

Abstract
Exposure to low to moderate arsenic (As) levels has been associated with type 2 diabetes (T2D) and other chronic diseases in American Indian communities. Prenatal exposure to As may also increase the risk for T2D in adulthood, and maternal As has been associated with adult offspring metabolic health measurements. We hypothesized that T2D-related outcomes in adult offspring born to women exposed to low to moderate As can be evaluated utilizing a maternally-derived molecular biosignature of As exposure. Herein, we evaluated the association of maternal DNA methylation with incident T2D and insulin resistance (Homeostatic model assessment of insulin resistance [HOMA2-IR]) in adult offspring. For DNA methylation, we used 20 differentially methylated cytosine-guanine dinucleotides (CpG) previously associated with the sum of inorganic and methylated As species (ΣAs) in urine in the Strong Heart Study (SHS). Of these 20 CpGs, we found six CpGs nominally associated (p < 0.05) with HOMA2-IR in a fully adjusted model that included clinically relevant covariates and offspring adiposity measurements; a similar model that adjusted instead for maternal adiposity measurements found three CpGs nominally associated with HOMA2-IR, two of which overlapped the offspring adiposity model. After adjusting for multiple comparisons, cg03036214 remained associated with HOMA2-IR (q < 0.10) in the offspring adiposity model. The odds ratio of incident T2D increased with an increase in maternal DNA methylation at one HOMA2-IR associated CpG in the model adjusting for offspring adiposity, cg12116137, whereas adjusting for maternal adiposity had a minimal effect on the association. Our data suggests offspring adiposity, rather than maternal adiposity, potentially influences the effects of maternal DNAm signatures on offspring metabolic health parameters. Here, we have presented evidence supporting a role for epigenetic biosignatures of maternal As exposure as a potential biomarker for evaluating risk of T2D-related outcomes in offspring later in life.
Keywords: American Indians, Arsenic, Diabetes, Epigenetics, Insulin resistance, DNA methylation, Strong Heart Study
1. Introduction:
Arsenic (As), a ubiquitous environmental toxicant in water and food (Chung et al., 2014), contributes to adverse health outcomes in human populations (Hong et al., 2014), and is considered a major global health concern. Long-term exposure to low to moderate As in drinking water, which is common in the rural Western US, has been associated with metabolic conditions in American Indian (AI) communities, including type 2 diabetes (T2D), the metabolic syndrome, and insulin resistance (Kuo et al., 2015; Grau-Perez et al., 2017; Spratlen et al., 2018; Spratlen et al., 2018). Arsenic is known to readily cross the placenta, may alter fetal development, and underlie disease risk later in life (Punshon et al., 2015). Growing experimental evidence shows that prenatal and early-life As exposure may contribute to adverse offspring health into adulthood, such as increased metabolic disease risk (Navas-Acien et al., 2019; Rodriguez et al., 2020; Rodriguez et al., 2016; Huang et al., 2018). These experimental findings are consistent with findings in the Strong Heart Study (SHS), a population-based cohort study in American Indian communities in the Southwest and the Great Plains, where maternal As exposure was associated with adult offspring T2D risk and insulin resistance (Tinkelman et al., 2020).
Epigenetic modifications, including DNA methylation (DNAm), are plausible molecular mechanisms linking prenatal and early life exposures to future disease risk, given these epigenetic marks are responsive to environmental factors, with long-lasting effects on gene expression (i.e., cellular phenotypes) (Godfrey et al., 2015). Due to improvements in methodology for acquiring and interpreting epigenetic data in large-scale studies, the utility of epigenetic information, including DNAm modifications, has gained prominence as a potential molecular biomarker of health and disease (García-Giménez et al., 2017). However, assessing long-term disease risk in those exposed to adverse environmental influences during sensitive windows of development remains a challenge. To identify those at risk for adverse metabolic health later in life—due, in part, to environmental exposure during development—minimally invasive strategies for identifying stable biomarkers amenable to the clinical setting will be invaluable. For instance, parental DNAm in buccal cells has predicted preterm birth in offspring (Winchester et al., 2022), and blood-based DNAm markers in mothers has been associated with offspring birthweight (Kheirkhah Rahimabad et al., 2021). Whether maternal DNAm signatures of As exposure can be used as a biosignature for evaluating offspring metabolic health outcomes into adulthood has yet to be examined.
The SHS provides a unique opportunity to identify potential epigenetic biosignatures of As exposure on risk for T2D-related outcomes across generations. Indeed, As exposure remained relatively constant over many decades in the SHS communities prior to 2006 and residential mobility is low. Thus, urinary As measurements obtained in mothers at baseline in 1989–1991 likely reflect long periods of exposure (Navas-Acien et al., 2009), including during pregnancy and fetal development, and, thus may have lasting effects on offspring health. Prior work by our group identified an association between maternal As exposure with adult offspring metabolic health (Tinkelman et al., 2020). Arsenic exposure has also been associated with blood-based DNAm markers in the SHS (Bozack et al., 2020), including at genes involved in glucose metabolism and As-induced oxidative stress. However, whether these blood-based epigenetic signatures of exposure are related to offspring T2D-related phenotypes is unknown. This study sought to explore blood-based DNAm signatures of maternal As exposure as a biomarker to assess T2D-related outcomes in adult offspring of mothers exposed to variable levels of As, likely covering sensitive windows of development (i.e., pregnancy and early life). Utilizing previously identified differentially methylated cytosine-guanine dinucleotides (CpGs) associated with As measurements from the SHS cohort at baseline (1989–1991) (Bozack et al., 2020), we examined an association of As-associated DNAm signatures in mothers with adverse metabolic phenotypes in adult offspring who were part of the SHS family expansion, the Strong Heart Family Study (SHFS).
2. Methods:
2.1. Study population
The SHS is a National Heart, Lung, and Blood Institute funded prospective cohort of men and women aged 45–74 years old from 13 AI communities spanning North and South Dakota, Oklahoma, and Arizona who were recruited and examined in 1989–1991 (Visit 1) with follow-up examinations every 3–4 years (Lee et al., 1990). The SHFS recruited family members of original SHS participants with at least 5 siblings (96 families) between 1998 and 1999 (Visit 3 pilot) or 2001 to 2003 (Visit 4). In the present study, participants from SHS and SHFS were excluded based on specific criteria (Fig. 1). From all participants in the SHS at Visit 1 (n = 4,549), we excluded data from one tribe that requested no further participation, participants who did not meet criteria for urine metals analysis (e.g., insufficient urine), male participants, and women without DNAm data collected or low-quality DNAm data. From the remaining 1,361 female SHS participants, we excluded those without offspring in the SHFS or whom had offspring with missing outcome (fasting glucose, Homeostasis Model Assessment of Insulin Resistance [HOMA2-IR], incident T2D) and covariate measurements (urinary total As, BMI, waist circumference, smoking status), as well as mothers missing covariate measurements (total urinary As, BMI, waist circumference, smoking status, fasting glucose) and estimated glomerular filtration rate (eGFR). In a similar approach, from SHFS participants at baseline (Visit 3 and Visit 4; n = 3,838), the same tribe as previously mentioned declined participation in the SHFS (n = 919). We further excluded participants who had prevalent diabetes to control for the effects of diabetes treatment regiments on metabolic measurements used in this study. We then excluded those with insufficient urine for metals analysis or whose SHS parent was excluded from urine metals analysis. Finally, SHFS participants who had a mother in SHS that was not excluded based on our previously mentioned criteria were retained (n = 1,720); an in-depth exclusion criteria for SHFS can be found elsewhere (Spratlen et al., 2018; Tinkelman et al., 2020). Subsequently, we excluded offspring missing relevant study variables (total urinary As, fasting glucose, HOMA2-IR, BMI, diabetes status at Visit 5, and waist circumference) or offspring whose mothers were missing relevant study variables (total urinary As, BMI, waist circumference, and fasting glucose). Finally, of the remaining 466 SHFS offspring participants, we excluded those whose mothers did not have DNAm data collected at Visit 1, retaining a total of 226 SHFS offspring paired to 119 mothers for DNAm analyses. The SHS protocol was approved by institutional review boards (IRBs), participating tribal communities, and the respective area Indian Health Service IRBs, and all participants provided informed consent.
Fig. 1. Study exclusion criteria.
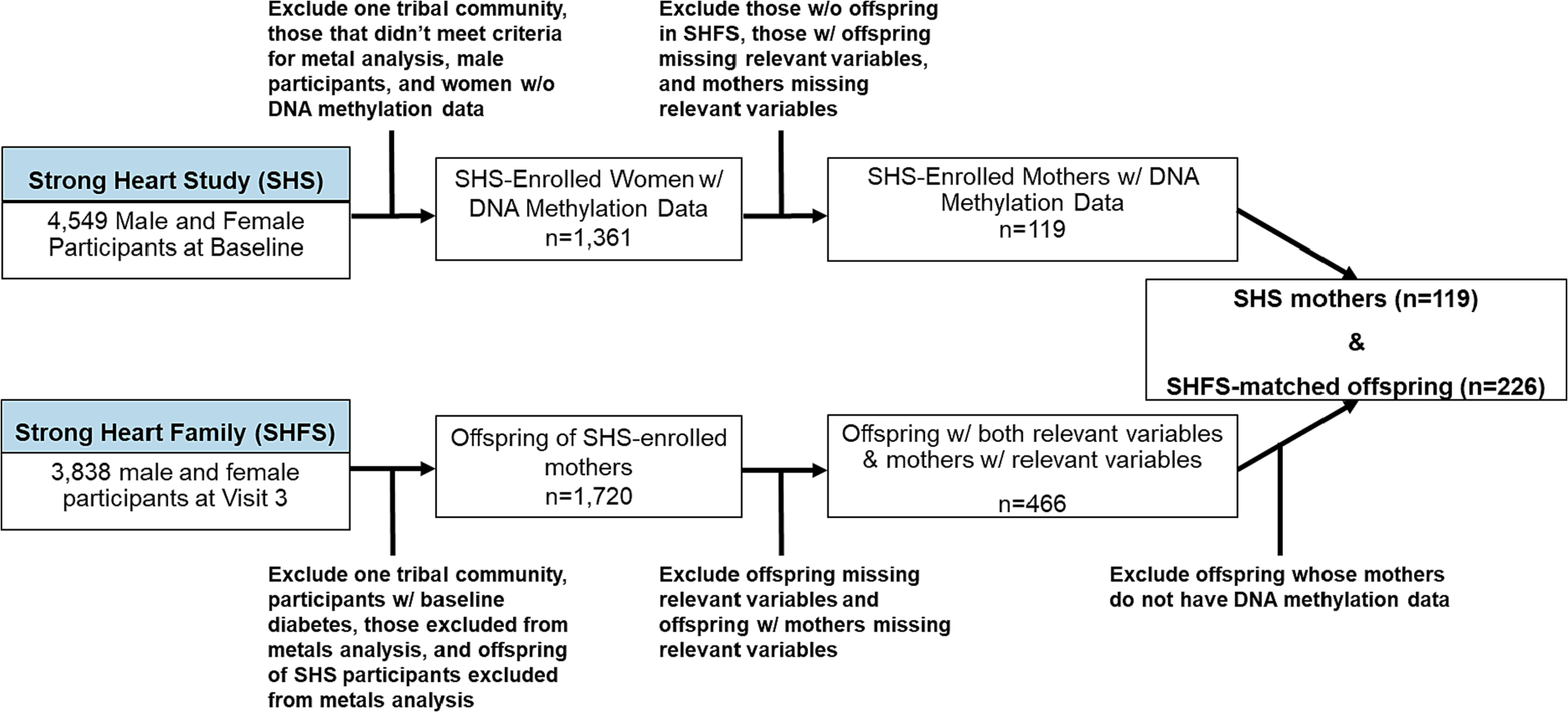
Flowchart represents exclusion criteria for the DNA methylation analysis of Strong Heart Study (SHS) mothers and matched Strong Heart Family Study (SHFS) offspring participants for this study.
2.2. Data collection
At SHS baseline (Visit 1) and follow-up visits, physical examinations and interviews were conducted by trained staff following standard procedures. Participants reported sociodemographic information (age, sex, study site, education), smoking status (never, former, and current), and medical history during interviews. Anthropometric measures were collected during physical examinations. Fasting blood samples (12 h) and spot urine samples were collected and stored according to standardized procedures (Lee et al., 1990). Maternal measurements were based on Visit 1 (1989–1991), baseline measurements for offspring were based either on Visit 3 (1998–1999) or Visit 4 (2001–2003), and outcome data for offspring were based on Visit 5 (2006–2009).
2.3. Urine arsenic measurements
Analytical methods and quality control for urinary As measurements are described in detail elsewhere (Scheer et al., 2012). Spot urine samples were collected in polypropylene tubes, frozen within 2 h of collection, and shipped on dry ice to MedStar Health Research Institute (Washington, DC, USA), where they were stored at < −70°C. In 2009–2010, aliquots of up to 1.0 mL from stored urine samples were shipped on dry ice to the Trace Element Laboratory at Graz University (Graz, Austria). Arsenic species (arsenite, arsenate, methylarsonate [MMA], and dimethylarsinate [DMA]) were measured using the Agilent z high-performance liquid chromatography coupled with Agilent 7700x inductive coupled plasma mass spectrometry (Agilent Technologies, Santa Clara California, USA). The LOD for inorganic As (iAs), MMA, and DMA was 0.1 μg/L (Scheer et al., 2012). No samples were below the LOD for these species. Arsenobetaine, a non-toxic As species typically found in seafood, was low in SHS participants (median: 0.65 μg/g creatinine), suggesting minimal seafood intake in this study population. Urinary creatinine was measured at the National Institute of Diabetes and Digestive and Kidney Diseases Epidemiology and Clinical Research Branch Laboratory (Phoenix, Arizona, USA) utilizing automated alkaline picrate methods on a rapid flow analyzer. Total urinary iAs concentrations (μg/L) were divided by urinary creatinine (g/L) concentration to account for inter-individual variability in urine dilution. The sum of iAs and methylated As species (MMA and DMA) were considered as the measure of exposure of inorganic As exposure, hereafter reported as total As (ΣAs).
2.4. Fasting glucose, HOMA2-IR, and T2D measurements
Blood samples collected after a 12-hour fast were obtained to measure lipids, glucose, insulin, and other metabolic markers in SHS and SHFS participants. Plasma samples were measured for fasting glucose and insulin by MedStar Research Institute (Washington, DC, USA) following standardized methods as previously described (Lee et al., 1990; Howard et al., 1995). The corrected Homeostasis Model Assessment of Insulin Resistance, HOMA2-IR, an improved computing model employed as a surrogate measure for beta cell function and insulin resistance, was obtained from calculations applied to fasting insulin and fasting glucose concentrations (Levy et al., 1998). T2D was defined as either a self-reported physician diagnosis, self-reported use of insulin or oral diabetic treatment, or a fasting plasma glucose concentration ≥ 126 mg/dL. By design for this study, no SHFS participants had prevalent T2D at baseline.
2.5. DNA methylation quantification, preprocessing and normalization
The quantification and processing of DNAm from blood samples provided by SHS participants at Visit 1 have been described in detail previously (Bozack et al., 2020). Briefly, leukocytes were isolated from blood samples and purified for genomic DNA. DNA was bisulfite-converted and hybridized to Illumina’s Infinium MethylationEPIC BeadChip (850K; Illumina, Inc., San Diego, CA, USA), which significantly improves on its predecessor, the HumanMethylation450 BeadChip, in genome coverage, reproducibility, accuracy and reliability (Pidsley et al., 2016). Samples were randomized within and across sample plates to control for batch effects, internal controls and replicate controls were included in each plate, and standard quality control probes were integrated using Illumina’s GenomeStudio software (Illumina, Inc.). IDAT files were generated in six batches and combined employing the R package minfi (Aryee et al., 2014). Quality control assessment and normalization was performed on both samples and CpG probes. Normalization utilized single sample noob normalization method within the minfi package. In total, 26 samples were removed that did not pass quality control and normalization (Fig. 1). ComBat in the sva package (version 3.36.0) was used to correct for batch effects (Leek et al., 2012). Failed probes based on Illumina’s recommendations were removed (detection p-value > 0.01 in 5 % of individuals or more). CpG probes residing on sex chromosomes, enriched at single-nucleotide polymorphisms (SNPs) and cross-reactive CpG probes were also removed. To correct for probe-type bias, Representative Concentration Pathway (RCP) normalization was performed. Houseman’s projection method was performed to estimate blood immune cell-type proportions in each sample (Houseman et al., 2012). DNAm data was available across 788,753 CpGs from 2,325 participants in the SHS, including the 119 mothers that were selected for this study. DNAm values (β-values) were generated for the remaining CpG loci based on the ratio of methylated CpG probe intensity and total CpG probe methylation intensity (methylated + unmethylated probe intensities), yielding DNA methylation proportions from 0.0 (unmethylated) to 1.0 (fully methylated).
Previously, our group identified 20 differentially methylated CpG positions (DMP) associated with urinary ΣAs in SHS participants at a significant threshold (q < 0.05) (Bozack et al., 2020), using the limma package in R (Ritchie et al., 2015). Here, β-values were logit transformed to M–values (M = log2[β-value/1 – β-value]), as they better satisfy parametric modeling assumptions for differential methylation analyses as compared to β-values, which tend to be heteroscedastic at the highly methylated or unmethylated CpGs (Du et al., 2010). Linear regression modeling for differential methylation analysis was adjusted for sex, age, BMI, smoking status, education (<high school, high school graduate or GED, > high school), study site (Arizona, North and South Dakota, or Oklahoma), eGFR, and Houseman’s cell type proportions. False-discovery rates (FDR) were accounted for using the Benjamini and Hochberg method for multiple comparisons and significance was determined at q < 0.05. Sensitivity analyses were performed to test for effect modification of covariates, including sex, smoking status, study site, and T2D status. Importantly, sensitivity analysis stratified by sex found the same 20 ΣAs-associated CpGs achieved nominal significance with ΣAs (p < 0.05), suggesting sex did not have a significant effect on the resulting relationship between urinary ΣAs and DNAm. This is relevant, as in the current study, we sought to solely employ female DNAm signatures of ΣAs exposure (i.e., mothers) to identify a relationship with offspring metabolic outcomes. These findings support that the 20 ΣAs-associated CpGs can be used in our study that includes an all-female (i.e., maternal) population.
2.6. Statistical analyses
Descriptive statistics for both maternal (n = 119) and offspring (n = 226) variables were calculated as: median (1st quartile, 3rd quartile) for continuous variables, and counts (frequency [%]) for categorical variables. Urinary ΣAs (iAs, MMA, and DMA) (μg/L) was adjusted for urine dilution using creatinine concentrations (g/L).
We used generalized estimating equations (GEE) with an independent correlation structure to examine the associations between maternal ΣAs-associated DNAm and the following offspring variables: HOMA2-IR (continuous), and incident T2D (binary). All regression models were performed between maternal methylation of ΣAs-associated CpGs (M–value) at SHS baseline (Visit 1) as the exposure variable and continuous or binary variables at SHFS Visit 5 (2006–2009) as the outcome of interest. Continuous outcome variables (i.e., HOMA2-IR) were log transformed to satisfy linear regression model assumptions, and the resulting β coefficients were exponentiated to delineate the relative change in the outcome variable (geometric mean ratios [GMR]). For the binary outcome, we employed GEE with a logit link to perform logistic regression analysis, and resulting coefficients were exponentiated to an odds ratio. GEE were used to account for clustered data based on familial relationships (i.e., mother–child pairs, and siblings) (Homish et al., 2010), given the flexibility of GEE in handling related data, such as shared exposures of siblings and DNAm data derived from paired mothers. To account for multiple comparisons, the Benjamini-Hochberg method was employed within each outcome at a threshold q < 0.10 for significance. To assist with interpretation of DNAm M–values, which are not as easily interpretable as β-values (percentage of DNAm at a specific CpG locus), DNAm data was mean-centered and scaled by subtracting the sample mean from every value of the predictor variable and running the model on the centered data. Results were interpreted as a change in the GMR or odds ratio (OR) of the outcome variable for every standard deviation in the predictor variable (i.e., ΣAs-associated CpG locus methylation). Likewise, for HOMA2-IR, we included the absolute change in HOMA2-IR for each model to assist in interpretation (e.g., change in HOMA-2IR scores vs GMR of HOMA2-IR). Four separate models were employed in our analyses, including an unadjusted model, a model adjusting for a priori selected variables (Model 1), a model including variables from Model 1 and offspring adiposity (Model 2), and a model including variables from Model 1 and maternal adiposity (Model 3). Covariates selected for Model 1, and subsequently included into Models 2 and 3, included offspring sex, age, smoking status (never, ever, current), and log2 transformed ΣAs (μg/g creatinine) at the baseline offspring visit (Visit 3 or 4); and maternal age, smoking status, log2 transformed ΣAs (μg/g creatinine), log2 transformed fasting glucose (mg/dL) at baseline (Visit 1). Model 2 further included maternal waist circumference (cm) at baseline and Model 3 included offspring waist circumference (cm) at baseline. Likewise, we performed the same analyses with offspring and maternal BMI (kg/m2) to determine which measure of adiposity was a better clinical indicator for including when assessing offspring T2D risk. We chose to adjust for offspring and maternal adiposity measurements separately to determine whether the relationship between maternal DNAm signatures and offspring metabolic parameters were influenced by maternal or offspring adiposity.
3. Results
3.1. Participant characteristics
The age (IQR) at enrollment was 54.4 (49.3, 61.6) years for the SHS mothers (n = 119) (Visit 1) and 40.4 (35.5, 47.2) years for SHFS offspring (Visit 3 or 4; n = 226) (Table 1). Of the SHFS offspring participants, 82 were male (36.2 %). BMI was similar between mothers and offspring at baseline time points, 30.9 (27.2, 35.2) kg/m2 and 30.4 (26.8, 34.9) kg/m2, respectively. Among the mothers, 44 (37.0 %) were never smokers, 32 (26.9 %) were former smokers, and 43 (36.1 %) were current smokers. Among the offspring, 78 (34.5 %) were never smokers, 55 (24.3 %) were former smokers, and 93 (41.2 %) were current smokers. 44 out of 119 (37.0 %) mothers had prevalent T2D at baseline, whereas 42 out of 104 (40.4 %; 15 participants were missing data) had diabetes at follow-up, including 6 new cases. No offspring had T2D at baseline by design, however, at follow-up there were 41 cases of incident T2D (41/226; 18.1 %). The median fasting glucose concentration was 111.0 (99.0, 168.0) mg/dL for mothers and 94.0 (87.0, 103.0) mg/dL for offspring at their respective baseline visits, and at follow-up it was 113.0 (98.0, 171.0) mg/dL and 94.0 (86.0, 106.3) mg/dL, respectively. Median (IQR) HOMA2-IR scores were higher in mothers compared to offspring at baseline (3.7 (2.4, 5.5) vs 1.35 (1.0, 2.5)) and follow-up (3.5 (1.9, 6.0) vs 1.6 (0.9, 2.7)). Median (IQR) ΣAs concentrations in mothers were also higher than offspring (7.3 (5.0, 13.8) vs 4.6 (3.0, 8.4) μg/g creatinine).
Table 1.
SHS and SHFS Participant Baseline Characteristics.
| SHS Mothers (n = 119) | SHFS Offspring (n = 226) | |
|---|---|---|
|
| ||
| Age (Years) | 54.4 (49.3, 61.6) | 40.4 (35.5, 47.2) |
| Sex (Male) | – | 82 (36.2 %) |
| Smoking Status | ||
| Never | 44 (37.0 %) | 78 (34.5 %) |
| Ever | 32 (26.9 %) | 55 (24.3 %) |
| Current | 43 (36.1 %) | 93 (41.2 %) |
| Waist Circumference (cm) | 105.0 (98.0, 116.0) | 100.0 (92.0, 111.0) |
| BMI (kg/m^2) | 30.9 (27.2, 35.2) | 30.4 (26.8, 34.9) |
| Diabetes Status (Diabetic) | 44 (37.0 %) | 0 (0.0 %) |
| Follow-up Diabetes Status | 41 (41.2 %) | 41 (18.1 %) |
| Fasting Glucose (mg/dL) | 111.0 (99.0, 168.0) | 94.0 (87.0, 103.0) |
| Follow-up Fasting Glucose (mg/dL) | 113.0 (98.0, 171.0) | 94.0 (86.0, 106.3) |
| HOMA2-IR | 3.7 (2.4, 5.5) | 1.5 (1.0, 2.5) |
| Follow-up HOMA2-IR | 3.5 (1.9, 6.0) | 1.6 (0.9, 2.7) |
| Total Arsenic (μg/g creatinine) | 7.3 (5.0, 13.8) | 4.6 (3.0, 8.4) |
Continuous variables: median (1st, 3rd Quartile); categorical variables: count (frequency %). Diabetes status based on criteria: currently taking diabetes medication, impaired fasting glucose (≥126 mg/dL), 2-hr post-prandial plasma glucose ≥ 200 mg/dL, or HbA1c ≥ 6.5 %. BMI: Body mass index, HOMA2-IR: Homeostatic Model Assessment for Insulin Resistance.
3.2. Maternal arsenic-associated DNA methylation and insulin resistance
We estimated the GMR and absolute difference in offspring HOMA2-IR scores for one SD increase in each of the candidate CpG locus methylation states (M–value), and found several maternal ΣAs-associated CpGs overlapped multiple models (Fig. 2.a) and were nominally associated with offspring HOMA2-IR in different models (Fig. 1.b–g; Supplementary Table 1). In the unadjusted model, three CpGs were nominally associated with HOMA2-IR (cg12106731 tagged to the TSS of neurochondrin [NCDN]/KIAA0319L, cg07021906 tagged to an intronic region of SLC7A5, also known as large amino transporter 1 [LAT1], and cg03036214 tagged proximal to an exon of carbonic anhydrase 12 [CA12]). These CpG remained differentially associated with HOMA2-IR after base adjustment for offspring and maternal factors (Model 1) and with further adjustment for maternal waist circumference (Model 2). Further adjustment for offspring waist circumference (instead of maternal waist circumference) (Model 3) resulted in six CpGs nominally associated with HOMA2-IR. Two of them overlapped with the CpGs found in the unadjusted model, Model 1 and Model 2 (cg07021906 and cg03036214). The CpGs exclusively found in model 3 were cg08059112 (tagged to the 5′ UTR of the leucine-rich repeat and Ig domain containing 3 [LINGO3]), cg14595618 (tagged intragenically to hexokinase 1 [HK1]), cg12116137 (tagged proximal to an exon of pre-mRNA processing factor 8 [PRPF8]), and cg03497652 (tagged intragenically/proximal to an exon of ankyrin repeat and sterile alpha motif domain containing 3 [ANKS3]). All the nominally significant GMRs of HOMA2-IR and maternal DNAm were positive, indicating higher methylation levels in the mother were associated with higher levels of HOMA2-IR in the offspring. After FDR-correction, cg03036214 (CA12) in Model 3 remained associated with HOMA2-IR at q < 0.10 (Supplementary Table 1). Finally, we compared these results to our group’s previous observations identifying an association between maternal urinary ΣAs and offspring adulthood HOMA-2-IR (Tinkelman et al., 2020). Previously, we found that the GMR of offspring HOMA2-IR per IQR increase in maternal urinary ΣAs ranged between 1.00 and 1.04 in five different covariate-adjusted models. Using maternal ΣAs-associated DNAm signatures, we found the GMR of adult offspring HOMA2-IR per standard deviation increase in maternal DNAm ranged between 1.13 and 1.19, depending on CpG of interest and covariates included in the model.
Fig. 2. Maternal ΣAs-associated CpGs associated with offspring HOMA2-IR.
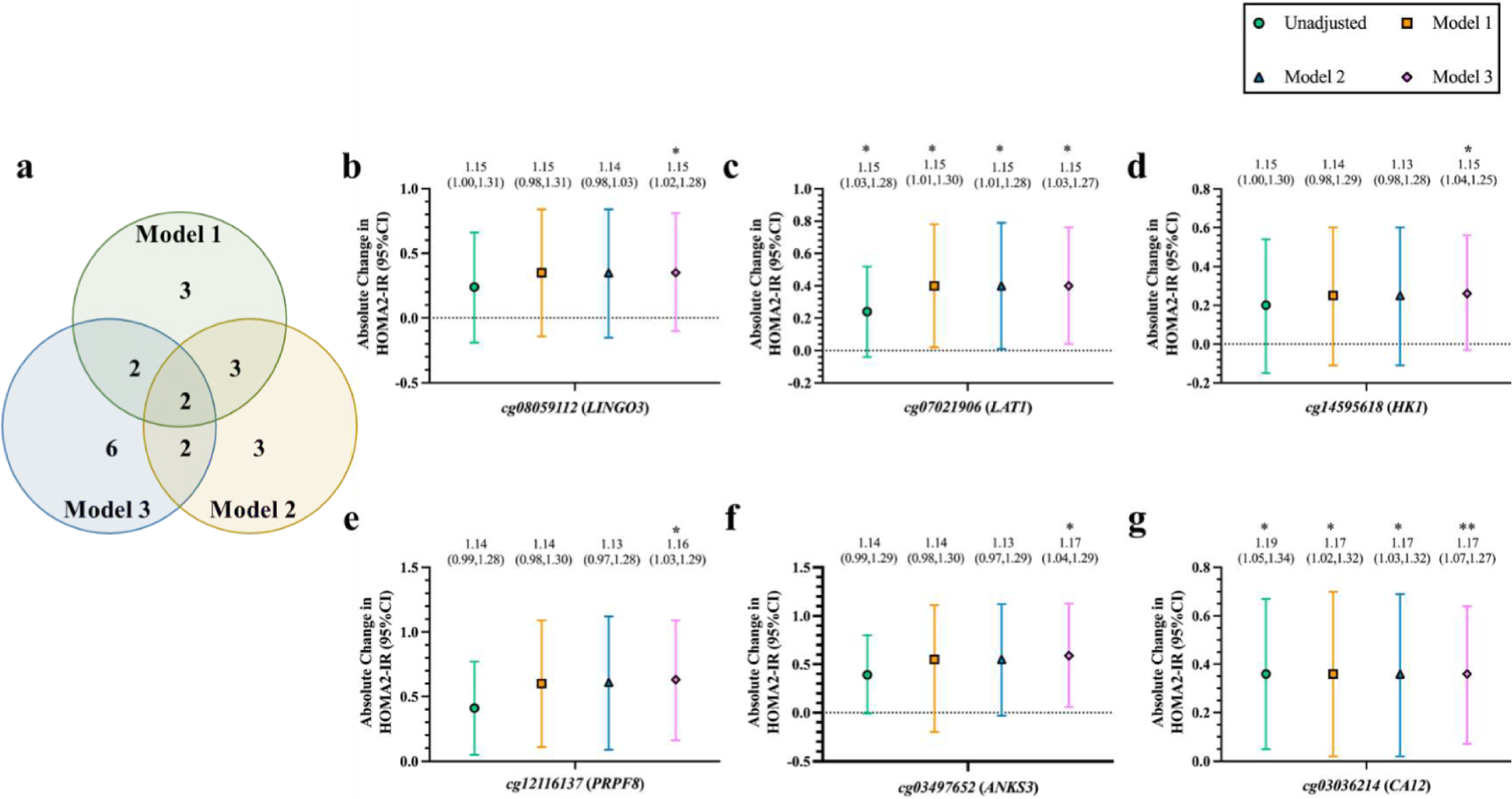
a. Venn diagram illustrates the number of CpGs overlapping each GEE model. b-f. Absolute difference in HOMA2-IR (95 % CI) in offspring at Visit 5 for every standard deviation change in baseline maternal DNA methylation (M–value) at specific CpG loci, including b) cg08059112 (LINGO3), c) cg07021906 (LAT1), d) cg14595618 (HK1), e) cg12116137 (PRPF8), f) cg03497652 (ANKS3), and g) cg03036214 (CA12). Values above each graph represent the GMR of offspring HOMA2-IR scores (95 % CI) per standard deviation change in baseline maternal DNA methylation; calculated by exponentiating the β-coefficients of each model using GEE on log-transformed HOMA2-IR scores. Asterisks represent unadjusted (for multiple comparisons), significant (p < 0.05) GMRs of HOMA2-IR per change in maternal DNA methylation. GEE: Generalized estimating equation; HOMA2-IR: Homeostatic Model Assessment for Insulin Resistance; CI: Confidence interval; LINGO3: leucine rich repeat and Ig domain containing 3; LAT1: large amino acid transporter 1 (SLC7A5); HK1: hexokinase 1; PRPF8: pre-mRNA processing factor 8; ANKS3: ankyrin repeat and sterile alpha motif domain containing 3; CA12: carbonic anhydrase 12. Significance taken at nominal p < 0.05* and p < 0.01 **. Model 1: adjusted for offspring sex, age, smoking status, and log(Σas), and for maternal age, smoking status, log(Σas), and log(fasting glucose). Model 2: Model 1 and further adjusted for maternal waist circumference. Model 3: Model 1 and further adjusted for offspring waist circumference.
3.3. Maternal arsenic-associated DNA methylation and type 2 diabetes risk
Insulin resistance is a hallmark characteristic of T2D pathogenesis and related cardiometabolic complications. Because our data suggests maternal ΣAs-associated CpG methylation is associated with offspring insulin resistance, we posited that this subset of CpG loci associated with offspring HOMA2-IR may contain an underlying biosignature capable of assessing T2D risk in adult offspring, as measured by incident T2D. In unadjusted logistic regression models, the OR of incident T2D in the adult offspring per SD in the M–value of each CpG ranged from 1.01 to 1.30, and none of them were nominally significant (Table 2). The OR remained similar with the base adjustment (Model 1) and with adjustment for maternal waist circumference (Model 2). However, adjustment for offspring waist circumference (Model 3) showed a non significant increase in the OR for cg03497652 (ANKS3) to 1.24 (95 %CI 0.83, 1.86) and for cg12116137 (PRPF8) to 1.45 (0.97, 2.18). Finally, our group’s previous observations identifying an increased OR of incident T2D for an IQR increase in maternal urinary ΣAs ranged between 1.18 (0.94, 1.49) to 1.43 (1.16, 1.78) in different covariate-adjusted models (Tinkelman et al., 2020), which was comparable to results utilizing ΣAs-associated DNAm signatures at cg03497652 and cg12116137 in Model 3.
Table 2.
Odds ratio (95% CI) of incident diabetes in adult offspring by maternal ΣAs-associated DNA methylation.
| (Incident diabetes: 41/226 SHFS Participants) | |||||
|---|---|---|---|---|---|
|
| |||||
| Unadjusted | Model 1 | Model 2 | Model 3 | ||
|
| |||||
| CpG | Gene(s) | OR (95 % CI) | OR (95 % CI) | OR (95 % CI) | OR (95 % CI) |
| cg14595618 | HK1 | 1.01 (0.70,1.46) | 0.96 (0.68,1.36) | 0.94 (0.66,1.34) | 1.02 (0.69,1.52) |
| cg08059112 | LINGO3 | 1.11 (0.81,1.54) | 0.99 (0.71,1.39) | 0.98 (0.70,1.38) | 1.03 (0.72,1.47) |
| cg03036214 | CA12 | 1.17 (0.86,1.59) | 1.07 (0.77,1.48) | 1.06 (0.76,1.47) | 1.15 (0.79,1.69) |
| cg07021906 | SLC7A5 (LAT1) | 1.27 (0.95,1.71) | 1.12 (0.79,1.59) | 1.11 (0.78,1.57) | 1.17 (0.80,1.73) |
| cg03497652 | ANKS3 | 1.06 (0.74,1.52) | 1.10 (0.76,1.59) | 1.08 (0.74,1.58) | 1.24 (0.83,1.86) |
| cg12116137 | PRPF8 | 1.30 (0.93,1.82) | 1.30 (0.90,1.88) | 1.27 (0.86,1.88) | 1.45 (0.97,2.18) |
GEE were used to account for family clustering. β-coefficients were exponentiated (shown here) to odds ratio (95% CI).
Model 1: adjusted for offspring sex, age, smoking status, and log(ΣAs) at baseline, and for maternal age, smoking status, log(ΣAs), and log(fasting glucose) at baseline.
Model 2: Model 1 adjustment, plus further adjustment for maternal waist circumference.
Model 3: Model 1 adjustment, plus further adjustment for offspring waist circumference.
4. Discussion
The SHS and SHFS prospective cohorts provide a unique opportunity to gain insight into the potential effects of adverse environmental exposures, such as As, on health across generations. Our results sought to expand and potentially improve upon maternal ΣAs measurements for assessing offspring metabolic health by investigating DNAm signatures of ΣAs exposure (Bozack et al., 2020) in mothers as a feasible molecular biosignature of this relationship. Leveraging twenty CpGs previously associated with As exposure in SHS-enrolled mothers – CpGs of which did not associate with metabolic health outcomes within mothers (Bozack et al., 2020) – we observed nominally significant positive relationships between differential methylation for 3 of these CpGs and insulin resistance (i.e., HOMA2-IR) in the offspring in unadjusted and adjusted models that included adjustment for maternal central adiposity. In the model adjusted for offspring central adiposity, we observed the DNAm states of six CpGs were associated with insulin resistance, including two CpGs that overlapped with the unadjusted model, an adjusted model, and the adjusted model that included maternal adiposity. Among these six CpGs, two of them were non-significantly associated with an increased risk of T2D after adjustment for offspring central adiposity. Despite the limited sample size, our results support the utility of ΣAs exposure-associated DNAm signatures in the maternal lineage as a potential epigentic biomarker to assess the long-term metabolic health risk in offspring from mothers exposed to variable levels of ΣAs.
Further examination of the six maternal-derived ΣAs-associated CpG loci associated with adult offspring HOMA2-IR found each to have some biological relevance to T2D pathogenesis. For instance, for the ubiquitous isoenzyme mediating the catalysis of glucose to glucose-6-phosphate as the initiating steps in canonical glycolysis, HK1 harbored the intragenic ΣAs-associated CpG in mothers, cg14595618. Research in diabetic rodent models have demonstrated changes in HK1 gene expression in response to increased glucose concentrations (Henningsen et al., 2003), which may act through epigenetic mechanisms (El-Osta et al., 2008). Hexokinase activity facilitates the activation of the NLRP3 inflammasome (Wolf et al., 2016). It is plausible epigenetic regulation of HK1 may contribute to shifts in inflammatory phenotypes, which have been linked to insulin resistance (Odegaard and Chawla, 2012). Further, differential methylation at a specific CpG locus of HK1 was found to be associated with adverse lung function in children (Everson et al., 2019), suggesting its role in early childhood health.
A member of the carbonic anhydrase family, CA12, contained an intragenic ΣAs-associated CpG, cg03036214. Carbonic anhydrases are ubiquitous zinc metalloenzymes that are involved in the reversible hydration of CO2 to , which is a required substrate for hepatic gluconeogenesis (Ismail, 2018). These metal enzymes are also altered in people with impaired fasting glucose (Biswas and Kumar, 2012). Carbonic anhydrases have also been proposed as potential therapeutic targets for T2D-related microvascular (Weiwei and Hu, 2009) and macrovascular diseases (Torella et al., 2014). Further, carbonic anhydrases have been investigated as viable biomarkers to characterize type 1 diabetes and T2D (Ghosh et al., 2016).
Cg03497652 is situated proximal to exon 9 of ANKS3. Changes in Anks3 levels have been linked to altered cellular and mitochondrial metabolism in mice (Schlimpert et al., 2018). Notably, a previous epigenome-wide association study observed cg03497652 was differentially methylated in those with T2D compared with healthy controls (Walaszczyk et al., 2018), supporting a role for this CpG locus in metabolic disease risk. That cg03497652 was associated with HOMA2-IR and incident T2D in all models, it may be a feasible molecular marker for assessing insulin resistance and T2D risk. Given that although other clinical variables—such as those incorporated as covariates in the models used in this study—strengthened the association between maternal DNAm and offspring metabolic parameters, a strong association was still present in our unadjusted model, suggesting a clinically feasible marker that would bypass the need for other clinical variables for evaluating metabolic disease risk.
Cg07021906 is an intragenic CpG between exons 9 and 10 of SLC7A5, also known as LAT1. An amino acid transporter, LAT1 supplies the key amino acids required for amino acid metabolism. Previous evidence has revealed its expression levels are altered under hyperglycemic and hyperinsulinemic conditions via inhibition of AMP-activated protein kinase, suggesting a role for LAT1 in aberrant energy metabolism in hyperglycemic conditions (Yamamoto et al., 2017). Likewise; LAT1 plays an active role in metabolic processes involved in T-cell differentiation and effector function (Sinclair et al., 2013), facilitating the metabolic demands of T-cells to propagate a pro-inflammatory response; which; if persistent, has been linked to T2D pathogenesis (Jagannathan-Bogdan et al., 2011; Shoelson et al., 2006). On the other hand; in vitro studies using a human placental cell line found LAT1 has a protective role against oxidative stress and inflammation induced by metal toxicity (Granitzer et al., 2021). Taken together; depending on the cellular context, LAT1 may have a fundamental role in environmental toxicant-induced oxidative damage and inflammation.
A core component of the spliceosome, PRPF8, contained a ΣAs-associated CpG within exon 24, cg12116137. Intragenic CpGs—within or proximal to exons—facilitate alternative splicing, increasing the complexity of the transcriptome and subsequent proteome (Shayevitch et al., 2018; Maunakea et al., 2013). Aberrant mRNA splicing—potentially induced by altered DNAm at environmentally responsive loci—may be involved in the expression of atypical isoforms that induce cellular phenotypes contributing to disease pathogenesis (Kim et al., 2018). Indeed, mis-splicing of mRNA caused by dysfunctional PRPF8 isoforms have been linked to chronic diseases, including myeloid neoplasms (Kurtovic-Kozaric et al., 2015) and insulin resistance (Sánchez-Ceinos et al., 2021). In general, aberrant alternative splicing has also been associated with T2D-related macrovascular diseases (Cornelius et al., 2021). Taken together, ΣAs-associated epigenetic alterations to spliceosome factors, such as PRPF8, may have adverse effects on proper splicing of downstream targets, contributing to diabetic phenotypes.
Harbored within the 5′ UTR of LINGO3 is cg08059112. Members of the Lingo family of genes are expressed during early mouse development, and Lingo3 is ubiquitously expressed across embryonic tissues of the developing embryo (Haines and Rigby, 2008). A SNP within a member of the LINGO family, LINGO2 (rs10968576), has previously been associated with an almost two-times higher likelihood of pediatric-onset T2D (Miranda-Lora et al., 2019). These findings suggest a fundamental role for LINGO members in early development, and its aberrant expression in early life, including during pregnancy, may contribute to adverse health of the child later in life.
Regarding the two models that were adjusted for measures of adiposity in mothers (Model 2) or offspring (Model 3), we found offspring adiposity to be more influential than maternal adiposity on the association between maternal DNAm signatures of ΣAs exposure and offspring metabolic health. In the study by Tinkelman et al., the association of maternal ΣAs exposure with offspring metabolic health was attenuated after adjustment for offspring adiposity, supporting the potential role of offspring adiposity as a mediator of the relationship between maternal As and offspring metabolic health (Tinkelman et al., 2020). With DNAm signatures related to ΣAs exposure, we also found a potential influence by offspring adiposity, although in this case the association between DNAm and offspring metabolic factors became actually stronger. While the interpretation of these findings are speculative at this point, it is possible that the influence of offspring adiposity could be related to adiposity reprogramming occurring early in life that may act through differing mechanisms between maternal urinary As and DNAm on offspring metabolic health later in life. Early life adiposity has been linked to insulin resistance and metabolic syndrome into adulthood (Liang et al., 2015). Prior evidence suggests exposure to adverse environmental influences during prenatal and early childhood development may increase adiposity and metabolic disease later in life through epigenetic mediators (Nugent and Bale, 2015). Further, maternal obesity may induce offspring adiposity through epigenetic alterations (Yang et al., 2013). Taken together, it is plausible that ΣAs-induced alterations to the maternal epigenome during pregnancy may influence fetal development via epigenetic programming towards adipogenic phenotypes in offspring that may underlie metabolic disease risk later in life.
The Developmental Origins of Health and Disease (DOHaD) hypothesis posits adverse environmental exposure during developmental windows may affect offspring health later in life, likely by reprogramming fetal phenotypes without altering genotype (Gillman, 2005). Consistent with DOHaD, exposure to As during pregnancy may have lasting effects on offspring health (Young et al., 2018), such as to T2D later in life (Navas-Acien et al., 2019). Although urinary As measurements have a relatively short half-life, previous evidence in the SHS has shown that urinary As measures spanning a decade were relatively constant, consistent with stable levels in drinking water and supporting the use of urinary As as an indicator of long-term exposure in the SHS (Navas-Acien et al., 2009). These findings suggest that SHS-participating mothers were potentially exposed to As during sensitive timeframes, such as pregnancy and early childhood, which may have had an impact on fetal development in offspring, ultimately, contributing to metabolic disease risk later in life. However, capturing As exposure during these sensitive windows is not always feasible. Epigenetic modifications—known to alter phenotype and not genotype—are relatively stable, reflect environmental influences, and may be involved in altered fetal reprogramming in response to exposures during development, having a lasting effect on an offspring’s short and long-term health (Nugent and Bale, 2015). Prenatal exposures have been shown to alter genome-wide DNAm in both maternal and fetal blood (Kile et al., 2012), and the expression of epigenetic regulatory genes in fetal placenta (Winterbottom et al., 2019), including at genes related to T2D (Rojas et al., 2015). Exposure-induced changes to the maternal epigenome may be similarly imparted to offspring via in utero exposure, altering fetal programming. Developing stable epigenetic markers of exposure may prove a clinically viable option to assess As exposure-associated metabolic disease risk of children born to exposed mothers. Because maternal ΣAs levels may represent better indicators of offspring metabolic health than offspring ΣAs (Tinkelman et al., 2020), maternal ΣAs-associated epigenetic biosignatures may show even more promise as an accurate measure for evaluating metabolic disease risk in offspring born to chronically exposed mothers; long before clinical and subclinical disease manifestation, which will prove indispensable for preventative health measures.
Our results should be interpreted with caution due to several limitations. Despite observing a nominal association between maternal DNAm signatures of ΣAs exposure and offspring insulin resistance, all but one of the CpGs remained significant after adjustment for multiple comparisons. Another limitation was the small sample size, given our inclusion criteria, including the modest number of mothers with DNAm data and paired diabetes-free children enrolled in the subsequent SHFS (Lai et al., 2003). Future work should seek to validate these results in larger populations, including additional studies in American Indian communities. Although participants in this study represent geographically distinct American Indian communities throughout the U.S., replicating these results in other diverse cohorts will provide compelling broader generalizability. Whether ΣAs-associated DNAm patterns in blood are representative of target tissues relevant to metabolic disease pathogenesis is unknown. Blood, however, provides a much more accessible resource that circumvents the need for target tissues directly involved in disease pathogenesis and is suitable for a wide range of clinical applications (O’Neill et al., 2016). Although DNAm marks and ΣAs measurements are relatively stable, we could not ascertain the stability of DNAm changes during pregnancy and early life, given SHS and SHFS participants were recruited after those windows. Our study, however, has multiple strengths. We employed robust ΣAs-associated DNAm signatures collected in a modest sample size of American Indian participants and used strict criteria for inclusion in this association study. The prospective nature of our study allowed us to investigate follow-up disease outcomes, such as incident T2D, in offspring. This is the first study that has employed molecular patterns as a proxy of ΣAs exposure in mothers to identify T2D and related metabolic risk factors in adult offspring.
5. Conclusions
In this study, we found that ΣAs-associated DNA methylation signatures in the maternal lineage were associated with insulin resistance, as measured by HOMA2-IR, and non-significantly associated with type 2 diabetes mellitus in adult offspring from AI communities across the Southwest and the Great Plains. Our results suggest offspring adiposity moderates the relationship between maternal DNA methylation signatures and adult offspring diabetes-related outcomes. These results support the utility of epigenetic biomarkers for assessing metabolic disease risk of offspring to mothers who may have been exposed to As, especially during labile windows of development, and provide a molecular signature for disease risk and prevention. Future studies should confirm these findings, validate in other diverse cohorts, and elucidate the mechanisms by which epigenetic phenomena may be inherited or contribute to long-term health trajectories in offspring exposed to As during sensitive windows of development.
Supplementary Material
Acknowledgements
The authors would like to thank the Strong Heart Study and Strong Heart Family Study participants and staff, without your help, none of this work would have been made possible. This study was supported by the National Institute of Environmental Health Sciences grant: 5T32ES007322 (CKD). The Strong Heart Study has been funded in whole or in part with federal funds from the National Heart, Lung, and Blood Institute, National Institute of Health, Department of Health and Human Services, under contract numbers 75N92019D00027, 75N92019D00028, 75N92019D00029, & 75N92019D00030. The study was previously supported by grants: R01HL109315, R01HL109301, R01HL109284, R01HL109282, R01HL109319 and R01HL090863, by cooperative agreements: U01HL41642, U01HL41652, U01HL41654, U01HL65520, and U01HL65521 and by grants from the National Institute of Environmental Health Sciences: P42ES033719, R01ES032638, P30ES009089, and past grant R01ES021367.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Christian K. Dye: Conceptualization, Writing – review & editing. Arce Domingo-Relloso: . Allison Kupsco: . Naomi E. Tinkelman: . Miranda J. Spratlen: . Anne K. Bozack: . Maria Tellez-Plaza: . Walter Goessler: . Karin Haack: . Jason G. Umans: Conceptualization, Methodology, Investigation, Supervision. Andrea A. Baccarelli: Investigation, Supervision. Shelley A. Cole: Conceptualization, Methodology, Investigation, Supervision. Ana Navas-Acien: Conceptualization, Methodology, Formal analysis, Writing – original draft, Writing – review & editing, Supervision, Funding acquisition.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.envint.2023.107774.
Data availability
The authors do not have permission to share data.
References
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, 2014. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30 (10), 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas UK, Kumar A, 2012. Study on the changes of carbonic anhydrase activity in insulin resistance and the effect of methylglyoxal. J. Pak. Med. Assoc. 62, 417–421. [PubMed] [Google Scholar]
- Bozack AK, Domingo-Relloso A, Haack K, Gamble MV, Tellez-Plaza M, Umans JG, Best LG, Yracheta J, Gribble MO, Cardenas A, Francesconi KA, Goessler W, Tang W-Y, Fallin MD, Cole SA, Navas-Acien A, 2020. Locus-Specific Differential DNA Methylation and Urinary Arsenic: An Epigenome-Wide Association Study in Blood among Adults with Low-to-Moderate Arsenic Exposure. Environ. Health Perspect. 128 (6), 067015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JY, Yu SD, Hong YS, 2014. Environmental source of arsenic exposure. J. Prev. Med. Public Health 47, 253–257. 10.3961/jpmph.14.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius VA, Fulton JR, Margariti A, 2021. Alternative Splicing: A Key Mediator of Diabetic Vasculopathy. Genes (Basel) 12 (9), 1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM, 2010. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 11 (1) 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M, 2008. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 205 (10), 2409–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson TM, Zhang H, Lockett GA, Kaushal A, Forthofer M, Ewart SL, Burrows K, Relton CL, Sharp GC, Henderson AJ, Patil VK, Rezwan FI, Arshad SH, Holloway JW, Karmaus W, 2019. Epigenome-wide association study of asthma and wheeze characterizes loci within HK1. Allergy Asthma Clin. Immunol. 15 (1) 10.1186/s13223-019-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Giménez JL, Seco-Cervera M, Tollefsbol TO, Romá-Mateo C, Peiró-Chova L, Lapunzina P, Pallardó FV, 2017. Epigenetic biomarkers: Current strategies and future challenges for their use in the clinical laboratory. Crit. Rev. Clin. Lab. Sci. 54 (7–8), 529–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh C, Mandal S, Banik GD, Maity A, Mukhopadhyay P, Ghosh S, Pradhan M, 2016. Targeting erythrocyte carbonic anhydrase and (18)O-isotope of breath CO2 for sorting out type 1 and type 2 diabetes. Sci. Rep. 6 (1) 10.1038/srep35836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman MW, 2005. Developmental origins of health and disease. N. Engl. J. Med. 353, 1848–1850. 10.1056/NEJMe058187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey KM, Costello PM, Lillycrop KA, 2015. The developmental environment, epigenetic biomarkers and long-term health. J. Dev. Orig. Health Dis. 6, 399–406. 10.1017/S204017441500121X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granitzer S, Widhalm R, Forsthuber M, Ellinger I, Desoye G, Hengstschläger M, Zeisler H, Salzer H, Gundacker C, 2021. Amino Acid Transporter LAT1 (SLC7A5) Mediates MeHg-Induced Oxidative Stress Defense in the Human Placental Cell Line HTR-8/SVneo. Int. J. Mol. Sci. 22 (4), 1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau-Perez M, Kuo C-C, Gribble MO, Balakrishnan P, Jones Spratlen M, Vaidya D, Francesconi KA, Goessler W, Guallar E, Silbergeld EK, Umans JG, Best LG, Lee ET, Howard BV, Cole SA, Navas-Acien A, 2017. Association of Low-Moderate Arsenic Exposure and Arsenic Metabolism with Incident Diabetes and Insulin Resistance in the Strong Heart Family Study. Environ. Health Perspect. 125 (12), 127004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines BP, Rigby PW, 2008. Expression of the Lingo/LERN gene family during mouse embryogenesis. Gene Expr. Patterns 8, 79–86. 10.1016/j.modgep.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Henningsen C, Zahner G, Thaiss F, 2003. High glucose induces type 1 hexokinase gene expression in isolated glomeruli of diabetic rats and in mesangial cells. Nephron Physiol. 93, 67–75. 10.1159/000069555. [DOI] [PubMed] [Google Scholar]
- Homish GG, Edwards EP, Eiden RD, Leonard KE, 2010. Analyzing family data: A GEE approach for substance use researchers. Addict. Behav. 35, 558–563. 10.1016/j.addbeh.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YS, Song KH, Chung JY, 2014. Health effects of chronic arsenic exposure. J. Prev. Med. Public Health 47, 245–252. 10.3961/jpmph.14.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT, 2012. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 13 (1) 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard BV, Lee ET, Cowan LD, Fabsitz RR, Howard WJ, Oopik AJ, Robbins DC, Savage PJ, Yeh JL, Welty TK, 1995. Coronary heart disease prevalence and its relation to risk factors in American Indians. The Strong Heart Study. Am. J. Epidemiol. 142 (3), 254–268. [DOI] [PubMed] [Google Scholar]
- Huang MC, Douillet C, Dover EN, Styblo M, 2018. Prenatal arsenic exposure and dietary folate and methylcobalamin supplementation alter the metabolic phenotype of C57BL/6J mice in a sex-specific manner. Arch. Toxicol. 92, 1925–1937. 10.1007/s00204-018-2206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail IS, 2018. The Role of Carbonic Anhydrase in Hepatic Glucose Production. Curr. Diabetes Rev. 14, 108–112. 10.2174/1573399812666161214122351. [DOI] [PubMed] [Google Scholar]
- Jagannathan-Bogdan M, et al. , 2011. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J. Immunol. 186, 1162–1172. 10.4049/jimmunol.1002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirkhah Rahimabad P, Arshad SH, Holloway JW, Mukherjee N, Hedman A, Gruzieva O, Andolf E, Kere J, Pershagen G, Almqvist C, Jiang Y.u., Chen S.u., Karmaus W, 2021. Association of Maternal DNA Methylation and Offspring Birthweight. Reprod. Sci. 28 (1), 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile ML, Baccarelli A, Hoffman E, Tarantini L, Quamruzzaman Q, Rahman M, Mahiuddin G, Mostofa G, Hsueh Y-M, Wright RO, Christiani DC, 2012. Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord blood leukocytes. Environ. Health Perspect. 120 (7), 1061–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Pham MHC, Ko KS, Rhee BD, Han J, 2018. Alternative splicing isoforms in health and disease. Pflugers Arch. 470, 995–1016. 10.1007/s00424-018-2136-x. [DOI] [PubMed] [Google Scholar]
- Kuo C-C, Howard BV, Umans JG, Gribble MO, Best LG, Francesconi KA, Goessler W, Lee E, Guallar E, Navas-Acien A, 2015. Arsenic Exposure, Arsenic Metabolism, and Incident Diabetes in the Strong Heart Study. Diabetes Care 38 (4), 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, Nakashima M, Hsi ED, Yoshida K, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Ogawa S, Boultwood J, Makishima H, Maciejewski JP, Padgett RA, 2015. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia 29 (1), 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, King TM, Moye LA, Wei Q, 2003. Sample size for biomarker studies: more subjects or more measurements per subject? Ann. Epidemiol. 13, 204–208. 10.1016/s1047-2797(02)00261-2. [DOI] [PubMed] [Google Scholar]
- Lee ET, et al. , 1990. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. Am. J. Epidemiol, 132, 1141–1155, 10.1093/oxfordjournals.aje.a115757. [DOI] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD, 2012. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JC, Matthews DR, Hermans MP, 1998. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21, 2191–2192. 10.2337/diacare.21.12.2191. [DOI] [PubMed] [Google Scholar]
- Liang Y, Hou D, Zhao X, Wang L, Hu Y, Liu J, Cheng H, Yang P, Shan X, Yan Y, Cruickshank JK, Mi J, 2015. Childhood obesity affects adult metabolic syndrome and diabetes. Endocrine 50 (1), 87–92. [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Cui K, Zhao K, 2013. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Lora AL, Molina-Díaz M, Cruz M, Sánchez-Urbina R, Martínez-Rodríguez NL, López-Martínez B, Klünder-Klünder M, 2019. Genetic polymorphisms associated with pediatric-onset type 2 diabetes: A family-based transmission disequilibrium test and case-control study. Pediatr. Diabetes 20 (3), 239–245. [DOI] [PubMed] [Google Scholar]
- Navas-Acien A, Umans JG, Howard BV, Goessler W, Francesconi KA, Crainiceanu CM, Silbergeld EK, Guallar E, 2009. Urine arsenic concentrations and species excretion patterns in American Indian communities over a 10-year period: the Strong Heart Study. Environ. Health Perspect. 117 (9), 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Acien A, Spratlen MJ, Abuawad A, LoIacono NJ, Bozack AK, Gamble MV, 2019. Early-Life Arsenic Exposure, Nutritional Status, and Adult Diabetes Risk. Curr. Diab. Rep. 19 (12) 10.1007/s11892-019-1272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent BM, Bale TL, 2015. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front. Neuroendocrinol. 39, 28–37. 10.1016/j.yfrne.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A, 2012. Connecting type 1 and type 2 diabetes through innate immunity. Cold Spring Harb. Perspect. Med. 2 (3), a007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill S, Bohl M, Gregersen S, Hermansen K, O’Driscoll L, 2016. Blood-Based Biomarkers for Metabolic Syndrome. Trends Endocrinol. Metab. 27, 363–374. 10.1016/j.tem.2016.03.012. [DOI] [PubMed] [Google Scholar]
- Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, Van Djik S, Muhlhausler B, Stirzaker C, Clark SJ, 2016. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17 (1) 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punshon T, Davis MA, Marsit CJ, Theiler SK, Baker ER, Jackson BP, Conway DC, Karagas MR, 2015. Placental arsenic concentrations in relation to both maternal and infant biomarkers of exposure in a US cohort. J. Eposure Sci. Environ. Epidemiol. 25 (6), 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D.i., Hu Y, Law CW, Shi W, Smyth GK, 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez KF, Ungewitter EK, Crespo-Mejias Y, Liu C, Nicol B, Kissling GE, Yao H-C, 2016. Effects of in Utero Exposure to Arsenic during the Second Half of Gestation on Reproductive End Points and Metabolic Parameters in Female CD-1 Mice. Environ. Health Perspect. 124 (3), 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez KF, Mellouk N, Ungewitter EK, Nicol B, Liu C, Brown PR, Willson CJ, Yao H-C, 2020. In utero exposure to arsenite contributes to metabolic and reproductive dysfunction in male offspring of CD-1 mice. Reprod. Toxicol. 95, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas D, Rager JE, Smeester L, Bailey KA, Drobná Z, Rubio-Andrade M, Stýblo M, García-Vargas G, Fry RC, 2015. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol. Sci. 143 (1), 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Ceinos J, et al. , 2021. Impaired mRNA splicing and proteostasis in preadipocytes in obesity-related metabolic disease. Elife 10, 10.7554/eLife.65996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer J, Findenig S, Goessler W, Francesconi KA, Howard B, Umans JG, Pollak J, Tellez-Plaza M, Silbergeld EK, Guallar E, Navas-Acien A, 2012. Arsenic species and selected metals in human urine: validation of HPLC/ICPMS and ICPMS procedures for a long-term population-based epidemiological study. Anal. Methods 4 (2), 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlimpert M, Lagies S, Budnyk V, Müller B, Walz G, Kammerer B, 2018. Metabolic Phenotyping of Anks3 Depletion in mIMCD-3 cells - a Putative Nephronophthisis Candidate. Sci. Rep. 8 (1) 10.1038/s41598-018-27389-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayevitch R, Askayo D, Keydar I, Ast G, 2018. The importance of DNA methylation of exons on alternative splicing. RNA 24, 1351–1362. 10.1261/rna.064865.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB, 2006. Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801. 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, Cantrell DA, 2013. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14 (5), 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratlen MJ, Grau-Perez M, Umans JG, Yracheta J, Best LG, Francesconi K, Goessler W, Balakrishnan P, Cole SA, Gamble MV, Howard BV, Navas-Acien A, 2018. Arsenic, one carbon metabolism and diabetes-related outcomes in the Strong Heart Family Study. Environ. Int. 121, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratlen MJ, Grau-Perez M, Best LG, Yracheta J, Lazo M, Vaidya D, Balakrishnan P, Gamble MV, Francesconi KA, Goessler W, Cole SA, Umans JG, Howard BV, Navas-Acien A, 2018. The Association of Arsenic Exposure and Arsenic Metabolism With the Metabolic Syndrome and Its Individual Components: Prospective Evidence From the Strong Heart Family Study. Am. J. Epidemiol. 187 (8), 1598–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkelman NE, Spratlen MJ, Domingo-Relloso A, Tellez-Plaza M, Grau-Perez M, Francesconi KA, Goessler W, Howard BV, MacCluer J, North KE, Umans JG, Factor-Litvak P, Cole SA, Navas-Acien A, 2020. Associations of maternal arsenic exposure with adult fasting glucose and insulin resistance in the Strong Heart Study and Strong Heart Family Study. Environ. Int. 137, 105531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torella D, Ellison GM, Torella M, Vicinanza C, Aquila I, Iaconetti C, Scalise M, Marino F, Henning BJ, Lewis FC, Gareri C, Lascar N, Cuda G, Salvatore T, Nappi G, Indolfi C, Torella R, Cozzolino D, Sasso FC, 2014. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 3 (2), e000434. 10.1161/JAHA.113.000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walaszczyk E, Luijten M, Spijkerman AMW, Bonder MJ, Lutgers HL, Snieder H, Wolffenbuttel BHR, van Vliet-Ostaptchouk JV, 2018. DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: a systematic review and replication in a case-control sample of the Lifelines study. Diabetologia 61 (2), 354–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwei Z, Hu R, 2009. Targeting carbonic anhydrase to treat diabetic retinopathy: emerging evidences and encouraging results. Biochem. Biophys. Res. Commun. 390, 368–371. 10.1016/j.bbrc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- Winchester P, Nilsson E, Beck D, Skinner MK, 2022. Preterm birth buccal cell epigenetic biomarkers to facilitate preventative medicine. Sci. Rep. 12, 3361. 10.1038/s41598-022-07262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbottom EF, Moroishi Y, Halchenko Y, Armstrong DA, Beach PJ, Nguyen QP, Capobianco AJ, Ayad NG, Marsit CJ, Li Z, Karagas MR, Robbins DJ, 2019. Prenatal arsenic exposure alters the placental expression of multiple epigenetic regulators in a sex-dependent manner. Environ. Health 18 (1). 10.1186/s12940-019-0455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, Underhill DM, 2016. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 166 (3), 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Sawa R, Wake I, Morimoto A, Okimura Y, 2017. Glucose-mediated inactivation of AMP-activated protein kinase reduces the levels of L-type amino acid transporter 1 mRNA in C2C12 cells. Nutr. Res. 47, 13–20. 10.1016/j.nutres.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Yang Q-Y, Liang J-F, Rogers CJ, Zhao J-X, Zhu M-J, Du M, 2013. Maternal obesity induces epigenetic modifications to facilitate Zfp423 expression and enhance adipogenic differentiation in fetal mice. Diabetes 62 (11), 3727–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JL, Cai L, States JC, 2018. Impact of prenatal arsenic exposure on chronic adult diseases. Syst. Biol. Reprod. Med. 64, 469–483. 10.1080/19396368.2018.1480076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors do not have permission to share data.