

Abstract
The Trp-cage miniprotein is one of the smallest systems to exhibit a stable secondary structure and fast-folding dynamics, serving as an apt model system to study transient intermediates with both experimental and computational analyses. Previous spectroscopic characterizations that have been done on Trp-cage have inferred a single stable intermediate on a pathway from folded to unfolded basins. We aim to bridge the understanding of Trp-cage structural folding dynamics on microsecond-time scales, by utilizing time-resolved X-ray solution scattering to probe the temperature-induced unfolding pathway. Our results indicate the formation of a conformationally extended intermediate on the time scale of 1 μs, which undergoes complete unfolding within 5 μs. We further investigated the atomistic structural details of the unfolding pathway using a genetic algorithm to generate ensemble model fits to the scattering profiles. This analysis paves the way for direct benchmarking of theoretical models of protein folding ensembles produced with molecular dynamics simulations.
Graphical Abstract

It is established that a protein’s three-dimensional structure largely determines its function. The structure–function relationship has been a central area of scientific research for decades.1,2 Now, physiologically relevant protein structures have been known to be more heterogeneous, including intrinsically disordered proteins (IDPs), comprising of dynamic states and flexible regions where functionality requires not a single three-dimensional structure but an ensemble of structures.3 Resolving these conformational ensembles is a major challenge that, when overcome, would reveal insights in IDP folding/unfolding mechanisms and the functional roles of disordered intermediates.4 External perturbations–like temperature, pH, and ion concentration changes–are some factors that play key roles in cellular signaling pathways, further governing protein structural dynamics. Studying these stimuli driven protein structural dynamics is paramount in revealing functional mechanisms. Beyond elucidating fundamental structural relationships, the connections of non-native structures and their dynamics are essential for preventing diseases and guiding discovery of new treatments.5 To understand disordered protein structures and their interplay within the ensemble, we turn to model systems and structural motifs that can reveal generalizable insights.
One such model system is the 20-residue synthetic peptide TC5b, which adopts the “Trp-cage” structural motif. This Trpcage miniprotein contains a dense web of long-range tertiary contacts around a hydrophobic core surrounding, giving it an atypically high contact order for its relatively short peptide sequence. Trp-Cage’s secondary structure consists of an -helix (residues 2–9), a 310-helix (residues 11–14), and a C-terminal polyproline II helix to complete the packing around the central Trp-6 residue.6 Extensive unfolding thermodynamics studies, carried out by Streicher and Makatadze, demonstrated a two-state model across a broad range of temperatures, from 2 to 97 °C, through the use of differential scanning calorimetry and circular dichroism spectroscopy.7 They additionally noted the broad melting transition of Trpcage while attributing this behavior to the structural heterogeneity of the protein. At equilibrium, the Trp-cage miniprotein was not completely folded even at temperatures approaching the freezing point of water.7 More complex kinetic models emerged from another work with UV resonance Raman spectroscopy, where Asher and co-workers proposed the existence of intermediate conformations along the unfolding pathway of Trp-cage.8 This was further supported by Perczel et al. with NMR and electronic circular dichroism spectroscopies to also propose intermediate conformations.9 By contrast, a time-resolved temperature-jump (T-jump) spectroscopy study by Hagen and co-workers observed two-state cooperative folding within 4 μs.10 The debate about its folding mechanism and lack of consensus from experimental work made Trp-cage a popular model system for molecular dynamics (MD) simulation studies.11,12 Zhou conducted an extensive exploration of the free energy landscape of the TC5b Trp-cage with replica exchange molecular dynamics (REMD) simulations. This work identified an intermediate state and postulated the Asp-9 to Arg-16 salt bridge as a key step in establishing the natively folded conformation.13 In Juraszek and Bolhuis’ work, several different intermediate states were uncovered via the reordering of tertiary contacts, salt bridge, and -helix formation.14 Today, Trp-cage remains a well-known intrinsically disordered protein (IDP), often used as a model system for advanced Markov state modeling studies and machine learning guided computational studies.15,16 Despite the previous work, there still exists a gap in bridging the knowledge between the theoretical models and dynamics extracted from time-resolved experiments.
Validation of the simulation results remains as an ongoing challenge, not only for the TC5b Trp-cage unfolding mechanisms, but also for other protein folding problems.1 Few experimental methods can track protein folding dynamics on time scales shorter than milliseconds and offer relatively low structural resolution.17 For instance, T-jump time-resolved vibrational spectroscopy methods observe the transient conformations of specific amines and carbonyls, yet concrete global information is lacking.18,19 An emerging technique, time-resolved X-ray solution scattering (TRXSS), has been shown to capture protein folding dynamics on nanosecond to milliseconds time scales while avoiding spectroscopic probes or biochemical modifications.20–28 In this work, we implement T-jump TRXSS to capture the transient conformational states of Trp-cage. To bring detailed structural insights to the measured X-ray solution scattering signals, we then implemented ensemble modeling of the TRXSS-derived species states from MD simulations, giving a global representation of the structural heterogeneity.
To capture steady state temperature-dependent structural changes, small-angle X-ray solution scattering (SAXS) profiles of Trp-cage were measured from 15 to 70 °C, in 5 °C intervals, shown in Figure 1. The signal intensity at wider angles, from 0.025 to 0.5 Å−1, decreases with increasing temperature, indicative of global loss in secondary and tertiary structure. These equilibrium measurements confirmed the broad temperature-induced unfolding of Trp-cage, in alignment with previous spectroscopic studies.7 From singular value decomposition (SVD) analysis of this temperature series, only two significant species were apparent (see Supporting Information, SI, note S2). These correspond to the folded and unfolded species, which is consistent with the findings of Streicher and Makhatdze.7 Using their model, with a melting transition midpoint at 43.9 °C, we derived the species associated scattering profiles of the folded and unfolded Trp-cage equilibrium states (Figure 1). Guinier analysis of the species-associated scattering profiles resulted in apparent radii of gyration equal to 8.3 ± 0.2 Å for the folded state and 11.3 ± 0.3 Å for the unfolded state. Kratky plots of this equilibrium temperature series demonstrates the trend of predominantly globular and folded protein in at low temperatures transitioning toward disordered protein signal at high temperatures, through loss in peak prominence around 0.25 Å−1 (see Figure S1 in the SI).
Figure 1.
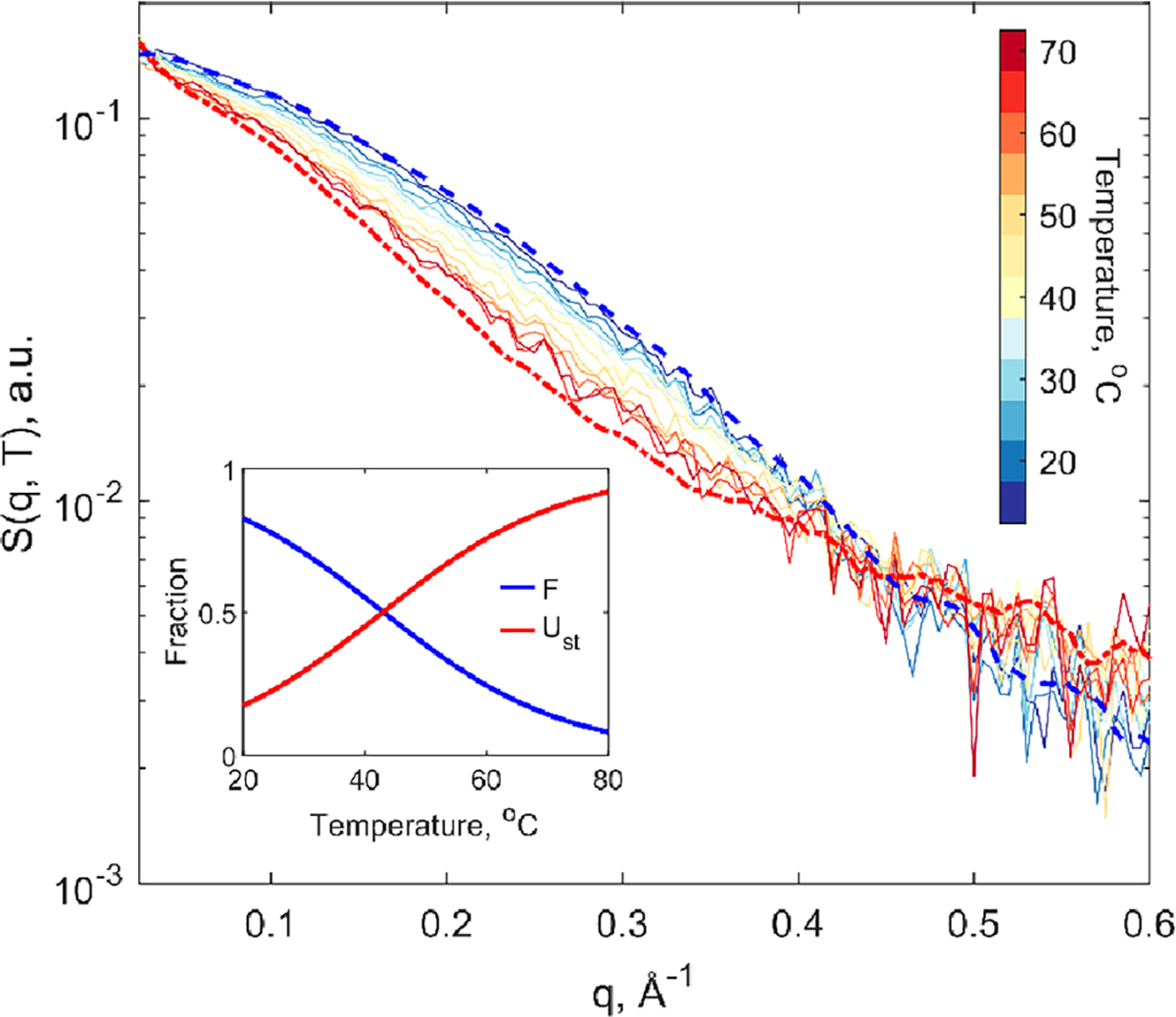
Temperature series of equilibrium small-angle X-ray solution scattering pattern, S(q), of Trp-cage from 15 to 70 °C. The smoothed species associated scattering patterns for the fully folded and unfolded states are respectively shown in blue and red dashed lines. (Inset) relative population fractions of the equilibrium folded (F) and unfolded (Ust) species-associated protein scattering signals.
Temperature-jump TRXSS experiments were performed at BioCARS 14-ID-B beamline at the Advanced Photon Source (APS) at Argonne National Laboratory to study the transient unfolding dynamics of Trp-cage. A detailed description of the experimental procedures can be found in the SI. Briefly, this experiment was conducted from an initial temperature of 30 °C, where the temperature-dependent unfolding was induced by a 7 ns infrared laser pulse, resulting in a 13 °C T-jump (estimated by comparing the time-resolved solvent response to its temperature-dependent character between , see Figure S5 in the SI). TRXSS difference signals, , were obtained by subtracting the scattering patterns obtained at after the T-jump from those before the perturbation. To produce the final protein associated TRXSS signatures, the transient buffer heating artifacts were removed by fitting the wide-angle region with experimentally determined buffer heating signals (SI, note S3).
The resulting protein associated TRXSS time series, from 300 ns to 10 μs, is shown in Figure 2a. We highlight two observable characteristics in the scattering difference curves that evolved with time. At shorter time delays, a positive difference signal was measured at the lowest detectable q region (0.02 Å−1 to 0.03 Å−1). This feature decayed with the increased prominence of a broadly negative difference signal (up to 0.4 Å−1) at longer time delays. This transition between two shapes of curves and the nonzero difference scattering signal at the earliest reported time delay implies the population of an unfolding intermediate species by 300 ns. To further investigate the time course of the signal, we integrated the absolute values of the time series in , shown in Figure 2b. The relative loss of SAXS intensity across this time-dependent scattering signal confirmed an unfolding structural trend within 10 μs of the temperature jump. This observed time-dependent signal was corroborated by our SVD analysis.
Figure 2.
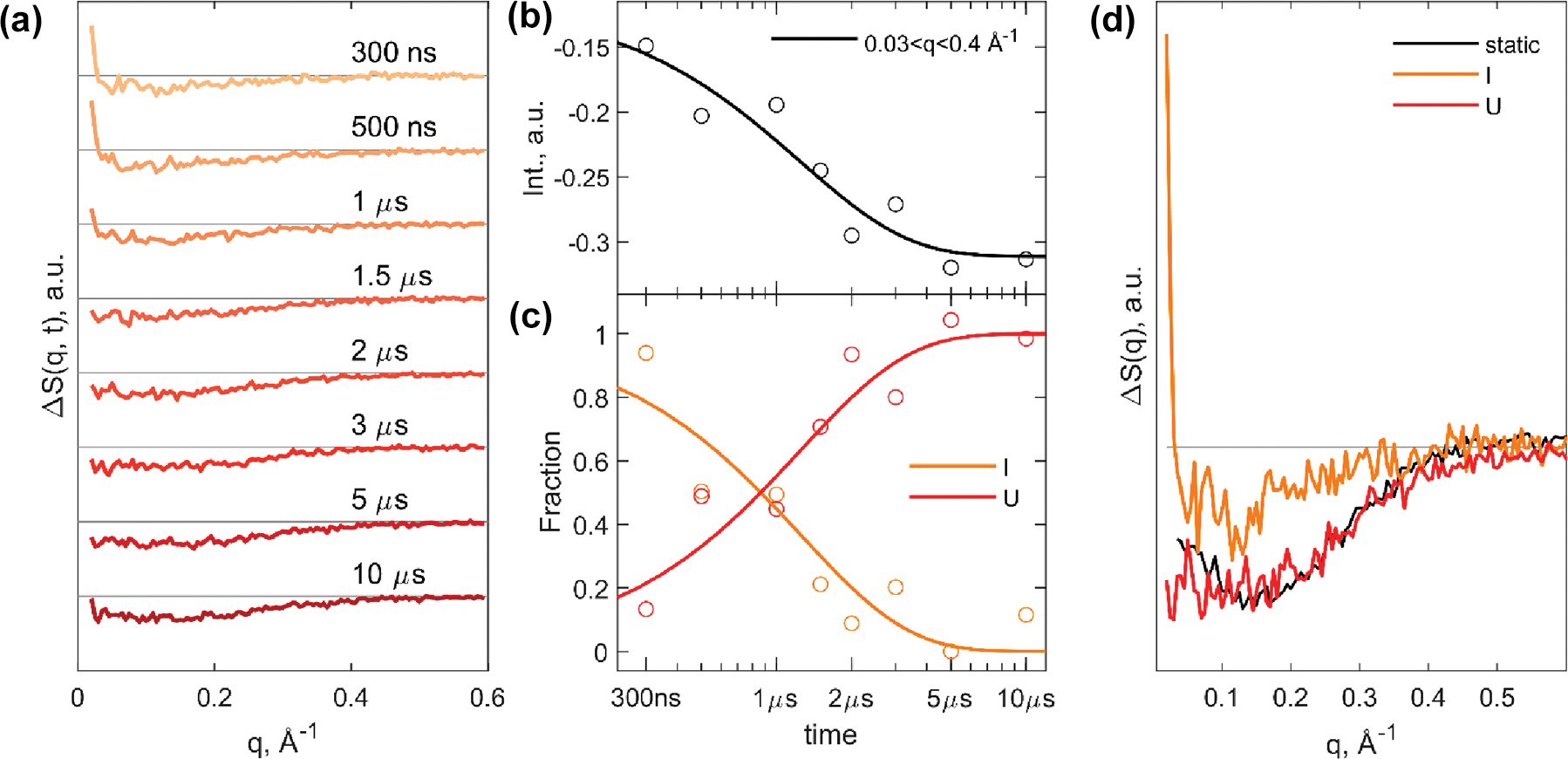
TRXSS time series and analysis results of Trp-cage unfolding. (a) Time series of scattering difference profiles from 300 ns to 10 μs. (b) Integrated difference signal at each time delay plotted with kinetic model from global analysis. (c) Species-associated population dynamics of the intermediate (I) and unfolded (U) states. (d) Species-associated scattering differences corresponding to the kinetic model from global analysis with comparison of unfolded species-associated difference profile (Ust) from static temperature series.
Global analysis was performed on the T-jump TRXSS time series to extract the species associated protein signals according to the proposed kinetic model, using established data analysis procedures.21,25–27,29 Based on the SVD analysis of the time series present in the SI (Figure S5), two distinct species were observed during unfolding. Thus, for global analysis we fitted the data using the following kinetic model:
where represents the folded species, is an intermediate species populated quicker than 300 ns, and is the tentative unfolded species. The time constant of the transition between and was determined from the fitting to be 1.2 ± 0.2 μs. The best fit model is in good agreement with the data, indicating of an adequate model (SI, note Figure S5a). The extracted time dependent populations and species associated difference (SAD) signals for and species are shown in Figure 2c and d. The SAD profile for matched the difference profile from the equilibrium temperature-dependent studies, confirming the assignment of to the final unfolded state in the kinetic model. A key characteristic of the resulting intermediate SAD curve is the positive difference signal at low q, which is attributed to an increase in forward scattering in the transient intermediate. This type of signal response has been discussed in previous work to be a result of the increase in solvent-accessible surface area of a protein, inducing a rearrangement of the overall solvation shell.30–32
The transient TRXSS SAD profiles were scaled and added onto the folded equilibrium species associated scattering pattern to generate reconstructed species associated scattering patterns for the transient species (Figure 3a). The and reconstructed species associated scattering patterns showed less small-angle X-ray scattering signal intensity compared to that of , which is consistent with less secondary and tertiary structural content. The , determined from the Guinier analysis of the reconstructed scattering patterns of the transient species, were 10.5 ± 1.0 Å for the intermediate and 10.3 ± 1.0 Å for the unfolded species. These empirically larger imply that was more unfolded than the . To extract more information about the conformations within these ensemble species, a Bayesian inference Fourier Transform method was used to calculate the electron pair distance distribution function of the folded state and the two reconstructed transient states (Figure 3b).33 The pair distribution function (PDF) of the showed a compact conformation with the electron density peak around 10 Å. By comparison, the PDF of exhibits a less intense main peak at 10 Å with some of the electron density shifted to longer distances. Subsequently, the ensemble has the broadest distribution of electron density with even less intense peak at 10 Å and a distinct shoulder between 20 and 30 Å. The stepwise broadening of the PDFs suggests the sequential unfolding pathway from to to .
Figure 3.
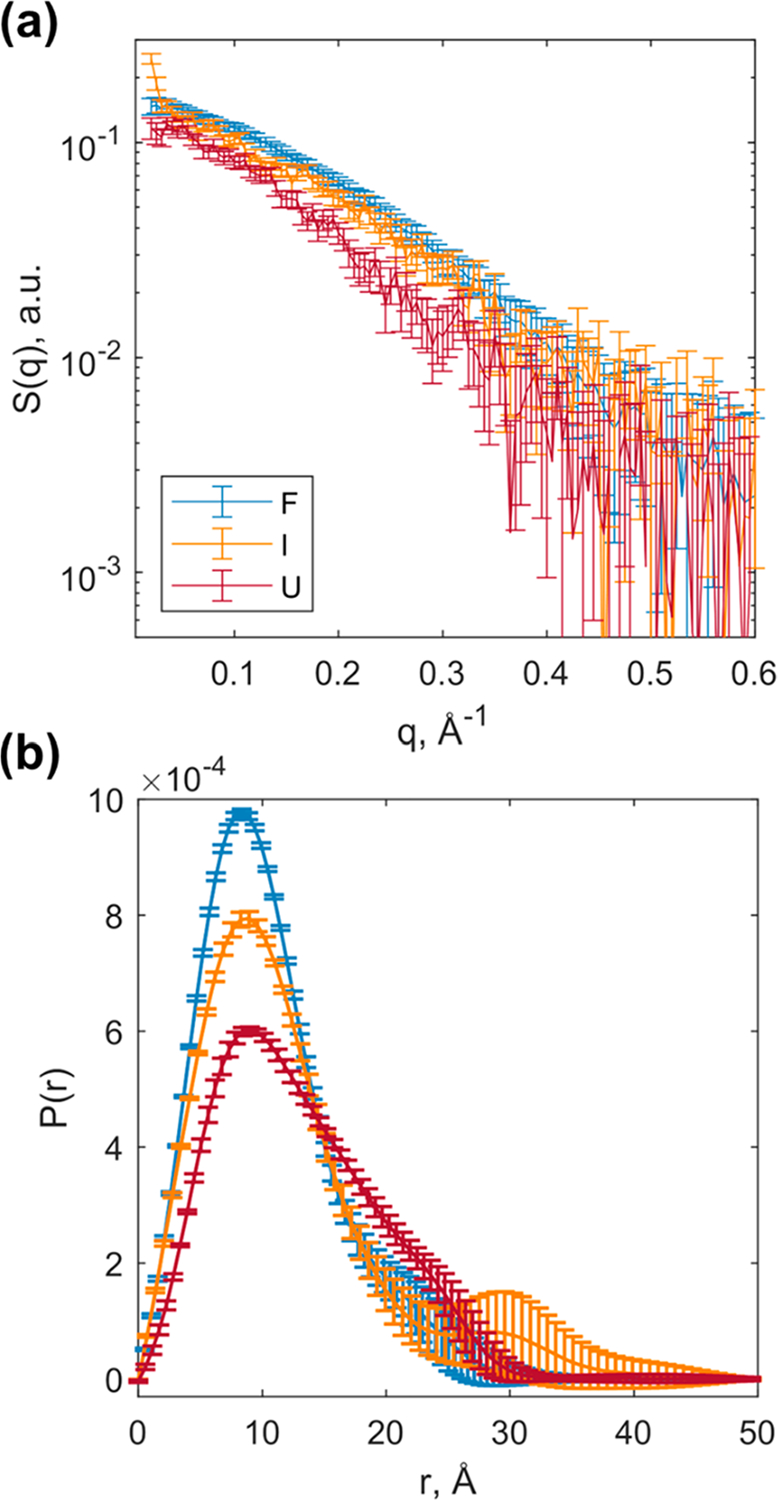
Kinetically observed unfolding states. (a) Species associated reconstructed SAXS patterns corresponding to each state. (b) Pair distribution function of each state.
To gain further insight into the structural conformation of Trp-cage in each of the kinetic ensemble species, we turn to data modeling using MD simulations. Due to the disordered nature of the Trp-cage system, the analysis must represent each species as a mixture of flexible structures accounting for ensemble heterogeneity. To accomplish this, we adapted the ensemble optimization method (EOM) with MD simulations to reveal the best fit structural candidates within each species, treating , and as heterogeneous ensembles as described in the literature; see the SI.28,30,34 An initial data set of Trp-cage candidate structures, distributed with Boltzmann ensemble weights, were generated with replica exchange solute tempering MD simulations (see the SI for details). Each of the simulation frames, containing atomic resolution structural information, was used to calculate theoretical X-ray scattering curves corresponding to individual conformation (see the SI).35 Then, a genetic algorithm was used to select and refine a group of individual structures whose average X-ray scattering signal best matched the experimentally observed ensemble signal. Ten EOM runs were executed and accumulated for each of the three experimentally observed states (Figures S9 and S10). histograms of the ten aggregate genetic algorithm results for each species are displayed in Figure 4a–c. The refined structures from EOM showed that larger Trp-cage structures were more populated in the and states. The ensemble histogram is dominated by mostly native structures while EOM accounted for the heterogeneity of the system by including smaller fractions of nonnative “unfolded” structures. Both and contain extended structures with calculated demonstrating the ensemble X-ray scattering signals indeed captured transient Trp-cage unfolding.
Figure 4.
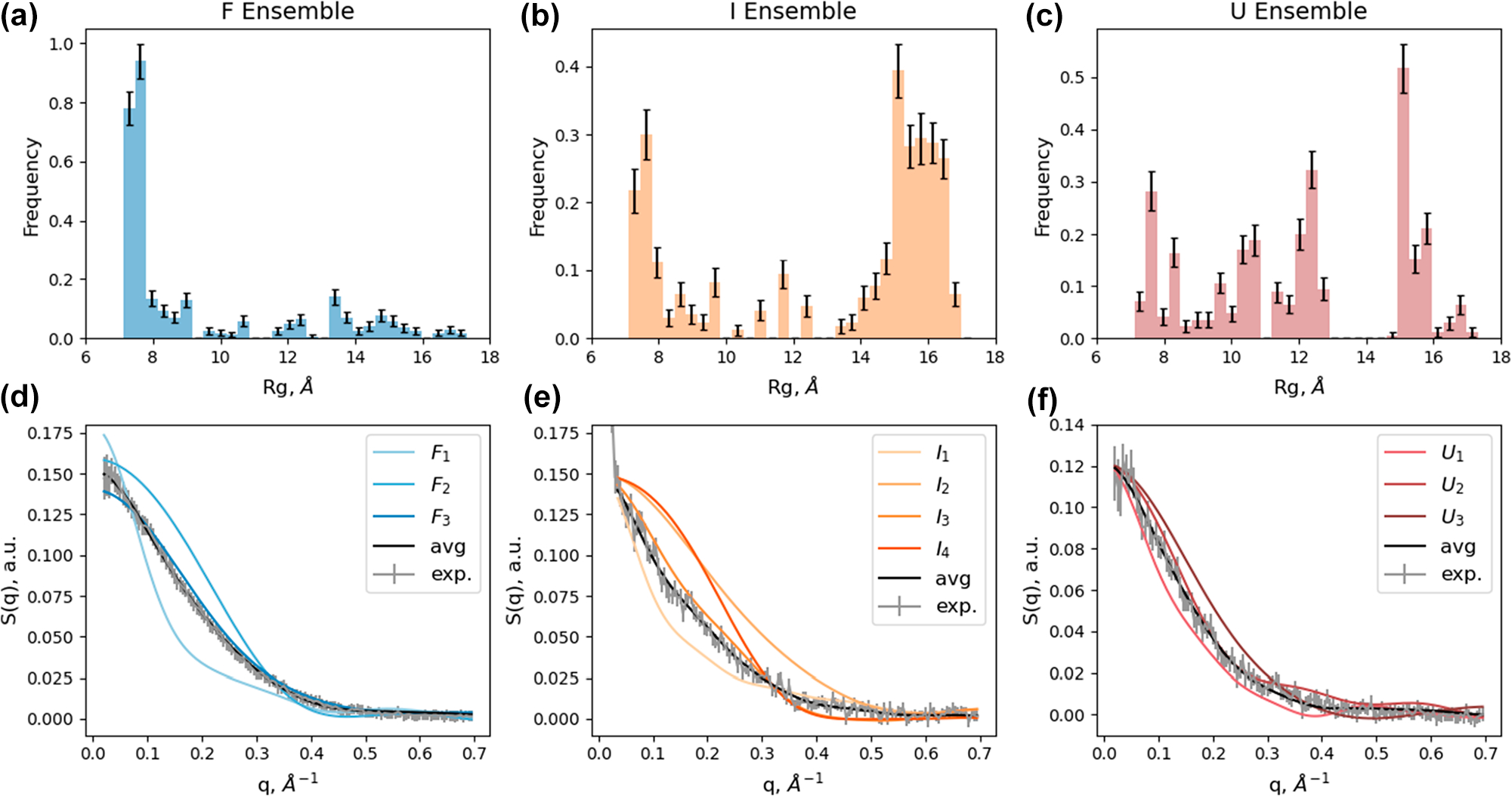
Histograms of Rg for genetic algorithm results of reconstructed ensemble species and linear combination fit results of reconstructed experimental species associated scattering patterns. (a–c) Rg histograms of the average genetic algorithm results for each species associated scattering pattern with standard errors: F, I, and U, respectively. (d–f) Linear combination fit results for clustered representative structures for each species (F, I, and U, respectively); average theoretical ensemble scattering shown in black; experimentally reconstructed species associated scattering signal shown in gray.
After clustering the aggregated EOM conformations into representative structural classes (based on an average rmsd threshold greater than 6 Å of heavy atoms, refer to the SI for details), the individual theoretical scattering patterns of those representative conformations were linearly fit to the experimentally measured signals (Figure 4d–f) to extract corresponding population fractions (Figure 5a). The representative structures, obtained from ensemble fitting and subsequent clustering analysis, are labeled with their associated experimental reference ensemble and a numerical subscript. Their corresponding calculated values are plotted in Figure 5b.
Figure 5.
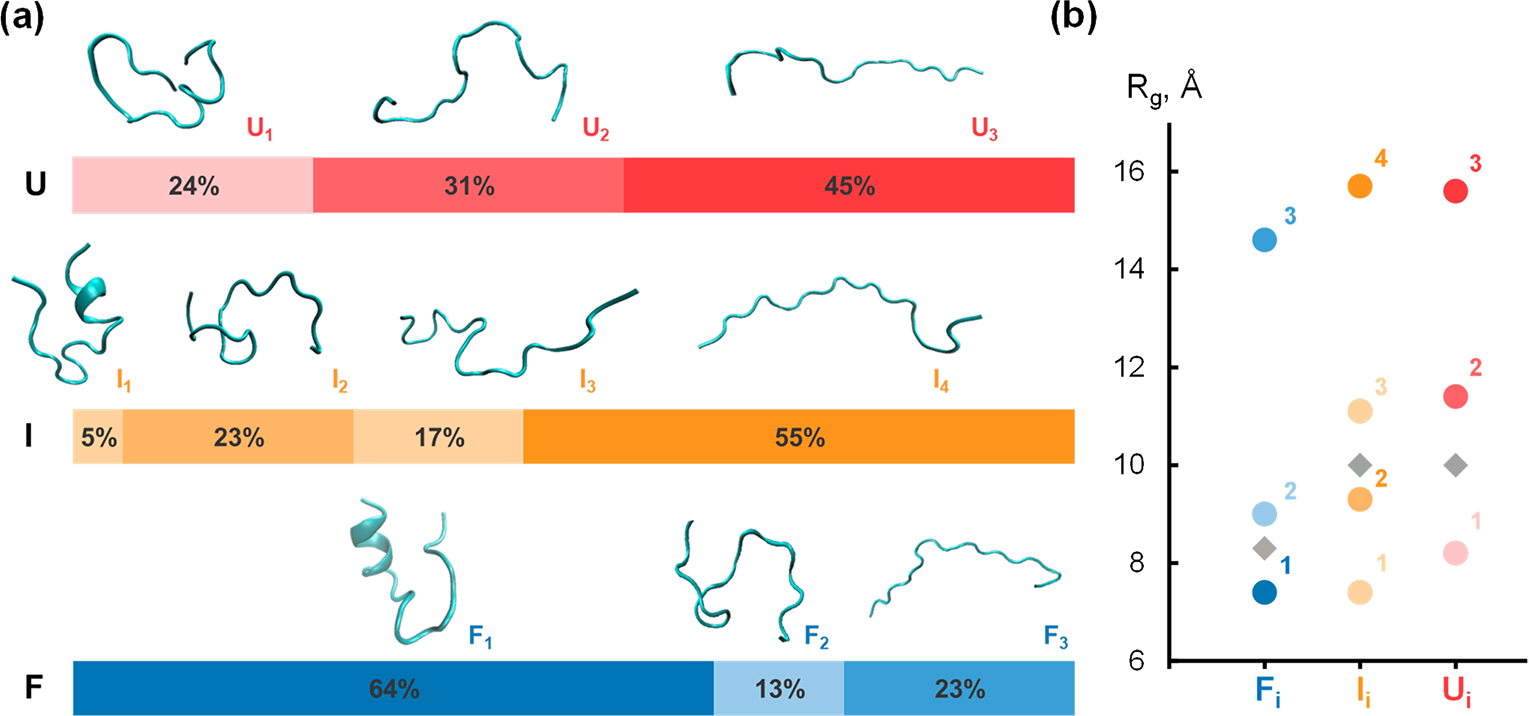
Conformational heterogeneity of each state shown by structural model fitting. (a) Representative conformations of Trp-cage obtained from structural clustering shown with associated species ensemble population fraction from linear combination fitting. (b) Structural similarities between extracted conformations shown by Rg comparison plot. Experimentally observed ensemble Rg plotted as gray diamonds.
The diversity of representative structures, which fitted to each ensemble experimentally observed species, agrees well with the intrinsic disorder and conformational heterogeneity of Trp-cage, as determined by previous studies. The initial state of the time-resolved experiment was the ensemble, a mixture of native-like folded structures and relatively unfolded structures Å, and . The most compact structure in , has the smallest of 7.4 Å and represents most of that ensemble, consistent with the histogram representation in Figure 4a. After the T-jump, the system is first perturbed into the I ensemble, in which we found only 5% of folded structures (, 7.4 Å) that were consistent with the natively folded Trp-cage. The remainder was a mixture of compact structure and extended unfolded intermediates, and . The final evolution of the system into the ensemble species confirmed the dynamic heterogeneity of Trp-cage at high temperatures. This species contains predominantly extended unfolded conformations , and , while maintaining a population of a smaller , structure class. We emphasize the importance of considering the EOM analysis when studying Trp-cage unfolding because the true ’s are more varied than just the experimentally observed ensemble average (Figure 2b).
The compact conformations in these ensembles support the transient observation and evolution of the molten globule state in Trp-cage unfolding. , and are compact due to tertiary contact association, yet lack secondary structure; therefore, we assign these structural clusters to the molten globule state. Piana and co-workers simulated a T-jump in a Trp-cage unfolding computational study and reported that Trp-cage’s molten globule state acts as a kinetic trap, that extends the unfolding time.36 Although the molten globule was observed in each of the transient species, our TRXSS study and structural analysis was not able directly assess the connectivity of structural class to others. However, it is likely that upon T-jump, the initial population of molten globule transitioned into the more unfolded structures while the molten globule of the transient states ( and ) was populated by the unfolding of initially folded structures . Furthermore, we attribute and to be kinetically populated short-lived intermediates that are conformations explored by Trp-cage upon unfolding and transition to more compact structures within 2 μs. This result compares well with the Markov state model (MSM) proposed by Piana and co-workers.36 Ferguson and co-workers used MSM to study a close Trp-cage variant (TC10b) and described the molten globule state as the metastable transition between various high-energy unfolded structures.15
This intermediate species from this T-jump TRXSS study differed from the ones proposed in the transient 2D-IR spectroscopic study by Woutersen and co-workers.18 The submicrosecond intermediate state in their work was characterized by conformations with unique structural changes in the 310-helix; however, such structures are not spatially resolved with X-ray solution scattering and would be classified within the structures similar to and . Additionally, Gai and co-workers reported observing the hydrophobic core intermediate effects of the 310-helix in the TC5b Trp-cage at temperatures below 15 °C; thus, this meant that we would likely not observe an early unfolding intermediate from our initial temperature of 30 °C.19 The intermediate species observed by TRXSS was a dynamic state comprised of molten globule and unfolded Trp-cage structures. supports a single hydrophobic collapse intermediate with little -helical content, rather than those involving the initial breakage of the Asp9-Arg16 salt bridge with two separate hydrophobic lobes.14,37 By comparing the representative conformations and considering structural similarities, for instance looking at structures with similar across the three transient ensembles, we posit that these similar conformations are connected to one another. Future analysis would be conducted with Markov state modeling, incorporated with TRXSS analysis, to validate the observed transient intermediate species and mechanism of unfolding.
In summary, we conducted a time-resolved X-ray solution scattering experiment to observe conformational evolution during the unfolding process of the intrinsically disordered Trp-cage miniprotein. Through kinetic modeling of the X-ray scattering time-series signals, we were able to derive species-associated X-ray scattering patterns, which represent each ensemble state in the unfolding dynamics. Leveraging the EOM and genetic algorithm to fit MD simulation data to our experimental observations, we were able to understand the heterogeneous composition of the , , and states. Structural analysis with MD simulation results was especially appropriate for studying Trp-cage unfolding since X-ray solution scattering was able to collect global electron density information, which was aptly sampled by our REST2 trajectory. Because SAXS allows for global flexibility in the ensemble, the dynamic presence of a molten globule state and other short-lived intermediates ( and ) are apparent in the unfolding process of Trp-cage. The results from this work demonstrate the strengths of revealing heterogeneous protein folding dynamics through combined X-ray solution scattering and molecular dynamics simulation methods.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health (NIH), under Contract No. R01-GM115761. A.M.C. and D.J.H. acknowledge support from the NIH/National Institute of General Medical Sciences (NIGMS) sponsored Molecular Biophysics Training Program at Northwestern University (T32GM140995). This research used resources of the APS, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of BioCARS was also supported by the NIH-NIGMS under grant number R24GM111072. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Time-resolved setup at Sector 14 was funded in part through a collaboration with Philip Anfinrud (NIH/NIDDK). Optical equipment used for IR beam delivery at BioCARS was purchased with support from the Fraser lab at University of California San Francisco. We acknowledge Robert W. Henning (BioCARS) for his assistance in performing the TRXSS experiments. We would also like to acknowledge Guy Macha (BioCARS) for his assistance in designing the sample holder.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.2c03680.
Experimental methods, details of TRXSS fitting and analysis procedures, REST2MD simulation details, EOM genetic algorithm, and clustering analysis procedures (PDF)
Contributor Information
Arnold M. Chan, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Adam K. Nijhawan, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Darren J. Hsu, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Denis Leshchev, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States.
Dolev Rimmerman, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States.
Irina Kosheleva, Center for Advanced Radiation Sources, The University of Chicago, Chicago, Illinois 60637, United States.
Kevin L. Kohlstedt, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Lin X. Chen, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States; Chemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, Illinois 60439, United States
REFERENCES
- (1).Gelman H; Gruebele M Fast protein folding kinetics. Q. Rev. Biophys. 2014, 47 (2), 95–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gruebele M The Fast Protein Folding Problem. Annu. Rev. Phys. Chem. 1999, 50 (1), 485–516. [DOI] [PubMed] [Google Scholar]
- (3).Wright PE; Dyson HJ Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16 (1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bhowmick A; Brookes DH; Yost SR; Dyson HJ; Forman-Kay JD; Gunter D; Head-Gordon M; Hura GL; Pande VS; Wemmer DE; et al. Finding Our Way in the Dark Proteome. J. Am. Chem. Soc. 2016, 138 (31), 9730–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Marinko JT; Huang H; Penn WD; Capra JA; Schlebach JP; Sanders CR Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis. Chem. Rev. 2019, 119 (9), 5537–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Neidigh JW; Fesinmeyer RM; Andersen NH Designing a 20-residue protein. Nat. Struct. Biol. 2002, 9 (6), 425–430. [DOI] [PubMed] [Google Scholar]
- (7).Streicher WW; Makhatadze GI Unfolding Thermodynamics of Trp-Cage, a 20 Residue Miniprotein, Studied by Differential Scanning Calorimetry and Circular Dichroism Spectroscopy. Biochemistry 2007, 46 (10), 2876–2880. [DOI] [PubMed] [Google Scholar]
- (8).Ahmed Z; Beta IA; Mikhonin AV; Asher SA UV–Resonance Raman Thermal Unfolding Study of Trp-Cage Shows That It Is Not a Simple Two-State Miniprotein. J. Am. Chem. Soc. 2005, 127 (31), 10943–10950. [DOI] [PubMed] [Google Scholar]
- (9).Rovó P; Farkas V; Hegyi O; Szolomájer-Csikós O; Tóth GK; Perczel A Cooperativity network of Trp-cage miniproteins: probing salt-bridges. J. Pept. Sci. 2011, 17 (9), 610–619. [DOI] [PubMed] [Google Scholar]
- (10).Qiu L; Pabit SA; Roitberg AE; Hagen SJ Smaller and Faster: The 20-Residue Trp-Cage Protein Folds in 4 μs. J. Am. Chem. Soc. 2002, 124 (44), 12952–12953. [DOI] [PubMed] [Google Scholar]
- (11).Barua B; Lin JC; Williams VD; Kummler P; Neidigh JW; Andersen NH The Trp-cage: optimizing the stability of a globular miniprotein. Protein Eng. Des. Sel. 2008, 21 (3), 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Snow CD; Zagrovic B; Pande VS The Trp Cage: Folding Kinetics and Unfolded State Topology via Molecular Dynamics Simulations. J. Am. Chem. Soc. 2002, 124 (49), 14548–14549. [DOI] [PubMed] [Google Scholar]
- (13).Zhou R Trp-cage: Folding free energy landscape in explicit water. Proc. Natl. Acad. Sci. U.S.A. 2003, 100 (23), 13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Juraszek J; Bolhuis PG Sampling the multiple folding mechanisms of Trp-cage in explicit solvent. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (43), 15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sidky H; Chen W; Ferguson AL High-Resolution Markov State Models for the Dynamics of Trp-Cage Miniprotein Constructed Over Slow Folding Modes Identified by State-Free Reversible VAMPnets. J. Phys. Chem. B 2019, 123 (38), 7999–8009. [DOI] [PubMed] [Google Scholar]
- (16).Shin K; Tran DP; Takemura K; Kitao A; Terayama K; Tsuda K Enhancing Biomolecular Sampling with Reinforcement Learning: A Tree Search Molecular Dynamics Simulation Method. ACS Omega 2019, 4 (9), 13853–13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nijhawan AK; Chan AM; Hsu DJ; Chen LX; Kohlstedt KL Resolving Dynamics in the Ensemble: Finding Paths through Intermediate States and Disordered Protein Structures. J. Phys. Chem. B 2021, 125 (45), 12401–12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Meuzelaar H; Marino KA; Huerta-Viga A; Panman MR; Smeenk LEJ; Kettelarij AJ; van Maarseveen JH; Timmerman P; Bolhuis PG; Woutersen S Folding Dynamics of the Trp-Cage Miniprotein: Evidence for a Native-Like Intermediate from Combined Time-Resolved Vibrational Spectroscopy and Molecular Dynamics Simulations. J. Phys. Chem. B 2013, 117 (39), 11490–11501. [DOI] [PubMed] [Google Scholar]
- (19).Culik RM; Serrano AL; Bunagan MR; Gai F Achieving Secondary Structural Resolution in Kinetic Measurements of Protein Folding: A Case Study of the Folding Mechanism of Trp-cage. Angew. Chem., Int. Ed. 2011, 50 (46), 10884–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cammarata M; Levantino M; Schotte F; Anfinrud PA; Ewald F; Choi J; Cupane A; Wulff M; Ihee H Tracking the structural dynamics of proteins in solution using time-resolved wideangle X-ray scattering (vol 5, pg 881, 2008). Nat. Methods 2008, 5 (11), 988–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rimmerman D; Leshchev D; Hsu DJ; Hong J; Kosheleva I; Chen LX Direct Observation of Insulin Association Dynamics with Time-Resolved X-ray Scattering. J. Phys. Chem. Lett. 2017, 8 (18), 4413–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hsu DJ; Leshchev D; Kosheleva I; Kohlstedt KL; Chen LX Unfolding bovine -lactalbumin with T-jump: Characterizing disordered intermediates via time-resolved x-ray solution scattering and molecular dynamics simulations. J. Chem. Phys. 2021, 154 (10), 105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Thompson MC; Barad BA; Wolff AM; Sun Cho H; Schotte F; Schwarz DMC; Anfinrud P; Fraser JS Temperature-jump solution X-ray scattering reveals distinct motions in a dynamic enzyme. Nat. Chem. 2019, 11 (11), 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Orädd F; Ravishankar H; Goodman J; Rogne P; Backman L; Duelli A; Nors Pedersen M; Levantino M; Wulff M; Wolf-Watz M; et al. Tracking the ATP-binding response in adenylate kinase in real time. Sci. Adv. 2021, 7 (47), No. eabi5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rimmerman D; Leshchev D; Hsu DJ; Hong J; Abraham B; Henning R; Kosheleva I; Chen LX Probing Cytochrome c Folding Transitions upon Phototriggered Environmental Perturbations Using Time-Resolved X-ray Scattering. J. Phys. Chem. B 2018, 122 (20), 5218–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rimmerman D; Leshchev D; Hsu DJ; Hong J; Abraham B; Henning R; Kosheleva I; Chen LX Revealing Fast Structural Dynamics in pH-Responsive Peptides with Time-Resolved X-ray Scattering. J. Phys. Chem. B 2019, 123, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rimmerman D; Leshchev D; Hsu DJ; Hong J; Abraham B; Kosheleva I; Henning R; Chen LX Insulin hexamer dissociation dynamics revealed by photoinduced T-jumps and timeresolved X-ray solution scattering. Photochem. Photobiol. Sci. 2018, 17 (7), 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kim TW; Lee SJ; Jo J; Kim JG; Ki H; Kim CW; Cho KH; Choi J; Lee JH; Wulff M; et al. Protein folding from heterogeneous unfolded state revealed by time-resolved X-ray solution scattering. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (26), 14996–15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hsu D; Leshchev D; Rimmerman D; Hong J; Kelley MS; Kosheleva I; Zhang X; Chen LX X-ray Snapshots Reveal Conformational Influence on Active Site Ligation During Metalloprotein Folding. Chem. Sci. 2019, 10, 9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bernadó P; Mylonas E; Petoukhov MV; Blackledge M; Svergun DI Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. J. Am. Chem. Soc. 2007, 129 (17), 5656–5664. [DOI] [PubMed] [Google Scholar]
- (31).Cho HS; Schotte F; Dashdorj N; Kyndt J; Henning R; Anfinrud PA Picosecond Photobiology: Watching a Signaling Protein Function in Real Time via Time-Resolved Small- and Wide-Angle X-ray Scattering. J. Am. Chem. Soc. 2016, 138 (28), 8815–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Riback JA; Bowman MA; Zmyslowski AM; Knoverek CR; Jumper JM; Hinshaw JR; Kaye EB; Freed KF; Clark PL; Sosnick TR Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science 2017, 358 (6360), 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hansen S BayesApp: a web site for indirect transformation of small-angle scattering data. J. Appl. Crystallogr. 2012, 45 (3), 566–567. [Google Scholar]
- (34).Lee SJ; Kim Y; Kim TW; Yang C; Thamilselvan K; Jeong H; Hyun J; Ihee H Reversible molecular motional switch based on circular photoactive protein oligomers exhibits unexpected photo-induced contraction. Cell Rep. Phys. Sci. 2021, 2 (8), 100512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schneidman-Duhovny D; Hammel M; Tainer JA; Sali A Accurate SAXS Profile Computation and its Assessment by Contrast Variation Experiments. Biophys. J. 2013, 105 (4), 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Marinelli F; Pietrucci F; Laio A; Piana S A Kinetic Model of Trp-Cage Folding from Multiple Biased Molecular Dynamics Simulations. PLoS Comput. Biol. 2009, 5 (8), No. e1000452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Shao Q; Shi J; Zhu W Enhanced sampling molecular dynamics simulation captures experimentally suggested intermediate and unfolded states in the folding pathway of Trp-cage miniprotein. J. Chem. Phys. 2012, 137 (12), 125103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.