

Abstract
Cellular stress, arising from accumulation of unfolded proteins, occurs frequently in rapidly proliferating cancer cells. This cellular stress, in turn, activates the unfolded protein response (UPR), an interconnected set of signal transduction pathways that alleviate the proteostatic stress. The UPR is implicated in cancer cell survival and proliferation through upregulation of pro-tumorigenic pathways that ultimately promote malignant metabolism and neoangiogenesis. Here, we reviewed mechanisms of signaling crosstalk between the UPR and angiogenesis pathways, as well as transmissible ER stress and the role in tumor growth and development. To characterize differences in UPR and UPR-mediated angiogenesis in malignancy, we employed a data mining approach using patient tumor data from The Cancer Genome Atlas (TCGA). The analysis of TCGA revealed differences in UPR between malignant samples versus their non-malignant counterparts.
Keywords: Unfolded protein response, IRE1α, XBP1, ATF6, PERK, Tumor microenvironment
1. Introduction
Unfolded protein response (UPR) is an essential cellular reaction to stresses in the endoplasmic reticulum (ER), an organelle responsible for production, folding, and modifications of proteins necessary for proper functioning. In the ER lumen, the unique ionic and oxidative milieu coupled with the presence of molecular chaperones and enzymes such as Protein Disulfide Isomerase (PDI), Peptidyl-prolyl Isomerase, and ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 2 (SERCA2) permit proper folding and maturation of ER translocated proteins. A host of enzymes enable additions and modifications of glycosidic moieties onto proteins. Cumulatively, ER functions to ensure the bonafide maturation of proteins prior to their advancement through the secretory pathway. An increase in unfolded or misfolded proteins within the ER triggers the UPR to enhance ER’s folding capacity and slow protein production, which favors tumor cell survival. This comprises the adaptive UPR. Paradoxically, under conditions of persistent ER stress, the UPR activates cell death pathways. This comprises the pro-apoptotic UPR. Indeed, the roles of ER stress and UPR in tumorigenesis are well recognized and previously reviewed [2,3]. Here, we focus on dissecting the signaling crosstalk between UPR and angiogenesis in malignancy.
Perturbations and stresses to the ER, including accumulation of aberrant proteins and microenvironmental changes such as hypoxia and disruption of the oxidative milieu, trigger a host of signal transduction pathways. ER stress is sensed by multiple ER-localized proteins, including the transmembrane proteins Inositol Requiring Enzyme 1 α (IRE1α), Protein Kinase RNA (PKR)-like ER Kinase (PERK) and Activating Transcription Factor 6 (ATF6).
IRE1α:
IRE1α, also known as Endoplasmic Reticulum to Nucleus Signaling 1 (ERN1), is an ER transmembrane protein present either as an inactive monomeric form, or an active dimeric or multimeric form [4]. Dimerization of IRE1α, in response to ER stress, results in transautophosphorylation of the luminal domains and activation of the endoribonuclease domain in the cytosolic portion [5]. The endoribonuclease activity results in excision of a 26-nucleotide intronic sequence from the XBP1 (X-Box Binding Protein 1) mRNA, resulting in the formation of spliced XBP1 (XBP1s) [5]. The resulting XBP1 protein product acts as a critical transcription factor, and regulates the expression of important UPR regulating proteins, resulting in increased ER/Golgi biogenesis and ER protein folding capacity. In another mechanism termed regulated IRE1-dependent decay (RIDD), activated IRE1α endoribonuclease activity may degrade various mRNAs and microRNAs (miRNAs). RIDD serves to decrease mRNA levels to reduce protein folding in the ER. The targeted mRNAs contain the common consensus CUGCAG sequence. The IRE1 arm also functions in the proapoptotic UPR through induction of caspase-2, caspase-8, or BAX/BAK-dependent apoptosis. RIDD may also activate inflammasome-mediated apoptosis.
PERK:
PERK, also known as Eukaryotic Translation Initiation Factor 2 Alpha Kinase 3 (EIF2AK3), is also a transmembrane protein. The UPR leads to its oligomerization and autophosphorylation. Interestingly, PERK mediated phosphorylation of Eukaryotic Translation Initiator Factor-2 (eIF2α) at serine 51 blocks protein synthesis [6], and limits the influx of nascent proteins into the ER. Simultaneously, phosphorylated eIF2α initiates translation of mRNAs that contain an open reading frame in the 5′ untranslated region [7–10]. One of these mRNAs is ATF4, which encodes a transcription factor [8,9]. ATF4 activity, resulting from cellular stress and insult, initiates an adaptive-protective program through upregulation of chaperone synthesis, amino acid import, and redox homeostasis. However, in response to sustained stress, ATF4 functions to promote an apoptotic program by activating the proapoptotic transcription factor CCAAT- Enhancer-Binding Protein Homologous Protein (CHOP; also known as DNA damage inducible Transcript 3 (DDIT3) [11]. Of note, CHOP may also activate Protein Phosphatase 1 Regulatory Subunit 15A (PPP1R15A, also known as GADD34), which dephosphorylates eIF2α, leading to translational recovery. The result is increased proteostatic stress. CHOP promotes ER stress induced apoptosis by activating DR5 (death receptor 5), and modulating NOXA (Phorbol-12-myristate-13-acetate-induced protein 1), BIM (Bcl-2-like protein 11; also referred to as Bcl-2 Interacting Mediator of cell death), and PUMA (BCL2 binding component 3), which induce protein synthesis further increasing proteostatic stress. The thiol oxidoreductase ERp57 and protein disulfide isomerase A1 play important roles in PERK activation, which was elucidated in HCT116 colon carcinoma cells [12]. Loss of ERp57 leads to oxidation of protein disulfide isomerase A1, and activation of PERK [12]. On the other hand, loss of protein disulfide isomerase A1 reduces PERK signaling, and sensitizes cells to hypoxia and ER stress [12].
ATF6:
ATF6 is also an ER transmembrane protein which traffics to Golgi complex in response to ER stress. During ER stress, the full length AFT6 (ATF6p90) moves from ER to Golgi complex. ERp18, a single domain member of the protein disulfide isomerase family, interacts with ATF6 under conditions of ER stress and promotes ATF6 transport to the Golgi complex [13]. Golgi resident proteases site-1 protease (S1P, also known as MBTPS1 for Membrane Bound Transcription Factor Peptidase, Site 1) and site-2 protease (S2P, also known as MBTPS2 for Membrane Bound Transcription Factor Peptidase, Site 2) cleave and release the cytosolic domain of ATF6, generating a fragment called ATF6p50 [5]. Of note, ATF6p50 contains a basic leucine zipper (bZIP) transcription factor domain, that upregulates select UPR genes, including ER chaperones and enzymes that promote protein folding, maturation, and secretion, as well as promoting ER/Golgi biosynthesis.
Binding immunoglobulin Protein (BiP), a member of the heat shock protein 70 family, is a molecular chaperone which associates with and coordinately regulates IRE1α and ATF6. Under conditions of no stress, BiP binds and prevents PERK and IRE1α dimerization. However, in response to ER stress, BiP dissociates from PERK and IRE1α, leading to their homodimerization and activation.
A model of directing binding of misfolded proteins to UPR sensors is another mechanism of UPR activation. Studies suggest that following BiP dissociation, IRE1 can bind misfolded proteins [14,15]. A similar direct mechanism has also been shown for PERK activation [16].
UPR has also been shown to attenuate translation of proteins that aid in translational recovery (including ER chaperones, PDI, SERCA2, amino acid importers), and upregulate anti-oxidative responses (glutathione synthesis), which ultimately favor cell survival. However, prolonged activation due to excessive unfolded protein load in the ER may lead to cell cycle arrest or activation of apoptotic programs mediated by transcriptional activation of CHOP, c-JUN N-terminal kinase (JNK), or caspase-12. Interestingly, caspase-12-mediated cell death occurs in response to UPR, but not via death receptor or mitochondrial pathways. For example, IRE1 activation may induce caspase-12 activation and cell death [17]. However, Caspase-12 is not present or functional in a majority of humans due to a single nucleotide polymorphism that introduces a premature stop codon. Only a fraction of African descendants expresses full-length caspase-12, and therefore such mechanisms may not be broadly applicable to human UPR and cancer. However, this is not the case in mice, and therefore mouse is a useful model organism for studying caspase-12 biology. ER stress arising from decreased glucose has also been reported to promote cell death via caspase-12 [18].
The UPR and ER stress play important roles in tumor development, due to the restricted supply of nutrients and oxygen, which in turn restrict the ability of cancer cells to generate proteins sufficient for rapid cell cycle rate [19]. Ma and Hendershot put forth the idea that the activation of UPR in tumors is apparently due to their inadequate vascularization, resulting in hypoxia and nutrient deficiency and therefore altering the physiology of protein secretion [19]. In the same work, the link between UPR activation in cancer cells and sensitivity to chemotherapeutic agents was discussed [19]. Previously, the relationship between ER stress and angiogenesis has been discussed, with a special emphasis on retinopathy, atherosclerosis, and cancer, in general [20]. Using a data mining approach combined with principal component analysis (PCA), here we focus on the signaling crosstalk between UPR and angiogenesis in malignancies. We also discuss the role of exosomes in UPR/angiogenesis, transmissible ER stress, and how currently used anti-cancer drugs may modulate UPR. We found that malignancies cluster distinctly compared to their non-malignant counterparts with regard to the expression of UPR genes, suggesting fundamental differences in the regulation of UPR and ER stress.
2. UPR signaling activates angiogenesis
UPR and Pro-inflammatory Pathways: Activation of NF-κB is well recognized as a major promoter of cancer hallmarks, including secretion of multiple pro-inflammatory and pro-angiogenic mediators, including IL-1, IL-6, and IL-8, which facilitate tumor growth and metastasis [21–24]. HIF-1α, ATF4, XBP1, and ATF6 are capable of binding to the VEGF promoter, thereby heightening VEGF expression [25–27]. VEGF secretion is induced by the UPR in human tumor cell lines, including neuroblastoma and medulloblastoma [26]. In medulloblastoma cell line, UPR activation induced VEGF, FGF2, angiogenin, and IL8 as measured by RT-PCR [28]. In the same model, VEGF mRNA stability was increased by UPR activation through activation of AMP kinase and increased secretion of VEGF [28]. In endothelial cells, ATF4 and XBP1 induce IL8, which in turn stimulates VEGF [29–31]. NF-κB, activated by various stressors, including infection and inflammation, serves as a stress sensor. Interestingly. the UPR, elicited in response to ER stress, also stimulates NF-κB activation and contributes to the pro-tumorigenic functions of the UPR. In mouse embryonic fibroblasts, IRE1, PERK, and IKK are required for full activation of NF-κB in response to ER stress [32]. ER stress activates NF-κB in an IRE1α-dependent mechanism [33]. Activated IRE1α interacts with tumor necrosis factor receptor associated factor 2 (TRAF2), leading to NF-κB [33] and JNK activation [34]. ATF6 is also shown to activate NF-κB in part through AKT phosphorylation [35]. ER stress also leads to Ca2+ release from ER resulting in oxidative stress and NF-κB activation [36]. Under conditions of glucose deprivation and hypoxia, A549/8 carcinoma cells and U87 glioma cells increase expression of VEGF [37]. This hypoglycemia/hypoxia induced VEGF induction is eliminated when the cells are transfected with a dominant negative IRE1 construct, suggesting 1) a link between activation of the UPR and angiogenesis effector genes and 2) the IRE1 activation may occur upstream of HIF-1a activation [37]. The latter is supported by the observation that dominant negative IRE1 carrying cells express less HIF-1a under conditions of hypoxia and hypoglycemia [37]. Consistently, dominant negative IRE1 carrying cells formed smaller tumors with decreased vascularity in vivo [37]. In a model of glioma, inhibition of IRE1 resulted in downregulation of several proangiogenic factors, including VEGF, IL1B, IL6, and IL8 [38]. In vivo, tumors expressing decreased IRE1 had decreased angiogenesis and tumor perfusion and a decreased growth rate [38]. In triple negative breast cancer (TNBC), inhibition of IRE1 reduced tumor growth in vivo, and synergized with anti-VEGF therapy, which is largely ineffective as monotherapy [39]. This suggests that IRE1 promotes angiogenesis and resistance to anti-angiogenics in TNBC.
PERK is widely considered tumor-protective and therefore a therapeutic target in hypoxic tumors, since activation of PERK increases glutathione synthesis to counter reactive oxygen species (ROS) encountered during hypoxia ([40] [41]). However, in squamous carcinoma T-Hep3 cells, which display low level PERK-eIF2a signaling, inducing PERK reduces proliferation, and therefore it may be possible that targeting PERK facilitate proliferation of slow-cycling tumor cells [42].
3. Angiogenic signaling activates UPR mediators
While several reports have focused on describing how UPR promotes angiogenesis, very few studies discussed on how increased angiogenesis promotes the UPR signaling. Angiogenesis, a key hallmark of cancer, is intimately linked to other hallmarks, including invasion, metastasis and sustained proliferation [43]. Classically, cancer is associated with an angiogenic switch that is mediated by changes in relative balance of inducers and inhibitors of angiogenesis, which activates otherwise quiescent vasculature to sprout new vessels [44]. Angiogenesis functions to increase nutrient and oxygen delivery to the tumor, resulting in its growth, hematogenous dissemination and metastasis.
VEGF is a critical proangiogenic factor secreted by tumor cells by activating autocrine and paracrine signaling. VEGF is upregulated in tumor cells primarily due to activation of HIF-1α because of hypoxia in the tumor microenvironment. Treatment of endothelial cells with VEGF activates ER stress sensors PERK, IRE1, and ATF6 in the absence of ER stress and protein misfolding [45]. VEGF induced ER signaling was independent of dissociation of BiP from PERK, IRE1, and ATF6. Rather, VEGF signals through PLCY and mTORC1 to activate the ER stress sensors, resulting in increased endothelial cell survival and angiogenesis [45]. VEGF promotes survival through ATF6- and PERK-mediated phosphorylation of AKT by mTORC2 [45].
In mouse embryonic fibroblasts, toll-like receptor (TLR) induces IRE1 activation through TRAF6 [46]. Loss of TRAF6 decreases IRE1 phosphorylation and XBP1s, but not IRE1 levels. Restoration of XBP1 partially restores pro-inflammatory and pro-angiogenic cytokines TNF-α and IL-6, suggesting that XBP1 functions to increase pro-inflammatory and pro-angiogenic cytokines [46]. These findings also suggest that angiogenic signaling through TRAF6 has the potential to trigger UPR pathways through IRE1.
The UPR couples angiogenic and survival signaling in cancer under various stress conditions, including hypoxia, high glucose dependence, ER stress, and amino acid (nutrient) deficiency, ultimately advancing tumor development and metastasis through neoangiogenesis.
4. Hypoxia and UPR crosstalk
Both ER stress and UPR are also involved in hypoxia and hypoxia-dependent cellular responses. Reduced tissue oxygen concentration is a hallmark of tumorigenesis. The high proliferation rate of tumor cells creates a hypoxic TME that increases oxygen demand. High energy demand of tumor cells enhances glycolytic flux particularly in harsh hypoxic environments [47]. Within the tumor, hypoxia, glucose and amino acid deprivation, and acidosis, among others, result in accrual of unfolded and misfolded proteins and initiation of UPR [48,49]. In tumor cells, the PERK/ATF4 arm of the UPR is activated in response to glucose deprivation and stimulates pro-angiogenic mediators including VEGF, FGF2, and IL6 [50]. In tumors, hypoxia also promotes pro-survival gene expression as part of adaptation to cellular stress [51]. Activation of HIF-1α upregulates target gene expression to enhance angiogenesis, metastasis, and metabolic reprogramming [52]. Accompanying this is VEGF-mediated neoangiogenesis, which promotes oxygen and nutrient delivery to the primary tumor and metastatic foci in secondary locations. In triple-negative breast cancer, XBP1 assembles a transcriptional complex with HIF-1α leading to hypoxic gene expression. Additionally, blockade of XBP1 reduced tumor growth, reduced relapse, and inhibited CD44high/CD24low cells, which represent tumor stem cells [53]. Under conditions of hypoxia, the PERK/eIF2α/ATF4 arm of the UPR, induces the metastasis-associated gene LAMP3 at both mRNA and protein levels, independent of HIF-1α [54]. The PERK/ATF4 arm also regulates autophagy through MAP1LC3B and ATG5, especially in hypoxic tumor regions [55]. Blockade of the tumor protective UPR-mediated autophagy sensitized tumors to hypoxia and irradiation and reduced cell viability [55].
Severe hypoxia triggers translocation of ATF6 into the nucleus [56,57]. It has been shown that ER stress and hypoxia-induced pathways intersect, with binding of HIF-1α, ATF4, XBP1, or ATF6 to the VEGF promoter [25], thereby heightening VEGF expression [26,27]. Interestingly, VEGF secretion depends on ER stress induced chaperone oxygen-regulated protein 150 (ORP150) [58]. Immunoprecipitation experiments also demonstrated that ORP150 binds the ER stress sensors [58]. The HIF-1α/VEGF axis has also been shown to be negatively regulated. For example, in breast cancer, ER stress is shown to activate transcription of microRNA-153 (miR-153) via the IRE1α/XBP1 axis, and increased miR153, in turn, limits both IRE1α activity and angiogenesis [59]. These seemingly polar responses demonstrate the complexity of UPR in modulating angiogenic responses. This also indicates heterogeneity in responses elicited from activation of ER stress sensor proteins ATF4 and IRE1α.
Binding of VEGF to VEGFR2 promotes VEGFR2 internalization in endothelial cells [1,60] (Fig. 1). Internalized VEGFR2 then heterodimerizes with IRE1α at the ER membrane, resulting in the activation of the XBP1 splicing complex [60]. It is important to note that these findings are quite unique and should be interpreted with care, especially since retrograde transport of receptor tyrosine kinases has been rarely described. XBP1 then activates the Akt/GSK/β-catenin axis to drive cell proliferation and growth by increasing VEGF transcription, translation, and secretion [1,20]. Thus, VEGF secreted by both tumor cells and endothelial cells act synergistically to promote vascularization via paracrine and autocrine mechanisms [61]. As reported, this phenomenon occurs in endothelial cells, however, it is unknown whether this holds true for malignancy.
Fig. 1.
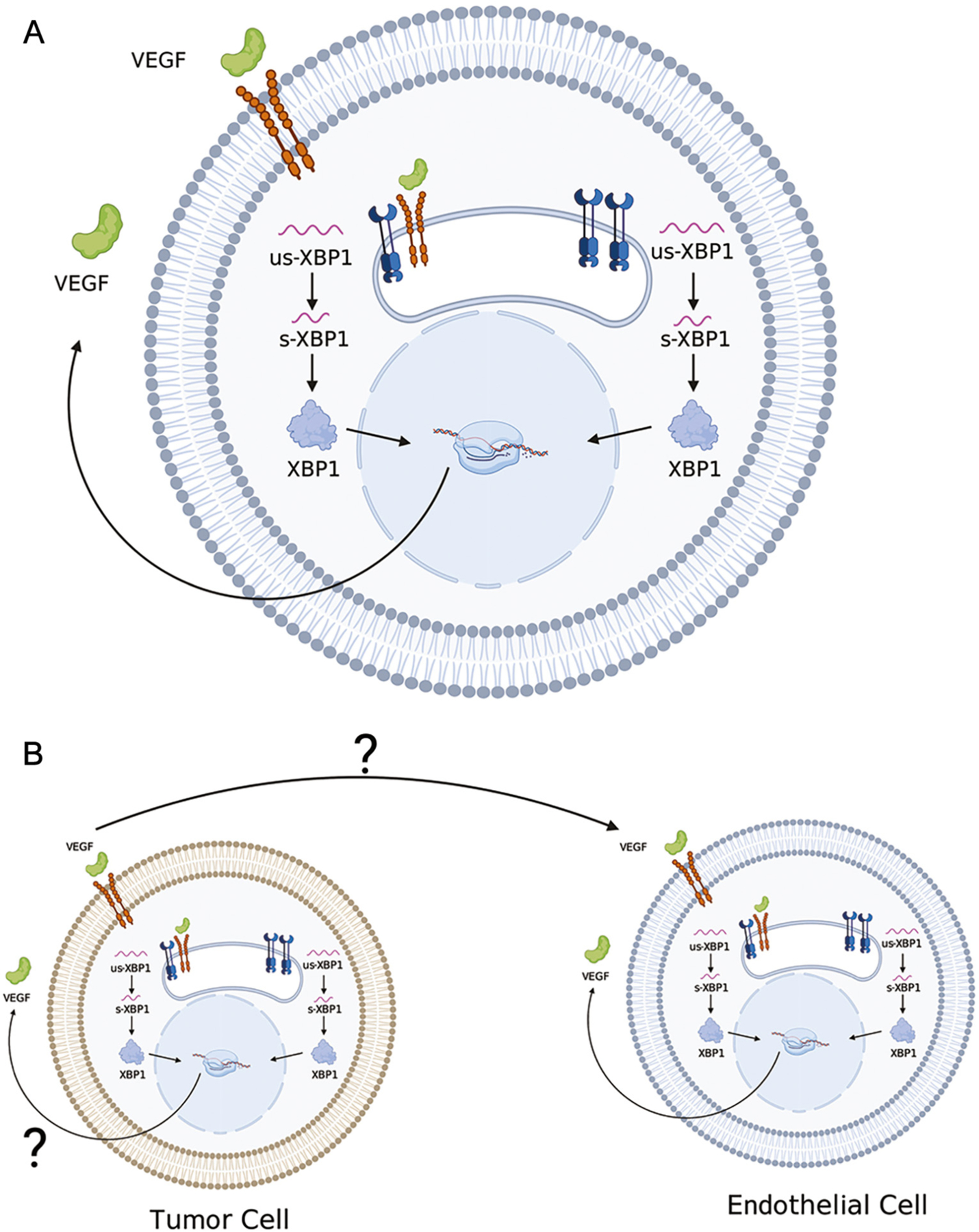
VEGF-IRE1α interactions in UPR. (A) In endothelial cells, binding of VEGF to the VEGF2R causes internalization of VEGFR2 [1]. Internalized VEGFR2 then heterodimerizes with IRE1α at the ER membrane and activates the XBP-1 splicing complex [1]. XBP1 also activates the Akt/GSK/bcatenin axis to drive cell proliferation and growth, and increased VEGF transcription, translation and secretion [1]. VEGF via membrane VEGFR2 activates signaling pathways that rapidly enhance angiogenesis. (B) Both tumor cell-derived VEGF and endothelial cell-derived VEGF may act via paracrine and autocrine mechanisms to promote vascularization (65, 66).
Nevertheless, we postulate that cancer cells secrete VEGF, transmit ER stress via exosomes to endothelial cells, activate a VEGF/VEGFR2/XBP1 signaling cycle within endothelial cells, and ultimately promote neoangiogenesis and metastasis (illustrated in Fig. 1).
VEGFR2 is overexpressed across various tumors, including breast cancer [62], cervical cancer [63], non-small cell lung cancer [64], and glioblastoma [65], among others. Therefore, it is plausible that activation of an autocrine VEGF/VEGFR2/XBP1 signaling could occur within tumor cells, resulting in reduced apoptosis due to activation of pro-survival pathways by the UPR, resulting in increased angiogenesis through XBP1-induced VEGF expression and secretion.
VEGF also induces angiogenesis and cell survival through the PERK and ATF6 arms of the UPR [20]. The binding of VEGF to VEGFR2 results in the activation of the PLCγ-mTORC1 cascade, leading to PERK and ATF6 activation, endothelial cell survival and angiogenesis [20,45]. Interestingly, this signaling event occurs in the absence of ER stress and canonical activation of the UPR [45]. This suggests a significant crosstalk between the UPR machinery and VEGF-induced signaling, and that the activation of UPR proteins occurs in response to pro-tumorigenic processes or microenvironmental conditions in the absence of ER stress.
In addition to angiogenesis, HIF-1α activation also plays a critical role in tumor metabolism. HIF-1α promotes metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis. Cancer cells require high energy levels, and preferentially utilize aerobic glycolysis to induce bioenergetically efficient mitochondrial oxidative phosphorylation (Warburg hypothesis) [66]. Through this metabolic shift, ATP production is less efficient per molecule of glucose. Therefore, cancer cells enhance the glycolytic pathway to meet ATP demands. Notably, glucose-6-phosphate produced in glycolysis can enter the pentose phosphate pathway to generate nucleotides for rapid cell proliferation. These cells also generate large amounts of lactate (necessary to regenerate NAD+ for continued glycolytic activity) that acidify the local microenvironment to enhance tumor invasion. Importantly, the increased uptake of glucose in cancer cells enhances the availability of carbon skeletons which can be used to generate amino acids [67,68]. In particular, the pyruvate dehydrogenase complex can generate acetyl-coA, the precursor for ketogenic amino acids. Gluconeogenic amino acids can be generated from a host of tricarboxylic acid cycle (TCA cycle) intermediates, including fumarate and oxaloacetate [69,70].
Altered metabolism and metabolic signaling in cancer are critical in meeting cellular requirements for protein synthesis and energy demand. In cancer, these changes result in the accumulation of raw biomolecules and carbon skeletons which enhance protein synthetic activity, in accordance with enhanced secretory phenotype of certain cancer cells [71,72]. As the flux through the secretory pathway increases, molecular chaperones become less available, thereby increasing the misfolding of protein products. At the same time, hypoxia exacerbates this condition by increasing ER stress through inhibition of proper protein folding. This results in a robust activation of the UPR to upregulate expression of molecular chaperones necessary for protein folding. Concomitantly, UPR also increases nutrient and oxygen delivery through neoangiogenesis. Thus, the tumor cell can mitigate hypoxia to a variable degree by enhancing the ER protein folding capacity through upregulation of molecular chaperones. Combined with an enhanced nutrient (e.g., glucose and amino acids) delivery, the tumor cell can enhance the secretion of protein products. However, the factors that limit a tumor cell’s ability to increase protein folding remain to be fully elucidated.
Cellular stress related to dysregulated protein metabolism may activate the Amino Acid Response (AAR) and/or the UPR. The AAR is activated in response to low amino acid availability extracellularly. AAR results in the in phosphorylation of eIF2α through activation of Eukariotic Translation Initiation Factor Alpha Kinase 4, also known as GCN2 for General Control Nondepressible 2 [73]. The ATF4/PERK arm of the UPR is classically activated in response to unfolded proteins intracellularly (endogenous stress) [74]. The AAR and UPR both converge on the phosphorylation of eIF2α resulting in a global halt of protein translation, with the exception of ATF4, whose translation is upregulated. ATF4 transcriptionally regulates target genes which function in angiogenesis.
5. Transmissible ER stress
Further, the TME can serve as a transmitter of ER stress to neighboring cells, resulting in the perpetuation of ER stress-induced pro-tumorigenic responses, a phenomenon related to transmissible ER stress (TERS) [75,76]. TERS corresponds to release of proteotoxic entities by stressed cells that act on target neighbor cells. Macrophages cultured in conditioned media from tumor cells under ER-stress tumor cells promoted ER stress in those macrophages [77]. Spliced XBP1 mRNA, the product of ER stress mediated IRE1 activation, has been shown accumulate in exosomes. TME mediated transmission of ER stress also decreases immune capacity, such as decreased cytotoxic T cell response, a hallmark commonly observed in cancers [78]. In fact, pancreatic cancer derived exosomes induce apoptosis of T-lymphocytes through induction of ER stress via p38 MAPK induced CHOP, and upregulation in pro-inflammatory and pro-angiogenic cytokine expression, including IL-1α and IL-1β [79].
TERS may also correspond to transmission of stress signaling components produced downstream of ER stress in the originating stressed cell, and subsequently transmitted, such as in extracellular vesicle/exosomes to target cells, where de novo signaling is elicited. In addition to soluble factors [77], exosomes are important constituents of the TME. In fact, exosomes are shown to play a role in UPR activation [80], while ER stress itself increases formation of exosomes [81] (Fig. 2). These membrane-bound structures released by tumor cells greatly influence metastatic, angiogenic, and proliferative properties of cells within the TME [75]. We previously showed that the quantity of exosomes are increased in malignant cells [82]. We also showed that malignant cell-derived exosomes contain oncoproteins and microRNAs [83]. Interestingly, ER stress increases the production and release of exosomes in cancer via PERK- and IRE1α-dependent mechanisms [75].
Fig. 2.
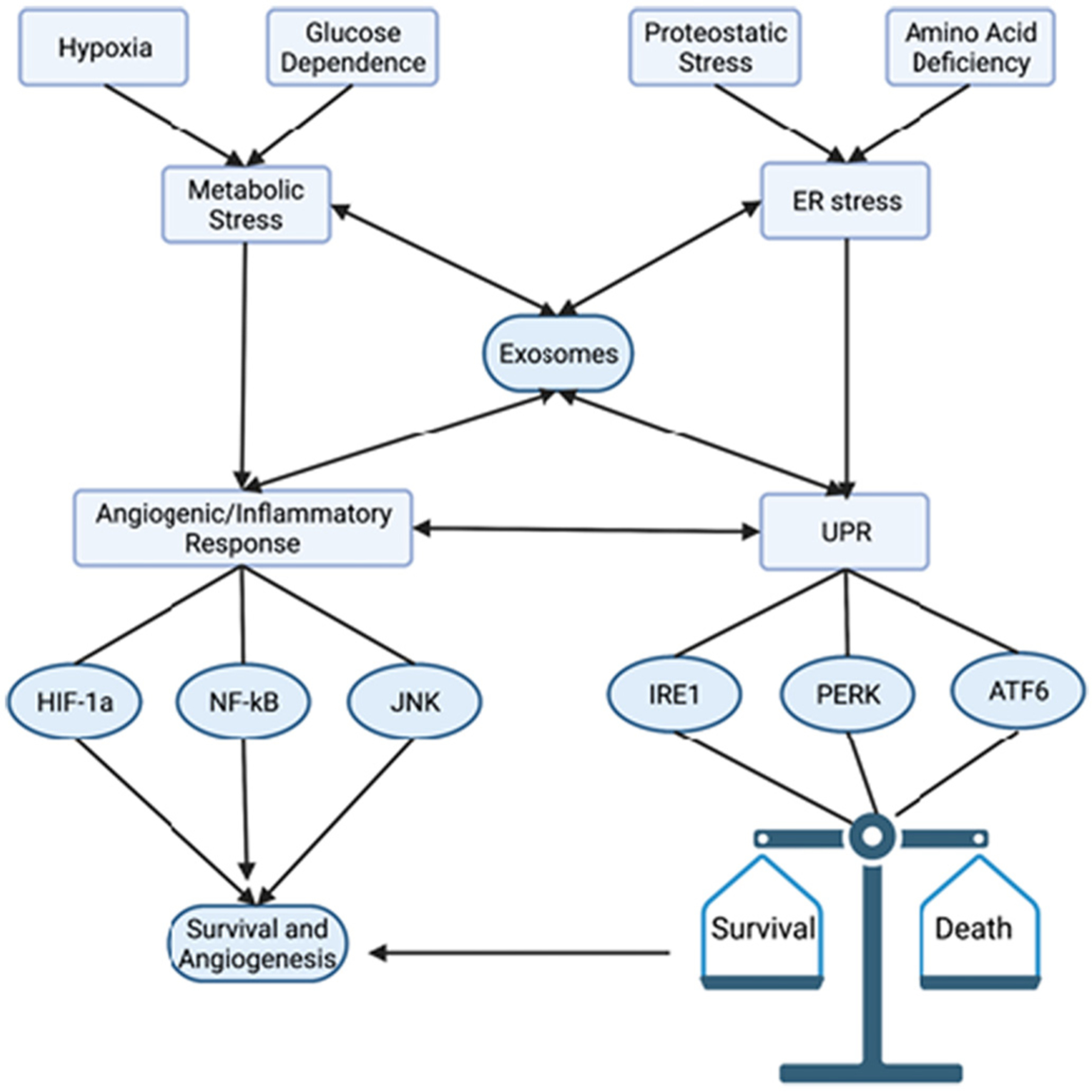
ER stress and Angiogenesis in Cancer. Due to their rapid growth, cancer cells encounter several cellular stresses including hypoxia, low availability of glucose, proteostatic stress, and amino acid deficiencies. These stresses activate a generalized inflammatory response, leading to HIF-1a, NF-kB, and JNK (among other pro-inflammatory transcription factors) activation. This increases angiogenesis, nutrient delivery, and initiates survival signaling. Proteostatic (ER) stress and amino acid deficiency lead to activation of the UPR, which may lead to cell survival or death. Importantly, these cellular stress events may be communicated between cancer cells and non-malignant stromal cells in various mechanisms involving exosomes (discussed in text). Typically, the pro-survival outcome of the UPR corresponds to increased angiogenic signaling and exhibits significant synergy and crosstalk with the generalized inflammatory response.
ER stress markers GRP78, ATF6, PERK, and IRE1 were shown to be upregulated in hepatocellular carcinoma (HCC) and negatively correlated with overall survival [84]. Further, increased expression of these ER stress markers was correlated with increased macrophage recruitment and PD-L1 expression in HCC tissue [84], both properties associated with poor anti-tumor immune response [85,86]. Indeed, TERS has been shown to alter anti-tumor immune responses. For instance, TERS can activate TAMs which induce inflammation while promoting immunosuppression of the tumor microenvironment that inhibits anti-tumor T cells [48,77].
Exosomes derived from ER stressed liver cancer cells also activated JAK2/STAT3 to increase expression of pro-inflammatory and pro-angiogenic cytokines such as TNF-α, MCP-1, and IL-6 in macrophages [87]. This suggests that the anti-cancer efficacy of JAK/STAT inhibitors may be due to the inhibitory effect on ER stress induced angiogenesis and inflammation. In breast cancer, ER stress induced exosomal miR-27a-3p also promoted tumor progression and blocked anti-tumor immunity via upregulation of PD-L1 through the PI3K signaling pathway, which is also critical in driving breast cancer angiogenesis through inducing HIF-1α and VEGF expression [88]. In fact, EVs from human umbilical cord mesenchymal stromal cells have shown to protect cardiac cells from ischemia/reperfusion injury via PI3K-mediated suppression of ER stress [89]. Exposure of human umbilical vein endothelial cells (HUVEC) to HeLa cell-derived exosomes decreased levels of zonula occludens-1 (ZO-1) and Claudin-5, which are tight junction proteins responsible for the structural integrity of the endothelial layer [90]. This resulted in significantly increased permeability of the endothelial layer. These changes were attributed to PERK activity, since knockdown of PERK prevented HeLa-exosomes induced decrease in ZO-1 and Claudin 5. These findings suggest that UPR signaling plays a critical role not only in driving angiogenesis, but also inducing changes in the vasculature including altered permeability which may facilitate tumor dissemination [90]. Also, bladder cancer-derived extracellular vesicles (EVs) are 5 shown to induce the UPR in non-malignant cells, resulting in their transformation and acquisition of oncogenic properties, including genomic instability, loss of cell-cell contact inhibition, and invasiveness. These findings suggest that tumor cells have characteristic ER stress and UPR signatures that favor the pro-tumorigenic and pro-survival UPR outcome and block the CHOP-mediated cell death UPR, and that EV-mediated transmission of this signaling can promote oncogenesis, tumor growth, and invasion [91].
Mesenchymal stem cell (MSC)-derived exosomes are shown to protect beta cells against hypoxia-induced apoptosis by alleviating ER stress through miRNA-21 (miR-21) activity. Hypoxia induces important UPR genes including GRP78, GRP94, p-eIF2α and CHOP as well as p38 MAPK signaling, which were downregulated by exposure to MSC exosomes. While this phenomenon was studied mostly in diabetes, hypoxia-induced ER stress and UPR activation also occur in cancer, and it is not unlikely that tumor interaction with MSCs [92] may modulate the UPR via exosomes. In human epidermal growth factor receptor 2-positive (HER2+) gastric cancer, ER stress was shown to induce resistance to trastuzumab (a monoclonal antibody that targets HER2) through upregulation of miR-301a-3p, which in turn downregulated LRIG1 (leucine-rich repeats and immunoglobulin-like domains containing protein 1) and subsequent activation of insulin-like growth factor 1 receptor (IGF-1R) and fibroblast growth factor receptor 1 (FGFR1) expression, resulting in trastuzumab resistance [93].
Transmissible ER stress may also play a role in response to chemotherapy and immunotherapy. Cancer cachexia, a wasting syndrome seen in states of chronic inflammation due to significantly increased levels of pro-inflammatory cytokines such as TNF-α, confers a suboptimal response to chemotherapy. However, increased chemokine levels contribute to higher immune cell infiltration that may exert anti-tumor effects. In fact, the UPR is central to a robust anti-tumor immune response since it leads to calreticulin transport to the cell surface where it facilitates phagocytosis of tumor cells by macrophages. Transmitted ER stress also increases production of pro-inflammatory and pro-angiogenic cytokines, such as IL-6, which lead not only to resistance to cellular stress but an adaptive increase in vascularization to alleviate nutrient deprivation and stress. It has also been proposed that this transmitted ER stress may serve as a rescue mechanism for the tumor to alleviate cellular stress, all while increasing the immunosuppressive nature of the TME [94].
On the other hand, the efficacy of certain anti-cancer treatments may be improved by activation of the pro-apoptotic arm of the UPR. For example, PPZ023, a novel Peroxisome proliferator-activated receptor gamma (PPARɣ) agonist was recently shown to exert powerful anti-cancer effects by suppressing tumor growth and in overcoming radioresistance through the activation of the PERK-eIF2α-CHOP axis [95]. Cell death was blocked by an inhibitor of NADPH oxidase (Nox), which is a critical generator of ROS, suggesting the contribution of oxidative stress to cell death [95]. A novel herbal extract (JI017) has been shown to increase oxidative stress through ROS and calcium, while also increasing markers of ER stress including p-PERK, p-eIF2α, ATF4, and CHOP, via activation of both exosomal GRP78 and cell lysate GRP78, resulting in cellular death in breast cancer cell lines [96]. Further, ROS inhibitors diphenyleneiodonium and N-acetyl cysteine blunt apoptosis and excessive ER stress through suppression of Nox4, suggesting oxidative stress as a major contributor to cell death. JI017 also reduced resistance to paclitaxel in breast cancer through the blockade of the epithelial to mesenchymal transition (EMT) as well as a decrease in HIF-1α, a potent driver of hypoxia-induced angiogenesis [96]. JI017 has also been shown to induce apoptosis via the NOX4-PERK-CHOP axis in ovarian cancer [97].
6. UPR and angiogenesis pathways differ in malignant vs non-malignant tissue
Data from The Cancer Genome Atlas (TCGA) was accessed through GEPIA (http://gepia.cancer-pku.cn). We analyzed the expression of genes involved in UPR (Fig. 3), angiogenesis (Fig. 4), or both (Fig. 5) in tumors and non-malignant counterparts with available data. Gene ontology enrichment analysis was performed on each gene list to verify whether gene signatures represent.
Fig. 3.
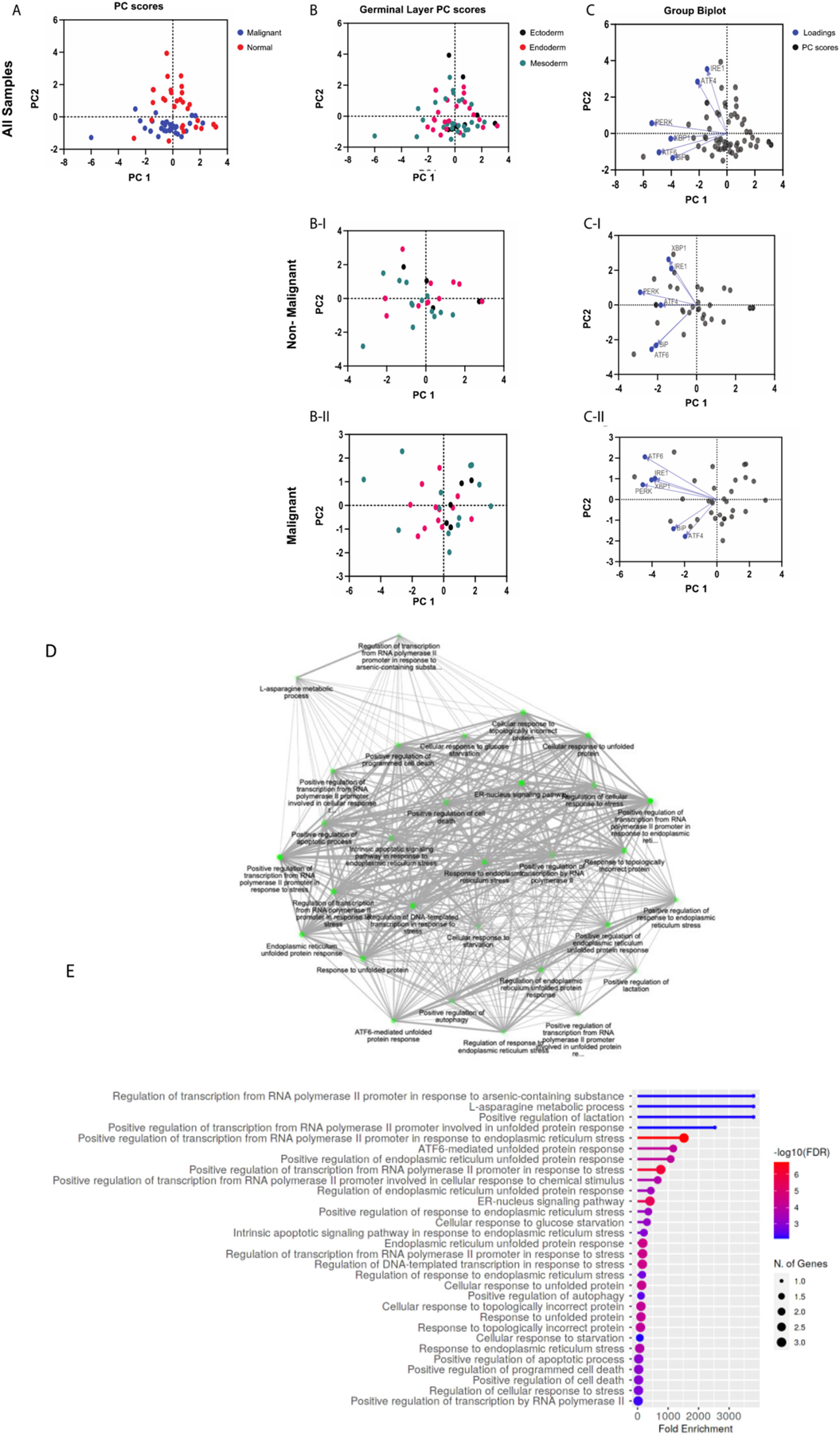
Principal Component Analysis of UPR genes. Gene expression data from The Cancer Genome Atlas (TCGA) was accessed through GEPIA (http://gepia.cancer-pku.cn). Gene expression levels are the median of the cohort, and both tumor samples and matched normal samples were analyzed. (A) PC score plot for malignant versus normal samples (normal samples = red dots; malignant samples = blue dots). PC score plots for non-malignant (B–I) and malignant (B-II) samples based on embryological origin (germinal layer; ectoderm = black dots, endoderm = red dots, mesoderm = green dots). Biplots with PC scores (black dots) and loadings (blue dots) for genes from non-malignant (C–I) and malignant (C-II) samples. Gene ontology enrichment analysis was performed on each gene list to verify that the gene signatures represented UPR pathways (Shiny GO v0.741; bioinformatics.sdstate.edu) (D and E).
Fig. 4.
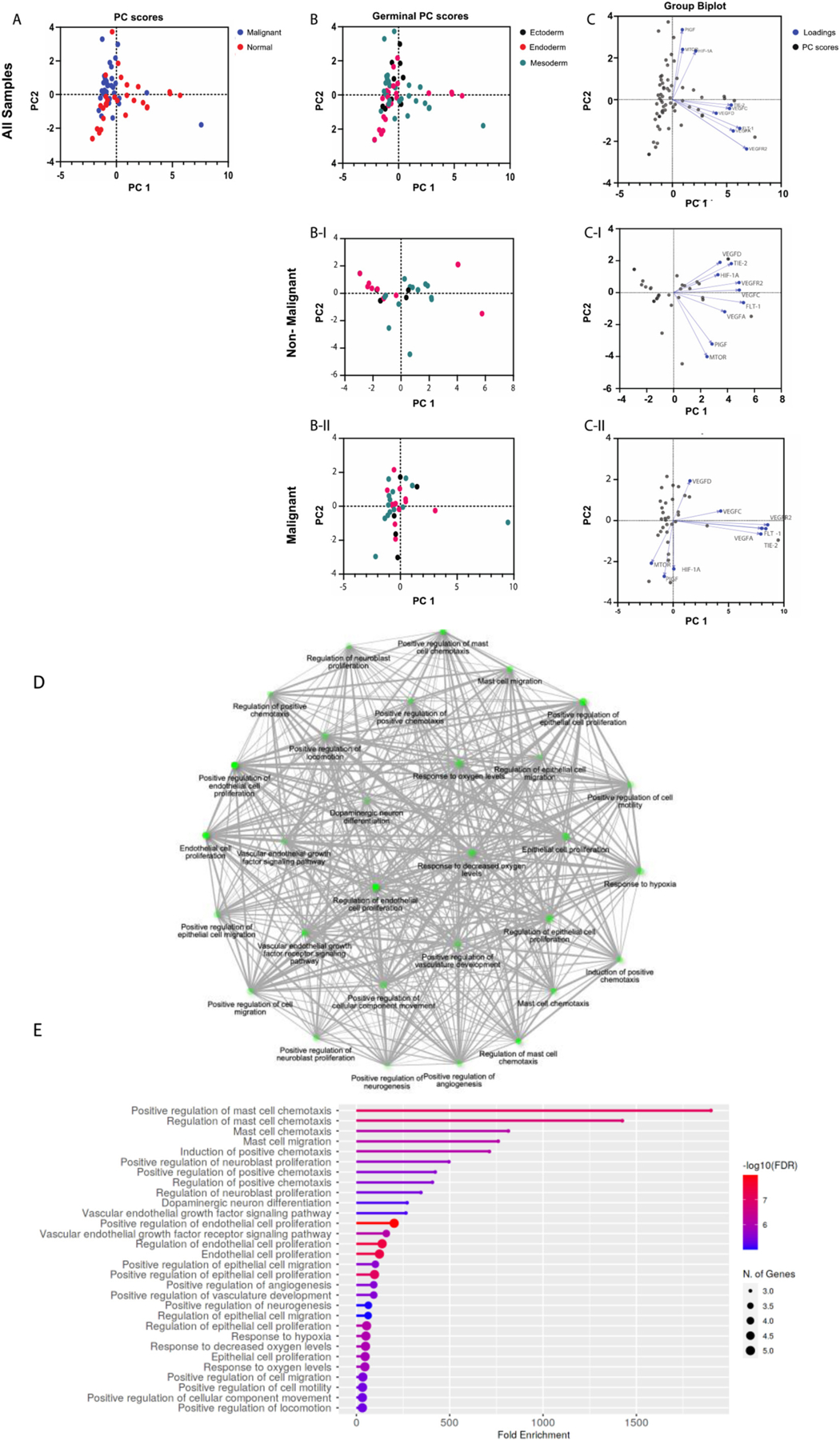
Principal Component Analysis of Angiogenesis genes. Gene expression data from The Cancer Genome Atlas (TCGA) was accessed through GEPIA (http://gepia.cancer-pku.cn). Gene expression levels are the median of the cohort, and both tumor samples and matched normal samples were analyzed. (A) PC score plot for malignant versus normal samples (normal samples = red dots; malignant samples = blue dots). PC score plots for non-malignant (B–I) and malignant (B-II) samples based on embryological origin (germinal layer; (germinal layer; ectoderm = black dots, endoderm = red dots, mesoderm = green dots). Biplots with PC scores (black dots) and loadings (blue dots) for genes from non-malignant (C–I) and malignant (C-II) samples. Gene ontology enrichment analysis was performed on each gene list to verify that the gene signatures represented angiogenesis pathways (Shiny GO v0.741; bioinformatics.sdstate.edu) (D and E).
Fig. 5.
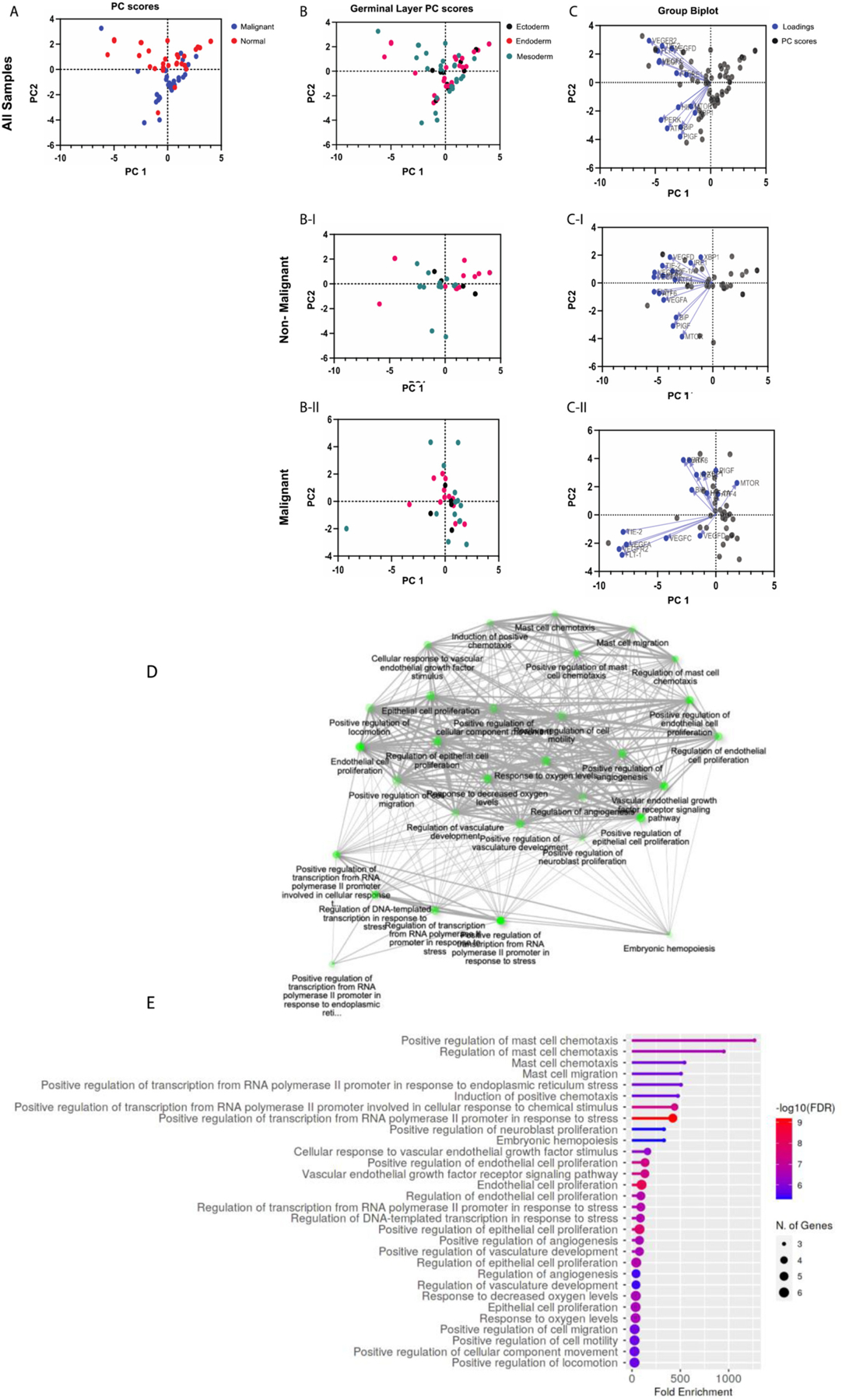
Principal Component Analysis of UPR and Angiogenesis genes. Gene expression data from The Cancer Genome Atlas (TCGA) was accessed through GEPIA (http://gepia.cancer-pku.cn). Gene expression levels are the median of the cohort, and both tumor samples and matched normal samples were analyzed. (A) PC score plot for malignant versus normal samples (normal samples = red dots; malignant samples = blue dots). PC score plots for non-malignant (B–I) and malignant (B-II) samples based on embryological origin (germinal layer; ectoderm = black dots, endoderm = red dots, mesoderm = green dots)). Biplots with PC scores (black dots) and loadings (blue dots) for genes from non-malignant (C–I) and malignant (C-II) samples. Gene ontology enrichment analysis was performed on each gene list to verify that the gene signatures represented UPR and angiogenesis pathways (Shiny GO v0.741; bioinformatics.sdstate.edu) (D and E).
UPR and/or pro-angiogenic pathways (Shiny GO v0.741; bioinformatics.sdstate.edu). Results of pathway enrichment analysis are shown in Fig. 3 D–E for UPR genes, Fig. 4 D–E for angiogenesis genes, and Fig. 5 D–E for both. Gene expression levels are the median of the cohort, and both tumor samples and matched normal samples were analyzed (Fig. 3A, B-I, B-II, C-I, and C-II).
We utilized GraphPad Prism for the analysis. Dimensionality reduction was achieved through principal component analysis (PCA) performed on the data to identify trends and clusters. The PCA process performed by GraphPad Prism includes standardization of the data such that each variable has a mean of zero and standard deviation of 1, and a variance of 1. Principal components and their eigenvalues were calculated and selected for analysis. When analyzed for UPR genes, malignant and normal samples cluster distinctly, along principal component 2 (PC2). When analyzed for both UPR and angiogenesis genes, the distinct clustering between was diminished (Fig. 3 C-I, C-II, and Fig. 4 C-I, C-II). When analyzed for angiogenesis genes alone, there was a poor differentiation between malignant and normal samples. In all 3 analyses, no notable clustering was identified based on embryological germinal layer (Fig. 3 B, 4 B, 5 A–D). Biplots were constructed to identify clustering of genes. Among the UPR genes (Fig. 3) in nonmalignant samples, XBP1 and IRE1 are highly correlated (their vectors form a ~ 0-degree angle), ATF4 and PERK are highly correlated, and ATF6 and BiP are highly correlated.
Interestingly, this patterning of clustering seems to mirror the UPR signaling axes (Fig. 3 C-I). In malignant samples, however, this distinct clustering is altered: PERK and ATF4 seem to be minimally correlated (their vectors form a ~ 90-degree angle) and ATF6 and BiP have minimal correlation (Fig. 3 C-II). Among angiogenesis genes (Fig. 4), PIGF and mTOR were highly correlated in both non-malignant and malignant samples. However, in contrast to non-malignant samples, in malignant samples HIF-1α was highly correlated with PIGF and mTOR. Since mTOR regulates HIF-1α, it is interesting to speculate that this interaction in enhanced in malignancy. Further, in malignant samples, VEGFR2, FLT-1, TIE-2, and VEGFA showed a tighter correlation compared to non-malignant samples (Fig. 4 C-II). Among the combined UPR and angiogenesis genes (Fig. 5), malignant samples displayed two distinct clusters: a) a cluster with TIE-2, VEGFA, VEGFR2, and FLT-1, and b) a cluster with UPR genes and notably HIF-1α (Fig. 5 C-II).
PCA is a powerful tool for dimensionality reduction of multidimensional datasets and identification of trends. We grouped the gene expression data from all malignancies in the malignant group. While this facilitates identification of global trends in gene expression, we do acknowledge that it obscures meaningful dissection of UPR and angiogenesis pathways in a specific cancer and consider it as a weakness of the approach. Nevertheless, it is important to note that malignant and non-malignant samples cluster distinctly with respect to UPR and pro-angiogenesis genes. This confirms and strengthens the possibility that UPR and angiogenesis pathways are perturbed or altered in cancer, and therefore, could be targeted as potential therapies in cancer.
Drugs Modulating UPR and Angiogenesis in Cancer
An impressive battery of pharmacologic modulators of ER stress and the UPR have been developed and reviewed extensively elsewhere [98–100]. Here, we focus on notable, selected clinical drugs to emphasize the intersection of the UPR/ER stress and angiogenesis in cancer. Sunitinib, a small molecule, inhibits IRE1α kinase activity by binding to the ATP-binding pocket, which is necessary for IRE1α ribonuclease activity [101,102]. It has also been shown to inhibit XBP1 splicing in vivo [101,102]. However, there is a mixed evidence as to whether sunitinib inhibits or activates the IRE1α RNase activity [103,104], and how this influences downstream effectors, such as XBP1, especially in vivo remains unknown and needs further investigation [102,105]. The mechanism of action of sunitinib emphasizes complex regulation of the IRE1α complex, and suggests that potential inhibitors of IRE1α-mediated activity need to be targeted and selective, as it is still possible for a putative inhibitory drug to inhibit phosphorylation but yet activate RNase activity [102]. Notably, sunitinib also inhibits multiple receptor tyrosine kinases, including VEGFR1–3, PDGFRA, PDGFRB, Kit, FLT-4, and CSF-1R, accounting for its ability to inhibit angiogenesis and stabilize the vascular endothelium in cancer [106]. Therefore, the anti-angiogenic effects of sunitinib may be a synergistic combination of its effects on angiogenic cytokine receptor expression, the UPR and ER stress. Sunitinib is currently FDA approved for treatment of renal cell carcinoma, gastrointestinal stromal tumors and pancreatic neuroendocrine tumors [107].
Sorafenib is an FDA approved small molecule inhibitor of intracellular tyrosine and serine/threonine kinases. It acts via two mechanisms: i) inhibition of kinase activity, resulting in decreased RAF, PDGF, and VEGF receptor signaling, and ii) inhibition of vasolin containing protein (VCP, also known as p97 or Cdc48p in yeast), which is an AAA+ ATPase regulating endoplasmic reticulum–associated degradation (ERAD) [102,108]. VCP functions in the ERAD pathway to expel misfolded proteins from the ER for proteasomal degradation [102,108]. Sorafenib treatment results in accumulation of proteins within the ER and activation of the UPR pathway to induce cancer cell death [102]. This mechanism is analogous to the use of bortezomib for the treatment of multiple myeloma. Bortezomib, a 26S proteasome inhibitor, results in accumulation of excess protein within the ER. As proteostatic stress increases, the pro-apoptotic arm of the UPR, involving PERK, ATF4 and CHOP, is activated resulting in the death of myeloma cells.
It is worth mentioning that Sorafenib is approved primarily for its anti-angiogenic properties. Sorafenib demonstrates high efficacy in renal cell carcinoma and hepatocellular carcinoma [109,110]. Since both cancers rely heavily on hematogenous routes for metastasis, they are sensitive to anti-angiogenic drugs. In fact, bevacizumab, an anti-VEGF antibody, is FDA approved for both cancers [111–113]. Since UPR mediators regulate pro-tumorigenic angiogenesis, drugs developed as UPR modulators or anti-angiogenics might have trans-pathway effects, which explain their efficacy and multimodal mechanisms of action.
The preclinical drugs STF-083010, 4u8C, MKC-3946, and Toyocamycin have all been shown to inhibit splicing of XBP1 [114–117]. MG-132 is the preclinical analog of bortezomib; both share a common mechanism of action of inhibiting the 26S proteosome [118–120]. Eeyarestatin, ML240, and DBeQ are preclinical drugs which inhibit VCP and therefore prevent ERAD, causing increased proteostatic stress and reduced tumor cell proliferation [121–123].
7. Conclusion and future directions
Triggered primarily by proteostatic stress, the UPR is an adaptive orchestrated signaling cascade that serves the alleviate the inciting stress. As we discussed, the UPR has significant crosstalk with other stress-induced pathways, particularly angiogenesis, with several examples of UPR-mediated angiogenesis, as well as angiogenesis-mediated ER stress sensor activation. Exosome-mediated communication represents an additional mechanism by which ER stress may be transmitted, with important effects in the context of tumor angiogenesis. Importantly, under conditions of severe stress, the UPR has the paradoxical ability to initiate cell death. While this makes modulating the UPR attractive translationally, it also complicates the therapeutic strategy since promoting the UPR may also increase pro-tumorigenic properties such as angiogenesis. Therefore, the critical need is development of a strategy to modulate the UPR in cancer to prevent angiogenic crosstalk and simultaneously promote the pro-death functions of the UPR. While we focus on angiogenesis here, it is well recognized that activation of UPR in cancer promotes many other hallmarks of cancer.
Clinically, many anti-cancer drugs have been shown to exert multimodal secondary mechanisms of action, including modulation of UPR. Therefore, it is critical to determine whether the current anti-cancer drugs and preclinical drugs also modulate UPR. Through these investigations, one can elucidate the clinical role of UPR signaling in tumor growth and metastasis.
In this comprehensive review, we discussed the following novel ideas, which may help guide future preclinical, pharmaceutical, and clinical investigations:
Based on gene expression analysis from TCGA, the UPR is fundamentally different between malignancies and their non-malignant counterparts. However, our analysis was unable to identify a role for embryologic origin in this difference.
We discuss the intersection between pro-tumorigenic metabolic and angiogenic pathways and UPR signaling, concluding that the UPR machinery contains key mediators which are utilized by cancer cells to drive tumor growth and metastasis.
We discuss recent updates in transmissible ER stress, particularly through exosomes, and its role in driving tumor growth by regulating angiogenesis and vascular permeability.
We connect malignant metabolism to tumor microenvironment and ER stress, and explain the metabolic origins and consequences of ER stress in cancer.
We briefly discuss key anti-cancer drugs with UPR-modulating activity, and connect their clinical applications to tumor metastatic and angiogenic behavior through UPR signaling.
These novel insights may trigger a new angle in translational UPR research that will ultimately lead to development of more effective treatments for malignancy and overall survival.
Acknowledgments
This project is supported through grants from the Alliance of Cardiovascular Researchers (to AI, KW, RI and to EA). Additionally, RI received a grant for this study from the Elsa U. Pardee Foundation. Also, this work was supported in part by a grant from the National Institutes of Health (P30GM145498) to RI. BC is a recipient of the Department of Veterans Affairs Senior Research Career Scientist award (BX004016), and is supported by the U.S. Department of Veterans Affairs, Office of Research and Development-Biomedical Laboratory Research and Development (ORD-BLRD) Service Award BX005845 and NIDDK R01 DK130243. The authors would like to specially thank and express their gratitude to Hamid Izadpanah for the graphical and art works.
Abbreviations:
- Akt
AKT Serine/Threonine Kinase
- ATF
Activating Transcription Factor
- BiP
Binding immunoglobulin protein
- bZIP
Basic leucine zipper
- CHOP
CCAAT- enhancer-binding protein homologous protein
- COP-II
Coat protein II
- CSF-1R
Colony Stimulating Factor 1 Receptor
- EIF2α
Eukaryotic translation initiator factor-2 α
- EIF2AK3
Eukaryotic Translation Initiation Factor 2 Alpha Kinase 3
- ERAD
Endoplasmic reticulum–associated degradation
- ERN1
Endoplasmic Reticulum to Nucleus Signaling 1
- ER
Endoplasmic Reticulum
- ER+ Breast Cancer
Estrogen receptor+ (ER+) breast cancer
- EVs
Extracellular Vesicles
- FLT-1
Fms Related Receptor Tyrosine Kinase 1
- GSK
Glycogen Synthase Kinase
- HIF-1α
Hypoxia Inducible Factor α
- IRE1α
Inositol Requiring Enzyme 1 α
- Kit
KIT Proto-Oncogene, Receptor Tyrosine Kinase
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- mTOR
Mechanistic Target Of Rapamycin Kinase
- OXPHOS
Oxidative phosphorylation
- PDI
Protein Disulfide Isomerase
- PDGFRa
Platelet Derived Growth Factor Receptor Alpha
- PERK
Protein Kinase RNA (PKR)-like ER Kinase
- PIGF
Phosphatidylinositol Glycan Anchor Biosynthesis Class F
- PLCg
Phospholipase Cg
- S1P
site-1 protease
- S2P
site-2 protease
- SERCA2a
ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2 + Transporting 2
- TCA cycle
Tricarboxylic acid cycle
- TIE-2
Tyrosine Kinase With Immunoglobulin Like And EGF Like Domains 2
- TME
Tumor microenvironment
- TNBC
Triple negative breast cancer
- UPR
Unfolded Protein Response
- VCP
Vasolin Containing Protein
- VEGFA
Vascular Endothelial Growth Factor A
- VEGFR2
Vascular Endothelial Growth Factor Receptor 2
- XBP1
X-Box Binding Protein 1
- XBP1s
Spliced XBP1
Footnotes
Author disclosure statement
Authors have declared no conflict of interest.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data will be made available on request.
References
- [1].Zeng L, Xiao Q, Chen M, Margariti A, Martin D, Ivetic A, Xu H, Mason J, Wang W, Cockerill G, Mori K, Li JY, Chien S, Hu Y, Xu Q, Vascular endothelial cell growth-activated XBP1 splicing in endothelial cells is crucial for angiogenesis, Circulation 127 (16) (2013) 1712–1722. [DOI] [PubMed] [Google Scholar]
- [2].Chevet E, Hetz C, Samali A, Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis, Cancer Discov. 5 (6) (2015) 586–597. [DOI] [PubMed] [Google Scholar]
- [3].Urra H, Dufey E, Avril T, Chevet E, Hetz C, Endoplasmic reticulum stress and the hallmarks of cancer, Trends Cancer 2 (5) (2016) 252–262. [DOI] [PubMed] [Google Scholar]
- [4].Li H, Korennykh AV, Behrman SL, Walter P, Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering, Proc. Natl. Acad. Sci. U. S. A 107 (37) (2010) 16113–16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hetz C, Papa FR, The unfolded protein response and cell fate control, Mol. Cell 69 (2) (2018) 169–181. [DOI] [PubMed] [Google Scholar]
- [6].Walter P, Ron D, The unfolded protein response: from stress pathway to homeostatic regulation, Science 334 (6059) (2011) 1081–1086. [DOI] [PubMed] [Google Scholar]
- [7].Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG, Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast, Cell 68 (3) (1992) 585–596. [DOI] [PubMed] [Google Scholar]
- [8].Vattem KM, Wek RC, Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells, Proc. Natl. Acad. Sci. U. S. A 101 (31) (2004) 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lu PD, Harding HP, Ron D, Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response, J. Cell Biol 167 (1) (2004) 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yaman I, Fernandez J, Liu H, Caprara M, Komar AA, Koromilas AE, Zhou L, Snider MD, Scheuner D, Kaufman RJ, Hatzoglou M, The zipper model of translational control: a small upstream ORF is the switch that controls structural remodeling of an mRNA leader, Cell 113 (4) (2003) 519–531. [DOI] [PubMed] [Google Scholar]
- [11].Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I, The role of the PERK/eIF2alpha/ATF4/CHOP Signaling pathway in tumor progression during endoplasmic reticulum stress, Curr. Mol. Med. 16 (6) (2016) 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kranz P, Neumann F, Wolf A, Classen F, Pompsch M, Ocklenburg T, Baumann J, Janke K, Baumann M, Goepelt K, Riffkin H, Metzen E, Brockmeier U, PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR), Cell Death Dis. 8 (8) (2017) e2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oka OB, van Lith M, Rudolf J, Tungkum W, Pringle MA, Bulleid NJ, ERp18 regulates activation of ATF6alpha during unfolded protein response, EMBO J. 38 (15) (2019) e100990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gardner BM, Walter P, Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response, Science 333 (6051) (2011) 1891–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K, Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins, J. Cell Biol 179 (1) (2007) 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang P, Li J, Tao J, Sha B, The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization, J. Biol. Chem 293 (11) (2018) 4110–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Szegezdi E, Fitzgerald U, Samali A, Caspase-12 and ER-stress-mediated apoptosis: the story so far, Ann. N. Y. Acad. Sci 1010 (2003) 186–194. [DOI] [PubMed] [Google Scholar]
- [18].Nakagawa T, Yuan J, Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis, J. Cell Biol 150 (4) (2000) 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ma Y, Hendershot LM, The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4 (12) (2004) 966–977. [DOI] [PubMed] [Google Scholar]
- [20].Binet F, Sapieha P, ER Stress and Angiogenesis, Cell Metab. 22 (4) (2015) 560–575. [DOI] [PubMed] [Google Scholar]
- [21].Mohankumar K, Francis AP, Pajaniradje S, Rajagopalan R, Synthetic curcumin analog: inhibiting the invasion, angiogenesis, and metastasis in human laryngeal carcinoma cells via NF-kB pathway, Mol. Biol. Rep 48 (8) (2021) 6065–6074. [DOI] [PubMed] [Google Scholar]
- [22].Batlle R, Andres E, Gonzalez L, Llonch E, Igea A, Gutierrez-Prat N, Berenguer-Llergo A, Nebreda AR, Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38alpha through TGF-beta and JNK signaling, Nat. Commun 10 (1) (2019) 3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tam SY, Law HK, JNK in tumor microenvironment: present findings and challenges in clinical translation, Cancers (Basel) 13 (9) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Taniguchi K, Karin M, NF-kappaB, inflammation, immunity and cancer: coming of age, Nat. Rev. Immunol. 18 (5) (2018) 309–324. [DOI] [PubMed] [Google Scholar]
- [25].Sengupta S, Sharma CG, Jordan VC, Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells, Horm. Mol. Biol. Clin. Invest 2 (2) (2010) 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pereira ER, Frudd K, Awad W, Hendershot LM, Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF), J. Biol. Chem 289 (6) (2014) 3352–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F, Transcriptional regulation of VEGF-A by the unfolded protein response pathway, PLoS One 5 (3) (2010) e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pereira ER, Liao N, Neale GA, Hendershot LM, Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response, PLoS One 5 (9) (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ, The unfolded protein response is an important regulator of inflammatory genes in endothelial cells, Arterioscler. Thromb. Vasc. Biol 26 (11) (2006) 2490–2496. [DOI] [PubMed] [Google Scholar]
- [30].Martin D, Galisteo R, Gutkind JS, CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex, J. Biol. Chem 284 (10) (2009) 6038–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ, Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids, Proc. Natl. Acad. Sci. U. S. A 103 (34) (2006) 12741–12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tam AB, Mercado EL, Hoffmann A, Niwa M, ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK, PLoS One 7 (10) (2012) e45078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH, Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and downregulation of TRAF2 expression, Mol. Cell. Biol 26 (8) (2006) 3071–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D, Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1, Science 287 (5453) (2000) 664–666. [DOI] [PubMed] [Google Scholar]
- [35].Grootjans J, Kaser A, Kaufman RJ, Blumberg RS, The unfolded protein response in immunity and inflammation, Nat. Rev. Immunol 16 (8) (2016) 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Oyadomari S, Mori M, Roles of CHOP/GADD153 in endoplasmic reticulum stress, Cell Death Differ. 11 (4) (2004) 381–389. [DOI] [PubMed] [Google Scholar]
- [37].Drogat B, Auguste P, Nguyen DT, Bouchecareilh M, Pineau R, Nalbantoglu J, Kaufman RJ, Chevet E, Bikfalvi A, Moenner M, IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo, Cancer Res. 67 (14) (2007) 6700–6707. [DOI] [PubMed] [Google Scholar]
- [38].Auf G, Jabouille A, Guerit S, Pineau R, Delugin M, Bouchecareilh M, Magnin N, Favereaux A, Maitre M, Gaiser T, von Deimling A, Czabanka M, Vajkoczy P, Chevet E, Bikfalvi A, Moenner M, Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma, Proc. Natl. Acad. Sci. U. S. A 107 (35) (2010) 15553–15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Harnoss JM, Le Thomas A, Reichelt M, Guttman O, Wu TD, Marsters SA, Shemorry A, Lawrence DA, Kan D, Segal E, Merchant M, Totpal K, Crocker LM, Mesh K, Dohse M, Solon M, Modrusan Z, Rudolph J, Koeppen H, Walter P, Ashkenazi A, IRE1alpha disruption in triple-negative breast cancer cooperates with antiangiogenic therapy by reversing ER stress adaptation and Remodeling the tumor microenvironment, Cancer Res. 80 (11) (2020) 2368–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rouschop KM, Dubois LJ, Keulers TG, van den Beucken T, Lambin P, Bussink J, van der Kogel AJ, Koritzinsky M, Wouters BG, PERK/eIF2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS, Proc. Natl. Acad. Sci. U. S. A 110 (12) (2013) 4622–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bu Y, Diehl JA, PERK integrates oncogenic Signaling and cell survival during cancer development, J. Cell. Physiol 231 (10) (2016) 2088–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA, Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival, Cancer Res. 68 (9) (2008) 3260–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ, Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer, Oncogene 34 (29) (2015) 3760–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hanahan D, Folkman J, Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis, Cell 86 (3) (1996) 353–364. [DOI] [PubMed] [Google Scholar]
- [45].Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T, VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress, Mol. Cell 54 (4) (2014) 559–572. [DOI] [PubMed] [Google Scholar]
- [46].Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, Zhang Z, Lin Z, Gui M, Li X, Zhang T, Kong Q, Li H, Chen S, Chen A, Kaufman RJ, Yang WL, Lin HK, Zhang D, Perlman H, Thorp E, Zhang K, Fang D, Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis, EMBO J. 32 (18) (2013) 2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC, Pyruvate kinase M2 is a phosphotyrosine-binding protein, Nature 452 (7184) (2008) 181–186. [DOI] [PubMed] [Google Scholar]
- [48].Mahadevan NR, Zanetti M, Tumor stress inside out: cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment, J. Immunol 187 (9) (2011) 4403–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li X, Zhang K, Li Z, Unfolded protein response in cancer: the physician’s perspective, J. Hematol. Oncol 4 (2011) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, Polverini PJ, The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway, Cancer Res. 72 (20) (2012) 5396–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ding L, Yan J, Zhu J, Zhong H, Lu Q, Wang Z, Huang C, Ye Q, Ligand-independent activation of estrogen receptor alpha by XBP-1, Nucleic Acids Res. 31 (18) (2003) 5266–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dengler VL, Galbraith M, Espinosa JM, Transcriptional regulation by hypoxia inducible factors, Crit. Rev. Biochem. Mol. Biol 49 (1) (2014) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH, XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway, Nature 508 (7494) (2014) 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mujcic H, Rzymski T, Rouschop KM, Koritzinsky M, Milani M, Harris AL, Wouters BG, Hypoxic activation of the unfolded protein response (UPR) induces expression of the metastasis-associated gene LAMP3, Radiother. Oncol 92 (3) (2009) 450–459. [DOI] [PubMed] [Google Scholar]
- [55].Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG, The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5, J. Clin. Invest 120 (1) (2010) 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Le QT, Denko NC, Giaccia AJ, Hypoxic gene expression and metastasis, Cancer Metastasis Rev. 23 (3–4) (2004) 293–310. [DOI] [PubMed] [Google Scholar]
- [57].Koumenis C, Wouters BG, “translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways, Mol. Cancer Res 4 (7) (2006) 423–436. [DOI] [PubMed] [Google Scholar]
- [58].Ozawa K, Tsukamoto Y, Hori O, Kitao Y, Yanagi H, Stern DM, Ogawa S, Regulation of tumor angiogenesis by oxygen-regulated protein 150, an inducible endoplasmic reticulum chaperone, Cancer Res. 61 (10) (2001) 4206–4213. [PubMed] [Google Scholar]
- [59].Liang H, Xiao J, Zhou Z, Wu J, Ge F, Li Z, Zhang H, Sun J, Li F, Liu R, Chen C, Hypoxia induces miR-153 through the IRE1alpha-XBP1 pathway to fine tune the HIF1alpha/VEGFA axis in breast cancer angiogenesis, Oncogene 37 (15) (2018) 1961–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Anspach L, Tsaryk R, Seidmann L, Unger RE, Jayasinghe C, Simiantonaki N, Kirkpatrick CJ, Prols F, Function and mutual interaction of BiP-, PERK-, and IRE1alpha-dependent signalling pathways in vascular tumours, J. Pathol 251 (2) (2020) 123–134. [DOI] [PubMed] [Google Scholar]
- [61].Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML, Autocrine VEGF signaling is required for vascular homeostasis, Cell 130 (4) (2007) 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yan JD, Liu Y, Zhang ZY, Liu GY, Xu JH, Liu LY, Hu YM, Expression and prognostic significance of VEGFR-2 in breast cancer, Pathol. Res. Pract 211 (7) (2015) 539–543. [DOI] [PubMed] [Google Scholar]
- [63].Prasad CB, Singh D, Pandey LK, Pradhan S, Singh S, Narayan G, VEGFa/VEGFR2 autocrine and paracrine signaling promotes cervical carcinogenesis via beta-catenin and snail, Int. J. Biochem. Cell Biol 142 (2022), 106122. [DOI] [PubMed] [Google Scholar]
- [64].Devery AM, Wadekar R, Bokobza SM, Weber AM, Jiang Y, Ryan AJ, Vascular endothelial growth factor directly stimulates tumour cell proliferation in non-small cell lung cancer, Int. J. Oncol 47 (3) (2015) 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Michaelsen SR, Staberg M, Pedersen H, Jensen KE, Majewski W, Broholm H, Nedergaard MK, Meulengracht C, Urup T, Villingshoj M, Lukacova S, Skjoth-Rasmussen J, Brennum J, Kjaer A, Lassen U, Stockhausen MT, Poulsen HS, Hamerlik P, VEGF-C sustains VEGFR2 activation under bevacizumab therapy and promotes glioblastoma maintenance, Neuro-Oncology 20 (11) (2018) 1462–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science 324 (5930) (2009) 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pavlova NN, Thompson CB, The emerging hallmarks of cancer metabolism, Cell Metab. 23 (1) (2016) 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Vettore L, Westbrook RL, Tennant DA, New aspects of amino acid metabolism in cancer, Br. J. Cancer 122 (2) (2020) 150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Grasmann G, Smolle E, Olschewski H, Leithner K, Gluconeogenesis in cancer cells - repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 1872 (1) (2019) 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kiesel VA, Sheeley MP, Coleman MF, Cotul EK, Donkin SS, Hursting SD, Wendt MK, Teegarden D, Pyruvate carboxylase and cancer progression, Cancer Metab. 9 (1) (2021) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, Hada A, Arai M, Wakatsuki T, Matsubara O, Yamamoto N, Yamamoto M, Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis, J. Hepatol 38 (5) (2003) 605–614. [DOI] [PubMed] [Google Scholar]
- [72].Zhuang L, Scolyer RA, Lee CS, McCarthy SW, Cooper WA, Zhang XD, Thompson JF, Hersey P, Expression of glucose-regulated stress protein GRP78 is related to progression of melanoma, Histopathology 54 (4) (2009) 462–470. [DOI] [PubMed] [Google Scholar]
- [73].Zhang F, Zeng QY, Xu H, Xu AN, Liu DJ, Li NZ, Chen Y, Jin Y, Xu CH, Feng CZ, Zhang YL, Liu D, Liu N, Xie YY, Yu SH, Yuan H, Xue K, Shi JY, Liu TX, Xu PF, Zhao WL, Zhou Y, Wang L, Huang QH, Chen Z, Chen SJ, Zhou XL, Sun XJ, Selective and competitive functions of the AAR and UPR pathways in stress-induced angiogenesis, Cell Discov. 7 (1) (2021) 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sbodio JI, Snyder SH, Paul BD, Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease, Proc. Natl. Acad. Sci. U. S. A 113 (31) (2016) 8843–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Obacz J, Avril T, Rubio-Patino C, Bossowski JP, Igbaria A, Ricci JE, Chevet E, Regulation of tumor-stroma interactions by the unfolded protein response, FEBS J. 286 (2) (2019) 279–296. [DOI] [PubMed] [Google Scholar]
- [76].Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, Willert K, Lin JH, Zanetti M, Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells, Sci. Signal 10 (482) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M, Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells, Proc. Natl. Acad. Sci. U. S. A 108 (16) (2011) 6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M, Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming, PLoS One 7 (12) (2012) e51845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shen T, Huang Z, Shi C, Pu X, Xu X, Wu Z, Ding G, Cao L, Pancreatic cancer-derived exosomes induce apoptosis of T lymphocytes through the p38 MAPK-mediated endoplasmic reticulum stress, FASEB J. 34 (6) (2020) 8442–8458. [DOI] [PubMed] [Google Scholar]
- [80].Ludwig N, Yerneni SS, Razzo BM, Whiteside TL, Exosomes from HNSCC promote angiogenesis through reprogramming of endothelial cells, Mol. Cancer Res 16 (11) (2018) 1798–1808. [DOI] [PubMed] [Google Scholar]
- [81].Kanemoto S, Nitani R, Murakami T, Kaneko M, Asada R, Matsuhisa K, Saito A, Imaizumi K, Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress, Biochem. Biophys. Res. Commun 480 (2) (2016) 166–172. [DOI] [PubMed] [Google Scholar]
- [82].Alt EU, Barabadi Z, Pfnur A, Ochoa JE, Daneshimehr F, Lang LM, Lin D, Braun SE, Chandrasekar B, Izadpanah R, TRAF3IP2, a novel therapeutic target in glioblastoma multiforme, Oncotarget 9 (51) (2018) 29772–29788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kruger S, Abd Elmageed ZY, Hawke DH, Worner PM, Jansen DA, Abdel-Mageed AB, Alt EU, Izadpanah R, Molecular characterization of exosome-like vesicles from breast cancer cells, BMC Cancer 14 (2014) 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, Wang F, Li X, Liu Q, Li Y, Guo Z, Gao B, Wei W, Wang H, Sun G, Endoplasmic reticulum stress causes liver cancer cells to release Exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages, Hepatology 70 (1) (2019) 241–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X, Tumor-associated macrophages: recent insights and therapies, Front. Oncol 10 (2020) 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wu Y, Chen W, Xu ZP, Gu W, PD-L1 distribution and perspective for cancer immunotherapy-blockade, knockdown, or inhibition, Front. Immunol 10 (2019) 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].He C, Hua W, Liu J, Fan L, Wang H, Sun G, Exosomes derived from endoplasmic reticulum-stressed liver cancer cells enhance the expression of cytokines in macrophages via the STAT3 signaling pathway, Oncol. Lett 20 (1) (2020) 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yao X, Tu Y, Xu Y, Guo Y, Yao F, Zhang X, Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages, J. Cell. Mol. Med 24 (17) (2020) 9560–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhang C, Wang H, Chan GCF, Zhou Y, Lai X, Lian M, Extracellular vesicles derived from human umbilical cord mesenchymal stromal cells protect cardiac cells against hypoxia/Reoxygenation injury by inhibiting endoplasmic reticulum stress via activation of the PI3K/Akt pathway, Cell Transplant. 29 (2020), 963689720945677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lin Y, Zhang C, Xiang P, Shen J, Sun W, Yu H, Exosomes derived from HeLa cells break down vascular integrity by triggering endoplasmic reticulum stress in endothelial cells, J. Extracell Vesicles 9 (1) (2020), 1722385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wu CH, Silvers CR, Messing EM, Lee YF, Bladder cancer extracellular vesicles drive tumorigenesis by inducing the unfolded protein response in endoplasmic reticulum of nonmalignant cells, J. Biol. Chem 294 (9) (2019) 3207–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ahn SY, The role of MSCs in the tumor microenvironment and tumor progression, Anticancer Res. 40 (6) (2020) 3039–3047. [DOI] [PubMed] [Google Scholar]
- [93].Guo J, Zhong X, Tan Q, Yang S, Liao J, Zhuge J, Hong Z, Deng Q, Zuo Q, miR-301a-3p induced by endoplasmic reticulum stress mediates the occurrence and transmission of trastuzumab resistance in HER2-positive gastric cancer, Cell Death Dis. 12 (7) (2021) 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yamagata AS, Freire PP, Are cachexia-associated tumors transmitTERS of ER stress? Biochem. Soc. Trans 49 (4) (2021) 1841–1853. [DOI] [PubMed] [Google Scholar]
- [95].Kim TW, Hong DW, Kang CM, Hong SH, A novel PPAR ligand, PPZ023, overcomes radioresistance via ER stress and cell death in human non-small-cell lung cancer cells, Exp. Mol. Med 52 (10) (2020) 1730–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kim TW, Ko SG, The herbal formula JI017 induces ER stress via Nox4 in breast cancer cells, Antioxidants (Basel) 10 (12) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kim T, Ko SG, JI017, a complex herbal medication, induces apoptosis via the Nox4-PERK-CHOP Axis in ovarian cancer cells, Int. J. Mol. Sci 22 (22) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ojha R, Amaravadi RK, Targeting the unfolded protein response in cancer, Pharmacol. Res 120 (2017) 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rivas A, Vidal RL, Hetz C, Targeting the unfolded protein response for disease intervention, Expert Opin. Ther. Targets 19 (9) (2015) 1203–1218. [DOI] [PubMed] [Google Scholar]
- [100].Khanna M, Agrawal N, Chandra R, Dhawan G, Targeting unfolded protein response: a new horizon for disease control, Expert Rev. Mol. Med 23 (2021) e1. [DOI] [PubMed] [Google Scholar]
- [101].Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH, Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response, EMBO J. 30 (5) (2011) 894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hetz C, Chevet E, Harding HP, Targeting the unfolded protein response in disease, Nat. Rev. Drug Discov 12 (9) (2013) 703–719. [DOI] [PubMed] [Google Scholar]
- [103].Makhov P, Naito S, Haifler M, Kutikov A, Boumber Y, Uzzo RG, Kolenko VM, The convergent roles of NF-kappaB and ER stress in sunitinib-mediated expression of pro-tumorigenic cytokines and refractory phenotype in renal cell carcinoma, Cell Death Dis. 9 (3) (2018) 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Prischi F, Nowak PR, Carrara M, Ali MM, Phosphoregulation of Ire1 RNase splicing activity, Nat. Commun 5 (2014) 3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Doultsinos D, Carlesso A, Chintha C, Paton JC, Paton AW, Samali A, Chevet E, Eriksson LA, Peptidomimetic-based identification of FDA-approved compounds inhibiting IRE1 activity, FEBS J. 288 (3) (2021) 945–960. [DOI] [PubMed] [Google Scholar]
- [106].Griffioen AW, Mans LA, de Graaf AMA, Nowak-Sliwinska P, de Hoog C, de Jong TAM, Vyth-Dreese FA, van Beijnum JR, Bex A, Jonasch E, Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients, Clin. Cancer Res 18 (14) (2012) 3961–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Blumenthal GM, Cortazar P, Zhang JJ, Tang S, Sridhara R, Murgo A, Justice R, Pazdur R, FDA approval summary: sunitinib for the treatment of progressive well-differentiated locally advanced or metastatic pancreatic neuroendocrine tumors, Oncologist 17 (8) (2012) 1108–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A, Donati D, O’Brien T, Jackson PK, Isacchi A, Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death, Nat. Chem. Biol 9 (9) (2013) 548–556. [DOI] [PubMed] [Google Scholar]
- [109].Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, Liang C, Booth B, Chidambaram N, Morse D, Sridhara R, Garvey P, Justice R, Pazdur R, Sorafenib for the treatment of advanced renal cell carcinoma, Clin. Cancer Res 12 (24) (2006) 7271–7278. [DOI] [PubMed] [Google Scholar]
- [110].Lang L, FDA approves sorafenib for patients with inoperable liver cancer, Gastroenterology 134 (2) (2008) 379. [DOI] [PubMed] [Google Scholar]
- [111].McDermott DF, George DJ, Bevacizumab as a treatment option in advanced renal cell carcinoma: an analysis and interpretation of clinical trial data, Cancer Treat. Rev 36 (3) (2010) 216–223. [DOI] [PubMed] [Google Scholar]
- [112].Summers J, Cohen MH, Keegan P, Pazdur R, FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma, Oncologist 15 (1) (2010) 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Casak SJ, Donoghue M, Fashoyin-Aje L, Jiang X, Rodriguez L, Shen YL, Xu Y, Jiang X, Liu J, Zhao H, Pierce WF, Mehta S, Goldberg KB, Theoret MR, Kluetz PG, Pazdur R, Lemery SJ, FDA approval summary: atezolizumab plus bevacizumab for the treatment of patients with advanced Unresectable or metastatic hepatocellular carcinoma, Clin. Cancer Res 27 (7) (2021) 1836–1841. [DOI] [PubMed] [Google Scholar]
- [114].Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP, The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule, Proc. Natl. Acad. Sci. U. S. A 109 (15) (2012). E869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC, Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma, Blood 117 (4) (2011), 1311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kawamura T, Tashiro E, Yamamoto K, Shindo K, Imoto M, SAR study of a novel triene-ansamycin group compound, quinotrierixin, and related compounds, as inhibitors of ER stress-induced XBP1 activation, J. Antibiot (Tokyo) 61 (5) (2008) 303–311. [DOI] [PubMed] [Google Scholar]
- [117].Lee AH, Iwakoshi NN, Glimcher LH, XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response, Mol. Cell. Biol 23 (21) (2003) 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Kardosh A, Golden EB, Pyrko P, Uddin J, Hofman FM, Chen TC, Louie SG, Petasis NA, Schonthal AH, Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib, Cancer Res. 68 (3) (2008) 843–851. [DOI] [PubMed] [Google Scholar]
- [119].Mujtaba T, Dou QP, Advances in the understanding of mechanisms and therapeutic use of bortezomib, Discov. Med 12 (67) (2011) 471–480. [PMC free article] [PubMed] [Google Scholar]
- [120].Suraweera A, Munch C, Hanssum A, Bertolotti A, Failure of amino acid homeostasis causes cell death following proteasome inhibition, Mol. Cell 48 (2) (2012) 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Brem GJ, Mylonas I, Bruning A, Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression, Gynecol. Oncol 128 (2) (2013) 383–390. [DOI] [PubMed] [Google Scholar]
- [122].Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ, Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways, Proc. Natl. Acad. Sci. U. S. A 108 (12) (2011) 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ, Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase, ChemMedChem 8 (2) (2013) 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.