

Abstract
The Trans-Proteomic Pipeline mass spectrometry data analysis suite has been in continual development and refinement since its first tools PeptideProphet and ProteinProphet were published twenty years ago. The current release provides a large complement of tools for spectrum processing, spectrum searching, search validation, abundance computation, protein inference, and more. Many of the tools include machine-learning modeling to extract the most information from datasets and build robust statistical models to compute the probabilities that derived information is correct. Here we present the latest information on the many TPP tools, and how TPP can be deployed on various platforms from personal Windows laptops to Linux clusters and expansive cloud computing environments. We describe tutorials on how to use TPP in a variety of ways and describe synergistic projects that leverage TPP. We conclude with plans for continued development of TPP.
Keywords: mass spectrometry, proteomics, Trans-Proteomic Pipeline, computational proteomics, PeptideProphet, ProteinProphet, 6.1.0 Parhelion
Introduction
Mass spectrometry (MS) is the predominant technology for high-throughput analysis to define the protein and post-translational complement of complex biological samples, providing context to biology. As with most high-throughput platforms, data analysis software is crucial for the proper interpretation of the complex data produced by modern instruments. For MS in particular, the proper and complete interpretation of the complex and sometimes multiplexed spectra produced has been an especially challenging problem given the complexity of multitudes of protein sequence possibilities. The primary workhorse of such analysis is the so-called “search engine” that associates mass spectra with proposed peptide interpretations1, and the past 25 years have seen a continual development of faster and more sensitive algorithms. However, even with the most recent search engine developments, a substantial amount of post-search-engine processing by additional software is required to yield a final set of identifications of high sensitivity and specificity and accurate abundance information2.
Twenty years ago it was standard practice to apply somewhat arbitrary sequence search engine score thresholds (e.g., SEQUEST3 xcorr > 1.5 for 1+ peptide-spectrum matches (PSMs), xcorr > 2.0 for 2+ PSMs, xcorr > 2.5 for 3+ PSMs, etc.) to search results, perhaps manually looking at some spectra near the thresholds to keep some and discard some, with minimal grasp of the error rates present in the final results. Diligent researchers would spend a few days or weeks looking at spectra, subjectively discarding some and keeping others given the comparatively small size of the data obtained. This dramatically changed in 2003 with the advent of PeptideProphet4 and ProteinProphet5, which applied Bayesian machine learning techniques to the various search engine scores to model the correct and incorrect assignment distributions and then use these models to assign a probability of being correct based on these learned models. With these tools it became possible to validate search engine results on large-scale datasets in a short order, enabling users to select probability thresholds based on a selected tolerable false discovery rate (FDR). A few years later, these two software tools plus some quantitative tools were bundled together and made fully interoperable via the open XML-based formats pepXML and protXML to become the Trans-Proteomic Pipeline6 (TPP). Since then, the TPP has been in continual development, with the enhancement of the founding tools as well as the addition of many more tools for different aspects processing data-dependent acquisition (DDA) proteomics data, resulting in a complete suite of tools for processing the increasingly larger datasets from start to finish. From its inception, the TPP has been and will always be free and open-source software, allowing anyone to use it without cost and to inspect its source code, alter the source code for their own needs, or even incorporate parts of it into their own products.
In this article, we provide a brief overview of TPP and describe important additions in the past few years since we last provided a comprehensive description of the TPP7. In addition to the primary flagship tools with which most regular TPP users are familiar, we also briefly summarize the many minor tools that can be used for simple data tidying and transformation operations. We also describe the many platforms on which the TPP can be deployed, including Windows notebooks or desktops, Linux clusters, and various cloud computing platforms. We conclude with a few major applications and synergistic projects that leverage the power of TPP.
Overview of major tools
By design the TPP is a collection of separate tools that each specialize in one particular data analysis task. They are all made interoperable via the mzML8, pepXML, and protXML formats, where each tool will get its input from one or more of these formats and write its results to one of these formats, except for the conversion tools at the beginning or end of the pipeline. As a collection of individual tools, they are easily amenable to pipelining in a very flexible manner to support a huge variety of combinations and workflows, and a custom program may easily be inserted into the pipeline to support technology development. A summary of the major tools is depicted in Figure 1.
Figure 1.
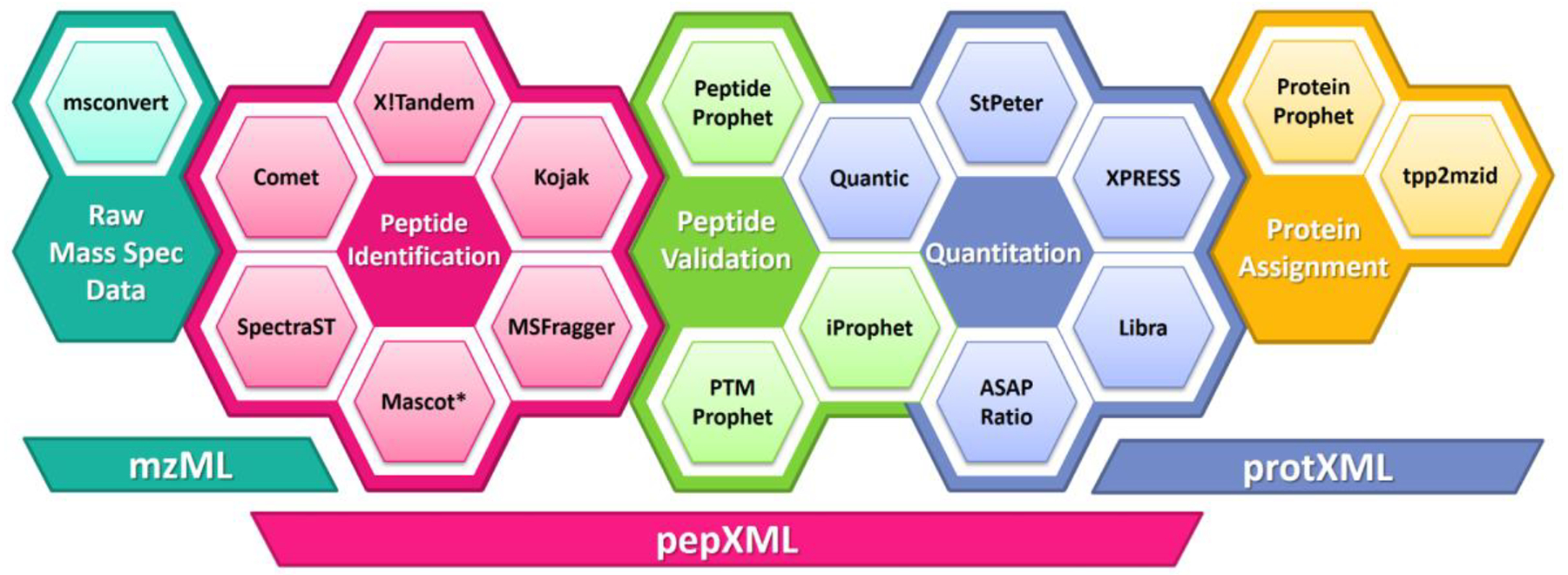
Overview of the software tools and data formats of the TPP. Tools are grouped into broad categories for which there are often alternatives, and underpinned by a set of XML open formats. Most of the depicted tools come bundled with the TPP, but a few such Mascot are external tools whose output can be used by the TPP.
Format conversion
There is a dizzying array of formats commonly used in MS-based proteomics9, but the TPP tools primarily use three: mzML, pepXML, and protXML. The first step is often to convert the raw vendor format to mzML. The TPP distributions have bundled the ProteoWizard10 msconvert tool for many years to provide this functionality, although a substantial drawback is that msconvert can only perform vendor-format conversion when running on the Windows platform, which complicates pipeline analyses on Linux and other platforms.
This has recently been ameliorated by some new tools that can convert vendor formats on non-Windows platforms. The ThermoRawFileParser tool11 can convert Thermo Fisher instrument RAW files to mzML in a platform-independent manner, although it depends on a functional Mono (https://www.mono-project.com/) dependency, which is not always trivial to configure. The TPP does not directly bundle ThermoRawFileParser because of these complex dependencies, but if it is available on the TPP instance, then it can be used. Bruker Corporation makes available dynamic libraries that enable other tools to provide reader or converter functionality for its TDF format (used by timsTOF type instruments). The Python-based TIMSCONVERT tool12 uses the Bruker library to perform file conversion of timsTOF data to mzML. This is also not bundled in the TPP download, but can be used with the TPP if installed separately (it is included in TPP cloud-computing images and containers, see below). Certain other vendor-supplied converters, such as the SCIEX converter may be downloaded separately and used with TPP. Whereas some tool suites provide a table of compatibility of various components between different spectral file formats (e.g. https://fragpipe.nesvilab.org/ for FragPipe), the TPP requires that all other formats first be converted to the standard format mzML (which can be performed for all mainstream formats), and all TPP tools are compatible with mzML.
Although the TPP does not use the Proteomics Standards Initiative13–15 (PSI) standard format mzIdentML16 internally, the final output of a TPP analysis may be exported to mzIdentML 1.217 via the TPP tool tpp2mzid for deposition to ProteomeXchange18–20 repositories or for use with other mzIdentML-supporting tools. In the other direction, users may use the ProteoWizard10 idconvert tool (bundled with TPP) to convert externally acquired mzIdentML files to pepXML for further TPP processing.
Search engines
The heart of MS proteomics DDA data continues to be the “search engine”, a tool that interprets mass spectra to determine the peptide or peptides that yielded them. The most common type of search engine continues to be the sequence search engine, which uses a reference input list of protein sequences, usually in FASTA format, to define the search space of possible peptide sequences. Spectral library search engines and de novo search engines, which are less common, are also available with TPP and are described further below.
The sequence search engine most commonly used with TPP is still Comet21, which is actively maintained and advancing with new functionality22. A versatile staple among search algorithms and an optimized variation of the original SEQUEST3 algorithm, it works very well with spectra from any instrument and its scoring algorithm is extremely robust and produces scores that work very well with TPP validation tools. TPP also bundles a slightly customized version of X!Tandem23 that continues to be a good choice. A newer tool MSFragger24 is faster in comparison to the current choices of search algorithms included in the TPP, especially when run on large numbers of spectra at one time, due to its fragment-indexing approach. It is not bundled directly in TPP 6.1.0, but is fully supported when installed separately or is available in pre-built TPP containers and images. In future releases, MSFragger will be included within the TPP package.
For spectral library searching, SpectraST25 has been a TPP tool since its original development. With this approach, new spectra are matched against a library of previously identified spectra in the form of a spectral library. This approach is much faster, more sensitive, and more specific than sequence database searching, although is only as good as the reference spectral library provided. There is renewed interest in spectral libraries because of data-independent acquisition (DIA) approaches being increasingly deployed and therefore the quality and coverage of libraries is very important26 and likely to improve in the coming years, aided by a new standard spectral library format being developed by the PSI27 (https://psidev.info/mzSpecLib). Furthermore, the approach is complementary to sequence searching since new data can be searched with both SpectraST and a sequence search engine and then merged into a combined, better result with iProphet (described below). SpectraST is capable not just of searching new data, but also of creating high-quality libraries28 based on the output of TPP searches as well as generating decoy libraries29.
Novor,30 although not bundled with TPP for licensing reasons, is a very fast and capable de novo sequence search engine. If Novor is installed on the machine running TPP, it can be executed via the TPP interface, its output converted to pepXML, and then integrated with downstream TPP tools. Since the scores are not very amenable to modeling, and there is no target-decoy approach to guide the models, an effective approach is to create a sequence database from the Novor output, and then run Comet or another search engine with the new sequences included in the sequence FASTA file, and then let the search engine-score modeling provide downstream probability values and FDR control.
The TPP provides robust support for alternative MS data searching including Kojak31 (and Hoopmann et al., submitted, this issue) for standard or cleavable MS2-based crosslinking techniques, and Magnum32 for open modification database searching. Crosslinking-based MS analyses are employed to elucidate protein-protein interactions and facilitate protein structure and topology predictions. MS2-based crosslinking methodologies are similar to those employed by standard shotgun MS2-based methods, but require specialized algorithms for data analysis. Kojak is designed to identify two independent peptides covalently bonded with a crosslinker and fragmented in a single MS2 scan event. Magnum, on the other hand, is specialized in identification of non-peptide masses that are bound to peptides. The tool is capable of identifying xenobiotic mass adducts, in addition to PTMs that were uncharacterized in the search parameters. Both tools support the pepXML file format, making results easily parsed, sorted, explored, and visualized within the TPP graphical user interface.33 TPP 6.1.0 provides a guided pipeline interface supporting these tools, that include detailed instructions on how to set all parameters and batching of the data files for fast, automated analyses.
Although TPP primarily focuses on DDA workflows, it does support library-free analysis of DIA data via the DISCO tool. DISCO reads mzML files containing the instrument-produced spectra and uses signal processing approaches to isolate the fragment ions in the multiplexed MS2 spectra that correlate with precursors in the MS1 and write the results to new mzML files that may then be searched with standard DDA search engines and downstream tools, including target-decoy analysis. This provides a comprehensive analysis of DIA data without the need for building a spectral library first. It is somewhat less sensitive than library approaches, but not limited by currently available libraries, and is complementary to library-based approaches. The most recent version of DISCO in TPP 6.2.0 also supports ion-mobility mass spectrometry such as from the Bruker timsTOF, and allows the extraction of precursors and spectra acquired with an ion mobility dimension.
The TPP also supports other search engines not mentioned here to varying degrees. Figure 2 provides a support matrix of the extent of support for various search engines by TPP tools. Over a dozen sequence search engines are supported, and then a few additional tools for open mass searching, crosslinking analysis, spectral library search, and de novo search analysis. Some tools are bundled with the TPP, whereas others must be downloaded separately (often due to licensing restrictions).
Figure 2.
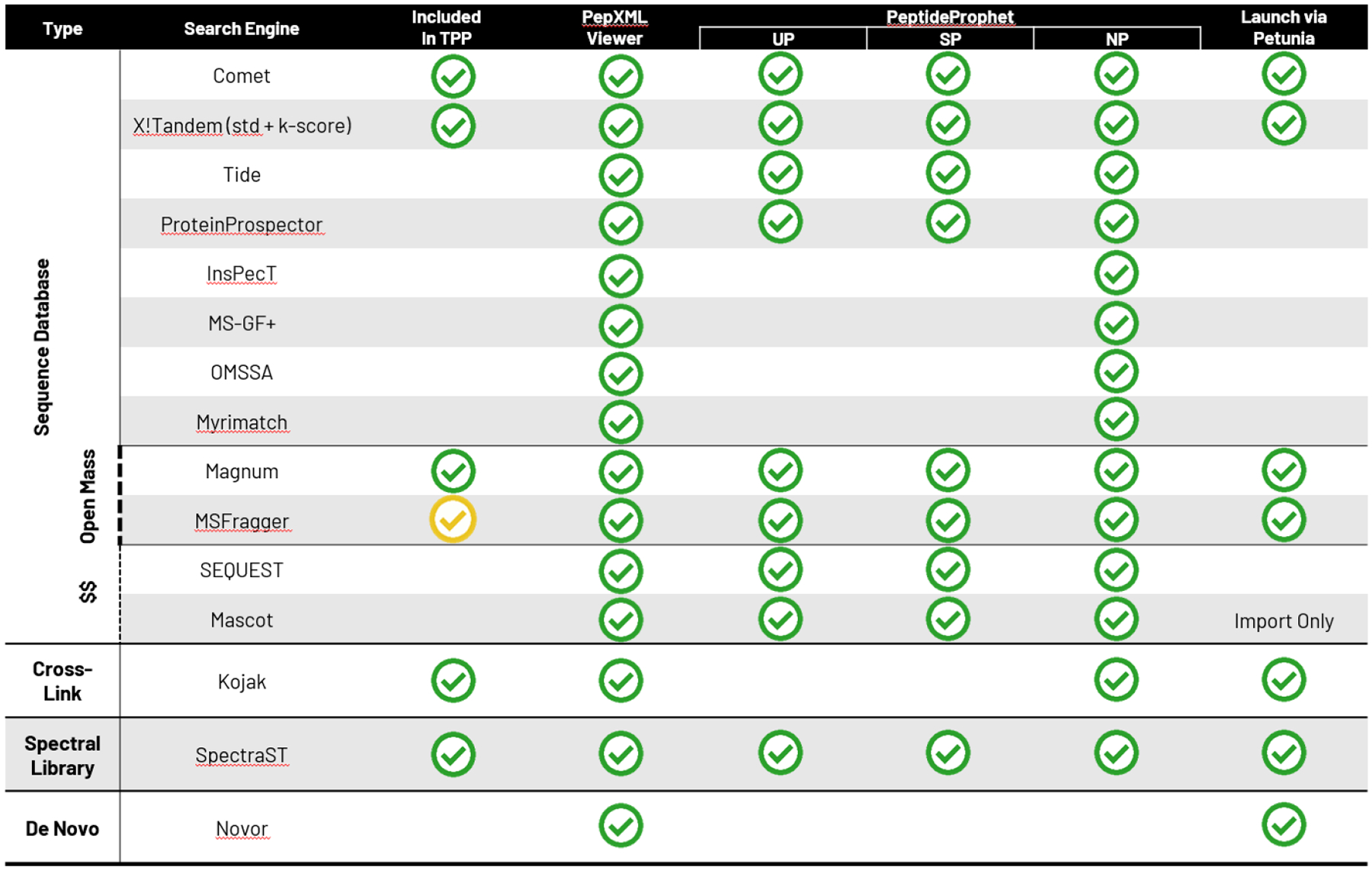
Summary of support of many search engines by TPP components. Different components of the TPP are listed going across at the top. Search engines are listed as rows; “$ $” denotes commercial offerings A green checkmark indicates support by that component of the search engine. Under PeptideProphet, UP means unsupervised parametric model, SP means supervised parametric, and NP means non-parametric model is supported.
Validation tools
The heart of the TPP is its validation tools, originally conceived and published 20 years ago, but periodically updated since then with new features, better accuracy, and improved speed via multithreading. In most cases datasets should be subjected to each of PeptideProphet, iProphet, PTMProphet, and ProteinProphet in turn as described below.
PeptideProphet5 is designed to model the search engine output scores and other attributes of each PSM to compute a machine-learning Bayesian model of the correct and incorrect distribution of PSMs, with or without the aid of a decoy database. The model is refined based on other attributes such as the mass delta, the number of missed proteolytic cleavages, the number of protease-specific termini, the precursor charge, delta from predicted retention time, and the number of variable mass modifications. PSMs that are more like the correct-PSM distribution are boosted, and PSMs that are more like the incorrect-PSM distribution are penalized. The models are learned from each dataset, which can vary widely. Each PSM is assigned a probability of being correct based on the learned model, and output tables and ROC plots are provided so that the user may select a probability threshold that meets their FDR or sensitivity/specificity requirements.
The iProphet tool34 has two primary functions. First, it can combine multiple output pepXML files of PeptideProphet from multiple analyses of the input spectra, typically via different search engines. For example, if a dataset is searched with both Comet and SpectraST, each set of output results is first modeled separately by PeptideProphet (since the search engine scores are different) to produce PSM probabilities, and then combined into a single set of results by iProphet based on those probabilities. The second main functionality of iProphet is to further refine all the input PeptideProphet probabilities based on other corroborating evidence within the whole dataset. iProphet applies models for the number of search engines agreeing on the result, the number of replicate spectra of a precursor ion, the number of different charge states for the same peptidoform, the number of different peptidoforms for the same peptide sequence, and the number of PSMs for each protein sequence. In each case, multiple lines of corroborating evidence boost probabilities, and lack thereof reduces probabilities, as learned by the Bayesian machine-learning model for each dataset. If any of the above attributes provide no discriminating power between correct and incorrect, there is simply no boost/penalty to the probabilities. In nearly all cases, iProphet will markedly improve the overall discrimination between correct and incorrect in any dataset and yield more identifications above any selected tolerable FDR than without. Output modeling results and probabilities are added to the pepXML file.
PTMProphet35 is designed to compute probabilities that variable mass modifications on the PSMs are correctly localized via Bayesian models. PTMProphet performs this task by reading the pepXML output of iProphet (or PeptideProphet) and the mzML files from which the identifications are derived, and then consider all the allowed permutations of mass modifications to assign a probability for each possible site. The most common use case is for phosphorylation-enriched datasets, where the location of one or more phosphate groups is permuted to all possibilities for each PSM and probabilities computed based on the spectral evidence. Probabilities that are all identical on a PSM (e.g., 0.5, 0.5 for two potential sites, 0.33, 0.33, 0.33 for three potential sites) indicate no discriminating evidence of location in the PSM, while probabilities near 0.0 indicate confidence that the PTM is not at a particular site, and probabilities near 1.0 indicate high confidence that the PTM is at that site. Use of decoy amino acids when searching and processing with PTMProphet is encouraged.36 PTMProphet is not limited to phosphorylation, but may be applied to any mass modifications (e.g., acetylation on n-termini or lysine, oxidation on M or W or H, or deamidation on N or Q) separately or in combination. As with iProphet, the PTMProphet model results are added to the pepXML in a cumulative manner.
The final prophet in the pantheon is ProteinProphet5, which takes as input the output from any tool that writes pepXML with probabilities (typically PeptideProphet, iProphet, with or without PTMProphet) and writes protXML as output, providing protein inference information based on the input PSMs and their probabilities. Corroborating information is again learned via Bayesian models to provide robust probabilities for each protein in the most parsimonious list of which have been truly detected in the sample. Protein entries which are not needed to explain the detected peptides are labeled as “subsumed” and grouped with the proteins that best explain the available evidence. The output of the models enables users to select a threshold that meets their tolerance of false discoveries.
TPP Quantitation tools
Measuring the abundance of proteins is a crucial part of most proteomics experiments and TPP provides several different tools for quantitation based on the experimental workflow, label-free, isobaric labeling, or isotopic labeling. The StPeter tool37 is designed for label-free datasets. It reads as input a protXML file as well as the corresponding pepXML and mzML files, deriving abundance measurements based on quantitation of the most abundant identified peaks in the MS2 spectra. PSM, peptidoform, and peptide abundances are rolled up to protein-level metrics using the SIn approach38 and written to protXML for further use. The StPeter2Matrix tool takes a series of protXML files from different samples and produces a quantitation matrix where each column is a sample.
The Libra tool39 is designed for isobaric labeling datasets, such as iTRAQ40 and TMT41. It reads as input a protXML file as well as the corresponding pepXML and mzML files, deriving abundance measurements based on the reporter ions in each spectrum. Isotopic corrections are applied based on batch-to-batch variation spec sheets provided with labeling kits. While originally published for the 4-channel iTRAQ reagents, Libra has been periodically extended to support the ever-growing multiplexing possibilities and now supports up to 32 reporter ions per spectrum, and is not limited to any specific set of reagent tags. Libra supports both MS2-based workflows as well as MS3-based workflows to reduce contamination from co-fragmented ions.
ASAPRatio42 is designed for isotopic labeling datasets such as biological incorporation SILAC43 or chemical approaches such as dimethylation44,45 or older ICAT46 and similar approaches to labeling samples. ASAPRatio also reads as input a protXML file as well as the corresponding pepXML and mzML files, deriving abundance measurements based on the MS1 profiles of the precursors. In addition to the automatic first pass quantitation, ASAPRatio has a manual-inspection mode whereby outlier signals can be inspected to see if the measurements seem accurate or if interfering ions have contaminated the measurements, allowing the user to adjust quantitation or discard individual measurements. Corrected abundance measures are automatically rolled back up to the protein level.
The XPRESS tool46 can operate in either isotopic mode like ASAPRatio (but with fewer features) or in label-free mode, measuring the MS1-area-under-the-curve label-free quantitation (for peptides only) for each identified PSM. Although ASAPRatio or StPeter are preferred, it is useful in this circumstance. Future revisions will combine these attributes into a single tool to simplify the data analysis process.
Quantic is a fragment spectrum level quantification tool with several different applications. First, Quantic can be used to quantify the fragment ion intensities of identified PSMs in a given pepXML file. Second, Quantic has an ANNOTATE mode to provide a single string annotation of matched fragments and their intensities. Third, Quantic can be used to quantify data from cleavable tag linkers that can be measured in DDA or DIA modes. For DIA data or data collected with wider isolation windows, Quantic has DIAMODE options that implements an isotopic correction allowing it to recover underlying quantities of measured peptides.
Additional TPP tools
Extending beyond the major tools described above, the TPP is distributed with dozens of additional minor tools that perform specific tasks within workflows or can be used for custom analysis. Many operate with the major TPP formats, or in some cases, support other formats. Most of these tools are listed in Table 1, including their formal name, publication describing them if any, input format(s), output format, whether available in the GUI (or CLI only), and a very brief synopsis. Additional information for each tool may be found either in the TPP GUI if supported there, or via the command-line usage statement.
Table 1.
Overview of many of the more minor TPP tools not described elsewhere in the article, along with their input formats, output formats, whether they are accessible with the GUI (or only the command-line), and a brief description.
| Name | Input | Output | GUI | Synopsis |
|---|---|---|---|---|
| Analysis Tools | ||||
| reSpect47 | pepXML, mzML | mzML | Y | Writes out new mzML files wherein the chimeric spectra with confident peptide identifications have had their MS2 peaks for those peptides subtracted to leave behind the peaks of co-eluting precursors, which may be re-subjected to searching to identify these co-eluting precursors. |
| RefreshParser | pepXML, FASTA | pepXML | Y | Refreshes protein-mapping information within a pepXML file by remapping all the peptide sequences therein to a new FASTA database, updating protein identifiers and protease specificity information. |
| ProphetModels.pl and ProtProphModels.pl | pepXML and protXML, respectively | same as input | Y | Generate analysis models, including decoy analysis, suitable for visualization |
| compareProts.pl | tsv | tsv, html | Y | Compare ProteinProphet results of two or more files. When two files are compared with -D3 option enabled, an HTML page will be generated with a graphic comparing protein quantities. |
| compareProts_ClusterHM.pl | tsv | tsv,html | Y | Compare ProteinProphet results of two or more files. When -D3 option is enabled an HTML page will be generated with a graphic comparing protein quantities in a self-organizing heatmap. |
| DidIScanThat | mzML, mzXML | Y | Lists the subset of MS2 spectra that match a provided precursor mass, potentially constrained further by charge state, retention time, and mass tolerance | |
| RTCatalog | Y | Creates a retention time catalog of peptidoforms (with mass modifications) based on one or more input pepXML files | ||
| RTCalc | Y | Computes retention times for a list of input peptidoforms based on a retention time catalog or estimated via a model computed from a catalog. | ||
| Hardklör | mzML, mzXML | Computes a list of MS1 features from a mzML, mzXML or RAW file | ||
| SPACEPro48 | Y | Computes an extensive set of protease digestion efficiency metrics to aid in evaluation of sample digestion | ||
| assess_swathlib.pl | various | Y | Analyze and repair DIA spectral ion libraries in OpenSWATH, Spectronaut, or PeakView formats | |
| mzML Utilities | ||||
| MikesMagicalMzMLShrinker | mzML | mzML | Y | Rewrites mzML files using a variety of space-saving techniques to reduce their file sizes |
| MzXML2Search | mzXML | various | Y | Convert mzXML files to other open formats (such as MGF) |
| indexmzXML | mzML or mzXML | mzML or mzXML | Y | Recomputes the file index or mzML or mzXML files |
| readmzXML | mzML or mzXML | Extracts and prints a single spectrum from an mzML or mzXML file given a filename and scan number | ||
| Database Tools | ||||
| subsetdb | FASTA | FASTA | Extracts a subset of entries in a FASTA file | |
| digestdb | FASTA | tsv | Digest a FASTA file following user-specified rules | |
| checkdb | FASTA | FASTA | Reads an input FASTA file and flags various potential problems that might cause downstream issues with some search engines and TPP tools, such as non-ascii characters in descriptions, excessive description lengths, etc. | |
| decoyFastaGenerator.pl | FASTA | FASTA | Y | Creates a set of decoy sequences for an input FASTA file, using the de Bruijn method49 (recommended) |
| decoyFASTA | FASTA | FASTA | Y | Creates a set of decoy sequences using a sequence reversing approach |
| translateDNA2AA-FASTA | FASTA (DNA) | FASTA (AA) | Y | This program takes in a FASTA formatted nucleotide database as input and creates an output database of translated amino acid sequences. Sequences are translated in all six reading frames |
| promast.pl50 | pepXML, text file | pepXML, tsv | Y | Maps a list of input peptides (text file list or pepXML) to a pre-indexed reference database of protein sequences, previously created by clips |
| clips.pl50 | FASTA, PEFF | Y | Creates an indexed database of proteins suitable for use with ProteoMapper, based on an input FASTA file or PEFF51 file. | |
| Interfaces | ||||
| MADCAPS | FASTA | HTML | Y | Interactive protein sequence workbench, which enables the visualization of multiple sequence alignment, enzymatic digestion, experimental coverage, and more |
| Pep3D | mzML, mzXML | HTML | Y | Interactive visual “gel-like” display of MS run data with links to individual MS/MS spectra. Can also overlay search and validation results. |
| TPPcel | various | HTML | Y | General utility for the display and comparison of tabular data, e.g., search parameters |
| ploTPP | various | HTML | Y | Basic interactive data graphing workbench with overplot ability |
| tpp_models.pl | pepXML, protXML | HTML | y | Produce “dashboard” view of TPP file, including visual representation of analytical models, parameters used, etc. |
| Miscellaneous | ||||
| xinteract | pepXML | pepXML, protXML | Y | Allows the user to chain multiple tools such as PeptideProphet, iProphet, ProteinProphet, StPeter with a single command |
| Lib2HTML | splib | HTML | Y | Produces an HTML representation of a spectral library, with links to all spectra and annotations |
| updatepaths.pl | pepXML | pepXML | Y | Updates the absolute file paths of input files (such as pepXML, mzML, FASTA, PEFF) in pepXML files, which is necessary for pepXML files that are moved to a new location or computer. |
| FetchDataset | URL | Y | Downloads a dataset given a URL and automatically unpacks the file in a specified location | |
| TPPTray | NA | NA | A Windows widget that can sit in the applications tray (lower right corner) to allow the user to start and stop the web server and provide other functions | |
Readers interested in additional details of new features to these many tools as well as more description of new utilities are encouraged to read through the release notes of recent releases of the TPP, most specifically http://tools.proteomecenter.org/wiki/index.php?title=TPP:6.0.0_Release_Notes for the major 6.0.0 release and http://tools.proteomecenter.org/wiki/index.php?title=TPP:6.1.0_Release_Notes for the more modest 6.1.0 release, which still includes over 100 additions and fixes.
Although the TPP supports many different workflows, the TPP does not provide complete support for affinity purification (AP-MS) experiments. The TPP may be used to process the raw data into identification and quantification results, and then those results would need to be exported to an external tool such as SAINT v2,52 SAINTexpress,53 or SAINTq54 for significance assessment. In addition, other popular programs such as the Mass spectrometry interaction STatistics (MiST),55 and comPASS56 can be used with the output of the TPP analysis, as well as other established mass spectrometry-based protein:protein interaction programs with or without some output file structure optimization.57
TPP Anywhere
One of the major advantages of TPP is its ability to be deployed in a wide variety of environments, from personal interactive use on Windows laptops, on extensive large Linux compute clusters, to automated use within workflow engines or cloud computing environments. All tools have a command-line interface, originally developed under Linux, but equally at home and natively compiled under Windows. Many TPP users have written custom scripts that string together TPP command-line invocations to easily reproduce their analysis with one command to simplify analyses and foster reproducibility.
While the command-line interfaces are appreciated by many users, others prefer a graphical user interface (GUI), which is provided by the TPP GUI called Petunia. The GUI is web-based with client-side JavaScript to enrich the interface, allowing users to use the TPP from any web browser on any platform. Petunia has the advantages that the same exact GUI is available on a modest Windows laptop, a powerful expandable Linux server shared by a research group, or a remote cloud computing instance running on Amazon Web Services (AWS).
Installation on the Windows machine is typically a simple process of downloading and running the installer executable. Installation on Linux or other type of UNIX machine is not too difficult when following the installation tutorials provided on the TPP web site, but the many different distributions of Linux can provide some challenges with getting the dependencies sorted out and typically requires some level of administrator privileges.
The hardware requirements for TPP tools are the same on all operating systems and are generally modest (16 GB of memory) for modestly sized data sets. Processing large data sets does require more memory for certain steps. For a data set of one million spectra, PeptideProphet and iProphet will use approximately 12 GB and 24 GB of memory, respectively. Most TPP tools can take advantage of computers with multiple cores and thus run faster on such machines, but will work perfectly fine, although slower, on single-core machines.
A simpler method of using the TPP on Linux machines with Docker (https://www.docker.com/) already installed (or if it can be installed) is to use our prebuilt Docker images. Docker is a containerization system that allows complex sets of tools to be pre-configured by the maintainer and then trivially run by anyone with the Docker system installed. Thousands of bioinformatics tools are available via Docker, often as part of the BioContainers58,59 infrastructure. The TPP Docker container uses the BioContainers scheme to provide Docker users with the ability to run the TPP, either in command-line mode via docker run commands or via the GUI by launching the web server in the container, with no installation required. Docker even runs under Windows and one can run the Linux version of the TPP under Windows when Docker is installed there.
One drawback of Docker is that it usually requires administrator privileges to run, which often makes it unsuitable for shared computing clusters, where security is a concern. The Singularity (https://sylabs.io/) container system solves this problem by allowing container execution with ordinary user privileges, and is thus preferred in shared computing situations. As of TPP 6.1.0, there is now a distributed Singularity container for the TPP and related tools. A tutorial demonstrates the use of the TPP with singularity.
The most popular cloud computing platform is Amazon Web Services (AWS), providing a tremendous variety of computing and storage functionality at an attractive cost. One of the popular ways of using AWS for computing is via the Elastic Compute Cloud (EC2) service, which allows users to launch instances of computers (with nearly any operating system) remotely on the cloud and use them for whatever they wish. An easy way to launch such an instance is via an Amazon Machine Image (AMI), which provides a pre-configured template for starting one or more instances of a computer. For each version of the TPP, we provide a free AMI running Ubuntu Linux with the TPP already pre-installed and configured along with several other useful tools. The AMIs can be launched by anyone with an AWS account.
For anyone wishing to launch a cloud instance running TPP with zero installation, we have developed the TPP Web Application60 (TWA), available at http://tools.proteomecenter.org/twa whereby anyone with AWS credentials may launch a TPP instance of their choosing via a web browser from anywhere, use it or share it for as long as they wish, and shut it down when not in use.
In order to truly leverage the power of AWS, for example to launch 100 cloud computing instances to process hundreds to thousands of MS run files at scale, we have developed the AMZTPP60 system. It enables users to process any number of MS runs in a highly configurable manner, automatically uploading the data to AWS storage, automatically launching and terminating worker instances, and copying the data back to the local computer in a highly configurable manner. The system is complex, but can be installed locally with some effort or used trivially via TWA.
Training and Tutorials
We have developed over 20 tutorials61 for learning about and using aspects of the TPP. The tutorials cover a range of topics from installation of TPP on Windows and Linux and Docker to using many of the individual tools described above. A full listing of current tutorials is available at http://www.tppms.org/tutorials/.
Key related projects
Continued TPP development influences and is influenced by several other major projects. First, TPP is the primary processing engine behind PeptideAtlas62,63, a resource that reprocesses thousands of publicly accessible datasets, mostly via ProteomeXchange Consortium18–20 data repositories, and makes the ensemble results available to the community. PeptideAtlas has reprocessed datasets for dozens of species, and also produces specialized builds for important subtypes of data for certain species, e.g., builds composed entirely of human blood plasma datasets64–66, or phospho-enriched datasets. Raw data files are downloaded from the repositories, converted to mzML, processed with one or more search engines, and validated with the tools described above to carefully control the FDR across all datasets in a build, so that the final combined FDR across all datasets is approximately 1% at the protein level, and much lower at the peptide and PSM levels. PeptideAtlas is made possible by the automatable, high throughput processing of the TPP. The TPP is made more robust by being applied to process public datasets from laboratories from around the world (and fixed or improved when issues are encountered).
One of the goals of the Human Proteome Project67–69 is to secure confident detections via mass spectrometry of all predicted human proteins and has relied on PeptideAtlas and TPP to provide this compendium since its inception in 2012. In order to ensure the highest quality and minimal false positives, a set of guidelines, known as the HPP Mass Spectrometry Data Interpretation Guidelines 3.070 were developed to guide the publication of new detections as well as the PeptideAtlas and TPP process for reanalysis of datasets.
The TPP developers have participated in the PSI since 2006, and were instrumental in the development of mzML and many other PSI standards. mzML is the recommended format for TPP input spectra, although the legacy mzXML71 is still supported. While the TPP still maintains its own internal pepXML and protXML formats for operational efficiency, import and export to mzIdentML are fully supported via tpp2mzid and idconvert as described above. Comet and the TPP were one of the first tools to implement the PSI Extended FASTA Format (PEFF) standard51, demonstrating its use in detecting known mass modifications and single amino acid variants72. The ProteoMapper tool50 supports PEFF files as input as well. The TPP is also one of the first tools to support the Universal Spectrum Identifier (USI) standard73 with its ProForma 2.074 peptidoform notation. The TPP spectrum viewer accepts USIs for viewing and assists users with determining USIs in their own datasets.
Conclusion
We have described the many components of the latest version 6.1.0 of TPP, which has been under continual development since its original tools were published in 2002. Although several TPP tools have been re-written to be faster and superior to the previous versions, and nearly all tools have been extended and optimized since their original release and subsequent publication, the core capabilities are ever present and overlaid with modern processing tools to deal with new workflow demands. Development in the TPP ecosystem will continue in the future, with further extension of existing tools as well as the addition of new tools as demanded by users. Special areas of focus include the enhancement of quantitation tools to keep up with evolving quantitative techniques, as well as bundling and development of post-quantitation tools such as batch-effect correction, imputation, clustering, and visualization. We will also extend the DISCO workflow to merge the library-free and library-based analyses to leverage the best of both approaches. We will continue to make the TPP more robust to ensure that the true number of false positives is within the estimates, and develop an analysis assistant that will help users set parameters correctly and advise them when they seem suboptimal for the dataset being analyzed. We will add additional integration with external knowledge bases via the continually expanding web services provided by those resources. Finally, we will continue to focus on the flexibility of deployment of the TPP so that it may be used anywhere from laptop to cloud, to bring the computation wherever the data may be located.
Figure 3.
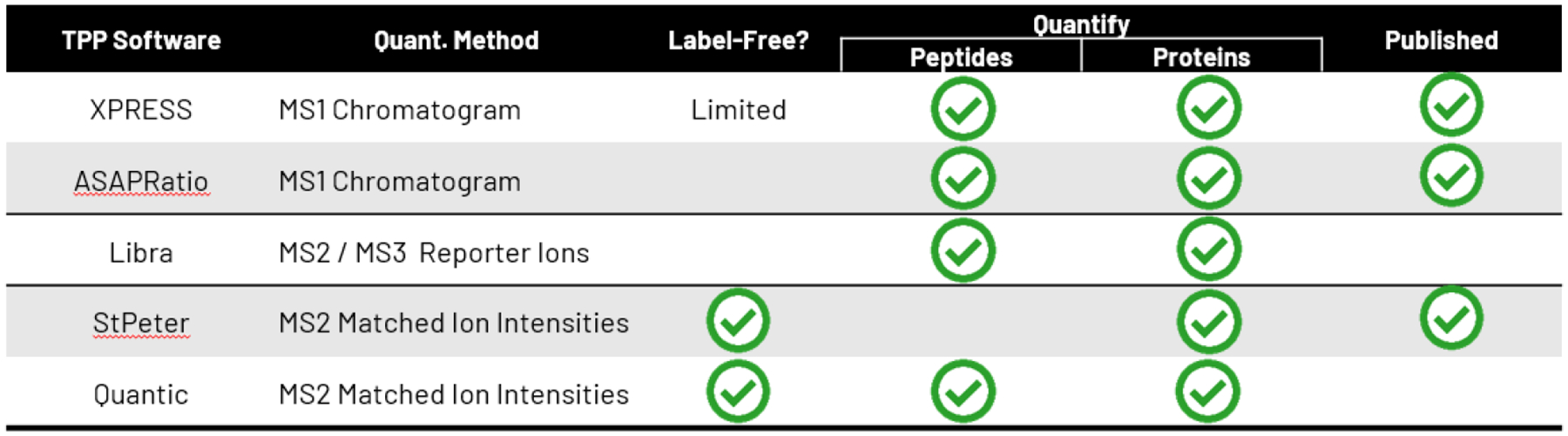
Summary of applications of the TPP quantitation tools. Component tools are listed as rows. The columns are supported applications and other attributes. A green checkmark indicates support by that component.
Acknowledgements
This work was funded in part by the National Institutes of Health grants from the National institute General Medical Sciences R01 GM087221, R24 GM127667, the National Heart, Lung, and Blood Institute R01 HL133135, the Office of the Director S10 OD026936, the National Institute on Aging U19 AG023122, and the National Science Foundation awards 1920268, DBI-1933311, and IOS-1922871.
Footnotes
The authors declare no competing financial interest.
References
- (1).Eng JK; Searle BC; Clauser KR; Tabb DL A Face in the Crowd: Recognizing Peptides through Database Search. Mol. Cell. Proteomics MCP 2011, 10 (11), R111.009522. 10.1074/mcp.R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nesvizhskii AI A Survey of Computational Methods and Error Rate Estimation Procedures for Peptide and Protein Identification in Shotgun Proteomics. J. Proteomics 2010, 73 (11), 2092–2123. 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Eng JK; McCormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- (4).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem 2002, 74 (20), 5383–5392. [DOI] [PubMed] [Google Scholar]
- (5).Nesvizhskii AI; Keller A; Kolker E; Aebersold R A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem 2003, 75 (17), 4646–4658. [DOI] [PubMed] [Google Scholar]
- (6).Keller A; Eng J; Zhang N; Li X; Aebersold R A Uniform Proteomics MS/MS Analysis Platform Utilizing Open XML File Formats. Mol. Syst. Biol 2005, 1, 2005.0017. 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Deutsch EW; Mendoza L; Shteynberg D; Slagel J; Sun Z; Moritz RL Trans-Proteomic Pipeline, a Standardized Data Processing Pipeline for Large-Scale Reproducible Proteomics Informatics. Proteomics Clin. Appl 2015, 9 (7–8), 745–754. 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Martens L; Chambers M; Sturm M; Kessner D; Levander F; Shofstahl J; Tang WH; Römpp A; Neumann S; Pizarro AD; Montecchi-Palazzi L; Tasman N; Coleman M; Reisinger F; Souda P; Hermjakob H; Binz P-A; Deutsch EW MzML--a Community Standard for Mass Spectrometry Data. Mol. Cell. Proteomics MCP 2011, 10 (1), R110.000133. 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Deutsch EW File Formats Commonly Used in Mass Spectrometry Proteomics. Mol. Cell. Proteomics MCP 2012, 11 (12), 1612–1621. 10.1074/mcp.R112.019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak M-Y; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol 2012, 30 (10), 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hulstaert N; Shofstahl J; Sachsenberg T; Walzer M; Barsnes H; Martens L; Perez-Riverol Y ThermoRawFileParser: Modular, Scalable, and Cross-Platform RAW File Conversion. J. Proteome Res 2020, 19 (1), 537–542. 10.1021/acs.jproteome.9b00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Luu GT; Freitas MA; Lizama-Chamu I; McCaughey CS; Sanchez LM; Wang M TIMSCONVER: A Workflow to Convert Trapped Ion Mobility Data to Open Data Formats. Bioinforma. Oxf. Engl 2022, btac419. 10.1093/bioinformatics/btac419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Orchard S; Hermjakob H; Apweiler R The Proteomics Standards Initiative. Proteomics 2003, 3 (7), 1374–1376. 10.1002/pmic.200300496. [DOI] [PubMed] [Google Scholar]
- (14).Deutsch EW; Orchard S; Binz P-A; Bittremieux W; Eisenacher M; Hermjakob H; Kawano S; Lam H; Mayer G; Menschaert G; Perez-Riverol Y; Salek RM; Tabb DL; Tenzer S; Vizcaíno JA; Walzer M; Jones AR Proteomics Standards Initiative: Fifteen Years of Progress and Future Work. J. Proteome Res 2017, 16 (12), 4288–4298. 10.1021/acs.jproteome.7b00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Deutsch EW; Vizcaíno JA; Jones AR; Binz P-A; Lam H; Klein J; Bittremieux W; Perez-Riverol Y; Tabb DL; Walzer M; Ricard-Blum S; Hermjakob H; Neumann S; Mak TD; Kawano S; Mendoza L; Van Den Bossche T; Gabriels R; Bandeira N; Carver J; Pullman B; Sun Z; Hoffmann N; Shofstahl J; Zhu Y; Licata L; Quaglia F; Tosatto SC; Orchard SE The Proteomics Standards Initiative at Twenty Years: Current Activities and Future Work; preprint; Chemistry, 2022. 10.26434/chemrxiv-2022-ksqg1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jones AR; Eisenacher M; Mayer G; Kohlbacher O; Siepen J; Hubbard SJ; Selley JN; Searle BC; Shofstahl J; Seymour SL; Julian R; Binz P-A; Deutsch EW; Hermjakob H; Reisinger F; Griss J; Vizcaíno JA; Chambers M; Pizarro A; Creasy D The MzIdentML Data Standard for Mass Spectrometry-Based Proteomics Results. Mol. Cell. Proteomics MCP 2012, 11 (7), M111.014381. 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vizcaíno JA; Mayer G; Perkins S; Barsnes H; Vaudel M; Perez-Riverol Y; Ternent T; Uszkoreit J; Eisenacher M; Fischer L; Rappsilber J; Netz E; Walzer M; Kohlbacher O; Leitner A; Chalkley RJ; Ghali F; Martínez-Bartolomé S; Deutsch EW; Jones AR The MzIdentML Data Standard Version 1.2, Supporting Advances in Proteome Informatics. Mol. Cell. Proteomics MCP 2017, 16 (7), 1275–1285. 10.1074/mcp.M117.068429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vizcaíno JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Ríos D; Dianes JA; Sun Z; Farrah T; Bandeira N; Binz P-A; Xenarios I; Eisenacher M; Mayer G; Gatto L; Campos A; Chalkley RJ; Kraus H-J; Albar JP; Martinez-Bartolomé S; Apweiler R; Omenn GS; Martens L; Jones AR; Hermjakob H ProteomeXchange Provides Globally Coordinated Proteomics Data Submission and Dissemination. Nat. Biotechnol 2014, 32 (3), 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang M; Ishihama Y; Bandeira N; Hermjakob H; Vizcaíno JA The ProteomeXchange Consortium in 2017: Supporting the Cultural Change in Proteomics Public Data Deposition. Nucleic Acids Res. 2017, 45 (D1), D1100–D1106. 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Deutsch EW; Bandeira N; Sharma V; Perez-Riverol Y; Carver JJ; Kundu DJ; García-Seisdedos D; Jarnuczak AF; Hewapathirana S; Pullman BS; Wertz J; Sun Z; Kawano S; Okuda S; Watanabe Y; Hermjakob H; MacLean B; MacCoss MJ; Zhu Y; Ishihama Y; Vizcaíno JA The ProteomeXchange Consortium in 2020: Enabling “big Data” Approaches in Proteomics. Nucleic Acids Res. 2020, 48 (D1), D1145–D1152. 10.1093/nar/gkz984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eng JK; Jahan TA; Hoopmann MR Comet: An Open-Source MS/MS Sequence Database Search Tool. Proteomics 2013, 13 (1), 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- (22).Eng JK; Deutsch EW Extending Comet for Global Amino Acid Variant and Post-Translational Modification Analysis Using the PSI Extended FASTA Format. Proteomics 2020, 20 (21–22), e1900362. 10.1002/pmic.201900362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Craig R; Beavis RC TANDEM: Matching Proteins with Tandem Mass Spectra. Bioinforma. Oxf. Engl 2004, 20 (9), 1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- (24).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat. Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lam H; Deutsch EW; Eddes JS; Eng JK; King N; Stein SE; Aebersold R Development and Validation of a Spectral Library Searching Method for Peptide Identification from MS/MS. Proteomics 2007, 7 (5), 655–667. 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- (26).Midha MK; Campbell DS; Kapil C; Kusebauch U; Hoopmann MR; Bader SL; Moritz RL DIALib-QC an Assessment Tool for Spectral Libraries in Data-Independent Acquisition Proteomics. Nat. Commun 2020, 11 (1), 5251. 10.1038/s41467-020-18901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Deutsch EW; Perez-Riverol Y; Chalkley RJ; Wilhelm M; Tate S; Sachsenberg T; Walzer M; Käll L; Delanghe B; Böcker S; Schymanski EL; Wilmes P; Dorfer V; Kuster B; Volders P-J; Jehmlich N; Vissers JPC; Wolan DW; Wang AY; Mendoza L; Shofstahl J; Dowsey AW; Griss J; Salek RM; Neumann S; Binz P-A; Lam H; Vizcaíno JA; Bandeira N; Röst H Expanding the Use of Spectral Libraries in Proteomics. J. Proteome Res 2018, 17 (12), 4051–4060. 10.1021/acs.jproteome.8b00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lam H; Deutsch EW; Eddes JS; Eng JK; King N; Stein SE; Aebersold R Development and Validation of a Spectral Library Searching Method for Peptide Identification from MS/MS. Proteomics 2007, 7 (5), 655–667. 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- (29).Lam H; Deutsch EW; Aebersold R Artificial Decoy Spectral Libraries for False Discovery Rate Estimation in Spectral Library Searching in Proteomics. J. Proteome Res 2010, 9 (1), 605–610. 10.1021/pr900947u. [DOI] [PubMed] [Google Scholar]
- (30).Ma B Novor: Real-Time Peptide de Novo Sequencing Software. J. Am. Soc. Mass Spectrom 2015, 26 (11), 1885–1894. 10.1007/s13361-015-1204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hoopmann MR; Zelter A; Johnson RS; Riffle M; MacCoss MJ; Davis TN; Moritz RL Kojak: Efficient Analysis of Chemically Cross-Linked Protein Complexes. J. Proteome Res 2015, 14 (5), 2190–2198. 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Riffle M; Hoopmann MR; Jaschob D; Zhong G; Moritz RL; MacCoss MJ; Davis TN; Isoherranen N; Zelter A Discovery and Visualization of Uncharacterized Drug-Protein Adducts Using Mass Spectrometry. Anal. Chem 2022, 94 (8), 3501–3509. 10.1021/acs.analchem.1c04101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hoopmann MR; Mendoza L; Deutsch EW; Shteynberg D; Moritz RL An Open Data Format for Visualization and Analysis of Cross-Linked Mass Spectrometry Results. J. Am. Soc. Mass Spectrom 2016, 27 (11), 1728–1734. 10.1007/s13361-016-1435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shteynberg D; Nesvizhskii AI; Moritz RL; Deutsch EW Combining Results of Multiple Search Engines in Proteomics. Mol. Cell. Proteomics MCP 2013, 12 (9), 2383–2393. 10.1074/mcp.R113.027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res 2019, 18 (12), 4262–4272. 10.1021/acs.jproteome.9b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ramsbottom KA; Prakash A; Riverol YP; Camacho OM; Martin M-J; Vizcaíno JA; Deutsch EW; Jones AR Method for Independent Estimation of the False Localization Rate for Phosphoproteomics. J. Proteome Res 2022, 21 (7), 1603–1615. 10.1021/acs.jproteome.1c00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hoopmann MR; Winget JM; Mendoza L; Moritz RL StPeter: Seamless Label-Free Quantification with the Trans-Proteomic Pipeline. J. Proteome Res 2018, 17 (3), 1314–1320. 10.1021/acs.jproteome.7b00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Griffin NM; Yu J; Long F; Oh P; Shore S; Li Y; Koziol JA; Schnitzer JE Label-Free, Normalized Quantification of Complex Mass Spectrometry Data for Proteomic Analysis. Nat. Biotechnol 2010, 28 (1), 83–89. 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pedrioli PGA; Raught B; Zhang X-D; Rogers R; Aitchison J; Matunis M; Aebersold R Automated Identification of SUMOylation Sites Using Mass Spectrometry and SUMmOn Pattern Recognition Software. Nat. Methods 2006, 3 (7), 533–539. 10.1038/nmeth891. [DOI] [PubMed] [Google Scholar]
- (40).Ross PL; Huang YN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin DJ Multiplexed Protein Quantitation in Saccharomyces Cerevisiae Using Amine-Reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics MCP 2004, 3 (12), 1154–1169. 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- (41).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Johnstone R; Mohammed AKA; Hamon C Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem 2003, 75 (8), 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- (42).Li X-J; Zhang H; Ranish JA; Aebersold R Automated Statistical Analysis of Protein Abundance Ratios from Data Generated by Stable-Isotope Dilution and Tandem Mass Spectrometry. Anal. Chem 2003, 75 (23), 6648–6657. 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- (43).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteomics MCP 2002, 1 (5), 376–386. 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- (44).Hsu J-L; Huang S-Y; Chow N-H; Chen S-H Stable-Isotope Dimethyl Labeling for Quantitative Proteomics. Anal. Chem 2003, 75 (24), 6843–6852. 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- (45).Jung J; Jeong K; Choi Y; Kim SA; Kim H; Lee JW; Kim VN; Kim KP; Kim J-S Deuterium-Free, Three-Plexed Peptide Diethylation for Highly Accurate Quantitative Proteomics. J. Proteome Res 2019, 18 (3), 1078–1087. 10.1021/acs.jproteome.8b00775. [DOI] [PubMed] [Google Scholar]
- (46).Han DK; Eng J; Zhou H; Aebersold R Quantitative Profiling of Differentiation-Induced Microsomal Proteins Using Isotope-Coded Affinity Tags and Mass Spectrometry. Nat. Biotechnol 2001, 19 (10), 946–951. 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Shteynberg D; Mendoza L; Hoopmann MR; Sun Z; Schmidt F; Deutsch EW; Moritz RL ReSpect: Software for Identification of High and Low Abundance Ion Species in Chimeric Tandem Mass Spectra. J. Am. Soc. Mass Spectrom 2015, 26 (11), 1837–1847. 10.1007/s13361-015-1252-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kailash V; Mendoza L; Moritz RL; Hoopmann MR SPACEPro: A Software Tool for Analysis of Protein Sample Cleavage for Tandem Mass Spectrometry. J. Proteome Res 2021, 20 (4), 1911–1917. 10.1021/acs.jproteome.0c00928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Moosa JM; Guan S; Moran MF; Ma B Repeat-Preserving Decoy Database for False Discovery Rate Estimation in Peptide Identification. J. Proteome Res 2020, 19 (3), 1029–1036. 10.1021/acs.jproteome.9b00555. [DOI] [PubMed] [Google Scholar]
- (50).Mendoza L; Deutsch EW; Sun Z; Campbell DS; Shteynberg DD; Moritz RL Flexible and Fast Mapping of Peptides to a Proteome with ProteoMapper. J. Proteome Res 2018, 17 (12), 4337–4344. 10.1021/acs.jproteome.8b00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Binz P-A; Shofstahl J; Vizcaíno JA; Barsnes H; Chalkley RJ; Menschaert G; Alpi E; Clauser K; Eng JK; Lane L; Seymour SL; Sánchez LFH; Mayer G; Eisenacher M; Perez-Riverol Y; Kapp EA; Mendoza L; Baker PR; Collins A; Van Den Bossche T; Deutsch EW Proteomics Standards Initiative Extended FASTA Format. J. Proteome Res 2019, 18 (6), 2686–2692. 10.1021/acs.jproteome.9b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Choi H; Larsen B; Lin Z-Y; Breitkreutz A; Mellacheruvu D; Fermin D; Qin ZS; Tyers M; Gingras A-C; Nesvizhskii AI SAINT: Probabilistic Scoring of Affinity Purification-Mass Spectrometry Data. Nat. Methods 2011, 8 (1), 70–73. 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Teo G; Liu G; Zhang J; Nesvizhskii AI; Gingras A-C; Choi H SAINTexpress: Improvements and Additional Features in Significance Analysis of INTeractome Software. J. Proteomics 2014, 100, 37–43. 10.1016/j.jprot.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Teo G; Koh H; Fermin D; Lambert J-P; Knight JDR; Gingras A-C; Choi H SAINTq: Scoring Protein-Protein Interactions in Affinity Purification - Mass Spectrometry Experiments with Fragment or Peptide Intensity Data. Proteomics 2016, 16 (15–16), 2238–2245. 10.1002/pmic.201500499. [DOI] [PubMed] [Google Scholar]
- (55).Jäger S; Cimermancic P; Gulbahce N; Johnson JR; McGovern KE; Clarke SC; Shales M; Mercenne G; Pache L; Li K; Hernandez H; Jang GM; Roth SL; Akiva E; Marlett J; Stephens M; D’Orso I; Fernandes J; Fahey M; Mahon C; O’Donoghue AJ; Todorovic A; Morris JH; Maltby DA; Alber T; Cagney G; Bushman FD; Young JA; Chanda SK; Sundquist WI; Kortemme T; Hernandez RD; Craik CS; Burlingame A; Sali A; Frankel AD; Krogan NJ Global Landscape of HIV-Human Protein Complexes. Nature 2011, 481 (7381), 365–370. 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sowa ME; Bennett EJ; Gygi SP; Harper JW Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell 2009, 138 (2), 389–403. 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Armean IM; Lilley KS; Trotter MWB Popular Computational Methods to Assess Multiprotein Complexes Derived from Label-Free Affinity Purification and Mass Spectrometry (AP-MS) Experiments. Mol. Cell. Proteomics MCP 2013, 12 (1), 1–13. 10.1074/mcp.R112.019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).da Veiga Leprevost F; Grüning BA; Alves Aflitos S; Röst HL; Uszkoreit J; Barsnes H; Vaudel M; Moreno P; Gatto L; Weber J; Bai M; Jimenez RC; Sachsenberg T; Pfeuffer J; Vera Alvarez R; Griss J; Nesvizhskii AI; Perez-Riverol Y BioContainers: An Open-Source and Community-Driven Framework for Software Standardization. Bioinforma. Oxf. Engl 2017, 33 (16), 2580–2582. 10.1093/bioinformatics/btx192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bai J; Bandla C; Guo J; Vera Alvarez R; Bai M; Vizcaíno JA; Moreno P; Grüning B; Sallou O; Perez-Riverol Y BioContainers Registry: Searching Bioinformatics and Proteomics Tools, Packages, and Containers. J. Proteome Res 2021, 20 (4), 2056–2061. 10.1021/acs.jproteome.0c00904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Slagel J; Mendoza L; Shteynberg D; Deutsch EW; Moritz RL Processing Shotgun Proteomics Data on the Amazon Cloud with the Trans-Proteomic Pipeline. Mol. Cell. Proteomics MCP 2015, 14 (2), 399–404. 10.1074/mcp.O114.043380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Deutsch EW; Mendoza L; Shteynberg DD; Sun Z; Hoopmann MR; Moritz RL The Tutorials of the Trans-Proteomic Pipeline, Chapter 13 in Processing Metabolomics and Proteomics Data with Open Software: A Practical Guide; Winkler R, Ed.; New Developments in Mass Spectrometry; Royal Society of Chemistry: Cambridge, pp 333–344, 2020. 10.1039/9781788019880. [DOI] [Google Scholar]
- (62).Desiere F; Deutsch EW; Nesvizhskii AI; Mallick P; King NL; Eng JK; Aderem A; Boyle R; Brunner E; Donohoe S; Fausto N; Hafen E; Hood L; Katze MG; Kennedy KA; Kregenow F; Lee H; Lin B; Martin D; Ranish JA; Rawlings DJ; Samelson LE; Shiio Y; Watts JD; Wollscheid B; Wright ME; Yan W; Yang L; Yi EC; Zhang H; Aebersold R Integration with the Human Genome of Peptide Sequences Obtained by High-Throughput Mass Spectrometry. Genome Biol. 2005, 6 (1), R9. 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R The PeptideAtlas Project. Nucleic Acids Res. 2006, 34 (Database issue), D655–658. 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Deutsch EW; Eng JK; Zhang H; King NL; Nesvizhskii AI; Lin B; Lee H; Yi EC; Ossola R; Aebersold R Human Plasma PeptideAtlas. Proteomics 2005, 5 (13), 3497–3500. 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- (65).Schwenk JM; Omenn GS; Sun Z; Campbell DS; Baker MS; Overall CM; Aebersold R; Moritz RL; Deutsch EW The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res 2017, 16 (12), 4299–4310. 10.1021/acs.jproteome.7b00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Deutsch EW; Omenn GS; Sun Z; Maes M; Pernemalm M; Palaniappan KK; Letunica N; Vandenbrouck Y; Brun V; Tao S-C; Yu X; Geyer PE; Ignjatovic V; Moritz RL; Schwenk JM Advances and Utility of the Human Plasma Proteome. J. Proteome Res 2021, 20 (12), 5241–5263. 10.1021/acs.jproteome.1c00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Legrain P; Aebersold R; Archakov A; Bairoch A; Bala K; Beretta L; Bergeron J; Borchers CH; Corthals GL; Costello CE; Deutsch EW; Domon B; Hancock W; He F; Hochstrasser D; Marko-Varga G; Salekdeh GH; Sechi S; Snyder M; Srivastava S; Uhlén M; Wu CH; Yamamoto T; Paik Y-K; Omenn GS The Human Proteome Project: Current State and Future Direction. Mol. Cell. Proteomics MCP 2011, 10 (7), M111.009993. 10.1074/mcp.M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Adhikari S; Nice EC; Deutsch EW; Lane L; Omenn GS; Pennington SR; Paik Y-K; Overall CM; Corrales FJ; Cristea IM; Van Eyk JE; Uhlén M; Lindskog C; Chan DW; Bairoch A; Waddington JC; Justice JL; LaBaer J; Rodriguez H; He F; Kostrzewa M; Ping P; Gundry RL; Stewart P; Srivastava S; Srivastava S; Nogueira FCS; Domont GB; Vandenbrouck Y; Lam MPY; Wennersten S; Vizcaino JA; Wilkins M; Schwenk JM; Lundberg E; Bandeira N; Marko-Varga G; Weintraub ST; Pineau C; Kusebauch U; Moritz RL; Ahn SB; Palmblad M; Snyder MP; Aebersold R; Baker MS A High-Stringency Blueprint of the Human Proteome. Nat. Commun 2020, 11 (1), 5301. 10.1038/s41467-020-19045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Omenn GS; Lane L; Overall CM; Paik Y-K; Cristea IM; Corrales FJ; Lindskog C; Weintraub S; Roehrl MHA; Liu S; Bandeira N; Srivastava S; Chen Y-J; Aebersold R; Moritz RL; Deutsch EW Progress Identifying and Analyzing the Human Proteome: 2021 Metrics from the HUPO Human Proteome Project. J. Proteome Res 2021, 20 (12), 5227–5240. 10.1021/acs.jproteome.1c00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Deutsch EW; Lane L; Overall CM; Bandeira N; Baker MS; Pineau C; Moritz RL; Corrales F; Orchard S; Van Eyk JE; Paik Y-K; Weintraub ST; Vandenbrouck Y; Omenn GS Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J. Proteome Res 2019, 18 (12), 4108–4116. 10.1021/acs.jproteome.9b00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Pedrioli PGA; Eng JK; Hubley R; Vogelzang M; Deutsch EW; Raught B; Pratt B; Nilsson E; Angeletti RH; Apweiler R; Cheung K; Costello CE; Hermjakob H; Huang S; Julian RK; Kapp E; McComb ME; Oliver SG; Omenn G; Paton NW; Simpson R; Smith R; Taylor CF; Zhu W; Aebersold R A Common Open Representation of Mass Spectrometry Data and Its Application to Proteomics Research. Nat. Biotechnol 2004, 22 (11), 1459–1466. 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- (72).Wippel HH; Santos MDM; Clasen MA; Kurt LU; Nogueira FCS; Carvalho CE; McCormick TM; Neto GPB; Alves LR; da Gloria da Costa Carvalho M; Carvalho PC; Fischer J. de S. da G. Comparing Intestinal versus Diffuse Gastric Cancer Using a PEFF-Oriented Proteomic Pipeline. J. Proteomics 2018, 171, 63–72. 10.1016/j.jprot.2017.10.005. [DOI] [PubMed] [Google Scholar]
- (73).Deutsch EW; Perez-Riverol Y; Carver J; Kawano S; Mendoza L; Van Den Bossche T; Gabriels R; Binz P-A; Pullman B; Sun Z; Shofstahl J; Bittremieux W; Mak TD; Klein J; Zhu Y; Lam H; Vizcaíno JA; Bandeira N Universal Spectrum Identifier for Mass Spectra. Nat. Methods 2021, 18 (7), 768–770. 10.1038/s41592-021-01184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).LeDuc RD; Deutsch EW; Binz P-A; Fellers RT; Cesnik AJ; Klein JA; Van Den Bossche T; Gabriels R; Yalavarthi A; Perez-Riverol Y; Carver J; Bittremieux W; Kawano S; Pullman B; Bandeira N; Kelleher NL; Thomas PM; Vizcaíno JA Proteomics Standards Initiative’s ProForma 2.0: Unifying the Encoding of Proteoforms and Peptidoforms. J. Proteome Res 2022, 21 (4), 1189–1195. 10.1021/acs.jproteome.1c00771. [DOI] [PMC free article] [PubMed] [Google Scholar]