

PURPOSE
SJMB03 (ClinicalTrials.gov identifier: NCT00085202) was a phase III risk-adapted trial that aimed to determine the frequency and clinical significance of biological variants and genetic alterations in medulloblastoma.
PATIENTS AND METHODS
Patients 3-21 years old were stratified into average-risk and high-risk treatment groups based on metastatic status and extent of resection. Medulloblastomas were molecularly classified into subgroups (Wingless [WNT], Sonic Hedgehog [SHH], group 3, and group 4) and subtypes based on DNA methylation profiles and overlaid with gene mutations from next-generation sequencing. Coprimary study end points were (1) to assess the relationship between ERBB2 protein expression in tumors and progression-free survival (PFS), and (2) to estimate the frequency of mutations associated with WNT and SHH tumors. Clinical and molecular risk factors were evaluated, and the most robust were used to model new risk-classification categories.
RESULTS
Three hundred thirty eligible patients with medulloblastoma were enrolled. Five-year PFS was 83.2% (95% CI, 78.4 to 88.2) for average-risk patients (n = 227) and 58.7% (95% CI, 49.8 to 69.1) for high-risk patients (n = 103). No association was found between ERBB2 status and PFS in the overall cohort (P = .74) or when patients were stratified by clinical risk (P = .71). Mutations in CTNNB1 (96%), DDX3X (37%), and SMARCA4 (24%) were most common in WNT tumors and PTCH1 (38%), TP53 (21%), and DDX3X (19%) in SHH tumors. Methylome profiling classified 53 WNT (17.4%), 48 SHH (15.7%), 65 group 3 (21.3%), and 139 group 4 (45.6%) tumors. A comprehensive clinicomolecular risk factor analysis identified three low-risk groups (WNT, low-risk SHH, and low-risk combined groups 3 and 4) with excellent (5-year PFS > 90%) and two very high-risk groups (high-risk SHH and high-risk combined groups 3 and 4) with poor survival (5-year PFS < 60%).
CONCLUSION
These results establish a new risk stratification for future medulloblastoma trials.
INTRODUCTION
Most children with medulloblastoma survive with multimodal therapy that combines maximal surgical resection, craniospinal irradiation (CSI), and chemotherapy. However, the impact of treatment-related morbidities on quality of life in survivors remains high,1 and about a quarter of patients still die of their disease. Thus, an important aspect of clinical trials must be to mitigate toxicity. However, determining which component of therapy to modify, and which patients are most likely to benefit, remains a challenge.
CONTEXT
Key Objectives
The SJMB03 Protocol was designed to explore and define the clinical impact of biological differences found within medulloblastoma in a patient population treated on a clinically defined risk-stratified protocol.
Knowledge Generated
This study places the outcome of 330 prospectively treated patients with medulloblastoma in the context of clinical risk features, molecular subgroups, driver mutations, focal amplifications, and chromosomal aberrations and includes the latest methylation subtypes. Considering these data, we propose a novel integrated clinical and molecular risk-stratification model.
Relevance
This sophisticated risk model identifies about 40% of patients with newly diagnosed medulloblastoma who have a > 95% cure rate on SJMB03 treatment, about 20% who have an 80% cure rate, and 40% with an unsatisfactory cure rate ranging between 30% and 60%. Thus, this study lays the framework for an improved risk-stratification model and provides insight necessary for the development of less toxic and more efficacious therapy for childhood medulloblastoma.
Traditionally, patients have been risk-stratified based on clinical characteristics to reduce the deleterious effects of therapy. Those with subtotally resected tumors (defined as residual disease of > 1.5 cm2) and metastatic spread of disease (M+) are classified as high risk, whereas those with gross totally resected (GTR) or near totally resected tumors (NTR; defined as residual tumor ≤ 1.5 cm2) and no evidence of metastatic disease (M0) are classified as average risk. High-risk patients receive a maximal dose of CSI with additional radiation therapy (RT) delivered to the posterior fossa, or tumor bed, and to sites of gross metastatic disease and higher or more frequent doses of chemotherapy.1-3 By contrast, average-risk patients receive a lower CSI dose with additional RT delivered only to the posterior fossa, or tumor bed, and adjuvant chemotherapy. Using this general approach, approximately 80% of average-risk and 60% of high-risk presentations are cured.1-5 Yet, despite similar outcomes, treatment approaches differ by the treating center or trial, and an optimized standard regimen is lacking.
Recent literature has concluded that biological differences have a major impact on outcome. Patients with histologically defined large cell/anaplastic (LC/A) medulloblastoma exhibit inferior outcomes, whereas patients with desmoplastic nodular (D/N) variants have favorable outcomes.6,7 Aberrant protein expression, such as ERBB2, or gene amplifications, such as MYC or MYCN, have been proposed as independent risk factors that negatively impact outcome.8-11 Recently, four molecular subgroups (Wingless [WNT], Sonic Hedgehog [SHH], group 3, and group 4) have been identified, with distinct genomic and demographic characteristics that exhibit different outcomes.12,13 Additionally, further division of these molecular subgroups into discrete subtypes has increased biologic dimensions for clinical consideration.14,15
SJMB03 (ClinicalTrials.gov identifier: NCT00085202) was a multi-institutional phase III clinical trial that emerged during a transformative era when new biologic information was rapidly accumulating. The impetus behind the trial was two-fold: (1) to improve on a promising, short dose-intense therapeutic regimen from our prior study, SJMB96 (ClinicalTrials.gov identifier: NCT00003211),1 and (2) to initiate a large biology-based trial to assess the clinical utility of molecular features in a uniformly treated patient cohort.
Herein we present the results of the medulloblastoma cohort from the SJMB03 trial, including patient outcomes according to ERBB2 protein expression status, clinical risk-based classification, and molecular features. Additionally, we identify new molecular risk features and present the first prospective outcome data based on novel medulloblastoma subtypes.
PATIENTS AND METHODS
Patients and Patient Samples
SJMB03 was a phase III clinical trial for patients 3-21 years old newly diagnosed with medulloblastoma, supratentorial primitive neuroectodermal tumor, or atypical teratoid rhabdoid tumor (Data Supplement, online only). Eligibility criteria were similar to SJMB96.1
Patients were stratified postoperatively into two risk groups (average risk and high risk). Patients with M0 status (as indicated by magnetic resonance imaging [MRI] of the brain or spine and cytological examination of CSF) and a surgical GTR or NTR on postoperative MRI (R0) were assigned to average risk, whereas those with M+ status (M1-3; as indicated by MRI of the brain or spine and/or cytological examination of CSF) or residual tumor of > 1.5 cm2 on postoperative MRI (R+) were assigned to high risk. Toxicity was monitored and graded according to Common Terminology Criteria for Adverse Events version 3.0.
Diagnoses were histologically confirmed by central pathology review. Medulloblastomas were assigned to classic, D/N, or LC/A histologic variants. Molecular analyses were done on tumor specimens and matched blood samples. Protein expression of ERBB2 was detected from fresh-frozen tumor samples using immunoblotting.8 DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumor using the Maxwell rapid sample concentrator (RSC) DNA FFPE kit (#AS1450, Promega, Madison, WI) and quantified using Qubit (Thermo Fisher Scientific, Waltham, MA) and from patient-matched peripheral WBCs using the DNeasy kit (Qiagen, Germantown, MD). RNA was extracted from snap-frozen medulloblastomas using STAT-60, and mRNA expression profiles were generated using the U133 Plus 2.0 microarray (Affymetrix, Santa Clara, CA).16 DNA methylation–based classification was performed using methods described previously.17 For prediction of subtypes, we implemented a random forest classifier trained on reference data sets. DNA copy number variants were inferred using the Conumee R package.18 Tumor and germline DNA exomes were captured using the SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA) platform. All next-generation sequencing (NGS) data were processed with either germline paired or unpaired analysis pipelines and curated manually to ensure consistent germline and/or somatic mutation calling. Genomic datasets included in this study can be freely explored using the online St Jude Cloud pediatric genomic data resource.19
SJMB03 was approved by the St Jude Institutional Review Board and institutional review boards at participating institutions. Patients, parents, or guardians provided written informed consent.
Treatment
Patients with average-risk disease received CSI (23.4 Gy) followed by focal RT to the primary tumor bed (total dose 55.8 Gy; 1.8 Gy daily fraction). Patients with high-risk disease received CSI (36 [M0-1]-39.6 [M2-3] Gy) followed by focal RT to the primary tumor (total dose 55.8-59.4 Gy; 1.8 Gy daily fraction). The clinical target volume margin surrounding the postoperative tumor bed was 1.0 cm for both risk groups. Areas of macroscopic metastatic disease > 0.5 cm received additional RT (total dose 50.4-59.4 Gy). Twenty-nine patients received proton-beam RT.
After a 6-week rest, patients began four cycles of high-dose chemotherapy consisting of vincristine (1.0 mg/m2 once daily [max dose 2.0 mg] on day −4 and 6), cisplatin (75 mg/m2 once daily on day −4), and cyclophosphamide (2 g/m2 once daily on days −3 and −2). Each cycle was followed by stem-cell or bone-marrow rescue on day 0. Filgrastim (5 µg/kg once daily per day) was given on day +1 until absolute neutrophil count (ANC) > 2,000 on 2 consecutive days. The planned duration of chemotherapy was 16 weeks (4 weeks per cycle).
Treatment continued until completion, disease progression, withdrawal of consent, or unacceptable toxicity. Disease assessments were done at defined intervals until 72 months from diagnosis.
Statistical Analysis
All eligible patients who initiated RT were included. Coprimary study end points were (1) to assess the relationship between ERBB2 protein expression and progression-free survival (PFS), and (2) to estimate the frequency of mutations associated with WNT and SHH tumors. The planned sample size to detect a 15% higher PFS at 2 years for ERBB2-negative subjects compared with ERBB2-positive subjects was 123 with 5% type I error and 80% power, taking into account the expected imbalance in the ERBB2 positivity rate between the two risk groups.
Patients with adequate tissue for methylation profiling were included in the subgroup-specific outcome analyses. Secondary biological end points aimed to describe alterations in oncogenes and tumor suppressor genes and to evaluate the relationship of these molecular features with subtype, clinical characteristics, and clinical outcome. Outcome distributions were estimated using the method of Kaplan and Meier. Log-rank tests or stratified log-rank tests were used for the outcome comparisons. Cox proportional hazard models were used to estimate coefficients of risk factors in forest plots. A Fisher's exact test was used to compare distributions of categorical variables among patient groups. Statistical analyses were done using R 3.6.0.
RESULTS
Study Patients
Between September 2003 and June 2013, 413 of 416 screened patients were enrolled. Pathology review confirmed 330 (80%) as medulloblastoma: 227 (69%) average risk and 103 (31%) high risk. Tolerance of therapy was very similar to what was previously described.1 Toxicities attributed to treatment that occurred in at least 5% of patients and any study-related deaths not from disease progression are listed in the Data Supplement.
Three hundred five (94%) medulloblastomas underwent DNA methylation-based classification, assigning patients to WNT (n = 53), SHH (n = 48), group 3 (n = 65), and group 4 (n = 139) subgroups. Two hundred ninety-three (89%) had NGS, including 145 with matched germline DNA (Fig 1). RNA gene expression array analysis was conducted on 149 patients. Demographics and baseline characteristics are listed in Table 1.
FIG 1.
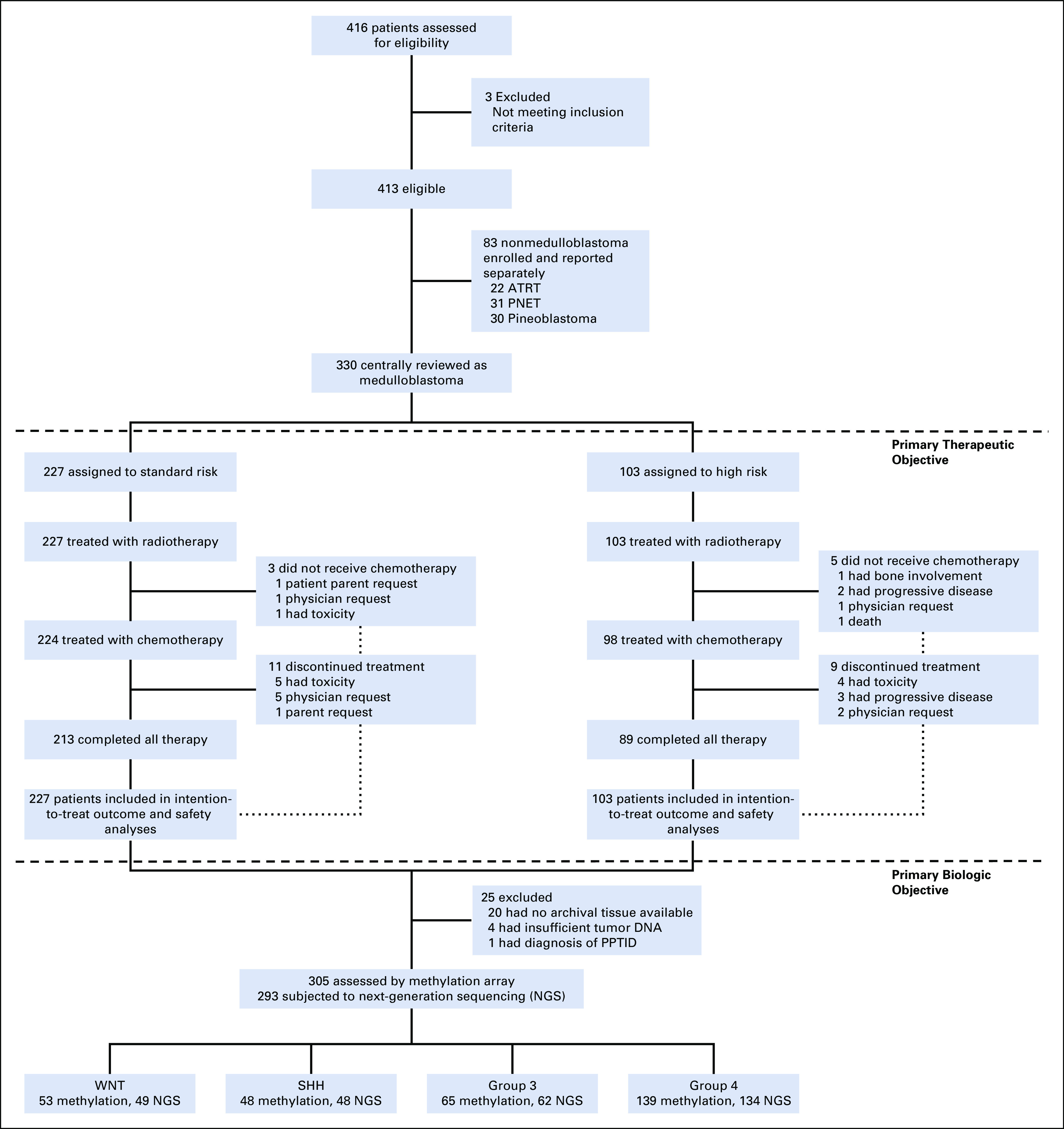
CONSORT diagram of the patients enrolled on the Protocol and details of patients with medulloblastoma who were treated and molecularly profiled. NGS, next-generation sequencing; SHH, Sonic Hedgehog; WNT, Wingless.
TABLE 1.
Demographic and Baseline Characteristics of the Patients

Outcomes by ERBB2 Expression Status
The primary analysis was conducted on 120 eligible patients (84 average risk and 36 high risk). Seventy-five patients were ERBB2-positive and 45 patients ERBB2-negative. There was no difference in PFS by ERBB2 status (71.8% [95% CI, 62.3 to 82.8] ERBB2-positive v 70.6% [95% CI, 58.3 to 85.4] ERBB2-negative; P = .74) in the overall cohort (Fig 2A) or in the context of clinical risk (P = .71; Fig 2B).
FIG 2.
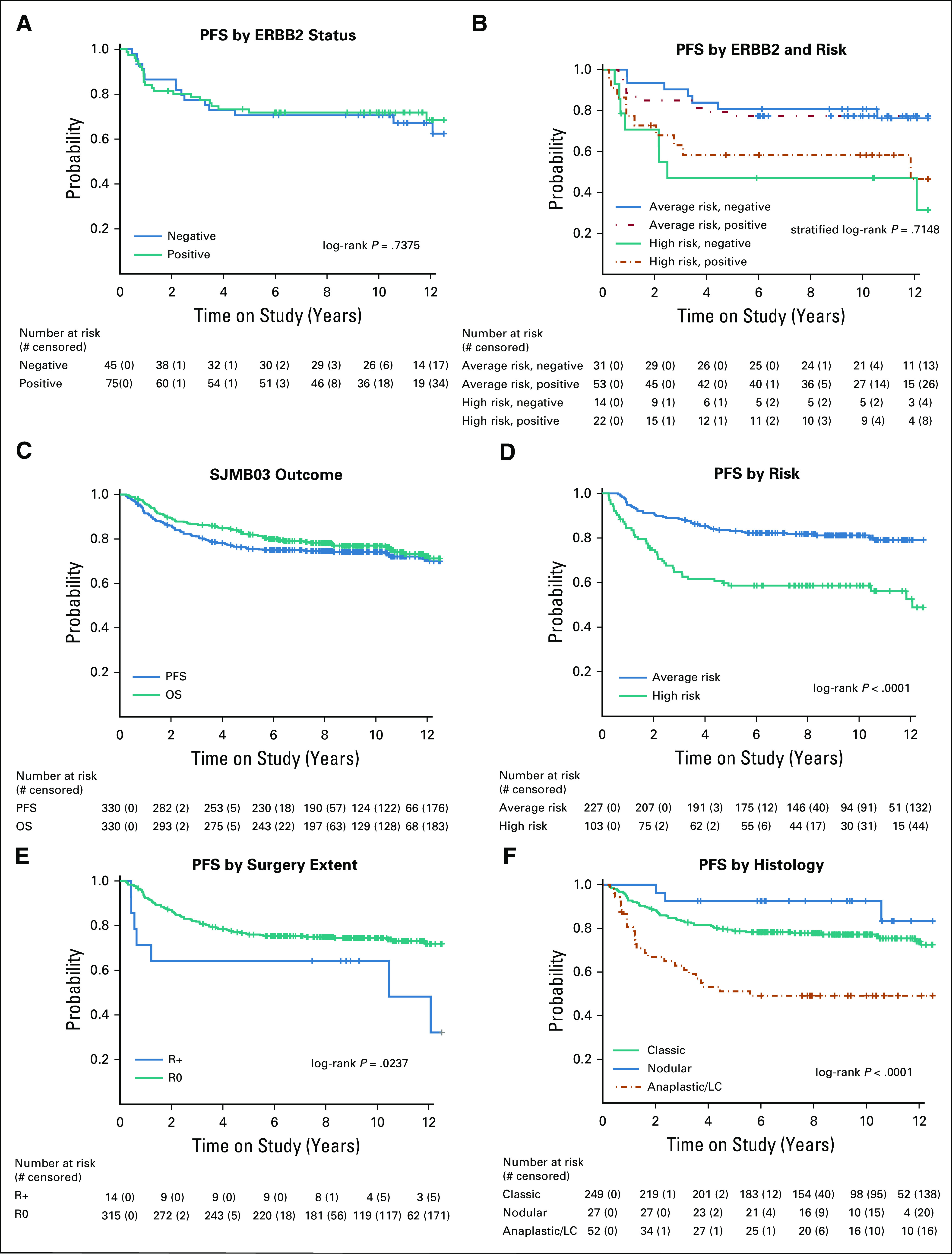
Survival analysis by ERBB2 and clinical risk: PFS (A) by ERBB2 status, (B) ERBB2 and risk, (C) PFS and OS of all patients with medulloblastoma, (D) PFS by risk, (E) by extent of resection, and (F) by histology. OS, overall survival; PFS, progression-free survival.
Outcomes by Clinical Risk Groups and Individual Clinical Risk Factors
Based on a median follow-up of 8.75 years (interquartile range, 4.30 to 11.03), the 5-year PFS was 75.6% (95% CI, 71.1 to 80.4) and the 5-year OS was 82.3% (95% CI, 78.2 to 86.5) for the overall cohort (N = 330; Fig 2C). The 5-year PFS was 83.2% (95% CI, 78.4 to 88.2) for average-risk and 58.7% (95% CI, 49.8 to 69.1) for high-risk patients (P < .0001; Fig 2D). Five-year PFS based on metastatic status was 82.9% (95% CI, 78.1 to 87.9) for M0 and 58.8% (95% CI, 49.9 to 69.4) for M+ patients (P < .0001) (Data Supplement).
At enrollment, 315 (96%) patients had R0 disease and 14 (4%) had R+ disease. R0 subjects had better PFS compared with R+ subjects (P = .0237; Fig 2E). No difference in PFS was observed between patients who underwent a GTR versus NTR (P = .89; Data Supplement), and this result held when metastatic status was incorporated (P = .34; Data Supplement).
Outcomes by Histopathology
Histology was significantly associated with PFS (P < .0001; Fig 2F). Patients with D/N histology experienced the most favorable outcomes with 5-year PFS of 92.6% (n = 27, 95% CI, 83.2 to 1.00), followed by those with classic histology (n = 249; 5-year PFS of 78.6%, 95% CI, 73.7 to 83.9). Patients with LC/A histology had inferior outcomes (n = 52; PFS of 51.1% [95% CI, 39.1 to 66.9]). The impact of histology on PFS was maintained when patients were stratified by clinical risk (P = .0003; Data Supplement).
Outcomes by Molecular Subgroup
Subgroup representation varied by risk group (Fig 3A). DNA methylation subgroups, putative driver gene alterations, and chromosomal gains and losses are summarized by molecular subgroup in Figures 3B and 3C.
FIG 3.
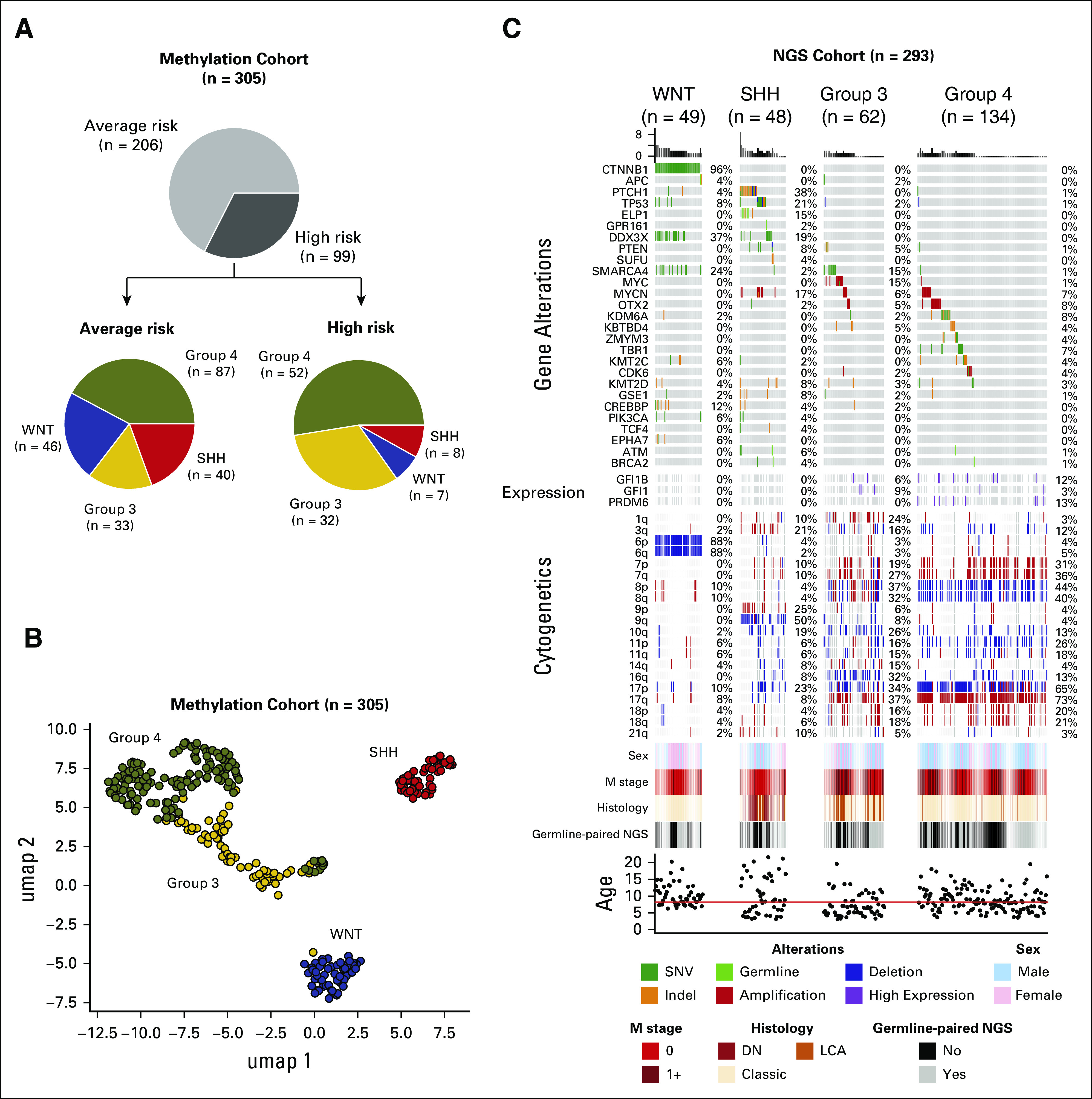
Genomic landscape of pediatric patients with medulloblastoma: (A) Clinical risk (AR and HR) and distribution of molecular subgroups within each risk category, (B) UMAP scatter plot based on weighted Pearson correlation of DNA methylation beta values showing the distribution of consensus medulloblastoma subgroups (n = 305), and (C) oncoprint summarizing recurrently altered genes, frequent cytogenetic events, and sample annotation according to medulloblastoma subgroup (n = 293). AR, average risk; DN, desmoplastic nodular; HR, high risk; LCA, large cell anaplastic; NGS, next-generation sequencing; SHH, Sonic Hedgehog; UMAP, uniform manifold approximation and projection; WNT, Wingless.
Among average-risk patients, 5-year PFS rates were 100% for the WNT subgroup, 77.5% (95% CI, 65.6 to 91.6) for SHH, 66.7% (95% CI, 52.4 to 84.9) for group 3, and 87.3% (95% CI, 80.5 to 94.6) for group 4 (P = .001; Fig 4A). Among high-risk patients, 5-year PFS rates were 100% for the WNT subgroup, 25% (95% CI, 7.5 to 83.0) for SHH, 40.6% (95% CI, 26.7 to 61.8) for group 3, and 68.1% (95% CI, 56.3 to 82.3) for group 4 (P = .0017; Fig 4B). Stratification by clinical risk within subgroups demonstrated inferior outcomes for high-risk patients in each subgroup except WNT (Figs 4C-4F).
FIG 4.
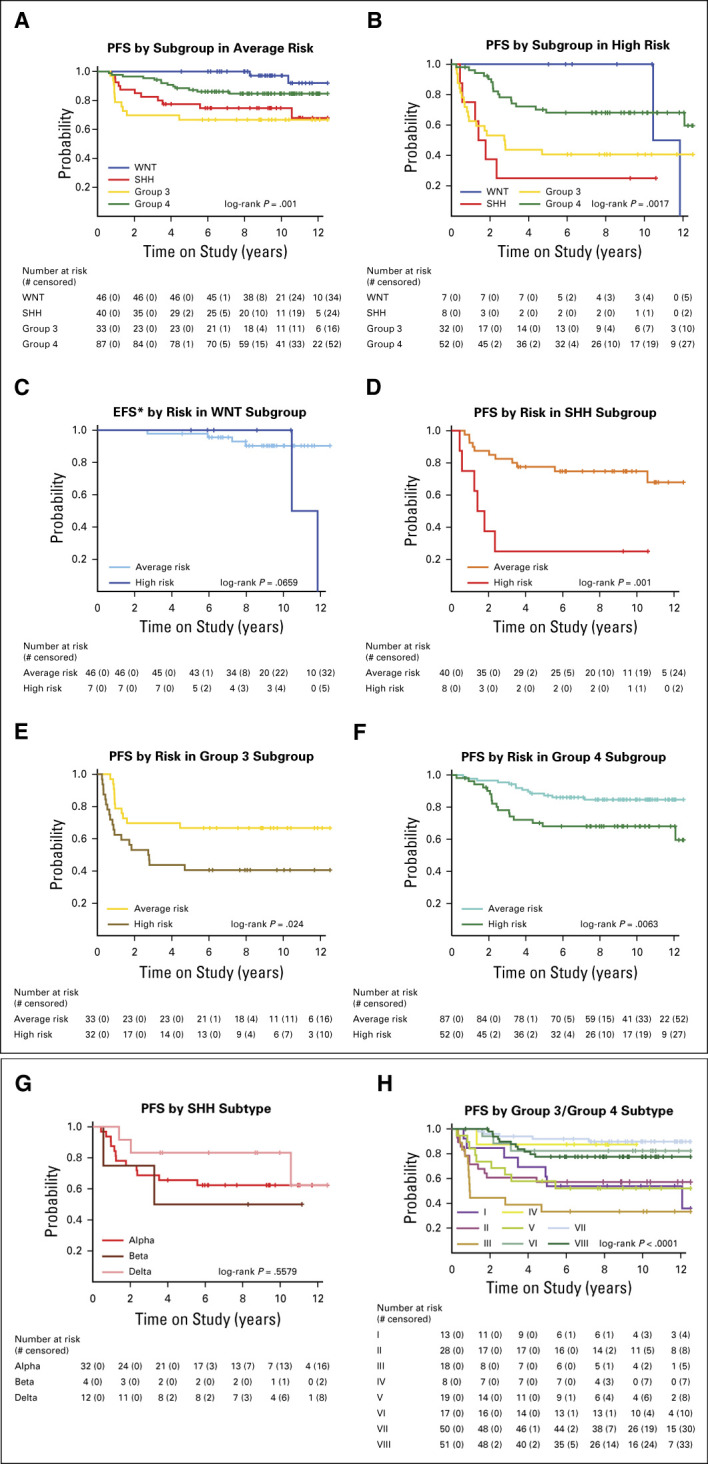
Survival analysis by molecular subgroup and subtype: PFS (A) by subgroup in average risk, (B) by subgroup in high risk, (C) EFS by risk in the WNT subgroup, (D) by risk in the SHH subgroup, (E) by risk in the group 3 subgroup, (F) by risk in the group 4 subgroup, (G) by SHH subtype, and (H) by group 3 or group 4 subtype. EFS, event-free survival; PFS, progression-free survival; SHH, Sonic Hedgehog; WNT, Wingless.
WNT Subgroup
The majority (45 of 53, 85%) of WNT tumors were midline and within the fourth ventricle. The remainder (8 of 53, 15%) extended from the fourth ventricle to the cerebellar pontine angle. Seven (13%) were high risk, six with metastatic disease.
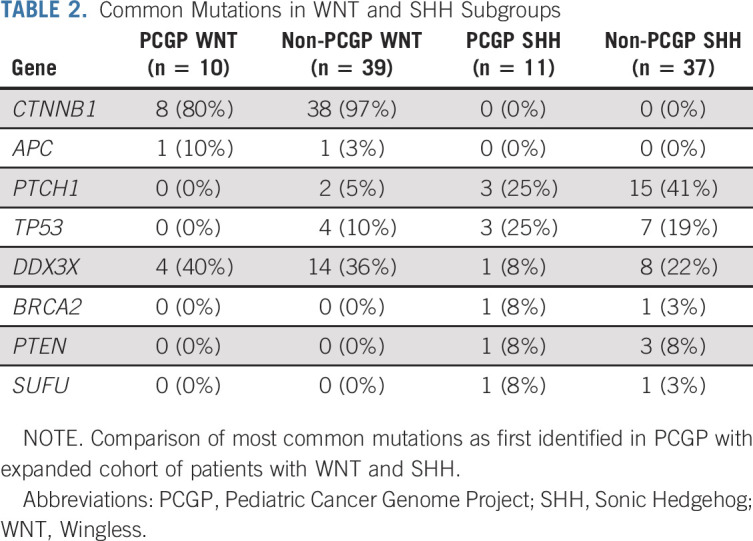
The most frequent mutations were CTNNB1 (96%), DDX3X (37%), SMARCA4 (24%), and CREBBP (12%) (Fig 3C). Monosomy chromosome 6 was detected in 89% of WNT tumors. Mutational frequencies were similar between the initial Pediatric Cancer Genome Project (PCGP) WNT cohort (n = 10) and the expanded (non-PCGP) WNT cohort (n = 39), except for TP53 mutations that were not detected in the initial cohort (Table 2, all P > .10).
TABLE 2.
Common Mutations in WNT and SHH Subgroups
Although 5-year PFS and OS was 100%, four late deaths occurred in patients with WNT. One patient developed pulmonary fibrosis and died 8.3 years from diagnosis. Additionally, four patients with WNT developed second malignancies at a median time of 7.0 years from diagnosis (range, 2.7-10.5 years) and three died. One had a confirmed germline APC mutation (Data Supplement).
SHH Subgroup
The majority of SHH tumors (35 of 48, 73%) were located within a cerebellar hemisphere, whereas the remaining (13 of 48, 27%) were midline. Eight (17%) were high risk, all with M+ disease. Fourteen (29%) had a disease recurrence. The pattern of recurrence was local in six (43%), distant in seven (50%), and combined local and distant in one (7%). One second malignancy, a fatal high-grade glioma in a patient with Li-Fraumeni syndrome, was observed (Data Supplement).
The most commonly altered genes were PTCH1 (38%), TP53 (21%), DDX3X (19%), and MYCN (17%) (Fig 3C). Common chromosomal alterations included 9q loss (53%), 17p loss (26%), and 10q loss (21%) (Fig 3C). Mutational frequencies were similar between the initial PCGP SHH cohort (n = 11) and the expanded (non-PCGP) SHH cohort (n = 37) (Table 2, all P > .10).16 Mutations in ELP1 (7 in 48, 15%) were exclusive to SHH tumors, five of which were confirmed to be germline (Fig 3C).20
In univariable analyses, M+ disease, LC/A histology, TP53 mutation, MYCN amplification, GLI2 amplification, and chromosome 17p loss were all associated with poor PFS, whereas chromosome 14q loss was equivocal (P = .06) (Data Supplement).
Group 3 Subgroup
Thirty-three group 3 patients (51%) were average risk and 32 (49%) high risk. All except one high-risk patient were metastatic. Twenty-six (40%) experienced disease recurrence: 21 (81%) distant (16 leptomeningeal, two isolated to the CSF, and three extra-CNS [bone = 2; liver = 1]), four (15%) local, and one (4%) combined local and distant. No second malignancies were observed.
Frequently mutated genes included SMARCA4 (15%) and KBTBD4 (5%). Fifteen percent harbored MYC amplification and 6% MYCN amplification. GFI1 and GFI1B overexpression was observed in 15% of tumors evaluated by gene expression array. Common chromosomal alterations included isochromosome 17q (27%), chromosome 16q loss (43%), and chromosome 10q loss (33%) (Fig 3C).
In univariable analyses, patients with M+ disease and/or MYC amplification experienced a worse outcome compared with patients lacking these features (Data Supplement). No association with outcome was detected based on histology, MYCN amplification, chromosome 11 loss, chromosome 17 gain, or isochromosome 17q (Data Supplement).
Group 4 Subgroup
Eighty-seven (63%) group 4 patients were average-risk and 52 (37%) high-risk. All high-risk patients were metastatic. Disease recurred in 24 (17%) patients: 20 (83%) distant (17 in the CNS and 3 extra-CNS [bone marrow, soft tissue lower extremity, and femur]), three (13%) local, and one (4%) combined local and distant. Three patients developed second malignancies: an atypical meningioma, a fatal esthesioneuroblastoma, and a fatal high-grade glioma (Data Supplement).
The most common genetic abnormalities were amplification of OTX2 (8%) and MYCN (7%) and mutations in KDM6A (8%) and TBR1 (7%) (Fig 3C). Frequent chromosomal alterations included isochromosome 17q (55%), chromosome 8 loss (39%), chromosome 7 gain (31%), chromosome 11 loss (14%), and chromosome 18 gain (20%).
In univariable analyses, M+ disease was associated with a worse outcome (Data Supplement). No association with outcome was detected for histology, MYCN amplification, chromosome 11 loss, chromosome 17 gain, or isochromosome 17q (Data Supplement).
Outcomes by Molecular Subtype
Methylation analysis further classified the SHH subgroup into known subtypes21,22: SHHα (n = 32), SHHβ (n = 4), SHHδ (n = 12), and SHHγ (n = 0) (Data Supplement). SHHα subtype tumors (67%) harbored recurrent genetic alterations in PTCH1, ELP1, TP53, MYCN, and GLI2 and were characterized by frequent isochromosome 9, chromosome 10q loss, and chromosome 17p loss (Data Supplement). There was no significant difference in outcome among SHH subtypes (P = .5579; Fig 4G).
Group 3 and group 4 subgroup patients were further classified into eight subtypes based on methylation analysis14: subtype I (n = 13), subtype II (n = 28), subtype III (n = 18), subtype IV (n = 8), subtype V (n = 19), subtype VI (n = 17), subtype VII (n = 50), and subtype VIII (n = 51) (Data Supplement). Subtypes II, III, and IV were nearly exclusively made up of group 3 tumors, subtypes VI, VII, and VIII were predominantly group 4 tumors, and subtypes I and V contained a mixture of both subgroups (Data Supplement). Recurrent genetic events by subtype included OTX2 amplification (subtype I), MYC amplification (subtype II), SMARCA4 mutation (subtypes II and III), MYCN amplification (subtypes V and VI), and KDM6A, ZMYM3, KMT2C, and TBR1 mutations (subtype VIII) (Data Supplement). Age, histology, sex, and metastatic status differed among group 3 and group 4 subtypes (Data Supplement). There were also differences in PFS among group 3 and group 4 patients by subtype (P < .0001; Fig 4H). Subtype III patients had the worst outcomes, with a 5-year PFS of 33.3% (95% CI, 17.3 to 64.1). Subtype VII patients had the best outcomes, with a 5-year PFS of 92.0% (95% CI, 84.7 to 99.8).
Molecularly Informed Risk Classification
We analyzed potential risk factors and modeled outcomes based on pertinent risk features. No modeling was performed on the WNT subgroup patients because of their excellent outcome. Since metastatic status was consistently associated with an inferior outcome for all non-WNT subgroups, metastatic disease was retained as a high-risk feature and additional risk factors were analyzed.
For the SHH subgroup, TP53 mutation, LC/A histology, MYCN amplification, GLI2 amplification, and chromosome 17p loss were all significantly associated with an increased risk in univariable models (Fig 5A, Data Supplement). Hence, when these risk factors were combined together with metastatic disease, the difference in PFS of SHH patients with (n = 25) versus without (n = 22) any of these features was significant (hazard ratio [HR] = 23.2 [95% CI, 3.0 to 176.0] P < .0001) and identified a new high-risk and a new low-risk group, respectively (Fig 5B).
FIG 5.
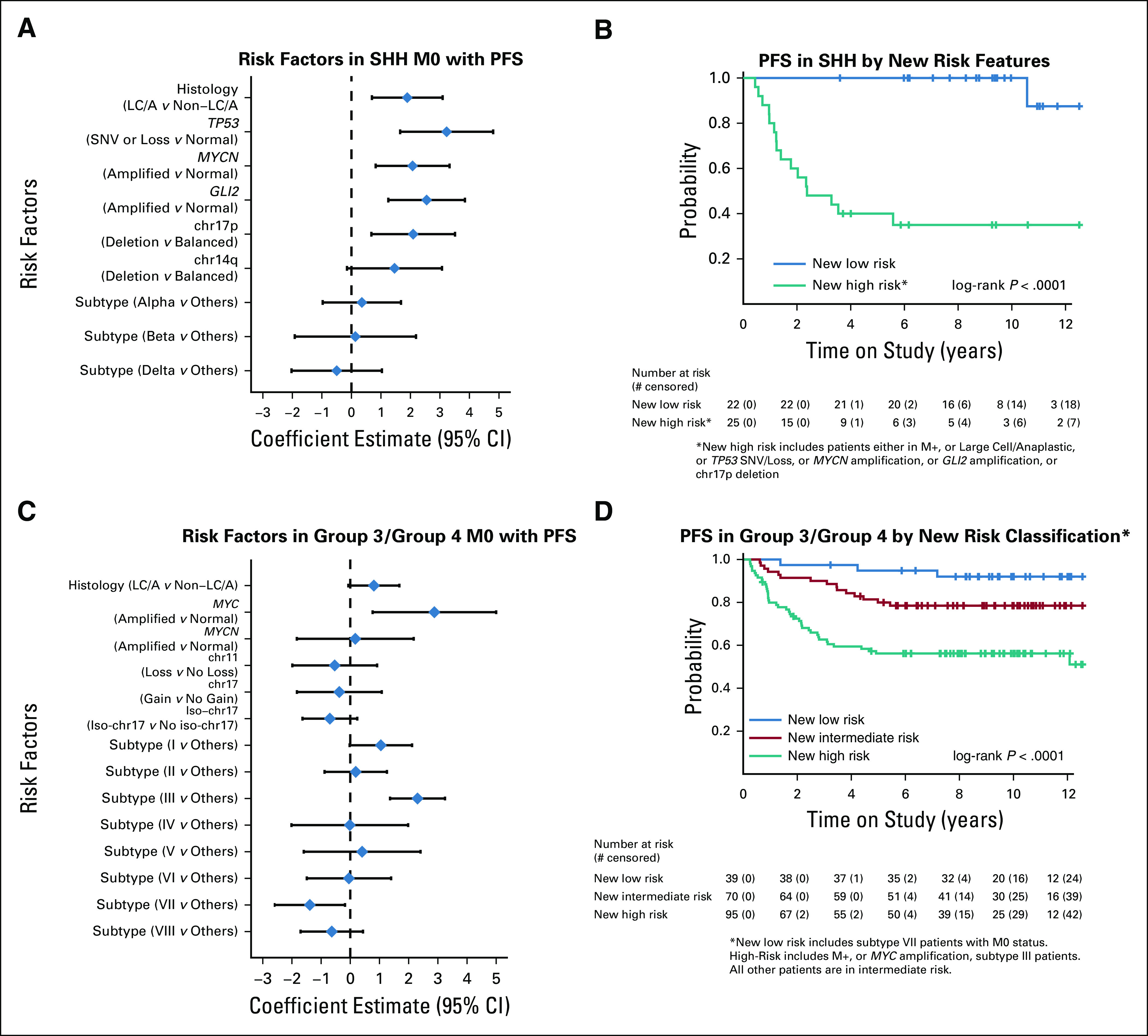
Risk factor modeling and survival analysis by new risk classification: (A) Forest plot displaying Log-HR and 95% CI estimates for clinical and demographic risk factors for PFS for M0 SHH medulloblastoma estimated by univariable Cox models. (B) Kaplan-Meier PFS estimates by proposed new risk classification for SHH medulloblastoma where new high risk includes patients with any of the following: M+, large cell/anaplastic, TP53 SNV/Loss, MYCN amplification, GLI2 amplification, or chr17p deletion; the new low risk includes patients who lack all these risk factors. (C) Forest plot displaying Log-HR and 95% CI estimates for clinical and demographic risk factors for PFS of the combined cohort of group 3 and group 4 M0 medulloblastoma estimated by univariable Cox models. (D) Kaplan-Meier PFS estimates by proposed new risk classification for the combined cohort of group 3 and group 4 medulloblastoma where new low risk is limited to M0 subtype VII patients and new high risk includes patients who are M+, have MYC amplification, or have subtype III tumors. All other patients are in new intermediate risk. HR, hazard ratio; PFS, progression-free survival; SHH, Sonic Hedgehog; SNV, single nucleotide variant.
Group 3 and group 4 tumors were combined for this analysis, given the challenges associated with their confident discrimination and overlapping biology (Fig 3B).14,23 Subtype III and MYC amplification were adverse risk factors, whereas subtype VII was associated with a favorable outcome (Fig 5C). MYCN amplification, chromosome 11 loss, chromosome 17 gain, and isochromosome 17q were not associated with adverse risk. Thus, three risk groups were identified: a low-risk group (patients with M0 and subtype VII), an intermediate-risk group (patients with M0 and subtype not within III or VII), and a high-risk group (patients with M+ disease or subtype III or MYC amplified) (HR = 0.3 [95% CI, 0.09 to 1.1] low- v intermediate-risk group; HR = 2.6 [95% CI, 1.4 to 4.7] high- v intermediate-risk group; P < .0001) (Fig 5D).
DISCUSSION
In this study, outcomes for children treated with SJMB03 therapy were as favorable as those from previous prospective studies.1-5 Importantly, these results were achieved using the smallest radiation clinical target volume margin (1 cm) and the lowest cumulative doses of vincristine (8 mg/m2) and cisplatin (300 mg/m2) of any contemporary trial. Conversely, relative to contemporary trials, SJMB03 therapy used a high dose of CSI (36 [M0-1] – 39.6 [M2-3] Gy) for high-risk patients and a high cumulative dose of cyclophosphamide (16 g/m2). Still, none of these variations produced a significant difference in outcome. Taken together, this suggests that differences in outcome between contemporary medulloblastoma therapies are marginal. As such, we reason that continued modifications to clinically defined risk-stratified therapy, without regard for molecular features, will not result in any measurable benefit. Hence, this study looked to define and inform on outcome by both clinical and molecular features.
Informatively, ERBB2 protein expression did not impact the outcome of patients with medulloblastoma overall or in the context of clinical risk, in spite of preliminary data that suggested ERBB2 overexpression was an adverse risk factor.8,11
Traditional risk features such as presence of metastatic disease, LC/A histology, and amplification of MYC and MYCN were confirmed to negatively impact survival. Also, our extent of resection analysis supported maximal surgical resection, but the lack of difference in outcome between NTR and GTR reinforced recent findings that resection of small residual tumors that carry a high neurologic risk is of little to no benefit.24
However, most importantly, we showed methylation-based subgrouping and subtyping yielded a more precise risk profile and revealed that many of these traditional features are inextricably linked with the molecular composition of the tumor.
SJMB03 treatment achieved excellent tumor control in patients with WNT. This result substantiates the findings of many other studies,1,25,26 and the superb outcome of > 50 patients treated uniformly on a clinical trial across multiple institutions reinforces a deintensification strategy for this subgroup. Radiation reduction has already begun for patients with WNT in three currently accruing clinical trials (SJMB12, ClinicalTrials.gov identifier: NCT01878617; PNET5, ClinicalTrials.gov identifier: NCT02066220; and ACNS1422, ClinicalTrials.gov identifier: NCT02724579).
Patients with SHH had the most dichotomous outcomes of all subgroups. Although survival was significantly different by clinical risk, there were other features that, when factored in post hoc, subdivided the SHH subgroup into very low-risk and very high-risk categories. Previous publications have postulated that molecular events such as TP53 mutation, LC/A histology, chromosome 17p loss, and GLI2 and MYCN amplifications are interconnected,27,28 making it impossible in this relatively small cohort to determine whether one high-risk feature outperformed another. Nevertheless, patients with any of these features, including metastatic disease, had an extremely poor prognosis, whereas patients without had a remarkably favorable outcome.
For group 3 and group 4 medulloblastomas, we took multiple approaches to analyzing outcomes and risk factors. Consistent with previous reports, patients with group 3 tumors had a significantly worse outcome than group 4 in both the average-risk and high-risk categories.26,29 MYC amplification was prognostic in group 3, but these events typically occurred among patients with metastatic disease, rendering MYC genomic status no more prognostic than metastatic status. Upon post hoc introduction of additional previously implicated risk features, we were unable to identify additional low- or high-risk features that could better stratify these patients. LC/A histology, MYCN amplification, chromosome 11 loss, chromosome 17 gain, and isochromosome 17q were not prognostic in either subgroup. Thus, these findings led us to evaluate a joint clinical and subtype-driven approach that resulted in the identification of three new risk groups among combined group 3 and group 4 patients: low (patients with M0 and subtype VII), intermediate (patients with M0 and subtype I, II, IV, V, VI, and VIII), and high (patients with M+ disease or subtype III or MYC amplified). These groups will enable subsequent clinical trials to test judicious dose reductions on the lowest risk group (ie to cyclophosphamide and CSI), optimize therapy for the intermediate-risk patients by harmonizing the lowest doses of agents from contemporary trials to mitigate long-term sequela, and advance needed experimental therapeutics on the highest risk patients.
Nonetheless, caution should be used before adopting these stratifications. Although SJMB03 enrolled a relatively large cohort, the survival modeling remains limited by small numbers and tumor heterogeneity, especially now that medulloblastoma consists of four molecular subgroups and 12 subtypes of disease.14 Thus, these models need to be validated on independent trial cohorts.
In conclusion, this study demonstrates the limitations of clinically defined risk stratification and highlights the power and potential of employing a combined molecular and clinical risk stratification to improve medulloblastoma therapy for all.
ACKNOWLEDGMENT
The authors thank Emily Walker and Granger Rideout from the Hartwell Center for assistance with methylation and NGS of our tumor samples; Sujuan Jia from the Diagnostic Biomarkers Shared Resource for nucleic acids extraction; Matthew Lear from the Biorepository for assistance in archiving and providing the study material; Amanda Johnson, Keri Meyer, Molly Clarke, Leigh Ann Christy, Lenzie Denzin, Kate Lyons, Annemarie McClellan, Tabatha Doyle, Aksana Vasilyeva, and Dana Wallace for providing excellent research support and data management for the study; and, especially, the children and families who were treated on the study.
SUPPORT
Supported by the American Lebanese Syrian Associated Charities, St Jude Children's Research Hospital, NCI Cancer Center Grant (P30CA021765), Sontag Foundation (P.A.N.), Alexander and Margaret Stewart Trust (P.A.N.), American Association for Cancer Research (P.A.N.), The Brain Tumor Charity (P.A.N., G.W.R.), St. Baldrick's Foundation (P.A.N.), and Musicians Against Childhood Cancer (A.G.).
CLINICAL TRIAL INFORMATION
A.G., G.W.R., D.W.E., and P.A.N. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Amar Gajjar, Giles W. Robinson, Thomas E. Merchant, Stewart J. Kellie, Richard Cohn, Matthew J. Krasin, Clinton F. Stewart, Richard J. Gilbertson, Arzu Onar-Thomas, Paul A. Northcott
Financial support: Amar Gajjar, Giles W. Robinson, Paul A. Northcott
Collection and assembly of data: Amar Gajjar, Giles W. Robinson, Kyle S. Smith, Thomas E. Merchant, Murali Chintagumpala, Anita Mahajan, Eric Bouffet, Ute Bartels, Tim Hassall, Thomas Robertson, Wayne Nicholls, Sridharan Gururangan, Kristin Schroeder, Greg Wheeler, Jordan R. Hansford, Stewart J. Kellie, Geoffrey McCowage, Richard Cohn, Michael J. Fisher, Matthew J. Krasin, Alberto Broniscer, Ruth G. Tatevossian, Brent A. Orr, Geoff Neale, Frederick Boop, Stefan M. Pfister, Richard J. Gilbertson, David W. Ellison
Data analysis and interpretation: Amar Gajjar, Giles W. Robinson, Kyle S. Smith, Tong Lin, Thomas E. Merchant, Anita Mahajan, Jack Su, Eric Bouffet, Tal Schechter, Sridharan Gururangan, Michael Sullivan, Jordan R. Hansford, Ivo Buchhalter, Brent A. Orr, Paul Klimo, Frederick Boop, Ashok Srinivasan, Stefan M. Pfister, Richard J. Gilbertson, Arzu Onar-Thomas, David W. Ellison, Paul A. Northcott
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03)
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Amar Gajjar
Consulting or Advisory Role: Roche/Genentech
Research Funding: Genentech, Kazia Pharmaceutical, QED Therapeutics Inc
Giles W. Robinson
Consulting or Advisory Role: Lilly, Roche/Genentech
Research Funding: Novartis, Genentech/Roche, Genentech/Roche
Thomas E. Merchant
Travel, Accommodations, Expenses: Philips Healthcare
Eric Bouffet
Consulting or Advisory Role: Novartis
Research Funding: Roche, Bristol-Myers Squibb
Jordan R. Hansford
Consulting or Advisory Role: Bayer
Geoffrey McCowage
Consulting or Advisory Role: Biogen
Research Funding: Novartis, Merck
Travel, Accommodations, Expenses: BIOGEN
Michael J. Fisher
Honoraria: AstraZeneca
Research Funding: AstraZeneca, Array BioPharma, Exelixis
Travel, Accommodations, Expenses: AstraZeneca, SpringWorks
Alberto Broniscer
Consulting or Advisory Role: QED Therapeutics, Inc
Frederick Boop
Employment: Semmes Murphey Clinic
Stefan M. Pfister
Research Funding: Lilly, Bayer, Roche, PharmaMar, Pfizer
Patents, Royalties, Other Intellectual Property: patent on utilizing DNA methylation profiling for tumor classification
Richard J. Gilbertson
Research Funding: AstraZeneca/MedImmune
Patents, Royalties, Other Intellectual Property: Patent together with AstraZeneca/MedImmune and the University of Cambridge for the C11orf95-RELA diagnostic antibody
Arzu Onar-Thomas
Consulting or Advisory Role: Roche
Research Funding: Novartis, Apexigen, Pfizer, Celgene, Merck, Novocure
Travel, Accommodations, Expenses: Roche
David W. Ellison
Patents, Royalties, Other Intellectual Property: Sole inventor of U.S. Patent No. 9,005,907 issued April 14, 2015 “Methods and Compositions for Typing Molecular Subgroups of Medulloblastoma”, 62627291/S88435 1190US.P1 February 2018 “Epigenetic Histone Regulation Mediated by CXorf67” published August 2019 as WO 2019/155387
No other potential conflicts of interest were reported.
REFERENCES
- 1. Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 2. Jakacki RI, Burger PC, Zhou T, et al. Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: A Children's Oncology Group Phase I/II study. J Clin Oncol. 2012;30:2648–2653. doi: 10.1200/JCO.2011.40.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. von Bueren AO, Kortmann RD, von Hoff K, et al. Treatment of children and adolescents with metastatic medulloblastoma and prognostic relevance of clinical and biologic parameters. J Clin Oncol. 2016;34:4151–4160. doi: 10.1200/JCO.2016.67.2428. [DOI] [PubMed] [Google Scholar]
- 4. Lannering B, Rutkowski S, Doz F, et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: Results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. 2012;30:3187–3193. doi: 10.1200/JCO.2011.39.8719. [DOI] [PubMed] [Google Scholar]
- 5. Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202–4208. doi: 10.1200/JCO.2006.06.4980. [DOI] [PubMed] [Google Scholar]
- 6. Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas: A Pediatric Oncology Group study. Cancer. 2002;94:552–560. doi: 10.1002/cncr.10189. [DOI] [PubMed] [Google Scholar]
- 7. McManamy CS, Pears J, Weston CL, et al. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17:151–164. doi: 10.1111/j.1750-3639.2007.00058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gajjar A, Hernan R, Kocak M, et al. Clinical, histopathologic, and molecular markers of prognosis: Toward a new disease risk stratification system for medulloblastoma. J Clin Oncol. 2004;22:984–993. doi: 10.1200/JCO.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 9. Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400–1407. doi: 10.1200/JCO.2010.30.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eberhart CG, Kratz J, Wang Y, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441–9. doi: 10.1093/jnen/63.5.441. [DOI] [PubMed] [Google Scholar]
- 11. Gilbertson RJ, Clifford SC, MacMeekin W, et al. Expression of the ErbB-neuregulin signaling network during human cerebellar development: Implications for the biology of medulloblastoma. Cancer Res. 1998;58:3932–3941. [PubMed] [Google Scholar]
- 12. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Northcott PA, Shih DJ, Peacock J, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma T, Schwalbe EC, Williamson D, et al. Second-generation molecular subgrouping of medulloblastoma: An international meta-analysis of group 3 and group 4 subtypes. Acta Neuropathol. 2019;138:309–326. doi: 10.1007/s00401-019-02020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi: 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hovestadt V, Remke M, Kool M, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013;125:913–916. doi: 10.1007/s00401-013-1126-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The St Jude Cloud. https://pecan.stjude.cloud/proteinpaint/study/MB-SJMB03
- 20. Waszak SM, Robinson GW, Gudenas BL, et al. Germline elongator mutations in sonic hedgehog medulloblastoma. Nature. 2020;580:396–401. doi: 10.1038/s41586-020-2164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31:737–754 e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hovestadt V, Ayrault O, Swartling FJ, et al. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat Rev Cancer. 2020;20:42–56. doi: 10.1038/s41568-019-0223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hovestadt V, Smith KS, Bihannic L, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–79. doi: 10.1038/s41586-019-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thompson EM, Hielscher T, Bouffet E, et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016;17:484–495. doi: 10.1016/S1470-2045(15)00581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ellison DW, Onilude OE, Lindsey JC, et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951–7957. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 26. Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kool M, Jones DT, Jager N, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shih DJ, Northcott PA, Remke M, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32:886–896. doi: 10.1200/JCO.2013.50.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]