

PURPOSE
Although the majority of patients with relapsed or refractory large B-cell lymphoma respond to axicabtagene ciloleucel (axi-cel), only a minority of patients have durable remissions. This prospective multicenter study explored the prognostic value of circulating tumor DNA (ctDNA) before and after standard-of-care axi-cel for predicting patient outcomes.
METHODS
Lymphoma-specific variable, diversity, and joining gene segments (VDJ) clonotype ctDNA sequences were frequently monitored via next-generation sequencing from the time of starting lymphodepleting chemotherapy until progression or 1 year after axi-cel infusion. We assessed the prognostic value of ctDNA to predict outcomes and axi-cel–related toxicity.
RESULTS
A tumor clonotype was successfully detected in 69 of 72 (96%) enrolled patients. Higher pretreatment ctDNA concentrations were associated with progression after axi-cel infusion and developing cytokine release syndrome and/or immune effector cell–associated neurotoxicity syndrome. Twenty-three of 33 (70%) durably responding patients versus 4 of 31 (13%) progressing patients demonstrated nondetectable ctDNA 1 week after axi-cel infusion (P < .0001). At day 28, patients with detectable ctDNA compared with those with undetectable ctDNA had a median progression-free survival and OS of 3 months versus not reached (P < .0001) and 19 months versus not reached (P = .0080), respectively. In patients with a radiographic partial response or stable disease on day 28, 1 of 10 patients with concurrently undetectable ctDNA relapsed; by contrast, 15 of 17 patients with concurrently detectable ctDNA relapsed (P = .0001). ctDNA was detected at or before radiographic relapse in 29 of 30 (94%) patients. All durably responding patients had undetectable ctDNA at or before 3 months after axi-cel infusion.
CONCLUSION
Noninvasive ctDNA assessments can risk stratify and predict outcomes of patients undergoing axi-cel for the treatment of large B-cell lymphoma. These results provide a rationale for designing ctDNA-based risk-adaptive chimeric antigen receptor T-cell clinical trials.
INTRODUCTION
Axicabtagene ciloleucel (axi-cel, KTE-C19, Yescarta) is approved for the treatment of chemotherapy-refractory large B-cell lymphoma (LBCL). Despite an overall response rate of 83% and a complete response (CR) rate of 58%, many patients experience progression resulting in a 2-year progression-free survival (PFS) of approximately 40%.1 Serial imaging with positron emission tomography and computed tomography is the current standard of care to assess response and to detect early asymptomatic relapse after chimeric antigen receptor (CAR) T-cell therapy. However, the clinical utility of positron emission tomography and computed tomography early after CAR T-cell therapy is limited in approximately 40% of patients who achieve stable disease (SD) or partial response (PR) at 1 month after therapy.2 Of these patients, approximately 40% will deepen into a CR over time, whereas the remainder progress.1,3 Clinically validated tools that could identify the patient's trajectory earlier could lead to the development of earlier interventions, which might further improve outcomes.
CONTEXT
Key Objective
This prospective multicenter study seeks to determine the utility of circulating tumor DNA (ctDNA) monitoring to risk stratify and assess treatment response for patients with large B-cell lymphoma undergoing chimeric antigen receptor (CAR) T-cell therapy.
Knowledge Generated
We find that ctDNA identifies patients with large B-cell lymphoma at risk for disease relapse after treatment with axicabtagene ciloleucel and at risk of developing higher-grade cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome. Moreover, ctDNA predicts disease progression with added value to standard positron emission tomography and computed tomography scans.
Relevance
ctDNA appears to predict progression after CAR-T therapy and should be incorporated as an integral biomarker in future trials of consolidation therapy after CAR-T to prevent relapse.
Circulating tumor DNA (ctDNA) is an emerging biomarker to risk stratify and assess treatment response to chemotherapy for patients with LBCL and other malignancies.4-10 B-cell malignancies have a unique marker of clonality as a result of variable, diversity, and joining gene segments (VDJ) recombination in the immunoglobulin genes. These unique tumor-associated VDJ clonotype sequences can be noninvasively detected from the cell-free DNA in the plasma of patients with LBCL with high sensitivity using next-generation sequence–based assays.11 In our pilot study of six patients enrolled on the pivotal ZUMA-1, ctDNA dynamics after axi-cel therapy were analyzed.8 The molecular response as determined by ctDNA mirrored the patient's clinical course, and similar results have been reported.12 Durably responding patients rapidly developed and sustained undetectable ctDNA, whereas those patients who eventually relapsed continued to have persistently detectable ctDNA. On the basis of these results, a prospective multi-institutional study was designed to evaluate ctDNA-based assessments before and after axi-cel infusion as a tool for risk stratification and efficacy monitoring in relapsed or refractory LBCL.
METHODS
Patient Eligibility
Eligible patients were ≥ 18 years old; diagnosed with diffuse LBCL, transformed follicular lymphoma, or primary mediastinal B-cell lymphoma; enrolled before receiving lymphodepleting chemotherapy for standard-of-care axi-cel; had archival formalin-fixed paraffin-embedded (FFPE) tissue available for clonotype identification; and had measurable PET-avid disease. Patients unable to reach day 28 because of death, progression, or lost to follow-up were excluded from the primary analysis, as prespecified by the study protocol. The key end points were to determine (1) the ability of ctDNA-based assessments at the time of starting lymphodepletion (pre-LD) to predict PFS of patients with LBCL undergoing axi-cel infusion with at least a 6-month follow-up and (2) the association of ctDNA-based minimal residual disease (MRD) assessments on day 28 to predict PFS of patients with LBCL undergoing axi-cel therapy. A PFS event was defined as disease relapse or death. A progressor was defined as a participant who experienced a PFS event. A durable responder was defined as a participant who did not experience a PFS event with at least a 6-month follow-up.
Study Assessments
Clonotypes were identified via polymerase chain reaction amplification of IgH-VDJ, IgH-DJ, and Igkappa or lambda regions using universal consensus primers from archival FFPE samples or from the initial plasma sample. A clonotype was considered acceptable for tracking using a previously validated method.13 All identified clonotypes were tracked with the highest concentration clonotype used for analysis. CFD tubes were determined to be the optimal tube for analyte stability (Data Supplement online only). Approximately 8.5 mL of blood was collected in a CFD tube (Roche Diagnostics, Indianapolis, IN) before lymphodepletion and 0, 7, 14, 21, 28, 56, 90, 180, and 365 days after axi-cel infusion. Samples were sent overnight at ambient temperature to Adaptive Biotechnologies (Seattle, WA) for processing. Plasma was isolated within 5 days of collection. ctDNA levels were subsequently determined from isolated plasma via next-generation sequence as described.13 Clinicians did not have these data available at the time of clinical decision making. PET-CT scans were obtained preapheresis and 1, 3, 6, and 12 months after axi-cel infusion. Responses were assessed by Lugano criteria.2 Cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) were graded as previously described.14,15
Patients were enrolled at Stanford University, Moffitt Cancer Center, and University of Maryland Medical Center. The study was approved by each institution's Institutional Review Board, and written informed consent was obtained from all patients before sample collection per the Declaration of Helsinki.
Statistical Analysis
PFS and overall survival (OS) were determined by Kaplan-Meier analysis and significance by log-rank test. Follow-up was calculated from the time of axi-cel infusion until death or last time of contact. The comparison between two groups was conducted using a two-tailed Mann-Whitney test. Significances were calculated using a Fisher exact test between two categorical variables or as otherwise specified. The area under the receiver operating characteristic curve, 95% CI, and P values were determined using Wilson-Brown method.16 Statistical analysis and plots were generated using Prism 8.4.1 (GraphPad) and SPSS Statistics 21 (IBM).
RESULTS
Patient Characteristics
Between January 2018 and May 2019, 72 eligible patients undergoing standard-of-care axi-cel were enrolled in the study. The patient characteristics and overall response rate (84.7%), CR rate (63.9%), median PFS (10.3 months), duration of response (not reached [NR], 79.5% at 1 year), and median OS (NR, 71.2% at 1 year) for these patients were similar to those enrolled in ZUMA-1 (Table 1, Data Supplement). The rates of CRS and ICANS were also similar to those of ZUMA-1. Any grade versus grade 3 or higher of CRS and ICANS occurred in 91.7% versus 5.6% and 55.6% versus 27.7% of patients, respectively. The median age was 62 years (range, 19-79 years), 60% were male, and the median lines of therapy before axi-cel infusion was 3 (range, 1-7). Sixty-four patients were included in the primary analysis after eight patients were excluded from the primary analysis, as prespecified by the study protocol (CONSORT diagram, Data Supplement). The patient characteristics of the enrolled patients and primary analysis were similar. After a median follow-up of 10.9 months, 31 patients had PFS events (progressors)—30 because of disease relapse and 1 because of treatment-related death (Pneumocystis jirovecii pneumonia at 4.5 months after axi-cel infusion). The characteristics of durable responders and progressors are shown in Table 1. Younger patients and those with elevated pre-LD lactate dehydrogenase (LDH) were significantly more likely to progress following axi-cel treatment. These patients are included in previously published data.3
TABLE 1.
Patient Demographics

Baseline ctDNA and ctDNA Dynamics After Treatment
Sixty-nine (96%) of the 72 enrolled patients had adequate DNA to permit tumor clonotype tracking. The tumor clonotype(s) were identified from FFPE samples for 65 patients (93%), whereas the remaining 4 (7%) patients had their tumor clonotype(s) identified from the initial plasma sample when the archival FFPE sample could not identify a clonotype. Additional details regarding these clonotypes are available (Data Supplement).
We evaluated whether pre-LD ctDNA concentration would correlate with the international prognostic index and with patient characteristics reflecting tumor burden. Patients with an international prognostic index between 0 and 2 versus three or higher had a median ctDNA concentration of 14 (range 0-3,200) lymphoma genomes per mL of plasma (LG/mL) versus 187 (0-17,903) LG/mL (P = .0143). Patients with stage I or II versus stage III or IV disease had a ctDNA concentration of 5 (0-3,200) LG/mL versus 133 (0-17,903) LG/mL (P = .0196). Patients with normal versus elevated LDH had a median ctDNA concentration of 7 (0-13,270) LG/mL versus 529 (1-17,903) LG/mL (P < .0001). Additionally, patients who did not versus did receive bridging therapies had a higher median pre-LD ctDNA concentration of 14 (0-2,032) LG/mL versus 187 (0-17,903) LG/mL and P = .0310. There was a significant correlation between pre-LD LDH and ctDNA (Spearman correlation 0.6855, P < .0001). However, high pre-LD ctDNA was a significant and independent predictor from elevated pre-LD LDH via a likelihood ratio chi-square test of Cox proportional hazard analyses (Data Supplement).
ctDNA was monitored until progression, death, or 1 year after therapy. The ctDNA dynamics over the first 90 days for the 33 durably responding patients and 31 progressing patients, including representative examples of each, are shown in Figure 1. Durably responding patients had lower pre-LD ctDNA compared with progressing patients, 8 (0-1,327) LG/mL versus 581 (0-17,903) LG/mL, P < .0001. By day 7, 70% of the durably responding patients (23 of 33) had no detectable ctDNA, and by day 90, all durably responding patients had undetectable ctDNA thereafter. By contrast, progressing patients had a nadir median ctDNA concentration of 6 (0-2,945) LG/mL on day 28, which started to rise thereafter. Most progressing patients demonstrated a reduction in ctDNA concentration. Twenty-four (77%) patients demonstrated at least 1-log reduction and 16 (52%) achieved a 2-log reduction in ctDNA concentration at the nadir compared with baseline. Moreover, 12 (39%) progressing patients had at least 1 undetectable ctDNA sample between day 7 and day 21. Interestingly, 12 patients—nine durably responding and three progressing patients—had no detectable pre-LD ctDNA despite having PET-avid disease at the time of enrollment. In all three progressing patients, ctDNA eventually became detectable (on day 0, 7, and 90, respectively), whereas ctDNA remained undetectable at all time points in the durably responding patients. None of these 12 patients started with elevated LDH, and five patients received bridging chemotherapy.
FIG 1.
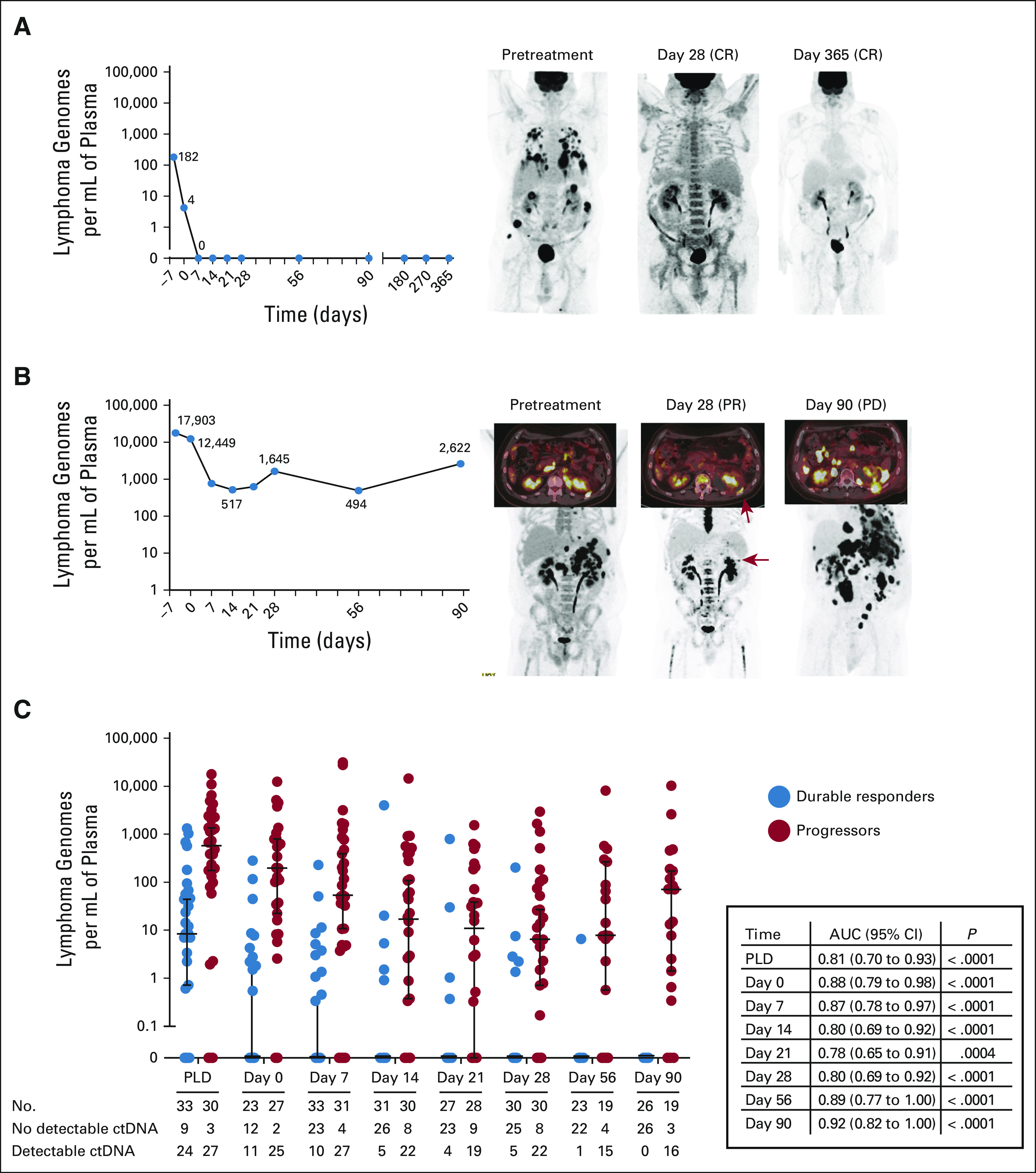
ctDNA dynamics during axi-cel therapy. The patient shown in (A) demonstrated a durable CR and no detectable ctDNA starting from day 7 after axi-cel infusion. By contrast, the patient in (B) demonstrated a PR as shown by PET-CT on day 28 after axi-cel infusion, followed by PD as shown on the PET-CT scan on day 90. The patient has persistently detectable ctDNA after axi-cel infusion. (C) The concentration of ctDNA of durably responding (blue) and progressing (red) patients is shown at each time point starting from PLD until day 90. The median concentration of ctDNA (black horizontal bar) and 95% CI are shown. The number of samples available and whether there is detectable or no detectable ctDNA are shown below. The AUC with the 95% CI and P value is shown for each time point. The four patients who relapsed on day 28 were excluded at the day 56 and day 90 time points. AUC, area under the receiver operating characteristic curve; axi-cel, axicabtagene ciloleucel; CR, complete response; ctDNA, circulating tumor DNA; PD, progressive disease; PET-CT, positron emission tomography-computed tomography; PLD, prelymphodepletion; PR, partial response.
We evaluated the pre-LD ctDNA concentration as a risk factor for CAR T-cell relapse or progression. Patients with the ctDNA concentration below 10 LG/mL or between 10 and 100 LG/mL demonstrated a similar 1-year PFS of 78% and 77% and a similar 1-year OS of 90% and 91%, respectively (Fig 2). These patients had a significantly better PFS and OS compared with patients with a ctDNA above 100 LG/mL. Patients with both a pre-LD ctDNA concentration between 100 and 1,000 LG/mL and above 1,000 LG/mL had a median PFS of 3 months, whereas the median OS was 19 months and 7.4 months, respectively.
FIG 2.
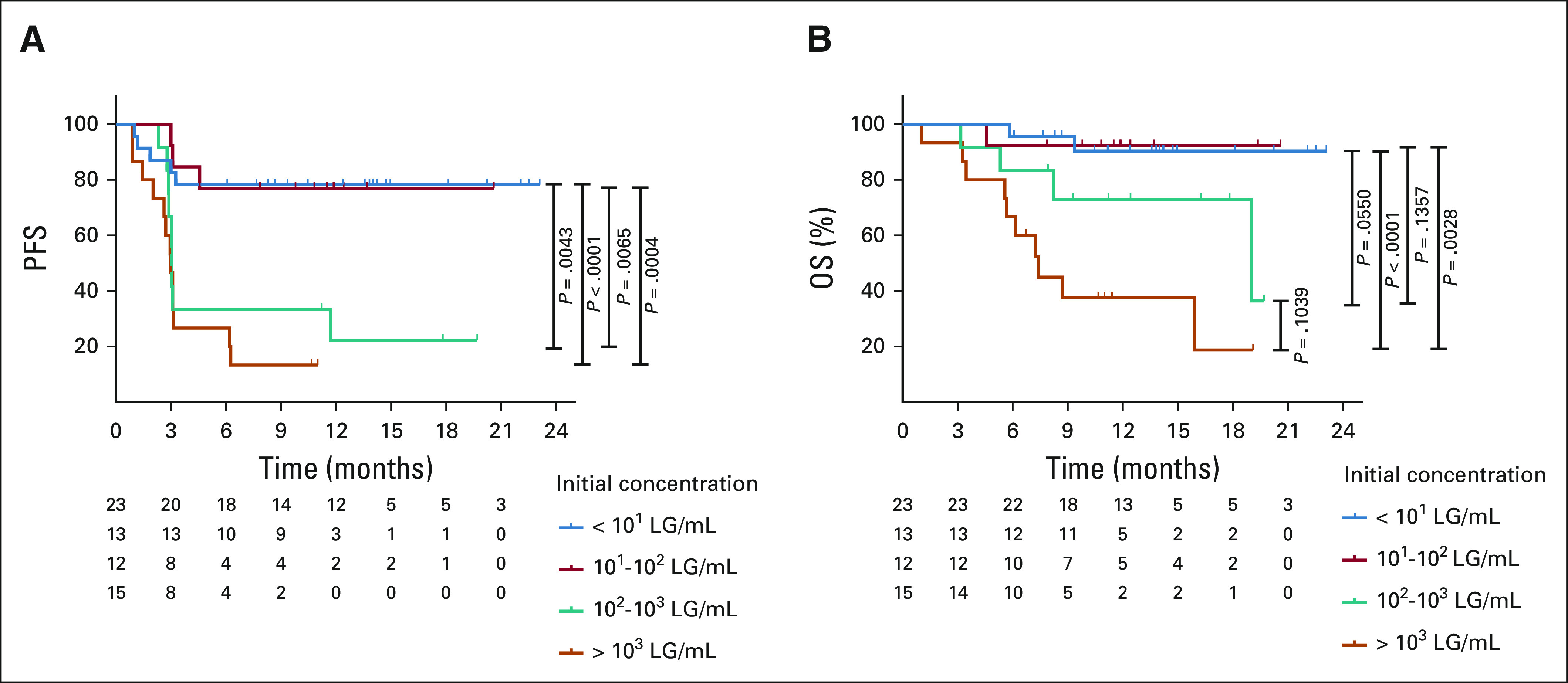
Clinical outcomes by initial ctDNA concentration. The (A) PFS and (B) OS are shown by initial ctDNA concentration from below 10 (blue), between 10 and 100 (red), between 100 and 1,000 (green), and more than 1,000 (orange) LG/mL. ctDNA, circulating tumor DNA; LG/mL, lymphoma genomes per mL of plasma; OS, overall survival; PFS, progression-free survival.
We evaluated pre-LD ctDNA levels as a risk factor for developing CRS and ICANS. Patients who developed no CRS (n = 3) or grade 1 CRS (n = 17) versus grade 2 (n = 39) or grade 3 CRS (n = 1) had a significantly lower median ctDNA concentration of 15 LG/mL (0-6,543) versus 139 LG/mL (0-17,903), P = .0485. Patients who developed no ICANS (n = 27) or grade 1 ICANS (n = 9) versus grade 2 (n = 9), grade 3 (n = 12), or grade 4 (n = 4) had a significantly lower median ctDNA concentration of 38 LG/ml (0-6,543) versus 214 LG/ml (0-17,903), P = .0340.
Circulating Tumor DNA Surveillance After CAR T Therapy
ctDNA was monitored for MRD in concordance with radiographic and clinical staging starting on day 28. A patient was considered MRD-positive (MRD+) if any ctDNA was detected. Sixty patients had an available sample for analysis on day 28, which included 30 durably responding patients and 30 progressing patients. 25 of 30 (83%) durably responding patients were MRD-negative (MRD−; Fig 1). By contrast, 22 of 30 (73%) progressing patients were MRD+ (Figs 1 and 3).
FIG 3.

PET-CT and MRD status in progressing patients. This swimmer plot shows the MRD status and the clinical response on the basis of PET-CT imaging in the 31 patients who had a PFS event. The MRD status, positive (brown), negative (green), and unknown (purple), is shown in addition to the clinical status of PD (purple diamond), SD (open circle), PR (open diamond), and CR (purple circle). aOne patient died in remission because of treatment-related Pneumocystis jirovecii pneumonia; this death is considered a PFS event. axi-cel, axicabtagene ciloleucel; CR, complete response; MRD, minimal residual disease; PD, progressive disease; PET-CT, positron emission tomography-computed tomography; PFS, progression-free survival; PR, partial response; SD, stable disease.
All 30 patients with progression had at least one MRD+ sample before relapse (Fig 3). However, 1 patient was MRD+ at day 28, became MRD− at day 56, and remained MRD− at day 90 concurrently with relapse. In this case, the patient was considered to have progressed when the fluorodeoxyglucose (FDG) avidity of a left gluteal muscle lesion increased from a standard uptake valuemax of 3.0 at day 28 to a standard uptake valuemax of 5.3 at day 90 (no change in the tumor size occurred). This result prompted the local oncologist to begin treatment with pembrolizumab and rituximab. No biopsy was performed to confirm disease relapse; biopsies confirmed relapses in 20 of 30 patients with progression. At the time of data cutoff, this patient has not progressed after receiving this subsequent palliative treatment. The treating physicians were not aware of the contemporary ctDNA status at the time of clinical decision making for any patient during this study.
We compared MRD status on day 28 with PET-CT–based response since PET-CT imaging is frequently obtained on day 28 after axi-cel infusion in routine clinical practice.1,3 All four patients with progressive disease on day 28 were MRD+ concurrently and had a median OS of 3.2 months. After excluding these patients, we found a significant difference in median PFS and OS for those with MRD+ versus MRD− disease or for those with FDG-avid disease (PR or SD) compared with those with a CR (Fig 4). Patients who were MRD+ on day 28 versus MRD− had a median PFS of 3.03 months versus NR (P < .0001) and a median OS of 19.0 months versus NR (P = .0080). MRD+ versus MRD− patients on day 21 (including those who relapsed on day 28) or on day 56 (excluding day 28 relapses) also had inferior outcomes (Data Supplement). The PFS and OS on the basis of PET-CT assessment on day 28 are also shown in Figure 4. Patients who had a PR or SD on day 28 compared with those who achieved a CR on day 28 had a median PFS of 3.1 months versus NR (P = .0018) and a median OS of 19.0 months versus NR (P = .0033). The PFS and OS of patients who achieved a CR on day 28 were similar to MRD− patients (Fig 4).
FIG 4.
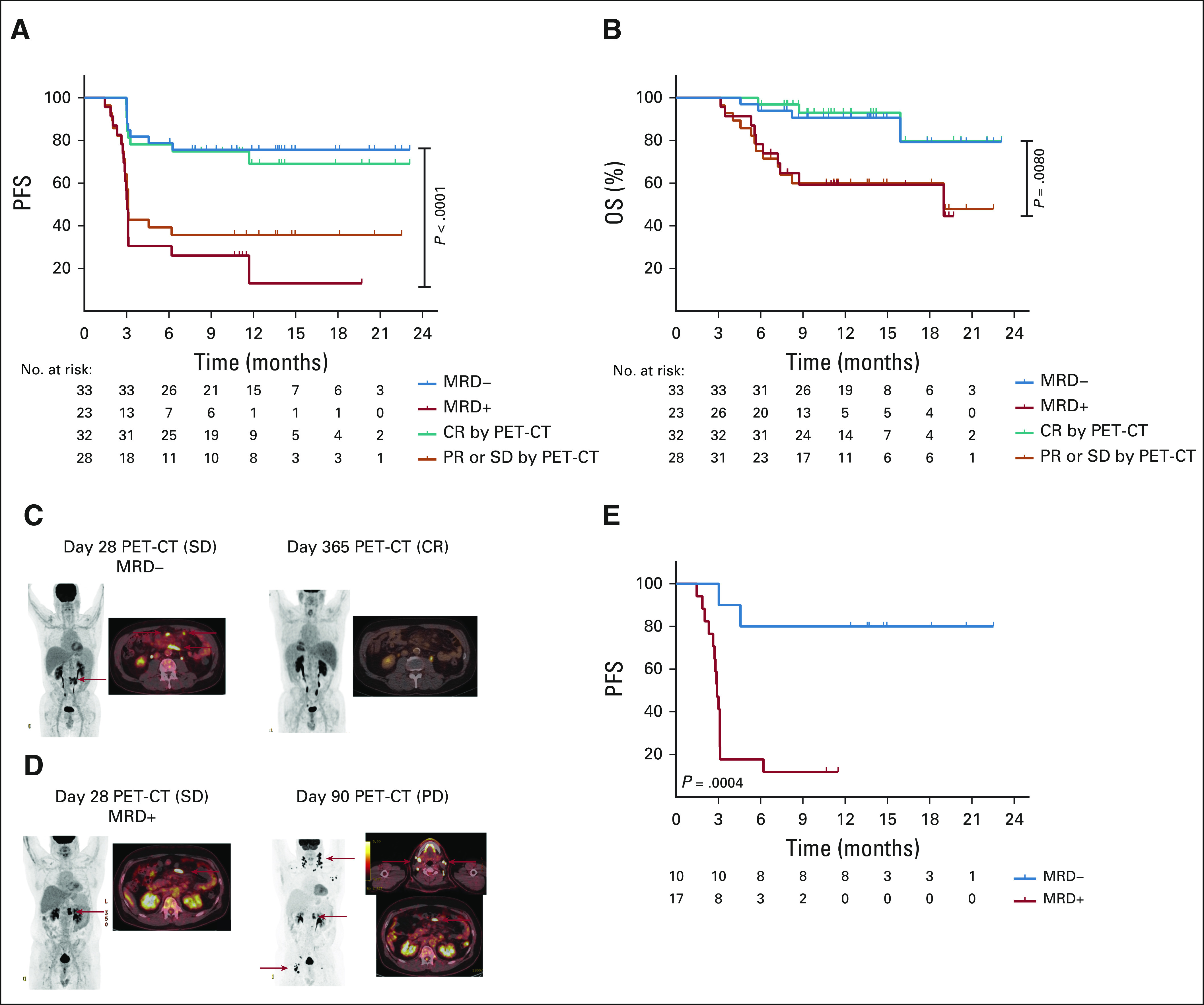
Outcomes on the basis of PET-CT and MRD status. (A) PFS and (B) OS on the basis of the MRD status on day 28, negative (blue) versus positive (red), and clinical status as determined by PET-CT imaging on day 28, CR (green) versus PR or SD (orange). Patients who were MRD− compared with those who were MRD+ had a longer median PFS of NR versus 3 months (P < .0001) and a longer median OS of NR versus 19 months (P = .0080). Patients who had FDG-avid disease on day 28 compared with those who achieved a CR had a median PFS of 3.1 months versus NR (P = .0018) and a median OS of 19 months versus NR (P = .0033). (C and D) Representative patients with SD as determined by the PET-CT on day 28 who are MRD− or MRD+ achieve a durable CR or relapse by day 90, respectively. (E) PFS from day 28 was examined by Kaplan-Meier analysis and log-rank test in patients with a SD or PR on day 28 by radiographic criteria, who were MRD− (blue) or MRD+ (red). Patients who were MRD− had a significantly longer PFS than those who were MRD+ with a median PFS of NR versus 2.9 months (P = .0004). CR, complete response; FDG, fluorodeoxyglucose; MRD, minimal residual disease; NR, not reached; OS, overall survival; PD, progressive disease; PET-CT, positron emission tomography-computed tomography; PFS, progression-free survival; PR, partial response; SD, stable disease.
Next, we evaluated outcomes by MRD status for those who had a CR on day 28 and a concomitant available MRD sample (n = 29). The PFS and OS were not significantly different between MRD+ (n = 6) and MRD− (n=23) patients in CR (Data Supplement). Nine patients with a radiographic CR on day 28 experienced relapse—six were MRD− and three were MRD+ concurrently—and ultimately, these patients became MRD+ at or before relapse (Fig 3).
Finally, we evaluated outcomes by MRD status for those who had a radiographic PR or SD on day 28. 16 of the 24 (66.7%) patients with a PR and two of the four patients with SD on the PET-CT on day 28 experienced progression (Fig 3). For the 27 patients who had an available MRD sample, MRD status was highly predictive for subsequent relapse (Figs 4C-4E). Two of 10 MRD− patients with a PR or SD on day 28 experienced a PFS event because of relapse (n = 1) and death from Pneumocystis jirovecii pneumonia (n = 1), whereas 15 of 17 MRD+ patients experience a PFS event. MRD− patients with a PR or SD on day 28 compared with MRD+ patients had a significantly longer median PFS, NR versus 2.9 months (P = .0004). Moreover, the MRD status for patients with a PR or SD on day 28 had a sensitivity, specificity, positive predictive value, and negative predictive value of 94%, 82%, 88%, and 90%, respectively.
DISCUSSION
This multicenter prospective study demonstrates that early after axi-cel therapy, peripheral blood ctDNA assessments can predict for progression events with added value to standard PET-CT scans. We show that lymphoma-specific clonotype(s) can be identified in the majority (96%) of patients permitting ctDNA tracking. Key baseline patient characteristics are associated with outcomes after axi-cel infusion for LBCL including tumor burden, LDH, performance status, and inflammatory markers.3,17-19 Patients with pre-LD ctDNA concentrations above 100 LG/mL have a median PFS of 3 months. By contrast, patients below 100 LG/mL experience an approximately 80% PFS and 90% OS at 12 months post-axi-cel. Grade 2 or higher CRS and ICANS were also associated with higher pre-LD ctDNA.
Relapse after axi-cel was associated with persistent detection of ctDNA 1 month after axi-cel infusion or beyond. ctDNA-based assessments were particularly prognostic for patients with a PR or SD on day 28. For patients with a PR or SD on day 28, the MRD status accurately predicted whether progression would occur in 23 of 27 (85%) patients. MRD+ patients with a PR or SD on day 28 have a high risk (positive predictive value of 88%) of relapsing.
At the time of starting lymphodepleting chemotherapy, 12 patients had no detectable ctDNA, despite having FDG-avid disease by PET-CT before axi-cel manufacturing. Notably, three patients later developed detectable ctDNA preceding disease relapse. The nine other patients remained in a durable remission with no detectable ctDNA. These findings are consistent with previous studies in newly diagnosed DLBCL, where 8%-18% of patients had undetectable ctDNA before starting initial immunochemotherapy and nearly all patients developed detectable ctDNA concurrently with relapse.5,20
Our current study had limitations. We were unable to detect clonotypes in 4% of patients. This study assessed ctDNA for 1 year after axi-cel infusions and does not address the role of ctDNA surveillance thereafter. A fresh biopsy was not required at the time of enrollment nor at relapse to ensure that FDG-avid lesions were not a false-positive. ctDNA was not measured at the time of apheresis (as many patients were enrolled after apheresis), and therefore, the impact of bridging therapies on ctDNA is unknown. Patients were permitted to receive bridging treatment without confirming persistent disease thereafter and therefore might have achieved MRD remissions before axi-cel infusion. No validation cohort of patients was available to confirm our findings.
Despite these limitations, the data suggest that serial ctDNA monitoring may be a noninvasive approach to assess progression after axi-cel. Twenty-nine of 30 patients had detectable ctDNA concurrently with progression. The patient with undetectable ctDNA concurrently with progression, which was based on a relatively small increase in the FDG avidity of a single lesion, did not have a biopsy to prove relapse. We speculate that this asymptomatic patient was actually in remission when they started new therapy, particularly since the treating physician was not aware of the contemporary ctDNA status at the time of clinical decision making. Our data suggest for MRD-negative patients, such as this, with a small, mildly avid lesion (particularly when obtaining a biopsy would be challenging), close follow-up using serial imaging or ctDNA monitoring may be appropriate instead of starting systemic therapy.
Given that nearly all patients became MRD+ at or before the time of disease relapse, serial ctDNA-based assessments that reflex to PET-CT imaging at the time of MRD positivity may be a viable alternative to serial PET-CT imaging in asymptomatic patients. However, a well-conducted clinical trial would be needed to confirm this hypothesis. Furthermore, serial ctDNA monitoring is likely an informative tool to assess alternative CAR therapies in LBCL. Since each CAR therapy has unique expansion and persistence characteristics, additional studies will determine how to best use ctDNA-based assessments for each CAR therapy.
In conclusion, to our knowledge, this is the first study to comprehensively evaluate ctDNA dynamics prospectively in patients undergoing CAR therapy. ctDNA-based surveillance of patients with LBCL undergoing axi-cel may be a useful adjunct to radiographic assessments of disease status. Future studies may leverage this approach to identify patients requiring consolidative therapies following CAR therapy.
ACKNOWLEDGMENT
The authors would like to thank our patients for their participation in this study.
Matthew J. Frank
Employment: Roche/Genentech
Stock and Other Ownership Interests: Roche/Genentech
Ilan Kirsch
Employment: Adaptive Biotechnologies
Stock and Other Ownership Interests: Adaptive Biotechnologies
Allison P. Jacob
Employment: Adaptive Biotechnologies
Stock and Other Ownership Interests: Adaptive Biotechnologies
Chelsea D. Mullins
Employment: Adaptive Biotechnologies
Stock and Other Ownership Interests: Adaptive Biotechnologies
Lik Wee Lee
Employment: Adaptive Biotechnologies
Crystal L. Mackall
Stock and Other Ownership Interests: Lyell Immunopharma, Alimera Sciences, Vor Pharmaceuticals, Apricity Health, Syncopation Life Sciences
Consulting or Advisory Role: Bryology, Vor Biopharma, Apricity Health, TPG, Alimera Sciences, PACT Pharma, Nektar, Lyell Immunopharma, NeoImmuneTech, Syncopation Life Sciences, NeoImmuneTech, Bristol Myers Squibb, Immatics
Research Funding: Lyell Immunopharma
Patents, Royalties, Other Intellectual Property: I am an inventor on numerous patents related to chimeric antigen receptor therapeutics and received royalties from NIH for the CD22-CAR patent licensed to Juno therapeutics
Travel, Accommodations, Expenses: NeoImmuneTech, Roche, Nektar
Other Relationship: Lyell Immunopharma
Michael D. Jain
Consulting or Advisory Role: Kite/Gilead, Novartis, Bristol Myers Squibb, Takeda
Travel, Accommodations, Expenses: Kite/Gilead
Saurabh Dahiya
Consulting or Advisory Role: Kite/Gilead, Atara Biotherapeutics
Frederick L. Locke
Consulting or Advisory Role: Novartis, Celgene, GammaDelta Therapeutics, Calibr, WUGEN Inc, Alimera Sciences, Cellular Biomedicine Group, Gerson Lehrman Group, EcoR1 Capital, Amgen, Bluebird Bio, Bristol Myers Squibb, Iovance Biotherapeutics, Legend Biotech, Cowen
Research Funding: Kite, a Gilead company, Alimera Sciences, Novartis
Patents, Royalties, Other Intellectual Property: Double Mutant Survivin Vaccine. US010414810B2, CAR T Cells with Enhanced Metabolic Fitness. Serial Number: 62/939727, Methods of Enhancing CAR T Cell Therapies. Serial Number: 62/892292, Evolutionary Dynamics of Non-Hodgkin Lymphoma CAR-T cell therapy. Serial Number: 62/879534
Travel, Accommodations, Expenses: Kite, a Gilead company
David B. Miklos
Consulting or Advisory Role: Kite, a Gilead company, Adaptive Biotechnologies, Novartis, Juno/Celgene, Pharmacyclics, Janssen, Precision Biosciences, Alimera Sciences, Miltenyi Biotec
Research Funding: Pharmacyclics, Novartis, Roche/Genentech, Kite, a Gilead company, Adaptive Biotechnologies, Alimera Sciences, Precision Biosciences
Patents, Royalties, Other Intellectual Property: Patent held with Pharmacyclics supporting Ibrutinib for cGVHD (no royalty claim).
Travel, Accommodations, Expenses: Kite, a Gilead company, Pharmacyclics/Janssen, Adaptive Biotechnologies, Juno Therapeutics, Miltenyi Biotec, Novartis
No other potential conflicts of interest were reported.
Listen to the podcast by Dr Reagan at jcopodcast.libsynpro.com
DISCLAIMER
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
SUPPORT
Supported by the National Cancer Institute of the National Institutes of Health under Award No. K08CA248968 (M.J.F.). The T32 training grant HL007952-18 supported N.M.H. CRC support at Maryland was provided by Gary Jobson Endowed Professorship in Oncology. This research was supported by P01 CA049605 and 5P30CA124435. C.L.M. is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program.
M.J.F. and N.M.H. are cofirst authors. S.D., F.L.L., and D.B.M. are cosenior authors.
DATA SHARING STATEMENT
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/JCO.21.00377.
AUTHOR CONTRIBUTIONS
Conception and design: Matthew J. Frank, Nasheed M. Hossain, Lik Wee Lee, Saurabh Dahiya, Frederick L. Locke, David B. Miklos
Financial support: David B. Miklos
Administrative support: Aaron P. Rapoport, Frederick L. Locke
Provision of study materials or patients: Aaron P. Rapoport, Michael D. Jain, Frederick L. Locke, David B. Miklos
Collection and assembly of data: Matthew J. Frank, Nasheed M. Hossain, Ali Bukhari, Erin Dean, Jay Y. Spiegel, Gursharan K. Claire, Allison P. Jacob, Chelsea D. Mullins, Lik Wee Lee, Katherine A. Kong, Juliana Craig, Aaron P. Rapoport, Michael D. Jain, Saurabh Dahiya, Frederick L. Locke, David B. Miklos
Data analysis and interpretation: Matthew J. Frank, Nasheed M. Hossain, Ali Bukhari, Jay Y. Spiegel, Ilan Kirsch, Chelsea D. Mullins, Lik Wee Lee, Crystal L. Mackall, Aaron P. Rapoport, Frederick L. Locke, David B. Miklos
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Monitoring of Circulating Tumor DNA Improves Early Relapse Detection After Axicabtagene Ciloleucel in Large B-Cell Lymphoma: Results of a Prospective Multi-Institutional Trial
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Matthew J. Frank
Employment: Roche/Genentech
Stock and Other Ownership Interests: Roche/Genentech
Ilan Kirsch
Employment: Adaptive Biotechnologies
Stock and Other Ownership Interests: Adaptive Biotechnologies
Allison P. Jacob
Employment: Adaptive Biotechnologies
Stock and Other Ownership Interests: Adaptive Biotechnologies
Chelsea D. Mullins
Employment: Adaptive Biotechnologies
Stock and Other Ownership Interests: Adaptive Biotechnologies
Lik Wee Lee
Employment: Adaptive Biotechnologies
Crystal L. Mackall
Stock and Other Ownership Interests: Lyell Immunopharma, Alimera Sciences, Vor Pharmaceuticals, Apricity Health, Syncopation Life Sciences
Consulting or Advisory Role: Bryology, Vor Biopharma, Apricity Health, TPG, Alimera Sciences, PACT Pharma, Nektar, Lyell Immunopharma, NeoImmuneTech, Syncopation Life Sciences, NeoImmuneTech, Bristol Myers Squibb, Immatics
Research Funding: Lyell Immunopharma
Patents, Royalties, Other Intellectual Property: I am an inventor on numerous patents related to chimeric antigen receptor therapeutics and received royalties from NIH for the CD22-CAR patent licensed to Juno therapeutics
Travel, Accommodations, Expenses: NeoImmuneTech, Roche, Nektar
Other Relationship: Lyell Immunopharma
Michael D. Jain
Consulting or Advisory Role: Kite/Gilead, Novartis, Bristol Myers Squibb, Takeda
Travel, Accommodations, Expenses: Kite/Gilead
Saurabh Dahiya
Consulting or Advisory Role: Kite/Gilead, Atara Biotherapeutics
Frederick L. Locke
Consulting or Advisory Role: Novartis, Celgene, GammaDelta Therapeutics, Calibr, WUGEN Inc, Alimera Sciences, Cellular Biomedicine Group, Gerson Lehrman Group, EcoR1 Capital, Amgen, Bluebird Bio, Bristol Myers Squibb, Iovance Biotherapeutics, Legend Biotech, Cowen
Research Funding: Kite, a Gilead company, Alimera Sciences, Novartis
Patents, Royalties, Other Intellectual Property: Double Mutant Survivin Vaccine. US010414810B2, CAR T Cells with Enhanced Metabolic Fitness. Serial Number: 62/939727, Methods of Enhancing CAR T Cell Therapies. Serial Number: 62/892292, Evolutionary Dynamics of Non-Hodgkin Lymphoma CAR-T cell therapy. Serial Number: 62/879534
Travel, Accommodations, Expenses: Kite, a Gilead company
David B. Miklos
Consulting or Advisory Role: Kite, a Gilead company, Adaptive Biotechnologies, Novartis, Juno/Celgene, Pharmacyclics, Janssen, Precision Biosciences, Alimera Sciences, Miltenyi Biotec
Research Funding: Pharmacyclics, Novartis, Roche/Genentech, Kite, a Gilead company, Adaptive Biotechnologies, Alimera Sciences, Precision Biosciences
Patents, Royalties, Other Intellectual Property: Patent held with Pharmacyclics supporting Ibrutinib for cGVHD (no royalty claim).
Travel, Accommodations, Expenses: Kite, a Gilead company, Pharmacyclics/Janssen, Adaptive Biotechnologies, Juno Therapeutics, Miltenyi Biotec, Novartis
No other potential conflicts of interest were reported.
REFERENCES
- 1. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J Clin Oncol. 2014;32:3059–3068. doi: 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: Results from the US lymphoma CAR T consortium. J Clin Oncol. 2020;38:3119–3128. doi: 10.1200/JCO.19.02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kurtz DM, Scherer F, Jin MC, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018;36:2845–2853. doi: 10.1200/JCO.2018.78.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roschewski M, Dunleavy K, Pittaluga S, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015;16:541–549. doi: 10.1016/S1470-2045(15)70106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scherer F, Kurtz DM, Diehn M, et al. High-throughput sequencing for noninvasive disease detection in hematologic malignancies. Blood. 2017;130:440–452. doi: 10.1182/blood-2017-03-735639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7:1394–1403. doi: 10.1158/2159-8290.CD-17-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hossain NM, Dahiya S, Le R, et al. Circulating tumor DNA assessment in patients with diffuse large B-cell lymphoma following CAR T-cell therapy. Leuk Lymphoma. 2019;60:503–506. doi: 10.1080/10428194.2018.1474463. [DOI] [PubMed] [Google Scholar]
- 9. Logan AC, Gao H, Wang C, et al. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc Natl Acad Sci USA. 2011;108:21194–21199. doi: 10.1073/pnas.1118357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Logan AC, Zhang B, Narasimhan B, et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia. 2013;27:1659–1665. doi: 10.1038/leu.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boyd SD, Marshall EL, Merker JD, et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Transl Med. 2009;1:12ra23. doi: 10.1126/scitranslmed.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng Q, Han G, Puebla-Osorio N, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med. 2020;26:1878–1887. doi: 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ching T, Duncan ME, Newman-Eerkes T, et al. Analytical evaluation of the clonoSEQ assay for establishing measurable (minimal) residual disease in acute lymphoblastic leukemia, chronic lymphocytic leukemia, and multiple myeloma. BMC Cancer. 2020;20:612. doi: 10.1186/s12885-020-07077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown LD, Cai TT, DasGupta A. Interval estimation for a binomial proportion. Stat Sci. 2001;16:101–133. [Google Scholar]
- 17. Locke FL, Rossi JM, Neelapu SS, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020;4:4898–4911. doi: 10.1182/bloodadvances.2020002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dean EA, Mhaskar RS, Lu H, et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020;4:3268–3276. doi: 10.1182/bloodadvances.2020001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faramand R, Jain M, Staedtke V, et al. Tumor microenvironment composition and severe cytokine release syndrome (CRS) influence toxicity in patients with large B-cell lymphoma treated with axicabtagene ciloleucel. Clin Cancer Res. 2020;26:4823–4831. doi: 10.1158/1078-0432.CCR-20-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kurtz DM, Green MR, Bratman SV, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125:3679–3687. doi: 10.1182/blood-2015-03-635169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/JCO.21.00377.