

Abstract
Extracellular matrix in the form of patches and locally injected hydrogels have long been used as therapies in animal models of disease. Here, we report the safety and feasibility of an intravascularly infused extracellular matrix as a biomaterial for the repair of tissue in animal models of acute myocardial infarction, traumatic brain injury and pulmonary arterial hypertension. The biomaterial consists of decellularized, enzymatically digested and fractionated ventricular myocardium, localizes to injured tissues by binding to leaky microvasculature, and is largely degraded in about 3 days. In rats and pigs with induced acute myocardial infarction followed by intracoronary infusion of the biomaterial, we observed substantially reduced left ventricular volumes and improved wall-motion scores, as well as the differential expression of genes associated with tissue repair and inflammation. Delivering pro-healing extracellular matrix by intravascular infusion post-injury may provide translational advantages for the healing of inflamed tissues ‘from the inside out’.
Extracellular matrices (ECM) derived from decellularized tissues have shown promising results as tissue engineering scaffolds as an acellular strategy for regenerative medicine1–5. In particular, decellularized ECM can be processed via enzymatic digestion into inducible hydrogels that can be injected for minimally-invasive delivery to tissues6–9. These degradable hydrogels are pro-survival, immunomodulatory, and promote vascularization, among a host of other pro-regenerative functions2,10–12.
Current decellularized ECM biomaterials are limited to surgically implanted patches or localized injections. We aimed to develop a formulation of ECM that could be delivered via intravascular infusion to target leaky vasculature, allowing for less invasive, more evenly distributed, and potentially earlier delivery since tissue access is not a concern. Acute delivery following injury could reduce cell death, promoting tissue salvage and maintenance. In terms of translation, intravascular infusion could easily be performed at the time of intervention using conventional techniques for difficult to access tissues (i.e. heart, brain, and lungs). This delivery modality is not amenable to current ECM hydrogels because of their physical properties, which would not allow for passage into the gaps of leaky endothelium at the site of injury or disease. In this study, we show development of a ECM formulation for intravascular infusion and proof-of-concept for targeting damaged tissue in models of acute myocardial infarction (MI), traumatic brain injury (TBI), and pulmonary arterial hypertension (PAH). We further demonstrated safety, efficacy, and translational relevance using small and large animal acute MI models.
RESULTS
Fabrication and characterization of infusible ECM
A decellularized myocardial ECM hydrogel was selected as a starting material (Fig. 1a–d) for its demonstrated efficacy in preclinical models7–10,13,14 and translational proof-of-concept in a Phase I clinical trial15. While this material retains the complexity of the myocardial ECM14 and is injectable, it is a translucent suspension (Fig. 1d), which contains large submicron particulate, which would be too large to pass into the gaps of leaky vasculature at sites of acute injury and/or inflammation16,17.
Figure 1:
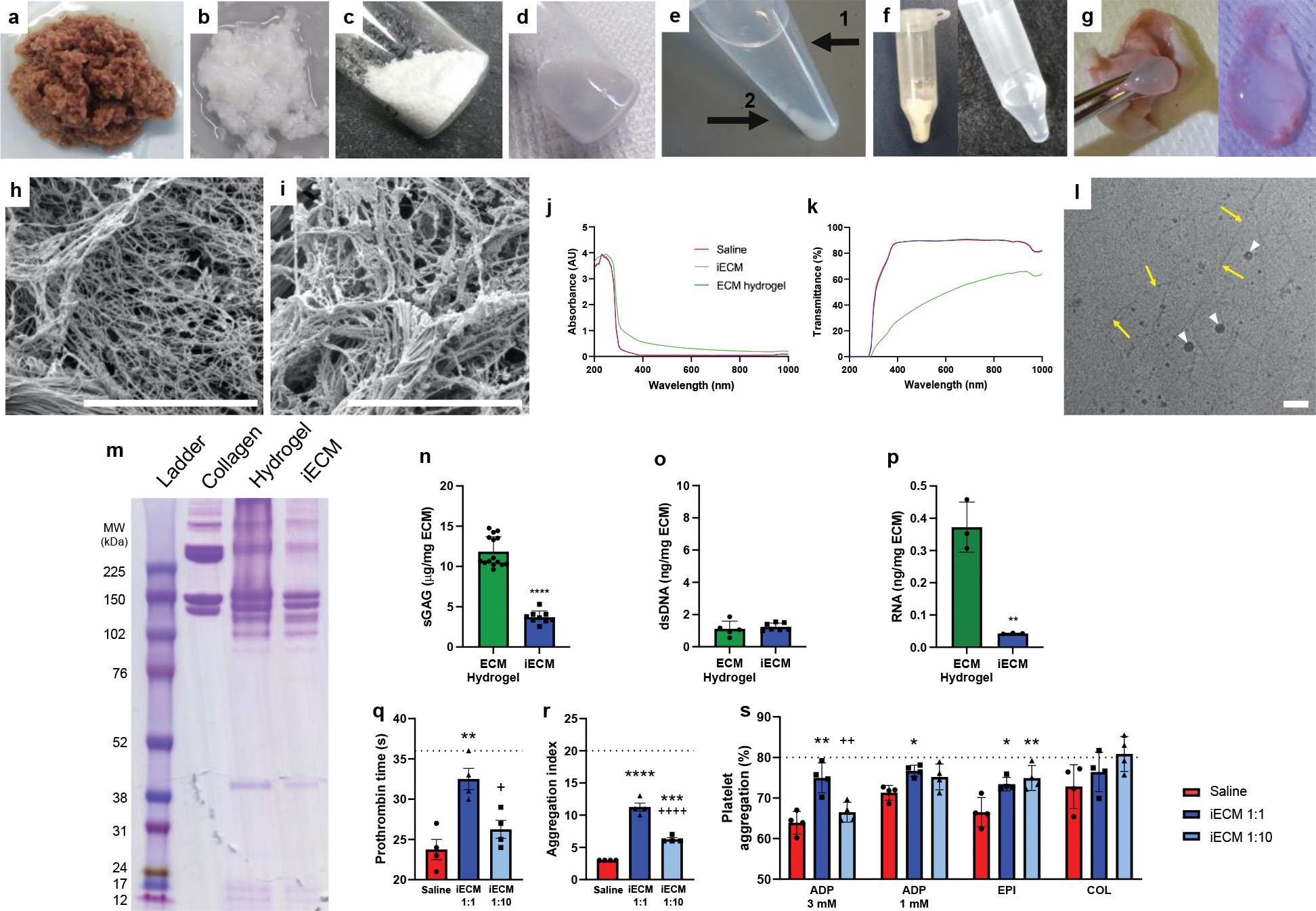
Generation and architecture of infusible extracellular matrix (iECM). a, Isolated left ventricular myocardium is cut into pieces. b, Decellularized ECM after continuous agitation in 1% sodium dodecyl sulfate. c, Lyophilized and milled ECM. d, Liquid digested ECM hydrogel. e, Fractionated ECM hydrogel after centrifugation; (1) supernatant low molecular weight fraction (iECM) and (2) high molecular weight pellet. f, Lyophilized (left) and resuspended (right) iECM. g, Subcutaneous injection and gelation of iECM. Scanning electron microscopy images showing nanofibrous architecture of ECM hydrogel (h) and iECM (i) following in situ gelation. Scale bar is 5 μm. j-p, Characterization of iECM. j-k, Optical measurements of iECM vs liquid ECM hydrogel. iECM showed minimal differences in optical properties from saline whereas the ECM hydrogel had increased absorbance and decreased transmittance. j, Absorbance sweep of iECM, ECM hydrogel, and saline. k, Calculated transmittance of iECM, ECM hydrogel, and saline. l, Cryogenic transmission electron microscopy image of iECM in solution showing nanoscale fibrillar architecture (yellow arrows). White arrowheads indicate ice. Scale bar is 100 nm. m, Polyacrylamide gel electrophoresis of ladder (Full-Range RPN800E, lane 1), collagen (lane 2), ECM hydrogel (lane 3), and iECM (lane 4), showing depletion of high molecular weight (>200 kDa) peptides/proteins in iECM. n, Sulfated glycosaminoglycan (sGAG) content was significantly lower in iECM (n=9, 9 vials sampled across 2 batches) vs ECM hydrogel (n=15, 15 vials sampled across 2 batches). ****p<0.0001. o, Double stranded DNA (dsDNA) content was not significantly different between iECM (n=7, 7 vials sampled across 2 batches) and ECM hydrogel (n=5, 5 vials sampled across 2 batches). p, Total RNA content was significantly lower in iECM (n=3 batches) vs. ECM hydrogel (n=3 batches). **p=0.002 q-s, Hemocompatibility of infusible extracellular matrix (iECM) with human blood and platelet rich plasma (n=4 all groups). q, Prothrombin time. **p=0.004 compared to saline; +p=0.02 compared to iECM 1:1. r, Red blood cell aggregation index. ****p<0.0001 compared to saline; ***p<0.001 compared to saline; ++++p<0.0001 compared to iECM 1:1. s, Platelet aggregation following addition of agonists: adenosine diphosphate (ADP; 3mM: **p=0.002 compared to saline, ++p=0.009 compared to iECM 1:1; 1 mM: *p=0.02 compared to saline), epinephrine (EPI; *p=0.02 compared to saline; **p<0.007 compared to saline), collagen (COL). Standard ranges for each parameter are indicated below dashed lines and data are biological replicates. Data are mean ± SEM. n-p were evaluated with a two tailed unpaired t-test. q-s were evaluated with one way ANOVA with Tukey post-hoc test.
To generate an intravascularly infusible ECM, an optimized centrifugation, dialysis, and sterile filtration protocol was developed to remove the high molecular weight peptides/proteins from the liquid ECM hydrogel (Fig. 1e). The supernatant was isolated from the high molecular weight pellet and dialyzed, yielding the low molecular weight fraction, infusible ECM (iECM), which could then be sterile filtered, lyophilized for long-term storage, and resuspended for injection/infusion (Fig. 1f). While the iECM was capable of gelation in tissue after subcutaneous injection (Fig. 1g), it did not gel when incubated at body temperature in vitro like the ECM hydrogel counterpart.7 Scanning electron imaging of the materials demonstrated that both the ECM hydrogel (Fig. 1h) and iECM (Fig. 1j) have a nanofibrous architecture (Fig. 1h) following subcutaneous injection as previously reported for ECM hydrogels;18 the fiber diameters were not statistically different (ECM hydrogel: 72.4 ± 1.4 nm, iECM: 70.1 ± 1.4 nm, p=0.24) between the two materials.
Across most wavelengths tested, the optical properties of iECM were nearly identical to saline, whereas the liquid ECM hydrogel showed increased absorbance and decreased transmittance (Fig. 1j,k), reinforcing the absence of large particulate in iECM. To determine whether the iECM contained any smaller particulate, we performed cryogenic transmission electron microscopy (cryo-TEM), which showed the dominant nanoscale structure in solution is a fibrillar architecture. These nanofibers were approximately 10 nm in width and 200–500 nm in length (Fig. 1l).
When comparing iECM to the ECM hydrogel, gel electrophoresis showed a depletion of high molecular weight proteins (>200 kDa), which were likely removed during the fractionation process (Fig. 1m). Sulfated glycosaminoglycan content was significantly decreased (Fig. 1n), whereas double stranded DNA content was not different (Fig. 1o). Total RNA content was significantly decreased in the iECM (Fig. 1o), and electropherograms showed that the iECM had little RNA larger than 25 nt as compared to the ECM hydrogel, which had a peak at 400 nt (Supplementary Fig. 1).
Infusible ECM is hemocompatible
As iECM was derived from the ECM hydrogel, it was expected that iECM would be hemocompatible. While it is counterintuitive to think that decellularized ECM could be hemocompatible since exposed ECM initiates clotting in vivo, we previously demonstrated that the ECM hydrogel was hemocompatible9. iECM was tested with human blood using the highest likely scenario of 1:1 iECM to human blood and a physiologically relevant concentration of 1:10 iECM, given the immediate dilution with blood following infusion. Nearly all prothrombin times, red blood cell aggregation indices, as well as platelet aggregation with agonists fall within standard physiological ranges (Fig. 1q–s)19,20. Fibrinogen and platelet concentrations were unaffected by the addition of iECM (Supplementary Fig. 2).
ECM infusions target regions of injury in multiple preclinical models
Three preclinical models were selected to cover diverse conditions in which leaky vasculature is observed, specifically acute MI, TBI, and PAH, which represent both acute and chronic injuries while also covering ischemic, traumatic, and pressure overload injuries. iECM was delivered intravascularly in all models with simulated intracoronary infusion in the rat MI model and intravenous (IV) infusion in the mouse TBI and rat PAH models. In the acute MI model (Fig. 2a–h), iECM was localized to the infarcted region (Fig. 2a,b, Supplementary Fig. 3). Minimal material was observed in the neighboring myocardium (Fig. 2c), and no material was seen in the remote myocardium (Fig. 2d), including the septum and right ventricle. In the TBI model (Fig. 2i–n), material was also localized to the region of trauma (Fig. 2i,j) and no material was observed in remote brain (Fig. 2k). In the PAH model (Fig. 2o–s), material was localized throughout the lungs (Fig. 2o–p), as the PAH model affects the entire organ. All results observed histologically were also confirmed following near-infrared fluorescent scans (Fig. 2g,h, 2n, 2s). Biodistribution analysis suggested excess iECM accumulates in the liver and kidneys (Supplementary Fig. 4). As controls, saline or a small tagged peptide (trilysine) control were infused and showed no retention in target organs (Fig. 2e,f, 2l, 2q, Supplementary Fig. 5), with the majority going to the kidneys (Supplementary Fig. 4).
Figure 2:
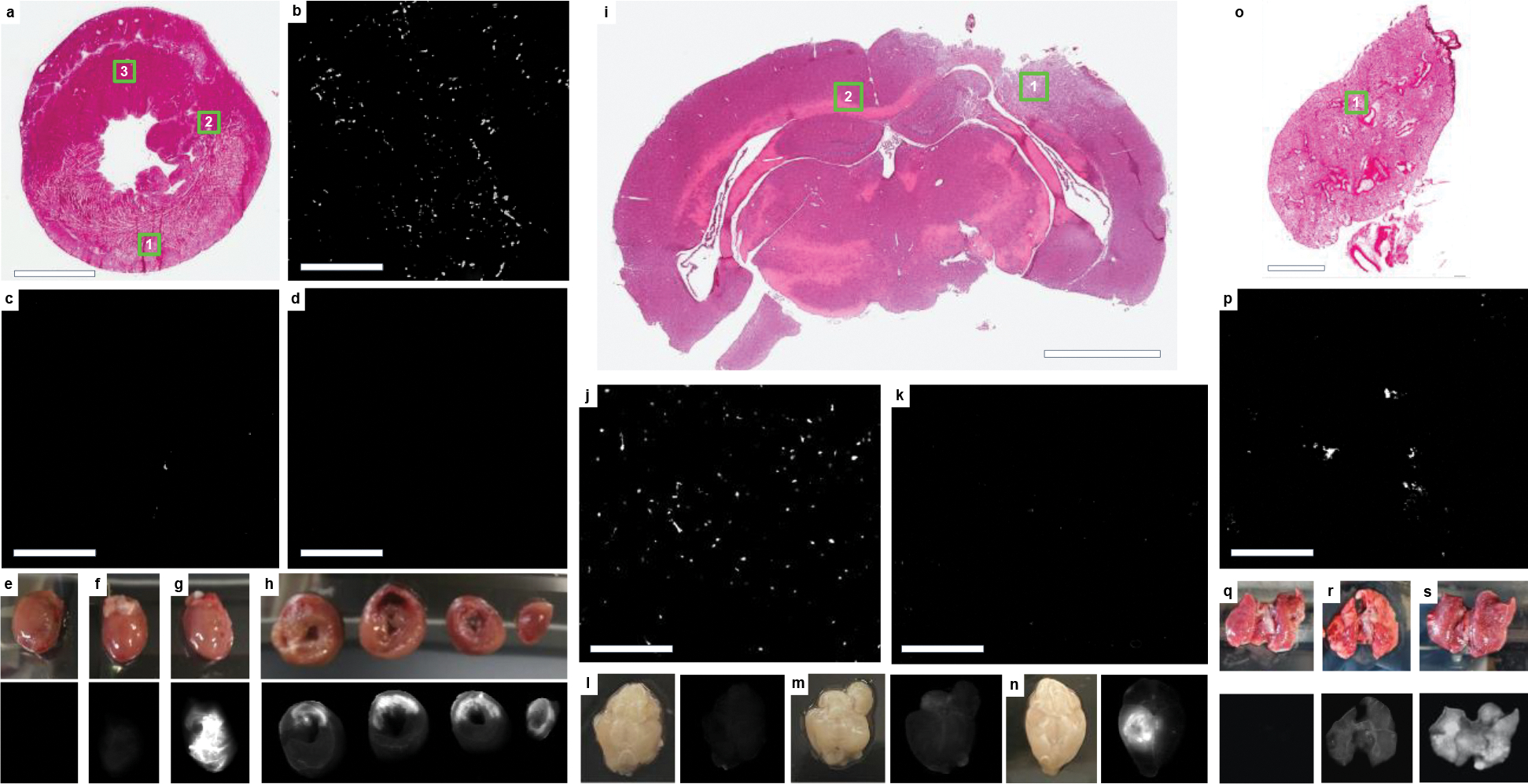
iECM infusions target injured tissues. a, H&E short axis section of an acute MI heart following iECM infusion, scale bar 3 mm. b-d, Fluorescence images for locations shown in insets in a of infarcted myocardium (b, 1), neighboring myocardium (c, 2), and remote myocardium (d, 3), scale bars are 200 μm. e-h, Heart near-infrared scans of saline without dye (e), trilysine conjugated with VivoTag®-S 750 (VT750) (f), and iECM conjugated with VT750 (g). h, Short axis slices of an iECM infused heart from the base to the apex (left to right), infarct regions oriented in the upper portion of the slices. iECM tracking using VT750 24 hours following infusion and MI procedure. i, H&E section of a brain following traumatic brain injury (TBI) and iECM infusion. j-k, Fluorescence images for locations shown in insets in i of injured brain (j, 1) or remote brain (k, 2). Near-infrared scans of brain infused with saline without dye following TBI (l), healthy brain infused with iECM conjugated with VT750 (m), and brain infused with iECM conjugated with VT750 following TBI (n). o, H&E section of a lung following monocrotaline injury, a model of pulmonary arterial hypertension (PAH), and iECM infusion. p, Fluorescence image for location shown in inset in o of injured lung (1). Lung near-infrared scans of lung infused saline without dye following PAH (q), healthy lung infused with iECM conjugated with VT750 (r), and lung infused with iECM conjugated with VT750 following PAH (s).
Across all models, iECM was distributed specifically to injured tissues, suggesting leaky vasculature is required for material retention. To confirm this, iECM infusions were performed in healthy animals without MI (Supplementary Fig. 6), TBI (Fig. 2m), or PAH (Fig. 2r) as well as in rats with a chronic MI, which does not have leaky vasculature (Supplementary Fig. 6). All of these animals showed minimal matrix retention in the respective tissue; however, material was observed in the kidneys and urine, suggesting almost immediate elimination (Supplementary Fig. 6).
Subsequent experiments were performed only in MI models to evaluate the translational potential of iECM for this initial application. iECM showed a dose-dependent retention and distribution in the rat MI model, increasing with iECM concentrations from 6 to 10 mg/ml, but with no appreciable increase from 10 to 12 mg/ml (Supplementary Fig. 7). However, due to manufacturing limitations, specifically filtration, 10 mg/ml was the highest concentration that we could routinely produce. Furthermore, evaluating hearts at multiple timepoints between 2 hours and 7 days post-infusion showed that iECM degraded over approximately 3 days following infusion (Supplementary Fig. 8).
Infusible ECM binds to inflamed endothelium, reduces vascular leakage, and accelerates vascular healing
For safety evaluation, we wanted to confirm that iECM was not blocking the lumen of blood vessels. We confirmed that iECM did not block arterioles (Fig. 3a), but we then observed that the iECM aggregates were colocalized with the microvasculature network, specifically endothelial cells (Fig. 3b). To understand the ratio of endothelial cells that contain labelled iECM, a signal intensity/area image analysis in the infarct area was performed, demonstrating the ratio of percent area endothelial cell signal to percent area iECM signal was 7.3 ± 2.4. Upon further observation, confocal imaging suggested that material was present within the gaps between endothelial cells, resembling a fibrous matrix, and in some areas, coating the inner lining, but not blocking the lumen of the microvasculature (Fig. 3c–g), leading to the hypothesis that iECM infusions were reducing pathological tissue permeability by filling in the gaps of leaky vasculature. In line with this hypothesis, iECM infusions significantly decreased leakage of tagged bovine serum albumin (BSA) into the infarct (Fig. 3h–j), suggesting iECM reduced tissue permeability. We then further assessed how vascular leakage was affected as the material degrades, finding that that the BSA signal was still decreased at 1 and 3 days following iECM infusion, suggesting the iECM accelerates vascular healing and closure. By 7 days, saline animals had decreased to similar levels as the vascular barrier is restored. While the presence of the iECM did not affect neutrophil emigration at 1 day (Fig. 3k), it did decrease macrophage density in the infarct at 3 days post-MI (Fig. 3l).
Figure 3:
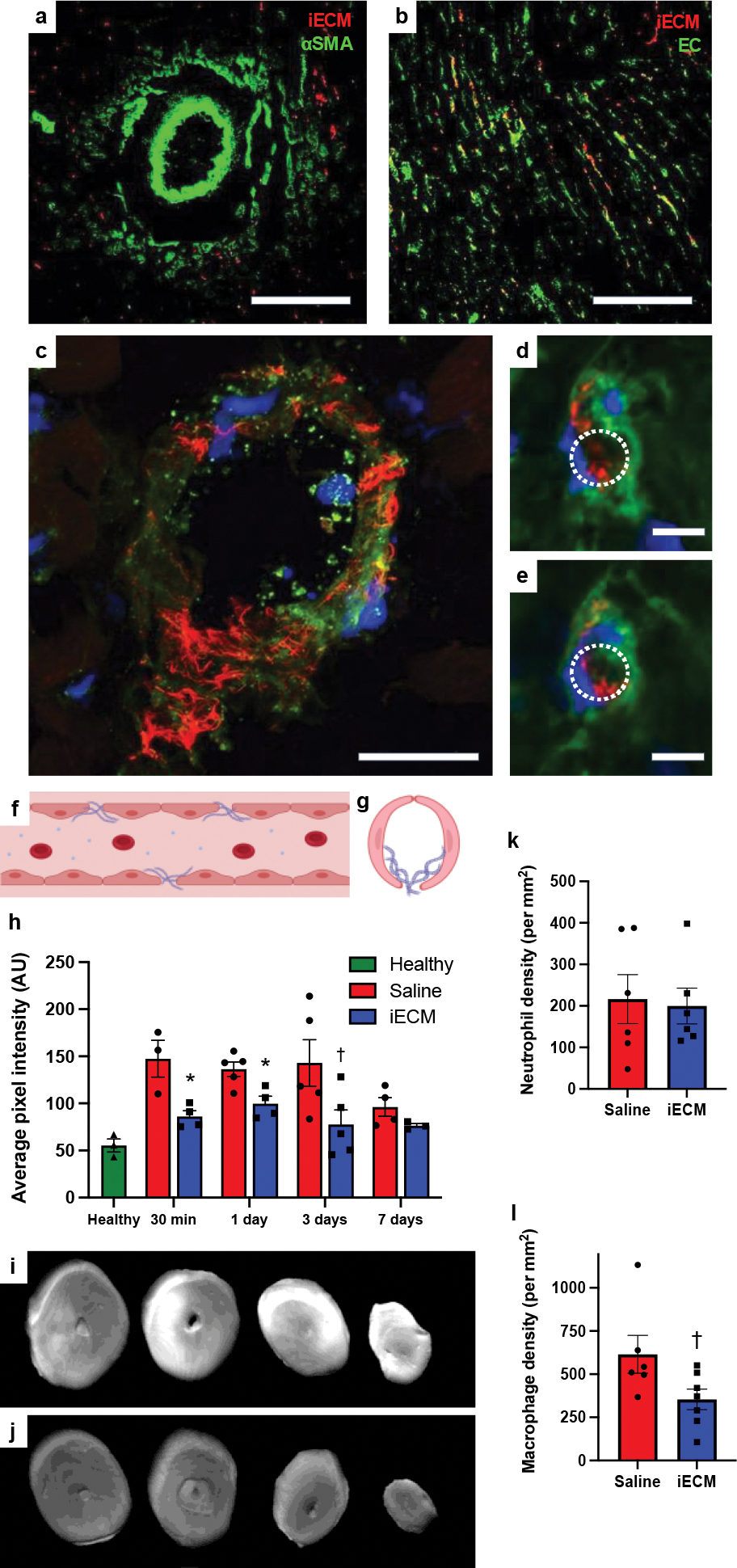
iECM colocalizes with the microvasculature, and reduces vascular permeability and macrophage density after MI. iECM was pre-labeled and visualized in red in all parts of Fig. 3. a, Staining for arterioles using anti-alpha smooth muscle actin (αSMA) in green. (b-e), Staining for endothelial cells using isolectin in green. Scale bar is 200 μm for a-b. c, iECM does not block the lumen of an arteriole but fills in the gaps in the endothelium. Scale bar is 25 μm. d,e, Representative sequential z-stack images of a capillary showing that iECM binds to the endothelium while not blocking it. Lumen traced with dotted white lines. Scale bar is 5 μm. f, Diagram of longitudinal axis view of iECM fibers binding in a leaky vessel. g, Diagram of short axis view of iECM binding in a capillary. h, Quantified cardiac tissue signal intensity following MI, intracoronary infusion, and IV BSA, suggesting iECM infusions decrease tissue permeability and accelerate vascular healing. BSA signal was measured at 30 min (n=3 saline, n=4 iECM, *p=0.02) and 1 (n=5 saline, n=4 iECM, *p=0.01), 3 (n=5 saline, n=5 iECM, †p=0.055), and 7 (n=4 saline, n=3 iECM) days after infusion in the MI model, and in a healthy animals (n=3) 30 min after infusion. i-j, Fluorescent scans of hearts 30 min following MI, intracoronary infusion of saline (i) or iECM (j), and IV infusion of fluorescent bovine serum albumin (IV BSA). (k) Neutrophil (n=6 saline, n=6 iECM) and (l) macrophage density (n=6 saline, n=7 iECM, †p=0.05) in the infarct area of rats one and three post-MI and infusion, respectively. Data are mean ± SEM. All data are biological replicates. Data were evaluated with a two tailed unpaired t-test. f,g created with Biorender.com
To further confirm that iECM can bind to damaged and inflamed endothelium, we used an in vitro adhesion flow assay21 where fluorescently labeled iECM was mixed in red blood cells and plasma before flowing over a coverslip with a human umbilical vein endothelial cell (HUVEC) monolayer. HUVECs were either physically damaged with a scalpel score to expose the underlying cell secreted ECM and/or inflamed with low or high doses of tumor necrosis factor-alpha (TNF-α).21 At both low (100 s−1) and high shear (1000 s−1) rates, iECM bound to areas where the ECM of the HUVEC culture had been exposed by scalpel score or had been highly inflamed via TNF-α treatment (Fig. 4a–d). We next evaluated whether iECM would facilitate platelet adhesion. In this case, iECM was added to whole blood with fluorescently labelled platelets before flowing over a HUVEC monolayer. While the control and iECM groups were not statistically significant via 2-way-ANOVA (p=0.09) given the high variability in magnitude between patient blood donors, at the low shear rate (100 s−1), iECM did result in an increase in platelet adhesion for every individual blood donor (Fig. 4e).
Figure 4:
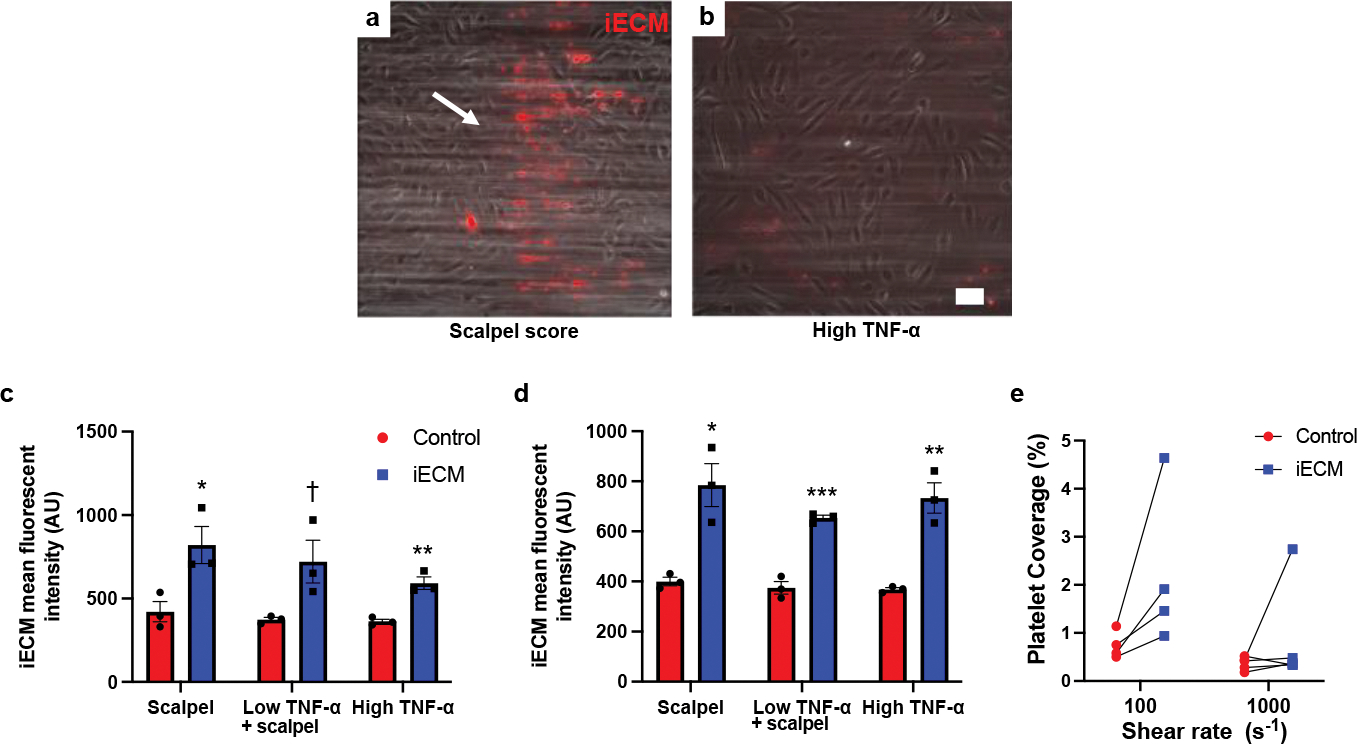
iECM binds to injured endothelial cells and facilitates platelet adhesion in vitro. Representative images of iECM binding to either scalpel score (white arrow region) damaged HUVECs (a) or strongly TNF-α inflamed HUVECs (b) under flow conditions with red blood cells and plasma in an in vitro adhesion flow assay Scale bar is 100 μm. iECM binding to injured (scalpel) and/or inflamed (TNF-α) HUVECs was significantly increased over controls at both low (100s−1) (c) and high (1000s−1) (d) shear rates (n=3 all groups). Low shear (c): scalpel: *p=0.03, low TNF-α + scalpel: †p=0.055, high TNF-α: **p=0.004. High shear (d): scalpel: *p=0.01, low TNF-α + scalpel: ***p<0.001, high TNF-α: **p=0.004. Under flow conditions with whole blood, iECM increased platelet adhesion for all 4 blood donors at low shear rates (e). Data are mean ± SEM. All data are biological replicates. Data were evaluated with a two tailed unpaired t-test.
Infusible ECM mitigates negative left ventricular remodeling in a rat acute MI model
To test the efficacy of iECM in an acute MI model, rats were infused with iECM or saline following occlusion-reperfusion (Fig. 5a–c). At 24 hours following infusions, iECM significantly decreased left ventricular (LV) volumes, both end-systolic (ESV, Fig. 5d) and diastolic volumes (EDV, Fig. 5e), compared to the saline control alongside a trending increase in ejection fraction (EF, Fig. 5f). We confirmed that this was not a result of differences in infarct size between groups (Extended Data Fig. 1a–d) suggesting similar initial injuries. This significant decrease in LV volumes of iECM animals was maintained at 5 weeks post-infusion as well. For comparison, healthy animals had an ESV, EDV, and EF of 61 ± 4 μL, 282 ± 3 μL, and 78.3 ± 1.2 %, respectively.
Figure 5:
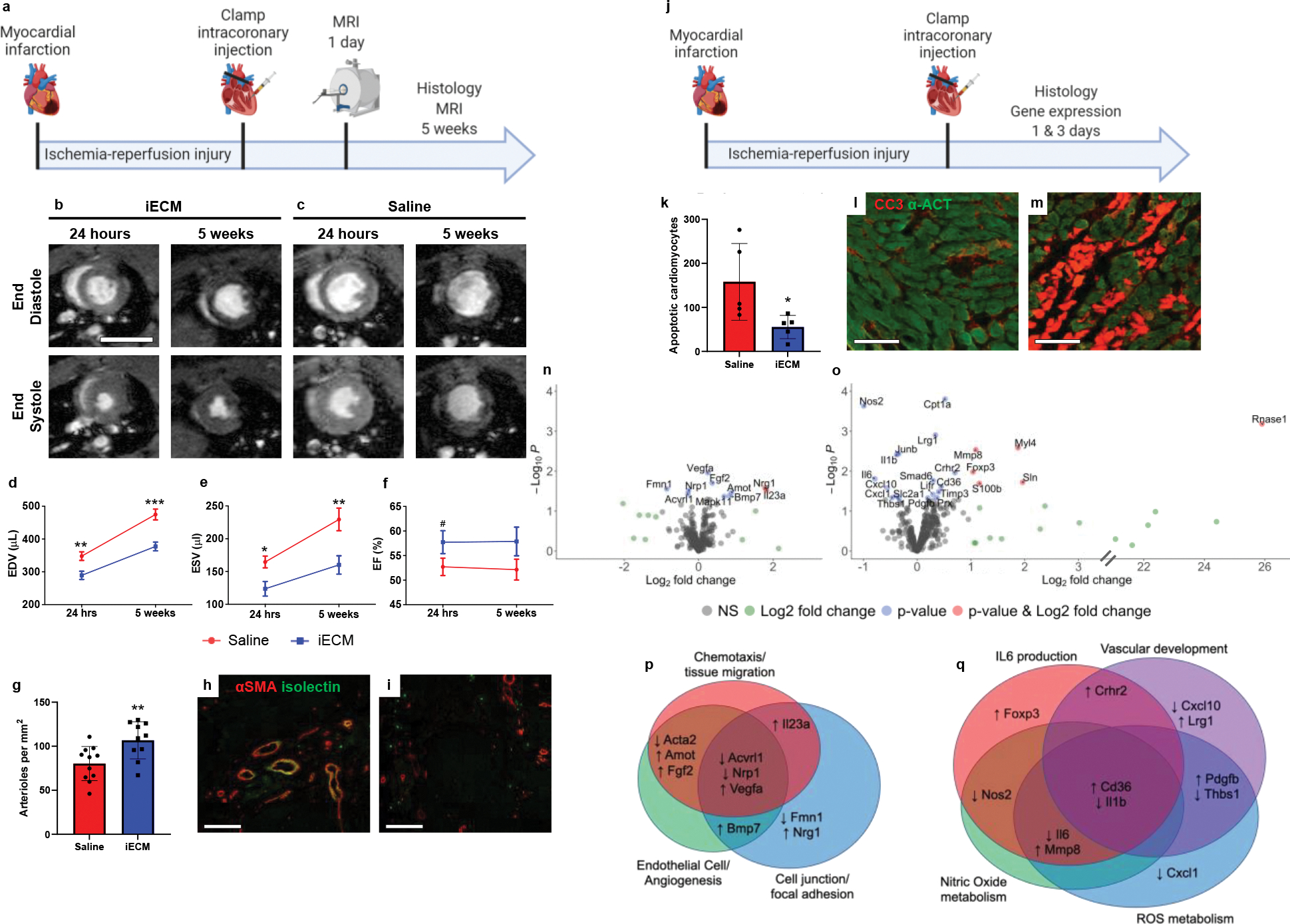
Infusible extracellular matrix (iECM) significantly improved cardiac function post-MI. a, Timeline of survival study. b-c, Representative magnetic resonance images at 24 hours and 5 weeks post-injection of iECM (b) or saline (c). Scale bar 10 mm. d-f, iECM infusions preserve left ventricle volumes at 24 hours and 5 weeks post-MI, end diastolic volume (d, EDV, n=11 saline, n=10 at 24 hrs and n=11 at 5 weeks iECM, **p=0.005 at 24 hrs, ***p<0.001 at 5 weeks), end systolic volume (e, ESV, n=11 saline, n=10 at 24 hrs and n=11 at 5 weeks iECM, *p=0.01 at 24 hrs, **p=0.006 at 5 weeks), ejection fraction (f, EF, n=11 saline, n=10 at 24 hrs and n=11 at 5 weeks iECM, #p=0.1 at 24 hrs). g, Infarct arteriole density increased with iECM infusions at 5 weeks post-infusion (n=11 saline, n=10 iECM, **p=0.008), promoting neovascularization. Representative images of iECM (h) and saline (i) infused hearts, alpha smooth muscle actin (αSMA) in red for arterioles and isolectin in green for endothelial cells. Scale bar 250 μm. j, Timeline of acute mechanisms of repair study. Mechanisms of repair were evaluated through histology (k-m) and gene expression (n-q) analyses. k, Number of cardiomyocytes undergoing apoptosis significantly decreased in the border zone of iECM infused hearts at 3 days post-infusion (n=5 saline, n=5 iECM, *p=0.04). Representative images of iECM (l) and saline (m) infused hearts, cleaved caspase 3 (CC3) for apoptosis in red, and alpha actinin (α-ACT) for cardiomyocytes in green. Scale bar 100 μm. Volcano plots showing differential gene expression from NanoString analysis at 1 day (n) and 3 days (o) after iECM infusion. Genes related to chemotaxis/tissue migration, endothelial cells/angiogenesis, and cell junction/focal adhesion proteins are differentially expressed 1 day (p) following iECM infusions and genes related to Interleukin 6 (IL6), vascular development, nitric oxide metabolism, and reactive oxygen species (ROS) metabolism are differentially expressed at 3 days (q) following infusion. Data are mean ± SEM. All data are biological replicates. Data in d-f, g, and k were evaluated with a two tailed unpaired t-test. a,j created with Biorender.com.
Infusible ECM is pro-survival and promotes tissue repair
As ECM hydrogels have previously been shown to promote tissue repair, including increasing vascularization, being pro-survival, and modulating inflammation10,22, we hypothesized that iECM would likewise have similar pro-reparative effects. At 5 weeks post-infusion, iECM significantly increased infarct vascularization over controls (Fig. 5g–i). Infarct fibrosis and interstitial fibrosis were not significantly different between groups (Extended Data Fig. 1e–g). At 3 days post-infusion (Fig. 5j), iECM significantly decreased the number of cardiomyocytes undergoing apoptosis in the border zone region (Fig. 5k–m). We also observed significant differential expression of 11 and 23 genes in a Nanostring nCounter custom cardiac codeset at 1 day and 3 days post-infusion, respectively (Fig. 5n,o). Gene Ontology (GO) pathway enrichment analysis suggested pathways including angiogenesis, cell-substrate, reactive oxygen species (ROS) and nitric oxide (NO) metabolism, and interleukin 6 (IL6) and other cytokine signaling as illustrated by the Venn diagram of differentially expressed genes in Figure 4p,q. The pathway distribution and full gene list are in Supplementary Tables 1 and 2. Correlation matrices and the top 30 pathways from GO analysis can be viewed in Extended Data Figure 2 and Supplementary Tables 3 and 4, respectively. To further evaluate the ability of iECM to modulate endothelial cell metabolism and survival, rat cardiac endothelial cells were subjected to hydrogen peroxide incubation in vitro with or without iECM, and changes in redox metabolism and cell survival were measured. iECM significantly increased the percent difference in reduction in alamarBlue (Extended Data Fig. 3a) and cell viability (Extended Data Fig. 3b). Given these changes and our previous findings that the original ECM hydrogel acts as an ROS scavenger through free thiols,23 we also assessed the ability of the iECM to reduce incubated hydrogen peroxide concentration over time, and quantified free thiols in the iECM, showing a significant decrease in hydrogen peroxide compared to phosphate buffered saline (Extended Data Fig. 3c) and the presence of free thiols (Extended Data Fig. 3d).
Infusible ECM is amenable to intracoronary infusion with a clinically-relevant catheter
Given the promising results in the small animal acute MI model, we tested if iECM could also be delivered using a clinically-relevant infusion catheter in a large animal model. To ensure that iECM would pass through a catheter, complex viscosity was measured (Fig. 6a), which was higher than saline, but an order of magnitude lower than the liquid ECM hydrogel (Fig. 6a) and facilitated iECM passing through a catheter with an internal diameter of 0.36 mm. To assess clinical feasibility, a balloon infusion catheter was used to induce MI and infuse tagged iECM (Fig. 6b). As seen in the small animal model, iECM was observed in the infarct region in the porcine MI model (Fig 5c,d). Likewise, iECM was distributed throughout the infarcted myocardium and was not observed in the neighboring or remote myocardium (Supplementary Fig. 9). Satellite organs did not show any signs of material, or acute ischemia or inflammation (Supplementary Fig. 10, Supplementary Table 5). iECM was observed to similarly line endothelial cells of the infarcted myocardium in the pig model (Fig. 6e,f). No signal was observed in the infarct region with a peptide control (Supplementary Fig. 9).
Figure 6:
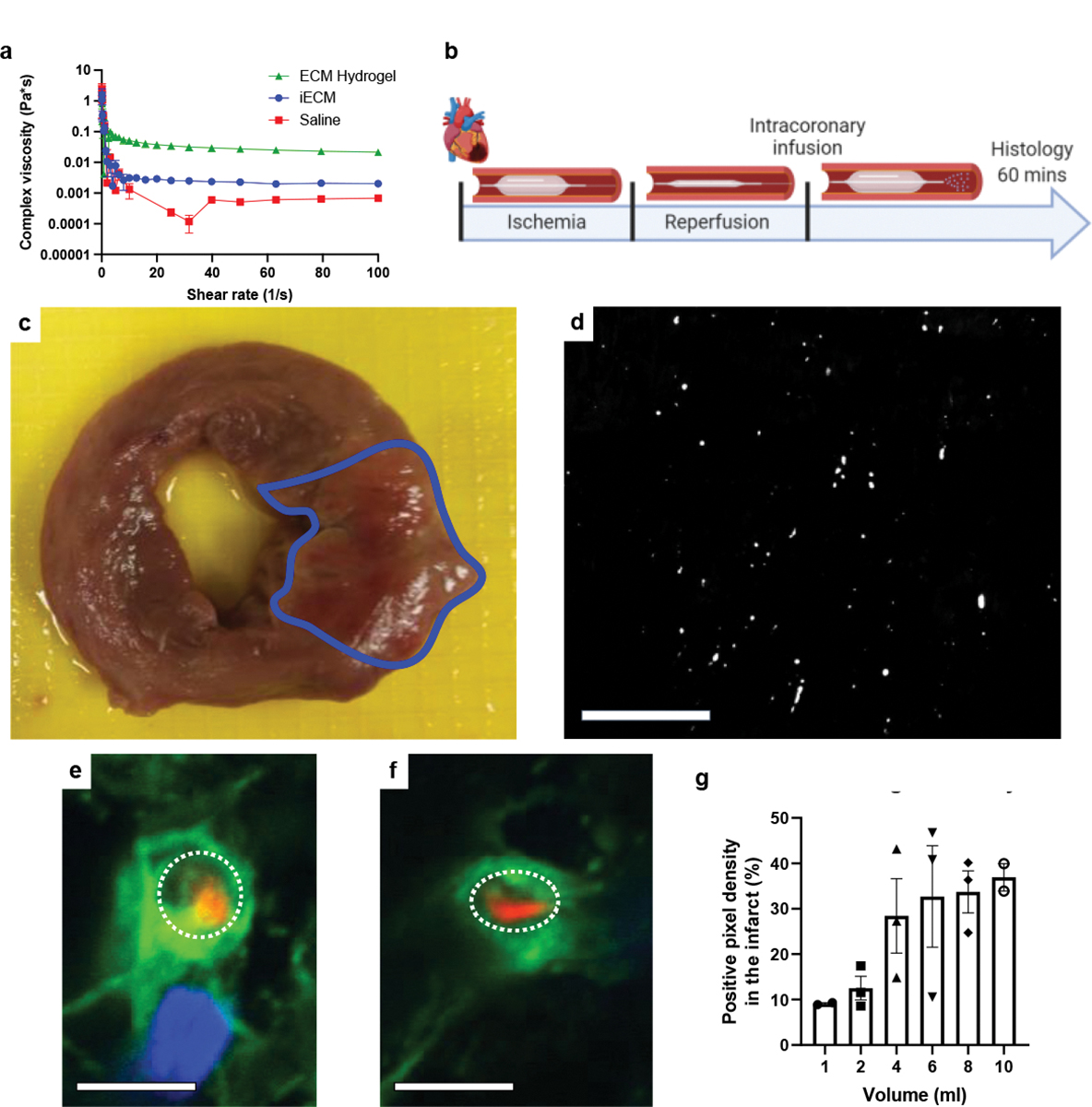
iECM infusions are amenable to intracoronary infusion with a balloon infusion catheter. a, iECM complex viscosity is lower than its full ECM hydrogel counterpart, suggesting potential for catheter delivery. Note, error bars are too small to be visible for most measurements. N=3 all groups. b, Timeline of catheter feasibility study. c, Macroscopic short-axis view of a pig heart post-MI and iECM infusion. Infarct outlined in blue. d, Fluorescent image of iECM distribution and retention in a histological section from the same heart. Scale bar 200 μm. e,f, Representative confocal images of iECM (red) and endothelial cells (green) from iECM infused pigs. iECM lines the endothelial cells of infarcted myocardium, as similarly observed in the rat MI model. Lumen traced with dotted white lines; Scale bars 5 μm. g, iECM infusions showed a plateau effect with increasing infusion volumes, as suggested by quantified signal density in the infarct (n=2 for 1 and 10 mL, n=3 for 2 to 8 mL). Data are mean ± SEM. All data are biological replicates. b created with Biorender.com.
Infusible ECM mitigates negative left ventricular remodeling in a porcine acute MI model
To optimize material retention, volumes ranging from 1 to 10 mL were infused following MI, demonstrating a plateau effect at 4 mL, suggesting that any additional infused iECM would not be retained, as the microvasculature may be saturated (Fig. 6g). Subsequently, pigs underwent MI, iECM or saline infusion, and serial echocardiography (echo) (Fig. 7, Extended Data Fig. 4). Immediately post-MI, EDV, ESV, EF, and infarct angle, a measure of infarct area (Extended Data Fig. 4h), were not significantly different between groups, suggesting a similar injury between groups. EDV, ESV, and EF were measured over time (Fig. 7b,d,f) showing iECM significantly reduced the expansion in EDV from immediately post-MI to 8 weeks later compared to saline (Fig. 7c,h) with a similar trending improvement in the change in ESV (Fig. 7e) and significant improvement in the change in LV systolic dimension from post-MI to 8 weeks post-MI (Extended Data Fig. 4e). iECM led to an approximate 15 and 10 mL decrease in the change in ED and ES volumes (Fig. 7c,e), respectively, compared to saline. There was also a significant improvement in the change in fractional shortening (Extended Data Fig. 4g), although the change in EF was not significantly different (Fig. 7g).
Figure 7:
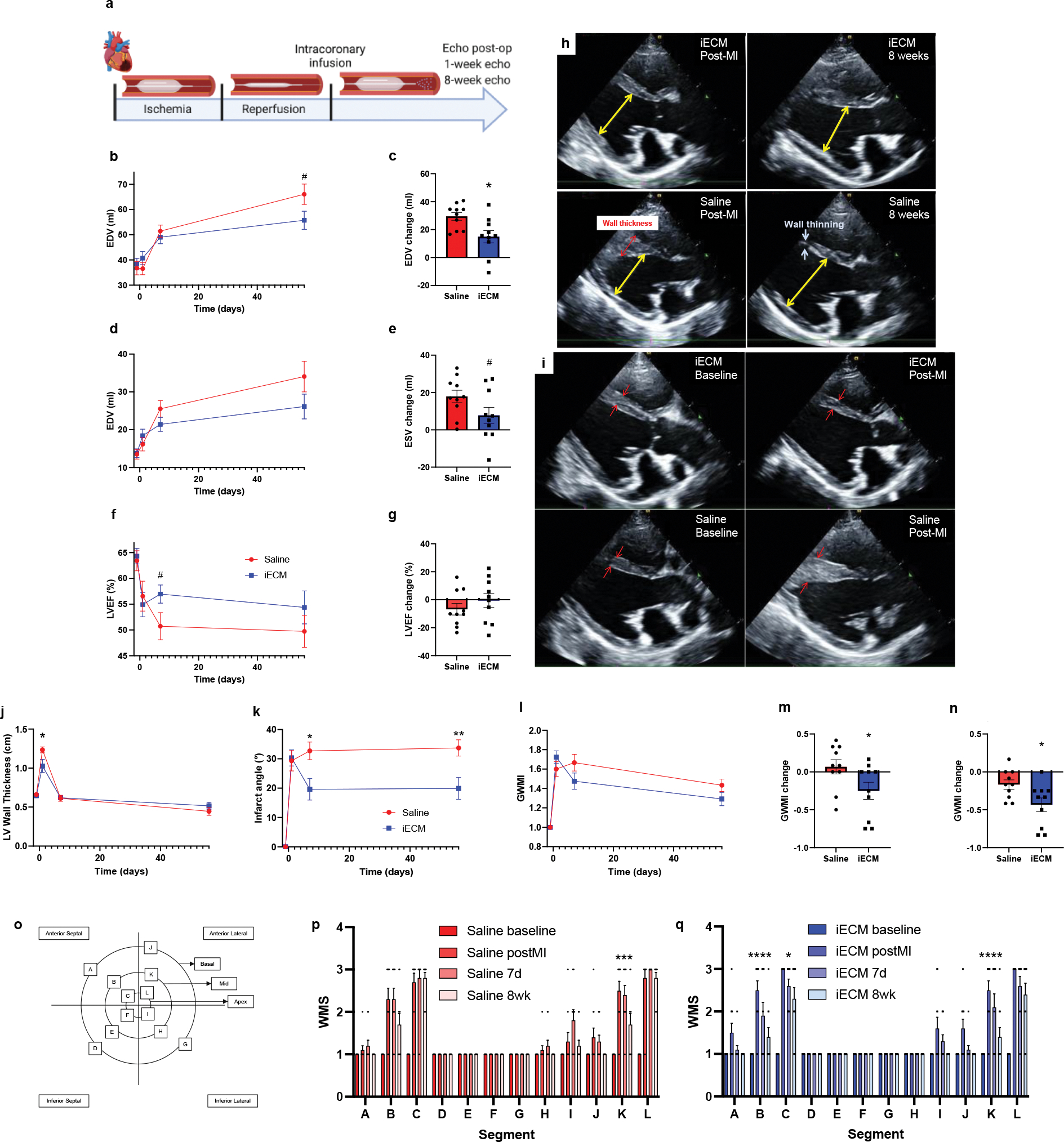
iECM infusions mitigate negative left ventricular remodeling in pig acute MI model. a, Timeline of survival study following induced MI and iECM or saline infusion. End diastolic volume (EDV), end systolic volume (ESV), ejection fraction (EF), left ventricular (LV) wall thickness, infarct angle, global wall motion index (GWMI), and wall motion scores (WMS) were measured before MI (baseline), post-MI, 7 days, and 8 weeks post-MI (n=10 all groups). B-g, Changes in EDV, ESV, and EF over time (b,d,f) and at 8 weeks post post-MI relative to immediately post-MI (c,e,g), suggest iECM infusions mitigate negative left ventricular remodeling (b: #p=0.07, c: *p=0.01, e: #p=0.08 compared to saline). Representative echocardiography images at end diastole show how iECM infusions mitigate negative LV remodeling from post-MI to 8 weeks post-MI (h) and mitigate acute increases in wall thickness post-MI (i). Red arrows indicate wall thickness, yellow arrows are used for LV end diastolic measurement. Changes in MI wall thickness (j, *p=0.04), infarct angle (k, *p=0.01, **p=0.008), GWMI (l-n; m: *p=0.045, n: *p=0.03 ), and WMS segments (o-q) over time, showing how iECM infusions preserve LV wall thickness and motion and mitigate infarct expansion. o, Wall motion score diagram used for scoring wall motion. p, Saline wall motion scores (Segment K: ***p<0.001 post-MI vs. 8 weeks). q, iECM wall motion scores (Segments B and K: ****p<0.0001, Segment C: *p=0.01 post-MI vs. 8 weeks). Data are mean ± SEM. All data are biological replicates. Data in b-n were evaluated with a two tailed unpaired t-test. Data in p and q were evaluated with one way ANOVA with Tukey post-hoc test. a created with Biorender.com.
Shortly post-MI and iECM infusion, LV wall thickness was significantly decreased (Fig. 7i,j), suggesting a potential reduction in edema, while infarct angle was significantly reduced at 7 days and 8 weeks post-MI (Fig. 7k) compared to saline. Global wall motion index scores also showed similar improvements in regional function (Fig. 7l) with significant differences in the change from post-MI to 7 days (Fig. 7m) and 8 weeks (Fig. 7n). This was attributed to significant improvements in infarcted wall segments B, C, and K and trending improvement in infarct wall segment L over time, whereas corresponding improvements were not observed for saline infused hearts (Fig. 7o–q). Electrocardiogram (ECG) and Holter monitoring analysis showed no differences in arrythmias between groups.
Discussion
In this study, we developed a formulation of decellularized ECM for intravascular infusion as an acellular strategy for regenerative medicine. We demonstrated feasibility of delivering iECM intravascularly to target injured and diseased tissue with leaky vasculature, showing broad applicability in acute MI, TBI, and PAH models. iECM was hemocompatible and we found no evidence of embolization, demonstrating the material can be safely delivered intravascularly. Furthermore, in small and large animal acute MI models, therapeutic efficacy was observed, indicating the potential for clinical translation. The material was only observed in injured tissues with leaky vasculature, demonstrating that iECM retention and therapeutic efficacy is conserved across species and injury models.
Our initial hypothesis was that iECM would pass through the gaps in the endothelial cell layer in the leaky vasculature and into the injured tissue as was previously seen with an infusible alginate material24. Instead, we found that iECM coats and/or fills in the gaps between endothelial cells (Fig. 3), specifically in regions of damaged tissues, and therefore represents a alternative type of targeted delivery for pro-healing biomaterials. Histology showed that the material was observed between endothelial cells, and using an in vitro model, we found that the material binds to injured and inflamed human endothelium (Fig. 4a–d), particularly in areas where underlying ECM had been exposed. However, in vivo we did observe some areas where the iECM coated endothelial cells. This may be a result of binding to exposed ECM and then assembly of the iECM, or binding to cell receptors. In vivo, we found this localization to inflamed endothelium led to a near immediate reduction in vascular permeability in the rat model (Fig. 3h–j). Post-MI, increased vascular permeability leads to tissue edema, which is known to contribute to cardiac cell dysfunction and death25,26. As observed in the pig MI model, wall thickness was significantly reduced post-MI, suggesting a barrier to fluid and/or small proteins. As myocardial edema begins during ischemia and quickly expands following reperfusion27, acute iECM infusions could prevent tissue edema and further tissue damage from reperfusion injury. Tissue edema is also observed in numerous pathologies with leaky vasculature, including TBI28,29 and PAH30,31, suggesting iECM could be a therapeutic approach for such conditions. In addition to the acute reduction in vascular permeability, we also observed a reduction at 1 and 3 days post-infusion (Fig. 3h), times when the iECM has already largely degraded (Supplementary Fig. 8d,f), indicating that not only can the iECM physically block the gaps in leaky vasculature immediately following infusion, but can also help to accelerate vascular healing and closure as it degrades. In vitro, we found that iECM increases platelet adhesion (Fig. 4e) at lower shear rates, which could play a beneficial role in vessel healing in vivo as these shear rates are physiologically mimetic of venous flow. Importantly, we did not observe any clotting or embolization in the vasculature in vivo. Platelet aggregation and adherence to vascular walls and exposed ECM is one of the first steps in a cascade of events that lead to repairing damaged endothelial barriers.32,33 This suggests a mechanism by which the iECM can modulate inflammatory responses in vivo, such as the observed reduction in macrophage density, and may work in concert with platelet activity to increase binding and accelerate vascular repair.32,33 Taken together, this suggests that dosing will be most effective very acutely post-MI (within hours), and therapeutic efficacy will likely plateau once the exposed ECM sites are saturated. Indeed, we saw that there are diminishing returns in terms of iECM retention with increasing dosage volumes in the pig model.
To demonstrate the therapeutic potential of iECM we extensively tested it in both small and large animal MI models, including the use of clinically relevant catheter procedures in the latter. iECM significantly reduced both end-diastolic and end-systolic volumes one day after MI and infusion in rats. This difference was maintained, but did not further improve out to 5 weeks post-infusion (Fig 4d,e), essentially creating a better baseline. This is in line with the observed degradation time of iECM, approximately 3 days, where there is an initial therapeutic effect. Therapeutic efficacy was also observed in the porcine model where iECM mitigated an increase in EDV from post-MI to 8 weeks post-MI, reinforcing that iECM infusions reduce negative LV remodeling. Significant improvements in infarct angle and wall motion were also observed in the porcine model, further suggesting localized therapeutic efficacy and improvements in regional function. While iECM significantly improved the change in fractional shortening in the porcine model, there were no changes in EF. Likewise in the rat model there were only trending improvements in EF. However, EF can be misleading as a measure of cardiac contractility34 and function given that parallel decreases in volumes, which would indicate lessened negative LV remodeling, can lead to a similar EF, a phenomenon has been previously seen in heart failure trials35,36. In patients, ≥10 mL reductions in LV volumes are considered clinically significant, which we observed in the porcine model despite its slightly smaller size compared to patients. Nevertheless, there was increased variability in the porcine model compared to the rat model, which may be explained by the variability in retention that we observed with the 4 mL dose (Fig. 6g) and warrants evaluation of higher doses that had more consistent retention (i.e. 8 and 10 mL; Fig. 6g) in future studies.
In terms of biological activity, ECM hydrogels have been shown to create a pro-remodeling versus a pro-inflammatory environment, leading to increases in vascularization and cell survival7,10,37. While part of this activity has been attributed to matrix bound nanovesicles, which contain microRNAs,38,39 we found little total RNA content after the processing of iECM unlike in the ECM hydrogel, suggesting bioactivity of the iECM is due to the ECM components. We found decreased numbers of apoptotic cardiomyocytes and increased arteriole density with the iECM infused rat hearts. Based on the finding that iECM binds to gaps between endothelial cells as opposed to entering the tissue, the mechanisms leading to improved survival and vascularization are likely different between the iECM and ECM hydrogels. From gene expression analyses, angiogenic, endothelial cells, and cell migration/recruitment pathways were expected and dominated the pathway analysis 1 day following iECM infusions. The focal adhesion and cell-substrate pathways may also suggest how the treated endothelial cells adapt in response to iECM infusions, as endothelial cells would be directly exposed immediately post-infusion. At 3 days post-infusion, ROS and NO metabolism and IL6 signaling emerged. In vitro analyses with rat cardiac endothelial cells further showed the potential of the iECM to impact redox metabolism and cell survival. This may be a result of the ROS scavenging ability of the iECM through free thiol content, which we found was similar to the original ECM hydrogel.23 IL6 downregulation at 3 days post-MI suggests that iECM infusions encourage a pro-regenerative switch to happen earlier, reducing negative LV remodeling40. This may also be impacted by the observed reduction in macrophages at 3 days.
Even though iECM is derived from the liquid form of an ECM hydrogel, it could be more appropriate to compare iECM to peptide therapeutics given its characteristics and delivery modality. This comparison to peptides is evident in the clearance of non-retained iECM, which like other peptide therapeutics41,42 appears to be rapidly excreted by the kidneys (Supplementary Fig. 4). However, iECM has advantages over traditional peptide therapeutics due to increased retention through potential binding in the gaps of leaky vasculature (Fig. 3c). This could shield the material from proteolysis, allowing for increased tissue retention and therapeutic effects over peptides therapeutics42. Another advantage of the iECM is the readily available and cost-effective starting material, porcine hearts. A current lab scale batch yields approximately 500 grams of iECM, which would provide approximately 8 to 9 human doses; larger batches are anticipated with GMP manufacturing. These could be stored frozen and rehydrated with sterile water immediately prior to use. While we have not evaluated long term stability of the iECM, we anticipate it will be greater than one year based on the original ECM hydrogel.
Interestingly, we observed that the iECM is present in solution in a fibrillar nanoscale morphology. Given the reduction in high molecular weight collagens, it is not surprising that the material will not gel with only a temperature stimulus (i.e. incubation at 37°C) as we have previously observed this with ECM hydrogels derived from tissues without high collagen content such as the brain and human umbilical cord.43,44 The ability to gel was only achieved when directly injected into solid tissue, potentially because of the pressurized environment; we have previously observed this phenomenon of inducing gelation in tissue, but not in vitro at 37°C for both naturally derived and synthetic hydrogels.43–45 While original ECM hydrogel was hemocompatible, likely because of the pepsin digestion processing and low concentration,9 the inability of the iECM to gel at 37°C provides an additional safety factor. To date, we found no evidence of clotting, emboli, or toxicity; however, continued evaluation of biocompatibility on the path to translation will be important given the increased risk with intravascular delivery.
ECM infusions are a versatile technology for the regenerative medicine and tissue engineering field. In this study, we chose to focus on the cardiac application of ECM infusions; however, infusible ECM could be generated from any type of decellularized tissue, and ECM infusions could be applied to many diseases or injuries where there is endothelial cell injury or dysfunction. Whereas traditional tissue engineering approaches have focused on scaffolds and/or cells to replace or repair damaged tissue, infusible ECM therapies could shift the focus towards treating the microvasculature and healing tissues from the inside out to improve tissue function.
METHODS
Materials
Unless specified, all materials were purchased from Sigma Aldrich.
Material production
Extracellular matrix (ECM) hydrogel was generated based on previously described protocols7,9,18. In brief, fresh hearts were harvested from adult Yorkshire farm pigs (30–45 kg), and the left ventricular (LV) myocardium was isolated. Major vessels and connective tissue were removed, and the remaining tissue was minced. Tissue was decellularized in 1% sodium dodecyl sulfate (SDS) in phosphate buffer saline (PBS) for 4–5 days, followed by 24 hours of water rinsing to remove detergent. The material was then lyophilized and milled into a fine powder, and subsequently partially enzymatically pepsin digested in 0.1 M hydrochloric acid at 10 mg/ml ECM powder and 1 mg/ml pepsin for at least 48 hours. The material was then neutralized with sodium hydroxide and buffered to match in vivo conditions, yielding liquid ECM hydrogel, capable of thermally induced gelation. ECM hydrogel was diluted to 6 mg/ml using PBS.
Next, the liquid ECM hydrogel was centrifuged at 15,000 RCF at 4°C for 45 minutes to separate the high and low molecular weight fractions. The supernatant (infusible ECM, iECM) was isolated from the high molecular weight pellet. The pellet was rinsed with PBS and centrifuged again to increase yield of iECM. To adjust the concentration and ratio of salts, iECM was dialyzed over 48 hours at 4°C in 0.5xPBS, 0.25xPBS, and then twice in deionized water (each solution was replaced approximately every 12 hours), and lyophilized. The iECM was then resuspended at a high concentration (16 mg/mL), passed through 0.22 μm syringe filters (Millipore) into sterile test tubes, lyophilized, weighed, and stored at −80°C for future use. The iECM was then resuspended to appropriate concentration in sterile water at least 30 minutes before injection while on ice.
Characterization assays
Characterization of iECM and its full ECM hydrogel counterpart was performed as previously described18. Protein size and distribution of iECM and ECM hydrogel were visualized against rat tail collagen I through SDS polyacrylamide gel electrophoresis (PAGE) NuPage Kit (Invitrogen). A 4–12% Bis-Tris gel (Invitrogen) and Full-Range RPN800E ladder (MilliPore Sigma) were used for PAGE. Protein bands were visualized using Imperial Protein Stain. For double stranded DNA (dsDNA) quantification, iECM and ECM hydrogel were digested using Proteinase-K, and DNA was isolated using a NucleoSpin kit (Macherey-Nagel). dsDNA was quantified using a PicoGreen fluorescent reporter (Life Technologies). RNA was isolated from ECM hydrogel and iECM following the RNeasy Mini Kit protocol. An initial Proteinase K (20 mg/mL) digest and a DNAse digest step were performed following manufacturer’s protocols. RNA concentration was determined by a NanoDrop 2000 Spectrophotometer, with the RNA integrity determined via an Agilent 2200 Tapestation. Sulfated glycosaminoglycans (sGAGs) were quantified using a 1,9-dimethylmethylene blue (DMMB) assay18 and compared against chondroitin sulfate standards. All values are reported relative to the dry mass of iECM or ECM hydrogel. Optical properties (absorbance) were measured via an absorbance scan on a Spark® microplate reader (Tecan) with 10 nm steps using 1:50 dilutions of iECM and ECM hydrogel (final concentrations of 0.2 mg/ml and 0.12 mg/ml, respectively) alongside saline as a control. Transmittance was calculated from absorbance using the equation, Transmittance = 10^(2-Absorbance). Complex viscosity of iECM (10 mg/mL, n=3), ECM hydrogel (6 mg/mL, n=3), and saline (n=3) were measured with a TA Instruments ARG2 Rheometer.
Gelation and scanning electron microscopy
To test gelation, 500 μL of iECM was resuspended to 10 mg/ml and subcutaneously injected in two locations on the back of a female Sprague-Dawley rat. 500 μL of ECM hydrogel (6 mg/mL) was also injected for comparison. Subcutaneous gels were harvested 5 minutes following injections, dissected from tissue, fixed in 4% paraformaldehyde and 4% glutaraldehyde, dehydrated in ethanol, transferred to isopropyl alcohol, critical point dried using an AutoSamdri 815A automated critical point drier (Tousimis, Rockville, MD), sputter coated (Leica SCD500, Leica, Vienna) with approximately 7 nm of iridium, and imaged at 10,000X under an FEI Quanta 250 scanning electron microscope (Thermo Fisher, Waltham, MA) at 3kV using the in-lens SE1 detector. Fiber diameter was measured manually from SEM images using ImageJ (NIH). Fibers were measured from SEM images of n=3 samples per material (iECM and ECM hydrogel). The fiber diameter was averaged over 100 measured fibers per material type.
Cryogenic transmission electron microscopy
Cryogenic transmission electron microscopy (cryo-TEM) samples were prepared via vitrification robot (Vitrobot Mark IV, Thermo Fisher Scientific, Inc.). Ultrathin Carbon film on Quantifoil R 2/2 200 mesh copper TEM grids were purchased from Ted Pella, Inc. TEM grids were surface plasma treated for 15 s with negative polarity prior to vitrification procedure using a PELCO easiGlow glow discharge cleaning system. For sample vitrification, 4 μL of iECM solution was pipetted onto the TEM grid placed inside the Vitrobot chamber, which was maintained at 4°C and 95% relative humidity. The grid was blotted immediately with filter paper to remove excess solution, with a blot time of 4 s, a blot force (the dimensionless Vitrobot parameter for controlling the blotting pressure) of 1, a wait time of 1 s, and a drain time of 1 s, and subsequently plunge-frozen in liquid ethane. The vitrified samples were transferred to and stored in liquid nitrogen. Samples were imaged using a Gatan Elsa Cryo-Transfer Holder (maintained at ≤−170 °C) loaded into a JEOL ARM300F GrandARM TEM (300 kV), equipped with cold field emission gun (15 μA). Images were recorded using a 4k × 4k Gatan One-View-IS camera.
Hemocompatibility
The interaction between iECM and human blood samples (N=4) was assessed at different concentrations of iECM to whole human blood or platelet rich plasma. A ratio of 1:1 (10 mg/ml iECM) represents the highest possible ratio between blood and iECM, whereas 1:10 (1 mg/ml) represents a more physiologically relevant dilution based on the volume of the coronary vasculature and relevant infusion rate (1 ml/min). Hemocompatibility was assessed as previously described46–48. Red blood cell aggregation was measured using 20 μl of blood with a photometric rheoscope (Myrenne Aggregometer, Myrenne Gmbh, Roetgen, Germany) within 4 hours of sampling after adjusting hematocrit to 45% with autologous plasma. The aggregometer measures changes in light transmission following red blood cell shearing, resulting in a decrease in optical signal from red blood cell aggregation. The aggregation time is the reciprocal of the slope (calculated between 0.5 and 2 s after shear has stopped). The aggregation index is the relative surface area above the curve calculated over the first 5 s. Platelet aggregation was measured with isolated platelet rich plasma on a lumi-aggregonometer (Chrono-log). Using the same dilutions as above for sample to platelet rich plasma, high concentration coagulation cascade agonists, including adenosine diphosphate (ADP), epinephrine (EPI), and collagen (COL) were added (1:200–1:1000 dilutions), and platelet aggregation was measured via absorbance (600–620 nm).
In vitro adhesion flow assay
Written, informed consent was obtained by all human blood donors prior to blood draw according to a University of Michigan Internal Review Board (IRB-MED)-approved protocol. Umbilical cords (Mott Children’s Hospital, Ann Arbor) were acquired via an IRB-MED-approved human transfer protocol. Human whole blood was drawn via venipuncture into acid citrate dextrose (ACD) as an anticoagulant. For experiments utilizing red blood cells (RBCs) in plasma, plasma was acquired by centrifuging whole blood at 2250g for 15 minutes. The buffy coat was discarded, and RBCs were isolated, and washed twice in phosphate buffered saline (PBS). Plasma and RBCs were reconstituted at 40% hematocrit.
Human umbilical vein endothelial cells (HUVEC) were isolated from umbilical cords using a modified collagenase perfusion technique.49 HUVEC were passaged and then seeded at a confluent density onto round glass coverslips coated with glutaraldehyde-crosslinked gelatin. HUVEC were utilized 7 days after seeding with media refreshed every 3 days.
For experiments mimicking acute vascular injury, HUVEC were ‘damaged’ manually via scalpel scores (perpendicular to blood flow) to expose the underlying cell secreted ECM as previously described.21 For experiments mimicking moderate inflammation, HUVEC were activated for 4 hours with 10 ng/mL TNF-α in cell media. For experiments mimicking severe inflammation, HUVEC were activated for 24 hours with 100 ng/mL TNF-α in cell media to increase vascular permeability.50 For experiments mimicking vascular damage with inflammation, HUVEC were first activated for 4 hours with 10 ng/mL TNF-α and then manually damaged using scalpel score to expose the ECM. Coverslips were then attached to a parallel plate flow chamber with a syringe pump utilized to control the volumetric flow rate of blood over the endothelium. Wall shear rates of 100s−1 (representative of slow-moving blood flow in human veins51) and 1000s−1 (representative of fast-moving blood flow in human arterioles and capillaries51) were chosen to examine the impact on iECM binding to the damaged or inflamed vasculature. Shear rate was varied by altering the volumetric flow rate according to the following equation where represents wall shear rate (s−1), represents volumetric flow rate (mL/s), represents channel height (0.0127 cm), and represents channel width (0.25 cm).
For experiments examining iECM adhesion to damaged or inflamed HUVEC, iECM-AF555 (or PBS for non-iECM controls) at a final concentration of 1 mg/mL was added to a mixture of isolated RBCs and plasma at 40% hematocrit immediately before flowing the mixture over the endothelium.
For experiments examining the impact of iECM on platelet adhesion, platelets in whole blood were stained 1 hour prior to flow using anti-CD41/61 FITC (Biolegend). iECM-AF555 (or PBS for non-IECM controls) was added to whole blood at a final concentration of 1 mg/mL immediately before flowing over a damaged HUVEC monolayer. After 5 minutes of laminar blood flow and rinsing, a digital camera was utilized to take 10 images of the endothelium, iECM-AF555 (TRITC filter), and adherent platelets (FITC filter; platelet adhesion experiments).
Metabolic Activity Assay
The evaluation of iECM protective properties against ROS was evaluated via change in redox metabolic activity of rat cardiac endothelial cells. In a 96 well plate, 10,000 cells per well were cultured in monolayer for 24 hours in Endothelial Growth Medium (EGM) (Cell Applications) with 1% penicillin-streptomycin. Media was then changed to either 250 μM hydrogen peroxide containing growth media or 250 μM hydrogen peroxide containing growth media with 1 mg/mL iECM for 6 hours. Cells were then incubated with a 10% alamarBlue solution for 4 hours. To evaluate changes in redox metabolism, 100 μL per sample was collected and recorded at 550/585 nm in a spectrophotometer, and percent difference in reduction in alamarBlue was calculated compared to cells in EGM only.
ROS Survival Assay
To assess the viability of rat cardiac endothelial cells, cells were cultured in the same conditions as the metabolic activity assay above. After incubation with 250 μM hydrogen peroxide containing growth media or 250 μM hydrogen peroxide containing growth media with 1 mg/mL iECM for 6 hours, cells were then stained with Calcein-AM (ThermoFisher) for 30 minutes and DAPI (Hoechst) for 10 minutes. Images were then acquired via an Axio Observer D1 (Carl Zeiss AG, Oberkochen, Germany). By using the multipoint function of the FIJI (FIJI is Just ImageJ) software, viability was quantified by counting the number of nuclei that colocalized with the Calcein-AM dyed cells. These values were then presented as a percentage compared to the number of nuclei of nontreated (healthy) Calcein-AM dyed cells.
ROS Scavenging Assay
To quantify the ROS scavenging ability of iECM, 150 μL of 166 μM H2O2 peroxide solution diluted in 1X PBS was applied to 100 μL of iECM (10 mg/mL), leading to a final 100 μM H2O2 peroxide concentration at baseline. A solution only control in 1x PBS was also performed. Samples were incubated at 37°C on a shaker plate set to 120 rpm and H2O2 content was determined by a Pierce™ Quantitative Peroxide Assay with a standard curve generated to quantify H2O2 concentration. Total thiol content of the iECM material was measured by ThermoFisher’s Thiol Fluorescence Detection Kit, with a standard curve of N-acetylcysteine generated to quantify total thiol content.
Animal surgeries
All procedures in this study were performed in accordance with the guidelines established by the Committee on Animal Research at the University of California, San Diego and the Association for the Assessment and Accreditation of Laboratory Animal Care. All surgeries were performed using aseptic conditions.
Small animal surgeries
To induce tissue injury, a rat ischemia-reperfusion model of myocardial infarction (MI)8,10, a mouse controlled cortical impact (CCI) model of traumatic brain injury (TBI)52–55, and a rat monocrotaline (MCT) pulmonary arterial hypertension (PAH) model56,57 were used as previously described. Adult female Sprague-Dawley rats (225–250 g), male Sprague-Dawley rats (200–225 g) or male C57 black 6 mice aged 8–10 weeks were anesthetized using isoflurane.
To allow for visualization, iECM or trilysine was conjugated with Alexa Fluor™ 568 N-hydroxysuccinimidyl ester to bind free amines (AF568, Invitrogen), as previously described58. AF568 was resuspended at 10 mg/ml in dimethyl sulfoxide, and AF568 was added to iECM or trilysine at a 1:100 volume dilution. For biodistribution, iECM and trilysine were conjugated with VivoTag®-S 750 Fluorochrome (VT750, PerkinElmer), similarly to AF568, as previously described59. VT750 was resuspended at 10 mg/ml in dimethyl sulfoxide, and VT750 was added to iECM or trilysine, at a 1:100 volume dilution.
To induce acute MI, the left main artery was accessed via a left thoracotomy and was ligated for 35 minutes with a suture. To simulate reperfusion, the suture was released to restore blood flow. To simulate intracoronary (IC) infusion, the aorta was clamped, and iECM, trilysine, or saline was injected into the LV lumen with a 30G needle, forcing the material into the coronary arteries which feed both the left and right side of the heart60,61. Infusions were performed within 10 minutes of reperfusion. To determine the effects of dose on retention, hearts were infused with 200 μl of 6, 10, or 12 mg/ml iECM and were harvested at 30 minutes post-infusion (n=2 rats per dose). To determine degradation, rats underwent MI and IC infusion of 200 μl of 10 mg/ml iECM+AF568, were harvested at 30 minutes, 1, 6, 12, and 24 hours and 2, 3, 4, 5, and 7 days post-infusion (n=2 rats per time point). Harvested hearts were rinsed with saline, embedded in Tissue Tek optimal cutting temperature (OCT) compound, and frozen for cryosectioning.
To determine if MI was necessary for iECM retention, IC infusions of 10 mg/ml iECM conjugated with AF568 were performed in healthy rats (no MI), and hearts, kidneys, lungs, liver, kidneys, and spleens were harvested after 1 hour post-infusion and assessed for material (n=3 rats). To assess if retention is possible in a chronic MI, rats underwent MI procedure four weeks prior to infusion. After four weeks post-MI, IC infusions were administered, consisting of either 10 mg/ml iECM conjugated with AF568 (n=2 rats), trilysine conjugated with AF568 (n=1 rat), or saline (n=1 rat). Hearts were harvested 1 hour post-infusion.
To determine MI biodistribution, rats underwent MI and IC infusion of 200 μl of 10 mg/ml iECM+VT750, trilysine+VT750, or saline without VT750 (n=2 rats per group). At 24 hours post-infusion, rats were perfused with saline to reduce background signal, and heart, lung, liver, brain, kidney, and spleen were harvested, rinsed with PBS, and stored at 4°C. Tissues were imaged within 24 hours using an Odyssey imaging system (Li-COR Biosciences).
To induce TBI, a midline incision was made to expose the skull in male mice, and a 4 mm diameter craniotomy performed over the right hemisphere between bregma and lambda. The CCI was applied to the exposed dura of the cortex with the ImpactOne (Leica Biosystems) fitted with a stainless steel 2 mm diameter probe at a velocity of 3 m/s and a 2 mm depth. The bone flap was replaced, and the scalp was sutured. To induce PAH, a single subcutaneous/intraperitoneal injection of MCT (60 mg/kg) was delivered into male rats. To confirm injury, rats were monitored weekly using echocardiography for changes in Tricuspid annular plane systolic excursion (TAPSE). To determine feasibility in TBI and PAH models, iECM was fluorescently tagged and infused via tail vein injection, and brains or lungs were harvested 1 hour post-infusion. For TBI, iECM (n=3) or saline (n=2) was infused 4 hours following TBI and imaged. Healthy brains (n=2) were also infused via tail vein infusion with iECM and imaged. For PAH, iECM (n=2) or saline (n=2) was infused 5 weeks following MCT injury. Healthy lungs (n=2) were also infused via tail vein infusion and imaged.
To assess vascular permeability, rats underwent MI and IC infusion of 200 μl of saline or iECM. After 30 minutes, or 1, 3, or 7 days (n=3–5 per group), rats underwent tail vein injection of Albumin from Bovine Serum (BSA), Alexa Fluor™ 680 conjugate (ThermoFisher Scientific, A34787). After an additional 30 minutes, hearts were harvested and imaged using an Odyssey Odyssey imaging system (Li-COR Biosciences). BSA injections were also performed in healthy rats (n=3)
To assess efficacy in the acute MI model, rats underwent MI and IC infusion of 200 μl of saline or iECM and then magnetic resonance imaging (MRI) 24 hours and 5 weeks post-infusion (iECM n=10 rats, saline n=11 rats). Animals were arbitrarily assigned with saline and iECM injections being performed each day of surgery. Hearts were harvested within 24 hours of the last imaging time point. Cardiac cine MRI were acquired using an 11.7T Bruker MRI System by Molecular Imaging Inc at the Sanford Consortium for Regenerative Medicine. Rats were anesthetized using isoflurane in oxygen during imaging. Respiratory and electrocardiogram-gated, cine sequences were acquired over contiguous heart axial slices. The following parameters were used: repetition time = 20 ms, echo time = 1.18 ms, flip angle = 30°, field of view = 40 mm2, data matrix size = 200 × 200. Eight or nine 1.5 mm, axial image slices were acquired with a total of 20 cine frames per image slice. ImageJ (NIH) was used to outline the endocardial surface at end diastole and end systole for each slice, defined as the minimum and maximum LV lumen area, respectively. Simpson’s method was used to calculate the end-diastolic volume (EDV) and end systolic volume (ESV). Ejection fraction (EF) was calculated as [(EDV-ESV)/EDV] x 100. Investigators were blinded to the groups during image acquisition and analyses. Healthy rats (n=3) were also imaged.
For gene expression analyses, rats underwent MI and IC infusion of 200 μl of saline or iECM, and hearts were harvested at 1 and 3 days post-infusion (saline 1 day n=5 rats; iECM 1 day, saline 3 day, and iECM 3 day n=6 rats per group). Hearts were sliced into six to seven approximately 1 mm coronal slices using a Rat Heart Slice Matrix (Zivic Instruments). Odd slices were used embedded in OCT for histology analyses, and the even slices were used for gene expression analyses. The LV free wall was isolated from even slices and flash-frozen.
Large animal surgeries
Female Yucatan mini-pigs (35–55 kg) were infarcted and infused using over-the-wire percutaneous transluminal coronary angioplasty (PTCA) dilatation catheters (Boston Scientific), as previously described24,62. iECM and trilysine were conjugated AF568 as described above. Pre-anesthesia, pigs were intramuscularly administered 3–5 mg/kg Telazol (Wyesth) or a combination of ketamine-xylazine (4:1, 5 ml). Animals were intubated and maintained under anesthesia using 1–2.5% isoflurane or ketamine-xylazine-atropine with 1 l/min oxygen throughout the remainder of the procedure. For each animal, a sheath was inserted into the right or left femoral artery, allowing catheter access to the artery and retrograde access to the LV through the aorta. Coronary angiography was performed to locate the left anterior descending (LAD) artery and to confirm patency using the left anterior oblique and/or anterior-posterior views, and the balloon was inflated in the LAD for 90 minutes to simulate MI. Visipaque contrast agent (GE Healthcare) was used to visualize the coronary vasculature using an OEC 9400 c-arm (OEC Medical Systems). The balloon was deflated for 30 minutes to restore blood flow, simulating an angioplasty procedure.
For feasibility testing, 4 or 6 ml of iECM or trilysine with AF568 (n=1 pig per group per volume) was infused using the same PTCA catheter. For dosing analysis, 1 (n=2), 2 (n=3), 4 (n=3), 6 (n=3), 8 (n=3), or 10 (n=2) ml of iECM with AF568 was infused following MI. The pigs from feasibility testing were also used for dosing analysis. For survival procedures, 4 ml of iECM (n=10) or saline (n=10) was infused. The balloon was inflated for 2 minutes, and during that time, up to 2 ml of solution was infused at a rate of approximately 1 ml per minute. The balloon was then deflated for 2 minutes. This cycle of balloon up, infuse 2 ml, and balloon down was repeated 2, 3, 4, or 5 times for a total infusion volume of 4, 6, 8, or 10 ml, respectively. ECG was monitored for signs of arrythmia and collected throughout. LAD patency was confirmed post-MI and post-infusion. For feasibility and dosing analyses, pigs were euthanized 1 hour post-infusion, and heart, lungs, liver, brain, kidney, and spleen were harvested. Hearts were perfused with saline, cut into approximately 1 cm x 1 cm x 2–3 cm rectangular pieces, and embedded in OCT compound for cryosectioning. Two approximately 1 cm x 1 cm x 2–3 cm pieces of each satellite organ (lungs, liver, brain, kidney, spleen) were embedded in OCT and frozen. Samples of each satellite organ were also fixed in 10% formalin for histopathological analysis.
For survival studies, pigs were given banamine (2.2 mg/kg) during recovery. Echocardiography (echo, Siemens Acuson Bonsai ultrasound system), 12-lead electrocardiography (ECG, GE MAC 1200), and Holter monitoring were performed before MI, following infusion, 7±1 days post-op, and 8 weeks ± 2 days post-op (pre-euthanasia), as previously described9. Echo and ECG were performed under anesthesia. Holter monitors were attached under anesthesia, pigs were then recovered from anesthesia, and monitors were removed following at least 24 hours, demonstrating at least 12 hours of continuous recording. Holter monitors were reattached if insufficient data (<12h) was collected. Five pigs died during the MI procedure. A standard two-dimensional echocardiogram (2-D Echo) was performed pre-MI, post-MI & infusion, 7 days post-MI and prior to euthanasia at 8 weeks post-MI using a commercially available instrument operating a broad band 1.5–3.5 mHz transducer with digital images stored on hard drive for subsequent playback and analysis. Attempts were made to record images in the standard long axis, short axis, and apical 4 and 2 and 3 chamber views. Depending upon the thoracic configuration of the animal, one or more views that depicted the entire perimeter of the left ventricle were possible. Images were optimized for porcine anatomy with minimal depth and highest frame rate whenever possible, and an acquisition length of 5 cardiac cycles. Once the optimal view was identified, a similar view containing the same landmarks was obtained on each subsequent examination, and single plane LV volumes and ejection fraction were derived using a modified Simpson rule method of discs. Holter recordings were sent to a blinded consultant that prepared full disclosure reports. ECG and Holter reports were evaluated for arrythmias.
Histological analyses
All histological and immunohistochemical (IHC) assessment was performed by investigators who were blinded to group assignment. Samples embedded in OCT compound were cryosectioned at 10 and/or 20 μm. Formalin fixed tissue samples were paraffin-embedded and sectioned. Hematoxylin and eosin (H&E)-stained sections were used to identify the infarct regions for fluorescent analyses and infarct size. H&E-stained sections of the pig satellite organs were sent to a histopathologist, who was blinded, and assessed for signs of ischemia and inflammation. Masson’s trichrome stain was used to quantify infarct size and interstitial fibrosis. Five evenly spaced slides spanning the infarct were used for trichrome analyses. Infarcts were manually traced in for infarct size. Slides stained with H&E and trichrome were mounted with Permount (Fisher Chemical) and scanned at 20x using an Aperio Scan Scope CS2 slide scanner (Leica Biosystems). Non-nuclear blue staining was measured using the ‘Positive Pixel Count V9’ algorithm in the ImageScope (Aperio) software for interstitial fibrosis analysis. Infarct size, infarct fibrosis and interstitial fibrosis was calculated for each animal, and averaged per group.
IHC was performed using antibodies for the following antigens: α-actinin (αACT, Sigma A7811, 1:700), CD68 (BioRad MCA341GA, 1:100), cleaved caspase 3 ASP175 5A1E (CC3, Cell Signaling 9664, 1:50), alpha smooth muscle actin (αSMA, Agilent M0851, 1:75), and myeloperoxidase (MPO, Sigma Aldrich N5787, 1:100). Unlabeled antibodies were visualized with secondary antibodies Alexa Fluor 488 or 568 (Life Technologies). Griffonia Simplicifolia Lectin I isolectin B4 (Vector Laboratories, 1:75) was pre-labeled with fluorescein. Nuclei were stained with Hoechst 33342 (Life Technologies). Following IHC, slides were mounted with Fluoromount (Sigma) and imaged at 10X or 20X on the Ariol DM6000 B microscope (Leica) or Carl Zeiss Observer D1.
For neutrophil quantification, MPO was used. Using tissue from 1 day post-infusion, 2 slides covering the infarct were stained. Neutrophils were identified by nuclei colocalization with MPO. Cells were automatically counted using a custom script in Matlab (Mathworks). For macrophage quantification, anti-CD68 stained slides (2 slides per heart) from 3 day post-infusion hearts were used. Three regions of interest were acquired from the infarcted region in each slide and quantified for CD68 positive cells using ImageJ. Neutrophils and macrophages per mm2 of infarct area were calculated for each animal, and averaged per group.
For infarct arteriole quantification, arterioles were identified by the following criteria: positive co-staining for isolectin and αSMA for endothelial cells and smooth muscle cells, respectively, a visible lumen, and an average Feret diameter (average of the minimum and maximum Feret diameters) of at least 10 μm. In rats, using tissue from 5 weeks post-infusion, 5 slides evenly spanning the infarct of each heart were stained. If possible, up to 5 images were captured across the infarct of each slide. Infarcts were identified by nuclei density, and arterioles were manually traced in ImageJ (NIH). Arteriole per mm2 of infarct area was calculated for each animal and averaged per group.
For cardiomyocyte apoptosis quantification in the border zone, slides from 3 days post-infusion were stained for αACT and CC3 to identify cardiomyocytes and apoptotic cells, respectively. The border zone was identified by expanding outside the infarct region 1 mm. The infarct was identified through an absence of αACT stain. Apoptotic cardiomyocytes were identified by positive co-staining for αACT and CC3. As CC3 could be observed on most cells in the border zone, a high threshold for CC3 expression was used. CC3 had to cover at least 50% of the cell body to be considered apoptotic. Two slides covering the infarct from each heart were used as a result of using the Slice Matrix. Analysis was manually performed in ImageJ (NIH). The number of apoptotic cardiomyocytes for each animal was measured and averaged per group.
For imaging of vessels and iECM, slides from 2, 12, and 24 hours post-infusion in rats and 1 hour post-infusion in pigs were stained with isolectin or αSMA to identify endothelial cells or smooth muscle cells, respectively. To analyze the colocalization of endothelial cells and iECM, images were taken at 10x magnification on the Carl Zeiss Observer D1. ImageJ (NIH) software was used to determine the percent area of each stain. For confocal microscopy analysis, slides were imaged using a Confocal Microscope LSM 780 (Zeiss).
RNA isolation and NanoString Multiplex Gene Expression Analysis
Hearts from 1 and 3 days post-infusion were used for gene expression analyses (see Small Animal Surgeries). The LV free wall was isolated and flash frozen for subsequent RNA extraction using the RNEasy Mini Kit (Qiagen). RNA concentrations were measured using a NanoDrop spectrophotometer (Thermo Scientific).
For screening of various pathways of interest, RNA samples were analyzed by NanoString nCounter® MAX Analysis System with nCounter® custom cardiac codeset (Rat) allowing for multiplexed assessment of 380 genes (see Supplementary Table 2 for full gene list). Samples were processed according to manufacturer instructions. In brief, RNA sample concentrations were measured on a Qubit 3.0 Fluorometer with a QubitTM RNA HS Assay kit. 70 μL of hybridization buffer was mixed with Immunology Panel Reporter CodeSet solution, and 8 μL of this master mix was mixed in a separate reaction vessel with 50–100 ng of RNA per tissue sample and RNA-free water up to 13 μL total. 2 μL of Capture ProbeSet was added to each vessel, mixed and placed on a thermocycler at 65°C for 16–48 hours before being maintained at 4°C for less than 24 hours. NanoString nCounter Prep Station performed automated fluidic sample processing to purify and immobilize hybridized sample to the cartridge surface. Digital barcode reads were analyzed by NanoString nCounter® Digital Analyzer. Results were analyzed by manufacturer nSolverTM Analysis Software 4.0 and custom R scripts. Outliers were detected and excluded based on NanoString’s methods for outlier detection.
Statistics
Data generated from dsDNA, sGAG, and RNA content assays, vascular permeability, in vitro adhesion flow, MRI, qPCR, echocardiography, and histological analyses were compared with a Student’s two-way t test. Data generated from hemocompatibility and individual segment wall motion scores were compared with a one-way ANOVA with Tukey post-hoc test. All data are presented as mean ± SEM. Significance was considered p < 0.05. Statistical analysis was performed in Prism (GraphPad).
Extended Data
Extended Data Fig. 1 |. Histological measurements post-infusion show no significant differences in infarct size or fibrosis between iECM and saline infused rats.
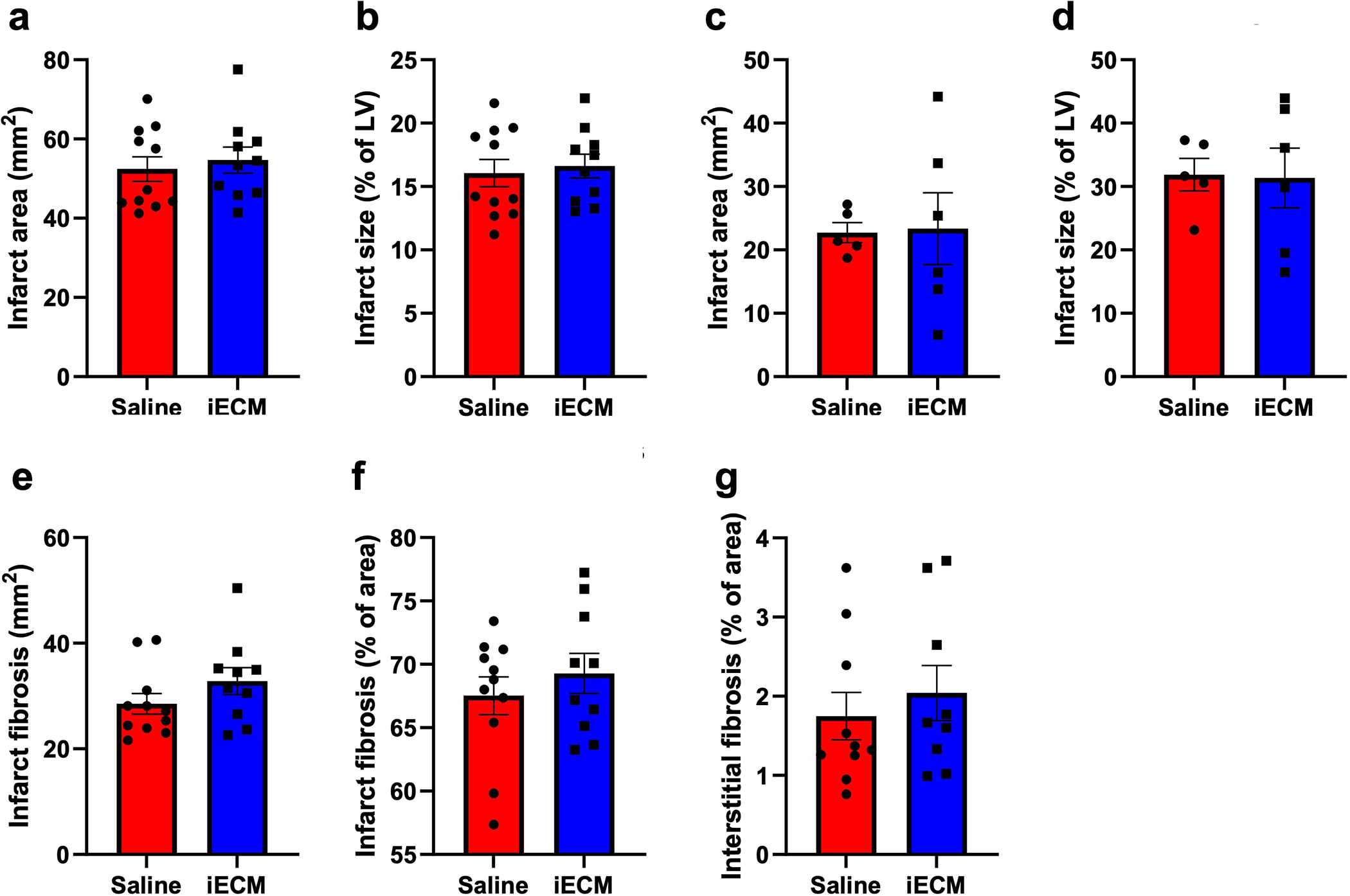
Infarct area at 5 weeks (a,b) and 3 days (c,d) post-infusion, reported as area (a,c) and percentage of the LV (b,d). Infarct fibrosis reported as area (e) and percentage of infarct area (f). g, Interstitial fibrosis of the remote myocardium reported as a percentage of area. N=5 for saline and n=6 for iECM for day 3 measurements, and n=10 for both groups for 5 week measurements. Data are mean ± SEM. Add data are biological replicates.
Extended Data Fig. 2 |. Correlation matrices for the NanoString nCounter data.
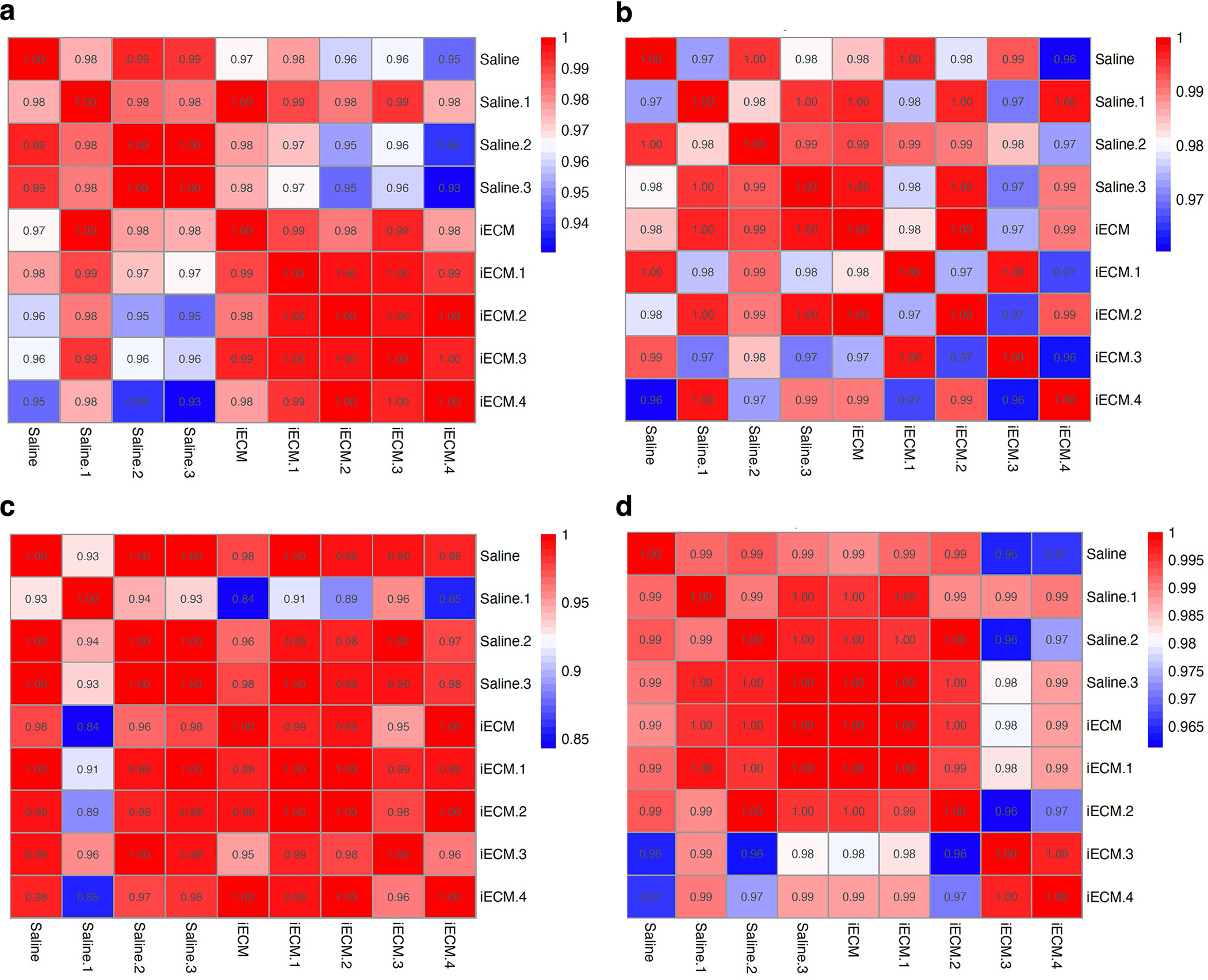
Correlation matrix for day 1 post-infusion significantly differentially expressed genes only (a) and all genes in the Nanostring nCounter custom cardiac codeset (b). Correlation matrix for day 3 post-infusion significantly differentially expressed genes (c) and all genes in the Nanostring nCounter custom cardiac codeset (d). The Nanostring panel used was a 380 gene custom panel designed to probe for differences in gene expression across a wide range of myocardial injury models.
Extended Data Fig. 3 |. Effect of iECM on metabolic activity and viability of rat cardiac endothelial cells, ROS scavenging, and thiol content.
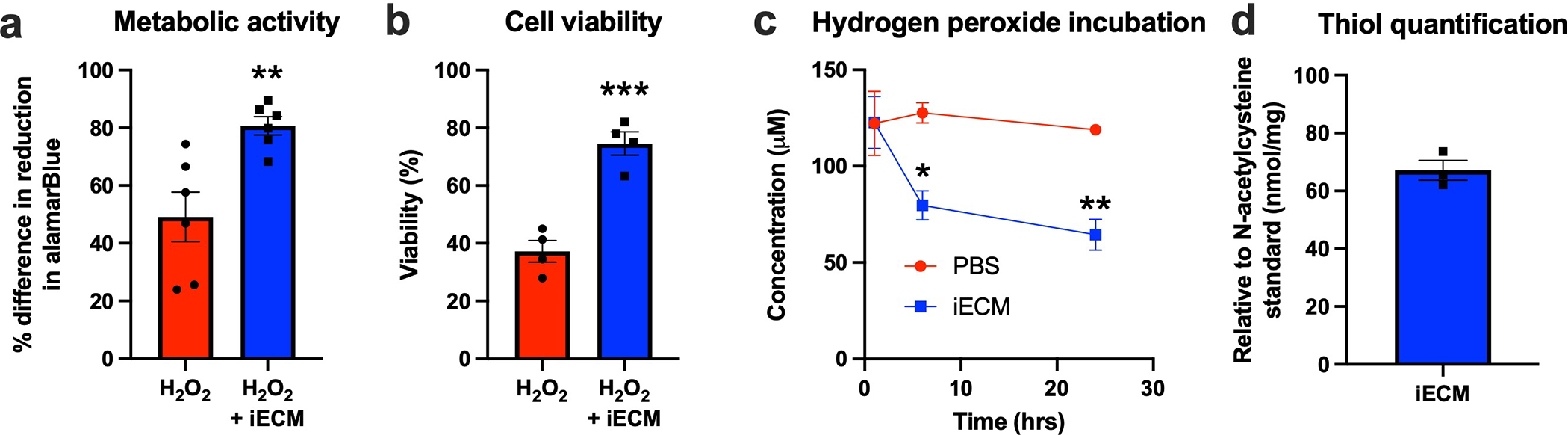
Percent difference in reduction in alamarBlue activity of rat cardiac endothelial cells in response to hydrogen peroxide with and without iECM, relative to controls with no hydrogen peroxide (a, n=6 both groups, **p=0.006). Percent viability as measured by Calcein-AM staining of rat cardiac endothelial cells in response to hydrogen peroxide with and without iECM, relative to controls with no hydrogen peroxide (b, n=4 both groups, ***p<0.001). Concentration of hydrogen peroxide was monitored following incubation with either PBS or iECM, showing a continuing decrease with iECM (c, *p=0.01, **p=0.003, n=3 for both groups at 1 and 24 hrs, n=2 at 6 hrs). Thiol content was determined compared to N-acetylcysteine standard in iECM per mg of material (d, n=3). Data are mean ± SEM. All data are biological replicates and were evaluated with a two tailed unpaired t-test.
Extended Data Fig. 4 |. Additional echocardiography results showing that iECM infusions mitigate negative LV remodeling in a pig acute MI model.
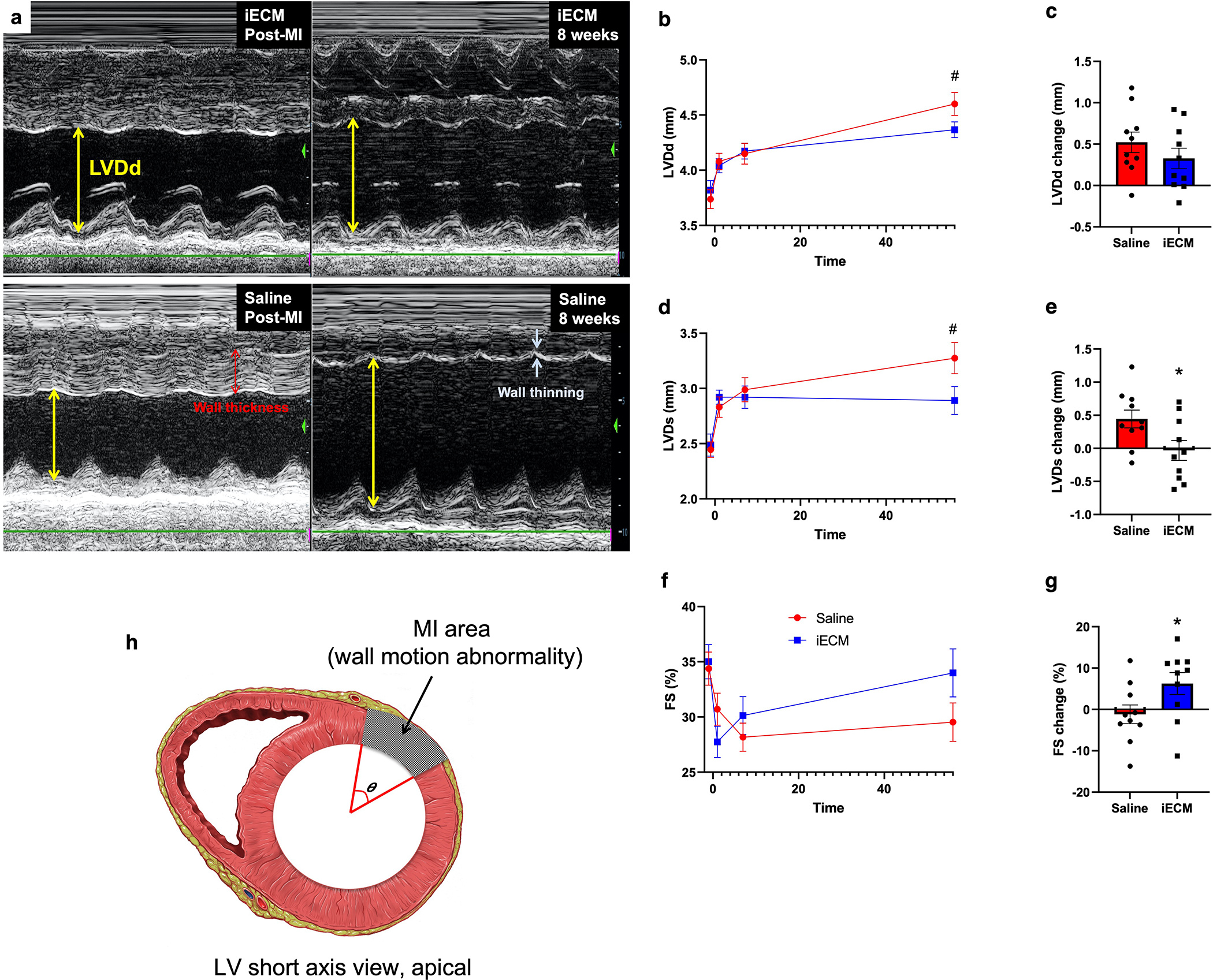
a, Representative M-mode echocardiographic images showing that iECM mitigates negative LV remodeling. Yellow arrows represent LV diastolic dimension, red arrows represent wall thickness, and white arrows represent wall thinning. b-g, LV diastolic dimension (LVDd, #p=0.08), LV systolic dimension (LVDs), and fractional shortening (FS) over time (b,d,f) and changes from post-MI to 8 weeks post-MI (c, e (*p=0.03), g (*p=0.047)). h, Diagram demonstrating how infarct angle was measured. N=10 all groups. Data are mean ± SEM. All data are biological replicates and were evaluated with a two tailed unpaired t-test.
Supplementary Material
Acknowledgements
KLC discloses support for the research described in this study from the NIH NHLBI [grant numbers R01HL113468, 1R01HL165232, and R43HL150917]. MTS, MD, JH, and RW disclose support for the publication of this study from the NIH NHLBI [grant number T32HL105373]. MD, JH, HS, and RW disclose support for the publication of this study from the NIH NHLBI [grant numbers F31HL152686, F31HL158212, F31HL152610, and F31HL137347]. MTS discloses support for the publication of this study from an AHA pre-doctoral fellowship. MAV discloses support for the publication of this study from the NSF [grant number DGE-1842165) and the Dr. John N. Nicholson Fellowship, which is awarded by Northwestern University. This work made use of the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-2025752), and the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern’s MRSEC program (NSF DMR-1720139). The authors would like to thank Jesse Placone for his confocal imaging assistance, Dr. Michael Davis for providing the rat cardiac endothelial cells, and Pamela Duran for her helpful comments during the manuscript editing process, and the University of California, San Diego Institutional Animal Care and Use Program veterinary staff for their assistance with large animal procedures and safety.
Footnotes
Competing interests
KLC and AND hold equity in Ventrix, Inc. KLC is a co-founder, consultant, and board member of Ventrix, Inc. The other authors declare no competing interests.
Code Availability
Custom scripts are available from the corresponding author (christman@eng.ucsd.edu).
Additional information
Supplementary information is available for this paper online.
Data Availability
The data sets generated and analyzed during the current study are available from the corresponding author (christman@eng.ucsd.edu).
References
- 1.Badylak SF The extracellular matrix as a biologic scaffold material. Biomaterials 28, 3587–3593 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Saldin LT, Cramer MC, Velankar SS, White LJ & Badylak SF Extracellular matrix hydrogels from decellularized tissues: Structure and function. Acta Biomaterialia 49, 1–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spang MT & Christman KL Extracellular matrix hydrogel therapies: In vivo applications and development. Acta Biomaterialia 68, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badylak SF, Freytes DO & Gilbert TW Extracellular matrix as a biological scaffold material: Structure and function. Vol. 5 1–13 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Christman KL Biomaterials for tissue repair. Science 363, 340–341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freytes DO, Martin J, Velankar SS, Lee AS & Badylak SF Preparation and rheological characterization of a gel form of the porcine urinary bladder matrix. Biomaterials 29, 1630–1637 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Singelyn JM, et al. Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering. Biomaterials 30, 5409–5416 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singelyn JM, et al. Catheter-deliverable hydrogel derived from decellularized ventricular extracellular matrix increases endogenous cardiomyocytes and preserves cardiac function post-myocardial infarction. Journal of the American College of Cardiology 59, 751–763 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seif-Naraghi SB, et al. Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction. Science translational medicine 5, 173ra125–173ra125 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wassenaar JW, et al. Evidence for Mechanisms Underlying the Functional Benefits of a Myocardial Matrix Hydrogel for Post-MI Treatment. Journal of the American College of Cardiology 67, 1074–1086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dziki JL & Badylak SF Immunomodulatory Biomaterials. Current Opinion in Biomedical Engineering (2018). [Google Scholar]
- 12.Sadtler K, et al. Proteomic composition and immunomodulatory properties of urinary bladder matrix scaffolds in homeostasis and injury. Seminars in Immunology 29, 14–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang RM & Christman KL Decellularized myocardial matrix hydrogels: In basic research and preclinical studies. Advanced Drug Delivery Reviews 96, 77–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz M, et al. Injectable myocardial matrix hydrogel mitigates negative left ventricular remodeling in a chronic myocardial infarction model. J Am Coll Cardiol: Basic to Translational Science 6, 350–361 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Traverse JH, et al. First-in-Man Study of a Cardiac Extracellular Matrix Hydrogel in Early and Late Myocardial Infarction Patients. JACC: Basic to Translational Science (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobbs SK, et al. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proceedings of the National Academy of Sciences of the United States of America 95, 4607–4612 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexis F, Pridgen E, Molnar LK & Farokhzad OC Factors affecting the clearance and biodistribution of polymeric nanoparticles. Vol. 5 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ungerleider JL, Johnson TD, Rao N & Christman KL Fabrication and characterization of injectable hydrogels derived from decellularized skeletal and cardiac muscle. Methods (San Diego, Calif.) 84, 53–59 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White MM & Jennings LK Platelet Protocols: Research and Clinical Laboratory Procedures, (1999).
- 20.Vayá A, et al. Erythrocyte aggregation determined with the Myrenne aggregometer at two modes (M0, M1) and at two times (5 and 10 sec). Vol. 29 119–127 (2003). [PubMed] [Google Scholar]
- 21.Banka AL & Eniola-Adefeso O Method article: an in vitro blood flow model to advance the study of platelet adhesion utilizing a damaged endothelium. Platelets, 1–8 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Gaetani R, et al. Cardiac-Derived Extracellular Matrix Enhances Cardiogenic Properties of Human Cardiac Progenitor Cells. Cell Transplantation 25, 1653–1663 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Wang RM, et al. Myocardial matrix hydrogel acts as a reactive oxygen species scavenger and supports a proliferative microenvironment for cardiomyocytes. Acta Biomater in press(2022). [DOI] [PubMed] [Google Scholar]
- 24.Leor J, et al. Intracoronary Injection of In Situ Forming Alginate Hydrogel Reverses Left Ventricular Remodeling After Myocardial Infarction in Swine. Journal of the American College of Cardiology 54, 1014–1023 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J & Barba I Myocardial edema: A translational view. Vol. 52 931–939 (Elsevier, 2012). [DOI] [PubMed] [Google Scholar]
- 26.Dongaonkar RM, Stewart RH, Geissler HJ & Laine GA Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Vol. 87 331–339 (Oxford University Press, 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thuny F, et al. Post-conditioning reduces infarct size and edema in patients with ST-segment elevation myocardial infarction. Journal of the American College of Cardiology 59, 2175–2181 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Habgood MD, et al. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. European Journal of Neuroscience 25, 231–238 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Higashida T, et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury: Laboratory investigation. Journal of Neurosurgery 114, 92–101 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Zhou C, Townsley MI, Alexeyev M, Voelkel NF & Stevens T Endothelial hyperpermeability in severe pulmonary arterial hypertension: Role of store-operated calcium entry. American Journal of Physiology - Lung Cellular and Molecular Physiology 311, L560–L569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugita T, et al. Lung vessel leak precedes right ventricular hypertrophy in monocrotaline-treated rats. Journal of Applied Physiology Respiratory Environmental and Exercise Physiology 54, 371–374 (1983). [DOI] [PubMed] [Google Scholar]
- 32.Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K & Deckmyn H Platelets at work in primary hemostasis. Blood Rev 25, 155–167 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Ruggeri ZM & Mendolicchio GL Adhesion mechanisms in platelet function. Circ Res 100, 1673–1685 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Konstam MA & Abboud FM Ejection Fraction: Misunderstood and Overrated (Changing the Paradigm in Categorizing Heart Failure). Circulation 135, 717–719 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jessup M, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124, 304–313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perin EC, et al. A Phase II Dose-Escalation Study of Allogeneic Mesenchymal Precursor Cells in Patients With Ischemic or Nonischemic Heart Failure. Circ Res 117, 576–584 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Ungerleider JL, et al. Extracellular Matrix Hydrogel Promotes Tissue Remodeling, Arteriogenesis, and Perfusion in a Rat Hindlimb Ischemia Model. JACC: Basic to Translational Science 1(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huleihel L, et al. Matrix-bound nanovesicles within ECM bioscaffolds. Sci Adv 2, e1600502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huleihel L, et al. Matrix-Bound Nanovesicles Recapitulate Extracellular Matrix Effects on Macrophage Phenotype. Tissue Eng Part A 23, 1283–1294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fontes JA, Rose NR & Čiháková D The varying faces of IL-6: From cardiac protection to cardiac failure. Vol. 74 62–68 (Academic Press, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fosgerau K & Hoffmann T Peptide therapeutics: current status and future directions. Drug Discovery Today 20(2015). [DOI] [PubMed] [Google Scholar]
- 42.Di L Strategic Approaches to Optimizing Peptide ADME Properties. AAPS Journal 17, 134–143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeQuach JA, Yuan SH, Goldstein LS & Christman KL Decellularized porcine brain matrix for cell culture and tissue engineering scaffolds. Tissue Eng Part A 17, 2583–2592 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ungerleider J, et al. Extracellular matrix hydrogel promotes tissue remodeling, arteriogenesis, and perfusion in a rat hindlimb ischemia model. J Am Coll Cardiol: Basic to Translational Science 1, 32–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grover GN, Braden RL & Christman KL Oxime Cross-Linked Injectable Hydrogels for Catheter Delivery. Advanced materials 25, 2937–2942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oronsky B, Oronsky N & Cabrales P Platelet inhibitory effects of the phase 3 anticancer and normal tissue cytoprotective agent, RRx-001. Journal of Cellular and Molecular Medicine 22, 5076–5082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jani VP, Yalcin O, Williams AT, Popovsky MA & Cabrales P Rat red blood cell storage lesions in various additive solutions. Clinical Hemorheology and Microcirculation 67, 45–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buono MJ, Krippes T, Kolkhorst FW, Williams AT & Cabrales P Increases in core temperature counterbalance effects of haemoconcentration on blood viscosity during prolonged exercise in the heat. Experimental Physiology 101, 332–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marin V, Kaplanski G, Gres S, Farnarier C & Bongrand P Endothelial cell culture: protocol to obtain and cultivate human umbilical endothelial cells. J Immunol Methods 254, 183–190 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Shamseddin A, et al. Resveratrol-Linoleate protects from exacerbated endothelial permeability via a drastic inhibition of the MMP-9 activity. Biosci Rep 38(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coenen DM, Mastenbroek TG & Cosemans J Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood 130, 2819–2828 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Mann AP, et al. A peptide for targeted, systemic delivery of imaging and therapeutic compounds into acute brain injuries. Nature Communications 7, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bharadwaj VN, Nguyen DT, Kodibagkar VD & Stabenfeldt SE Nanoparticle-Based Therapeutics for Brain Injury. Advanced Healthcare Materials 7(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romine J, Gao X & Chen J Controlled cortical impact model for traumatic brain injury. Journal of Visualized Experiments, 51781–51781 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osier N & Dixon CE The controlled cortical impact model of experimental brain trauma: Overview, research applications, and protocol. Vol. 1462 177–192 (Humana Press Inc., 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maarman G, Lecour S, Butrous G, Thienemann F & Sliwa K A comprehensive review: The evolution of animal models in pulmonary hypertension research; are we there yet?, Vol. 3 739–756 (Taylor and Francis Inc., 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gomez-Arroyo JG, et al. The monocrotaline model of pulmonary hypertension in perspective. Vol. 302 363–369 (American Physiological Society; Bethesda, MD, 2012). [DOI] [PubMed] [Google Scholar]
- 58.Wassenaar JW, Braden RL, Osborn KG & Christman KL Modulating in vivo degradation rate of injectable extracellular matrix hydrogels. J. Mater. Chem. B 4, 2794–2802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren Y, et al. Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Science Translational Medicine 4(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonios M, et al. Myocardial substrate and route of administration determine acute cardiac retention and lung biodistribution of cardiosphere-derived cells. Journal of Nuclear Cardiology 18, 443–450 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng K, et al. Magnetic enhancement of cell retention, engraftment, and functional benefit after intracoronary delivery of cardiac-derived stem cells in a rat model of ischemia/reperfusion. Cell transplantation 21, 1121–1135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Freyman T, et al. A quantitative, randomized study evaluating three methods of mesenchymal stem cell delivery following myocardial infarction. European Heart Journal 27, 1114–1122 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated and analyzed during the current study are available from the corresponding author (christman@eng.ucsd.edu).