

Abstract
The generation of islet-like endocrine clusters from human pluripotent stem cells (hPSCs) has the potential to provide an unlimited source of insulin-producing β cells for the treatment of diabetes. In order for this cell therapy to become widely adopted, highly functional and well-characterized stem cell-derived islets (SC-islets) need to be manufactured at scale. Furthermore, successful SC-islet replacement strategies should prevent significant cell loss immediately following transplantation and avoid long-term immune rejection. This review highlights the most recent advances in the generation and characterization of highly functional SC-islets as well as strategies to ensure graft viability and safety after transplantation.
Short Summary
Stem cell-derived islets (SC-islets) have the potential to provide an unlimited source of insulin-producing β cells for the treatment of diabetes. Here, we review the most recent developments associated with generating highly functional SC-islets, solving the immune rejection problem, and overcoming practical challenges associated with SC-islet transplantation.
A cell replacement therapy for treating type 1 diabetes
Diabetes as an ideal candidate for cell replacement therapy
Insulin is an essential regulator of energy metabolism throughout the body, directing the usage of carbohydrates, fats, and proteins.1–4 In particular, the binding of insulin to cell surface receptors facilitates the entry of glucose from the blood stream into many cell types within the body, most notably in muscle and fat, so that they can use it as energy. Insulin signals for excess glucose to be stored for future use as glycogen in the liver and muscle as well as converted to fat in adipocytes. In parallel, insulin signaling slows the breakdown of fats and proteins, as they are not needed much when glucose is plentiful. The amount of insulin circulating in the blood changes dynamically in response to the constantly changing energy needs of the body and the availability of each fuel source. Specialized endocrine cells called β cells, located in the islets of Langerhans within the pancreas, are responsible for this tightly regulated production and release of insulin. After a meal, for example, blood glucose levels rise as carbohydrates are broken down into glucose and absorbed into the blood stream. β cells can rapidly sense this increasing blood glucose level and secrete the appropriate amount of insulin in response, allowing cells throughout the body to utilize this glucose for energy production. Blood glucose levels fall as glucose enters cells, causing the β cells to slow their release of insulin. This reduction in insulin secretion in conjunction with an increase of the counter-regulatory hormone glucagon by α cells prevents blood glucose levels from dropping below a set threshold (~70 mg/dL in humans), as a minimum concentration is required for tissues of the central nervous system to function properly. By rapidly changing the amount of insulin and glucagon in circulation, β and α cells maintain blood glucose levels in a narrow optimal range.
In type 1 diabetes (T1D), β cells are selectively destroyed by an autoimmune process, resulting in the inability to produce and secrete insulin.5,6 Without insulin, glucose homeostasis and energy metabolism balance in the body are completely disrupted. Because many cell types can no longer import glucose for energy, they must switch to metabolizing free fatty acids that are liberated from the breakdown of triglycerides in adipocytes as their main energy source. Importantly, as the liver processes free fatty acids, it generates ketones that can also be used as an energy source by other tissues. While this process is normal during periods of fasting and low carbohydrate dieting, the complete absence of insulin signaling in T1D results in uninhibited fat breakdown and uncontrolled ketone production. The rapid buildup of ketones changes blood pH, resulting in the life-threatening condition called ketoacidosis.7,8 Thus, patients with T1D must inject exogenous insulin in order to survive and restore the necessary signaling pathways that properly regulate their energy metabolism.
While injecting insulin allows T1D patients to stay alive, replicating the precise insulin secretion dynamics of β cells can be difficult. Not only do patients need to consider what they are eating when calculating an insulin dose, but because insulin requirements are intimately linked to energy metabolism, other factors such as the intensity and duration of physical activity as well as stress levels can influence how much insulin the body requires at a given time. Incorrectly estimating the amount of insulin needed at a particular time can create both short and long-term issues. For example, not giving enough insulin will cause the blood glucose level of a patient to be higher than it should be. In the extreme, high glucose levels can change blood osmolarity and result in life-threatening dehydration.8 Moderately high glucose levels are not a serious issue in the short term, though they may make the patient feel suboptimal. Over the lifetime of the patient, however, this chronic elevation of blood glucose concentration can damage tissues throughout the body. Therefore, it is critically important to try to keep blood glucose levels as close to normal as possible to avoid long-term complications, such as cardiovascular, kidney, and eye disease.9,10 On the other hand, giving too much insulin is dangerous in the short term because it can cause blood glucose levels to dip below the normal lower limit. The central nervous system requires a constant supply of glucose to function, and a certain concentration in the blood is required to facilitate sufficient transport of glucose across the blood-brain barrier.11 Thus as blood glucose levels drop below this threshold, a person can start to act abnormally as their brain essentially starves. If blood glucose levels continue to fall, the person can lose consciousness and ultimately die.12 Consequently, patients taking insulin must try to navigate between these two extremes and estimate the amount of insulin their body needs based on their current metabolic state.
The treatment of T1D has made great strides in recent years. In particular, new tools such as insulin pumps and continuous glucose monitors have undoubtedly made living with diabetes easier and have helped many patients achieve better control over their blood glucose levels.13 Despite these advances, however, many patients still fail to achieve their target goals. And even if they do meet their blood glucose targets, the therapy can remain burdensome, as it still requires constant monitoring and adjustment. Because of the difficultly in replicating the precise insulin secretion dynamics of β cells with exogenous insulin injections, a potentially better treatment alternative consists of replacing the lost β cells with new ones, allowing these transplanted cells to monitor blood glucose levels and secrete the appropriate amount of insulin in response. Such a transplant would provide a “functional cure” for T1D patients, as they would no longer have to manage their blood glucose levels with insulin injections. Type 2 diabetics that rely on insulin injections may also benefit from such a transplant. Intriguingly, T1D is a potentially ideal candidate for cell replacement therapy. Because individual β cells can sense extracellular glucose changes and secrete insulin, there is less of a need for a complex working structure of multiple integrated cell types, as would be required in tissue engineering whole organs such as a heart or kidney. Rather, as long as the β cells secrete insulin properly in response to glucose stimulation and are transplanted in a location that facilitates adequate exchange of these molecules with the blood stream, such a cell replacement therapy could provide a functional cure for T1D.
Improvements in whole pancreas transplantation since the 1960s demonstrated that donor pancreatic tissue could restore glucose homeostasis in diabetic patients.14 Transplantation of pancreatic islets alone to restore β cell mass, however, had limited success until the development of the Edmonton Protocol in 2000, which demonstrated that infusion of islets into the liver via the portal vein was sufficient to restore glucose homeostasis.15,16 Their initial trial led to insulin independence of all seven patients 1 year after transplantation, vastly improving previous islet transplantation outcomes due to optimization of their immunosuppression regimen as well as benefiting from improvements in islet isolation techniques. Although all but one of these patients required supplemental insulin 10 years post-transplant, all recipients retained some level of graft function that provided substantial benefits to glucose control and elimination of severe hypoglycemic events.17 Subsequent transplantation studies continued to show improved patient outcomes, with islet transplantation approaching the success of whole pancreas transplantation without the corresponding surgical risk.18–23 Importantly, these remarkable studies demonstrated that the concept of replacing β cell mass by transplanting pancreatic islets was relatively safe and effective in treating T1D.
Recent clinical trials with stem cell-derived pancreatic tissue
While transplantation of human islets from deceased donors has demonstrated the feasibility of a cell replacement therapy for treating diabetes, there are a number of issues that limit its more widespread use. In particular, graft success increases when a high number of islets are transplanted, often resulting in the need for multiple donor pancreases. 15,20,24 Not only are the number of pancreases available for islet isolation limited, but transplanting cells from multiple genetic backgrounds per patient could exacerbate immune rejection and graft failure.25,26 Furthermore, the stress of purification from the pancreas can damage islets before transplantation.27 This sourcing problem could be circumvented by differentiating stem cells into β cells in vitro, generating an unlimited supply of insulin-producing cells for transplantation.28 These stem cell-derived islets (SC-islets) could be generated from a single cell source using a standardized process, and the resulting cell product could be well-characterized to allow for more predictable transplant outcomes. Furthermore, a stem cell-derived product could also be genetically engineered to have advantageous features not found in primary islets, such as resistance to stressors like hypoxia or being able to evade the host immune system.
Recent advances in stem cell technologies have led to the first human clinical trials using stem cell-derived pancreatic products. In particular, ViaCyte developed a stem cell-derived pancreatic endoderm cell population known as PEC-01, which they demonstrated to mature over several months in vivo into insulin-producing endocrine cells in rodent models.29–31 In conjunction, they developed several iterations of a retrievable macroencapsulation device to contain the transplanted cells. An initial 2014 human clinical trial (clinicaltrials.gov: NCT02239354) used the Encaptra device, which was designed to fully immunoprotect the cells using a cell-impermeable membrane.32 While the transplanted PEC-Encap product was well tolerated with few adverse effects, the trial was halted due to insufficient functional product engraftment.33 While some endocrine cells were observed, fibrosis around the capsule resulted in graft loss, and no insulin secretion from the device was detected.33,34 To overcome this issue, the newer PEC-Direct device contained openings in the membrane to allow for vascularization to enhance nutrient exchange and promote cell survival. Because host cells were able to penetrate the device, however, immunosuppression was required after transplantation. An ongoing clinical trial was started in 2017 at seven sites to test this device (clinicaltrials.gov: NCT03163511), and the first round of results have recently been published.33,35 They demonstrated glucose responsive C-peptide production 6-9 months post-transplant as the grafted cells matured from pancreatic progenitors into pancreatic endocrine cells. Upon graft removal, a majority of these graft-derived cells immunostained for the general endocrine marker chromogranin A (CHGA). Interestingly, while there were regions of C-peptide positive β-like cells, a majority of these endocrine cells stained for the α cell marker glucagon. Ductal cells and rare acinar cells were also observed. As intended, the porous design promoted the growth of host-derived blood vessels inside the device, and this phenomenon was more prominent in regions containing the graft-derived cells. In other regions of the device, however, host fibroblasts were the predominant cell type, resulting in the deposition of fibrotic tissue rather than host-derived vasculature. While the observed circulating C-peptide levels in these studies were too low to induce a measurable clinical benefit attributable to the transplanted cells, these clinical trials have demonstrated that facilitating host-derived vascularization into the graft promoted long-term endocrine cell survival and function in humans. In parallel, they highlight the importance of limiting the fibrotic response of the body to a transplanted device in order to ensure these outcomes. Importantly, these studies did not detect any serious safety concerns related to the transplanted cells, such as tumor formation.
In the ViaCyte clinical trial, the explanted grafts after months in vivo were observed to be highly heterogenous between patients since the cells were transplanted as pancreatic progenitors and allowed to finish differentiating in vivo.33,35 Ideally, the β cells could be terminally differentiated in vitro and capable of glucose-responsive insulin secretion before transplantation, which would allow for a more consistent cell population that would facilitate a relatively rapid restoration of blood glucose control. In 2014-15, several groups published protocols for generating stem cell-derived β (SC-β) cells that secreted insulin in response to glucose stimulation.36–38 These cells have been shown to improve glycemic control in diabetic mice, highlighting their potential utility in vivo.36,37,39–44 Recently, these SC-islets were also used to help treat diabetic non-human primates.45,46 While the primates did not achieve complete insulin independence, the transplanted SC-islets significantly reduced their insulin requirements and greatly improved their glycemic control, further illustrating their potential utility as a therapy. SC-islets have also now been transplanted into human T1D patients in a clinical trial by Vertex Pharmaceuticals, which began in 2021 (clinicaltrials.gov: NCT04786262). The first two patients were given half the anticipated target dose to assess the safety profile of their stem-cell derived product, VX-880. Furthermore, these cells were transplanted without an immune-protective device to avoid problems with nutrient exchange and fibrosis, and thus the patients required immunosuppressive drugs to ensure graft survival. While the initial findings have not yet been peer-reviewed, positive preliminary results reported in a press release have indicated that transplantation of these SC-islets improved glycemic control in these two T1D patients (https://www.businesswire.com/news/home/20220606005424/en/). The transplanted SC-islets took longer to improve glycemic control than in rodent models, however, and the reason for this delay has yet to be explored.
Remaining challenges for developing a widely used cell therapy
These promising initial clinical trial results highlight the great promise SC-islets hold for treating T1D. However, there are several remaining challenges to be addressed before this cell therapy can become a routine procedure (Figure 1). First, SC-β cells generated with current directed differentiation protocols are still not quite as functional as primary human islets, and their transcriptional and chromatin landscape remains immature. Furthermore, the current differentiation methodologies still produce off-target cell types, notably cells that resemble a type of intestinal endocrine called the enterochromaffin cell.47 The presence of these off-targets as well as the observed lower insulin secretion per cell compared to primary islets indicates that there is still room to improve these differentiation protocols. If the insulin secretion per cell can be increased further, fewer cells will be needed to cure a patient. Reducing the required cell number is significant in terms of both the graft volume that needs to be transplanted as well as cell production costs. To this end, significant progress has been made at improving the function of SC-islets when compared to the original protocols,39,40,44,48,49 and recent advances in single-cell sequencing technologies have allowed for unprecedented characterization of these cells to further elucidate ways to refine SC-β cell differentiation strategies.43,47,50,51
Figure 1. Key pillars of a successful SC-islet therapy for treating T1D.
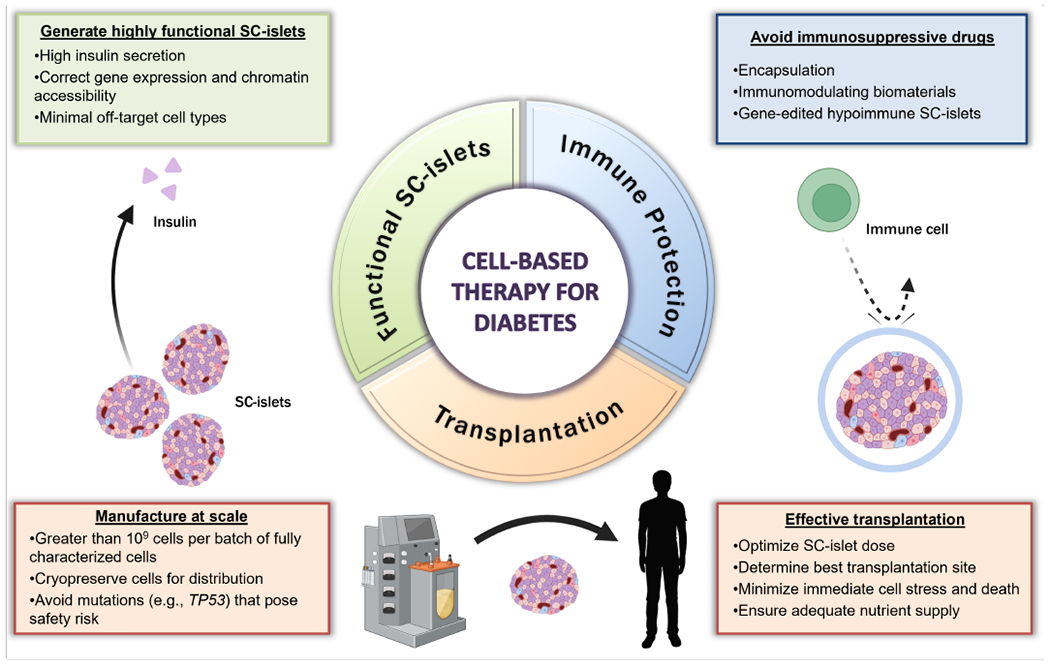
The remaining challenges limiting the application of SC-islets as a treatment option for diabetes cover a broad range of considerations. The first major component of a successful SC-islet therapy is the ability to produce a highly functional, uniform cell-based product for transplantation. Further optimization of differentiation protocols should aim limit the generation of off-target cell populations and improve the gene expression and chromatin accessibility profiles to match those of human adult islets. These advancements would ideally improve the amount of insulin secreted by SC-β cells and thus reduce the number of cells required per patient. Furthermore, improved transplant survival and a reduced, or eliminated, need for immunosuppressive drugs can be achieved by applying bioengineering strategies. Specifically, using immunomodulating biomaterials or genetically engineered hypoimmune SC-islets could maximize the efficacy of transplanted grafts while minimizing the immune response commonly associated with the transplantation of allogeneic materials. Finally, regulatory standards must be established to maximize the safety and efficacy of SC-islet transplantation. This includes improving methods for large-scale manufacturing of SC-islets with sufficient quality control characterizations, such as avoiding mutations that pose safety risks, and developing standardized methods for storage and distribution. Additionally, as these challenges are addressed and SC-islet products are improved, guidelines to optimize dosing, transplantation site, and graft survival must be established.
Secondly, there would ideally be a strategy to circumvent the need to take immunosuppressive drugs after transplantation. For the typical T1D patient that is able to achieve their blood sugar targets with insulin injections as indicated by their hemoglobin A1c levels, the potentially severe adverse side effects of life-long immunosuppression may not outweigh the benefits of further improved glycemic control. Thus, several strategies are currently being pursued to protect the transplanted SC-islets from immune attack. One attractive method is to encapsulate the cells in a device with finely controlled pore size that facilitates the diffusion of small molecules like glucose and insulin but blocks immune cells from infiltrating the device.52 Biomaterials may also be fabricated with instructive chemistry to induce local immune tolerance around the transplanted cells.53 Alternatively, the SC-islets themselves can be genetically engineered to both remove antigens that would signal them for destruction by the host immune system as well as add surface signaling molecules that induce immune tolerance.54,55
Lastly, there are several practical factors that need to be addressed for this cell-based therapy to become widely used in the clinic. Specifically, it is crucial to reduce the stress response within islets immediately following transplantation to reduce initial cell death, which can result in significant graft loss.56 Facilitating rapid vascularization is key in alleviating these stresses to promote cell survival and long-term health of the transplanted SC-islets. Genetically engineering the cells to be resistant to these non-ideal conditions could also aid in their survival during the period before vascularization can occur. Furthermore, developing large-scale manufacturing methods for generating SC-islets as well as methods for their cryopreservation and distribution will be crucial for the widespread adoption of this procedure. In the remainder of this review, we will be discussing the most recent advances in addressing these issues associated with 1) generating highly functional SC-islets, 2) solving the immune rejection problem, and 3) overcoming practical challenges associated with transplanting these cells.
Generating highly functional SC-islets
Strategies for improved SC-islet generation
The ongoing clinical trial led by Vertex Pharmaceuticals has indicated that existing protocols generate SC-islets that are capable of improving glycemic control in human T1D patients. However, further enhancing SC-islets to better mimic the functionality of primary adult islets would reduce the number of cells required for transplantation and make it easier to manufacture sufficient cell numbers. For example, conceptually, if insulin secretion per cell is doubled, then potentially only half as many cells are needed to cure a patient. Reducing graft volume facilitates an easier transplantation procedure, reduces nutrient exchange requirements at the transplantation site, and opens alternative transplantation site possibilities. Furthermore, fewer cells would need to be manufactured, which is significant in terms of both production costs and logistics for a cell-based therapy.
SC-islets are generated through a multistep differentiation process that attempts to mimic stages of embryonic development with the timed application of growth factors and small molecules, driving stem cells through several intermediate cell types on their way to becoming pancreatic endocrine cells (Figure 2).57 This differentiation methodology takes approximately a month or more to complete, and functional maturation can continue for months once the cells are transplanted in vivo.43,51 The developmental pathways targeted by this process can be very sensitive to changes in timing and signaling intensity, and thus small signaling changes at specific times can lead to large changes in cell fate selection. Consequently, many attempts to improve SC-islet generation consist of iterations with different combinations and timings of soluble factors. The latest generation of protocols are better at generating islet-like clusters consisting of mostly endocrine cells, though not all of these are β cells. A substantial fraction can be α cells along with some δ cells. Interestingly, the role or importance of these other endocrine cell types to β cell function within the context of SC-islets or if there is an optimum cell ratio to target during differentiation is unknown. Current protocols also produce a significant percentage of stem cell-derived enterochromaffin cells (SC-ECs), which resemble a type of intestinal endocrine.47 With less optimized protocols or cell lines, there is also the potential to retain progenitor cells in the final islet clusters as well as produce other endodermal cell types, such as those of hepatic origin.39,58 Consequently, increasing insulin secretion per cell in SC-islets can be achieved through improvements in either SC-β cell maturation or by decreasing the percentage of these off-target cell populations.
Figure 2. Growth factors and small molecules used during the multistage differentiation of hPSCs to SC-islets.
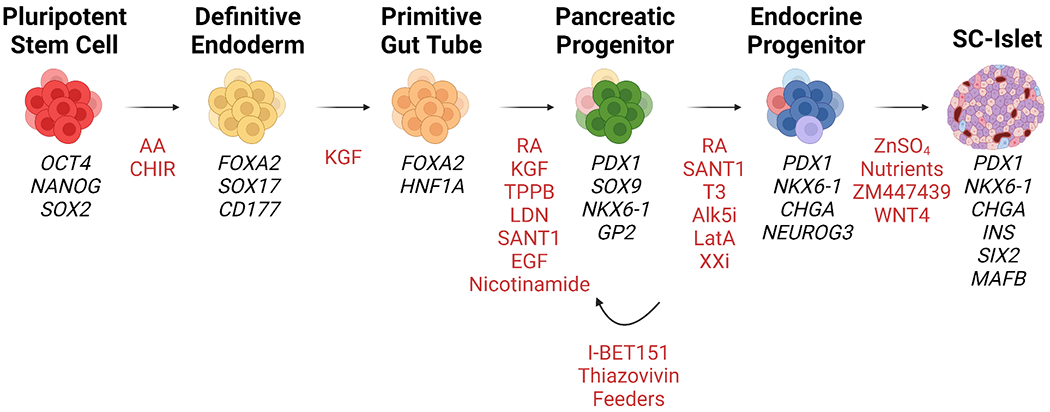
The generation of SC-islets from hPSCs is a multistage process that involves the temporal application of small molecules and growth factors (highlighted in red). At each stage, specific developmental pathways are targeted to mimic human pancreatic development. While multiple differentiation protocols have been reported, each follows the same general differentiation trajectory by first specifying hPSCs into definitive endoderm using Activin A (AA) and WNT pathway activation, most often with CHIR99021. Next, definitive endoderm cells are guided into a primitive gut tube state using keratinocyte growth factor (KGF), also known as FGF7. Notably, some protocols include additional compounds that target WNT (e.g., IWP249) and TGF-β signaling pathways to improve primitive gut tube specification. Generating pancreatic progenitor cells requires additional activation of protein kinase C with TPPB and retinoic acid signaling pathways while simultaneously inhibiting BMP signaling with LDN193189 and SHH signaling with SANT1. Epidermal growth factor (EGF) and nicotinamide have also been shown to improve the generation of PDX1+/NKX6-1+ pancreatic progenitors.63 Interestingly, it has been reported that the use of a selective BET inhibitor can maintain pancreatic progenitors cells in a proliferative and expandable state.184 This can potentially accelerate SC-islet manufacturing by providing an intermediate point from which to generate SC-islets. During the later stages of SC-islet differentiation, reported protocols begin to diversify both in nomenclature of cell populations and compounds applied. For example, latrunculin A (LatA) has been used to specify endocrine cells in planar culture.39 Additional nomenclature differences arise with some protocols describing the initial SC-islet product as “immature” and include an additional stage to generate “mature” SC-islets using an aurora kinase inhibitor (ZM447439)43 or WNT analogs67.
Since the original SC-islet protocol developments in 2014-15,36–38 there have been a number of reports that have significantly improved SC-islet function and characterized their maturation. The first significant advance established dynamic function in SC-islets.42,44 Biphasic insulin secretion is a well-known characteristic of human islet function,59,60 but SC-islets generated from early protocols notably lacked this feature. An enriched serum-free media combined with cluster resizing through a single-cell dispersion and aggregation technique generated SC-islet clusters that achieved biphasic insulin secretion kinetics resembling that of human islets.42 Notably, the exclusion of Alk5 inhibitor II in this media greatly improved insulin secretion. While this inhibitor is necessary during the previous endocrine induction step, the benefits of its removal in the subsequent maturation stage strongly indicated that TGF-β signaling was important for SC-β cell functional maturation. Similarly, enriched β-clusters (eBCs) generated by sorting INSGFP+ β-like cells and aggregating them into clusters demonstrated improved function.44 These eBCs were composed exclusively of stem cell-derived endocrine cells, with approximately 90% of the cells being C-peptide+/glucacon− and the remainder expressing multiple hormones. Mitochondrial respiratory function, mitochondrial energization, and mitochondrial membrane potential during glucose stimulation assays suggested metabolic maturation of eBCs as a driver of improved insulin secretion. Further adding to these improvements, single-cell dispersion and reaggregration of SC-islet clusters promoted enrichment of C-peptide+ endocrine cell types42,47 along with sorting based on the surface marker CD49a.47
The 3D architecture of human islets is known to be important for glucose responsiveness and insulin secretion.61 Consequently, early differentiation protocols for SC-islets were developed using 3D cell clusters, either from the outset of differentiation in suspension culture36,62 or prior to endocrine specification on an air-liquid interface37. Interestingly, PDX1+/NKX6-1+ pancreatic progenitors could be efficiently generated from hPSCs in traditional planar culture, but these protocols required 3D cell aggregation to efficiently generate endocrine cells from these progenitors.37,63 Recently, the state of the actin cytoskeleton and downstream yes-associated protein (YAP) signaling was found to be vital to the pancreatic progenitor program.39,64–66 In particular, a polymerized cytoskeletal state resulting from culture on tissue culture polystyrene inhibited NEUROG3-mediated endocrine induction in PDX1+/NKX6-1+ pancreatic progenitors.39 To circumvent the need for a 3D format during endocrine specification, a planar methodology was developed whereby the actin cytoskeleton was depolymerized with the compound latrunculin A during the first 24 hours of endocrine specification.39 This short treatment was sufficient to initiate robust endocrine specification of PDX1+/NKX6-1+ pancreatic progenitors in planar culture. These differentiated endocrine cells could subsequently be dispersed from the culture surface and aggregated into islet-like clusters for use in downstream assays. SC-islets generated with this approach demonstrated improved in vitro dynamic insulin secretion and faster reversal of severe diabetes in mice compared to cells generated with a suspension differentiation protocol. Importantly, this planar methodology was also more amenable to differentiating SC-islets from a wide range of stem cell lines with various genetic backgrounds.40,57
Several other unique approaches have been used to improve the function of SC-islets. For example, adhering to a strict feed/fast cycle activated circadian rhythm programs in maturing SC-islets.48 This feeding schedule triggered rhythmic transcription of genes involved in energy metabolism as well as genes related to the synthesis, transport, and release of insulin, resulting in improved SC-islet function. In several studies, modulation of WNT signaling at various time points in the differentiation protocol has been shown to improve endocrine cell fate selection and maturation. In one report, different subpopulations of hPSC-derived definitive endoderm displayed inverse activation of canonical and noncanonical WNT signaling.49 Specifically, definitive endoderm cells expressing CD177 were distinguished by increased noncanonical WNT signaling and their tendency to differentiate into a pancreatic lineage, whereas CD275-expressing cells upregulated canonical WNT signaling and specified a liver fate. Treatment with IWP2, a small molecule inhibitor of WNT ligand secretion, after endoderm specification promoted a pancreatic lineage. Furthermore, sorting the definitive endoderm population to enrich for CD177+ cells led to the generation of SC-islets with improved maturation and function. An alternative differentiation approach attempted to recreate multicellular interactions during pancreatic organogenesis by combining human adipose-derived stem cells and human umbilical vein endothelial cells with human iPSC-derived endocrine progenitors in a polysaccharide-based gel.67 Interestingly, these multicellular spheroids demonstrated that noncanonical WNT signaling drove maturation of the β-cells. Addition of exogenous WNT4 helped islet-like organoids achieve glucose-stimulated insulin secretion, implicating the need to promote noncanonical WNT signaling during SC-islet maturation. Conversely, another study demonstrated that inhibiting canonical WNT signaling during endocrine differentiation improved outcomes, further highlighting that the type and timing of WNT signaling during differentiation can drastically influence the specification and maturation of SC-islets.68
Utilizing a differentiation protocol that incorporated several of the improvements mentioned here, one study presented a comprehensive analysis of SC-islets compared to primary human islets.43 These SC-islets developed robust dynamic insulin secretion overtime in vitro, corresponding to changes in islet architecture but without changes in β cell mass. Furthermore, SC-β cells demonstrated transcriptional maturation over the 6-week culture period and had the correct ion channel profile and exocytosis machinery to support proper insulin secretion. Aspects of glucose metabolism, however, differed in SC-islets when compared with primary human islets. In particular, glucose processing was abnormal and not correctly coupled to mitochondrial respiration, which is normally key to canonically triggering insulin secretion in β cells. Similarly, another report demonstrated that defects of glucose metabolism in SC-β cells prevented proper insulin release rather than originating from any deficiencies in the exocytosis machinery.69 Treatment with cell-permeable metabolites to bypass this bottleneck in glycolysis resulted in greatly improved insulin secretion in SC-islets. These studies highlight that while current generation SC-islets can secrete significant amounts of insulin in response to glucose stimulation in vitro, fundamental differences still exist between SC-islets and primary human islets, resulting in the observed lower functional performance of SC-islets. Consequently, it is crucial to better characterize the cells generated with these protocols in order to inform the next generation of differentiation strategies.
Using single-cell sequencing to obtain new insights into SC-islet biology
In addition to profiling the functional characteristics of SC-β cells to assess their maturation state, insights into deficiencies in their gene expression and chromatin accessibility profile can reveal additional signaling pathway interactions that can be targeted to further improve SC-β cell lineage specification and maturation. The rise of single-cell sequencing technologies has steered much of the recent SC-islet research toward thoroughly characterizing their transcriptional and chromatin landscape. Single-cell RNA sequencing (scRNA-seq) has become one of the most widely used single-cell sequencing approaches, enabling high-throughput, multi-dimensional characterization of cellular diversity by transcriptionally profiling each individual cell.70 In parallel with the boom in scRNA-seq technology, the assay for transposase-accessible chromatin using sequencing (ATAC-seq) has facilitated robust and sensitive epigenomic profiling of open chromatin sites that are accessible for transcription.71 On the individual cell level, this ATAC-seq technology can use either single cells (scATAC-seq)72 or single nuclei (snATAC-seq).73 These technological advances have enabled new insights into the differentiation of stem cells into SC-islets, which is a transcriptionally dynamic process that often produces heterogeneous cellular populations. Furthermore, multimodal analysis through combined scRNA-seq and snATAC-seq using integrative computational methods has provided an even deeper understanding of SC-islet composition and maturation.
Recently, two separate studies utilized scRNA-seq to map transcriptional changes during SC-islet differentiation47,50 Despite the use of different pluripotent stem cell lines and protocols, both roadmaps observed well-known markers of pancreatic progenitors (e.g., PDX1, NKX6-1) during the middle stages of differentiation as well as islet-specific endocrine identity markers in their final cell populations (e.g., INS for β cells, GCG for α cells, SST for δ cells). These results validated well-established knowledge of pancreatic organogenesis that inspired early protocols for SC-islet differentiation. Interestingly, both scRNA-seq roadmaps detected significant cell diversification following the pancreatic progenitor stages. After sequencing 40,444 cells during the second half of SC-islet differentiation, the first study reported a mostly homogeneous population of PDX1+ progenitors at end of stage 3.47 In subsequent stages, high expression of PDX1 in combination with expression of NKX6-1 and PTF1A was predictive of endocrine induction potential. Notably, the non-endocrine cell populations of ductal-like, acinar-like, and mesenchymal-like cells were derived from populations that retained expression of cell cycle-associated genes. The second report sequenced 87,769 cells across 12 points during the differentiation of a stem cell line known to result in highly heterogeneous SC-islets.50 Using a semi-supervised method to construct a lineage tree of cell fate, it was demonstrated that divergent cell populations that arose during early stages did not significantly contribute to the off-target populations observed later. In contrast, the final non-endocrine off-target populations, including duct-like and pancreatic stellate-like cells, branched during pancreatic progenitor stages. 1,150 endocrine-specific switch genes were identified, and it was inferred that 656 pairs of transcription factor and target gene interactions potentially influenced endocrine specification. Of note, NOTCH signaling via HES1 was implicated as a driver of a non-endocrine fate.
One of the most interesting findings from these scRNA-seq studies was the identification of SC-EC cells, which are absent from primary human islets and resemble an endocrine cell type normally found in the intestines.47 These cells are characterized by the expression of enterochromaffin gene markers, such as TPH1 and LMX1A, as well as the secretion of serotonin rather than insulin. SC-β cells and off-target SC-EC cells appear to emerge from a common NEUROG3+ progenitor population.47 These cells have been subsequently identified in SC-islets generated from various differentiation protocols and stem cell lines by multiple groups.43,50,51 Despite distinguishable differences in gene expression between SC-EC and SC-β cells, the two populations appear to have a relatively similar overall transcriptional profile. Because SC-EC cells comprise a significant percentage of the final cell population in SC-islets but do not secrete insulin, differentiation efficiency and SC-islet insulin secretion on a per cell basis could be greatly improved by diverting these SC-EC cells toward a SC-β cell lineage during either the cell specification or maturation stages.
To gain further insights into the nature of these cells and other lineage fate decisions during SC-islet differentiation, several groups have recently implemented snATAC-seq analysis to ascertain the chromatin accessibility profiles of differentiating cells in combination with their transcriptional data obtained from scRNA-seq. Of note, these findings are currently published as pre-prints and still under peer-review due to the novelty of both snATAC-seq itself as well as the computational methods used to analyze the integrated scRNA-seq and snATAC-seq datasets. In one approach, a canonical correlation analysis algorithm was used to integrate chromatin accessibility and gene expression data from different cells in silico to generate pseudo-cells with matched epigenomic and transcriptomic information within differentiating SC-islets.74 In contrast, another study reported the simultaneous multiomic sequencing of RNA expression and chromatin accessibility from the same cell.75 Using canonical gene markers of islet cell types, both multiomic studies identified distinct populations representing SC-β, SC-α, SC-δ, and SC-EC cells. Notably, both studies also provided greater resolution of cellular heterogeneity by describing specific subpopulations of expected cell types. Specifically, one report identified two pancreatic progenitor populations based on differences in NKX6-1 expression and defined four distinct endocrine progenitor populations identified by the expression of NEUROG3, ARX, LMX1A, and RFX3.74 Similarly, the second study identified two SC-EC populations based on the integrated analysis of both mRNA and chromatin accessibility data.75
Both studies performed trajectory analysis using Monocle376 to characterize lineage selection during SC-islet differentiation. Previous analysis of scRNA-seq data alone had suggested that SC-β and SC-EC are distinct cell types that share a common progenitor lineage during in vitro differentiation of SC-islets,47,50 but examining the combined gene expression and chromatin accessibility data for each cell revealed a gradient of cell states between SC-EC and SC-β cells.75 Specifically, there appeared to be SC-β cell subpopulations that expressed TPH1, a known marker of SC-EC cells, and that had increased accessibility of binding sites for LMX1A, a transcription factor associated with the SC-EC cell fate. They also reported a potentially novel role for the chromatin remodeling transcription factor CTCF in regulating cell fate selection toward the enteroendocrine lineage. Additionally, the other study identified CDX2 as the earliest transcription factor expressed during SC-EC lineage specification.74 Interestingly, they also reported a novel CDX2+ β-cell precursor-like population in the human fetal pancreas that resembled SC-EC cells, leading them to suggest that SC-EC cells are in fact of pancreatic rather than intestinal origin. Collectively, these studies highlight the utility of single-cell multiomic analysis to gain greater resolution of cell identity during SC-islet differentiation. Importantly, both studies indicated that epigenetic regulators are important drivers of cell identity, particularly in the cell fate choice between SC-EC and SC-β cells.
Recent applications of single-cell sequencing technologies have not only improved our understanding of SC-islet composition but have also elucidated the transcriptional and epigenetic differences between stem cell-derived and human adult islets. SC-islets generated in vitro remain transcriptionally and functionally immature compared to adult human islets.44,77,78. The multiomic studies described here further demonstrated that SC-β cells were less similar to their primary counterparts than other stem cell-derived endocrine cell types. 74,75 Moreover, the chromatin accessibilities of primary adult islet cell types were much more restricted than those in SC-islet cell types, where the INS gene remained open in SC-α and SC-δ cells in addition to SC-β cells.75 Transplantation of SC-islets into mice improves their insulin secretion and glucose responsiveness, suggesting that SC-islet immaturity can be resolved.36,37,41–44 scRNA-seq analysis of transplanted SC-islets revealed that improved function corresponded with transcriptional maturation.43,51 Specifically, known markers of β cell maturation, such as INS, MAFA, IAPP, MNX1, and G6PC2, became more highly expressed in a greater proportion of SC-islet cells after transplantation. These transcriptional changes appeared to occur temporally, as some genes (e.g., G6PC2) increased in expression shortly after transplantation, while others (e.g., MAFA) took several months to upregulate. Interestingly, SC-EC cells also persisted after transplantation and exhibited increased expression of key gene markers of their identity, such as TPH1.51,75 Despite transcriptional improvements, SC-islets nonetheless had significantly reduced activity of metabolic pathways compared to primary islets.43 Furthermore, a multiomic analysis identified over 600 genes, 350 promoter regions, and 250 transcription factor binding motifs that increased after 6-months in vivo, while only a small fraction of these were also increased during in vitro maturation.75 These findings suggested that extended time in vivo tended to open chromatin regions associated with cell-specific identity and maturation, while in vitro culture methods remained relatively ineffective at promoting the same maturation effect. Overall, comparison of scRNA-seq and snATAC-seq data from transplanted SC-islets and primary islets has indicated that SC-islets become more similar to human islets overtime in vivo in terms of both their transcriptional and chromatin accessibility profiles, though some important differences still remain. These strategies for highly detailed characterization of SC-islet cell types will be invaluable for elucidating the mechanisms driving islet development and the factors that are currently limiting further SC-islet maturation.
Overcoming immune rejection
Encapsulation and immune-modulating biomaterials
As with the transplantation of any allogenic material, transplanted SC-islets are targeted by the host immune system as foreign, resulting in graft rejection. An additional complication is that T1D itself is an autoimmune disease that specifically targets β cells. Thus, even if autologous SC-islets were generated from patients using an iPSC intermediate, it is possible they would still be targeted by immune cells. This issue is currently addressed with the use of immunosuppressive drugs that are used for whole organ transplants, and the prescribed regimen has improved in recent years.79 While these drugs can successfully retain graft viability, they also can have serious side-effects, including an increased risk of infection and cancer as well as less severe but uncomfortable symptoms.80,81 For many patients, these adverse side-effects may not outweigh the benefits of increased glycemic control, and thus there has been much research focused on alternative methods for protecting the transplanted cells from the host immune system.
One of the most researched approaches is to encapsulate the cells in a biomaterial with finely tuned pore sizes that allow for the diffusion of small molecules like glucose and insulin but shield the transplanted SC-islets from contact with host immune cells. This field has been progressing for several decades and excellently reviewed in-depth elsewhere,52,82–84 and so the focus here will be on several recent examples to highlight the current status of these technologies. In the macroencapsulation strategy, many islets are put into a single device.85 These encapsulation devices are most often constructed of either a polymer film like polytetrafluoroethylene (PTFE) or an alginate hydrogel due to their excellent biocompatibility and ease of fabrication. Not only does the device membrane provide protection against contact with host immune cells, but it also prevents the graft cells from spreading to other parts of the body should any unwanted cell growth or tumor formation occur. Furthermore, the device can be easily retrieved if any other safety or efficacy concerns arise after transplantation. One popular option for islet macroencapsulation is the commercially available TheraCyte device, which is composed of a 0.4 μM inner PTFE membrane to block entry of host immune cells and an outer 5 μM PTFE membrane that promotes vascularization around the graft. While these devices have had success in preventing overt immune rejection of pancreatic tissue,86–89 these devices can be limited by a lack of proper nutrient exchange, particularly until vascularization has formed.90 As highlighted by the ViaCyte clinical trials with similar devices33,34 as well as other studies91,92, these macroencapsulation devices can also promote fibrosis around the implant, further preventing nutrient diffusion.85,93
New systems are being developed to combat these issues associated with nutrient delivery. For example, a PTFE macroencapsulation device that actively pumped fluid through a hollow fiber running through its center demonstrated enhanced nutrient exchange that promoted cell survival and allowed for a greater cell density to be successfully loaded into the device.94 Insulin secretion and blood glucose control was also enhanced when transplanted into diabetic rats. Interestingly, they also observed a decreased fibrotic response with their active fluid flow setup. While advancements need to be made to replace the external pump used to perfuse the device once implanted, this study highlights that relying on active fluid flow rather than passive diffusion for nutrient transport can greatly enhance the viability and performance of islets within a macroencapsulation device.94,95 Because oxygen is one of the most important of these nutrients to the metabolically active β cells, several oxygen delivery systems and oxygen generating materials have been incorporated into encapsulation device designs.92,96,97. In a recent iteration, CaO2 was integrated into a PDMS slab that was encased in an agarose hydrogel containing islets.98 The CaO2 reacted with the water in the hydrogel to generate oxygen that became available to the surrounding cells. In another example, Li2O2 particles were suspended in a perfluorocarbon oil and encased in silicone tubing.99 This core was surrounded by an islet-containing alginate hydrogel. The Li2O2 reacted with the CO2 waste generated from cellular respiration to generate O2, resulting in a self-sustaining source of oxygen within the hydrogel that was only limited by the amount of Li2O2 in the device. In both these examples, the oxygen generated by the device significantly enhanced the viability and function of the islets after transplantation.
One method to passively alter nutrient transport is to control the geometry of the device. For instance, a network of interconnected hydrophobic channels inside an islet-filled alginate hydrogel greatly increased the diffusion of oxygen throughout the device from the surrounding tissue.100 This design greatly enhanced islet survival upon transplantation into diabetic mice and allowed the device to be scaled up to 6.6 mm thick. Alternatively, increasing the surface-to-volume ratio and decreasing the distance of cells from the outer surface of the device can facilitate better mass transport to the encapsulated cells. For example, a durable polymer thread was coated in a thin alginate hydrogel layer containing islets, generating a long thread-like device with a high surface-to-volume ratio.101 Rat islets encapsulated within the device were able to survive and reverse diabetes in immune-competent diabetic mice. To further improve the mechanical strength of their device concept in a follow-up study, they developed an alternative design that placed the alginate hydrogel inside of an immunoprotective tube composed of a strong nanofibrous mesh that was electronspun from a medical grade thermoplastic silicone-polycarbonate-urethane.102
This concept of controlling device geometry to enhance nutrient transport can be further scaled down to the level of encapsulating individual islets in a strategy known as microencapsulation. One popular approach uses alginate microcapsules, and various iterations have been utilized.83 Interestingly, while decreasing the size of encapsulation devices down to the scale of individual islet microcapsules increases nutrient diffusion shortly after transplantation when compared to macroencapsulation devices, smaller capsule sizes have been shown to also induce a robust foreign body response and subsequent fibrosis.103 To this end, there has been some excellent recent work altering the surface chemistry of these microcapsules to minimize the fibrotic response. For example, a combinatorial biomaterial screen identified three different triazole-containing covalent modifications to alginate that significantly reduced the foreign body response in both rodents and non-human primates.104 One of these formulations, triazole–thiomorpholine dioxide alginate, was then used to encapsulate SC-β cells within 1.5 mm spheres.41 These encapsulated cells restored glycemic control in diabetic, immune-competent mice for at least 174 days without any signs of fibrosis. This formulation was subsequently shown to prevent fibrosis and facilitate cell survival of allogeneic islets in non-human primates.105 Another group added a zwitterioinic modification to alginate microcapsules to significantly reduce fibrosis due to its ability to resist protein adsorption and cell attachment.106 They then combined this zwitterionic concept with the previously discovered triazole modification to further improve the ability of the alginate microcapsules to resist fibrosis and as well as enhance their mechanical stability.107
These studies utilizing modified alginate chemistries point to an interesting shift in thinking whereby a biomaterial is not only thought of as a physical barrier to keep out immune cells but rather as a source of instructive signals to induce local immune tolerance, either by releasing molecules as a drug delivery system or by having a biofunctional immunomodulatory surface.84 By only targeting the area directly surrounding the implant, these strategies can increase the efficacy of the desired immune modulation while simultaneously avoiding systemic side-effects. For example, alginate microcapsules fabricated to release the immunomodulatory chemokine CXCL12 improved encapsulated SC-islet survival and reduced the fibrotic response in mice.108 Similarly, alginate microcapsules designed to release exosomes derived from umbilical cord mesenchymal stem cells were also shown to induce local immune tolerance by altering signaling in multiple immune cell types.109 The incorporation of the CSF1R inhibitor GW2580 in its crystallized form within alginate microcapsules allowed for its controlled release overtime, resulting in long-term reduction of fibrosis and significant improvements in islet transplantation outcomes in mice, even within the subcutaneous space.110 In an alternative approach, immune-modulating proteins such as programmed death-ligand 1 (PD-L1) or Fas ligand (FasL) can be immobilized onto the surface of biomaterials to dampen local T-cell adaptive immune responses. Both of these proteins play important roles in immune tolerance, as the binding of PD-L1 to PD-1 on effector T-cells reduces their proliferation and cytokine production, while the binding of FasL to the Fas receptor induces T-cell apoptosis. To this end, PD-L1111 or FasL112,113 proteins were immobilized onto the surface of poly(ethylene glycol) microgels, which were then mixed together with allogeneic islets and co-transplanted into either mice111,112 or non-human primates113. In conjunction with a transient rapamycin treatment, these functionalized microgels greatly improved islet survival without the need for long-term immunosuppression or encapsulation.
Engineering hypoimmune SC-islets
This concept of inducing local immunity can be pursued even further by engineering the transplanted cells themselves to hide from the immune system. Avoiding the use of biomaterials altogether eliminates the inherent diffusion barriers of encapsulation systems as well as circumvents the biomaterial-induced fibrotic response. For example, isolated mouse islets were engineered to transiently display FasL on their surface through a biotinylation technique.114,115 Allografts were able to survive indefinitely if also treated for the first 15 days with rapamycin, whereas those without FasL were rejected within 30 days. They demonstrated that FasL induced local immune tolerance by causing apoptosis in alloreactive T-cells and as well as promoting the development of regulatory T-cells (Tregs) for long-term maintenance. The development of local rather than systemic immune tolerance was further highlighted by the transplantation of a second set of unmodified, donor matched cells that survived when grafted to the original transplantation site but were rejected when transplanted to the other kidney of the same mouse. In another example, mesenchymal stromal cells were engineered using a lentiviral vector to overexpress PD-L1 and the cytotoxic T lymphocyte antigen 4 immunoglobulin (CTLA4-Ig) fusion protein. These gene-edited cells were co-transplanted with allogenic mouse islets to reverse diabetes significantly longer than without the genetic modifications.116 These accessory mesenchymal stromal cells reduced infiltration of CD4+ and CD8+ effector T-cells and increased the number of Tregs to induce local immune tolerance. This PD-L1 strategy was pushed even further by directly overexpressing PD-L1 in islet-like organoids made from hiPSCs using a lentiviral system.67 These engineered cells were found to evade the immune system in both immune-competent mice and in a humanized mouse model.
The immune response to allogeneic material leading to graft rejection is complex, as multiple cell types and pathways are involved.117 The main cause of rejection in allogeneic transplantation is T-cell recognition of human leukocyte antigens (HLAs). The HLA class I molecules A, B, and C are present on all nucleated cells and present antigens to CD8+ cytotoxic T-cells, while HLA class II proteins activate CD4+ helper T-cells. Thus, one successful strategy to eliminate the T-cell mediated adaptive immune response against allogeneic material is to remove HLAs from transplanted cells so that they cannot activate host T-cells. Deleting HLAs from cells, however, causes them to be targeted by cells of the innate immune system, such as natural killer (NK) cells and macrophages. Therefore, various strategies have been pursued to simultaneously eliminate both adaptive and innate immune responses. For example, in contrast to prior studies, it was recently reported that PD-L1 alone was not sufficient to protect against xeno- and allorejection in transplanted SC-islets.54 Instead, they reported HLA depletion facilitated improved cell survival. To further enhance the survival of these transplanted cells, the SC-islets were also engineered to secrete several factors to induce local immune tolerance, including IL-2 mutein to facilitate Treg expansion, as well as IL-10 and TGF-β to help the immunosuppressive function of T-regs. These cells reversed diabetes and survived for at least 8 weeks in an autoimmune diabetes mouse model. Overexpression of CD47 has also been successfully implemented to mitigate innate immune responses after HLA deletion.118 Using the CRISPR/Cas9 system in human iPSCs, the β2-microglbulin (B2M) and CIITA genes were deleted to remove HLA class I and class II molecules, respectively, while a lentiviral vector was used to induce overexpression of CD47. These gene-edited iPSCs could differentiate into endothelial cells and cardiomyocytes that demonstrated long-term survival in humanized mouse models without any immunosuppression.
In another strategy, the CRISPR/Cas9 system was used to selectively remove HLA-A, B, and C in hPSCs, and HLA class II molecules were eliminated by targeting the CIITA gene.119 In parallel, CRISPR/Cas9 was also used to overexpress PD-L1 to further suppress T-cells, HLA-G to modulate NK cells, and CD47 to inhibit macrophages. These cells could be differentiated into endothelial cells or vascular smooth muscle cells and demonstrated improved survival in co-cultures with T-cells, NK cells, and macrophages. Similarly, CRISPR/Cas9 was used to delete all HLA-A, B, and C genes in hPSCs with the exception of HLA-A2, which helped retain HLA-E expression.55 Importantly, HLA-E expression in hPSCs has been demonstrated to inhibit killing by NK cells.120 In addition, HLA class II molecules were also eliminated by removing the CIITA gene. These hPSCs could differentiate into SC-islets that demonstrated protection from T-cell mediated rejection and reduced NK immune responses in allogeneic humanized mice.55 Lastly, in a unique approach, scRNA-seq and a CRIPSR screen of SC-islets transplanted into a humanized mouse model revealed that SC-islets upregulated genes in the interferon (IFN) pathway during graft rejection.121 Knockout of CXCL10, which is induced by IFN signaling and appeared to be essential for early IFN-triggered immune responses in transplanted SC-islets, improved survival during allogeneic transplantation in humanized mice.
Further challenges associated with the clinical transplantation of SC-islets
Reducing stress responses within islets after transplantation
Early investigation of primary human islet transplants56 and animal studies122,123 demonstrated that 50% or more of the islet graft was lost during the first few days after transplantation. Because of this major cell loss during the immediate aftermath of transplantation, a high islet mass from multiple organ donors and several infusions was used in the clinic.124 Furthermore, primary islets often demonstrated progressive functional decline in the years following transplantation, necessitating patients to resume exogenous insulin injections overtime. Advances in islet isolation methodologies, immunotherapies, and transplantation techniques have improved islet transplantation outcomes in recent years, improving 5-year insulin independence rates to 50-80%.23 In particular, the amount of islet mass that is lost immediately following transplantation has been reduced closer to 25%, and thus fewer islets have been needed to achieve insulin independence.79,125 Discovering new ways to further address the acute stressors experienced by islets in the immediate aftermath of transplantation could further reduce the degree of β cell loss and dysfunction that impedes long-term restoration of normoglycemia.126
Due to the high metabolic demands of insulin production and secretion, β cells are particularly susceptible to stress. Perturbations in normal metabolism and environmental conditions can induce endoplasmic reticulum (ER) stress and initiate the unfolded protein response (UPR).127 While this pathway aims to restore protein homeostasis under mild stress conditions, it will induce apoptosis if the trigger is more severe. Inflammatory cytokines, hypoxia, and hyperglycemia can all induce an ER stress response in β cells that leads to cell dysfunction and apoptosis.127–129 This stress response is thought to be involved in the progression of both type 1 and type 2 diabetes.130 Hypoxic conditions and oxidative stress can also induce other stress pathways that result in β cell functional failure and apoptosis.131–134 Stress from a high glucose environment can cause downregulation of key islet genes and decreased function in human islets135 and animal models123, implicating that adequate glucose control is important for β cell health in the immediate aftermath of transplantation. Furthermore, while SC-islets appear to be generally more stress-resistant than isolated primary islets,136 they nevertheless respond to inflammatory cytokines with similar stress response pathways,137 highlighting the need to mitigate exposure to inflammation during transplantation.
Native islets are highly vascularized within the pancreas to receive the proper nutrients and high oxygen supply required for the production and secretion of large amounts of insulin. Consequently, primary islet transplantations have leveraged the portal vein for islet infusion to ensure an adequate initial blood supply. One consequence of this direct blood contact is an immune response called the instant blood-mediated inflammatory reaction (IBMIR).138–140 Within 15 minutes of contact with host blood, islets are encapsulated within a layer of platelets, reducing nutrient diffusion. Furthermore, leukocytes infiltrate the islets within an hour, leading to cell death. It is thought that a major cause of the initial islet death observed in the immediate aftermath of portal vein islet infusion is due to IBMIR. This initial cell death of transplanted cells can also cause release of molecules that initiate other immune responses, further exacerbating the issue by causing other transplanted cells to exhibit stress responses.141,142
A successful SC-islet transplantation strategy will need to adequately address these stressors, particularly those associated with hypoxia, nutrient deprivation, and inflammation immediately following the transplantation procedure. Similar to primary islet infusion, transplantation of SC-islets into mice has demonstrated that a significant portion of the cells can die shorty after transplantation due to the synergistic effects of nutrient deprivation and hypoxia.143 Consequently, many attempts to alleviate this issue have focused on modulating the graft environment by optimizing the transplantation site and promoting vascularization. In humans, several alternative transplantation sites to the liver have been proposed144, including the subcutaneous space145, intramuscular space146, and the omentum147,148. These sites require a less invasive procedure for transplantation and provide easier access to the graft so that it can be monitored and possibly removed if problems arise. These alternative sites may also potentially circumvent the intense IBMIR response that is observed with portal vein infusion.149 To date, however, these alternative sites have yet to lead to improved transplant outcomes when compared to portal vein infusion in humans.149 For SC-islet transplantations, site and dose have been shown to influence cure efficacy in mice, with placement underneath the highly-vascularized kidney capsule producing much better results than subcutaneous infusions.143,150 Similarly, mice transplant outcomes for SC-islets within a microencapsulation device were more favorable in the intraperitoneal cavity than in the subcutaneous space due to a much lower fibrotic response.102 In non-human primates, transplantation of SC-islets between the rectus abdominis muscle in the abdomen and its surrounding rectus sheath demonstrated better cell survival than in intramuscular or subcutaneous locations.46 Furthermore, this location promoted robust vascularization by week 12 and promoted an environment that allowed for SC-islet maturation.
Once an optimal transplantation site is chosen to minimize initial cell loss, other strategies can be pursued to further increase vascularization and nutrient transport. For example, implanting a device into the subcutaneous space one month prior to cell transplantation pre-vascularized the graft site and avoided the several week lag time that it takes for vascularization to occur.145,151 Incorporating vascular endothelial growth factor (VEGF)-releasing capsules into a similar pre-vascularization strategy152 or transplanting islets within VEGF-releasing hydrogels153 were shown to further improve vessel formation. Alternatively, islets embedded within sub-millimeter collagen cylinders coated with endothelial cells154 or islets combined with microvascular fragments155 were demonstrated to become rapidly vascularized and connected to the host vasculature when transplanted into rodents. Transplanting islets with either human umbilical cord perivascular cells156 or amniotic epithelial cells157 also improved islet engraftment and vascularization. To specifically improve oxygen levels at the transplantation site, oxygen generating materials have been inserted alongside the islet graft,96,98,99 while another strategy directly pumps oxygen into an encapsulation device92,97.
In contrast to modulating the transplantation environment, the SC-islets themselves can be altered to make them more resistant to different stressors. For example, to improve SC-islet resistance to low oxygen conditions following transplantation, stem cells were differentiated under hypoxic conditions.143 Transplanting these preconditioned SC-islets in combination with supplemental amino acids to mitigate nutrient deprivation drastically improved SC-islet survival within the first several weeks, highlighting how cell-targeted interventions can improve graft outcomes. One unique approach is to genetically engineer SC-islets to be resistant to different stressors, which has been largely unexplored in the context of SC-islets until recently. For example, SC-islets differentiated from iPSCs generated from a patient with Wolfram syndrome, which is characterized by ER stress, exhibited poor function and increased stress responses.40 Correcting this stress-causing pathogenic variant with CRISPR/Cas9, however, allowed this iPSC line to generate highly functional SC-islets that were capable of rapidly reversing severe diabetes in mice, illustrating that gene-editing strategies can be used to mitigate stress responses. Furthermore, a recent study manipulated normal stress responses in SC-islets to make them more resistant to transplantation conditions.136 Specifically, SC-islets exhibited dysfunction, apoptosis, and increased expression of genes related to stress and immune interaction when exposed to a variety of stressors, including a cytokine mix to mimic inflammatory stress, thapsigargin to induce ER stress, and high glucose to replicate metabolic stress. Lentiviral shRNA knockdown of XBP1, CDKN1A, NLRC5, and β2M decreased the level of apoptosis in SC-islets when treated with these stressors as well as downregulated the expression of genes associated with stress responses and immune interaction. Furthermore, these gene knockdowns decreased T-cell activation and T-cell induced apoptosis when SC-islets were co-cultured with peripheral blood mononuclear cells.
Long-term safety of the cell product
Differentiation of hPSCs to SC-islets poses potential challenges for ensuring their safety after transplantation. While optimized protocols used on research cell lines can often avoid these problems in animal models, special care is needed when protocols and cell lines are converted for clinical use in humans. In particular, not all stem cell lines differentiate with the same efficiency using the current generation of SC-islet protocols.57 In-depth characterization of the composition and genomic heterogeneity of the final cell population is especially important, particularly to identify subpopulations that may proliferate uncontrollably. Given the pluripotent nature of these stem cells and the complexity of the differentiation process, there is a risk that cells may differentiate into unintended or dangerous off-target cell types. In the context of SC-islet differentiation, SC-EC cells are one of the most common off-targets. Fortunately, while they may be detrimental to SC-β cell potency,47 no safety issues have been reported to date. Hepatic, mesenchyme, and pancreatic exocrine are also possible off-targets reported from SC-islet differentiations47,51,75 Of greatest concern is the potential for highly proliferative uncommitted progenitors or residual hPSCs to be present in the final SC-islet product that have the potential to form tumors.158–160 Interestingly, it was demonstrated that ensuring the pancreatic progenitor population expressed the glycoprotein GP2 prevented tumor formation upon transplantation, suggesting that dangerous cell populations in the final cell product can potentially be identified earlier in the differentiation.161 Additional refinements to the differentiation methodology can help eliminate specification of these off-target cell types in clinical-grade cell lines. Furthermore, thorough characterization of the final SC-islet cell product with flow cytometry and single cell sequencing technologies can identify differentiation batches that generate potentially harmful off-target cell types, such as uncommitted or highly proliferative cells, to ensure that these problematic cell populations are not transplanted into patients.
Accumulation of unsafe genetic variation, particularly oncogenic mutations, in the final cellular drug product can pose a significant safety risk for SC-islet transplantation. For example, a clinical trial in Japan for macular degeneration was paused due to genetic variation.162 Variants can be acquired through standard culture of the cells and reprogramming.163 Loss of function of the p53 protein is common in the majority of cancers,164 and six specific variants in TP53 have been identified in several hPSC lines.165 Furthermore, numerous karyotypic abnormalities have been observed in hPSCs.166 Substantial genomic variation has also been observed specifically in hiPSCs,167 particularly for BCOR mutations that are involved in many cancers.168 To further complicate this issue, reprogramming and genetic engineering itself have also been implicated in increasing genomic variation.169–171 Therefore, it is crucial to test the genomic status of cells at critical stages of product development, such as after reprogramming, genetic editing, creation of the master cell bank, and creation of the final cell product.
An additional safety precaution for an SC-islet therapy is the introduction of a safety switch to destroy the graft in the event of adverse outcomes. These often take the form of an inducible gene to encoded for a protein that kills the cell, such as inducible Caspase9172 or proliferation-induced constructs.173 This enables precise control over the timing and duration of the therapeutic effect to improve both safety and efficacy. These safety switches will most likely be particularly important when developing hypoimmune SC-islet strategies, as the cells are being purposefully designed to hide from the immune system and cannot be easily retrieved.
Scale-up and distribution
Development of a large-scale, cost-efficient manufacturing methodology for differentiating SC-islets will be necessary to make this therapy available to a substantial number of patients.174 While 2-5 x 106 SC-islet cells are sufficient to reverse hyperglycemia in mice,150 the required dose for humans is currently unknown and is dependent on SC-islet quality. Assuming a similar dose as with human islets from deceased donors,175,176 on the order of 109 cells will be required per patient. Even a relatively modest number of patients of about 1,000 will require on the order of one trillion cells to be produced. Current differentiation approaches will encounter challenges achieving these yields. SC-islet protocols that are performed completely in suspension culture36 benefit from the natural three-dimensional scaling of these systems. Large scale (>1,000 L) bioreactors for mammalian cell culture have been used in the biotechnology industry for decades.177 Notably, these bioreactors allow for real-time monitoring and culture of critical process parameters, such as dissolved oxygen, that will be critical for quality control during large-scale production. To-date, however, such large scales have not been extended to hPSCs, and the largest SC-islet differentiation that has been reported in literature was performed in 500 mL magnetic spinner flasks.36 A fundamental challenge to scaling hPSC differentiation in suspension culture is the fluid convection necessary to keep cells from settling, which is made more difficult due to hPSCs requiring either microcarriers or culture in aggregates.178 Critically, PSCs are sensitive to mechanical cues,179,180 and changes in shear stress due to increases in reactor volume as well as impeller speed and size will require careful optimization. Changes in these mechanical forces could alter crucial cell fate decisions during specific points in the differentiation protocol when the cells are highly sensitive to external signaling perturbations.
Alternatively, several versions of SC-islet protocols use a hybrid approach that employs traditional planar culture for some or all of the differentiation process and only aggregate the cells into clusters in later stages. For example, one popular methodology uses planar culture for making PDX1+/NKX6-1+ pancreatic progenitors and then clusters cells on an air-liquid interface for endocrine induction and β cell maturation.37 While this protocol is able to robustly generate functional SC-islets in a lab setting, this air-liquid interface culture system would be difficult to adapt to large-scale manufacturing. In contrast, another approach is able to fully differentiate SC-β cells in planar culture.39,57 The endocrine cells are then single-cell dispersed from the culture surface and aggregated into islet-like clusters in suspension culture. This hybrid methodology may encounter fewer optimization issues than a solely suspension-based approach during scale-up. Specifically, this SC-islet differentiation approach has been shown to scale proportionally to culture surface area,57 and it could potentially be scaled up further using cell stacks or hyperflask setups. Importantly, parameters such as shear stress that could influence cell signaling during early differentiation stages do not change in planar culture as surface area is increased. Once the cells are differentiated into endocrine cells, they are less susceptible to mechanically induced signaling changes, facilitating an easier transition to suspension culture for the final aggregation step. This methodology has been shown to produce more cells per unit media volume than a fully suspension approach,57 which is an important cost consideration since many of the differentiation factors are expensive. Furthermore, this approach has also been shown to be more amenable for differentiating a wide range of stem cell lines,57 which may be important when this protocol is adapted to clinical grade cell lines. Though this hybrid methodology seems to have a number of benefits, it is yet to be determined in practice whether a hybrid or fully suspension protocol will be the most effective strategy for generating trillions of SC-islet cells.
All well-established protocols require approximately one month of culture to achieve functional SC-islets, with additional time needed for SC-β cell maturation. Several studies, however, have demonstrated the ability to propagate the pancreatic progenitor population that is generated during the middle portion of the differentiation protocol.181,182 In particular, it was recently reported that defined culture media containing either the TGF-β pathway inhibitor SB431542183 or I-BET151,184 an inhibitor of the acetyl-lysine bromodomain-containing proteins, promoted the expansion of PDX1+/NKX6-1+ human pancreatic progenitors. Stable and robust expansion of hPSC-derived pancreatic progenitors that can be efficiently differentiated into functional SC-islets would reduce manufacturing time and costs. Furthermore, this population could be well-characterized and facilitate the generation of a master cell bank of high-quality pancreatic progenitors. Importantly, such a system could reduce variation in SC-islet differentiation efficiency between production runs when compared to starting with stem cells for every differentiation.
Once the SC-islets have been manufactured, a system will need to be developed for their efficient distribution. An attractive idea is to cryopreserve the SC-islets so that they can be used as an off-the-shelf product. To-date, however, freezing whole islets has been met with challenges.185 Better viability and post-thaw function has been achieved by dispersing islets, freezing them as single cells, and then reaggregating them into clusters upon thaw.186,187 A similar dispersion freezing method has been successfully used in the context of SC-islet transplantation into non-human primates.45,46 An optimized vitrification process was also recently developed to successfully freeze whole SC-islets,188 but scaling this process for the cell numbers needed for therapeutic use may be difficult. Future work will need to focus on scaling up these cryopreservation techniques while mitigating cell loss after thawing. Furthermore, other questions to be addressed relate to how the cells will actually be delivered to the clinic. For example, SC-islets frozen as single cells will need to be aggregated into clusters for several days before transplantation. This process could take place at the site of production, and the SC-islets could be shipped live overnight to the site of transplantation. Furthermore, it is possible that a recovery culture period may be necessary on-site before transplantation in order to maximize SC-islet survival and efficacy.
Concluding Remarks
The field has come a long way since the discovery of robust generation of definitive endoderm from hPSCs in 2005.189 Through the excellent contributions of many research groups, protocols were developed to drive these definitive endoderm cells through several intermediate stages, culminating in the generation of glucose-responsive β cells in 2014. Further refinement of these differentiation strategies induced further functional maturation of SC-islets, facilitating the acquisition of biphasic insulin secretion kinetics similar to that observed in primary human islets. These cells are able to rapidly cure severely diabetic mice and improve glycemic control in non-human primates, and continued progress has allowed the field to narrow in on the critical quality attributes of these cells (Figure 3).
Figure 3. Critical quality attributes of SC-islets.
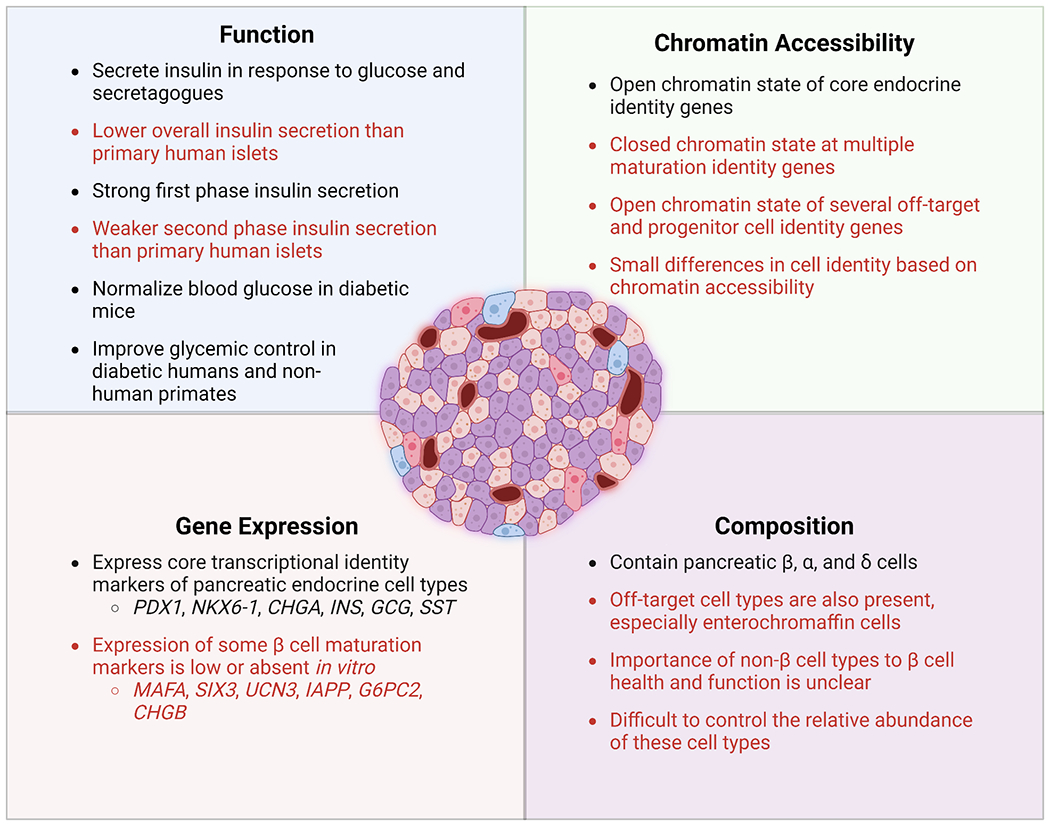
Despite making up a very small percentage of the pancreas, primary islets function to maintain blood glucose levels within a tightly regulated range. SC-islets have been able to replicate some of the key attributes of native islet function but fall short in some areas. Critical quality attributes that have been achieved in SC-islets are indicated in [black], while those that remain unresolved are indicated in [red].
As discussed throughout this review, the latest developments in SC-islet generation and immune protection have the potential to increase the efficacy and safety of the final cell product. For example, single-cell sequencing technologies have allowed for unprecedented characterization of SC-islets, providing insight into the identity and maturation state of the cells produced with these differentiation protocols. These data on the transcriptional and chromatin landscape of SC-islet cell types can guide the next generation of protocols for refining SC-islet production in an effort to increase insulin secretion per cell and eliminate off-target cell types. Progress has also been made with immunoprotective strategies, including tailored biomaterials that induced local immune tolerance as well as genetically engineered cells that evade the immune system. These approaches offer the potential for avoiding immunosuppressive drugs altogether, broadening the applicability of this cell therapy. Finally, further genetic engineering approaches to improve SC-islet resistance to the stressors they experience immediately after transplantation will likely be key to improving graft viability and reducing the number of cells needed for insulin independence.
Acknowledgements
We would like to thank Erika Brown for her assistance in creating the figures for this manuscript. N.J.H was supported by the JDRF (1-FAC-2023-1316-A-N). M.I. was supported by a Rita Levi-Montalcini Postdoctoral Fellowship from the Washington University School of Medicine Center of Regenerative Medicine and the Diabetes and Related Metabolic Diseases training grant in the Division of Endocrinology at Washington University School of Medicine (T32DK007120). J.R.M. was supported by the NIH (R01DK127497), Edward J. Mallinckrodt Foundation, and JDRF (3-SRA-2023-1295-S-B), and an NIH Human Islet Research Network (HIRN) Catalyst Award. Portions of the images were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
N.J.H. and J.R.M. are inventors on patents and patent applications related to SC-islets. J.R.M. is an employee and has stock or stock options in Sana Biotechnology. M.I. has stock in Vertex Pharmaceuticals.
References
- 1.Haeusler RA, McGraw TE, and Accili D (2018). Metabolic signalling: Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol 19, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tokarz VL, MacDonald PE, and Klip A (2018). The cell biology of systemic insulin function. J. Cell Biol 217, 2273–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saltiel AR (2021). Insulin signaling in health and disease. J. Clin. Invest 131, e142241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saltiel AR, and Kahn R (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806. [DOI] [PubMed] [Google Scholar]
- 5.Powers AC (2021). Type 1 diabetes mellitus: Much progress, many opportunities. J. Clin. Invest 131, e142242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiMeglio LA, Evans-Molina C, and Oram RA (2019). Type 1 Diabetes. Lancet 176, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perilli G, Saraceni C, Daniels MN, and Ahmad A (2013). Diabetic ketoacidosis: a review and update. Curr. Emerg. Hosp. Med. Rep 1, 10–17. [Google Scholar]
- 8.Chiasson JL, Aris-Jilwan N, Belanger R, Bertrand S, Beauregard H, Ekoe J-M, Fournier H, and Havrankova J (2003). Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ 168, 859–866. [PMC free article] [PubMed] [Google Scholar]
- 9.The Diabetes Control and Complications Trial Research Group (1993). The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med 329, 977–986. [DOI] [PubMed] [Google Scholar]
- 10.Nathan DM (2014). The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: Overview. Diabetes Care 37, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cryer PE (2007). Hypoglycemia, functional brain failure, and brain death. J. Clin. Invest 117, 868–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cryer PE (2012). Severe hypoglycemia predicts mortality in diabetes. Diabetes Care 35, 1814–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Latres E, Finan DA, Greenstein JL, Kowalski A, and Kieffer TJ (2019). Navigating two roads to glucose normalization in diabetes : automated insulin delivery devices and cell therapy. Cell Metab. 29, 545–563. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland DER, Gruessner RWG, Dunn DL, Matas AJ, Humar A, Kandaswamy R, Mauer SM, Kennedy WR, Goetz FC, Robertson RP, et al. (2001). Lessons learned from more than 1,000 pancreas transplants at a single institution. Ann. Surg 233, 463–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro J, Lakey JRT, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, and Rajotte RV (2000). Islet transplantation in seven patients with T1D using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med 343, 230–238. [DOI] [PubMed] [Google Scholar]
- 16.Bottino R, Knoll MF, Knoll CA, Bertera S, and Trucco MM (2018). The future of islet transplantation is now. Front. Med 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brennan DC, Kopetskie HA, and Sayre PH (2016). Long-term follow-up of the Edmonton Protocol of islet transplantation in the United States. Am. J. Transplant 16, 509–517. [DOI] [PubMed] [Google Scholar]
- 18.Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B, Oberholzer J, Odorico JS, Garfinkel MR, Levy M, et al. (2012). Improvement in outcomes of clinical islet transplantation: 1999-2010. Diabetes Care 35, 1436–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vantyghem MC, Chetboun M, Gmyr V, Jannin A, Espiard S, Le Mapihan K, Raverdy V, Delalleau N, MacHuron F, Hubert T, et al. (2019). Ten-year outcome of islet alone or islet after kidney transplantation in type 1 diabetes: A prospective parallel-arm cohort study. Diabetes Care 42, 2042–2049. [DOI] [PubMed] [Google Scholar]
- 20.Marfil-Garza BA, Imes S, Verhoeff K, Hefler J, Lam A, Dajani K, Anderson B, O’Gorman D, Kin T, Bigam D, et al. (2022). Pancreatic islet transplantation in type 1 diabetes: 20-year experience from a single-centre cohort in Canada. Lancet Diabetes Endocrinol. 10, 519–532. [DOI] [PubMed] [Google Scholar]
- 21.Moassesfar S, Masharani U, Frassetto LA, Szot GL, Tavakol M, Stock PG, and Posselt AM (2016). A comparative analysis of the safety, efficacy, and cost of islet versus pancreas transplantation in nonuremic patients with type 1 diabetes. Am. J. Transplant 16, 518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapiro AMJ, and Verhoeff K (2022). A spectacular year for islet and stem cell transplantation. Nat. Rev. Endocrinol 10. [DOI] [PubMed] [Google Scholar]
- 23.Verhoeff K, Marfil-Garza BA, and Shapiro AMJ (2021). Update on islet cell transplantation. Curr. Opin. Organ Transplant 26, 397–404. [DOI] [PubMed] [Google Scholar]
- 24.Hering BJ, Ballou CM, Bellin MD, Payne EH, Kandeel F, Witkowski P, Alejandro R, Rickels MR, and Barton FB (2022). Factors associated with favourable 5 year outcomes in islet transplant alone recipients with type 1 diabetes complicated by severe hypoglycaemia in the Collaborative Islet Transplant Registry. Diabetologia, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell PM, Senior PA, Salam A, LaBranche K, Bigam DL, Kneteman NM, Imes S, Halpin A, Ryan EA, and Shapiro AMJ (2007). High risk of sensitization after failed islet transplantation. Am. J. Transplant 7, 2311–2317. [DOI] [PubMed] [Google Scholar]
- 26.Rickels MR, Kearns J, Markmann E, Palanjian M, Markmann JF, Naji A, and Kamoun M (2006). HLA sensitization in islet transplantation. Clin. Transplant, 413–420. [PMC free article] [PubMed] [Google Scholar]
- 27.Paraskevas S, Maysinger D, Wang R, Duguid WP, and Rosenberg L (2000). Cell loss in isolated human islets occurs by apoptosis. Pancreas 20, 270–276. [DOI] [PubMed] [Google Scholar]
- 28.Siehler J, Blöchinger AK, Meier M, and Lickert H (2021). Engineering islets from stem cells for advanced therapies of diabetes. Nat. Rev. Drug Discov 20, 920–940. [DOI] [PubMed] [Google Scholar]
- 29.Kelly OG, Chan MY, Martinson LA, Kadoya K, Ostertag TM, Ross KG, Richardson M, Carpenter MK, D’Amour KA, Kroon E, et al. (2011). Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol 29, 750–756. [DOI] [PubMed] [Google Scholar]
- 30.Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, Gauvin R, Narayan K, Karanu F, O’Neil JJ, et al. (2012). Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 61, 2016–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. (2008). Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol 26, 443–452. [DOI] [PubMed] [Google Scholar]
- 32.Agulnick AD, Ambruzs DM, Moorman MA, Bhoumik A, Cesario RM, Payne JK, Kelly JR, Haakmeester C, Srijemac R, Wilson AZ, et al. (2015). Insulin-producing endocrine cells dfifferentiated in vitro from human embryonic stem cells function in macroencapsulation devices in vivo. Stem Cells Transl. Med 4, 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramzy A, Thompson DM, Ward-Hartstonge KA, Ivison S, Cook L, Garcia RV, Loyal J, Kim PTW, Warnock GL, Levings MK, et al. (2021). Implanted pluripotent stem-cell-derived pancreatic endoderm cells secrete glucose-responsive C-peptide in patients with type 1 diabetes. Cell Stem Cell 28, 2047–2061. [DOI] [PubMed] [Google Scholar]
- 34.Dolgin E. (2016). Encapsulating the problem. Nature 540, S60–62. [DOI] [PubMed] [Google Scholar]
- 35.Shapiro AMJ, Thompson D, Donner TW, Bellin MD, Hsueh W, Pettus J, Wilensky J, Daniels M, Wang RM, Brandon EP, et al. (2021). Insulin expression and C-peptide in type 1 diabetes subjects implanted with stem cell-derived pancreatic endoderm cells in an encapsulation device. Cell Reports Med. 2, 100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pagliuca FW, Millman JR, Gu M, Segel M, Dervort A. Van, Ryu JH, Peterson QP, Greiner D, and Melton DA (2014). Generation of functional human pancreatic ß-Cells in vitro. Cell 159, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol 32, 1121–1133. [DOI] [PubMed] [Google Scholar]
- 38.Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. (2015). Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 34, 1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, and Millman JR (2020). Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol 38, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, Morikawa S, Urano F, and Millman JR (2020). Gene-edited human stem cell–derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci. Transl. Med 12, eaax9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vegas AJ, Veiseh O, Gürtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, et al. (2016). Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat. Med 22, 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, and Millman JR (2019). Acquisition of dynamic function in human stem cell-derived β cells. Stem Cell Reports 12, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balboa D, Barsby T, Lithovius V, Saarimäki-Vire J, Omar-Hmeadi M, Dyachok O, Montaser H, Lund PE, Yang M, Ibrahim H, et al. (2022). Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol 40, 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, Li M. Ian, Nguyen VQ, Giacometti S, et al. (2019). Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol 21, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du Y, Liang Z, Wang S, Sun D, Wang X, Liew SY, Lu S, Wu S, Jiang Y, Wang Y, et al. (2022). Human pluripotent stem-cell-derived islets ameliorate diabetes in non-human primates. Nat. Med 28, 272–282. [DOI] [PubMed] [Google Scholar]
- 46.Luo Z, Zhan J, Wu S, Jiang Y, Lu Z, Wang Z, Yi LS, Zhang Y, Sun W, Fang X, et al. (2023). Implantation underneath the abdominal anterior rectus sheath enables effective and functional engraftment of stem-cell-derived islets. Nat. Metab 5, 29–40. [DOI] [PubMed] [Google Scholar]
- 47.Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH, Harb G, Quinn P, Poh Y, Sintov E, Gürtler M, et al. Charting cellular identity during human in vitro β - cell differentiation. Nature, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, Charlton J, Straubhaar JR, Meissner A, and Melton DA (2020). Circadian entrainment triggers maturation of human in vitro islets. Cell Stem Cell 26, 108–122.e10. [DOI] [PubMed] [Google Scholar]
- 49.Mahaddalkar PU, Scheibner K, Pfluger S, Sterr M, Beckenbauer J, Irmler M, Beckers J, Knöbel S, and Lickert H (2020). Generation of pancreatic beta cells from CD177+ anterior definitive endoderm. Nat. Biotechnol 38, 1061–1072. [DOI] [PubMed] [Google Scholar]
- 50.Weng C, Xi J, Li H, Cui J, Gu A, Lai S, Leskov K, Ke L, Jin F, and Li Y (2020). Single-cell lineage analysis reveals extensive multimodal transcriptional control during directed beta-cell differentiation. Nat. Metab 2, 1443–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Augsornworawat P, Maxwell KG, Velazco-Cruz L, and Millman JR (2020). Single-cell transcriptome profiling reveals β cell maturation in stem cell-derived islets after transplantation. Cell Rep. 32, 108067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Desai T, and Shea LD (2017). Advances in islet encapsulation technologies. Nat. Rev. Drug Discov 16, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samojlik MM, and Stabler CL (2021). Designing biomaterials for the modulation of allogeneic and autoimmune responses to cellular implants in Type 1 Diabetes. Acta Biomater. 133, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerace D, Zhou Q, Kenty JH, Boulanger KR, Li H, Melton DA, Gerace D, Zhou Q, Kenty JH, Veres A, et al. (2023). Engineering human stem cell-derived islets to evade immune rejection and promote localized immune tolerance. Cell Reports Med. 4, 100879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parent AV, Faleo G, Chavez J, Saxton M, Berrios DI, Kerper NR, Tang Q, and Hebrok M (2021). Selective deletion of human leukocyte antigens protects stem cell-derived islets from immune rejection. Cell Rep. 36, 109538. [DOI] [PubMed] [Google Scholar]
- 56.Eich T, Eriksson O, and Lundgren T (2007). Visualization of early engraftment in clinical islet transplantation by positron-emission tomography. N. Engl. J. Med 356, 2754–2755. [DOI] [PubMed] [Google Scholar]
- 57.Hogrebe NJ, Maxwell KG, Augsornworawat P, and Millman JR (2021). Generation of insulin-producing pancreatic β cells from multiple human stem cell lines. Nat. Protoc 16, 4109–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cuesta-Gomez N, Verhoeff K, Jasra IT, Pawlick R, Dadheech N, and Shapiro AMJ (2022). Characterization of stem-cell-derived islets during differentiation and after implantation. Cell Rep. 40, 111238. [DOI] [PubMed] [Google Scholar]
- 59.Wang Z, and Thurmond DC (2009). Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci 122, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rorsman P, Eliasson L, Renström E, Gromada J, Barg S, and Göpel S (2000). The cell physiology of biphasic insulin secretion. News Physiol. Sci 15, 72–77. [DOI] [PubMed] [Google Scholar]
- 61.Roscioni SS, Migliorini A, Gegg M, and Lickert H (2016). Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol 12, 695–709. [DOI] [PubMed] [Google Scholar]
- 62.Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. (2015). Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 34, 1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, Greiner DL, and Keller G (2015). Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Reports 4, 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mamidi A, Prawiro C, Seymour PA, Lichtenberg K.H. De, Jackson A, Serup P, and Semb H (2018). Mechanosignalling via Integrins Directs Fate Decisions of Pancreatic Progenitors. Nature 564, 114–118. [DOI] [PubMed] [Google Scholar]
- 65.Rosado-olivieri EA, Anderson K, Kenty JH, and Melton DA (2019). YAP inhibition enhances the differentiation of functional stem cell-derived insulin-producing β cells. Nat. Commun, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cebola I, Rodríguez-Seguí SA, Cho CHH, Bessa J, Rovira M, Luengo M, Chhatriwala M, Berry A, Ponsa-Cobas J, Maestro MA, et al. (2015). TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat. Cell Biol 17, 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshihara E, O’Connor C, Gasser E, Wei Z, Oh TG, Tseng TW, Wang D, Cayabyab F, Dai Y, Yu RT, et al. (2020). Immune-evasive human islet-like organoids ameliorate diabetes. Nature 586, 606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharon N, Vanderhooft J, Straubhaar J, Mueller J, Chawla R, and Zhou Q (2019). Wnt signaling separates the progenitor and endocrine compartments during pancreas development. Cell Rep. 27, 2281–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis JC, Alves TC, Helman A, Chen JC, Kenty JH, Cardone RL, Liu DR, Kibbey RG, and Melton DA (2020). Glucose response by stem cell-derived β cells in vitro is inhibited by a bottleneck in glycolysis. Cell Rep. 31, 107623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shapiro E, Biezuner T, and Linnarsson S (2013). Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet 14, 618–630. [DOI] [PubMed] [Google Scholar]
- 71.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, and Shendure J (2015). Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu H, Wang G, Kim D, Miller M, Goss G, Kovsky J, Harrington AR, Saunders D, Powers AC, Preissl S, et al. (2022). Improving stem cell-derived pancreatic islets using single-cell multiome-inferred regulomes. BioRxiv, 1–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Augswornworawat P, Hogrebe NJ, Ishahak M, Marquez E, Maestas M, Schmidt M, Veronese-Paniagua D, Gale S, Miller J, Velazco-Cruz L, et al. (2022). Defining the chromatin and transcriptional landscape of stem cell-derived islets. BioRxiv, 1–59. [Google Scholar]
- 76.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hrvatin S, O’Donnell CW, Deng F, Millman JR, Pagliuca FW, DiIorio P, Rezania A, Gifford DK, and Melton DA (2014). Differentiated human stem cells resemble fetal, not adult, β cells. Proc. Natl. Acad. Sci 111, 3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Melton D. (2021). The promise of stem cell-derived islet replacement therapy. Diabetologia 64, 1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rickels MR, and Paul Robertson R (2019). Pancreatic islet transplantation in humans: Recent progress and future directions. Endocr. Rev 40, 631–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blau J, Abegg M, Flegel W, Zhao X, Harlan D, and Rother K (2015). Long-term immunosupression after solitary islet transplantation is associated with preserved C-peptide secretion for more than a decade. Am J Transpl. 15, 2995–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ryan E, Paty B, Senior P, and Shapiro J (2004). Risks and side effects of islet transplantation. Curr. Diab. Rep 4, 304–309. [DOI] [PubMed] [Google Scholar]
- 82.Kharbikar BN, Mohindra P, and Desai TA (2022). Biomaterials to enhance stem cell transplantation. Cell Stem Cell 29, 692–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scharp DW, and Marchetti P (2014). Encapsulated islets for diabetes therapy: History, current progress, and critical issues requiring solution. Adv. Drug Deliv. Rev 67–68, 35–73. [DOI] [PubMed] [Google Scholar]
- 84.Samojlik MM, and Stabler CL (2021). Designing biomaterials for the modulation of allogeneic and autoimmune responses to cellular implants in Type 1 Diabetes. Acta Biomater. 133, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goswami D, Domingo-Lopez DA, Ward NA, Millman JR, Duffy GP, Dolan EB, and Roche ET (2021). Design considerations for macroencapsulation devices for stem cell derived islets for the treatment of type 1 diabetes. Adv. Sci 8, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kirk K, Hao E, Lahmy R, and Itkin-Ansari P (2014). Human embryonic stem cell derived islet progenitors mature inside an encapsulation device without evidence of increased biomass or cell escape. Stem Cell Res. 12, 807–814. [DOI] [PubMed] [Google Scholar]
- 87.Bruin JE, Rezania A, Xu J, Narayan K, Fox JK, O’Neil JJ, and Kieffer TJ (2013). Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia 56, 1987–1998. [DOI] [PubMed] [Google Scholar]
- 88.Motté E, Szepessy E, Suenens K, Stangé G, Bomans M, Jacobs-Tulleneers-Thevissen D, Ling Z, Kroon E, and Pipeleers D (2014). Composition and function of macroencapsulated human embryonic stem cell-derived implants: Comparison with clinical human islet cell grafts. Am. J. Physiol. - Endocrinol. Metab. Endocrinol. Metab 307, E838–E846. [DOI] [PubMed] [Google Scholar]
- 89.Kumagai-Braesch M, Jacobson S, Mori H, Jia X, Takahashi T, Wernerson A, Flodström-Tullberg M, and Tibell A (2013). The theracyte™ device protects against islet allograft rejection in immunized hosts. Cell Transplant. 22, 1137–1146. [DOI] [PubMed] [Google Scholar]
- 90.Rafael E, Wernerson A, Arner P, and Tibell A (1999). In vivo studies on insulin permeability of an immunoisolation device intended for islet transplantation using the microdialysis technique. Eur. Surg. Res 31, 249–258. [DOI] [PubMed] [Google Scholar]
- 91.Matveyenko AV, Georgia S, Bhushan A, and Butler PC (2010). Inconsistent formation and nonfunction of insulin-positive cells from pancreatic endoderm derived from human embryonic stem cells in athymic nude rats. Am. J. Physiol. - Endocrinol. Metab 299, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carlsson PO, Espes D, Sedigh A, Rotem A, Zimerman B, Grinberg H, Goldman T, Barkai U, Avni Y, Westermark GT, et al. (2018). Transplantation of macroencapsulated human islets within the bioartificial pancreas βAir to patients with type 1 diabetes mellitus. Am. J. Transplant 18, 1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anderson JM, Rodriguez A, and Chang DT (2008). Foreign body reaction to biomaterials. Semin. Immunol. 20, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang K, O’Cearbhaill ED, Liu SS, Zhou A, Chitnis GD, Hamilos AE, Xu J, Verma MKS, Giraldo JA, Kudo Y, et al. (2021). A therapeutic convection-enhanced macroencapsulation device for enhancing β cell viability and insulin secretion. Proc. Natl. Acad. Sci 118, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel SN, Ishahak M, Chaimov D, Velraj A, LaShoto D, Hagan DW, Buchwald P, Phelps EA, Agarwal A, and Stabler CL (2021). Organoid microphysiological system preserves pancreatic islet function within 3D matrix. Sci. Adv 7, eaba5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pedraza E, Coronel MM, Fraker CA, Ricordi C, and Stabler CL (2012). Preventing hypoxia-induced cell death in beta cells and islets via hydrolytically activated, oxygen-generating biomaterials. Proc. Natl. Acad. Sci 109, 4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barkai U, Weir G, Colton C, Ludwig B, Bornstein S, Brendel M, Neufeld T, Bremer C, Leon A, Evron Y, et al. (2013). Enhanced oxygen supply improves islet viability in a new bioartificial pancreas. Cell Transplant. 22, 1463–1476. [DOI] [PubMed] [Google Scholar]
- 98.Coronel MM, Liang JP, Li Y, and Stabler CL (2019). Oxygen generating biomaterial improves the function and efficacy of beta cells within a macroencapsulation device. Biomaterials 210, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang L-H, Ernst AU, Flanders JA, Liu W, Wang X, Datta AK, Epel B, Kotecha M, Papas KK, and Ma M (2021). An inverse-breathing encapsulation system for cell delivery. Sci. Adv 7, 5835–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang LH, Ernst AU, An D, Datta AK, Epel B, Kotecha M, and Ma M (2021). A bioinspired scaffold for rapid oxygenation of cell encapsulation systems. Nat. Commun 12, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.An D, Chiu A, Flanders JA, Song W, Shou D, Lu Y, Grunnet LG, and Winkel L (2017). Designing a retrievable and scalable cell encapsulation device for potential treatment of type 1 diabetes. Proc. Natl. Acad. Sci 115, E263–E272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang X, Maxwell KG, Wang K, Bowers DT, Flanders JA, Liu W, Wang LH, Liu Q, Liu C, Naji A, et al. (2021). A nanofibrous encapsulation device for safe delivery of insulin-producing cells to treat type 1 diabetes. Sci. Transl. Med 13, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, et al. (2015). Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat. Mater 14, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, et al. (2016). Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat. Biotechnol 34, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bochenek MA, Veiseh O, Vegas AJ, McGarrigle JJ, Qi M, Marchese E, Omami M, Doloff JC, Mendoza-Elias J, Nourmohammadzadeh M, et al. (2018). Alginate encapsulation as long-term immune protection of allogeneic pancreatic islet cells transplanted into the omental bursa of macaques. Nat. Biomed. Eng 2, 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Q, Chiu A, Wang LH, An D, Zhong M, Smink AM, de Haan BJ, de Vos P, Keane K, Vegge A, et al. (2019). Zwitterionically modified alginates mitigate cellular overgrowth for cell encapsulation. Nat. Commun 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu Q, Chiu A, Wang L, An D, Li W, Chen EY, Zhang Y, Pardo Y, McDonough SP, Liu L, et al. (2020). Developing mechanically robust, triazole-zwitterionic hydrogels to mitigate foreign body response (FBR) for islet encapsulation. Biomaterials 230, 119640. [DOI] [PubMed] [Google Scholar]
- 108.Alagpulinsa DA, Cao JJL, Driscoll RK, Sirbulescu RF, Penson MFE, Sremac M, Engquist EN, Brauns TA, Markmann JF, Melton DA, et al. (2019). Alginate-microencapsulation of human stem cell–derived β cells with CXCL12 prolongs their survival and function in immunocompetent mice without systemic immunosuppression. Am. J. Transplant 19, 1930–1940. [DOI] [PubMed] [Google Scholar]
- 109.Mohammadi MR, Rodriguez SM, Luong JC, Li S, Cao R, Alshetaiwi H, Lau H, Davtyan H, Jones MB, Jafari M, et al. (2021). Exosome loaded immunomodulatory biomaterials alleviate local immune response in immunocompetent diabetic mice post islet xenotransplantation. Commun. Biol 4, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Farah S, Doloff JC, Müller P, Sadraei A, Han HJ, Olafson K, Vyas K, Tam HH, Hollister-Lock J, Kowalski PS, et al. (2019). Long-term implant fibrosis prevention in rodents and non-human primates using crystallized drug formulations. Nat. Mater 18, 892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coronel MM, Martin KE, Hunckler MD, Barber G, O’Neill EB, Medina JD, Opri E, McClain CA, Batra L, Weaver JD, et al. (2020). Immunotherapy via PD-L1-presenting biomaterials leads to long-term islet graft survival. Sci. Adv 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Headen DM, Woodward KB, Coronel MM, Shrestha P, Weaver JD, Zhao H, Tan M, Hunckler MD, Bowen WS, Johnson CT, et al. (2018). Local immunomodulation with Fas ligand-engineered biomaterials achieves allogeneic islet graft acceptance. Nat. Mater 17, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lei J, Coronel MM, Yolcu ES, Deng H, Grimany-Nuno O, Hunckler MD, Ulker V, Yang Z, Lee KM, Zhang A, et al. (2022). FasL microgels induce immune acceptance of islet allografts in nonhuman primates. Sci. Adv 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yolcu ES, Zhao H, Bandura-Morgan L, Lacelle C, Woodward KB, Askenasy N, and Shirwan H (2011). Pancreatic islets engineered with SA-FasL protein establish robust localized tolerance by inducing regulatory T cells in mice. J. Immunol 187, 5901–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Woodward KB, Zhao H, Shrestha P, Batra L, Tan M, Grimany-Nuno O, Bandura-Morgan L, Askenasy N, Shirwan H, and Yolcu ES (2020). Pancreatic islets engineered with a FasL protein induce systemic tolerance at the induction phase that evolves into long-term graft-localized immune privilege. Am. J. Transplant 20, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang X, Wang K, Yu M, Velluto D, Hong X, Wang B, Chiu A, Melero-Martin JM, Tomei AA, and Ma M (2022). Engineered immunomodulatory accessory cells improve experimental allogeneic islet transplantation without immunosuppression. Sci. Adv 8, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bluestone JA, and Tang Q (2020). Solving the Puzzle of Immune Tolerance for β-Cell Replacement Therapy for Type 1 Diabetes. Cell Stem Cell 27, 505–507. [DOI] [PubMed] [Google Scholar]
- 118.Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thayer WO, Wahl A, Garcia JV, Reichenspurner H, et al. (2019). Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol 37, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han X, Wang M, Duan S, Franco PJ, Kenty JHR, Hedrick P, Xia Y, Allen A, Ferreira LMR, Strominger JL, et al. (2019). Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. U. S. A 116, 10441–10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, Prunkard D, Colunga AG, Hanafi LA, Clegg DO, et al. (2017). HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol 35, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sintov E, Nikolskiy I, Barrera V, Hyoje-Ryu Kenty J, Atkin AS, Gerace D, Ho Sui SJ, Boulanger K, and Melton DA (2022). Whole-genome CRISPR screening identifies genetic manipulations to reduce immune rejection of stem cell-derived islets. Stem Cell Reports 17, 1976–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Eich T, Eriksson O, Sundin A, Estrada S, Brandhorst D, Brandhorst H, Langstrom B,Nilsson B, Korsgren O, and Lundgren T (2007). Positron emission tomography: A real-time tool to quantify early islet engraftment in a preclinical large animal model. Transplantation 84, 893–898. [DOI] [PubMed] [Google Scholar]
- 123.Biarnés M, Montolio M, Nacher V, Raurell M, Soler J, and Montanya E (2002). β-cell death and mass in syngeneically transplanted islets exposed to short- and long-term hyperglycemia. Diabetes 51, 66–72. [DOI] [PubMed] [Google Scholar]
- 124.McCall M, and Shapiro AMJ (2012). Update on islet cell transplantation. Cold Spring Harb Perspect Med 2, a007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rickels MR, Liu C, Shlansky-Goldberg RD, Soleimanpour SA, Vivek K, Kamoun M, Min Z, Markmann E, Palangian M, Dalton-Bakes C, et al. (2013). Improvement in β-Cell secretory capacity after human islet transplantation according to the CIT07 protocol. Diabetes 62, 2890–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Emamaullee JA, and Shapiro AMJ (2007). Factors influencing the loss of β-cell mass in islet transplantation. Cell Transplant. 16, 1–8. [DOI] [PubMed] [Google Scholar]
- 127.Fonseca SG, Gromada J, and Urano F (2011). Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab 22, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen CW, Guan BJ, Alzahrani MR, Gao Z, Gao L, Bracey S, Wu J, Mbow CA, Jobava R, Haataja L, et al. (2022). Adaptation to chronic ER stress enforces pancreatic β-cell plasticity. Nat. Commun 13, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, and Mirmira RG (2012). Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eizirik DL, Pasquali L, and Cnop M (2020). Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat. Rev. Endocrinol 16, 349–362. [DOI] [PubMed] [Google Scholar]
- 131.Lazard D, Vardi P, and Bloch K (2012). Induction of beta-cell resistance to hypoxia and technologies for oxygen delivery to transplanted pancreatic islets. Diabetes. Metab. Res. Rev 28, 475–484. [DOI] [PubMed] [Google Scholar]
- 132.Drews G, Krippeit-Drews P, and Duïfer M (2010). Oxidative stress and beta-cell dysfunction. Pflugers Arch. Eur. J. Physiol 460, 703–718. [DOI] [PubMed] [Google Scholar]
- 133.Cantley J, Grey ST, Maxwell PH, and Withers DJ (2010). The hypoxia response pathway and β-cell function. Diabetes, Obes. Metab 12, 159–167. [DOI] [PubMed] [Google Scholar]
- 134.Kulkarni A, Muralidharan C, May SC, Tersey SA, and Mirmira RG (2022). Inside the β cell: molecular stress response pathways in diabetes pathogenesis. Endocrinology 164, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dai C, Kayton NS, Shostak A, Poffenberger G, Cyphert HA, Aramandla R, Thompson C, Papagiannis IG, Emfinger C, Shiota M, et al. (2016). Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J. Clin. Invest 126, 1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Leite NC, Pelayo GC, and Melton DA (2022). Genetic manipulation of stress pathways can protect stem-cell-derived islets from apoptosis in vitro. Stem Cell Reports 17, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Demine S, Schiavo AA, Marín-Cañas S, Marchetti P, Cnop M, and Eizirik DL (2020). Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nilsson B, Ekdahl KN, and Korsgren O (2011). Control of instant blood-mediated inflammatory reaction to improve islets of Langerhans engraftment. Curr. Opin. Organ Transplant 16, 620–626. [DOI] [PubMed] [Google Scholar]
- 139.Moberg L, Johansson H, Lukinius A, Berne C, Foss A, Källen R, Østraat, Salmela K, Tibell A, Tufveson G, et al. (2002). Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet 360, 2039–2045. [DOI] [PubMed] [Google Scholar]
- 140.Bennet W, Sundberg B, Groth C, Brendel MD, Brandhorst D, Brandhorst H, Bretzel RG, Elgue G, Larsson R, Nilsson B, et al. (1999). Incompatibility between human blood and isolated islets of Langerhans: a finding with implications for clinical intraportal islet transplantation? 48, 1907–1914. [DOI] [PubMed] [Google Scholar]
- 141.Sachet M, Liang YY, and Oehler R (2017). The immune response to secondary necrotic cells. Apoptosis 22, 1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.De Vos P, Lazarjani HA, Poncelet D, and Faas MM (2014). Polymers in cell encapsulation from an enveloped cell perspective. Adv. Drug Deliv. Rev 67–68, 15–34. [DOI] [PubMed] [Google Scholar]
- 143.Faleo G, Russ HA, Wisel S, Parent AV, Nguyen V, Nair GG, Freise JE, Villanueva KE, Szot GL, Hebrok M, et al. (2017). Mitigating Ischemic Injury of Stem Cell-Derived Insulin-Producing Cells after Transplant. Stem Cell Reports 9, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Merani S, Toso C, Emamaullee J, and Shapiro AMJ (2008). Optimal implantation site for pancreatic islet transplantation. Br. J. Surg 95, 1449–1461. [DOI] [PubMed] [Google Scholar]
- 145.Pepper AR, Gala-Lopez B, Pawlick R, Merani S, Kin T, and Shapiro AMJ (2015). A prevascularized subcutaneous device-less site for islet and cellular transplantation. Nat. Biotechnol 33, 518–523. [DOI] [PubMed] [Google Scholar]
- 146.Christoffersson G, Henriksnäs J, Johansson L, Rolny C, Ahlström H, Caballero-Corbalan J, Segersvärd R, Permert J, Korsgren O, Carlsson PO, et al. (2010). Clinical and experimental pancreatic islet transplantation to striated muscle: Establishment of a vascular system similar to that in native islets. Diabetes 59, 2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baidal DA, Ricordi C, Berman DM, Alvarez A, Padilla N, Ciancio G, Linetsky E, Pileggi A, and Alejandro R (2017). Bioengineering of an intraabdominal endocrine pancreas. N. Engl. J. Med 376, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Berman DM, Molano RD, Fotino C, Ulissi U, Gimeno J, Mendez AJ, Kenyon NM, Kenyon NS, Andrews DM, Ricordi C, et al. (2016). Bioengineering the endocrine pancreas: Intraomental islet transplantation within a biologic resorbable scaffold. Diabetes 65, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gamble A, Pepper AR, Bruni A, and Shapiro AMJ (2018). The journey of islet cell transplantation and future development. Islets 10, 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maxwell KG, Kim MH, Gale SE, and Millman JR (2022). Differential function and maturation of human stem cell-derived islets after transplantation. Stem Cells Transl. Med 11, 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pepper AR, Pawlick R, Bruni A, Wink J, Rafiei Y, O’Gorman D, Yan-Do R, Gala-Lopez B, Kin T, MacDonald PE, et al. (2017). Transplantation of human pancreatic endoderm cells reverses diabetes post transplantation in a prevascularized subcutaneous site. Stem Cell Reports 8, 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kasoju N, Pátíková A, Wawrzynska E, Vojtíţková A, Sedlačík T, Kumorek M, Pop-Georgievski O, Sticová E, Kříž J, and Kubies D (2020). Bioengineering a pre-vascularized pouch for subsequent islet transplantation using VEGF-loaded polylactide capsules. Biomater. Sci 8, 631–647. [DOI] [PubMed] [Google Scholar]
- 153.Weaver JD, Headen DM, Aquart J, Johnson CT, Shea LD, Shirwan H, and García AJ (2017). Vasculogenic hydrogel enhances islet survival, engraftment, and function in leading extrahepatic sites. Sci. Adv 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vlahos AE, Cober N, and Sefton MV (2017). Modular tissue engineering for the vascularization of subcutaneously transplanted pancreatic islets. Proc. Natl. Acad. Sci. U. S. A 114, 9337–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nalbach L, Roma LP, Schmitt BM, Becker V, Körbel C, Wrublewsky S, Pack M, Später T, Metzger W, Menger MM, et al. (2021). Improvement of islet transplantation by the fusion of islet cells with functional blood vessels. EMBO Mol. Med 13, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Forbes S, Bond AR, Thirlwell KL, Burgoyne P, Samuel K, Noble J, Borthwick G, Colligan D, McGowan NWA, Lewis PS, et al. (2020). Human umbilical cord perivascular cells improve human pancreatic islet transplant function by increasing vascularization. Sci. Transl. Med 12. [DOI] [PubMed] [Google Scholar]
- 157.Lebreton F, Lavallard V, Bellofatto K, Bonnet R, Wassmer CH, Perez L, Kalandadze V, Follenzi A, Boulvain M, Kerr-Conte J, et al. (2019). Insulin-producing organoids engineered from islet and amniotic epithelial cells to treat diabetes. Nat. Commun 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Millman JR, Tan JH, and Colton CK (2021). Mouse pluripotent stem vell differentiation under physiological oxygen reduces residual teratomas. Cell. Mol. Bioeng 14, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fong CY, Gauthaman K, and Bongso A (2010). Teratomas from pluripotent stem cells: A clinical hurdle. J. Cell. Biochem 111, 769–781. [DOI] [PubMed] [Google Scholar]
- 160.Lee MO, Moon SH, Jeong HC, Yi JY, Lee TH, Shim SH, Rhee YH, Lee SH, Oh SJ, Lee MY, et al. (2013). Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. U. S. A 110, 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aghazadeh Y, Sarangi F, Poon F, Nkennor B, McGaugh EC, Nunes SS, and Nostro MC (2022). GP2-enriched pancreatic progenitors give rise to functional beta cells in vivo and eliminate the risk of teratoma formation. Stem Cell Reports 17, 964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Garber K (2015). RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol 33, 890–891. [DOI] [PubMed] [Google Scholar]
- 163.Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, Lynch C, Harness JV, Lee S, Barrero MJ, et al. (2011). Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Muller PAJ, and Vousden KH (2013). P53 mutations in cancer. Nat. Cell Biol 15, 2–8. [DOI] [PubMed] [Google Scholar]
- 165.Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Taapken SM, Nisler BS, Newton MA, Sampsell-Barron TL, Leonhard KA, McIntire EM, and Montgomery KD (2011). Karyotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol 29, 313–314. [DOI] [PubMed] [Google Scholar]
- 167.Rouhani FJ, Zou X, Danecek P, Badja C, Amarante TD, Koh G, Wu Q, Memari Y, Durbin R, Martincorena I, et al. (2022). Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat. Genet 54, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Astolfi A, Fiore M, Melchionda F, Indio V, Bertuccio SN, and Pession A (2019). BCOR involvement in cancer. Epigenomics 11, 835–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, et al. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Talkowski ME, and Musunuru K (2014). Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 15, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Merkle FT, Neuhausser WM, Santos D, Valen E, Gagnon JA, Maas K, Sandoe J, Schier AF, and Eggan K (2015). Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 11, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Itakura G, Kawabata S, Ando M, Nishiyama Y, Sugai K, Ozaki M, Iida T, Ookubo T, Kojima K, Kashiwagi R, et al. (2017). Fail-safe system against potential tumorigenicity after transplantation of iPSC derivatives. Stem Cell Reports 8, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liang Q, Monetti C, Shutova MV, Neely EJ, Hacibekiroglu S, Yang H, Kim C, Zhang P, Li C, Nagy K, et al. (2018). Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature 563, 701–704. [DOI] [PubMed] [Google Scholar]
- 174.Wallner K, Pedroza RG, Awotwe I, Piret JM, Senior PA, Shapiro AMJ, and McCabe C (2018). Stem cells and beta cell replacement therapy: A prospective health technology assessment study. BMC Endocr. Disord 18, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Markmann JF, Deng S, Huang X, Desai NM, Velidedeoglu EH, Lui C, Frank A, Markmann E, Palanjian M, Brayman K, et al. (2003). Insulin independence following isolated islet transplantation and single islet infusions. Ann. Surg 237, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ryan EA, Lakey JRT, Paty BW, Imes S, Korbutt GS, Kneteman NM, Bigam D,Rajotte RV, and Shapiro AMJ (2002). Successful islet transplantation. Diabetes 51, 2148–2157. [DOI] [PubMed] [Google Scholar]
- 177.Nienow AW (2006). Reactor engineering in large scale animal cell culture. Cytotechnology 50, 9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kehoe DE, Jing D, Lock LT, Tzanakakis ES, and Ph D (2010). Scalable stirred-suspension bioreactor culture. Tissue Eng. Part A 16, 405–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Niebruegge S, Bauwens CL, Peerani R, Thavandiran N, Masse S, Sevaptisidis E, Nanthakumar K, Woodhouse K, Husain M, Kumacheva E, et al. (2009). Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol. Bioeng 102, 493–507. [DOI] [PubMed] [Google Scholar]
- 180.Ismadi MZ, Gupta P, Fouras A, Verma P, Jadhav S, Bellare J, and Hourigan K (2014). Flow characterization of a spinner flask for induced pluripotent stem cell culture application. PLoS One 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Trott J, Tan EK, Ong S, Titmarsh DM, Denil SLH, Giam M, Wong CK, Wang J, Shboul M, Eio M, et al. (2017). Long-term culture of self-renewing pancreatic progenitors derived from human pluripotent stem cells. Stem Cell Reports 8, 1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Konagaya S, and Iwata H (2019). Chemically defined conditions for long-term maintenance of pancreatic progenitors derived from human induced pluripotent stem cells. Sci. Rep 9, 6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nakamura A, Wong YF, Venturato A, Michaut M, Venkateswaran S, Santra M, Gonçalves C, Larsen M, Leuschner M, Kim YH, et al. (2022). Long-term feeder-free culture of human pancreatic progenitors on fibronectin or matrix-free polymer potentiates β cell differentiation. Stem Cell Reports 17, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ma X, Lu Y, Zhou Z, Li Q, Chen X, Wang W, Jin Y, Hu Z, Chen G, Deng Q, et al. (2022). Human expandable pancreatic progenitor–derived β cells ameliorate diabetes. Sci. Adv 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kojayan GG, Alexander M, Imagawa DK, and Lakey JRT (2018). Systematic review of islet cryopreservation. Islets 10, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rawal S, Harrington S, Williams SJ, Ramachandran K, and Stehno-Bittel L (2017). Long-term cryopreservation of reaggregated pancreatic islets resulting in successful transplantation in rats. Cryobiology 76, 41–50. [DOI] [PubMed] [Google Scholar]
- 187.Marquez-Curtis LA, Dai XQ, Hang Y, Lam JY, Lyon J, Manning Fox JE, McGann LE, MacDonald PE, Kim SK, and Elliott JAW (2022). Cryopreservation and post-thaw characterization of dissociated human islet cells. PLoS One 17, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhan L, Rao JS, Sethia N, Slama MQ, Han Z, Tobolt D, Etheridge M, Peterson QP, Dutcher CS, Bischof JC, et al. (2022). Pancreatic islet cryopreservation by vitrification achieves high viability, function, recovery and clinical scalability for transplantation. Nat. Med 28, 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, and Baetge EE (2005). Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol 23, 1534–1541. [DOI] [PubMed] [Google Scholar]