

Abstract
Ubiquitination-mediated protein degradation is associated with the development of pulmonary fibrosis. We and others have shown that Nedd4L plays anti-inflammatory and anti-fibrotic roles by targeting lysophosphatidic acid receptor 1 (LPAR1), p-Smad2/3, and β-catenin, and other molecules for their degradation in lung epithelial cells and fibroblasts. However, the molecular regulation of Nedd4L expression in lung fibroblasts has not been studied. In this study, we find that Nedd4L levels are significantly suppressed in lung myofibroblasts in IPF patients and in experimental pulmonary fibrosis, and in TGF-β1-treated lung fibroblasts. Nedd4L knockdown promotes TGF-β1-mediated phosphorylation of Smad2/3 and lung myofibroblast differentiation. Mechanistically, Nedd4L targets TGF-β receptor II (TβRII), the first key enzyme of TGF-β1-mediated signaling, for its ubiquitination and degradation. Further, we show that inhibition of transcriptional factor E2F rescues Nedd4L levels and mitigates experimental pulmonary fibrosis. Together, our data reveal insight into mechanisms by which E2F-mediated Nedd4L suppression contributes to the pathogenesis of lung fibrosis. This study provides evidence showing that upregulation of Nedd4L is a potential therapeutic strategy to treat fibrotic disorders including lung fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is an aging-associated, chronic, irreversible, and ultimately fatal lung disease, which is characterized by the excessive accumulation of differentiated myofibroblasts and extracellular matrix (ECM) deposition, resulting in scarring of the lungs.1–3 Repetitive lung epithelial cell injury contributes to the initiation phase of fibrosis, while abnormal activation of fibroblasts/myofibroblasts leads to the development and progression of pulmonary fibrosis (PF).4–6 TGF-β1 is a master regulator in the progression of PF. TGF-β1 binds to and activates the TGF-β receptor complex (TβRII activation of TβR1) to signal via canonical (Smad-mediated pathways) and non-canonical pathways to promote myofibroblast differentiation, activation, and survival.3,7–9 Because of its central role in fibroblast biology and the pathogenesis of fibrosis, TGF-β1 signaling has been targeted as a therapeutic strategy in human diseases including cancer and fibrosis.
Ubiquitination-mediated protein degradation is dependent on ubiquitin E3 ligases.10,11 Different linkages of polyubiquitin chains regulate distinct protein functions, such as degradation, cellular localization, protein-protein interactions, and enzyme activity. The ubiquitin E3 ligase Nedd4L has been shown to exhibit anti-fibrotic properties by promoting degradation of TβRI, p-Smad2/3, LPAR1, and ENaC.12–15 Recent studies demonstrated that reduced Nedd4L levels in lung epithelial cells in IPF patients showed that the reduction of Nedd4L promoted chronic lung epithelial injury and subsequent PF16 and identified β-catenin as a novel substrate of Nedd4L.17 Taken together, these studies suggest that Nedd4L plays an anti-fibrotic role by targeting multiple substrates. However, the role of Nedd4L in TβRII stability and molecular regulation of Nedd4L suppression have not been revealed.
The E2F family regulates cellular functions by acting as an activator or repressor of transcription.18–22 In this study, we revealed that E2F4 suppresses Nedd4L transcription. Additionally, the current study discovered new molecular mechanisms by which Nedd4L regulates TGF-β1-mediated signaling through ubiquitinating and degrading TβRII. This study provides molecular mechanistic evidence that restoring Nedd4L levels in the lungs may reduce the severity of PF.
Material and methods
Cells and reagents
Human lung fibroblast cell lines (Mrc5 and IMR90) were purchased from ATCC (Manassas, VA). Human primary lung fibroblasts (HLF) and lung myofibroblasts (IPF-LF) from adult normal human subjects and IPF patients were obtained from the Center for Organ Recovery and Education and Lung Transplantation at the University of Pittsburgh. The study was approved by the institutional Review Board at the University of Pittsburgh (STUDY18100070). Cells were cultured in Eagle’s Minimum Essential Medium (EMEM) containing 10% fetal bovine serum (FBS) in 5% CO2 cell culture incubator. V5 antibody, mammalian expressional plasmid pcDNA3.1/His-V5-topo, and Escherichia coli Top10 competent cells were purchased from Life technologies (Grand Island, NY). Nedd4L, TβRII, collagen I, ubiquitin, smurf1, Smad4, p-Smad2/3, and Smad2/3 antibodies were purchased from Cell Signaling (Danvers, MA). Fibronectin (FN), alpha-smooth muscle actin (α-SMA), E2F4 antibodies, immobilized protein A/G beads, and control IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HLM006474, bleomycin, and β-actin antibody were purchased from Sigma (St. Louis, MO). Human recombinant TGF-β1 was purchased from R&D systems (Minneapolis, MN). All materials used in the experiments are the highest grade commercially available.
Plasmid and siRNA transfection
Human Nedd4L and human TβRII cDNA were inserted into pCDNA3.1/V5-His-Topo vector or pCDNA3.1/HA-Topo vector. Plasmids and siRNAs transfections in fibroblast cells were performed using Lipo-Jet or PepMute siRNA transfection reagent (SignaGen Laboratories, Frederick, MD). Overexpression or downregulation of genes was confirmed by immunoblotting.
Immunoblotting and co-immunoprecipitation (co-IP)
Lung tissue or lung fibroblast cells were lysed in 1x lysis buffer (Cell signaling, Danvers, MA). Equal amounts of total protein (20 μg) were subjected to SDS-PAGE, transferred to nitrocellulose, and then incubated with primary and secondary antibodies, sequentially. For co-IP, 1 mg of protein in 1 ml of cell lysis buffer was incubated with a primary antibody overnight at 48C, followed by incubation with protein A/G beads for an additional 2 hour in room temperature. The beads were rinsed with PBS and lysis buffer 3 times. Proteins on the beads were eluted by boiling in 2x SDS sample buffer. Eluted samples were subjected to immunoblotting. Imaging was conducted using Azura Biosystems Imaging Station C600.
In vivo ubiquitination assay
Fibroblasts were collected in PBS and cell pellets will be obtained by centrifuge. Cell pellets were then resuspended with ubiquitin aldehyde and N-ethylmaleinimid (NEM) in 50−80 μl of 2% SDS lysis buffer, followed by heating at 100°C for 10 minutes. This procedure disrupts any non-covalent protein-protein interaction. The denatured cell lysates were then sonicated on ice, followed by 10-fold dilution with TBS. Cell lysates were subjected to IP and immunoblotting.
Immunofluorescence staining
Fibroblasts were cultured in glass-bottom dishes. After indicated treatment, cells were fixed with 3.7% formaldehyde for 20 minutes. Cells were blocked by 1% BSA in TBST buffer. Cells were then exposed to primary antibodies, followed by incubation with fluorescence-labeled secondary antibodies and 4′,6-diamindino-2-phenylindole (DAPI). Immunofluorescent cell imaging was conducted using a Nikon A1R confocal microscope.
Animals and flow cytometry
C57/BL6 mice were housed in the specific pathogen-free animal care facility at the Ohio State University in accordance with institutional guidelines and guidelines of the US National Institutes of Health. All animal experiments were approved by the Ohio State University Animal Resources Centers. Lentiviruses were administered to the lungs intratracheally. C57BL/6 mice (8−10 weeks, male and female) were given intratracheal (IT) bleomycin (2 mg/kg body weight) with or without intraperitoneal injection of HLM006474 as detailed in the Results section. Lung tissues were fixed for hematoxylin and eosin (H&E) and trichrome staining. Protein expression in lung tissues was examined by immunoblotting. Collagen levels were determined by measuring the hydroxyproline content. Lung tissues were minced and incubated in a dissociation medium containing collagenase and DNase I at 37°C for 20 minutes. The digested lung tissue mixture was filtered with 70 μm nylon mesh strainer to obtain a single-cell suspension. Nedd4L expression was analyzed with flow cytometry with antibodies specific to Nedd4L and cell-type markers.
Statistical analysis
Statistical analysis was carried out by 1 or 2-way analysis of variance or unpaired t test, with a P value of < 0.05 considered indicative of significance.
Results
Nedd4L levels are reduced in fibrotic lungs and isolated myofibroblast cells
We have shown that Nedd4L mediates the ubiquitin-proteasomal degradation of LPAR114, which has been implicated in lung injury and fibrosis. We hypothesized that Nedd4L activity may be associated with the pathogenesis of PF, and focused on investigation the role of Nedd4L in lung fibroblast activation because these cells are the ultimate effector cells of fibrosis. We reanalyzed microarray data from the Lung Genomics Research Consortium (http://www.lung-genomics.org/) including 160 patients with IPF and 108 donor control subjects. Consistent with recent studies16,17, Nedd4L mRNA levels in lungs were significantly lower in IPF cohorts compared to control subjects and the reduction of Nedd4L levels directly correlates with DLCO% and FVC% predicted (Fig. 1A–C). To further evaluate Nedd4L expression in a murine model of PF, Nedd4L levels in lungs from bleomycin-challenged mice were examined. IT injection of bleomycin (2 U/kg, 14 and 21 days) decreased Nedd4L protein levels in lungs (Fig. 1D–F). The Nedd4L levels showed an inverse correlation with fibronectin (FN) levels (Fig. 1G). Consistent with the findings from IPF patients, Nedd4L mRNA levels were also decreased in experimental fibrotic lungs (Fig. 1H), supporting a link between Nedd4L and fibroblast phenotypes in lung fibrosis. Nedd4L levels were decreased in lung fibroblasts/myofibroblasts from IPF patients compared to control subjects. Collagen I and α-SMA levels were examined as biomarkers for myofibroblasts from IPF patients (Fig. 1I). Nedd4L reduction was also observed in isolated fibroblasts from murine fibrotic lungs (Fig. 1J, K). The data indicates that in addition to epithelial cells, Nedd4L is also decreased in lung fibroblasts/myofibroblasts in IPF patients and experimental fibrotic lungs. Thus, we believe that Nedd4L reduction may have clinically relevant implications.
Fig 1. Nedd4L levels are reduced in fibrotic lungs and myofibroblast cells.
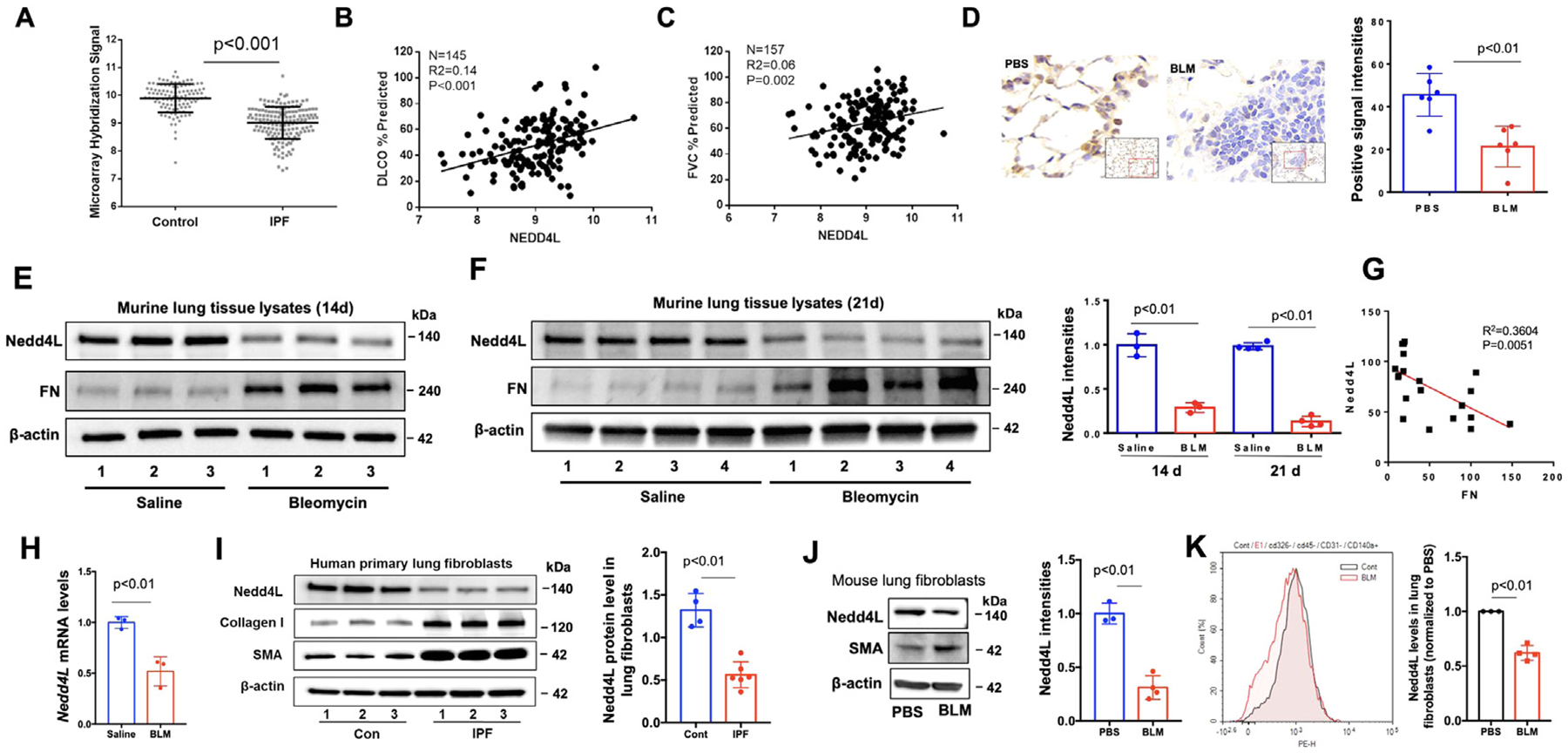
A. Nedd4L mRNA levels in controls (n=108) compared to IPF group (n=160). Mann Whitney test, P<0.001. B. Expression of Nedd4L in arbitrary units as a function of diffusing capacity of carbon monoxide (DLCO)% predicated (IPF patients, n=145). C. Expression of Nedd4L in arbitrary units as a function of FVC% predicated. (IPF patients, n=157). D. IHC staining of Nedd4L in lung tissues from IT PBS and IT bleomycin (BLM)-challenged mice. Nedd4L positive intensities were analyzed by Image J. E.F., Immunoblotting analysis of lung tissues of BLM-challenged mice. Nedd4L intensities were analyzed by Image J (n=3−4). G. A negative correlation between expression of Nedd4L and FN in lung tissues from BLM-challenged mice. H. Nedd4L mRNA levels from lungs of IT PBS- or IT BLM (2 U/kg, 14 d)-challenged mice were examined by realtime PCR. I. Representative immunoblots of lung fibroblasts from normal control subjects and IPF patients. Nedd4L intensities were analyzed by Image J (n=4−6). J. Representative immunoblots in isolated lung fibroblast cells from IT PBS and IT BLM-challenged mice (n=3−4). K. Flow cytometry analysis of Nedd4L in isolated lung fibroblasts (cd326−/cd45−/cd31−/cd140a+) from IT PBS (cont)- or IT BLM-challenged mice. Gray: Cont; red: IT BLM-treated cells. Shown are representative flow cytometry histograms. n=3−4, P<0.01.
Nedd4L ubiquitinates and targets TβRII for degradation, thereby attenuating lung fibroblast differentiation to myofibroblasts
It has been shown that Nedd4L regulates TGF-β1 signaling through targeting phosphorylated Smad2/3 and TβRI.12,13 We confirmed that overexpression of Nedd4L attenuates TGF-β1-induced phosphorylation of Smad2/3 and induction of collagen I, FN, and α-SMA. In contrast, downregulation of Nedd4L by siRNA transfection enhanced Smad2/3 phosphorylation and collagen expression (Fig. 2A–D), indicating that Nedd4L diminishes pro-fibrotic response in normal lung fibroblasts. Furthermore, we found that Nedd4L exhibited same phenomenon in A549 cells (Supple Fig. S1).
Fig 2. Nedd4L attenuates TGF-β1-induced pro-fibrotic responses through targeting TβRII in lung fibroblasts.
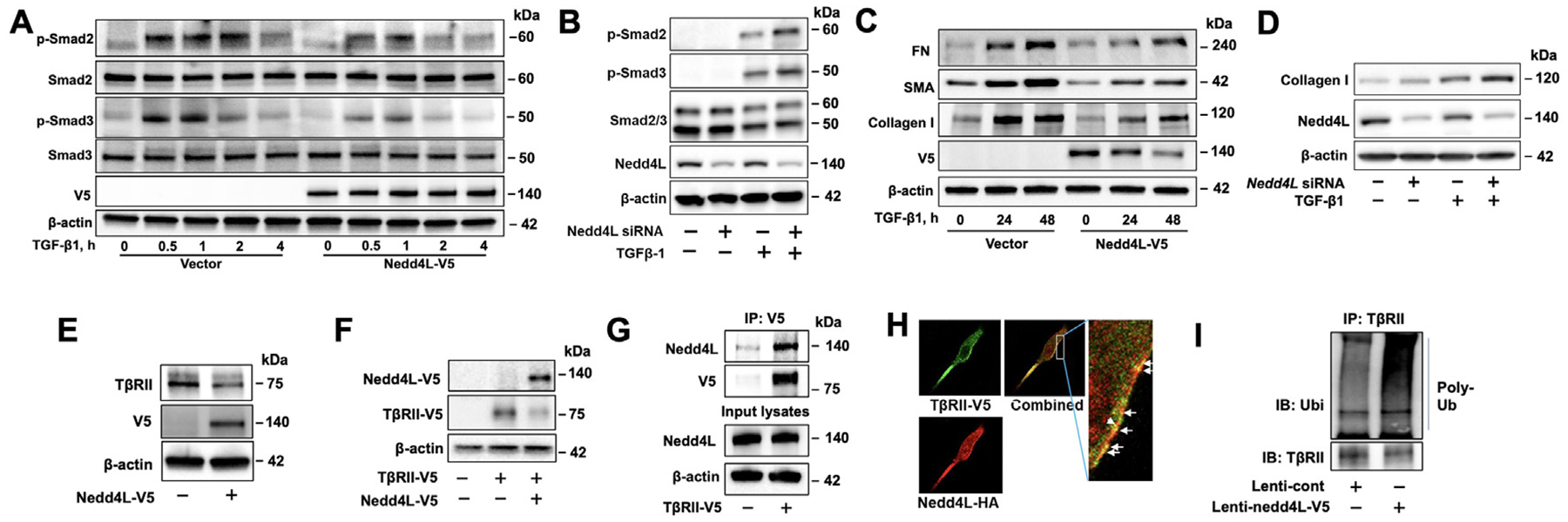
A. Immunoblotting analysis of TGF-β1 (10 ng/ml, 0–4 hour)-treated Nedd4L-V5-transfected HLF cells. B. Immunoblotting analysis of TGF-β1 (10 ng/ml, 15 minutes)-treated Nedd4L siRNA-transfected HLF cells. C. Immunoblotting analysis of TGF-β1 (10 ng/ml, 0, 24, and 48 hour)-treated Nedd4L-V5-transfected Mrc5 cells. D. Immunoblotting analysis of TGF-β1 (10 ng/ml, 24 hour)-treated Nedd4L siRNA-transfected HLF cells. E. Immunoblotting analysis of Nedd4L-V5-transfected Mrc5 cells. F. Immunoblotting analysis of Nedd4L-V5 and TβRII-V5-transfected Mrc5 cells. G. Immunoblotting analysis of Nedd4L and TβRII-V5 levels in the immunoprecipitated complex pulled down by a V5 antibody. H. Coimmunostaining of Nedd4L-HA and TβRII-V5 in double plasmid-transfected Mrc5 cells. Arrows indicate co-localization. I. Immunoblotting analysis of poly-ubiquitinated TβRII levels in the immunoprecipitated complex pulled down by a TβRII antibody in a denatured condition by an in vivo ubiquitination assay. Immunoblots are representative of three independent experiments.
We have shown that Nedd4L is localized on the inner plasma membrane;14 thus, we hypothesized that Nedd4L may target TβRII for its ubiquitination and degradation. We found that upregulation of Nedd4L reduced both endogenous and overexpressed TβRII (Fig. 2E, F). Association between the substrate and E3 ligase were confirmed by co-immunoprecipitation and co-immunofluorescence staining (Fig. 2G, H). The data from ubiquitination assay indicated that Nedd4L increased polyubiquitination of TβRII (Fig. 2I). TβRII directly binds to TGF-β1. Our study suggests that Nedd4L regulates TGF-β1-mediated fibrotic effects through targeting TβRII for its degradation.
Overexpression of Nedd4L ameliorates lung fibrosis in a bleomycin-induced model of PF
To elucidate the anti-fibrotic effect of Nedd4L in lung fibrosis, we delivered the Nedd4L-V5 gene to C57BL/6 mouse lungs using an intratracheal (IT) lentiviral vector delivery system. After 7 days, mice were challenged with IT single-dose BLM (2 U/kg). Mice were sacrificed after additional 3 weeks of IT BLM. After 7 days of lentiviral transduction, expression of Nedd4L-V5 in isolated lung fibroblast cells was confirmed by immunoblotting (Fig. 3A). Immunohistochemical staining with a V5 antibody showed that Nedd4L-V5 is detected in most cell types including lung epithelial cells (Fig. 3B). Lenti-Nedd4L-V5 attenuated IT BLM challenge-increased FN, collagen levels, and lung fibrosis with extension into the adjacent alveolar parenchyma in mouse lungs (Fig. 3C–F), indicating an anti-fibrotic effect of Nedd4L against PF.
Fig 3. Nedd4L overexpression dampens severity of experimental PF.
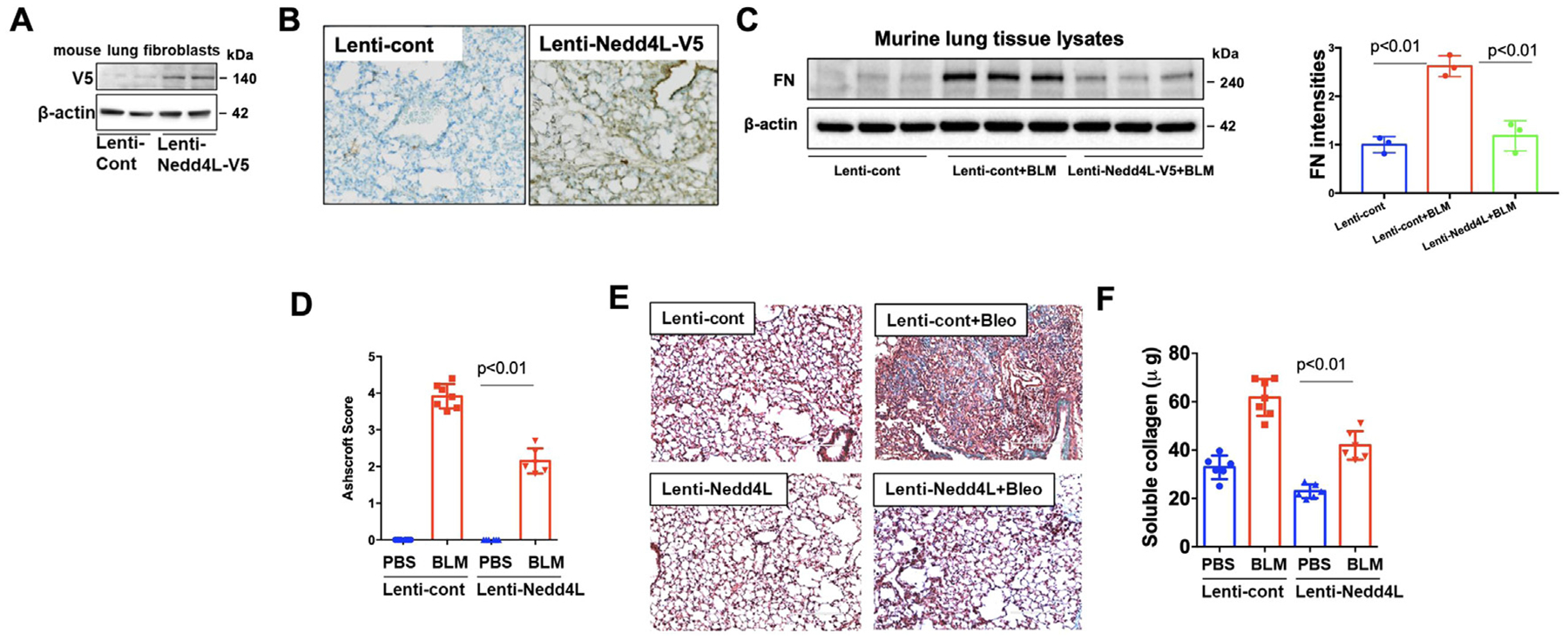
A. Immunoblotting analysis of isolated lung fibroblast cells from IT lenti-Nedd4L-V5-transducted mice. B. IHC staining of mouse lung tissues from lenti-control and lenti-Nedd4L-V5-transduced mice with a V5 antibody. n=3. C. Immunoblotting analysis of lung tissue lysates. FN intensities were analyzed by Image J. (n=3). D. Ashcraft score of lung histology. n=6−8. E. Masson trichrome staining of lung tissues. n=4−5. F. Collagen content in the right lung was analyzed by Sircol collagen assay. n=6−8.
Nedd4L suppression is induced by TGF-β1
TGF-β1 signaling is critical for the development of fibrosis.3,6 TGF-β1 treatment of Mrc5 and human lung fibroblast cells (HLF) decreased Nedd4L protein in a time- and dose-dependent manner (Fig 4A–C). TGF-β1 had no effect on levels of Smurf1, another TGF signaling-related E3 ligase (Fig. 4A–C). Similar to the changes in protein levels, Nedd4L mRNA levels were reduced by TGF-β1 in a dose- and time-dependent manner (Fig. 4D–F). Changes of mRNA levels are usually caused by modulation of gene transcription or mRNA stability. To investigate if TGF-β1 affects Nedd4L mRNA stability, HLF cells were treated with actinomycin D (an mRNA synthesis inhibitor) with or without TGF-β1. As shown in Fig. 4F, TGF-β1 did not alter actinomycin D-induced Nedd4L mRNA degradation, suggesting that TGF-β1 does not influence Nedd4L mRNA stability. To examine Nedd4L transcription, a 4.5 kbp proximal Nedd4L promoter region (p4500) was inserted into pGL3 vector. Luciferase assay showed that TGF-β1 treatment reduced Nedd4L promoter activity (Fig. 4G). Taken together, the data indicates that TGF-β1 reduces Nedd4L expression by inhibiting its transcription.
Fig 4. TGF-β1 suppresses Nedd4L transcription.
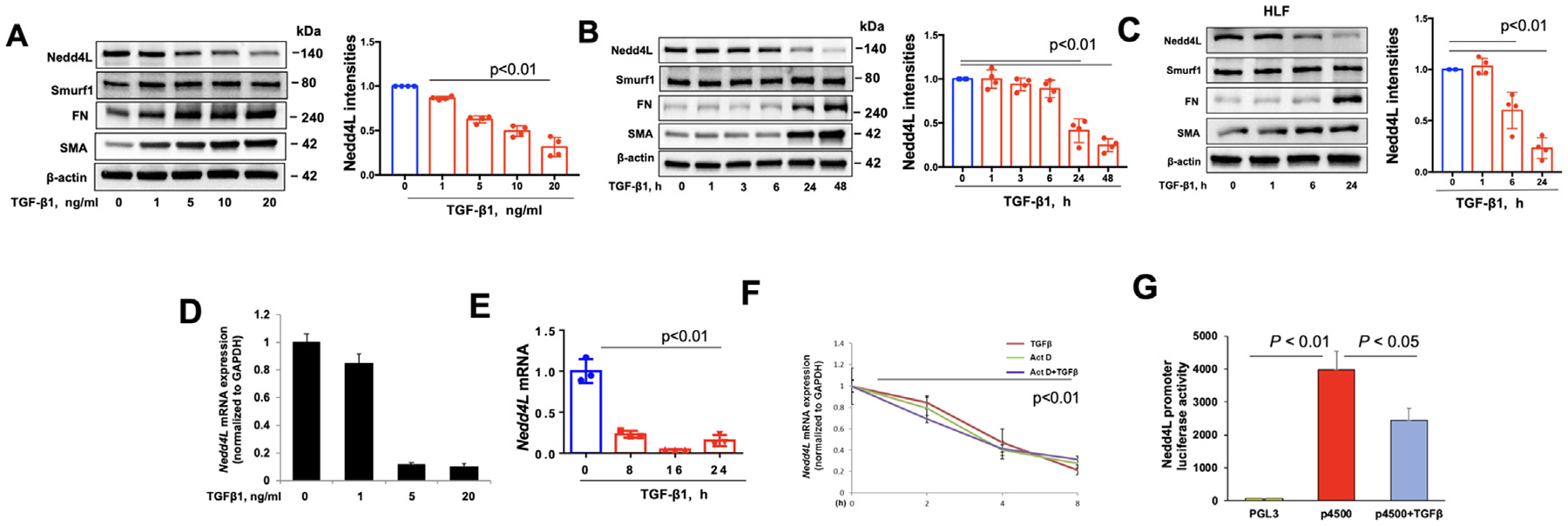
A-C. Immunoblotting analysis of TGF-β1 (10 ng/ml)-treated Mrc5 and HLF cells. Nedd4L intensities were analyzed by Image J (n=4). D.E. Realtime PCR analysis of TGF-β1 (10 ng/ml)-treated Mrc5 cells. n=3. F. Nedd4L mRNA levels were examined in HLF cells in response to actinomycin D (Act D), or TGF-β1+Act D, by real-time PCR (n=3). G. pGL3 empty vector or pGL3 vector containing Nedd4L promoter region (4.5 kbp length, p4500) were transfected into HLF cells for 48 h, and then cells were treated with TGF-β1 (10 ng/ml) for 6 h. Nedd4L promoter activity was assessed by luciferase assay (n=3).
Transcription factor E2F1/4 suppresses Nedd4L expression
The role of the canonical Smad pathway in TGF-β1-reduced Nedd4L was examined. Knockdown of Smad2, 3, and 4 had no effect on TGF-β1-reduced Nedd4L levels (Supple. Fig. S2A,B), suggesting the canonical Smad pathway is not involved in Nedd4L suppression. To screen which pathway regulates Nedd4L expression, we tested 30 non-canonical TGFβ signaling and transcriptional inhibitors. Nedd4L suppression was not altered by p38MAPK inhibitor (SB203580), mTOR inhibitor (rapamycin), MAPKK inhibitor (PD98059), HDAC inhibitor (TSA), acetyltransferase inhibitor (C646), DNA methylation inhibitor (AZA), or histone methyltransferase inhibitor (BIX-01294) (Fig. S1D–H). DNA damage-binding protein 2 (DDB2) has been shown to suppress Nedd4L expression in ovarian cancer; however, downregulation of DDB2 did not alter Nedd4L mRNA levels in TGF-β1-treated Mrc5 cells (Supple. Fig. S3). E2F inhibitor HLM006474 exhibited the maximal effect on attenuation of Nedd4L reduction (Fig. 5A). By analysis of Nedd4L promoter region, multiple E2F1 binding sites on the human Nedd4L promoter region were identified (data not shown). HLM006474 pretreatment attenuated TGF-β1-reduced Nedd4L levels in lung fibroblasts (Fig. 5B), suggesting that TGF-β1-reduced Nedd4L levels through activation of E2F. Among E2F isoforms, overexpression of E2F4 decreased Nedd4L abundance, while downregulation of E2F4 increased Nedd4L levels (Fig. 5C–E), suggesting that E2F4 suppresses Nedd4L transcription.
Fig 5. Inhibition of E2F4 rescues TGF-β1-rnduced Nedd4L expression.
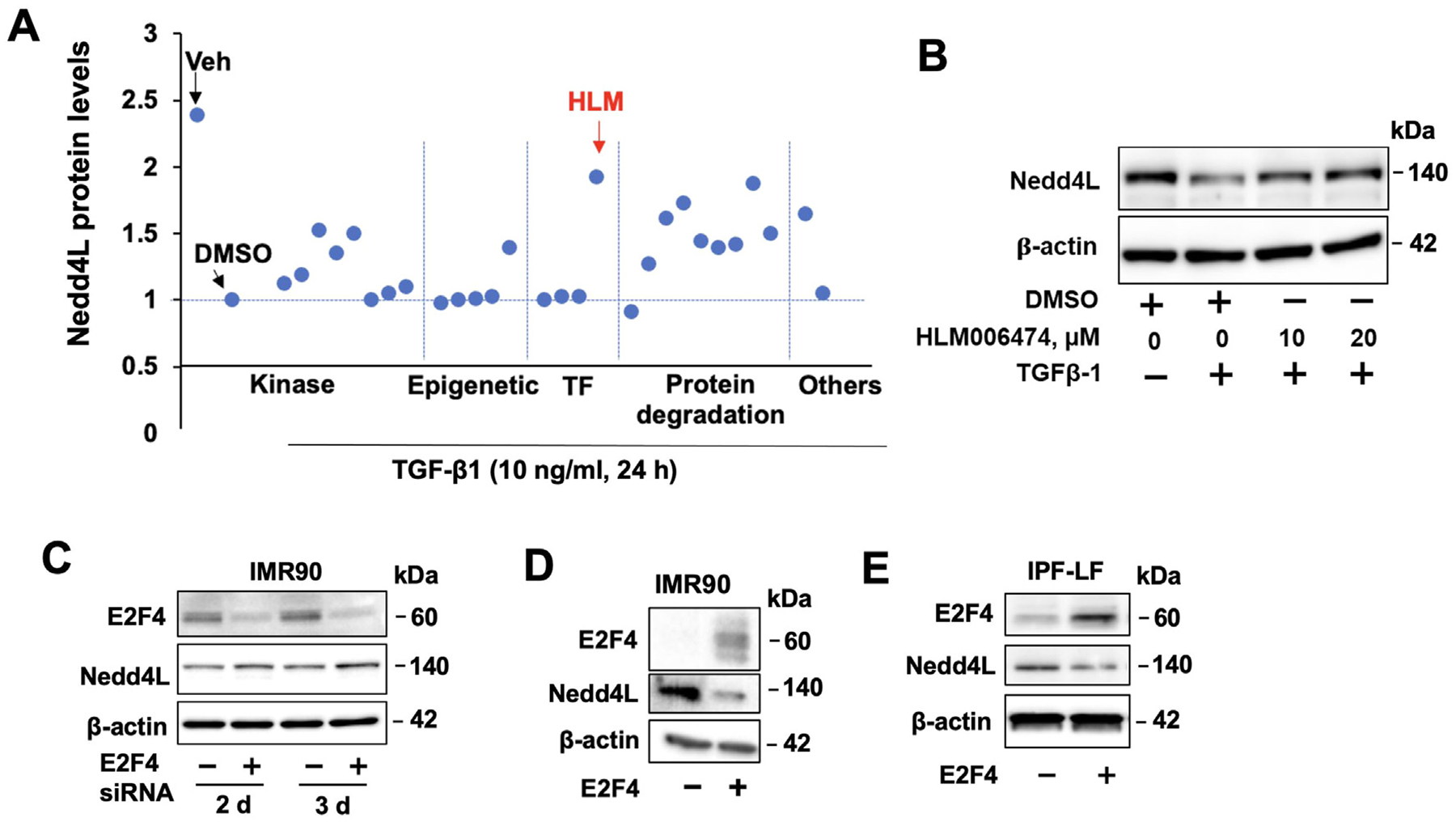
A. HLF cells were pretreated with various inhibitors prior to TGF-β1 treatment. Nedd4L expression was determined by immunoblotting and normalized to TGF-β1+DMSO. B. Immunoblotting analysis of HLF cells treated with HLM006474 with or without TGF-β1. C. Immunoblotting analysis of E2F4 siRNA-transfected HLF and IMR90 cells. D.E. Immunoblotting analysis of E2F4-transfected IMR90 and IPF-LF cells. Immunoblots are representative of three independent experiments.
Inhibitor of E2F dampens severity of PF
Next, we determined whether inhibition of E2F by HLM006474 promotes the de-differentiation of myofibroblasts to fibroblasts. TGF-β1-differentiated myofibroblast cells were treated with HLM006474 for 24 hour. HLM006474 significantly reduced FN and α-SMA levels in the differentiated cells (Fig. 6A, B). In IPF-LF cells, inhibition of E2F4 reduced FN levels, as well as cell migration (Fig. 6C, D). To examine the effect of HLM006474 on bleomycin-induced ECM accumulation, C57BL/6 mice were challenged with bleomycin (IT 2 U/kg body weight). Starting on day 9, HLM006474 (1 mg/kg body weight) was intraperitoneal (IP) injected at 2-day intervals. Mouse lungs were collected at day 21 (Fig. 7A). HLM006474 post-treatment dramatically reduced FN and collagen levels in mouse lung tissues (Fig. 7B). Histological analysis showed that HLM006474 significantly decreased collagen accumulation and severity of PF (Fig. 7C, D).
Fig6. HLM006474 attenuates TGF-β1-induced fibroblast differentiation.
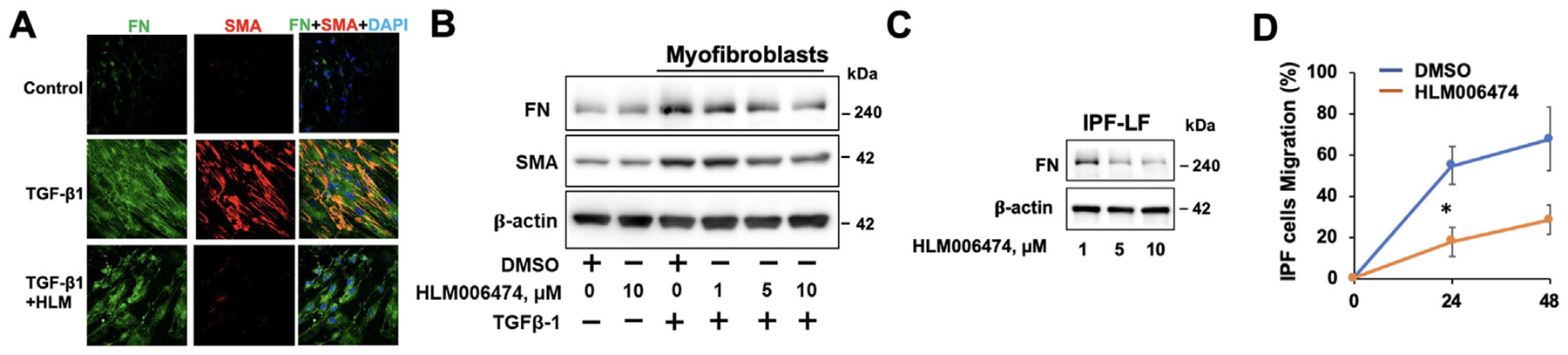
A. HLF cells were treated with TGF-β1 for 48 hour, and then cells were post-treated with HLM006474 for an additional 24 hour. FN and α-SMA were examined by immunofluorescence saining. B. Immunoblotting analysis of HLM006474-treated TGF-β1-induced lung myofibroblast cells. C. Immunoblotting analysis of HLM006474-treated IPF-LF cells. Immunoblots are representative of three independent experiments. D. IPF-LF cell migration was measured by a cell scratch assay. n=6, * P<0.01, compared to DMSO-treated cells.
Fig 7. HLM006474 diminishes experimental PF.
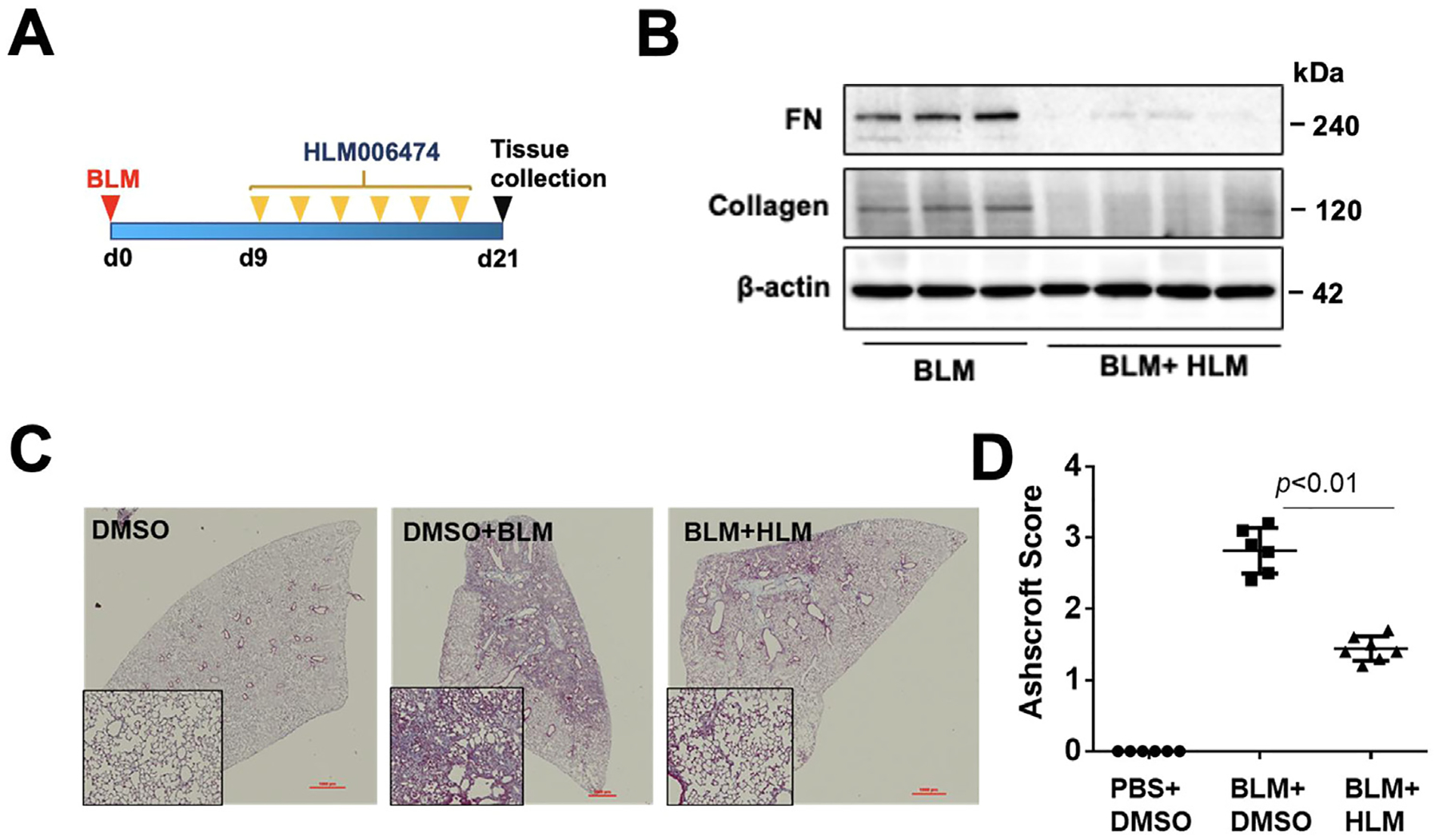
A. Scheme of experimental design. B. Immunoblotting analysis of lung tissues from day 21. BLM: bleomycin; HLM: HLM006474. n=3−4. C. Masson trichrome staining of lung tissues from day 21. Images are representative from 5−6 mice/group. D. Summary of the histopathological score from H&E staining. n=5−6.
Discussion
Nedd4L E3 ligase regulates various cellular functions through ubiquitinating multiple substrates. Here we demonstrate an anti-fibrotic effect of Nedd4L that is mediated by its ability to diminish TGF-β1 signaling. Importantly, Nedd4L expression in lung fibroblasts from IPF patients is reduced, and the extent of reduction correlates with markers of disease severity, suggesting that this reduction in Nedd4L contributes to the pathogenesis of pulmonary fibrosis. Supporting this role, we also show that Nedd4L is reduced in murine lung tissue following in vivo administration of bleomycin and murine fibroblasts isolated from fibrotic murine lungs. Consistently, Nedd4L is reduced by TGF-β1 via non-canonical E2F4-mediated gene suppression. Further, we show that TβRII is a novel target protein of Nedd4L ubiquitination. Collectively, this study suggests that prevention of Nedd4L suppression by inhibition of E2F represents a novel new therapeutic strategy for treatment of patients with IPF and other fibrotic lung diseases.
The ubiquitin-proteasome or lysosome systems regulate the majority of protein degradation. Abnormal expression of key enzymes in the protein degradation systems lead to the pathogenesis of human disorders. Nedd4L E3 ligase has been shown to be downregulated in lung tissues from IPF patients, consistent with our current finding. Kimura T. et al. reported that Nedd4L was reduced in lung epithelial cells in IPF patients. Repetitive lung epithelial injury and repair triggers initiation of fibrosis; however, myofibroblast differentiation is key step for the development of fibrosis. This study shows that Nedd4L levels are reduced in lung myofibroblasts from IPF patients, in an experimental animal model of PF, and in TGF-β1-differentiated myofibroblasts, indicating that Nedd4L reduction occurs in both lung epithelial and myofibroblast cells. This study also suggests that Nedd4L reduction contributes to both initiation and progression of PF.
Molecular regulation of Nedd4L has not been well studied. We screened multiple pathways and transcriptional factors and found that E2F4 potently suppressed Nedd4L in lung fibroblasts. The E2F family of transcription factors regulate cellular functions by modulating gene transcription.18–22 E2Fs, including E2F4, mediate TGF-β1-induced cell cycle arrest, apoptosis, and gene suppression in cancer cells.23–25 E2F has been shown to be novel fibrogenic genes that regulate cholestatic liver, renal, and pulmonary fibrosis.26–29 In our study, the E2F family inhibitor HLM006474 diminished lung fibrosis, supporting a key role for E2F family proteins in lung fibrosis. However, this broad-spectrum inhibitor may cause non-specific effects and cytotoxicity, which limits its therapeutic potential and highlights the need to develop specific inhibitors of E2F4 for interventional trials in fibrotic disorders.
Nedd4L targets multiple proteins in the TGF-β1 pathway, with prior studies demonstrating effects on TβRI and p-Smad2/3. This study reveals that TβRII is also a target protein for Nedd4L ubiquitination. Our previous study showed that TβRII stability is regulated by USP11-mediated deubiquitination. Here we show that Nedd4L ubiquitinates TβRII for its degradation. USP11 is increased in myofibroblasts30, while Nedd4L is reduced, suggesting that TβRII levels are regulated by the balance of Nedd4L and USP11. Together, Nedd4L/USP11-mediated TβRII stability may determine the activity TGF-β1 signaling pathways, thus, the development of PF. The interplay between Nedd4L and USP11 has not been studied. Our earlier study revealed that USP11 regulates E2F1 stability.30 Further, Qiao L, et al. reported that E2F1 increases USP11 transcription.31 The effect of USP11 on E2F4 stability has not been revealed. It is possible that E2F4 plays a role in the regulation of USP11 and Nedd4L expression.
Taken together, this study reveals that Nedd4L suppression in lung myofibroblast contributes to the development of PF, providing evidence that the development of a new strategy to rescue of Nedd4L may halt the pro-fibrotic progression of PF. E2F4 plays a central role in Nedd4L suppression in lung myofibroblast cells. Targeting E2F4 to rescue Nedd4L may be developed a new therapeutic strategy to treat IPF.
Supplementary Material
At A Glance Commentary
Li S, et al.
Background
The ubiquitin E3 ligase, Nedd4L, exhibits an anti-fibrotic property via targeting multiple substrates for their degradation; however, the regulation of Nedd4L expression in lung fibroblasts has not been studied. Mechanisms for regulation of TGF-β signaling pathway by Nedd4L have not been well revealed.
Translational Significance
Our study revealed that Nedd4L expression is suppressed in lung myofibroblasts, which are effector cells during lung fibrosis. E2F4 negatively regulates Nedd4L expression and contributes to pathogenesis of lung fibrosis. Further, we revealed that Nedd4L targets and ubiquitinates TGF-β receptor II, thus, attenuating TGF-β1 signaling in lung fibroblasts. This study provides evidence showing that upregulation of Nedd4L is a potential therapeutic strategy to treat fibrotic disorders including lung fibrosis.
Acknowledgment
This research was supported by National Institutes of Health, R01HL131665, R01HL136294, and R01HL157164 to Y.Z., R01GM115389 and R01HL151513 to J.Z. All the authors have read the journal’s authorship agreement and that the manuscript has been reviewed by and approved by all named authors.
Disclosures
D.J.K. reports collaborative research funding from Regeneron Pharmaceuticals in pulmonary hypertension, which is unrelated to this article. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
Abbreviations:
- IPF
idiopathic pulmonary fibrosis
- TGF-β1
transforming growth factor-β1
- TβRII
TGF-β receptor II
- ECM
extracellular matrix
- FN
fibronectin
- α-SMA
alpha-smooth muscle actin
- BLM
bleomycin
Footnotes
Supplementary materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.trsl.2022.10.002.
References
- 1.Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. The Journal of pathology 2013;229:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Therapeutic advances in respiratory disease 2010;4:367–88. [DOI] [PubMed] [Google Scholar]
- 3.Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proceedings of the American Thoracic Society 2012;9:130–6. [DOI] [PubMed] [Google Scholar]
- 4.Elmufdi F, Henke CA, Perlman DM, Tomic R, Kim HJ. Novel mechanisms and treatment of idiopathic pulmonary fibrosis. Discovery medicine 2015;20:145–53. [PubMed] [Google Scholar]
- 5.Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol 2003;284:F243–52. [DOI] [PubMed] [Google Scholar]
- 6.Walton KL, Johnson KE, Harrison CA. Targeting TGF-beta Mediated SMAD Signaling for the Prevention of Fibrosis. Frontiers in pharmacology 2017;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez IE, Eickelberg O. The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proceedings of the American Thoracic Society 2012;9:111–6. [DOI] [PubMed] [Google Scholar]
- 8.Hough C, Radu M, Dore JJ. Tgf-beta induced Erk phosphorylation of smad linker region regulates smad signaling. PloS one 2012;7:e42513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Y Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. The Journal of clinical investigation 2017;127:3675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.d’Azzo A, Bongiovanni A, Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 2005;6:429–41. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nature immunology 2012;13:651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao S Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Molecular cell 2009;36:457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuratomi G NEDD4–2 (neural precursor cell expressed, developmentally down-regulated 4–2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. The Biochemical journal 2005;386:461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J Destabilization of Lysophosphatidic Acid Receptor 1 Reduces Cytokine Release and Protects Against Lung Injury. EBioMedicine 2016;10:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang C, Kawabe H, Rotin D. The Ubiquitin Ligase Nedd4L Regulates the Na/K/2Cl Cotransporter NKCC1/SLC12A2 in the Colon. The Journal of biological chemistry 2017;292:3137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duerr J Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nature communications 2020;11:2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Yang Y, Yan H, Peng X, Zou J. NEDD4L-induced beta-catenin ubiquitination suppresses the formation and progression of interstitial pulmonary fibrosis via inhibiting the CTHRC1/HIF-1alpha axis. Int J Biol Sci 2021;17:3320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Helin K, Jin P, Nadal-Ginard B. Inhibition of in vitro myogenic differentiation by cellular transcription factor E2F1. Cell Growth Differ 1995;6:1299–306. [PubMed] [Google Scholar]
- 19.Wang J, Huang Q, Tang W, Nadal-Ginard B. E2F1 inhibition of transcription activation by myogenic basic helix-loop-helix regulators. J Cell Biochem 1996;62:405–10. [DOI] [PubMed] [Google Scholar]
- 20.Liew CW. Ablation of TRIP-Br2, a regulator of fat lipolysis, thermogenesis and oxidative metabolism, prevents diet-induced obesity and insulin resistance. Nature medicine 2013;19:217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Annicotte JS. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nature cell biology 2009;11:1017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto Y, Hayashi R, Kang D, Yoshida K. Acute loss of transcription factor E2F1 induces mitochondrial biogenesis in HeLa cells. J Cell Physiol 2006;209:923–34. [DOI] [PubMed] [Google Scholar]
- 23.Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002;110:19–32. [DOI] [PubMed] [Google Scholar]
- 24.Liu X TGF-beta induces growth suppression in multiple myeloma MM.1S cells via E2F1. Oncol Lett 2017;14:1884–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spender LC, Inman GJ. TGF-beta induces growth arrest in Burkitt lymphoma cells via transcriptional repression of E2F-1. The Journal of biological chemistry 2009;284:1435–42. [DOI] [PubMed] [Google Scholar]
- 26.Sheu CC. Gene Expression Changes Associated with Nintedanib Treatment in Idiopathic Pulmonary Fibrosis Fibroblasts: A Next-Generation Sequencing and Bioinformatics Study. J Clin Med 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X Overexpressing p130/E2F4 in mesenchymal stem cells facilitates the repair of injured alveolar epithelial cells in LPS-induced ARDS mice. Stem Cell Res Ther 2019;10:74. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Zhang Y E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr-1/SHP/EID1 network. Hepatology 2014;60:919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ouyang P Microarray analysis of differentially expressed genes in L929 mouse fibroblast cells exposed to leptin and hypoxia. Mol Med Rep 2017;16:181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacko AM. De-ubiquitinating enzyme, USP11, promotes transforming growth factor beta-1 signaling through stabilization of transforming growth factor beta receptor II. Cell death & disease 2016;7:e2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiao L The E2F1/USP11 positive feedback loop promotes hepatocellular carcinoma metastasis and inhibits autophagy by activating ERK/mTOR pathway. Cancer Lett 2021;514:63–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.