

Graphical abstract

Keywords: COVID-19, SARS-CoV-2, 3CL protease, Nirmatrelvir, Peptidomimetic inhibitors
Abstract
In this paper, a series of peptidomimetic SARS-CoV-2 3CL protease inhibitors with new P2 and P4 positions were synthesized and evaluated. Among these compounds, 1a and 2b exhibited obvious 3CLpro inhibitory activities with IC50 of 18.06 nM and 22.42 nM, respectively. 1a and 2b also showed excellent antiviral activities against SARS-CoV-2 in vitro with EC50 of 313.0 nM and 170.2 nM, respectively, the antiviral activities of 1a and 2b were 2- and 4-fold better than that of nirmatrelvir, respectively. In vitro studies revealed that these two compounds had no significant cytotoxicity. Further metabolic stability tests and pharmacokinetic studies showed that the metabolic stability of 1a and 2b in liver microsomes was significantly improved, and 2b had similar pharmacokinetic parameters to that of nirmatrelvir in mice.
1. Introduction
Since late 2019, the outbreak of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),1, 2 poses a severe threat to global public health and safety. Although vaccines have been widely and repeatedly vaccinated, the high variability and certain immune escape ability of SARS-CoV-2 makes the vaccines less effective.3, 4 Therefore, the development of antiviral agents that retain activity against various SARS-CoV-2 variants is still necessary to relieve the symptoms and reduce the hospitalization rate of patients.5
During the replication of coronaviruses, 3-chymotrypsin-like cysteine protease (3CL protease, 3CLpro) and papain-like protease (PLpro) are responsible for the cleavage of two polyprotein precursors (pp1a/pp1ab) into nonstructural proteins, which are essential for viral genome replication and transcription.6, 7, 8, 9 3CLpro gene sequences are highly conserved among coronaviruses that have been discovered, and have no homologous proteins in humans.10, 11, 12 3CLpro inhibitors can be classified as peptidomimetic and non-peptidomimetics,13 Peptidomimetic 3CLpro inhibitors were designed by mimicking peptide substrates. The active site of 3CLpro is highly conserved and usually consists of four subsites: S1′, S1, S2 and S4, which can be occupied by the P1′, P1, P2 and P4 portions of peptidomimetic inhibitors.14, 15 The reported chemical warheads of 3CLpro inhibitors at the P1′ position including nitrile, α,β-unsaturated ester, phthalhydrazido-methyl ketone, benzothiazolyl ketone, aldehyde, α-ketoamide, hydroxymethyl ketone, acrylamide, 2-butynamide and so on.16, 17 According to the specificity and catalytic mechanism of the substrate, the inhibitors can competitively bind 3CLpro with the natural substrate, further inactivating 3CLpro.18, 19 Therefore, 3CLpro has become one of the most important targets for anti coronavirus drug research.
Nirmatrelvir, a 3CLpro inhibitor, which is the active pharmaceutical ingredient of Paxlovid approved by the FDA for emergency use in the treatment of COVID-19.20 Several groups reported similar designs as nirmatrelvir at the same time, and some 3CLpro inhibitors containing dimethylcyclopropylproline or cyclopentylproline at the P2 position were synthesized. Kneller et al. reported that BBH-1, BBH-2 and NBH-2 exhibited comparable antiviral properties to that of nirmatrelvir in vitro (Fig.1 ).21 Xia et al. designed two 3CLpro inhibitors UAWJ9-36-1 and UAWJ9-36-3, which also contain γ-lactam ring at the P1 position, exhibited broad-spectrum anti-coronavirus activity (Fig.1).22 Qiao et al. reported that MI-09 and MI-30 showed excellent antiviral activity in cell-based assays and displayed good pharmacokinetic properties in rats (Fig.1).23
Fig. 1.

Chemical structures of protease inhibitors.
In this paper, we designed and synthesized a series of peptidomimetic SARS-CoV-2 3CLpro inhibitors based on the structure of nirmatrelvir, and evaluated their biological activities. Among these compounds, 1a and 2b showed excellent inhibitory potency against SARS-CoV-2 3CLpro and notable antiviral activity against SARS-CoV-2 in vitro, which provided lead compounds for the development of clinical drug candidates. The design of peptidomimetic SARS-CoV-2 3CLpro inhibitors is shown in Fig.2 .
Fig. 2.

Design of peptidomimetic SARS-CoV-2 3CLpro inhibitors.
2. Results and discussion
2.1. Chemistry
The synthetic route of compounds 1 (a–f) is shown in Scheme 1 . The tert-Butoxycarbonyl (Boc) group of commercially available 1–1 (a–f) was deprotected and then conjugated with N-Boc-l-tert-leucine to afford ester 1–2 (a–f), then 1–3 (a–f) were obained via alkaline hydrolysis. Compound 1–4 was prepared according to the method reported in Ref. 24. 1–4 was deprotected in the presence of HCl-Dioxane, and then coupled with 1–3 (a–f) using HATU as coupling reagent to generate 1–5 (a–f). Finally, the Boc group of 1–5 (a–f) was deprotected, and trifluoroacetic anhydride (TFAA) was used to achieve the trifluoroacetylation of amino group and dehydration of amide to nitrile in a one-pot reaction to obtain compounds 1 (a–f).
Scheme 1.

Synthesis of compounds 1 (a–f), reagents and conditions: (i) 4 N HCl-Dioxane, Dioxane, 40 ℃, 1 h; (ii) BOP, NMM, N,N-Dimethylformamide, DCM, 25 ℃, 10 h; (iii) LiOH·H2O, THF, H2O, 40 ℃, 2 h; (iv) 4 N HCl-Dioxane, Dioxane, 40 ℃, 1 h; (v) HATU, DIPEA, DCM, 25 ℃, 10 h; (vi) 4 N HCl-Dioxane, Dioxane, 40 ℃, 1 h; TFAA, Et3N, 25 ℃, 10 h.
The synthesis of compounds 2 (a–d) was similar to the above. Removal of the Boc group of 2–4 and then reacted with different acid chlorides or anhydrides to give 2–5 (a–d). Finally, 2–5 (a–d) were dehydrated with Burgess reagent to obtain compounds 2 (a–d). (Scheme 2 ).
Scheme 2.

Synthesis of compounds 2 (a–d), reagents and conditions: (i) BOP, NMM, N,N- Dimethylformamide, DCM, 25 ℃, 10 h; (ii) LiOH·H2O, THF, H2O, 40 ℃, 2 h; (iii) 4 N HCl-Dioxane, Dioxane, 40 ℃, 1 h; (iv) HATU, DIPEA, DCM, 25 ℃, 10 h; (v) 4 N HCl-Dioxane, Dioxane, 40 ℃, 1 h; (vi) Et3N, DCM, 0 ℃, 10 h; (vii) Burgess Reagent, DCM, 25 ℃, 10 h.
2.2. Biological assay
The use of nitrile warhead at P1′ is one of the key factors contributing to the excellent activity of nirmatrelvir. The nitrile warhead can react with the sulfhydryl group of the cysteine 145 residue at S1′ site to form a reversible covalent thioimidate adduct, which is important for the inhibitors to maintain antiviral activity.25 The S1 site of 3CLpro has an extremely high recognition specificity for glutamine residues, and nirmatrelvir adopts the γ-lactam ring as the P1 fragment. γ-lactam ring has the ability to mimic glutamine, it can penetrate into the S1 site and form a stable interaction with the residues at S1 site.26 Several groups reported that boceprevir had potent SARS-CoV-2 3CLpro inhibitory activity as well as cellular antiviral activity.27, 28 And X-ray crystal structure analysis revealed that the binding of boceprevir to the catalytically active side of SARS-CoV-2 3CLpro is the main mechanism of inhibition.29, 30 These results demonstrated that the dimethylcyclopropylproline of boceprevir has an important effect on antiviral activity, which provided the guidance for the design of new 3CLpro inhibitors.
Based on this, we firstly modified the P2 position of nirmatrelvir. In order to investigate the importance of bicyclic proline, a series of compounds with different bicyclic proline at P2 position were synthesized and evaluated (Table 1 ). The compound 1a, bearing cyclopentyl proline, displayed a similar inhibitory potency to that of nirmatrelvir at 0.1 μM (1a inhibition was 82.50%, Nirmatrelvir inhibition was 91.38%). 1b and 1c, bearing bridged bicyclic proline and spiro bicyclic proline, exhibited slightly lower inhibitory potency than that of 1a at 0.1 μM (1b inhibition was 47.38%, 1c inhibition was 54.47%). This indicated that cyclopentyl proline was more easily bound to the S2 pocket of 3CLpro.
Table 1.
The inhibitory activity, anti-viral potency and cytotoxicity against SARS-CoV-2 3CLpro of inhibitors.
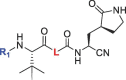 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cmpd. | R1 | L | SARS-CoV-2 3CLpro Inhibition (%), μM |
IC50 nM | EC50 nM | CC50 μM | SI | |||
| 0.02 | 0.1 | 1 | 10 | ||||||||
| 1 | 1a | 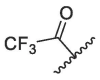 |
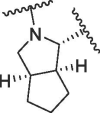 |
23.99 | 82.50 | 88.88 | 88.30 | 18.06 ± 2.35 | 313.0 ± 63.6 | >200 | >600 |
| 2 | 1b | |
 |
/ | 47.38 | 92.62 | 97.13 | 54.64 ± 12.98 | / | / | / |
| 3 | 1c | |
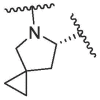 |
/ | 54.47 | 95.83 | 96.99 | 36.82 ± 1.99 | / | / | / |
| 4 | 1d | |
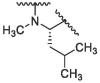 |
/ | / | 54.69 | 80.03 | / | / | / | / |
| 5 | 1e | 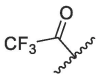 |
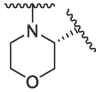 |
/ | / | / | 49.05 | / | / | / | / |
| 6 | 1f | |
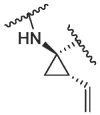 |
/ | / | / | 25.25 | / | / | / | / |
| 7 | 2a | 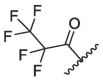 |
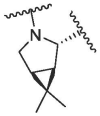 |
/ | −4.36 | 95.19 | 99.91 | / | / | / | / |
| 8 | 2b | 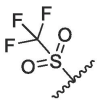 |
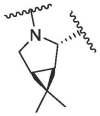 |
18.04 | 95.33 | 106.86 | 95.22 | 22.42 ± 2.78 | 170.2 ± 84.4 | >200 | >1500 |
| 9 | 2c | 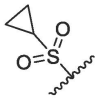 |
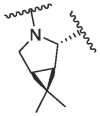 |
46.94 | 94.94 | 108.66 | 111.68 | 22.83 ± 0.21 | / | / | / |
| 10 | 2d | 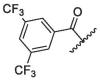 |
|
/ | / | / | 52.49 | / | / | / | / |
| 11 | Nirmatrelvir* | |
|
41.64 | 91.38 | 92.38 | 88.59 | 41.65 ± 2.37 | 579.5 ± 74.5 | / | / |
Note:/:No test.
*:reported compound.
IC50 and EC50 values are shown as means ± SD (n = 3).
The antiviral activity of tested compounds against the original SARS-CoV-2 stain in Vero E6 cells.
In the published 3CLpro inhibitors, isobutyl group was mostly adopted at P2 position.31, 32, 33 We speculate that the size and flexible conformation of the substituents at P2 position may influence their binding to S2 pocket. Therefore, isobutyl group and cyclopropyl vinyl group were introduced at the α-C of nitrogen atom (Table 1). The inhibitory activities of compound 1d (isobutyl group) and 1f (cyclopropyl vinyl group) were severely reduced compared to that of nirmatrelvir. In addition, compound 1e bearing hydrophilic morpholine at the P2 position showed poor inhibitory potency (10 μM inhibition was 49.05%, Table 1). This result was consistent with our prediction, indicating that polarity of substituents at P2 position had an important influence on the inhibitor activity.34
Generally, S4 pocket has a low specificity for the substrate, while P4 position may affect the metabolic stability of the inhibitors.35, 36 So, we replaced the P4 position with different groups in order to screen compounds with better biological activities. Firstly, we synthesized compound 2a by introducing pentafluoropropionyl at the P4 position of nirmatrelvir (Entry 7, Table 1). Unfortunately, The inhibitory activity of compound 2a was greatly reduced at low concentrations. A previous report revealed that the introduction of halogenated phenyl rings at the P4 position were able to form hydrophobic interactions with the S4 site.23 Inspired by this, we synthesized compound 2d by introducing a 3,5-bis (trifluoromethyl) phenyl group at the P4 position, however, the result showed 2d had a low inhibitory activity (Entry 10, Table 1).
According to the report, methanesulfonyl group at the P4 position can extend underneath Gln189 to improve the hydrogen bonding interactions. The introduction of the methanesulfonyl group was also able to improve the antiviral activity, human liver microsomal stability and oral absorption of the inhibitor.24 We introduced trifluoromethanesulfonyl and cyclopropylsulfonyl at the P4 position to synthesize compounds 2b and 2c (Table 1). The results showed that 2b and 2c had similar inhibitory activities to that of nirmatrelvir, the inhibition at 0.1 μM was 95.33% and 94.94%, respectively (Entry 8 and 9, Table 1). This indicates that sulfonyl group plays an important impact on the inhibitory activity.
The preliminary screening results showed that compounds 1a, 1b, 1c, 2b and 2c had good inhibitory activities against 3CLpro, we further tested IC50 and RNA copy inhibition of these compounds. The results showed that the IC50 of these five compounds were slightly better than that of nirmatrelvir (Table 1). The preliminary screening results showed that the RNA copy inhibition of 1a and 2b reached 70% and 98% at 1 μM. Therefore, we selected 1a and 2b for further research based on the above data.
We studied the cytotoxicity of 1a and 2b in Vero E6 cells, neither of the two compounds showed obvious cytotoxicity (Entry 1 and 8, Table 1). Subsequently, we tested the EC50 of these two compounds. The results showed that the EC50 of the two compounds were 313.0 nM and 170.2 nM, respectively, indicating that the antiviral activities were 2- and 4-fold that of nirmatrelvir (Table 1).
In addition, we studied the metabolic stability of 1a and 2b in human liver microsomes and mouse liver microsomes (Table 2 ). 1a showed moderate metabolic stability in human liver microsomes, but was susceptible to metabolic effects in mouse liver microsomes. 2b was stable in human liver microsomes, and also showed moderate metabolic stability in mouse liver microsomes. Compared with nirmatrelvir, the metabolic stability of these two compounds has been significantly improved. These results were consistent with our prediction, indicating that the introduction of sulfonyl group at P4 position can improve the metabolic stability of inhibitors.
Table 2.
Liver microsomal stability of 1a and 2b.
| Entry | Cmpd. | Liver microsomes | T1/2 (minute) | Clint (mL/min/kg) |
|---|---|---|---|---|
| 1 | 1a | HUMAN | 78.89 | 22.03 |
| MOUSE | 14.85 | 367.52 | ||
| 2 | 2b | HUMAN | 652.17 | 2.67 |
| MOUSE | 97.89 | 55.75 | ||
| 3 | Nirmatrelvir | HUMAN | 37.93 | 45.83 |
| MOUSE | 4.64 | 1175.45 |
Note:human, mouse liver microsomes from Xenotech.
Finally, we conducted pharmacokinetic (PK) studies on 1a and 2b in vivo. The two compounds showed favorable pharmacokinetic properties, with oral bioavailability of 22.80% and 23.09%, respectively (Table 3 ). When administered intravenously (i.v.) (10 mg/kg) and orally (p.o.) (20 mg/kg), 1a showed area under the curve (AUC) values of 2669 h*ng/mL and 1219 h*ng/mL, respectively, whereas 2b displayed AUC values of 4118.55 h*ng/mL and 1901.84 h*ng/mL, respectively. After p.o. administration, 1a showed peak blood concentration (Cmax) values of 1311 ng/mL, 2b displayed Cmax values of 2052.36 ng/mL. This series of pharmacokinetic parameters showed that 2b had similar Cmax and AUClast to nirmatrelvir, which indicated that it have the potential to develop into an oral drug.
Table 3.
Pharmacokinetics parameters of 1a and 2b after p.o and i.v. administration in mouse.
| Entry | Cmpd. | T1/2 |
Tmax |
Cmax |
AUClast |
AUCINF_obs |
CL_obs |
MRTINF_obs |
Vss_obs |
F |
|---|---|---|---|---|---|---|---|---|---|---|
| (h) | (h) | (ng/mL) | (h*ng/mL) | (h*ng/mL) | (mL/min/kg) | (h) | (mL/kg) | (%) | ||
| 1 | 1a (p.o.) | 1.00 | 0.42 | 1311.00 | 1219.00 | 1222.00 | / | 0.88 | / | 22.80 |
| 1a (i.v.) | 1.10 | / | / | 2669.00 | 2672.00 | 69.30 | 0.24 | 1011.00 | / | |
| 2 | 2b (p.o.) | 0.71 | 0.50 | 2052.36 | 1901.84 | 1904.90 | / | 0.80 | / | 23.09 |
| 2b (i.v.) | 0.35 | 0.08 | 10487.87 | 4118.55 | 4120.05 | 40.95 | 0.29 | 690.00 | / | |
| 3 | nirmatrelvir(p.o.) | 0.61 | 0.25 | 2521.64 | 2308.32 | 2311.94 | / | 0.85 | / | 34.59 |
| nirmatrelvir (i.v.) | 2.83 | 0.083 | 9626.87 | 3336.46 | 3361.32 | 50.06 | 0.39 | 1.18 | / |
Note:/:No test.
Table 3 report the pharmacokinetics parameters as the mean of 3 independent experiments.
3. Conclusion
As the COVID-19 pandemic and the constant variation of the virus, the development of drugs against SARS-CoV-2 is still of great significance. Based on the structure of nirmatrelvir, we synthesized a series of peptidomimetic SARS-CoV-2 3CLpro inhibitors and tested their biological activities. Among these inhibitors, 1a and 2b exhibited excellent enzyme inhibitory potency with IC50 of 18.06 nM and 22.42 nM, respectively. 1a and 2b also showed significant antiviral activities against SARS-CoV-2 in vitro with EC50 of 313.0 nM and 170.2 nM, respectively. The results of liver microsome stability tests showed that the metabolic stability of 1a and 2b was significantly improved compared to nirmatrelvir, 2b also showed similar pharmacokinetic properties to nirmatrelvir. All such results suggested 1a and 2b deserved further evaluation, and provided important clues for further optimization.
4. Experimental
4.1. Materials and methods
All commercially available chemicals and solvents were directly used without further purification. All reactions were monitored by thin layer chromatography (TLC) on silica gel plates (GF-254). High-resolution mass spectra (HRMS) were measured on an Agilent 1290–6545 UHPLC-QTOF LC/MS spectrometer. 1H NMR and 13C NMR data were recorded on a Bruker AVANCE III instrument (400 MHz) using TMS as an internal standard. Molecular mass was determined on a mass spectrometry (Waters (China) Co., Ltd.). All tested compounds exhibited purities of >95% as analyzed by HPLC (Dionex UltiMate 3000, Germany).
4.2. Synthetic procedures
4.2.1. Synthesis of ethyl(1S,3aR,6aS)-2-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl) octahydrocyclopenta[c]pyrrole-1-carboxylate (1-2a)
(S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (1.0 g, 4.3 mmol) and ethyl (1S,3aR,6aS)-octahydrocyclopenta[c]pyrrole-1-carboxylate 1-1a (1.0 g, 4.8 mmol) were dissolved in DCM/DMF (V:V = 1:1, 20 mL) solution. Then 4-Methylmorpholine (1.3 g, 13.0 mmol), BOP (2.3 g, 5.2 mmol) were added to the reaction at 25 °C and stirred for 10 h. Then DCM (70 mL) and 1 N HCl aqueous solution (8 mL) were added to the reaction. After separation, the organic phase was washed with H2O (10 mL), the organic phase was washed with saturated brine (10 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, PE/EA = 20/1–8/1) to give the compound 1-2a. Yield 51.8%; colourless oil; 1H NMR (400 MHz, DMSO‑d 6) δ 6.61 (d, J = 9.1 Hz, 1H), 4.14 (q, J = 4.2, 3.2 Hz, 2H), 4.09 (d, J = 7.1 Hz, 1H), 4.07–4.02 (m, 1H), 3.74 (qd, J = 10.3, 5.1 Hz, 2H), 2.76–2.65 (m, 1H), 2.58 (tt, J = 7.9, 4.1 Hz, 1H), 1.91–1.73 (m, 2H), 1.64 (dt, J = 14.5, 5.6 Hz, 1H), 1.59–1.49 (m, 2H), 1.48–1.38 (m, 1H), 1.37 (s, 9H), 1.17 (t, J = 7.1 Hz, 3H), 0.94 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 172.07, 170.62, 156.25, 78.66, 65.18, 60.84, 58.66, 53.67, 47.23, 43.16, 34.80, 32.63, 31.71, 28.57, 26.75, 25.09, 14.50. ESI-MS m/z 397.23 [M + H]+.
4.2.2. Synthesis of (1S,3aR,6aS)-2-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl) octahydrocyclopenta[c]pyrrole-1-carboxylic acid (1-3a)
Compound 1-2a (460 mg, 1.2 mmol), lithium hydroxide (201 mg, 4.8 mmol) and water (5 mL) were dissolved in THF (5 mL) solution at 40 °C and stirred for 2 h. Then 1 N HCl aqueous solution was added to the reaction to adjust the pH 2–3. EA (40 mL) was added to the solution and separation, the organic phase was washed with saturated brine (10 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, DCM/MeOH = 80/1–20/1) to give the compound 1-3a. Yield 99.2%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.35 (s, 1H), 6.58 (d, J = 9.2 Hz, 1H), 4.17–4.12 (m, 1H), 4.10 (d, J = 4.1 Hz, 1H), 3.73 (qd, J = 10.2, 4.8 Hz, 2H), 2.72–2.63 (m, 1H), 2.59 (tt, J = 7.8, 4.0 Hz, 1H), 1.90–1.83 (m, 2H), 1.78 (p, J = 7.2 Hz, 1H), 1.69–1.59 (m, 1H), 1.54 (dq, J = 8.2, 5.3, 4.3 Hz, 2H), 1.38 (s, 9H), 0.95 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 173.63, 172.41, 156.25, 78.63, 65.13, 58.60, 53.71, 47.27, 43.09, 34.85, 32.91, 31.87, 28.57, 26.78, 25.16. ESI-MS m/z 369.07 [M + H]+.
4.2.3. Synthesis of tert-butyl((S)-1-((1S,3aR,6aS)-1-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1-5a)
Compound 1–4 (298 mg, 1.1 mmol) and 4 N HCl in 1,4-dioxane (3 mL) were dissolved in DCM (6 mL), the reaction was stirred at 40 °C for 1 h. The reaction was concentrated to remove the solvent and dry HCl salt was obtained. Meanwhile, compound 1-3a (405 mg, 1.1 mmol) and HATU (646 mg, 1.7 mmol) were dissolved in DCM (8 mL), the reaction was stirred at 25 °C for 30 min. Then N, N-Diisopropylethylamine (310 mg, 2.4 mmol) and HCl salt were added to the reaction, the reaction was stirred at 25 °C for 10 h. Then DCM (30 mL) and 1 N HCl aqueous solution (3 mL) were added to the reaction. After separation, the organic phase was washed with H2O (5 mL), the organic phase was washed with saturated brine (5 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, DCM/MeOH = 150/1–20/1) to give the compound 1-5a. Yield 62.2%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.13 (d, J = 8.7 Hz, 1H), 7.54 (s, 1H), 7.21 (s, 1H), 7.01 (s, 1H), 6.58 (d, J = 9.1 Hz, 1H), 4.25 (ddd, J = 12.3, 8.7, 3.6 Hz, 1H), 4.15 (d, J = 4.4 Hz, 1H), 4.13–4.08 (m, 1H), 3.75 (d, J = 8.2 Hz, 1H), 3.68 (d, J = 11.4 Hz, 1H), 3.12 (t, J = 9.0 Hz, 1H), 3.08–2.98 (m, 1H), 2.72–2.60 (m, 1H), 2.45–2.34 (m, 1H), 2.14 (dt, J = 14.1, 8.0 Hz, 1H), 1.96 (dd, J = 15.2, 11.9 Hz, 1H), 1.79 (d, J = 7.0 Hz, 1H), 1.77 (s, 2H), 1.76 (d, J = 6.7 Hz, 1H), 1.69–1.62 (m, 3H), 1.60 (d, J = 12.4 Hz, 2H), 1.36 (s, 9H), 0.92 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.14, 174.02, 172.18, 170.40, 156.27, 78.55, 66.05, 58.79, 54.25, 50.88, 47.63, 43.24, 37.83, 34.83, 34.48, 32.09, 31.64, 28.59, 27.96, 26.84, 24.97. ESI-MS m/z 522.56 [M + H]+.
4.2.4. Synthesis of (1S,3aR,6aS)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-2-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)octahydrocyclopenta[c]pyrrole-1-carboxamide (1a)
Compound 1-5a (104 mg, 0.2 mmol) and TFA (1 mL) were dissolved in DCM (3 mL), the reaction was stirred at 25 °C for 2 h. The reaction was concentrated to remove the solvent. Then triethylamine (73 mg, 0.7 mmol), TFAA (93 mg, 0.4 mmol) and DCM (4 mL) were added to the reaction at 0 °C. The reaction was allowed to warm to 25 °C, and stirred for 10 h. Then DCM (10 mL) and 1 N HCl aqueous solution (2 mL) were added to the reaction. After separation, the organic phase was washed with H2O (2 mL), the organic phase was washed with saturated brine (2 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, DCM/MeOH = 100/1–25/1) to give the compound 1a. Yield 51.4%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.33 (d, J = 8.3 Hz, 1H), 8.92 (d, J = 8.6 Hz, 1H), 7.66 (s, 1H), 4.96 (ddd, J = 11.0, 8.6, 4.9 Hz, 1H), 4.54–4.49 (m, 1H), 4.03 (d, J = 5.0 Hz, 1H), 3.83 (dd, J = 10.4, 7.3 Hz, 1H), 3.63 (dd, J = 10.5, 3.3 Hz, 1H), 3.14 (tt, J = 9.2, 2.0 Hz, 1H), 3.04 (tdd, J = 9.2, 7.1, 1.5 Hz, 1H), 2.77–2.64 (m, 1H), 2.42 (ddd, J = 12.7, 7.3, 3.4 Hz, 1H), 2.21–2.15 (m, 1H), 2.14–2.10 (m, 1H), 2.10–2.05 (m, 1H), 1.84 (dd, J = 7.8, 4.8 Hz, 1H), 1.82–1.80 (m, 1H), 1.80–1.75 (m, 1H), 1.75–1.71 (m, 1H), 1.69 (q, J = 4.6, 3.9 Hz, 1H), 1.67–1.63 (m, 1H), 1.62–1.54 (m, 1H), 1.40–1.31 (m, 1H), 0.98 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.04, 172.12, 168.29, 157.12, 117.75, 114.89, 66.11, 58.46, 54.39, 47.86, 43.51, 38.12, 38.03, 37.18, 35.31, 34.72, 31.81, 31.43, 27.33, 26.69, 25.04. ESI-HRMS Calcd for C23H31F3N5O4 [M−H]-: 498.2334, found 498.2334. HPLC: t = 9.335 min, 99.6% purity.
4.2.5. Synthesis of methyl(1R,3S,4S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (1-2b)
Compound 1-2b was synthesized from compound 1-1b with N-Boc-l-tert-leucine using methods similar to the method described for the preparation of 1-2a. Yield 42.9%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 6.42 (d, J = 9.3 Hz, 1H), 4.55 (s, 1H), 4.24–4.18 (m, 1H), 3.87 (s, 1H), 3.61 (s, 3H), 2.61 (s, 1H), 1.85–1.78 (m, 1H), 1.74–1.68 (m, 1H), 1.67 (d, J = 4.4 Hz, 1H), 1.63 (s, 1H), 1.54–1.43 (m, 2H), 1.37 (s, 9H), 0.95 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 170.71, 168.84, 155.86, 78.60, 63.67, 58.78, 58.49, 52.13, 41.16, 35.57, 35.11, 31.23, 28.61, 27.56, 26.64. ESI-MS m/z 369.24 [M + H]+.
4.2.6. Synthesis of (1R,3S,4S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-2-azabicyclo[2.2.1]heptane-3-carboxylic acid (1-3b)
The compound 1-3b was synthesized from 1 to 2b using a procedure similar to that described for the preparation of 1-3a. Yield 98.0%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.31 (s, 1H), 6.38 (d, J = 9.3 Hz, 1H), 4.52 (s, 1H), 4.24–4.16 (m, 1H), 3.77 (s, 1H), 2.61 (s, 1H), 1.88–1.82 (m, 1H), 1.66 (q, J = 14.0, 10.3 Hz, 3H), 1.41 (s, 2H), 1.37 (s, 9H), 0.95 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 171.65, 168.77, 155.88, 78.56, 63.97, 58.76, 58.54, 41.12, 35.49, 35.17, 31.31, 28.62, 27.73, 26.69.
ESI-MS m/z 355.04 [M + H]+.
4.2.7. Synthesis of tert-butyl((S)-1-((1R,3S,4S)-3-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl) propan-2-yl)carbamoyl)-2-azabicyclo[2.2.1]heptan-2-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1-5b)
Compound 1-5b was synthesized from compound 1–4 with compound 1-3b using methods similar to the method described for the preparation of 1-5a. Yield 66.7%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.03 (d, J = 8.7 Hz, 1H), 7.54 (s, 1H), 7.26 (s, 1H), 7.01 (s, 1H), 6.48 (d, J = 9.3 Hz, 1H), 4.46 (s, 1H), 4.22 (dd, J = 29.0, 9.7 Hz, 2H), 3.86 (s, 1H), 3.08 (dt, J = 24.7, 8.9 Hz, 2H), 2.54 (s, 1H), 2.37 (q, J = 15.0, 8.8 Hz, 2H), 2.18–2.14 (m, 1H), 2.08 (d, J = 37.0 Hz, 1H), 1.94 (d, J = 8.6 Hz, 1H), 1.89 (s, 1H), 1.64 (s, 3H), 1.59 (d, J = 11.3 Hz, 1H), 1.55–1.44 (m, 1H), 1.37 (s, 9H), 0.94 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.22, 174.10, 172.46, 170.14, 155.99, 78.47, 64.95, 58.62, 50.88, 41.48, 37.92, 35.47, 35.07, 34.59, 31.33, 28.63, 28.04, 27.83, 26.78, 21.50. ESI-MS m/z 508.56 [M + H]+.
4.2.8. Synthesis of (1R,3S,4S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-2-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)-2-azabicyclo[2.2.1]heptane-3-carboxamide (1b)
The synthesis of 1b was achieved from 1 to 5b using a procedure similar to that described for the preparation of 1a. Yield 23.0%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.27 (d, J = 8.7 Hz, 1H), 8.85 (d, J = 8.6 Hz, 1H), 7.65 (s, 1H), 4.96 (ddd, J = 10.8, 8.6, 5.2 Hz, 1H), 4.67–4.52 (m, 2H), 3.77 (s, 1H), 3.21–2.98 (m, 2H), 2.39 (qd, J = 10.0, 4.3 Hz, 1H), 2.18–2.11 (m, 1H), 2.08 (d, J = 5.3 Hz, 3H), 1.76–1.62 (m, 4H), 1.62–1.53 (m, 1H), 1.36 (dd, J = 21.9, 10.1 Hz, 2H), 1.00 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.09, 169.93, 166.33, 157.28, 117.79, 114.93, 64.62, 58.67, 58.13, 41.60, 38.17, 37.24, 35.43, 35.36, 34.80, 31.15, 31.09, 27.90, 27.40, 26.61. ESI-HRMS Calcd for C22H29F3N5O4.
[M−H]-: 484.2177, found 484.2175. HPLC: t = 7.864 min, 96.1% purity.
4.2.9. Synthesis of methyl(S)-5-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-5-azaspiro[2.4]heptane-6-carboxylate (1-2c)
Compound 1-2c was synthesized from compound 1-1c with N-Boc-l-tert-leucine using methods similar to the method described for the preparation of 1-2a. Yield 82.9%; colourless oil; 1H NMR (400 MHz, DMSO‑d 6) δ 6.59 (d, J = 9.1 Hz, 1H), 4.48 (dd, J = 8.6, 5.4 Hz, 1H), 4.14–4.01 (m, 1H), 3.62 (s, 3H), 3.54 (d, J = 9.8 Hz, 1H), 3.36 (s, 1H), 2.17 (dd, J = 12.7, 8.6 Hz, 1H), 1.79 (dd, J = 12.7, 5.5 Hz, 1H), 1.37 (s, 9H), 0.95 (s, 9H), 0.57 (ttd, J = 11.7, 7.6, 6.7, 3.1 Hz, 4H). 13C NMR (101 MHz, DMSO‑d 6) δ 172.36, 170.30, 156.04, 78.65, 59.05, 55.29, 52.13, 37.00, 35.13, 28.63, 26.66, 21.72, 10.78, 9.75. ESI-MS m/z 369.28 [M + H]+.
4.2.10. Synthesis of (S)-5-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-5-azaspiro [2.4]heptane-6-carboxylic acid (1-3c)
The compound 1-3c was synthesized from 1 to 2c using a procedure similar to that described for the preparation of 1-3a. Yield 96.0%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.31 (s, 1H), 6.56 (d, J = 9.1 Hz, 1H), 4.39 (dd, J = 8.6, 5.4 Hz, 1H), 4.07 (t, J = 4.9 Hz, 1H), 3.63 (d, J = 9.8 Hz, 1H), 3.53 (d, J = 9.8 Hz, 1H), 2.14 (dd, J = 12.6, 8.6 Hz, 1H), 1.80 (dd, J = 12.7, 5.4 Hz, 1H), 1.37 (s, 9H), 0.95 (s, 9H), 0.66–0.46 (m, 4H). 13C NMR (101 MHz, DMSO‑d 6) δ 173.34, 170.17, 156.03, 78.61, 58.90, 55.36, 37.13, 35.20, 28.63, 26.71, 21.62, 10.87, 9.91. ESI-MS m/z 355.04 [M + H]+.
4.2.11. Synthesis of tert-butyl((S)-1-((S)-6-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-5-azaspiro[2.4]heptan-5-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1-5c)
Compound 1-5c was synthesized from compound 1–4 with compound 1-3c using methods similar to the method described for the preparation of 1-5a. Yield 55.9%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.07 (d, J = 8.6 Hz, 1H), 7.56 (s, 1H), 7.24 (s, 1H), 7.01 (s, 1H), 6.64 (d, J = 9.1 Hz, 1H), 4.45 (t, J = 7.5 Hz, 1H), 4.26 (ddd, J = 12.2, 8.6, 3.6 Hz, 1H), 4.05 (d, J = 8.9 Hz, 1H), 3.61 (d, J = 9.8 Hz, 1H), 3.54 (d, J = 9.8 Hz, 1H), 3.13 (t, J = 9.2 Hz, 1H), 3.05 (td, J = 9.2, 6.9 Hz, 1H), 2.48–2.39 (m, 1H), 2.19 (dt, J = 13.8, 7.7 Hz, 1H), 1.91 (dtd, J = 19.3, 13.0, 12.4, 5.5 Hz, 3H), 1.63 (dq, J = 11.8, 9.2 Hz, 1H), 1.49 (ddd, J = 14.1, 10.9, 3.4 Hz, 1H), 1.37 (s, 9H), 0.93 (s, 9H), 0.63 (dt, J = 11.7, 4.3 Hz, 1H), 0.59–0.46 (m, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.24, 174.04, 171.68, 170.19, 156.13, 78.56, 60.61, 58.83, 55.82, 50.97, 37.91, 37.62, 35.14, 34.61, 28.63, 26.78, 21.88, 11.59, 9.26. ESI-MS m/z 508.56 [M + H]+.
4.2.12. Synthesis of (S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-5-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)-5-azaspiro[2.4]heptane-6-carboxamide (1c)
The synthesis of 1c was achieved from 1 to 5c using a procedure similar to that described for the preparation of 1a. Yield 49.6%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.38 (d, J = 8.6 Hz, 1H), 8.87 (d, J = 8.6 Hz, 1H), 7.66 (s, 1H), 4.98 (ddd, J = 10.9, 8.5, 5.1 Hz, 1H), 4.48 (d, J = 8.7 Hz, 1H), 4.38 (t, J = 7.5 Hz, 1H), 3.68 (d, J = 9.8 Hz, 1H), 3.51 (d, J = 9.8 Hz, 1H), 3.20–3.11 (m, 1H), 3.06 (td, J = 9.3, 7.0 Hz, 1H), 2.44 (qd, J = 8.2, 6.2, 3.0 Hz, 1H), 2.21–2.10 (m, 2H), 2.00 (dd, J = 12.6, 8.1 Hz, 1H), 1.86 (dd, J = 12.6, 7.0 Hz, 1H), 1.77–1.65 (m, 2H), 0.99 (s, 9H), 0.73–0.48 (m, 4H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.12, 171.58, 167.98, 157.06, 117.77, 114.91, 60.41, 58.36, 55.92, 38.22, 37.60, 37.22, 35.54, 34.81, 27.42, 26.64, 21.92, 11.63, 9.22. ESI-HRMS Calcd for C22H29F3N5O4 [M−H]-: 484.2177, found 484.2175. HPLC: t = 8.150 min, 99.8% purity.
4.2.13. Synthesis of methyl N-((R)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-N-methyl-l-leucinate (1-2d)
Compound 1-2d was synthesized from compound 1-1d with N-Boc-l-tert-leucine using methods similar to the method described for the preparation of 1-2a. Yield 66.1%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 6.60 (d, J = 9.1 Hz, 1H), 5.25 (dd, J = 11.6, 4.2 Hz, 1H), 4.35 (t, J = 4.6 Hz, 1H), 3.61 (s, 3H), 2.98 (s, 3H), 1.75 (ddd, J = 14.9, 11.6, 3.7 Hz, 1H), 1.56 (ddd, J = 14.1, 10.1, 4.2 Hz, 1H), 1.47–1.38 (m, 1H), 1.36 (s, 9H), 0.96 (s, 9H), 0.86 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 172.89, 172.19, 156.32, 78.64, 56.70, 54.14, 52.38, 36.94, 34.50, 32.24, 28.53, 26.81, 24.34, 21.66. ESI-MS m/z 373.07 [M + H]+.
4.2.14. Synthesis of N-((R)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-N-methyl-l-leucine (1-3d)
The compound 1-3d was synthesized from 1 to 2d using a procedure similar to that described for the preparation of 1-3a. Yield 98.5%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.67 (s, 1H), 6.55 (d, J = 9.2 Hz, 1H), 5.20 (dd, J = 11.7, 4.1 Hz, 1H), 4.41–4.31 (m, 1H), 2.97 (s, 3H), 1.71 (td, J = 13.0, 11.7, 3.8 Hz, 1H), 1.62–1.48 (m, 2H), 1.36 (s, 9H), 0.96 (s, 9H), 0.82 (dd, J = 23.4, 6.5 Hz, 6H). 13C NMR (101 MHz, DMSO‑d 6) δ 173.42, 172.77, 156.27, 78.61, 56.66, 53.81, 37.07, 34.55, 31.97, 28.52, 26.86, 24.43, 21.69. ESI-MS m/z 359.04 [M + H]+.
4.2.15. Synthesis of tert-butyl((R)-1-(((S)-1-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)(methyl)amino)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1-5d)
Compound 1-5d was synthesized from compound 1–4 with compound 1-3d using methods similar to the method described for the preparation of 1-5a. Yield 41.2%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 7.89 (d, J = 8.2 Hz, 1H), 7.60 (s, 1H), 7.28 (s, 1H), 7.01 (s, 1H), 6.53 (d, J = 9.2 Hz, 1H), 5.16 (dd, J = 10.7, 5.0 Hz, 1H), 4.34 (d, J = 9.2 Hz, 1H), 4.19 (ddd, J = 11.8, 8.2, 4.1 Hz, 1H), 3.15 (t, J = 9.2 Hz, 1H), 3.09–3.04 (m, 1H), 3.01 (s, 3H), 2.21–2.13 (m, 1H), 2.13–2.03 (m, 1H), 2.01–1.92 (m, 1H), 1.67 (d, J = 3.6 Hz, 1H), 1.66–1.63 (m, 1H), 1.61 (dd, J = 7.7, 3.5 Hz, 1H), 1.54–1.48 (m, 1H), 1.48–1.43 (m, 1H), 1.36 (s, 9H), 0.93 (s, 9H), 0.86 (d, J = 6.4 Hz, 3H), 0.81 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.66, 173.89, 172.45, 171.28, 156.25, 78.63, 56.83, 53.92, 51.35, 38.17, 37.30, 34.64, 34.05, 31.73, 27.79, 26.86, 24.57, 23.78, 22.03. ESI-MS m/z 512.54 [M + H]+.
4.2.16. Synthesis of (S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-4-methyl-2-((R)-N,3,3-trimethyl-2-(2,2,2-trifluoroacetamido)butanamido)pentanamide (1d)
The synthesis of 1d was achieved from 1 to 5d using a procedure similar to that described for the preparation of 1a. Yield 42.7%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.30 (d, J = 8.7 Hz, 1H), 8.86 (d, J = 7.9 Hz, 1H), 7.72 (s, 1H), 4.99 (dd, J = 9.9, 5.8 Hz, 1H), 4.92 (ddd, J = 9.6, 7.9, 6.5 Hz, 1H), 4.81 (d, J = 8.8 Hz, 1H), 3.20–3.07 (m, 2H), 3.05 (s, 3H), 2.25 (qd, J = 9.3, 5.4 Hz, 1H), 2.16–2.05 (m, 2H), 1.81–1.74 (m, 1H), 1.73–1.67 (m, 1H), 1.63 (dt, J = 10.0, 4.7 Hz, 1H), 1.60–1.52 (m, 1H), 1.42–1.31 (m, 1H), 0.97 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.82 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 177.87, 171.21, 170.34, 157.35, 117.78, 114.92, 56.21, 54.73, 38.73, 37.54, 37.35, 35.50, 33.84, 32.62, 27.42, 26.69, 25.03, 23.39, 21.88. ESI-HRMS Calcd for C22H33F3N5O4 [M−H]-: 488.2490, found 488.2487. HPLC: t = 16.566 min, 96.0% purity.
4.2.17. Synthesis of methyl(S)-4-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl) morpholine-3-carboxylate (1-2e)
Compound 1-2e was synthesized from compound 1-1e with N-Boc-l-tert-leucine using methods similar to the method described for the preparation of 1-2a. Yield 59.5%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 5.07 (d, J = 3.4 Hz, 1H), 4.45 (dt, J = 13.6, 4.9 Hz, 1H), 4.24 (d, J = 3.3 Hz, 1H), 4.20 (d, J = 12.2 Hz, 1H), 4.08–3.96 (m, 1H), 3.83 (dd, J = 10.7, 7.6 Hz, 2H), 3.69 (s, 3H), 3.64 (s, 1H), 3.54 (dt, J = 16.2, 8.6, 7.2, 3.9 Hz, 1H), 1.39 (d, J = 5.0 Hz, 9H), 0.94 (d, J = 7.0 Hz, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 171.68, 170.39, 156.00, 78.53, 67.73, 66.49, 55.84, 52.76, 52.24, 44.43, 35.51, 28.60, 26.75. ESI-MS m/z 359.05 [M + H]+.
4.2.18. Synthesis of (S)-4-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)morpholine-3-carboxylic acid (1-3e)
The compound 1-3e was synthesized from 1 to 2e using a procedure similar to that described for the preparation of 1-3a. Yield 97.7%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.90 (s, 1H), 6.64 (d, J = 9.2 Hz, 1H), 4.94 (d, J = 3.7 Hz, 1H), 4.50–4.39 (m, 1H), 4.37–4.19 (m, 1H), 3.99 (d, J = 12.2 Hz, 1H), 3.88–3.78 (m, 1H), 3.50 (ddd, J = 15.8, 8.2, 3.2 Hz, 1H), 3.42–3.26 (m, 2H), 1.37 (d, J = 10.2 Hz, 9H), 0.92 (d, J = 13.9 Hz, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 172.45, 171.45, 155.97, 78.72, 67.95, 66.54, 56.93, 52.17, 44.45, 35.56, 28.61, 26.79. ESI-MS m/z 706.05 [2 M + NH4]+.
4.2.19. Synthesis of tert-butyl((S)-1-((S)-3-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl) carbamoyl)morpholino)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1-5e)
Compound 1-5e was synthesized from compound 1–4 with compound 1-3e using methods similar to the method described for the preparation of 1-5a. Yield 33.4%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 7.86 (d, J = 8.6 Hz, 1H), 7.62 (d, J = 11.9 Hz, 1H), 7.36 (d, J = 6.4 Hz, 1H), 7.08 (d, J = 6.8 Hz, 1H), 6.49 (d, J = 8.2 Hz, 1H), 4.77 (d, J = 3.4 Hz, 1H), 4.41 (d, J = 8.2 Hz, 1H), 4.38–4.30 (m, 1H), 4.27 (d, J = 11.5 Hz, 1H), 3.94 (d, J = 13.3 Hz, 1H), 3.80 (dd, J = 11.1, 3.4 Hz, 1H), 3.60 (ddt, J = 15.1, 12.2, 7.8 Hz, 1H), 3.47 (dt, J = 12.1, 3.1 Hz, 1H), 3.31 (s, 1H), 3.14 (dddd, J = 17.7, 14.8, 8.9, 3.6 Hz, 2H), 2.24–2.09 (m, 2H), 1.97 (tdd, J = 10.4, 7.7, 3.4 Hz, 1H), 1.67 (dddd, J = 12.9, 8.8, 6.1, 3.4 Hz, 1H), 1.55 (ddp, J = 15.6, 10.5, 5.8 Hz, 1H), 1.39 (d, J = 5.8 Hz, 9H), 0.90 (d, J = 27.6 Hz, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.72, 173.68, 171.55, 169.07, 155.98, 78.82, 68.53, 66.17, 55.38, 52.91, 51.16, 44.12, 38.20, 35.14, 34.44, 28.68, 27.86, 26.84. ESI-MS m/z 498.30 [M + H]+.
4.2.20. Synthesis of (S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-4-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)morpholine-3-carboxamide (1e)
The synthesis of 1e was achieved from 1 to 5e using a procedure similar to that described for the preparation of 1a. Yield 43.5%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.35 (d, J = 8.4 Hz, 1H), 8.59 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 5.2 Hz, 1H), 5.06–4.96 (m, 1H), 4.89 (d, J = 8.4 Hz, 1H), 4.72 (d, J = 3.7 Hz, 1H), 4.24 (dd, J = 12.2, 4.9 Hz, 1H), 4.05–3.94 (m, 1H), 3.84 (dt, J = 12.1, 5.9 Hz, 1H), 3.63–3.54 (m, 1H), 3.50–3.43 (m, 1H), 3.43–3.38 (m, 1H), 3.29–3.07 (m, 2H), 2.35–2.22 (m, 1H), 2.15 (dddt, J = 11.3, 9.2, 5.3, 2.6 Hz, 1H), 2.11–1.96 (m, 1H), 1.81 (dt, J = 13.5, 8.3 Hz, 1H), 1.76–1.63 (m, 1H), 0.97 (d, J = 20.4 Hz, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 177.85, 170.09, 169.07, 157.05, 117.77, 114.91, 68.06, 66.02, 55.70, 53.16, 44.26, 39.06, 37.61, 35.52, 35.05, 34.07, 27.51, 26.72. ESI-HRMS Calcd for C20H27F3N5O5 [M−H]-: 474.1970, found 474.1965. HPLC: t = 10.992 min, 99.2% purity.
4.2.21. Synthesis of methyl (1R,2S)-1-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanamido)-2-vinylcyclopropane-1-carboxylate (1-2f)
Compound 1-2f was synthesized from compound 1-1f with N-Boc-l-tert-leucine using methods similar to the method described for the preparation of 1-2a. Yield 71.7%; colourless oil; 1H NMR (400 MHz, DMSO‑d 6) δ 8.69 (s, 1H), 5.69–5.54 (m, 1H), 5.27 (dd, J = 17.2, 2.0 Hz, 1H), 5.09 (dd, J = 10.2, 2.0 Hz, 1H), 3.78 (t, J = 4.9 Hz, 1H), 3.57 (s, 3H), 2.10 (q, J = 8.7 Hz, 1H), 1.65 (dd, J = 7.9, 5.2 Hz, 1H), 1.39 (s, 9H), 1.28–1.22 (m, 1H), 0.89 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 171.64, 170.76, 155.70, 134.56, 118.11, 78.59, 62.18, 52.32, 34.66, 32.96, 28.59, 26.99, 22.89, 21.50. ESI-MS m/z 355.30 [M + H]+.
4.2.22. Synthesis of (1R,2S)-1-((S)-2-((tert-butoxycarbonylamino)-3,3-dimethylbutanamido)-2-vinylcyclopropane-1-carboxylic acid (1-3f)
The compound 1-3f was synthesized from 1 to 2f using a procedure similar to that described for the preparation of 1-3a. Yield 99.5%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.34 (s, 1H), 8.60 (s, 1H), 6.27 (d, J = 9.6 Hz, 1H), 5.76–5.60 (m, 1H), 5.26 (dd, J = 17.2, 2.0 Hz, 1H), 5.07 (dd, J = 10.3, 2.1 Hz, 1H), 3.86–3.72 (m, 1H), 2.04 (q, J = 8.8 Hz, 1H), 1.60 (dd, J = 7.8, 5.1 Hz, 1H), 1.38 (s, 9H), 1.27–1.15 (m, 1H), 0.88 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 171.94, 171.33, 155.65, 135.19, 117.51, 78.59, 62.09, 34.87, 32.85, 28.60, 27.01, 22.72, 21.49. ESI-MS m/z 698.67 [2 M + NH4]+.
4.2.23. Synthesis of tert-butyl((S)-1-(((1R,2S)-1-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl) propan-2-yl)carbamoyl)-2-vinylcyclopropyl)amino)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1-5f)
Compound 1-5f was synthesized from compound 1–4 with compound 1-3f using methods similar to the method described for the preparation of 1-5a. Yield 44.5%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.67 (s, 1H), 7.55 (s, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.16 (d, J = 15.0 Hz, 2H), 6.69 (d, J = 7.1 Hz, 1H), 5.60 (dt, J = 17.3, 9.7 Hz, 1H), 5.20 (dd, J = 17.2, 2.0 Hz, 1H), 4.99 (dd, J = 10.1, 2.1 Hz, 1H), 4.34–4.24 (m, 1H), 3.63 (d, J = 7.2 Hz, 1H), 3.13 (t, J = 9.1 Hz, 1H), 3.09–2.99 (m, 1H), 2.17–2.10 (m, 2H), 2.10–1.98 (m, 1H), 1.92 (s, 1H), 1.71–1.65 (m, 1H), 1.64–1.50 (m, 1H), 1.39 (s, 9H), 1.28 (d, J = 41.6 Hz, 1H), 1.07 (dd, J = 9.3, 5.0 Hz, 1H), 0.92 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.67, 173.58, 173.01, 172.45, 156.44, 135.42, 116.85, 78.96, 63.63, 51.43, 41.51, 38.01, 33.88, 32.13, 28.72, 28.07, 27.20, 21.50, 21.21. ESI-MS m/z 494.50 [M + H]+.
4.2.24. Synthesis of (1R,2S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-1-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanamido)-2-vinylcyclopropane-1-carboxamide (1f)
The synthesis of 1f was achieved from 1 to 5f using a procedure similar to that described for the preparation of 1a. Yield 41.2%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.21 (d, J = 7.9 Hz, 1H), 8.89 (s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.65 (s, 1H), 5.59 (ddd, J = 17.1, 10.3, 8.8 Hz, 1H), 5.21 (dd, J = 17.2, 2.0 Hz, 1H), 5.04 (dd, J = 10.3, 2.0 Hz, 1H), 4.98 (ddd, J = 9.9, 5.9, 4.2 Hz, 1H), 4.18–4.11 (m, 1H), 3.14 (t, J = 9.3 Hz, 1H), 3.06 (td, J = 9.2, 7.1 Hz, 1H), 2.28 (qd, J = 9.7, 6.2 Hz, 1H), 2.13 (t, J = 5.0 Hz, 1H), 2.11–2.09 (m, 1H), 2.09–2.06 (m, 1H), 2.04 (d, J = 8.8 Hz, 1H), 1.77–1.72 (m, 1H), 1.71–1.67 (m, 1H), 1.67–1.60 (m, 1H), 0.96 (s, 9H). 13C NMR (101 MHz, DMSO‑d 6) δ 177.92, 170.36, 169.10, 157.64, 134.75, 119.88, 117.47, 114.87, 61.85, 41.25, 38.85, 37.28, 34.73, 33.85, 32.06, 29.48, 27.44, 27.02, 21.22. ESI-HRMS Calcd for C21H27F3N5O4 [M−H]-: 470.2021, found 470.2017. HPLC: t = 6.600 min, 96.3% purity.
4.3. General synthetic procedure for the preparation 2(a–d)
4.3.1. Synthesis of methyl(1R,2S,5S)-3-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxylate (2–2)
Compound 2–1 (5.5 g, 26.7 mmol) and (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (5.6 g, 24.2 mmol) were dissolved in DCM/DMF (V:V = 1:1, 60 mL) solution. Then 4-Methylmorpholine (7.3 g, 72.3 mmol), BOP (11.8 g, 26.7 mmol) were added to the reaction at 25 °C and stirred for 10 h. Then DCM (60 mL) and 1 N HCl aqueous solution (10 mL) were added to the reaction. After separation, the organic phase was washed with H2O (40 mL), the organic phase was washed with saturated brine (20 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, PE/EA = 20/1 ∼ 8/1) to give the compound 2–2. Yield 63.0%; colourless oil; 1H NMR (400 MHz, DMSO‑d 6) δ 6.71 (d, J = 9.3 Hz, 1H), 4.21 (s, 1H), 4.09–4.03 (m, 1H), 3.93 (d, J = 10.4 Hz, 1H), 3.83–3.76 (m, 1H), 3.65 (s, 3H), 1.52 (dt, J = 8.6, 4.4 Hz, 1H), 1.41 (d, J = 7.6 Hz, 1H), 1.35 (s, 9H), 1.01 (s, 3H), 0.94 (s, 9H), 0.85 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 171.99, 170.67, 156.38, 78.70, 59.22, 52.37, 47.51, 34.49, 30.04, 28.52, 26.72, 19.42, 12.59. ESI-MS m/z 383.30 [M + H]+.
4.3.2. Synthesis of (1R,2S,5S)-3-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxylic acid (2–3)
Compound 2–2 (5.7 g, 14.9 mmol), lithium hydroxide (2.4 g, 59.2 mmol) and water (65 mL) were dissolved in THF (65 mL) solution at 40 °C and stirred for 2 h. Then 1 N HCl aqueous solution was added to the reaction to adjust the pH 2–3. EA (100 mL) was added to the solution and separation, the organic phase was washed with saturated brine (30 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, DCM/MeOH = 100/1–30/1) to give the compound 2–3. Yield 90.9%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 12.44 (s, 1H), 6.66 (d, J = 9.5 Hz, 1H), 4.13 (s, 1H), 4.08–4.03 (m, 1H), 3.91 (d, J = 10.4 Hz, 1H), 3.78 (tq, J = 8.8, 4.3, 3.8 Hz, 1H), 1.50 (dd, J = 7.6, 5.0 Hz, 1H), 1.39 (d, J = 7.6 Hz, 1H), 1.35 (s, 9H), 1.01 (s, 3H), 0.93 (s, 9H), 0.84 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 173.06, 170.52, 156.38, 78.67, 59.34, 59.02, 47.51, 34.56, 30.24, 28.53, 26.78, 21.49, 19.28, 12.66. ESI-MS m/z 369.30 [M + H]+.
4.3.3. Synthesis of tert-butyl((S)-1-((1R,2S,5S)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-3-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (2–4)
Compound 1–4 (3.7 g, 13.6 mmol) and 4 N HCl in 1,4-dioxane (4 mL) were dissolved in DCM (6 mL), the reaction was stirred at 25 °C for 2 h. The reaction was concentrated to remove the solvent and dry HCl salt was obtained. Meanwhile, compound 2–3 (5.0 g, 13.6 mmol) and HATU (7.7 g, 20.3 mmol) were dissolved in DCM (20 mL), the reaction was stirred at 25 °C for 30 min. Then N, N-Diisopropylethylamine (3.9 g, 29.9 mmol) and HCl salt were added to the reaction, the reaction was stirred at 25 °C for 10 h. Then DCM (40 mL) and 1 N HCl aqueous solution (10 mL) were added to the reaction. After separation, the organic phase was washed with H2O (10 mL), the organic phase was washed with saturated brine (10 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, DCM/MeOH = 150/1–20/1) to give the compound 2–4. Yield 45.1%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.23 (d, J = 8.7 Hz, 1H), 7.54 (s, 1H), 7.27 (s, 1H), 7.02 (s, 1H), 6.61 (d, J = 9.3 Hz, 1H), 4.34–4.20 (m, 2H), 4.07–3.98 (m, 1H), 3.89–3.77 (m, 2H), 3.12 (t, J = 9.2 Hz, 1H), 3.02 (td, J = 9.3, 7.0 Hz, 1H), 2.47–2.32 (m, 1H), 2.20–2.05 (m, 1H), 2.01–1.87 (m, 1H), 1.61 (dq, J = 11.9, 9.3 Hz, 1H), 1.53–1.48 (m, 1H), 1.46 (dt, J = 7.2, 3.5 Hz, 1H), 1.36 (s, 1H), 1.34 (s, 9H), 1.01 (s, 3H), 0.91 (s, 9H), 0.86 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.18, 174.02, 171.27, 168.88, 164.88, 136.53, 130.86, 130.53, 129.13, 124.93, 60.70, 58.51, 50.83, 48.10, 37.83, 35.33, 34.57, 31.08, 27.63, 27.08, 26.34, 19.06, 12.99. ESI-MS m/z 522.34 [M + H]+.
4.3.4. Synthesis of (1R,2S,5S)-N-((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-3-((S)-3,3-dimethyl-2-(2,2,3,3,3-pentafluoropropanamido)butanoyl)-6,6-dimethyl-3-azabicyclo [3.1.0] hexane-2-carboxamide (2-5a)
Compound 2–4 (450 mg, 0.9 mmol) and 4 N HCl in 1,4-dioxane (3 mL) were dissolved in DCM (6 mL), the reaction was stirred at 40 °C for 1 h. The reaction was concentrated to remove the solvent. Then triethylamine (261 mg, 2.6 mmol), Perfluoropropionic anhydride (294 mg, 0.9 mmol) and DCM (6 mL) were added to the reaction at 0 °C. The reaction was allowed to warm to 25 °C, and stirred for 10 h. Then DCM (10 mL) and 1 N HCl aqueous solution (3 mL) were added to the reaction. After separation, the organic phase was washed with H2O (5 mL), the organic phase was washed with saturated brine (5 mL), dried over anhydrous sodium sulphate, evaporated in vacuum and purified by column chromatography (silica gel, DCM/MeOH = 100/1 ∼ 20/1) to give the compound 2-5a. Yield 65.4%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.39 (d, J = 8.9 Hz, 1H), 8.29 (d, J = 8.8 Hz, 1H), 7.55 (s, 1H), 7.30 (s, 1H), 7.03 (s, 1H), 4.58–4.47 (m, 1H), 4.35–4.23 (m, 2H), 3.89 (dd, J = 10.3, 5.4 Hz, 1H), 3.66 (d, J = 10.4 Hz, 1H), 3.13 (t, J = 9.2 Hz, 1H), 3.03 (td, J = 9.3, 7.0 Hz, 1H), 2.15 (tt, J = 14.0, 7.7 Hz, 1H), 1.99–1.92 (m, 1H), 1.64 (dq, J = 11.8, 9.2 Hz, 1H), 1.52 (d, J = 13.4 Hz, 1H), 1.49 (d, J = 2.6 Hz, 1H), 1.47 (s, 1H), 1.43–1.32 (m, 1H), 1.01 (s, 3H), 0.98 (s, 9H), 0.81 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.11, 173.98, 172.44, 171.07, 167.52, 158.21, 157.96, 60.70, 58.37, 50.82, 48.18, 37.80, 35.28, 34.55, 30.99, 27.87, 27.48, 26.70, 26.32, 21.49, 18.98, 12.66. ESI-MS m/z 567.87 [M + H]+.
4.3.5. Synthesis of (1R,2S,5S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-3-((S)-3,3-dimethyl-2-(2,2,3,3,3-pentafluoropropanamido)butanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (2a)
Compound 2-5a (170 mg, 0.3 mmol) and Burgess reagent (127 mg, 0.5 mmol) were dissolved in DCM (5 mL), the reaction was stirred at 25 °C for 10 h. The reaction was purified by column chromatography (silica gel, DCM/MeOH = 150/1 ∼ 40/1) to give the compound 2a. Yield 42.5%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.40 (d, J = 8.8 Hz, 1H), 9.02 (d, J = 8.5 Hz, 1H), 7.67 (s, 1H), 4.98 (ddd, J = 10.9, 8.5, 5.1 Hz, 1H), 4.51 (d, J = 8.8 Hz, 1H), 4.16 (s, 1H), 3.92 (dd, J = 10.4, 5.5 Hz, 1H), 3.69 (d, J = 10.4 Hz, 1H), 3.22–3.12 (m, 1H), 3.05 (td, J = 9.3, 7.0 Hz, 1H), 2.20–2.14 (m, 1H), 2.11 (dd, J = 7.3, 4.1 Hz, 1H), 2.08 (dd, J = 8.5, 5.6 Hz, 1H), 1.77–1.72 (m, 1H), 1.72–1.67 (m, 1H), 1.57 (dd, J = 7.6, 5.3 Hz, 1H), 1.35–1.28 (m, 1H), 1.03 (s, 3H), 0.98 (s, 9H), 0.83 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 177.98, 171.14, 167.73, 158.30, 158.04, 157.78, 120.08, 60.51, 58.43, 48.08, 38.26, 37.21, 35.19, 34.61, 30.71, 27.70, 27.33, 26.67, 26.16, 19.23, 12.59. ESI-HRMS Calcd for C24H31F5N5O4 [M−H]-: 548.2302, found 548.2305. HPLC: t = 9.669 min, 99.0% purity.
4.3.6. Synthesis of (1R,2S,5S)-N-((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-3-((S)-3,3-dimethyl-2-((trifluoromethyl)sulfonamido)butanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (2-5b)
The compound 2-5b was synthesized from 2 to 4 using a procedure similar to that described for the preparation of 2-5a. Yield 21.0%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.63 (s, 1H), 8.30 (d, J = 9.0 Hz, 1H), 7.51 (s, 1H), 7.32 (s, 1H), 7.01 (s, 1H), 4.31 (s, 1H), 4.31–4.25 (m, 1H), 3.87 (s, 1H), 3.48 (d, J = 10.3 Hz, 1H), 3.16–3.07 (m, 1H), 3.01 (td, J = 9.3, 7.1 Hz, 1H), 2.46–2.35 (m, 1H), 2.13 (dt, J = 14.0, 8.0 Hz, 1H), 2.03–1.90 (m, 1H), 1.67 (dd, J = 11.0, 7.8 Hz, 1H), 1.64–1.54 (m, 1H), 1.53–1.50 (m, 1H), 1.48 (s, 1H), 1.38 (d, J = 7.7 Hz, 1H), 1.02 (s, 3H), 1.00 (s, 9H), 0.88 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.09, 173.99, 172.45, 171.06, 167.80, 63.18, 61.06, 50.66, 48.08, 37.71, 36.03, 34.73, 31.27, 28.01, 26.73, 26.33, 19.16, 12.94. ESI-MS m/z 554.11 [M + H]+.
4.3.7. Synthesis of (1R,2S,5S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-3-((S)-3,3-dimethyl-2-((trifluoromethyl)sulfonamido)butanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (2b)
The compound 2b was synthesized from 2 to 5b using a procedure similar to that described for the preparation of 2a. Yield 57.0%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.65 (d, J = 9.1 Hz, 1H), 9.08 (d, J = 8.6 Hz, 1H), 7.65 (s, 1H), 4.97 (ddd, J = 11.1, 8.6, 4.9 Hz, 1H), 4.18 (s, 1H), 3.97–3.84 (m, 2H), 3.50 (d, J = 10.4 Hz, 1H), 3.14 (t, J = 9.2 Hz, 1H), 3.03 (td, J = 9.2, 7.0 Hz, 1H), 2.39 (td, J = 10.4, 9.7, 3.9 Hz, 1H), 2.22–2.02 (m, 2H), 1.77–1.65 (m, 2H), 1.58 (dd, J = 7.8, 5.5 Hz, 1H), 1.33 (d, J = 7.7 Hz, 1H), 1.04 (s, 3H), 0.99 (s, 9H), 0.89 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 177.94, 171.08, 168.06, 120.09, 63.09, 60.90, 55.37, 48.02, 38.13, 37.14, 35.99, 34.78, 30.92, 28.24, 27.28, 26.66, 26.15, 19.41, 12.85. ESI-HRMS Calcd for C22H31F3N5O5S [M−H]-: 534.2003, found 534.2003. HPLC: t = 12.290 min, 97.3% purity.
4.3.8. Synthesis of (1R,2S,5S)-N-((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-3-((S)-2-(cyclopropanesulfonamido)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (2-5c)
The compound 2-5c was synthesized from 2 to 4 using a procedure similar to that described for the preparation of 2-5a. Yield 29.8%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.27 (d, J = 8.9 Hz, 1H), 7.52 (s, 1H), 7.30 (d, J = 2.1 Hz, 1H), 7.06–6.99 (m, 2H), 4.37–4.23 (m, 2H), 3.83 (dd, J = 10.1, 5.5 Hz, 1H), 3.80–3.75 (m, 1H), 3.66 (d, J = 10.3 Hz, 1H), 3.13 (d, J = 9.4 Hz, 1H), 3.11–3.03 (m, 1H), 3.01 (dd, J = 9.3, 7.2 Hz, 1H), 2.48–2.43 (m, 1H), 2.42–2.37 (m, 1H), 2.19–2.13 (m, 1H), 2.13–2.08 (m, 1H), 2.00–1.90 (m, 1H), 1.68–1.54 (m, 1H), 1.48 (qd, J = 8.0, 6.7, 3.5 Hz, 2H), 1.36 (d, J = 7.7 Hz, 1H), 1.01 (s, 3H), 0.97 (s, 9H), 0.90 (s, 3H), 0.89–0.84 (m, 2H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.14, 174.06, 171.31, 169.64, 61.18, 60.79, 50.73, 48.00, 37.73, 35.70, 34.59, 31.08, 27.99, 26.87, 26.44, 19.18, 13.37, 6.06, 5.07. ESI-MS m/z 526.37 [M + H]+.
4.3.9. Synthesis of (1R,2S,5S)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-3-((S)-2-(cyclopropanesulfonamido)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (2c)
The compound 2c was synthesized from 2 to 5c using a procedure similar to that described for the preparation of 2a. Yield 29.8%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 9.03 (d, J = 8.6 Hz, 1H), 7.64 (s, 1H), 7.07 (d, J = 9.5 Hz, 1H), 4.97 (ddd, J = 11.1, 8.6, 5.0 Hz, 1H), 4.16 (s, 1H), 3.85 (dd, J = 10.2, 5.6 Hz, 1H), 3.78 (d, J = 9.5 Hz, 1H), 3.68 (d, J = 10.3 Hz, 1H), 3.14 (d, J = 9.2 Hz, 1H), 3.12–3.05 (m, 1H), 3.02 (dd, J = 9.3, 7.0 Hz, 1H), 2.42 (qd, J = 10.2, 4.8 Hz, 1H), 2.20–2.15 (m, 1H), 2.15–2.09 (m, 1H), 2.09–2.03 (m, 1H), 1.77–1.71 (m, 1H), 1.70 (dd, J = 5.2, 2.4 Hz, 1H), 1.69–1.65 (m, 1H), 1.56 (dd, J = 7.7, 5.4 Hz, 1H), 1.29 (d, J = 7.7 Hz, 1H), 1.03 (s, 3H), 0.97 (s, 9H), 0.91 (s, 3H), 0.89–0.83 (m, 2H). 13C NMR (101 MHz, DMSO‑d 6) δ 177.99, 171.35, 169.88, 120.15, 61.11, 60.59, 47.92, 38.14, 37.14, 35.64, 34.68, 31.05, 28.21, 27.30, 26.82, 26.28, 19.43, 13.29, 6.07, 5.03. ESI-HRMS Calcd for C24H36N5O5S [M−H]-: 506.2443, found 506.2442. HPLC: t = 7.178 min, 98.3% purity.
4.3.10. Synthesis of (1R,2S,5S)-N-((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-3-((S)-2-(3,5-bis(trifluoromethyl)benzamido)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicyclo [3.1.0]hexane-2-carboxamide (2-5d).
The compound 2-5d was synthesized from 2 to 4 using a procedure similar to that described for the preparation of 2-5a. Yield 56.1%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.93 (d, J = 8.7 Hz, 1H), 8.46 (d, J = 1.7 Hz, 2H), 8.32–8.24 (m, 2H), 7.55 (s, 1H), 7.30 (d, J = 2.1 Hz, 1H), 7.07–7.01 (m, 1H), 4.68 (d, J = 8.7 Hz, 1H), 4.36–4.26 (m, 2H), 3.94 (dd, J = 10.3, 5.4 Hz, 1H), 3.81 (d, J = 10.3 Hz, 1H), 3.15 (t, J = 9.1 Hz, 1H), 3.09–3.01 (m, 1H), 2.46–2.32 (m, 1H), 2.23–2.12 (m, 1H), 1.94 (ddd, J = 13.5, 12.0, 3.8 Hz, 1H), 1.72–1.61 (m, 1H), 1.58–1.50 (m, 1H), 1.50–1.46 (m, 1H), 1.38 (d, J = 7.7 Hz, 1H), 1.04 (s, 9H), 1.00 (s, 3H), 0.84 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 179.18, 174.02, 171.27, 168.88, 164.88, 136.53, 130.86, 130.53, 129.13, 124.93, 60.70, 58.51, 50.83, 48.10, 37.83, 35.33, 34.57, 31.08, 27.91, 27.08, 26.34, 19.06, 12.99. ESI-MS m/z 662.11 [M + H]+.
4.3.11. Synthesis of (1R,2S,5S)-3-((S)-2-(3,5-bis(trifluoromethyl)benzamido)-3,3-dimethylbutanoyl)-N-((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (2d)
The compound 2d was synthesized from 2 to 5d using a procedure similar to that described for the preparation of 2a. Yield 40.9%; white solid; 1H NMR (400 MHz, DMSO‑d 6) δ 8.99 (d, J = 8.5 Hz, 1H), 8.92 (d, J = 8.6 Hz, 1H), 8.45 (s, 2H), 8.29 (s, 1H), 7.66 (s, 1H), 4.98 (ddd, J = 10.9, 8.5, 5.0 Hz, 1H), 4.66 (d, J = 8.6 Hz, 1H), 4.16 (s, 1H), 3.96 (dd, J = 10.3, 5.5 Hz, 1H), 3.83 (d, J = 10.4 Hz, 1H), 3.15 (t, J = 9.3 Hz, 1H), 3.05 (q, J = 8.6 Hz, 1H), 2.41 (td, J = 10.2, 9.6, 4.2 Hz, 1H), 2.21–2.05 (m, 2H), 1.80–1.64 (m, 2H), 1.57 (dd, J = 7.7, 5.3 Hz, 1H), 1.34–1.29 (m, 1H), 1.04 (s, 9H), 1.02 (s, 3H), 0.86 (s, 3H). 13C NMR (101 MHz, DMSO‑d 6) δ 178.01, 171.33, 169.06, 164.98, 136.51, 130.88, 130.55, 129.16, 124.93, 120.11, 60.50, 58.57, 48.01, 38.24, 38.15, 37.22, 35.24, 34.64, 30.81, 27.85, 27.04, 26.20, 19.31, 12.92. ESI-HRMS Calcd for C30H34F6N5O4 [M−H]-: 642.2520, found 642.2523. HPLC: t = 17.497 min, 97.4% purity.
4.4. Protein expression and purification
The cDNA of SARS-CoV-2 3CLpro (GenBank: MN908947.3) was cloned into the pGEX6p-1 vector. To obtain the SARS-CoV-2 3CLpro with authentic N and C terminals, four amino acids (AVLQ) were inserted between the GST tag and the full-length SARS-CoV-2 3CLpro, while eight amino acid (GPHHHHHH) were added to the C-terminal of SARS-CoV-2 3CLpro. The plasmid was then transformed into BL21 (DE3) cells for protein expression. The N terminal GST tag and four amino acids (AVLQ) was self-cleavable. The expressed protein with authentic N terminal was purified by a Ni-NTA column (GE Healthcare) and transformed into the cleavage buffer (150 mM NaCl, 25 mM Tris, pH 7.5) containing human rhinovirus 3C protease for removing the additional residues. The resulting protein sample was further passed through a size-exclusion chromatography (HiLoadTM 16/600 SuperdexTM 200 pg, GE Healthcare). The eluted protein samples were stored in a solution (10 mM Tris, pH 7.5) for the enzymatic inhibition assay.
4.5. Enzymatic inhibitory activity assay
A fluorescence resonance energy transfer (FRET) protease assay was applied to measure the inhibitory activity of compounds against SARS-CoV-2 3CLpro. The recombinant SARS-CoV-2 3CLpro at a concentration of 40 nM was mixed with serial dilutions of each compound in 80 μL of assay buffer (50 mM Tris–HCl, pH 7.3, 1 mM EDTA) and incubated for 10 min. The reaction was initiated by adding 40 μL of a fluorogenic substrate (Dacyl-KTSAVLQSGFRKME-Edans) at a final concentration of 10 μM. After that, the fluorescence signal at 340 nm (excitation)/490 nm (emission) was measured immediately every every 1 min for 10 min with a Bio-Tek SynergyH1 plate reader. The velocities of reactions with compounds added at various concentrations compared to the reaction added with DMSO were calculated and used to generate inhibition profiles. For each compound, experment in triplicate was performed to determine IC50 and SD values.
4.6. Anti-viral activity and cytotoxicity assays
The Vero E6 cell line was obtained from American Type Culture Collection (ATCC, Manassas, USA) and maintained in minimum Eagle’s medium (MEM; Gibco Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen, UK) in a humid incubator with 5% CO2 at 37 °C. The cytotoxicity of tested compounds on the Vero E6 cells were determined by CCK8 assays (Beyotime, China). A clinical isolate SARS-CoV-2 was propagated in the Vero E6 cells, and the viral titer was determined by 50% tissue culture infective dose (TCID50) using immunofluorescence assay. All the infection experiments were performed at biosafety level-3 (BSL-3).
Preseeded Vero E6 cells (5 × 104 cells/well) were incubated with different concentrations of compounds for 1 h in 48-well plate, and the virus was subsequently added (a multiplicity of infection of 0.01) to infect the cells for 2 h. After that, the virus–compound mixture was removed, and the cells were further cultured with a fresh compound-containing medium. At 24 h post infection, the cell supernatant was collected, and the viral RNA in the supernatant was submitted to quantitative real-time RT-PCR (qRT-PCR) analysis. DMSO was used in the controls. The half maximal effective concentration (EC50) values were calculated by GraphPad Prism 8.3.0 software.
Vero E6 cells were plated in the 96-well plates at a density of 2.5 × 103 cells per well for 16 h. Then the cells were incubated with the test articles at different cocentrations (0.5–200 µM) for another 24 h (n = 3). Cell viability was determined using the CCK8 assay kit after cells were incubated. The absorbance was measured by an automatic microplate reader (Biotek, Winooski, VT, USA) at OD450. The half cytotoxicity concentration (CC50) values for each compound were calculated by GraphPad Prism 8.3.0 software (GraphPad Software Inc., La Jolla, CA, USA).
4.7. Rat pharmacokinetics
Rat pharmacokinetics studies were done at Medicilon (Shanghai, China); tested male rats was administered via intravenous injection or oral administration. The blood was taken via submandibular vein or other suitable vein. Sample was placed in tubes containing K2-EDTA and stored on ice until centrifuged. The blood sample was centrifuged at 6800 g for 6 min at 2–8 °C within 1 h after collected and stored frozen at approximately −80 °C. An aliquot of 15 µL plasma sample was protein precipitated with 300 µL MeOH in which contains 100 ng/mL Warfarin (IS). The mixture was vortexed for 1 min and centrifuged at 18,000 g for 7 min. Transfer 300 µL supernatant to 96 well plates. An aliquot of 6 µL supernatant was injected for LC-MS/MS analysis. Standard set of parameters was calculated using noncompartmental analysis modules in FDA certified pharmacokinetic program Phoenix WinNonlin 7.0 (Pharsight, USA).
4.8. Liver microsomes metabolic stability
Human, mouse liver microsomes (from Xenotech) with final liver microsomal protein concentration of 0.5 mg/mL. 5 µL of 10 mM compound stock solution and reference solution were added to 95 µL ACN. 1.5 µL of 500 µM spiked solution and 18.75 µL of 20 mg/mL liver microsomes were added to 479.75 µL of K/Mg- buffer. NADPH stock solution (6 mM, 5 mg/mL) was prepared by dissolving NADPH into K/Mg-buffer. Dispense 30 µL of 1.5 µM spiking solution containing 0.75 mg/mL microsomes solution to the assay plates designated for different time points (0, 5, 15, 30, 45 min). Pre-incubate other plate at 37 °C for 5 min. For 0 min, added 150 µL of ACN containing IS to the wells before adding 15 µL of NADPH stock solution (6 mM). For other time points, added 15 µL of NADPH stock solution (6 mM) to the wells to start the reaction and timing. At 5 min, 15 min, 30 min, 45 min added 150 µL of ACN containing IS to the wells of corresponding plates, respectively, to stop the reaction. After quenching, shook the plates for 10 min (600 rpm) and then centrifuged at 6000 rpm for 15 min. 80 μL of the supernatant from each well was transferred to a 96-well sample plate containing 140 μL of pure water for LC/MS analysis.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was supported by National Key Research and Development Plan of China (2021YFC2300700 to L.K.Z., 2022YFC2303300 to L.K.Z.), Shanghai Institute of Materia Medica (Grant SIMM010203) and the Strategic Priority Research Program of Chinese Academy of Sciences (SIMM****).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2023.117316.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Wu F., Zhao S., Yu B., et al. Author correction: a new coronavirus associated with human respiratory disease in China. Nature. 2020;580:265–269. doi: 10.1038/s41586-020-2202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.L., Wang X.G., et al. Addendum: a pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;588:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levin E.G., Lustig Y., Cohen C., et al. Waning immune humoral response to BNT162b2 COVID-19 vaccine over 6 months. N Engl J Med. 2021;385:e84. doi: 10.1056/NEJMoa2114583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergwerk M., Gonen T., Lustig Y., et al. COVID-19 breakthrough infections in vaccinated health care workers. N Engl J Med. 2021;385:1474–1484. doi: 10.1056/NEJMoa2114583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirose Y., Shindo N., Mori M., et al. Discovery of chlorofluoroacetamide-based covalent inhibitors for severe acute respiratory syndrome coronavirus 2 3CL protease. J Med Chem. 2022;65:13852–13865. doi: 10.1021/acs.jmedchem.2c01081. [DOI] [PubMed] [Google Scholar]
- 6.Berry M., Fielding B.C., Gamieldien J. Potential broad spectrum inhibitors of the coronavirus 3CLpro: a virtual screening and structure-based drug design study. Viruses. 2015;7:6642–6660. doi: 10.3390/v7122963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. https:// doi.org/10.1126/ science.1085658. [DOI] [PubMed] [Google Scholar]
- 8.Usman B., Jennifer B., Adrian V.C., Leavitt S., Freire E., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. https://doi.org /10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 9.Jin Z., Du X., Xu Y., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 10.Lu R., Zhao X., Li J., et al. novel coronavirus: implications for virus origins and receptor binding. Lancet. 2019;395(2020):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanchan A., Gottfried J.P., Jeroen R.M., Stuart G.S., John Z., Rolf H. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong M.Y., Su H.X., Zhao W.F., Xie H., Shao Q., Xu Y.C. What coronavirus 3C-like protease tells us: from structure, substrate selectivity, to inhibitor design. Med Res Rev. 2021;3:1–34. doi: 10.1002/med.21783. https://doi.org /10.1002/med.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y.Z., Liang C.Y., Xin L., et al. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur J Med Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112711. https://www.sciencedirect.com/science/article/pii/S0223523420306838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai W., Zhang B., Jiang X.M., et al. Structure-based of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J., Lin C., Zhou X., Zhong F., Zeng P., Yang Y., Zhang Y., Yu B., Fan X., McCormick P.J., Fu R., Fu Y., Jiang H., Zhang J., Rozanne S.G. Structural basis of the main proteases of coronavirus bound to drug candidate PF-07321332. J. Virol. 2022;96:e0201321. doi: 10.1128/jvi.02013-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh A.K., Mishevich J.L., Mesecar A., Mitsuya H. Recent drug development and medicinal chemistry approaches for the treatment of SARS-CoV-2 infection and COVID-19. Chem Med Chem. 2022;17 doi: 10.1002/cmdc.202200440. https://chemistry-europe.onlinelibrary. wiley.com/doi/abs/10.1002/ cmdc.202200440 e202200440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan B., Joyce R., Tan H.Z., Hu Y.M., Wang J. SARS-CoV-2 main protease drug design, assay development, and drug resistance studies. Acc Chem Res. 2023;56:157–168. doi: 10.1021/acs.accounts.2c00735. https:// pubs. acs.org/doi/full/10.1021/acs.accounts.2c00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai W., Jochmans D., Xie H., et al. Design, synthesis, and biological evaluation of peptidomimetic aldehydes as broad-spectrum inhibitors against enterovirus and SARS-CoV-2. J Med Chem. 2022;65:2794–2808. doi: 10.1021/acs.jmedchem.0c02258. jmedchem.0c02258 https://doi.org/ 10.1021/acs. [DOI] [PubMed] [Google Scholar]
- 19.Gao K., Wang R., Chen J., Tepe J.J., Huang F., Wei G.W. Perspectives on SARS-CoV-2 main protease inhibitors. J Med Chem. 2021;64:16922–16955. doi: 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halford B. The path to Paxlovid. C&EN Global Enterprise 2022; 8: 405-407. <https:// doi.org/ 10.1021 /acscentsci.2c00369>. [DOI] [PMC free article] [PubMed]
- 21.Kneller D.W., Li H., Phillips G., et al. Covalent narlaprevir-and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat Commun. 2022;13:2268. doi: 10.1038/s41467-022-29915-z. https://wwwnature.53yu.com/articles/s41467-022-29915-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia Z.L., Sacco M., Hu Y.M., Ma C., Meng X.Z., Zhang F.S., Szeto T., Xiang Y., Chen Y., Wang J. Rational design of hybrid SARS-CoV-2 main protease inhibitors guided by the superimposed cocrystal structures with the peptidomimetic inhibitors GC-376, telaprevir, and boceprevir. ACS Pharmacol. Transl. Sci. 2021;4:1408–1421. doi: 10.1021/acsptsci.1c00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao J.X., Li Y.S., Zeng R., et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Owen D.R., Allerton C.M.N., Anderson A.S., et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y., Fang C., Zhang Q., et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell. 2022;13:689–693. doi: 10.1007/s13238-021-00883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabriele L.M., Alessia B., Antonino L., Martorana A. Targeting SARS-CoV-2 main protease for treatment of COVID-19: covalent inhibitors structure-activity relationship insights and evolution perspectives. J Med Chem. 2022;65:12500–12534. doi: 10.1021/acs.jmedchem.2c01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C.L., Sacco M.D., Hurst B., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. https://www.nature.com/articles/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konno S., Kobayashi K., Senda M., et al. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV–2 agents. J Med Chem. 2022;65:2926–2939. doi: 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- 29.Fu L.F., Ye F., Feng Y., et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. https://www.nature.com/articles/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kneller D.W., Galanie S., Phillips G., O’Neill H.M., Coates L., Kovalevsky A. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure. 2020;28:1313–1320.e3. doi: 10.1016/j.str.2020.10.007. https://www.sciencedirect.com/science/article/pii/S0969212620303798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L.L., Lin D.Z., Kusov Y., et al. α-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 32.Bai B., Belovodskiy A., Hena M., et al. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J Med Chem. 2022;65:2905–2925. doi: 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L., Lin D., Sun X., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thanigaimalai P., Konno S., Yamamoto T., et al. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: structure-activity relationship study. Eur J Med Chem. 2013;65:436–447. doi: 10.1016/j.ejmech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramos-Guzmán C.A., Ruiz-Pernía J.J., Tuñón I. Computational simulations on the binding and reactivity of a nitrile inhibitor of the SARS-CoV-2 main protease. Chem Commun. 2021;57:9096–9099. doi: 10.1039/D1CC03953A. [DOI] [PubMed] [Google Scholar]
- 36.Ngo S.T., Nguyen T.H., Tung N.T., Mai B.K. Insights into the binding and covalent inhibition mechanism of PF-07321332 to SARS-CoV-2 Mpro. RSC Adv. 2022;12:3729–3737. doi: 10.1039/D1RA08752E. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.