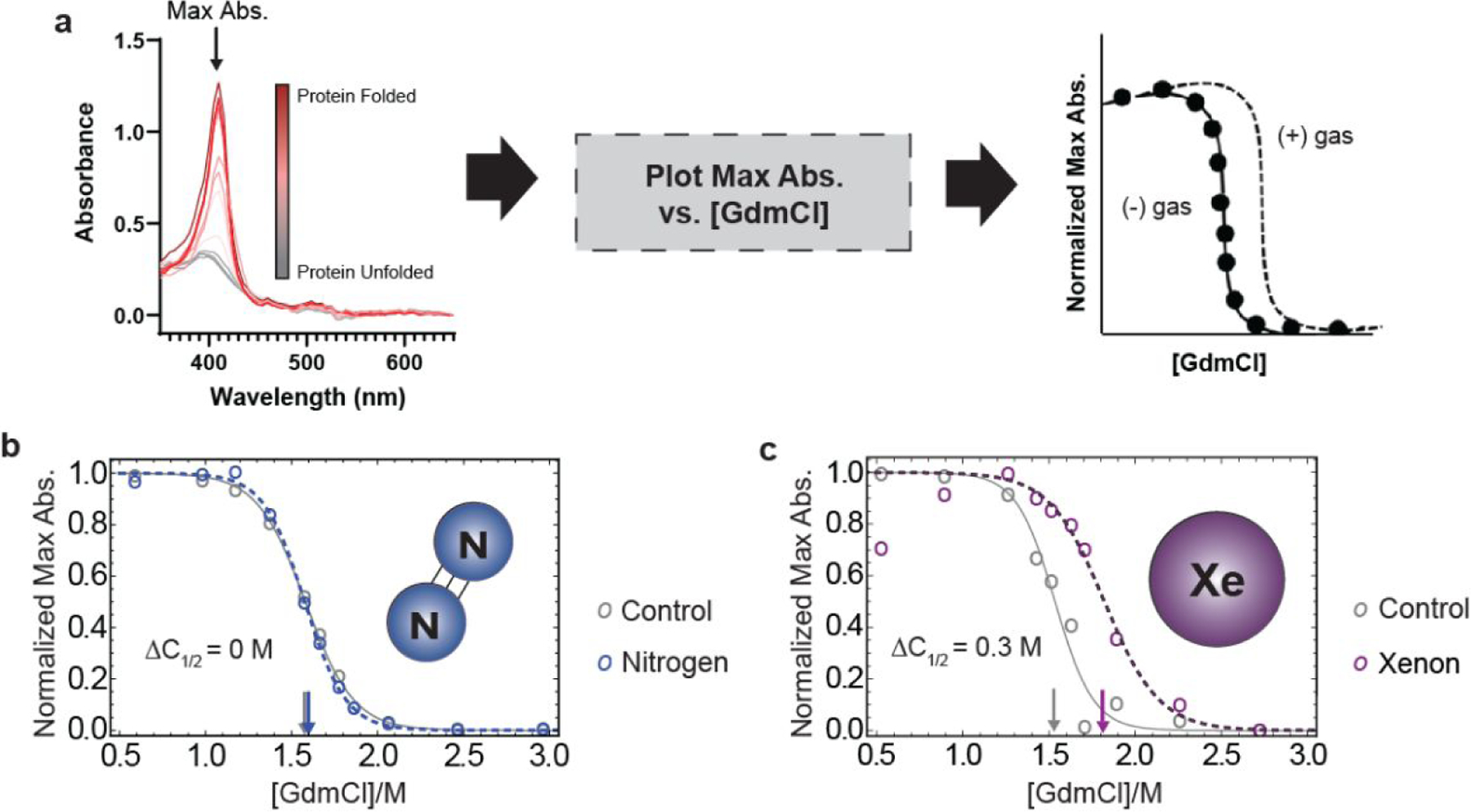
Detection and quantitation of MetMb-Xe interaction. (a) Workflow for obtaining protein stability curves of metmyoglobin from UV-Vis data; (b) representative protein stability curve from a single replicate of metmyoglobin after nitrogen purging shows no significant shift in stability (< 0.05 M ) and (c) representative protein stability curve from a single replicate of metmyoglobin after exposure to 1 atm xenon shows shift of 0.3 M , consistent with known binding affinity of xenon to metmyoglobin.
Large-Scale Analysis of Xenon-Protein Interactions in Yeast Lysates using SPROX and LiP.
The sample preparation protocol above was used in proteomic-scale surveys of xenon-protein binding in a yeast cell lysate using both SPROX and LiP. Prior to the SPROX and LiP analyses, the yeast cell lysate samples underwent a filtration step, performed under denaturing conditions (i.e., 8 M urea), to remove small molecule co-factors and metals, thereby eliminating competition of these ligands for potential Xe binding sites (Figure 4a). This protocol has been used previously to remove NAD from NAD-binding proteins.22 The SPROX analysis exploited a previously established one-pot, bottom-up proteomics workflow, and the LiP analysis utilized a previously reported semi-tryptic peptide enrichment strategy for proteolysis procedures (STEPP).23–24 The highly multiplexed workflows enabled the 5 biological replicates of each treatment condition to be analyzed in a single LC-MS/MS run per technique (see Figure 4b and c). The SPROX and LiP techniques provide complementary information about the ligand-induced changes in conformational states of proteins. As described further below, SPROX probes the more global unfolding/refolding properties of proteins by evaluating the ligand’s effect on a protein’s chemical denaturant-induced equilibrium unfolding/refolding reaction. In contrast, LiP probes the ligand’s effect on the more localized conformational properties associated with a protein’s native three-dimensional structure.
Figure 4.
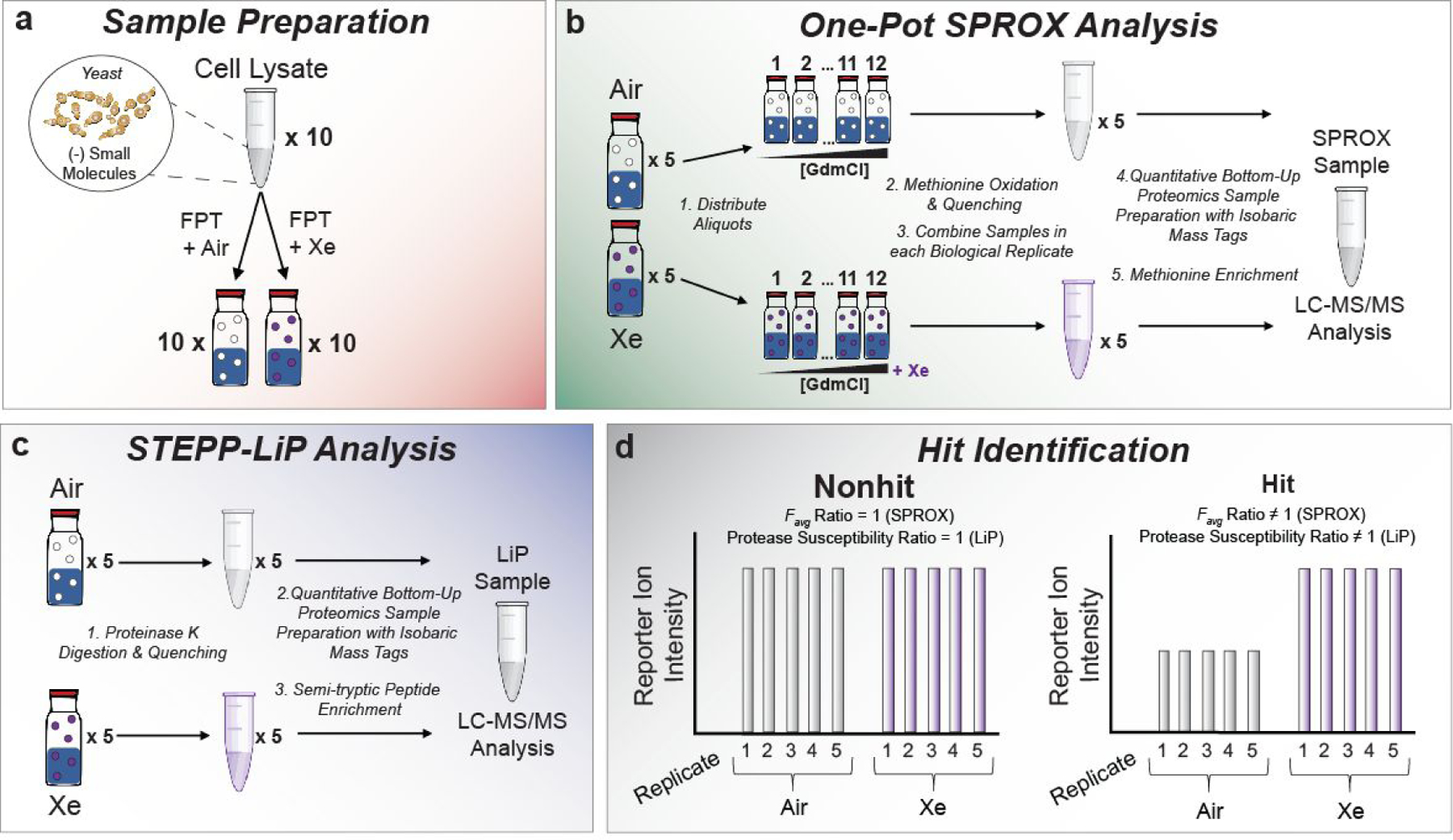
Experimental workflows used in this work. (a) the sample preparation steps for incubation of yeast lysates with xenon; (b) one-pot SPROX protocol to assay Xe-induced protein stability changes in proteins; (c) STEPP-LiP protocol to assay Xe-induced conformational changes in proteins; and (d) schematic representation of expected behavior of nonhit and hit proteins assayed in this work.
A key advantage of both the SPROX and LiP protocols employed here, over more conventional techniques for protein-ligand binding analysis, is that they are amenable to the analysis of relatively small amounts of unpurified proteins on the proteomic scale. However, unlike some experimental approaches for the characterization of protein-ligand binding interactions (e.g., X-ray crystallography, NMR spectroscopy), SPROX and LiP do not provide specific information about the location or stoichiometry of ligand binding. Like NMR spectroscopy, the SPROX and LiP experiments also probe the xenon-binding properties of proteins in the solution phase. This is in contrast to X-ray crystallographic studies where xenon-protein complexes are typically formed by subjecting protein crystals to high pressures of xenon.
One-pot SPROX.
In SPROX, a protein’s chemical denaturant-induced equilibrium unfolding/refolding properties are evaluated by quantifying the chemical denaturant dependence of a methionine oxidation reaction involving hydrogen peroxide. As proteins are unfolded at increasing concentrations of chemical denaturant, the methionine residues buried in hydrophobic regions of a protein’s native structure become exposed to solvent and more readily undergo oxidation. Ultimately, the relative abundances of the wild-type and oxidized methionine-containing peptides detected in the bottom-up proteomics readout are used to construct chemical denaturation curves for the protein to which they map. The relative amount of wild-type and oxidized methionine-containing peptides derived from the proteins in the denaturant-containing buffers will increase and decrease (respectively) as a function of denaturant concentration. The change in extent of oxidation is used to construct denaturation curves similar to those of metmyoglobin reported above (see Figure 3). The presence of a binding ligand (e.g., xenon) will typically increase the stability of a protein, and shift the midpoint of the protein’s unfolding curve to a higher denaturant concentration. In the one-pot SPROX approach, however, the denaturant containing samples are pooled into a single sample, which precludes the ability to generate denaturation curves for each treatment condition. Instead, the resulting measurement produces the average signal from all the denaturation points (i.e., an value). Direct (and indirect) ligand-binding interactions can be identified by looking for significant changes in a protein’s value between control and ligand-treated samples.
The one-pot SPROX workflow outlined in Figure 4b was applied to the proteins in a yeast cell lysate subject to the FPT protocol outlined above (Figure 2), using xenon as the binding ligand in one sample and air as a non-binding control in the other. A total of 5,187 wild-type methionine-containing peptides mapping to 1,505 yeast proteins were successfully identified and quantified in the one-pot SPROX analysis. From these assayed proteins, 36 peptide hits that mapped to 31 different proteins were identified with Xe-induced changes to their chemical denaturation behavior (Table 1, Supplementary Table 1, Supplementary Figure 3). Of the 36 hit peptides, 12 of them were stabilized (log2( ratio) > 0), and 24 of them were destabilized (log2( ratio) < 0) (Figure 5a).
Table 1.
Proteomic coverage and hit rate for each LC-MS/MS sample analyzed in this study.
| Experiment | Assayed Peptides (Proteins) | Hit Peptides (Proteins) | Hit Rate Peptide % (Protein %) |
|---|---|---|---|
| Yeast + Xe One-pot SPROX | 5187 (1505) | 36 (31) | 0.69 (2.06) |
| Yeast + Xe STEPP-LiP | 5116 (947) | 69 (60) | 1.34 (6.34) |
Figure 5.
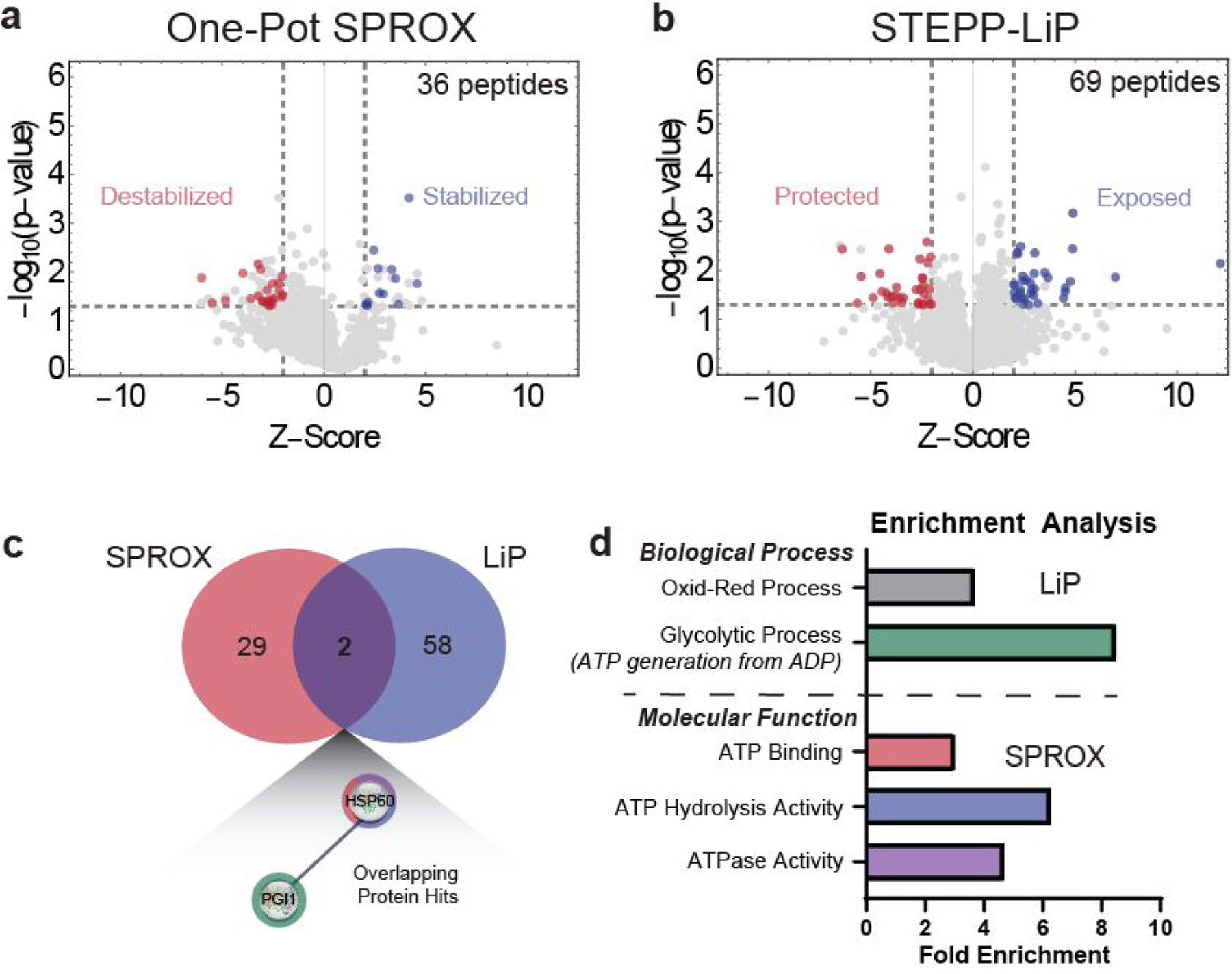
Analysis of protein stability and conformational changes in the presence of Xe using (a) one-pot SPROX and of protein conformational changes in the presence of Xe using (b) STEPP-LiP. Gray lines represent criteria used for the selection of differentially stabilized proteins or differentially cleaved proteins (|peptide log2(fold-change)| ≥ 2 STDEV from average log2(fold change), p ≤ 0.05). Red and blue dots represent hit proteins that passed the selection criteria for destabilization and stabilization in SPROX, or protection and exposure in LiP. (c) Venn diagram showing number of overlapping protein hits for SPROX and LiP techniques and (d) GO Biological Process and Molecular Function analysis of protein hits in SPROX and LiP experiments reveals the Xe-interactome is enriched in proteins with ATP-dependent activities.
The few protein-gas binding affinities that are known suggest that protein-gas binding interactions are relatively weak compared to other protein ligands such as small molecule drugs, enzyme co-factors, and other proteins. In turn, the low affinities for gas binding to proteins make the expected magnitude of the protein stability shifts relatively small. Although an absolute binding affinity of xenon to proteins cannot be obtained using the one-pot SPROX approach, the relative magnitude of the binding affinity can be ascertained by the log-2-transformed reporter ion ratio of the (−) and (+) Xe samples. These log-2-ratios ranged from 0.15–0.85 for the methionine-containing peptide hits in SPROX and averaged 0.42. This average ratio of 0.42 is close to the ratio of 0.34 that can be calculated from the metmyoglobin denaturation curves collected in this work (see Figure 3c). Therefore, it is likely that most of the protein hits identified here have protein-Xe dissociation constants in the 1–2 mM range, similar to that calculated for the model metmyoglobin-Xe interaction. While protein-Xe binding interactions with tighter binding affinities would be readily detected in the SPROX experiment, those with much lower affinities (i.e., values > ~10 mM) would not be detected in the experiment, as the expected shift in the denaturation curves and resulting log2( ratio) values would be small and within the experimental error.
STEPP-LiP.
In contrast to SPROX, which identifies global changes in a protein’s unfolding/refolding equilibrium upon ligand binding, LiP probes the more local ligand-induced conformational changes in a protein’s native three-dimensional structure. The premise of LiP is that ligand engagement and subsequent shifts in a protein’s conformational stability result in differential susceptibility to proteolytic digestion using a protease with broad specificity (e.g., proteinase ).16–17 When proteolysis is performed in the presence and absence of the target ligand, proteins whose native conformations are significantly perturbed can be identified by measuring ligand-induced differences in proteolytic cleavage of the polypeptide backbone at specific regions of a protein’s structure, which is referred to hereafter as protection or exposure.
The LiP workflow shown in Figure 4c was used to analyze the proteins in yeast cell lysates prepared with xenon and air using the FPT protocol in Figure 2. The LiP experiment yielded a total of 5,116 semi-tryptic peptide probes mapping to 947 yeast proteins. A total of 69 peptide hits mapping to 60 unique proteins with Xe-induced conformational changes were identified (Table 1, Supplementary Table 2, Supplementary Figure 4) using hit selection criteria identical to those used in SPROX. Of the 69 peptide hits, 34 peptides mapping to 30 proteins showed Xe-induced protection and 35 peptides mapping to 33 proteins showed Xe-induced exposure (Figure 5b). There were 2 overlapping protein hits, PGI1 and HSP60, in the one-pot SPROX and STEPP-LiP experiments (Figure 5c).
Bioinformatic analysis of protein hits from SPROX and LiP experiments.
The peptide hit rates of 0.69% and 1.34% observed in our SPROX and LiP experiments (respectively) were relatively low and only slightly higher than the expected false positive rates. False positive rates of 0.04% and 0.09% have been previously reported for SPROX and proteolysis experiments in a model protein-target discovery study using cyclosporine A (CsA) and the proteins in a yeast cell lysate.23 However, in order to enable the identification of more proteins with weaker binding affinities, our xenon binding study employed less stringent hit selection criteria than those utilized in reference 23. Application of the same hit selection criteria used in our xenon binding study to the SPROX and proteolysis model yeast data sets in reference 23 yields false positive rates of 0.69 and 0.48%, which match or are only slightly below the hit rates observed in our SPROX and LiP experiments.
The overlap in the differentially stabilized proteins detected in the SPROX and LiP experiments was relatively low, and only included 2 proteins. This is perhaps not surprising given that the two techniques probe different conformational properties of proteins (e.g., more global unfolding/refolding properties in SPROX and more local fluctuations in secondary structure in LiP). They also rely on different readouts (e.g., the detection and quantitation of methionine-containing tryptic peptides in SPROX and of semi-tryptic peptides in LiP.
To better understand the nature of the hit proteins, the 31 proteins with Xe-induced stability changes identified in the SPROX experiment and the 60 proteins with Xe-induced conformational changes identified in the LiP experiment were subject to a bioinformatics analysis to identify what biological pathways might be influenced by the xenon-interactome and what biophysical properties might drive noble gas binding to proteins. A GO-term analysis25–27 of molecular functions and biological processes of the protein hits identified in each technique revealed that the SPROX protein hits are enriched in both ATP binding proteins and proteins with ATPase or ATP hydrolysis activity, and the LiP protein hits are enriched in proteins involved in oxidation-reduction processes and glycolytic processes – more specifically, proteins that generate ATP from ADP (Figure 5d). Combining the data from both techniques resulted in enrichment of glycolytic proteins and ATP-binding proteins, suggesting these are specific classes of proteins that interact with xenon.
A total of 15 of the 31 xenon binding proteins identified in the SPROX experiments here are known ATP-binding proteins, and 6 of these 15 known ATP-binding proteins were identified as ATP-binding proteins in previously published studies using the SPROX methodology to identify the ATP-interactome in yeast.10, 28One of the 31 xenon binding proteins in this work, inorganic pyrophosphatase (IPP1), is not formally annotated as an ATP binding protein, but it was identified to have ATP-binding properties in our previous study using the SPROX methodology to identify the ATP-interactome in yeast.10 Also interesting is that IPP1 and all but one of the 15 xenon binding proteins identified in this work with previously annotated ATP-binding properties were destabilized in the presence of xenon. This xenon-induced destabilization is similar to the ATP-induced destabilizations we detected for a large number of the ATP-binding proteins identified in our SPROX experiments to characterize the ATP-interactome in yeast.10, 28 Indeed, all but one of the shared targets between the ATP- and Xe-interactome studies are destabilized. This suggests that ATP and Xe may interact with their protein targets by similar mechanisms (see Discussion).
Exploration of Xenon-Binding to Hit Proteins using Molecular Dynamics Simulations and Xenon Docking Analysis.
Because of the strong correlation observed between protein-Xe interactions and protein-ATP interactions, a molecular modeling strategy was used to explore potential Xe- binding interactions with annotated ATP-binding proteins. Four known ATP-binding proteins and one GTP-binding protein, all with known nucleotide-bound crystal structures, were subjected to molecular dynamics (MD) simulations and docking analysis. The four ATP-binding proteins included: two proteins, actin and endoplasmic reticulum chaperone BiP, that were identified as (destabilized) hits in SPROX (PDB IDs: 1YAG and 3QFU); one protein, phosphoglycerate kinase (3PGK), that was identified as having a xenon-induced exposure in LiP, and ATP-dependent RNA helicase EIF4A, that was not identified as a hit in either SPROX or LiP, but was co-crystallized with a LiP hit, eukaryotic initiation factor 4F subunit p150, with a xenon-induced protection. The non-hit GTP-binding protein was elongation factor 1-alpha (PDB ID: 1IJE).
MD simulations were carried out on each of the above five proteins using x-ray crystallographic data obtained for each protein both with and without a nucleotide ligand (e.g., ATP, ADP, AMP, or GDP). Xenon binding was assayed in all extracted MD snapshots using visual molecular dynamics (VMD) followed by docking analysis with Autodock4.29–30 Docking was performed under three protocols for each protein: (i) docking to extracted MD snapshots of the holo-protein, (ii) docking to extracted MD snapshots of the holo-protein with its nucleotide ligand removed prior to docking (to resemble an apo-structure), and (iii) docking to extracted MD snapshots of the apo-protein. Favored xenon binding sites were studied as a function of distance from the nucleotide binding site for each protein. The total number of times xenon was found to dock on each protein was counted over a 5 Å – 20 Å distance from the center of mass of each protein’s respective nucleotide ligand. The data from these simulations are shown in Figure 6.
Figure 6.
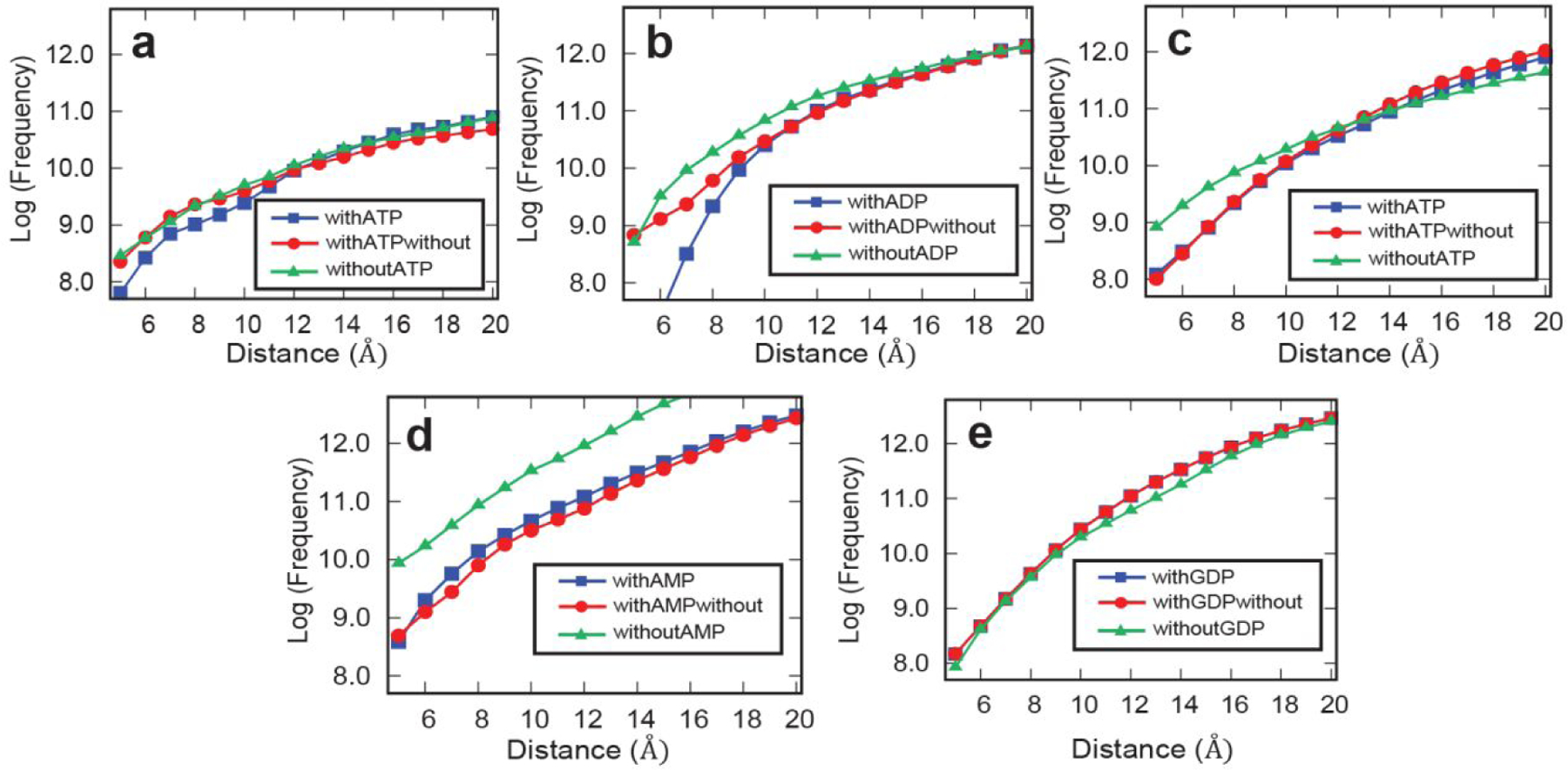
Xenon docking calculations with select protein hits and a protein nonhit. (a) actin (1YAG), (b) endoplasmic reticulum chaperone BiP (3QFU), (c) phosphoglycerate kinase (3PGK), (d) ATP-dependent RNA helicase EIF4A (2VSO), and (e) elongation factor 1-alpha (1IJE). Each curve represents one of the 3 cases in which binding of xenon to a protein was investigated. The blue curve represents the case where the nucleotide ligand was present, the green line represents the case where the MD simulations were done with the nucleotide ligand but removed before docking, and the red curve shows the case where the nucleotide ligand was not present. Distances are taken from the center of mass of the protein’s respective nucleotide ligand and are sampled in the range of 5 Å to 20 Å. The y-axis shows the log frequency of the number of times xenon was found at that distance.
In all four ATP-binding proteins studied, the frequency of xenon binding near the nucleotide was significantly higher in at least one of the apo-structures compared to the holo-structure, which is consistent with xenon binding in the nucleotide site of these proteins. The MD simulations on the GDP binding protein revealed the frequency of xenon binding was unchanged by the presence of nucleotide, consistent with the absence of a xenon binding interaction at the GDP site in this protein. In the case of the two SPROX hits (Figures 6a–b) higher docking frequencies were obtained with both the apo-structure and the holo-structure with the nucleotide removed. This contrasts with observations from the two LiP hits (Figures 6c–d), where only the MD simulations performed on the apo-crystal structure indicated a higher frequency of xenon binding.
The MD simulations and xenon docking experiments on the four proteins above support the hypothesis that xenon binds to the nucleotide binding site in these proteins. Previous results indicate that xenon has a computed binding free energy of −5.1 to −6.4 kcal/mol in sperm whale myoglobin D122N mutant (PDB ID: 1J52).31 Experimental xenon binding free energy values were determined in those studies to be 5.9 ± 0.2 kcal/mol, similar to their theoretical values, and to a previously calculated binding free energy of ATP to glutamyl-tRNA synthetase, which was predicted to be 7 kcal/mol.32 These previous findings indicate that xenon and ATP have nearly equal binding free energies, suggesting that xenon and ATP may have similar binding preferences for cavities in proteins, as found in our study.
DISCUSSION
Biological Activity.
There are currently many hypotheses developed to explain the mechanisms behind inert gas biological activities, with much focus on xenon due to its strikingly potent effects. These hypotheses fall into two broad categories, defined as biochemical or biophysical hypotheses, and they include a wide range of phenomena including: hypoxia, depression of metabolism, cell membrane stabilization, decreased ion permeability, and interference with ATP production.33 From this list, the biochemical hypotheses support the notion that xenon interacts with specific proteins, such as respiratory enzymes, while biophysical hypotheses imply that xenon interacts specifically with part of the cell, such as the cell membrane.34 The xenon-interactome revealed in the current study provides a rich data set to evaluate such hypotheses in the context of prior findings. Xenon’s biological effects were studied previously in numerous animal models (rat, dog, rabbit etc.)35–39, bacteria40, cancer cells41, kidney cells42, neurons43–44, and human volunteers.45–46 Although carried out in a different model system, our studies find protein targets that overlap with previously established modes of action for xenon, specifically xenon’s ability to inhibit or influence the activity of proteins with ATP-dependent activity.40, 47–50
The most notable therapeutic effects of xenon are its strong organoprotective properties, specifically on the brain39, 44, 51–52 and the heart.35–38 One hypothesis for how xenon mediates its cardioprotective effects is through direct interplay with kinase signaling pathways that lead to downstream cytoskeletal remodeling, as shown in myocardial cells.38, 53 Xenon has also been found to influence kinase signaling in other contexts.54 The results of our proteomic studies provide additional support for xenon’s effects on similar pathways, albeit in a different model system. We find a number of hit proteins from the kinase protein class (CDC19/PYK1, PGK1, PCK1, DAK1), as well as heat shock proteins that are readily phosphorylated by kinases (HSP60). Furthermore, we find significant changes in the stability of actin and in the conformational dynamics of an actin-associated protein (ABP1) from the yeast proteome. Actin is known to polymerize in response to xenon’s influence on kinase signaling pathways and heat shock proteins in myocardial cells.55–56
Other experiments that study xenon’s mode of action have implicated xenon’s direct interaction with, and influence over, the activity of multiple proteins with ATP-dependent activity, including Enterococcus hirae CopB ATPase40 and adenosine triphosphate-sensitive potassium () channels present in the brain.47–50 In our work, we find significant Xe-induced stability changes, or conformational changes, in multiple ATPase pumps including plasma membrane ATPase 1 (PMA1), a hydrogen ion pump; multiple subunits of the V-type ATPase (VMA1, VMA2), a proton pump present in vacuolar membrane vesicles; and the alpha subunit of ATP synthase (ATP1), a proton pump present in the mitochondrial membrane. PMA1 showed the strongest conformational changes of all the limited proteolysis protein hits, having one peptide with a fold change 12 z-scores from the average. Other ATPase pumps, particularly those identified as hits in SPROX, exhibited potent destabilizations (i.e., 3+ z-scores away from average) compared to other protein hits. These changes in stability have precedent, as xenon has been shown to decrease the sensitivity of similar proton pumps to ATP in a concentration-dependent manner.50 Electrolyte imbalance resulting from the changed activities of ion flow over the membrane has also been proposed as an anesthetic mechanism of action for xenon.57 Such past observations suggest that xenon may be able to interact allosterically or directly with the ATP sites of ion pumps, and thus alter their functions, leading to changes in ion flow.
Intermolecular Interactions.
We next consider the xenon-interactome identified by the SPROX and LiP techniques in the context of long-standing hypotheses that xenon and other anesthetic gases typically occupy small hydrophobic cavities in proteins, locking them in specific active or inactive conformations.34 A computational study supporting this hypothesis analyzed 348 xenon binding sites from 131 xenon-protein crystal structures in the Protein Data Bank (PDB) and used them to predict noble gas binding sites in proteins.58 The most favorable binding sites for Xe were found to be hydrophobic channels or small cavities with an average diameter of 4 Å. A more recent in silico reverse docking study involving 127,854 protein structures and five nonradioactive noble gases also revealed significant trends in the hydrophobic nature of noble gas binding sites across a wide range of protein structures.59There is some evidence in our study that xenon is attracted to hit proteins containing exposed hydrophobic cavities.
One key piece of evidence for xenon binding in such cavities originates from the most stabilized hit in SPROX, SIP18. This protein contains a phospholipid binding site that may be an attractive hydrophobic cavity for Xe. SIP18 was shown to be stabilized by Xe in the SPROX experiments as inferred by a decrease in methionine oxidation of multiple methionine-containing peptide probes (Supplementary Table 1), suggesting that xenon may interact directly with this protein.
Other evidence for xenon binding in hydrophobic cavities is supported by xenon’s attraction to ATP-binding proteins. ATP-binding sites often include non-polar and hydrophobic residues, such as valine and leucine, that favor protein-ATP binding.60 It has also been shown in both biophysical and theoretical studies that ATP molecules associate with lipid bilayers (primarily through their adenine ring), sharing a mechanism proposed for xenon’s biological effects.61–62 Xenon has been shown to exert biophysical changes on lipid bilayers, potentially effecting the function of membrane-bound proteins.63 In addition to the attraction of both Xe and ATP to hydrophobic sites in proteins, both ligands have high polarizabilities,4, 64 which could help to explain their preference for similar binding sites in proteins.
Another commonality between the protein-ATP interactions previously identified and protein-Xe interactions discovered in this work is the apparent and consistent destabilization of protein targets. Approximately half of the xenon protein targets identified by SPROX in this work were annotated as ATP-binding proteins, with the majority of those targets (93%) being destabilized in the presence of xenon (Supplementary Figure 3). Such destabilization was also frequently seen in previously reported ATP-interactome studies in yeast, where a large fraction of protein targets was destabilized by ATP.10, 28 The direct binding of a ligand to a protein’s native three-dimensional structure generally results in a ligand-induced stabilization in SPROX. However, the binding of ligands to non-native protein structures (for example protein folding intermediates) can produce ligand-induced destabilizations in energetics-based assays of protein-ligand binding interactions. In fact, this was shown to be the case with ATP and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from E. coli, in which ATP stabilized a partially unfolded intermediate of the protein’s unfolding/refolding pathway.14, 65 It is possible that ATP and xenon both bind preferentially to states other than their target protein’s native three-dimensional structure. The significant overlap of ATP and xenon targets, and similarities in the types of stability changes induced upon ligand binding, suggest that these two ligands target protein structures through similar mechanisms, and possibly similar binding pockets.
The observation that our protein hits are enriched with those that also bind ATP (e.g., kinases), or have ATPase dependent catalytic activities (e.g., proton pumps, molecular chaperones, heat shock proteins), adds to the evidence that there may be parallels in the molecular nature of protein-ATP and protein-Xe interactions. To further explore this hypothesis on a broader scale, our computational studies analyzed xenon localization in the presence or absence of ATP (and related ligands) across four protein hits. The results of these studies indicate that the presence of nucleotides has significant effects on the frequency of xenon docking to their pockets in hit proteins, but not in a nonhit GDP-binding protein which had no xenon-induced stability changes. This increased localization of xenon to adenine nucleotide sites may be explained by the high polarizabilities of ATP and xenon (although Xe’s polarizability is electronic in origin and ATP’s polarizability arises largely from nuclear motion), and therefore similarities in the types of pockets that attract their binding.4, 64 Previous computational studies have also revealed the proximity of noble gases to ligand binding pockets, indicating that noble gases have the potential to interfere with ligand binding and modulate protein function.59 Our observations provide further evidence for the likelihood of xenon to interact competitively with such sites in ATP-binding proteins, conceivably influencing the activity of the protein hits identified in this work.
The Protein Data Bank (PDB) currently includes X-ray crystallographic data on 131 protein crystals from ~100 unique proteins in the presence of xenon. Only 2 of the 131 xenon-protein complexes with X-ray crystallographic data in the PDB are yeast proteins. Neither one of these proteins were assayed in the LiP experiment (i.e., semi-tryptic peptides from these proteins were not detected). However, both of these proteins (MTF1 and POP2) were assayed in the SPROX experiment (i.e., methionine-containing peptides mapping to these proteins were detected). However, the detected peptides did not map to the xenon-binding domain of these proteins, which may explain why these proteins were not identified as a hit in SPROX.
Currently, there are 7 crystal structures of ATP binding proteins complexed with xenon in the PDB. Direct comparisons of our computational results to the X-ray crystallographic data on these 7 ATP-binding proteins complexed with xenon is difficult for several reasons. Not only are these ATP-binding proteins from species other than yeast, but the protein crystals used in these studies included a bound nucleotide or co-factor. Our SPROX and LiP experiments were performed on cell lysates with these nucleotides and co-factors removed. Additionally, xenon-containing protein crystals for X-ray crystallographic analyses are prepared by pressurizing protein crystals with xenon gas. Thus, the resulting xenon binding interactions may be different from those detected in our solution phase experiments.
Another major finding from our bioinformatic analyses of proteins with Xe-induced conformational changes was the statistical overrepresentation of proteins involved in glycolytic processes (Figure 6b, Supplementary Figure 4). Such metabolic enzymes rely on conformational changes induced by substrate binding to perform their functions. If xenon is able to interact directly with the hydrophobic cavities in these proteins, it is possible that it may lock them into specific nonnative conformations that inhibit enzymatic activity, thus leading to unique biological outcomes. Assaying such glycolytic proteins for xenon-induced functional changes is an area for future study, as the influence of xenon on protein function likely mediates its activity.
CONCLUSIONS
This study introduces an analytical methodology that can be used generally for the large-scale analysis of protein-gas binding interactions. There are few analytical techniques that can detect and quantify the binding constants of such interactions, both in solution and on the proteomic scale. The hit proteins identified in this study of inert gas binding properties are a first step toward understanding the biological activities and therapeutic effects of the noble gases.
The identification of the xenon-protein interactome in this work provides direct evidence that xenon interacts with specific proteins to affect their stabilities and conformations, and possibly their activities. Monitoring such stability changes revealed xenon’s effects on proteins involved in ATP-driven and glycolytic processes. How xenon mediates its biological effects through these pathways and protein classes is still under investigation; however, our protein hits add to the arsenal of information used to understand xenon’s important biological activities, and future studies may be used to link significant xenon-induced stability and conformational changes to changes in function of key protein hits found in this work. Further studies using this methodology could explore xenon protein targets in clinically relevant samples, such as lysates from myocardial cells or neurons. Such studies could also be expanded to other inert gases with known biological activities (e.g., argon). Furthermore, it is possible that the protein targets identified in this study may be used to motivate the synthesis of protein-like molecules for the detection, collection, and sensing of gases.
MATERIALS & METHODS
Materials.
The following materials were from Sigma Aldrich (St. Louis, MO): glucose, guanidine hydrochloride , S-methylmethanethiosulfonate (MMTS), urea, centrifugal filter units (Amicon Ultra, 0.5 mL, 10 MWCO), tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl), trifluoroacetic acid (TFA), triethylammonium bicarbonate buffer (TEAB, 1 M, pH 8.5), hydrogen peroxide (H2O2) (30% w/w), acetic acid, 2-mercaptoethanol, myoglobin from equine heart, proteinase from Tritirachium album, and phenylmethylsulfonyl fluoride (PMSF). The following materials were from ThermoScientific (Waltham, MA): acetonitrile (ACN, LC-MS grade), 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF), bestatin, E-64, leupeptin, pepstatin A, TMT10-Plex isobaric label reagent set and porcine pancreas trypsin (proteomics grade). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was from Santa Cruz Biotechnology (Dallas, TX). Phosphate-buffered saline 10X, Molecular Biology Grade (PBS, pH 7.4) was from Corning (Corning, NY). Yeast extract and peptone were from HiMedia (Mumbai, India). Adenine hemisulfate dihydrate was from MP Biomedicals (Santa Ana, CA). Macrospin columns (silica C18) and Pi3 Methionine reagent kit were from Nest Group (Southborough, MA). Research-grade xenon was from Airgas (Randor, PA). Screw top GC vials and caps were from Agilent Technologies (Santa Clara, CA).
Introduction of Xenon after Freeze-Pump-Thaw Degassing of Samples.
To ensure maximum levels of xenon in solution for gas binding analysis, a freeze-pump-thaw sample preparation was used to degas buffers and protein stocks. A series of denaturant buffers ranging from 0.5–3 M in PBS (pH 7.4) and a protein stock of metmyoglobin (5 mg/mL) were placed in 2 mL GC vials (290 μL each), capped, parafilmed, and frozen in liquid nitrogen. The vials were placed on a vacuum line setup with syringe needles piercing each septum to place the vials under an inert atmosphere. The headspace above each frozen sample in the vial was pumped off under dynamic vacuum. The samples were then allowed to thaw under static vacuum. After thawing, the samples were frozen again, and the freeze-pump-thaw process was performed a total of 3 times to guarantee the samples were properly degassed and under static vacuum. Xenon was then introduced to each degassed sample vial with a balloon full of pure xenon. After this freeze-pump-thaw method, it is assumed that each vial will be filled with 1 atm of xenon. The solubility of xenon in water at this pressure and at room temperature is ~ 5 mM.21
Quantitation of Xenon in Buffer using GC-MS Headspace Analysis.
To ensure that xenon was present in solution after freeze-pump-thaw degassing and introduction of gas with balloon, a GC-MS headspace analysis was performed after using sample preparation conditions in regular buffer (not containing denaturant). After 300 μL of PBS was degassed and incubated with xenon under the conditions described, it was incubated for 30 minutes on ice. The vial was then quickly uncapped, and the solution was transferred from the original GC vial to a new headspace vial with a crimp cap. Standards were prepared by using a syringe to inject known volumes of pure xenon into headspace vials containing the same volume of buffer. The standards were then vortexed to encourage proper mixing and solubilization of the gas. All standards and samples were sonicated at 35°C for 30 minutes in a water bath. Then, 10 μL of the headspace in each vial was injected into the GC for the detection and quantitation of xenon.
GC-MS headspace analysis of Xe-incubated buffers was performed using a Shimadzu GCMS-QP2010 gas chromatography mass spectrometer. The gas chromatograph was equipped with a Carboxen ® 1010 PLOT fused silica capillary column (30 m x 0.32 mm) for separation of inert gases. Instrument temperatures were 75°C for the initial column oven temperature and 210°C for the injection port. The ion source temperature was 200°C. Helium was used as the carrier gas and injections were conducted with a split ratio of 20. MS data were acquired from 10–150 m/z. The temperature program was as follows: 75 °C for 1 minute, 30°C/min ramp to 225°C, and a hold at 225 °C for 2 minutes. The total run time for each sample was 8 minutes.
Method Development with Metmyoglobin Model System.
A UV-Vis Analysis of metmyoglobin stability after incubation with gas was employed to detect ligand-induced stability changes upon binding of xenon. The absorbance of the heme in myoglobin decreases as the protein unfolds and the heme group is exposed to solvent, therefore, protein unfolding directly correlates with a reduction in absorbance of each sample. A xenon-containing stock solution of metmyoglobin (5 mg/mL) was distributed across the xenon-containing denaturant buffers ranging from 0.5–3 M (0.17 mg/mL, 10 μM protein final). The same was done with denaturants and protein stocks that did not undergo the freeze-pump-thaw and xenon introduction steps and served as control samples. Both control and xenon incubated samples sat on ice for 1 hr before analysis by UV-Vis spectroscopy. The absorbance of each sample was scanned from 350 nm to 650 nm. The maximum absorbance of each sample was plotted vs. [] to obtain protein stability curves of metmyoglobin in the presence and absence of xenon. Each sample was normalized to its pre- and post-transition baseline points to obtain final normalized protein stability curves. These curves were fit to eq 1. to extract midpoint values for estimations. In eq 1. is the estimated folding free energy of the protein, m is the estimated -value of the protein, [] is the concentration of denaturant, R is the ideal gas constant and T is the temperature in Kelvin.
| (eq 1.) |
Midpoint values for each protein unfolding curve were calculated by dividing the estimated folding free energy by the estimated -value () determined from the fitting. This entire process was also performed with metmyoglobin samples in the presence of nitrogen as a control system in which no ligand-induced stability changes should occur.
Estimation of Metmyoglobin-Xenon Interaction.
The value of metmyoglobin-xenon was determined by first calculating the binding free energy, , from the midpoint difference between the control and xenon UV-Vis protein denaturation curves using eq 2.
| (eq 2.) |
In eq 2, is the -value estimated for metmyoglobin from the steepness of its denaturation curves (~3.23 kcal/(mol*M)), and was the shift in the value upon xenon binding (). The dissociation constant was then calculated using eq 3,
| (eq 3.) |
where is the concentration of free ligand in the denaturant-containing buffers, is the ideal gas constant, is the temperature in Kelvin, and is the change in folding free energy determined in eq 2.
Cell Culture, Lysis & Ligand Incubation.
Yeast strain S288C was obtained from ATCC and cultured in YPAD medium (0.4 g of adenine sulfate, 10 g of yeast extract, 10 g of peptone and 20 g of glucose in 1 L of deionized (DI) water) according to standard protocols. Briefly, a yeast colony was incubated in 50 mL of YPAD medium at 30°C. Following an overnight incubation to reach an OD600 of ~1.6, a 20 mL portion of the culture was inoculated with the YPAD medium (1 L) to give an OD600 of ~ 0.3. The inoculated medium was incubated at 30°C until the OD600 of the solution was between 1.2 and 2.0. Fractions of the final YPAD medium (250 mL each) were centrifuged to generate yeast cell pellets.
Yeast cell pellets were lysed in PBS (1x, pH 7.4) containing the following protease inhibitors: 1 mM AEBSF, 20 μM leupeptin, 10 μM pepstatin A, 500 μM bestatin, and 15 μM E-64. Cell lysis was accomplished by mechanical disruption using glass beads (0.5 mm) with 25 s disruption and 1 min intervals on ice for a total of 15 cycles. The lysed cells were centrifuged at 14,000g for 10 min at 4°C. The initial total protein concentration in the supernatant from each cell lysate sample was determined by a Bradford assay. To remove small molecules, cell lysates were denatured in ~8 M urea at 37°C for 30 minutes. Samples were then buffer-exchanged and renatured in 10 kDa molecular weight cutoff filters with PBS (1x). The final protein concentration of each small molecule depleted lysate was determined by a Bradford assay and normalized to 2 mg/mL. The normalized lysates for each replicate were then divided into two equal aliquots; one aliquot underwent freeze-pump-thaw and xenon incubation steps to generate the (+) ligand sample, and the other aliquot underwent freeze-pump-thaw and air incubation steps to generate the (−) ligand sample. Lysates were equilibrated with or without the target gas for 30–45 minutes at 4°C for the SPROX experiments and 1 hr at for the limited proteolysis experiments.
One-Pot SPROX Analysis.
The (+) and (−) ligand samples were subjected to a one-pot SPROX analysis similar to that previously described.23 Aliquots of the (+) and (−) ligand samples were distributed into a series of 12 -containing buffers (PBS, pH 7.4), either xenon-incubated buffers or control buffers respectively, where the final concentrations of were equally spaced at 0.15 M intervals between 0.5 and 2.15 M . The expected concentration of xenon in the xenon incubated samples is ~5 mM based on GC-MS headspace analysis experiments, and the total amount of protein in each sample was 20 μg. The samples in the -containing buffers were incubated for 1 h at 4°C. Methionine oxidation was initiated by adding 4 μL of 30% (v/v) H2O2, and the oxidation was allowed to proceed for 3 min. The final concentration of H2O2 in each reaction was 0.98 M. Methionine oxidation was quenched by adding 250 μL of 1 M TCEP. Equal aliquots of the (+) ligand sample solutions were combined, as were equal aliquots of the (−) ligand samples. The above procedure was performed on 5 separate yeast lysates. This ultimately generated 5 (+) and (−) ligand sample pairs for a total of 10 samples.
The resulting 10 samples were subjected to the same iFASP protocol described previously.66 Each sample was transferred into a 10 kDa MWCO centrifugal filter unit. Buffer exchange was performed by adding 8 M urea in 0.1 M Tris-HCl (pH 8.5) followed by TCEP reduction, MMTS alkylation, digestion with trypsin, and TMT10-plex labeling according to manufacturer’s protocol. Labeled peptides were centrifuged through the filters after addition of 0.5 M NaCl. Equal volumes from each TMT10-plex labeled sample were combined into one final sample. This sample was enriched for methionine-containing peptides with the Pi3 Methionine reagent kit according to manufacturer’s protocol. After enrichment, the sample was transferred to a C18 Macrospin column for desalting prior to LC-MS/MS analysis.
STEPP-LiP Analysis.
The (+) and (−) ligand samples were also subjected to a limited proteolysis analysis similar to that previously described.23 Samples containing 100 μg of protein were reacted with 1 μg of proteinase for 5 minutes at . Proteinase digestion was quenched upon the addition of 5 mM PMSF. Samples were concentrated and desalted using C18 columns (Nest Group) and dried using a rotary evaporator. Dried samples were reconstituted in 100 μL of UA buffer (0.1 M Tris, ~8 M Urea, pH 8.5) and allowed to incubate for 30 minutes. Samples were then reacted with 1.5 mM TCEP for 1 hr at 30°C followed by 2.5 mM MMTS for 10 min at . The proteinase K-digested samples were then reacted with TMT10-plex isobaric mass tag labeling reagents upon addition of 0.5 units of each tag and incubation for 1.5 hr at . Labeling reactions were quenched with 5% hydroxylamine and all labeled samples were pooled into one tube. This final sample was dried, dissolved in 2% TFA, and desalted using C18 columns according to manufacturer’s protocol. Desalted samples were dried, reconstituted in 0.1 M TEAB (pH 8.5) and digested overnight at 37°C with trypsin at a ratio between 1:20 and 1:100 (w/w). NHS-activated agarose resin and 50 μL 0.5 M NaCl were added to the trypsin-digested samples, such that the resin to total peptide ratio was approximately 150:1 (w/w). The peptide mixture was allowed to react with the resin for 1.5 hr at . Samples were then directly acidified with 2% TFA, and added to C18 desalting columns. The desalted peptides were analyzed using LC-MS/MS analysis.
Quantitative LC-MS/MS Analysis.
The LC-MS/MS analyses were performed on a Thermo Easy nanoLC 1200 coupled to a Thermo Orbitrap Exploris 480 mass spectrometer system. The dried peptide material generated from the one-pot SPROX and STEPP-LiP experiments were reconstituted in 1% TFA, 2% acetonitrile in H2O, respectively. Aliquots of 1–2 μL (0.5–1 μg peptide) were injected in triplicate into the UPLC system. The peptides were first trapped on a Thermo Acclaim PepMap 100 75 μm x 2 cm, nanoViper 2Pk C18, 3 μm, 100 A trapping column. The analytical separation was performed using an PepMap RSLC C18 2 μm, 100 A, 75 μm x 25 cm column (Thermo); the column temperature was set to 45°C.
Peptide elution was performed using a 95 min linear gradient of 4–40 %B (80:20 acetonitrile:water, 0.1% formic acid) at a flow rate of 400 nL/min. The MS data was collected using a top 20 data-dependent acquisition (DDA) method which included MS1 at 120k and MS2 at 45k resolution. The MS1 normalized AGC target was set to 300%. For MS2, the normalized AGC target was set to 300% with a max injection time of 105 ms. The collision energy was set to 36%, and the scan range was 375–1500 m/z. The isolation window was 1.2 and the dynamic exclusion duration was 45 s. The raw MS data generated in this work has been uploaded to the PRIDE database (accession number PXD029353) and is available for confidential review using the account, reviewer_pxd029353@ebi.ac.uk, and the password, ZPcENeX6.
One-Pot SPROX Proteomic Data Analysis.
Proteome Discoverer 2.3 (Thermo) was used to search the raw LC-MS/MS files against the yeast proteins in the 2017–10-25 release of the UniProt Knowledgebase. The raw LC-MS/MS data generated in the SPROX experiments was searched using fixed MMTS modification on cysteine; TMT10-plex labeling of lysine side chains and peptide N-termini; variable oxidation of methionine; variable deamidation of asparagine and glutamine; and variable acetylation of the protein N-terminus. Trypsin (full) was set as the enzyme, and up to two missed cleavages were allowed. For peptide and protein quantification, reporter abundance was set as intensity, and normalization mode and scaling mode were each set as none. All other settings were left as the default values. Only methionine-containing peptides with protein/peptide FDR confidence labelled as “high” or “medium” (i.e., FDR < 1% or < 5%) were used for subsequent analyses. Peptides with any of the ten TMT-tag signal intensities equaling zero were removed.
For each condition (TMT-tag), a normalization factor was calculated from the average of all the intensities for that tag. The signal intensities used in the experiment were the reporter ion intensities from the wild-type (nonoxidized) methionine-containing peptides generated in Proteome Discoverer. For each identified wild-type methionine-containing peptide in the SPROX experiment a ratio of the observed reporter ion intensities in the (+) ligand sample to the (−) ligand sample was generated for each biological replicate. The resulting ratio was divided by the normalization factor for each of the 5 biological replicates. These final normalized protein stability ratios (fold changes) were then log-2-base transformed, averaged, and subjected to a Student’s two-tailed t-test comparing to a mean of zero. Hit peptides were identified based on three criteria: (i) the peptide must have a significantly altered log2(fold change) value (≥2σ deviation from mean log2(fold change) of all replicates), (ii) the log2(fold change) value must be significantly different from zero, as determined by the Student’s two-tailed t-test (p-value ≤ 0.05), and (iii) the observation of similar hit behaviors between wild-type and modified versions of the same peptide.
STEPP-LiP Proteomic Data Analysis.
Proteome Discoverer 2.3 (Thermo) was used to search the raw LC-MS/MS files against the yeast proteins in the 2017–10-25 release of the UniProt Knowledgebase. The raw LC-MS/MS data generated in the LiP experiments was searched using fixed MMTS modification on cysteine; TMT10-plex labeling of lysine side chains and peptide N-termini; variable oxidation of methionine; variable deamidation of asparagine and glutamine; and variable acetylation of the protein N-terminus. Trypsin (semi) was set as the enzyme, and up to three missed cleavages were allowed. For peptide and protein quantification, reporter abundance was set as intensity, and normalization mode and scaling mode were each set as none. All other settings were left as the default values. Only semi-tryptic peptides with protein/peptide FDR confidence labelled as “high” or “medium” (i.e., FDR < 1% or < 5%) were used for subsequent analyses. Peptides with any of the ten TMT-tag signal intensities equaling zero were removed.
For each condition (TMT-tag), a normalization factor was calculated from the average of all the intensities for that tag. The signal intensities used in the experiment were the reporter ion intensities from the semi-tryptic peptides generated in Proteome Discoverer. For each identified semi-tryptic peptide in the SPROX experiment a ratio of the observed reporter ion intensities in the (+) ligand sample to the (−) ligand sample was generated for each biological replicate. The resulting ratio was divided by the normalization factor for each of the 5 biological replicates. These final normalized protein stability ratios (fold changes) were then log-2-base transformed, averaged, and subjected to a Student’s two-tailed t-test comparing to a mean of zero. Hit peptides were identified based on three criteria: (i) the peptide must have a significantly altered log2(fold change) value (≥2σ deviation from mean log2(fold change) of all replicates), (ii) the log2(fold change) value must be significantly different from zero, as determined by the Student’s two-tailed t-test (p-value ≤ 0.05), and (iii) the observation of similar hit behaviors between wild-type and modified versions of the same peptide.
Computational Methods.
For each protein selected, molecular dynamics (MD) simulations were carried out using the native crystal structure both with and without the native ligand to sample the protein structure. Proteins were energy minimized and equilibrated using both NVT and NVP ensembles using the AMBER67 force field. The MD simulations were performed at 300 for 30 ns with an average pressure of 1 atm using a Langevin thermostat. The SHAKE algorithm68 was enabled to constrain all carbon-hydrogen bonds, allowing for longer step-sizes for our MD simulations, which were carried out in water using the TIP3P solvent model. A snapshot was extracted for every 2 picoseconds from trajectories.
Xenon binding was assayed to all extracted MD snapshots using VMD.29 For the case where we included the native ligand bound in the MD simulations, we removed the ligand and performed the MD both with and without ligand to explore how the molecule affected the binding of xenon to the respective pocket. Therefore, in total, there are three cases where the binding of xenon is investigated to its target protein. This computational approach was used on each of the five previously mentioned proteins.
While there are many tools that can be used to dock ligands to proteins69–70, the AutoDock430 toolbox was used because it was validated for the assessment of noble gas computational studies and was used to compute xenon gas binding sites and energetics.58 AutoDock was found to identify Xe- binding sites that are consistent with molecular dynamics simulations of xenon interactions for the NMDA receptors.71 The AutoDock4 software uses the AutoGrid and AutoDock packages. AutoGrid calculates grid maps of pairwise interaction energies for different atom types for a given ligand or protein structure. The grid map energies are calculated using only van der Waals and solvent-related energies.
AutoDock uses search algorithms to calculate the lowest energy terms of the outputted grid parameter files from AutoGrid. AutoGrid’s default center of mass was utilized to select the starting point for grid generation. Additionally, we always kept the grid size large enough to sample the entire protein is sampled for potential docking sites. Force field parameters for Xe were previously calculated and are as follows: Rii = 4.40Å, espii = 0.332 kcal/mol, vol = 12.000 Å3 and solpar = −0.00100.10 Here, Rii is the sum of the van der Waals radii, espii is the van der Waals well depth, vol is the atomic solvation volume, and solpar is the atomic solvation parameter.
The favored xenon binding positions were studied as a function of distance to the ATP binding site for each protein. The total number of times xenon was found to dock was counted for each protein over the distance range of 5 Å – 20 Å from the center of mass of each protein’s respective ligand. In the case where ligand was removed from the protein prior to docking, a cavity was defined by identifying all residues within a 5 Å radius from the center of mass of the site where the ligand was removed from the native structure. The center of mass of each residue was calculated and averaged to define an origin from which distances for xenon docking could be defined. The calculated energies were filtered by a threshold of 4KBT and we plotted the frequency of occurrence of these docked positions vs. distance for all proteins. It is assumed that xenon will not bind effectively if the binding energy is smaller than this threshold. Other energy thresholds and explored xenon binding sites were tested, and it was found that the distance to the ATP binding site is robust.
Supplementary Material
The Supporting Information is available free of charge via to Internet at http://pubs.acs.org.
Quantitation of Xe in Buffers using GC-MS Headspace analysis (Supplementary Figure 1), Additional trials of metmyoglobin-xenon and metmyoglobin-nitrogen denaturation curves (Supplementary Figure 2), STRING network map of one-pot SPROX hits split into clusters of stabilized and destabilized proteins (Supplementary Figure 3), STRING network map of STEPP-LiP hits split into clusters of exposed and protected proteins (Supplementary Figure 4), Complete list of protein stability hits identified in the one-pot SPROX experiment (Supplementary Table 1), Complete list of protein hits with Xe-induced conformational changes identified in the STEPP-LiP experiment (Supplementary Table 2).
Supplementary Dataset 2, search output, log2(fold change) calculations, and hit analysis from LC-MS/MS analyses of the TMT10-plex labeled STEPP-LiP sample.
Supplementary Dataset 1, search output, log2(fold change) calculations, and hit analysis from LC-MS/MS analyses of the TMT10-plex labeled one-pot SPROX sample.
Acknowledgments
This work was supported by DTRA grant, HDTRA1–18-0010, to S.L.C, D.N.B., and M.C.F. N.W. is grateful for fellowship support from the Duke Pharmacological Sciences Training Program (T32 GM007105). The authors also acknowledge Xujun Zheng and Cameron Brown for helpful conversations during the course of this work.
ABBREVIATIONS
- SPROX
Stability of Proteins from Rates of Oxidation
- LiP
Limited Proteolysis
- STEPP
Semi-Tryptic Peptide Enrichment strategy for Proteolysis Procedures
- NGs
noble gases
- FPT
Freeze-pump-thaw
guanidine hydrochloride
- MD
molecular dynamics
Footnotes
ASSOCIATED CONTENT
The authors declare no competing financial interest.
Raw proteomic data have been submitted to PRIDE via ProteomeXchange under the accession code PXD029353 and are available for confidential review using the account, reviewer_pxd029353@ebi.ac.uk, and the password, ZPcENeX6.
REFERENCES
- 1.Winkler DA; Thornton A; Farjot G; Katz I, The Diverse Biological Properties of the Chemically Inert Noble Gases. Pharmacol. Ther 2016, 160, 44–64. [DOI] [PubMed] [Google Scholar]
- 2.Rubin SM; Lee S-Y; Ruiz EJ; Pines A; Wemmer DE, Detection and Characterization of Xenon-Binding Sites in Proteins by 129Xe NMR Spectroscopy. J. Mol. Biol 2002, 322 (2), 425–440. [DOI] [PubMed] [Google Scholar]
- 3.Roose BW; Zemerov SD; Dmochowski IJ, Xenon-Protein Interactions: Characterization by X-Ray Crystallography and Hyper-CEST NMR. Methods Enzymol 2018, 602, 249–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quillin ML; Breyer WA; Griswold IJ; Matthews BW, Size versus Polarizability in Protein-Ligand Interactions: Binding of Noble Gases within Engineered Cavities in Phage T4 Lysozyme. J. Mol. Biol 2000, 302 (4), 955–977. [DOI] [PubMed] [Google Scholar]
- 5.Prangé T; Schiltz M; Pernot L; Colloc’h N; Longhi S; Bourguet W; Fourme R, Exploring Hydrophobic Sites in Proteins with Xenon or Krypton. Proteins 1998, 30 (1), 61–73. [PubMed] [Google Scholar]
- 6.Colloc’h N; Sopkova-de Oliveira Santos J; Retailleau P; Vivarès D; Bonneté F; Langlois d’Estainto B; Gallois B; Brisson A; Risso J-J; Lemaire M; Prangé T; Abraini JH, Protein Crystallography under Xenon and Nitrous Oxide Pressure: Comparison with In Vivo Pharmacology Studies and Implications for the Mechanism of Inhaled Anesthetic Action. Biophys. J 2007, 92 (1), 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colloc’h N; Marassio G; Prange T, Protein-Noble Gas Interactions Investigated by Crystallography on Three Enzymes - Implication on Anesthesia and Neuroprotection Mechanisms. In Current Trends in X-Ray Crystallography; InTech, 2011; pp 285–308. [Google Scholar]
- 8.Jafari R; Almqvist H; Axelsson H; Ignatushchenko M; Lundbäck T; Nordlund P; Molina DM, The Cellular Thermal Shift Assay for Evaluating Drug Target Interactions in Cells. Nat. Protoc 2014, 9 (9), 2100–2122. [DOI] [PubMed] [Google Scholar]
- 9.DeArmond PD; Xu Y; Strickland EC; Daniels KG; Fitzgerald MC, Thermodynamic Analysis of Protein–Ligand Interactions in Complex Biological Mixtures using a Shotgun Proteomics Approach. J. Proteome Res 2011, 10 (11), 4948–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran DT; Adhikari J; Fitzgerald MC, Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)-Based Strategy for Proteome-Wide Thermodynamic Analysis of Protein-Ligand Binding Interactions. Mol. Cellular Prot 2014, 13 (7), 1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.West GM; Tucker CL; Xu T; Park SK; Han X; Yates JR; Fitzgerald MC, Quantitative Proteomics Approach for Identifying Protein–Drug Interactions in Complex Mixtures using Protein Stability Measurements. PNAS 2010, 107 (20), 9078–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adhikari J; Fitzgerald MC, SILAC-Pulse Proteolysis: A Mass Spectrometry-Based Method for Discovery and Cross-Validation in Proteome-Wide Studies of Ligand Binding. J. Am. Soc. Mass Spectrom 2014, 25 (12), 2073–2083. [DOI] [PubMed] [Google Scholar]
- 13.Chang Y; Schlebach JP; VerHeul RA; Park C, Simplified Proteomics Approach to Discover Protein-Ligand Interactions. Protein Sci 2012, 21 (9), 1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu P-F; Kihara D; Park C, Energetics-Based Discovery of Protein-Ligand Interactions on a Proteomic Scale. J. Mol. Biol 2011, 408 (1), 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lomenick B; Hao R; Jonai N; Chin RM; Aghajan M; Warburton S; Wang J; Wu RP; Gomez F; Loo JA; Wohlschlegel JA; Vondriska TM; Pelletier J; Herschman HR; Clardy J; Clarke CF; Huang J, Target Identification using Drug Affinity Responsive Target Stability (DARTS). PNAS 2009, 106 (51), 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schopper S; Kahraman A; Leuenberger P; Feng Y; Piazza I; Müller O; Boersema PJ; Picotti P, Measuring Protein Structural Changes on a Proteome-Wide Scale using Limited Proteolysis-Coupled Mass Spectrometry. Nat. Protoc 2017, 12 (11), 2391–2410. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y; De Franceschi G; Kahraman A; Soste M; Melnik A; Boersema PJ; de Laureto PP; Nikolaev Y; Oliveira AP; Picotti P, Global Analysis of Protein Structural Changes in Complex Proteomes. Nat. Biotechnol 2014, 32 (10), 1036–1044. [DOI] [PubMed] [Google Scholar]
- 18.Tilton RF; Kuntz ID; Petsko GA, Cavities in Proteins: Structure of a Metmyoglobin Xenon Complex Solved to 1.9 .ANG. Biochemistry 1984, 23 (13), 2849–2857. [DOI] [PubMed] [Google Scholar]
- 19.Tilton RF; Singh UC; Weiner SJ; Connolly ML; Kuntz ID; Kollman PA; Max N; Case DA, Computational Studies of the Interaction of Myoglobin and Xenon. J. Mol. Biol 1986, 192 (2), 443–456. [DOI] [PubMed] [Google Scholar]
- 20.Ewing GJ; Maestas S, Thermodynamics of Absorption of Xenon by Myoglobin. J. Phys. Chem 1970, 74 (11), 2341–2344. [DOI] [PubMed] [Google Scholar]
- 21.Gevantman LH, Solubility of Selected Gases in Water. CRC, Handbook of Chemistry and Physics 1992, 82–83.
- 22.Zeng L; Shin W-H; Zhu X; Park SH; Park C; Tao WA; Kihara D, Discovery of Nicotinamide Adenine Dinucleotide Binding Proteins in the Escherichia coli Proteome Using a Combined Energetic- and Structural-Bioinformatics-Based Approach. J. Proteome Res 2017, 16 (2), 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabrera A; Wiebelhaus N; Quan B; Ma R; Meng H; Fitzgerald MC, Comparative Analysis of Mass-Spectrometry-Based Proteomic Methods for Protein Target Discovery Using a One-Pot Approach. J. Am. Soc. Mass Spectrom 2020, 31 (2), 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma R; Meng H; Wiebelhaus N; Fitzgerald MC, Chemo-Selection Strategy for Limited Proteolysis Experiments on the Proteomic Scale. Anal. Chem 2018, 90, 14039–14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashburner M; Ball CA; Blake JA; Botstein D; Butler H; Cherry JM; Davis AP; Dolinski K; Dwight SS; Eppig JT; Harris MA; Hill DP; Issel-Tarver L; Kasarskis A; Lewis S; Matese JC; Richardson JE; Ringwald M; Rubin GM; Sherlock G, Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet 2000, 25 (1), 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mi H; Muruganujan A; Ebert D; Huang X; Thomas PD, PANTHER Version 14: More genomes, a New PANTHER GO-Slim and Improvements in Enrichment Analysis Tools. Nucleic Acids Res 2019, 47 (D1), D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Gene Ontology C, The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res 2021, 49 (D1), D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geer MA; Fitzgerald MC, Characterization of the Saccharomyces cerevisiae ATP-Interactome using the iTRAQ-SPROX Technique. J. Am. Soc. Mass Spectrom 2016, 27 (2), 233–243. [DOI] [PubMed] [Google Scholar]
- 29.Humphrey W; Dalke A; Schulten K, VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- 30.Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ, AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem 2009, 30 (16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen J; Arkhipov A; Braun R; Schulten K, Imaging the Migration Pathways for O2, CO, NO, and Xe Inside Myoglobin. Biophys. J 2006, 91 (5), 1844–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L; Martinis SA; Luthey-Schulten Z, Capture and Quality Control Mechanisms for Adenosine-5’-Triphosphate Binding. J. Am. Chem. Soc 2013, 135 (16), 6047–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Aetiology of Compressed Air Intoxication and Inert Gas Narcosis. Arch. Surg 1967, 94 (6), 907–908. [Google Scholar]
- 34.Smith C; Spiess B, The Two Faces of Eve: Gaseous Anaesthesia and Inert Gas Narcosis. Diving Hyperb. Med 2010, 40, 68–77. [PubMed] [Google Scholar]
- 35.Preckel B; Ebel D; Müllenheim J; Frädorf J; Thämer V; Schlack W, The Direct Myocardial Effects of Xenon in the Dog Heart In Vivo. Anesth. Analg 2002, 94 (3), 545–551. [DOI] [PubMed] [Google Scholar]
- 36.Preckel B; Müllenheim J; Moloschavij A; Thämer V; Schlack W, Xenon Administration During Early Reperfusion Reduces Infarct Size After Regional Ischemia in the Rabbit Heart In Vivo. Anesth. Analg 2001, 91, 1327–32. [DOI] [PubMed] [Google Scholar]
- 37.Schwiebert C; Huhn R; Heinen A; Weber N; Hollmann M; Schlack W; Preckel B, Postconditioning by Xenon and Hypothermia in the Rat Heart In Vivo. Eur. J. Anaesthesiol 2010, 27, 734–9. [DOI] [PubMed] [Google Scholar]
- 38.Weber NC; Toma O; Wolter JI; Obal D; Müllenheim J; Preckel B; Schlack W, The Noble Gas Xenon Induces Pharmacological Preconditioning in the Rat Heart In Vivo via Induction of PKC-epsilon and p38 MAPK. Br. J. Pharmacol 2005, 144 (1), 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koziakova M; Harris K; Edge CJ; Franks NP; White IL; Dickinson R, Noble Gas Neuroprotection: Xenon and Argon Protect Against Hypoxic–Ischaemic Injury in Rat Hippocampus In Vitro via Distinct Mechanisms. Br. J. Anaesth 2019, 123 (5), 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrov E; Menon G; Rohde PR; Battle AR; Martinac B; Solioz M, Xenon-inhibition of the MscL Mechano-Sensitive Channel and the CopB Copper ATPase under Different Conditions Suggests Direct Effects on these Proteins. PLoS One 2018, 13 (6), e0198110–e0198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spaggiari S; Kepp O; Rello-Varona S; Chaba K; Adjemian S; Pype J; Galluzzi L; Lemaire M; Kroemer G, Antiapoptotic Activity of Argon and Xenon. Cell Cycle 2013, 12 (16), 2636–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rizvi M; Jawad N; Li Y; Vizcaychipi MP; Maze M; Ma D, Effect of Noble Gases on Oxygen and Glucose Deprived Injury in Human Tubular Kidney Cells. Exp. Biol. Med 2010, 235 (7), 886–891. [DOI] [PubMed] [Google Scholar]
- 43.Le Nogue D; Lavaur J; Milet A; Ramirez-Gil JF; Katz I; Lemaire M; Farjot G; Hirsch EC; Michel PP, Neuroprotection of Dopamine Neurons by Xenon against Low-Level Excitotoxic Insults is not Reproduced by Other Noble Gases. J. Neural Transm 2020, 127 (1), 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lavaur J; Le Nogue D; Lemaire M; Pype J; Farjot G; Hirsch EC; Michel PP, The Noble Gas Xenon Provides Protection and Trophic Stimulation to Midbrain Dopamine Neurons. J. Neurochem 2017, 142 (1), 14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saravanan P; Exley AR; Valchanov K; Casey ND; Falter F, Impact of Xenon Anaesthesia in Isolated Cardiopulmonary Bypass on Very Early Leucocyte and Platelet Activation and Clearance: A Randomized, Controlled Study. Br. J. Anaesth 2009, 103 (6), 805–810. [DOI] [PubMed] [Google Scholar]
- 46.Fahlenkamp AV; Coburn M; Rossaint R; Stoppe C; Haase H, Comparison of the Effects of Xenon and Sevoflurane Anaesthesia on Leucocyte Function in Surgical Patients: A Randomized Trial. Br. J. Anaesth 2014, 112 (2), 272–280. [DOI] [PubMed] [Google Scholar]
- 47.Bantel C; Maze M; Trapp S, Neuronal Preconditioning by Inhalational Anesthetics: Evidence for the Role of Plasmalemmal Adenosine Triphosphate–Sensitive Potassium Channels. Anesthesiology 2009, 110 (5), 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gruss M; Bushell TJ; Bright DP; Lieb WR; Mathie A; Franks NP, Two-Pore-Domain K+ Channels Are a Novel Target for the Anesthetic Gases Xenon, Nitrous Oxide, and Cyclopropane. Mol. Pharmacol 2004, 65 (2), 443. [DOI] [PubMed] [Google Scholar]
- 49.Li Q; Lian C; Zhou R; Li T; Xiang X; Liu B, Pretreatment With Xenon Protected Immature Rabbit Heart From Ischaemia/Reperfusion Injury by Opening of the mitoKATP Channel. Heart Lung Circ 2013, 22 (4), 276–283. [DOI] [PubMed] [Google Scholar]
- 50.Bantel C; Maze M; Trapp S, Noble Gas Xenon is a Novel Adenosine Triphosphate-Sensitive Potassium Channel Opener. Anesthesiology 2010, 112 (3), 623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lavaur J; Lemaire M; Pype J; Nogue DL; Hirsch EC; Michel PP, Xenon-mediated Neuroprotection in Response to Sustained, Low-level Excitotoxic Stress. Cell Death Discov 2016, 2 (1), 16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryu YK; Mintz CD, Neuroprotective Properties of Xenon in Traumatic Brain Injury. Crit. Care Med 2015, 43 (1), 250–251. [DOI] [PubMed] [Google Scholar]
- 53.Weber NC; Toma O; Damla H; Wolter JI; Schlack W; Preckel B, Upstream Signaling of Protein Kinase C-ε in Xenon-Induced Pharmacological Preconditioning: Implication of Mitochondrial Adenosine Triphosphate Dependent Potassium Channels and Phosphatidylinositol-Dependent Kinase-1. Eur. J. Pharmacol 2006, 539 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- 54.Fahlenkamp AV; Coburn M; Haase H; Kipp M; Ryang Y-M; Rossaint R; Beyer C, Xenon Enhances LPS-Induced IL-1β Expression in Microglia via the Extracellular Signal-Regulated Kinase 1/2 Pathway. J. Mol. Neurosci 2011, 45 (1), 48–59. [DOI] [PubMed] [Google Scholar]
- 55.Weber NC; Toma O; Wolter JI; Wirthle NM; Schlack W; Preckel B, Mechanisms of Xenon- and Isoflurane-Induced Preconditioning - a Potential Link to the Cytoskeleton via the MAPKAPK-2/HSP27 Pathway. Br. J. Pharmacol 2005, 146 (3), 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guay J; Lambert H; Gingras-Breton G; Lavoie JN; Huot J; Landry J, Regulation of Actin Filament Dynamics by p38 Map Kinase-Mediated Phosphorylation of Heat Shock Protein 27. J. Cell Sci 1997, 110 (3), 357. [DOI] [PubMed] [Google Scholar]
- 57.Bennett PB; Hayward AJ, Electrolyte Imbalance as the Mechanism for Inert Gas Narcosis and Anaesthesia. Nature 1967, 213 (5079), 938–939. [DOI] [PubMed] [Google Scholar]
- 58.Winkler DA; Katz I; Farjot G; Warden AC; Thornton AW, Decoding the Rich Biological Properties of Noble Gases: How Well Can We Predict Noble Gas Binding to Diverse Proteins? ChemMedChem 2018, 13 (18), 1931–1938. [DOI] [PubMed] [Google Scholar]
- 59.Winkler DA; Warden AC; Prangé T; Colloc’h N; Thornton AW; Ramirez-Gil JF; Farjot G; Katz I, Massive in Silico Study of Noble Gas Binding to the Structural Proteome. J. Chem. Inf. Model 2019, 59 (11), 4844–4854. [DOI] [PubMed] [Google Scholar]
- 60.Chauhan JS; Mishra NK; Raghava GPS, Identification of ATP Binding Residues of a Protein from its Primary Sequence. BMC Bioinform 2009, 10, 434–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia A; Pochinda S; Elgaard-Jørgensen PN; Khandelia H; Clarke RJ, Evidence for ATP Interaction with Phosphatidylcholine Bilayers. Langmuir 2019, 35 (30), 9944–9953. [DOI] [PubMed] [Google Scholar]
- 62.Ramkumar A; Leng X; Lybarger RZ; Petrache HI, Molecular Dynamics of Adenosine Triphosphate Interacting with Lipid Membranes. Biophys. J 2017, 112 (3, Supplement 1), 305a. [Google Scholar]
- 63.Booker RD; Sum AK, Biophysical Changes Induced by Xenon on Phospholipid Bilayers. Biochim. Biophys. Acta 2013, 1828 (5), 1347–1356. [DOI] [PubMed] [Google Scholar]
- 64.Abeyrathne CD; Halgamuge MN; Farrell PM; Skafidas E, An ab-initio Computational Method to Determine Dielectric Properties of Biological Materials. Sci. Rep 2013, 3 (1), 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu PF; Park C, Selective Stabilization of a Partially Unfolded Protein by a Metabolite. J. Mol. Biol 2012, 422 (3), 403–13. [DOI] [PubMed] [Google Scholar]
- 66.McDowell GS; Gaun A; Steen H, iFASP: Combining Isobaric Mass Tagging with Filter-Aided Sample Preparation. J. Proteome Res 2013, 12 (8), 3809–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Case D; Ben-Shalom I; Brozell S; Cerutti D; Cheatham III T; Cruzeiro V; Darden T; Duke R; Ghoreishi D; Gilson M, AMBER 2018; 2018. University of California, San Francisco: 2018. [Google Scholar]
- 68.Miyamoto S; Kollman PA, Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- 69.Lang PT; Brozell SR; Mukherjee S; Pettersen EF; Meng EC; Thomas V; Rizzo RC; Case DA; James TL; Kuntz ID, DOCK 6: Combining Techniques to Model RNA-Small Molecule Complexes. RNA 2009, 15 (6), 1219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coleman RG; Carchia M; Sterling T; Irwin JJ; Shoichet BK, Ligand Pose and Orientational Sampling in Molecular Docking. PLoS One 2013, 8 (10), e75992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu LT; Xu Y; Tang P, Mechanistic Insights into Xenon Inhibition of NMDA Receptors from MD Simulations. J. Phys. Chem. B 2010, 114 (27), 9010–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supporting Information is available free of charge via to Internet at http://pubs.acs.org.
Quantitation of Xe in Buffers using GC-MS Headspace analysis (Supplementary Figure 1), Additional trials of metmyoglobin-xenon and metmyoglobin-nitrogen denaturation curves (Supplementary Figure 2), STRING network map of one-pot SPROX hits split into clusters of stabilized and destabilized proteins (Supplementary Figure 3), STRING network map of STEPP-LiP hits split into clusters of exposed and protected proteins (Supplementary Figure 4), Complete list of protein stability hits identified in the one-pot SPROX experiment (Supplementary Table 1), Complete list of protein hits with Xe-induced conformational changes identified in the STEPP-LiP experiment (Supplementary Table 2).
Supplementary Dataset 2, search output, log2(fold change) calculations, and hit analysis from LC-MS/MS analyses of the TMT10-plex labeled STEPP-LiP sample.
Supplementary Dataset 1, search output, log2(fold change) calculations, and hit analysis from LC-MS/MS analyses of the TMT10-plex labeled one-pot SPROX sample.