

Abstract
During transcriptional elongation, RNA polymerases (RNAP) employ a stepping mechanism to translocate along the DNA template while synthesizing RNA. Optical trapping assays permit the progress of single molecules of RNA polymerase to be monitored in real time, at resolutions down to the level of individual base pairs. Additionally, optical trapping assays permit the application of exquisitely controlled, external forces on RNAP. Responses to such forces can reveal details of the load-dependent kinetics of transcriptional elongation and pausing. Traditionally, the bacterial form of RNAP from E. coli has served as a model for the study of transcriptional elongation using optical traps. However, it is now feasible to perform optical trapping experiments using the eukaryotic polymerase, RNAPII, as well. In this report, we describe the methods to perform optical trapping transcriptional elongation assays with both prokaryotic RNAP and eukaryotic RNAPII. We provide detailed instructions on how to reconstitute transcription elongation complexes, derivatize beads used in the assays, and perform optical trapping measurements.
Keywords: single-molecule, optical trapping, RNA polymerase, RNA polymerase II, transcription
1. Introduction
Transcription is the first and the most highly regulated step in gene expression. The molecular motor responsible for transcription, RNA polymerase is the direct or indirect target of all forms of transcriptional regulation. The multi-subunit RNA polymerases are structurally and functionally conserved across all three domains of life: bacteria, archaea and eukaryotes [1]. Prokaryotic RNAP is the best-studied of the multi-subunit RNA polymerases, and has traditionally served as a model to analyze transcription. However, over the past few years, driven by improvements in biochemical methodologies and in vitro transcription assays, it is now possible to study messenger RNA (mRNA) production by eukaryotic RNA polymerase II (RNAPII), as well [2–6].
The process of transcription may be subdivided, broadly, into three distinct phases: initiation, elongation, and termination. Transcription elongation complexes (TEC) are typically stable and processive, transcribing up to several thousands of basepairs before releasing from the DNA template [7,5]. However, the processive motion is irregular, because it is frequently interrupted by transcriptional pausing, as RNA polymerase enters into one or more off-pathway states. In both prokaryotes and eukaryotes, transcriptional pausing can serve, among other things, as an important gene regulatory mechanism, by controlling the overall speed of elongation, and thereby the rate of production of cellular mRNA [8–11].
The motion of RNAP during its elongation phase is a process ideally suited for optical trapping studies [12–16]. Translocation not only involves significant displacement of RNAP, but also the generation of force, and both these quantities can be measured directly with optical traps. Using an optical trapping assay, an external force can be applied between the transcription elongation complex (TEC) and the template DNA in a direction that either assists or hinders transcription. The directionality and magnitude of the applied load can also affect the elongation kinetics, and particularly the propensity to enter into, or exit from, transcriptional pause states [6,8–11].
In the original single-molecule assay for TEC motion, a transcriptionally-stalled RNAP from E. coli was attached directly to the coverglass surface in a flow cell, while the downstream end of its associated DNA template (typically, a few kb in length) was attached to a gold particle, to produce a “tethered particle assay,” where the Brownian motion of the bead was constrained by the length of its tether to the surface [17]. Subsequent transcription, which could be re-activated triggered by introducing the four ribonucleotides (NTPs) into the flow cell, would shorten the tether, and thereby reduce the extent of the Brownian motion, which could be monitored by video analysis. In a subsequent adaptation of this assay that considerably improved upon its temporal and spatial resolution, an optical trap was used to capture the diffusing, tethered bead (now made of polystyrene), and subsequently used to stretch out the DNA tether under constant tension, thereby allowing elongation to be scored in real-time by simply measuring the trapped bead displacement [13,14]. The main limitation of the surface-tethered assay is mechanical stability of the coverglass surface, which can be subject to significant drift and vibrational noise.
The advent of the “dumbbell assay” circumvented the limitations of the surface-tethered assay, by optically levitating the assay components above the coverglass surface. Owing to its enhanced mechanical stability, the dumbbell assay has achieved, with appropriate instrumentation, angstrom-level resolution (over a bandwidth of ~100 Hz), which is more than adequate to detect transcriptional motion down to the level of individual basepairs. The dumbbell assay has therefore become the method-of-choice for analyzing transcriptional elongation and pausing [12,11]. In the assay (shown in Fig. 1), a molecule of biotin-labelled RNAP (transcriptionally stalled on its DNA template) is attached to a small polystyrene bead via a biotin-avidin linkage, while the upstream end of the digoxigenin-labelled DNA template is attached to a slightly larger bead, via a digoxigenin-antibody linkage. The entire bead-RNAP-DNA-bead assembly is levitated above the coverglass surface by dual optical traps, and held under constant tension by the use of a force clamp (this clamp may be active or passive; see [18,19]). Upon the addition of four NTPs, the transcriptionally stalled RNAP actively elongates, and its subsequent motion is monitored by measuring extensional changes in the DNA, which drive displacement of the trapped beads.
Fig 1.
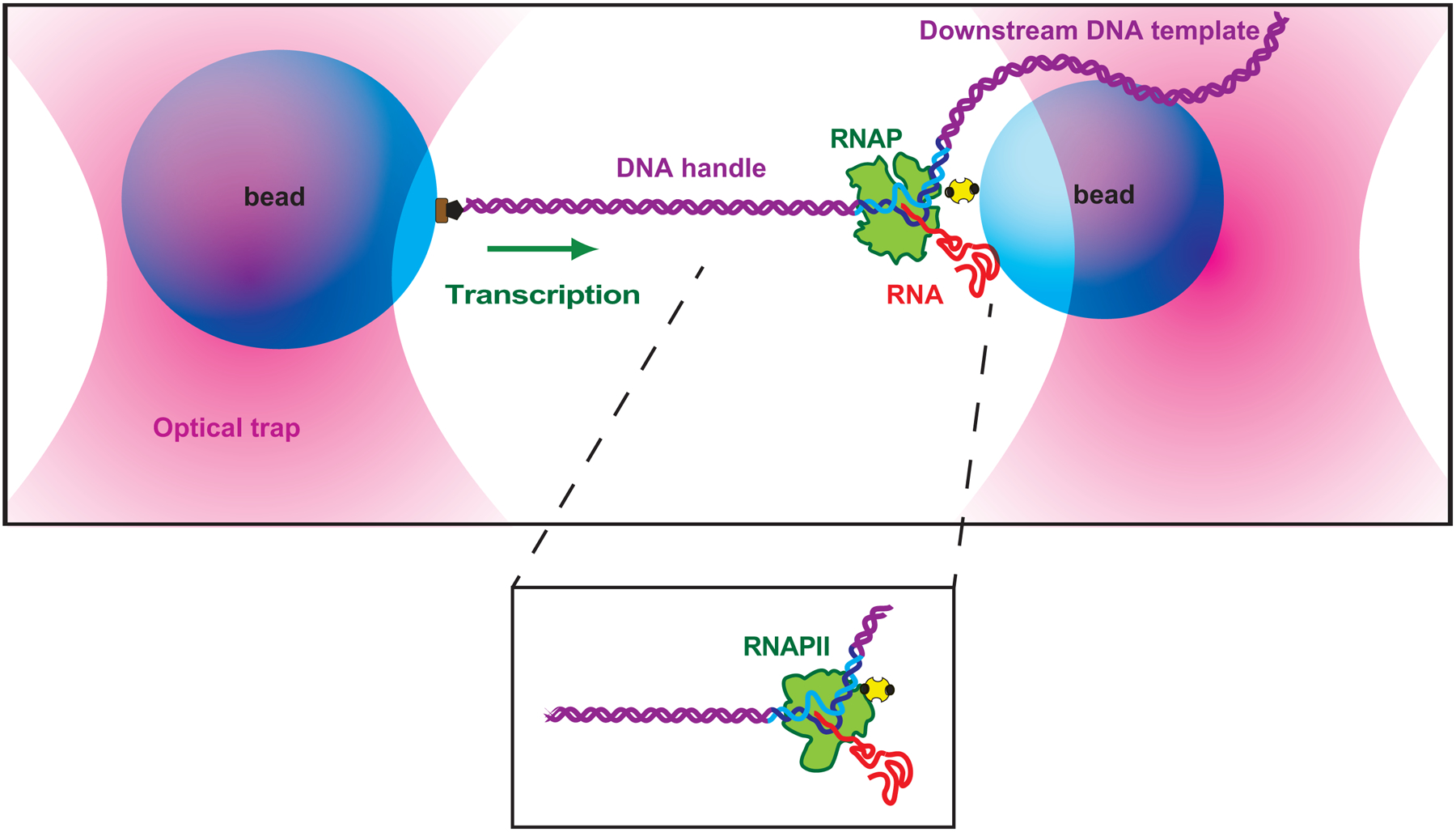
“dumbbell” tether (not to scale) is formed between two beads (blue) held in separate optical traps (pink), with one bead attached to RNAP (green) via an avidin–biotin linkage (yellow, black), and the other bead is attached to a upstream DNA handle (purple) via a digoxigenin linkage (brown, black). As transcription proceeds (green arrow indicates direction), the tether extension (inter-bead distance) increases. The same “dumbbell” geometry can be applied to study RNAPII TEC (see inset).
In this chapter, we describe protocols for performing two variants of the optical trapping dumbbell assay. The first is an assay designed to measure transcriptional elongation and pausing by E. coli RNAP under assisting loads [9]. The second is an assay designed to measure transcriptional elongation and pausing by Saccharomyces cerevisae RNAPII under assisting loads [4]. Although both the prokaryotic and eukaryotic versions of these assays exploit nearly identical experimental geometries, the procedures for assembling the enzyme complexes differ significantly. Transcriptionally stalled E. coli TECs can be reconstituted by the incubation of RNAP with a DNA template containing an appropriate promoter sequence, and the subsequent addition of a subset of the four NTPs, to walk it out to a known location near the promoter (Fig. 2a) [11]. An analogous strategy to prepare S. cerevisiae TEC is not feasible [5]: under single-molecule conditions, RNAPII molecules only rarely bind the promoter region and ultimately enter into a state of elongation. Instead, RNAPII typically requires the presence of at least five general transcription factors in order to bind the promoter and transition to a functional TEC [20]. General transcription factors may be omitted from RNAPII assays, however, by using a promoter-less approach: one that involves assembling an artificial transcription bubble that mimics the transcriptional complex configuration of the elongation phase, consisting of a DNA “scaffold” and a short RNA oligomer, to function as a nascent transcript (Fig. 2b) [4,21–23]. When appropriately produced, both RNAP and RNAPII elongation complexes produce reasonable density of tethers that can be optically trapped in the dumbbell assay. However, the RNAPII assay for S. cerevisiae is less efficient: Upon the addition of all four NTPs, the fraction of active TECs is much lower than for RNAP from E. coli.
Fig. 2.
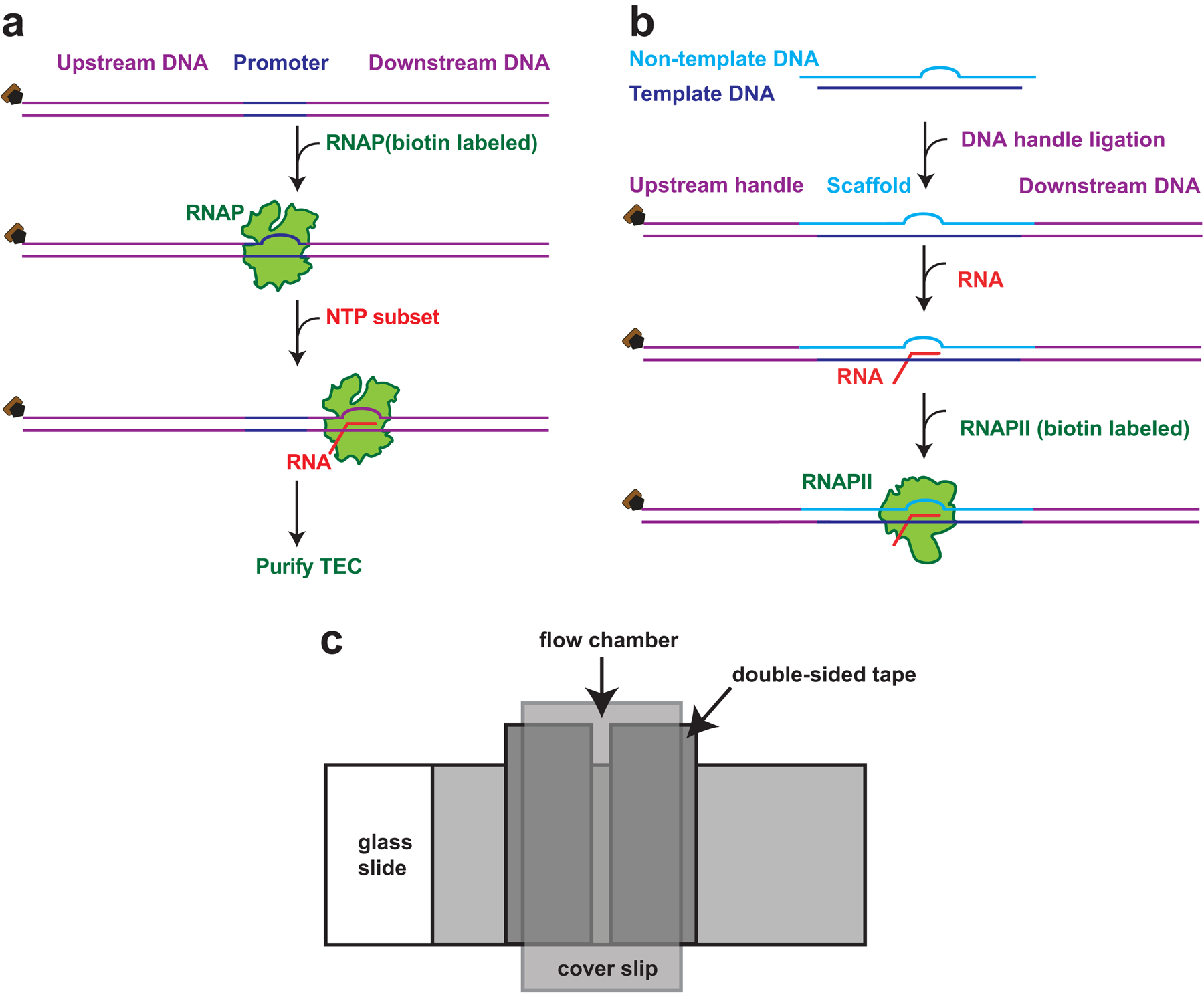
(a). Schematic representation of steps to assemble E. coli TEC. A DNA template (purple) containing a promoter sequence (yellow) is incubated with a RNAP and a subset of NTPs. RNAP is stalled at the nucleotide deficient site and forms a TEC, which can then be purified and used for optical trapping. (b). Schematic representation of steps for reconstituting RNAPII TEC. Non-template DNA (light blue) containing an 11 nt of non-complementary sequence anneals to a template DNA (dark blue) and forms a scaffold containing an artificial transcription bubble. The scaffold is ligated to an upstream DNA handle and a downstream DNA template (both in purple), and annealed to a 14 nt RNA fragment (red). Biotin labeled RNAPII (green) is incubated with the scaffold to forms the TEC. (c). Schematic diagram of a typical flow cell assembly. Two strips of double-sided tape, separated by 1.5–2 mm, are applied to a glass slide. A plasma-cleaned cover glass is then applied to the double-sided tape, allowing a flow chamber to be formed between glass slide and cover glass. The assembly is then pressed to seal the flow chamber.
2. Materials
2.1. Optical trapping assay for E. coli RNA polymerase transcription elongation
2.1.1. Preparation of DNA template
Microcentrifuge tube
Plasmid pALB3 (see Note 1)
Forward primer: 5ʹ-GCCCGACCGCTGCGCCTTATCC-3ʹ (see Note 2)
Reverse primer: 5ʹ-CGCCCAATACGCAAACCGCCTCT-3ʹ with a 5ʹ-digoxigenin modification (see Note 2)
Deoxynucelotide mix: 2.5 mM dATP, 2.5 mM dGTP, 2.5 mM dCTP, 2.5 mM dTTP
Deionized water
OneTaq polymerase kit (New England Biolabs)
Thermal cycler
Qiaquick PCR purification kit (Qiagen)
2.1.2. Preparation of RNA polymerase transcription elongation complex
ACG ribonucelotide mix: 25 μM ATP, 25 μM CTP, 25 μM GTP
31.5 mM initiating dinucleotide: ApU
1 mg/ml nuclease-free acetylated BSA
50% glycerol
Biotin-labeled E. coli RNAP
T7A1 buffer: 20 mM Tris-HCl, pH 8.0, 14 mM MgCl2, 20 mM NaCl, 14 mM 2-mercaptoethanol, 0.1 mM EDTA pH 8.0.
PHC column buffer: 50 mM HEPES, pH 8.0, 130 mM KCl, 4 mM MgCl2, 0.1 mM EDTA, pH 8.0, 20 μg/ml acetylated BSA, 4% glycerol
CL-6B sepharose (Sigma-Aldrich)
Geiger counter
2.1.3. Coupling anti-digoxigenin to carboxyl-polystyrene beads
Carboxyl-polystyrene beads, 0.915 μm average diameter (Bangs)
anti-digoxigenin antibody (Roche)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide·HCl (EDC)
N-Hydroxysulfosuccinimide (Sulfo-NHS)
1 M glycine
Tween-20
MES buffer: 100 mM MES, pH 5, 0.5% tween
Phosphate buffer: 100 mM NaH2PO4, pH 7.5, 0.5% tween
2-mercaptoethanol
Microcentrifuge
Tube rotator
Sonicator
2.1.4. Coupling avidin to carboxyl-polystyrene beads
Carboxyl-polystyrene beads, 0.6 μm average diameter (PolyMicrospheres)
5 bottles of 1 mg/ml Avidin-DN (Vector Laboratories)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide·HCl (EDC)
N-Hydroxysulfosuccinimide (Sulfo-NHS)
10 mg/ml biotin-X-cadaverin (Life Technologies; see Note 3)
1 M glycine
Tween-20
MES buffer: 100 mM MES, pH 5, 0.1% tween
Phosphate buffer: 100 mM NaH2PO4, pH 7.5, 0.1% tween
Microcentrifuge
Tube rotator
Sonicator
2.1.5. Flow cells preparation
Microscope cover glass (22 mm×40 mm×0.15 mm; Fisherbrand)
Plasma cleaner (Harrick Plasma PDC-001)
Ceramic rack for cover glass
Double-sided tape (0.5 inch width)
Microscope slides (1 mm thick, 25×75 mm; Thermo Scientific)
100% ethanol
2.1.6. Single-molecule optical tweezers assay
Anti-digoxigenin-coasted polystyrene beads, 0.915 μm average diameter
Avidin-coated polystyrene beads, 0.6 μm average diameter
E. coli transcription buffer: 50 mM HEPES pH 8.0, 130 mM KCl, 4 mM MgCl2, 0.1 mM EDTA, 1 mM DTT
Nuclease-free acetylated BSA
E. coli transcription buffer supplemented with 3 mg/ml BSA (see Note 4)
Trapping buffer: E. coli transcription buffer supplemented with 8.3 mg/ml glucose (Sigma), 46 U/ml glucose oxidase (Calbiochem), 94 U/ml catalase (Sigma Aldrich) (see Note 5)
1 mM ribonucleotide triphosphate solution (NTP): 1 mM ATP, 1 mM GTP, 1 mM CTP and 1 mM UTP
3 μM biotin-labeled E. coli RNAP
Home-built high-resolution dual-beam optical trap: the core components include 1064 nm ND:YVO4 laser (Spectra-Physics, T-Series), inverted microscope (Nikon), Nikon oil immersion objective (1.4 NA, APO, PLAN), eight-pole low-pass Bessel filter (Krohn-Hite), position-sensitive detector (Pacific Silicon Sensors) and acousto-optical deflectors (IntraAction) [15,16]. Refer to Fig. 3 for other optical components.
Fig. 3.
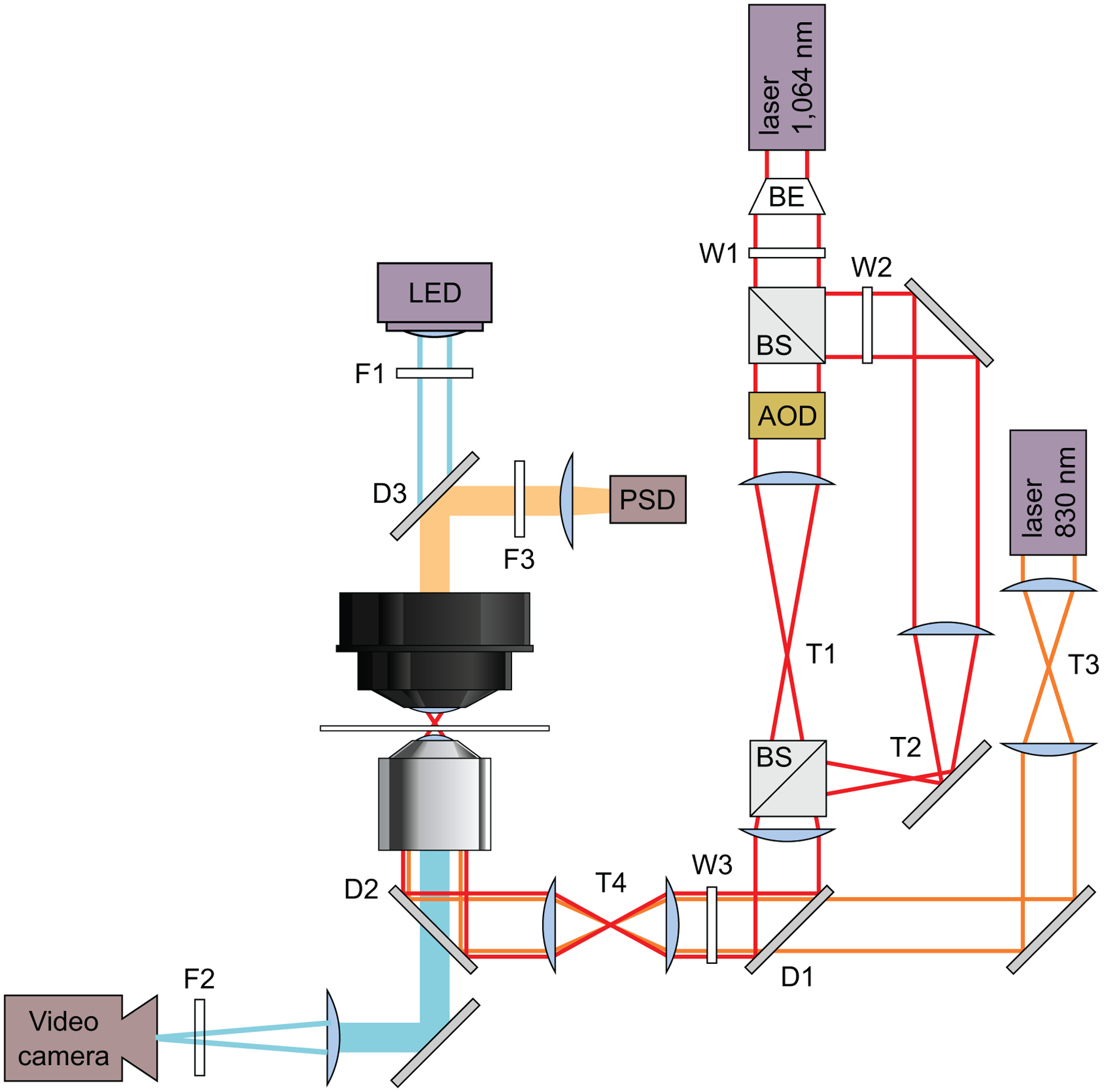
Trapping optics: Schematic optical layout of the dual-beam optical traps. Solid lines indicate lasers and light sources: 830 nm detection laser (orange), 1064 nm trapping laser (red), and illuminating LED (blue). Filled bars are emissions received by detectors: position sensitive detector (PSD), and a video camera. Trapping laser is expanded using a beam expander (BE), and then split by a polarizing beam splitting cube (BS). The strong trapping beam is then steered in position by acousto-optical deflectors (AOD), thus making it movable. A telescope T3, is used for manually steering and aligning detection beam position in the specimen plane. Similarly, telescope T4 is used for crude aligning both trapping and detection lasers in specimen plane. Filters (F), Wollaston prisms (W), pinholes (P), dichroic mirrors (D), mirrors (M), and telescopes (T) are labeled. In brief, F1 is used to adjust brightness of microscope illumination. F2 & F3 are IR filters (1064 nm) to shield video camera and PSD from the trapping laser.
2.2. Optical trapping assay for S. cerevisiae RNAPII transcription elongation
2.2.1. Preparation of DNA template
Microcentrifuge tube
Plasmid pPM172 (see Note 6)
Forward primer: 5ʹ-CACAGGCGCGCCCACGGG GTGAGCAGTCACG-3ʹ (see Note 7)
Reverse primer: 5ʹ-TGCGGCGGGAACACAACTGG-3ʹ (see Note 7)
Deoxynucelotide mix: 2.5 mM dATP, 2.5 mM dGTP, 2.5 mM dCTP, 2.5 mM dTTP
deionized water
OneTaq polymerase kit (New England Biolabs)
Thermal Cycler
Qiaquick PCR purification kit (Qiagen)
Restriction enzyme: DraIII (see Note 8)
UV spectrophotometer
2.2.2. Preparation of DNA handle
Microcentrifuge tube
Plasmid pRL702
Forward primer: 5ʹ-CAGCGGTAATTCCGAGCTGCA-3ʹ (see Note 9)
Reverse primer: 5ʹ-CGATTTCCAGCGCACGTTTGTC-3ʹ with a 5ʹ-digoxigenin modification (see Note 9)
10 mM Deoxynucelotide (dNTP) mix: 2.5 mM dATP, 2.5 mM dGTP, 2.5 mM dCTP, 2.5 mM dTTP
deionized water
OneTaq polymerase kit (New England Biolabs)
Thermal Cycler
Qiaquick PCR purification kit (Qiagen)
Restriction enzyme: StyI (see Note 10)
UV spectrophotometer
2.2.3. DNA scaffold assembly
Microcentrifuge tube
Template oligo: 5ʹ-GCTTTTCGCCTTGTCGGCTGCGCGTCGGTGGGTGTTTCCTGATGGCTGTTTGTTTCCTATAGC-3ʹ (see Note 11)
Non template oligo: 5ʹ-CAAGGCTATAGGAAACAATGTCGGTAGACGAAACACCCACCGACGCGCAGCCGACAAGGCGAAAAGCGGG-3ʹ (see Note 11)
Reconstitution Buffer (RB): 10 mM Tris-HCl, pH 7.9, 40 mM KCl
Deionized water
Thermal Cycler
T4 DNA ligase
T4 DNA ligase buffer
Agarose
TAE buffer: Tris-acetate-EDTA
1 kb plus DNA ladder (Life Technologies)
SYBR Gold stain (ThermoFisher Scientific)
2.2.4. Reconstitution of RNAPII transcription elongation complex
Yeast elongation buffer (YEB): 25 mM HEPES, pH 8.0, 130 mM KCl, 5 mM MgCl2, 0.15 mM EDTA, 1 mM DTT, 5% glycerol, 0.125 mg/ml BSA
2 μM RNA: 5ʹ-UUUUUACAGCCAUC-3ʹ
300 μM S. cerevisiae RNAPII
ACG ribonucelotide mix: 4 mM ATP, 4 mM CTP, 4 mM GTP
Liquid nitrogen
2.2.5. Single-molecule optical trapping assay
Anti-digoxigenin-coasted polystyrene beads, 0.915 μm average diameter (see subheading 2.1.3)
Avidin-coated polystyrene beads, 0.6 μm average diameter (see subheading 2.1.4)
S. cerevisiae transcription buffer: 20 mM HEPES (pH 8.0), 80 mM potassium acetate, 5 mM magnesium acetate, 0.1 mM EDTA, and 0.1 mM DTT
Nuclease-free BSA
S. cerevisiae transcription buffer supplemented with with 3 mg/ml BSA
Trapping buffer: S. cerevisiae transcription buffer supplemented with 8.3 mg/ml glucose (Sigma), 46 U/ml glucose oxidase (Calbiochem), 94 U/ml catalase (Sigma) (see Note 5)
1 mM ribonucleotide triphosphate solution (NTP): 1 mM ATP, 1 mM GTP, 1 mM CTP and 1 mM UTP
300 nM biotin-labeled S. cerevisiae RNAPII.
Home-built high-resolution dual-beam optical trap (see subheading 2.1.6 step 9)
3. Methods
3.1. Optical trapping assay for E. coli RNA polymerase transcription elongation
3.1.1. Preparation of DNA template
In a PCR microcentrifuge tube, prepare a 50 μl reaction mixture containing 0.2 ng/ml plasmid pALB3, 0.2 μM forward primer, 0.2 uM reverse primer, 0.8 mM of dNTP mix, 10 units of OneTaq DNA polymerase in 1X standard Taq buffer. (see Note 1–2)
Perform PCR amplification using an initial denaturation at 94 °C for 5 min and 34 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min, and extension at 72 °C for 6 s, followed by a final extension for 10 min at 72 °C.
Purify the 4750 bp DNA fragment using Qiaquick PCR purification kit. Determine the concentration of the DNA fragment by measuring UV absorbance using a spectrophotometer.
3.1.2. Reconstitution of RNA polymerase transcription elongation complex
Prepare a 200 μl transcription reaction mixture containing 2.5 μM ATP, 2.5 μM GTP, 2.5 μM CTP, 250 μM ApU, 10 nM DNA template, 20 μg/ml BSA, 1.5% glycerol, 30 μCi [α32P]-GTP, and 50 nM biotin-labeled E.coli RNAP, in T7A1 buffer.
Incubate the transcription reaction mixture at 37 °C for 20 min.
Apply 200 μl reaction mixture from step 1, to a 12 ml CL-6B sepharose column (pre-equilibrated with 100 ml PHC column buffer).
Elute the column with 4 ml PHC column buffer. Collect 300 μl fractions in microcentrifuge tubes.
Identify fractions containing TEC by measuring the radioactivity signal in each tube with a Geiger counter.
3.1.3. Coupling anti-digoxigenin to carboxyl-polystyrene bead
Remove polystyrene beads container from fridge, and vortex the container for 30 s. Sonicate the container in ice water for 3 min.
In a 1.5 ml microcentrifuge tube, add 150 μl polystyrene beads, and 150 μl MES buffer. Mix well by inverting the tube.
Centrifuge the beads at 8500 × g for 3 min. Remove the supernatant and resuspend the beads in 200 μl MES buffer. Repeat this step twice.
In a separate 1.5 ml microcentrifuge tube, add 900 μl MES buffer and 2.1 mg sulpho-NHS. Vortex the solution and add to the beads in step 3.
Add 5 mg EDC to the bead mixture. Vortex the tube to mix the contents, and then rotate the tube at room temperature for 30 min. (This reaction couples an amine-reactive group to the beads).
Quench the reaction by adding 1 μl 2-mercaptoethanol to the bead mixture. Vortex and rotate the tube at room temperature for 10 min.
Centrifuge the beads at 8500 × g for 5 min. Remove the supernatant and resuspend beads in 200 μl phosphate buffer. Repeat this step twice. After the last centrifugation, resuspend the beads in 500 μl phosphate buffer.
Separately, add 1 ml phosphate buffer to a microcentrifuge tube containing 200 μg anti-digoxigenin antibody. Dissolve the anti-dioxigenin antibody by rotating the tube at room temperature for 20 min.
Add the anti-digoxegenin antibody to the beads from step 7. Rotate the tube for 2 h at room temperature.
Quench the reaction by adding 30 μl 1 M glycine. Rotate the tube for 10 min at room temperature.
Centrifuge the bead mixture at 8500 × g for 6 min. Remove the supernatant and resuspend the beads in 200 μl phosphate buffer. Repeat this step four times.
Label and store the solution from step 11 on a rotator at 4 °C.(see Note 12)
Transfer 50 μl beads to a new microcentrifuge tube.
Centrifuge the beads at 8500 × g for 6 min. Remove supernatant and resuspend the beads in 600 μl phosphate buffer. Repeat this step four times. After the centrifugation, resuspend the beads in 50 μl phosphate buffer.
Sonicate the beads in ice water for 3 min. (see Note 13)
3.1.4. Coupling avidin to carboxyl-polystyrene beads
Remove polystyrene beads container from fridge, and vortex the container for 30 s. Sonicate the container in ice water for 3 min.
In a 15 ml tube, combine 200 μl beads, 200 μl 10 mg/ml biotin-X-cadaverin solution, 20 mg Sulfo-NHS, and 1.6 ml MES buffer. Mix by inverting the tube. Add 50 mg EDC. Vortex the falcon tube to mix the contents.
Rotate the tube for 16 h at room temperature.
Quench the reaction by adding 500 μl 1 M glycine. Rotate the tube for 15 min at room temperature.
Prepare five 500 μl aliquots of the beads in 1.5 ml microcentrifuge tubes.
Centrifuge the bead aliquots at 8500 × g for 5 min. Remove the supernatants and resuspend each pellet in 200 μl phosphate buffer. Repeat this step four times. After the last centrifugation, resuspend each bead pellet in 500 μl phosphate buffer. Sonicate the tube in ice water for 2 min.
Add each of the bead suspensions to a corresponding tube of 1 ml 1 mg/ml Avidin-DN. Mix quickly with a pipette and transfer each mixture to a 1.5 ml microcentrifuge tube. (see Note 14) Label and store the tubes on a rotator at 4 °C. Incubate, at least overnight, before the next step. (see Note 14)
Take one tube from step 7.
Centrifuge the beads at 8500 × g for 6 min. Remove supernatant and resuspend the beads in 600 μl phosphate buffer. Repeat the step four times. After the last centrifugation, resuspend the beads in 40 μl phosphate buffer. Sonicate the tube in ice water for 3 min. (see Note 13)
3.1.5. Flow cell preparation
Place a microscope cover glass in a ceramic cover glass rack.
Place the rack in the plasma cleaning chamber.
Start the vacuum pump. Adjust the air intake until pressure inside the chamber drops to 1 Torr. Expose the cover glass to plasma discharge for 5 min.
Use the cleaned cover glass immediately, or alternatively, store them in a closed container.
Use 100% ethanol to clean a designated bench surface.
Place a glass slide on the cleaned bench surface. On the glass slide, apply two parallel strips of double-sided tape separated by 1–2 mm.
Place a plasma-cleaned cover glass on the tape strips and press down to form seal. A chamber thus formed, can retain of 5–7 μl sample. See Fig. 2c for schematic diagram of a flow cell.
The assembled cover glass can be readily used or stored in closed container for future usage.
3.1.6. Single-molecule optical trapping assay
Add 2 μl anti-digoxigenin-coated polystyrene beads and 2 μl avidin-coated polystyrene beads in two separate 0.5 ml microcentrifuge tubes. Resuspend the beads in each tube in 60 μl E. coli transcription buffer supplemented with 3 mg/ml BSA. (see Note 16)
Centrifuge the beads at 8500 × g. Remove the supernatant and resuspend each bead pellet in 2 μl transcription buffer. Sonicate on ice water for 5 min at 50% intensity. After sonication, place the beads on ice.
In a microcentrifuge tube, mix, anti-digoxigenin-coated polystyrene beads, avidin-coated polystyrene beads and 2 μl TEC (from subheading 3.1.2). Incubate at room temperature for 40 min. (see Note 17) At this stage, the “dumbbell” (bead-RNAPII-DNA-bead complex) is formed (Fig. 1) and is ready for optical trapping.
In a 0.5 ml microcentrifuge tube, add 1 μl of the bead-RNAPII-DNA-bead complex and 30 μl E. coli trapping buffer. Mix gently with a pipette and keep on ice.
Fill a flow cell channel with 7 μl suspension from step 4.
Prepare 30 μl 1 mM NTP solution in E. coli trapping buffer. Place on ice. (see Note 18)
Mount the flow cell on the microscope stage. Adjust the objective and condenser to bring the surface of the cover glass into focus.
Use a customized LabView program to maneuver the optical trap to pick up a free-floating 0.6 μm bead, and perform position calibration. (see Note 19)
Flow 7 μl 1 mM NTP solution into the sample chamber.
Identify a candidate tether. (A typical candidate tether is represented by a 0.6 μm bead floating in the vicinity of a surface-adhered 0.915 μm bead.)
First, maneuver static optical trap to capture the floating 0.6 μm bead in the candidate tether, then maneuver the steerable optical trap to capture the surface-adhered 0.915 μm bead. Monitor the increase in tether extension while ramping up the force (up to a maximum force of 15 pN). Plot force parameterized by extension to generate a force extension curve (FEC). Fit the curve to a worm-like chain (WLC) model of DNA elasticity and determine the persistence length of the DNA (The WLC model provides a relation between force, extension and persistence length of DNA.) [24]. For a single tether, the appropriate persistence length is >20 nm and the tether extension is within 50 nm of expectation.
Upon finding an appropriate tether, turn on active force clamp program at the desired force (5–30 pN). (see Note 20)
Analyze the data using established methods using a customized program (see Note 21) [12,9,11,4]. See Fig. 4a) for representative traces.
Fig. 4.

Representative traces of transcription elongation are shown in panel (a) (RNAP) and (b) (RNAPII). Tether length is converted to RNAPII’s position along the DNA template and plotted as a function of time. The change in translocational position of RNAP with time shows transcription elongation which is occasionally interrupted by ubiquitous pauses (red arrows).
3.2. Optical trapping assay for S. cerevisiae RNAPII transcription elongation
3.2.1. Preparation of DNA template
In a PCR microcentrifuge tube, prepare a 50 ul reaction mixture containing 8 μl 10 ng/μl plasmid pPM172, 2.5 μl 10 μM forward primer, 2.5 μl 10 uM reverse primer, 1 μl 10 mM dNTP mix (10 mM each of dATP, dCTP, dTTP and dGTP), 5 μl 10X standard Taq reaction buffer and 10 units of OneTaq DNA polymerase. (see Note 6–7)
Perform PCR amplification using an initial denaturation at 94 °C for 30 s and 30 cycles of denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s, and extension at 65 °C for 5 min, followed by a final extension at 65 °C for 9 min.
Purify the 4750 bb DNA fragment using Qiaquick PCR purification kit.
Digest the purified DNA fragment with DraIII, following manufacturer’s protocol for 3 h at 37°C. (see Note 8)
Purify the digested DNA fragment using Qiaquick PCR purification kit. Determine the concentration of the DNA fragment by measuring UV absorbance using spectrophotometer.
3.2.2. Preparation of DNA handle
In a PCR microcentrifuge tube, prepare a 50 ul reaction mixture containing 8 μl of 10 ng plasmid pPM172, 2.5 μl 10 μM forward primer, 2.5 μl 10 uM reverse primer,1 μl 10 mM dNTP mix, 5 μl 10X standard Taq reaction buffer and 10 units of OneTaq DNA polymerase. (see Note 9)
Perform PCR amplification using an initial denaturation at 94 °C for 30 s and 30 cycles of denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s, and extension at 65°C for 3 min, followed by a final extension at 65 °C for 5 min.
Purify the 2700 bp DNA fragment using Qiaquick PCR purification kit.
Digest the purified DNA fragment with StyI for 3 h at 37 °C following manufacturer’s protocol. (see Note 10)
Purify the digested DNA fragment using Qiaquick PCR purification kit. Determine the concentration of the DNA fragment by measuring UV absorbance using a spectrophotometer.
3.2.3. DNA scaffold assembly
In a PCR microcentrifuge tube, mix 1 μl 5 μM template oligo, 1 μl 5 μM non-template oligo, 2 μl 5X RB and 6 μl water. (see Note 11)
Anneal the oligos in a thermal cycler by incubating the mixture at 45 °C for 5 min and then cooling down to 25 °C at the rate of 1 °C/min. This produces 5 μM scaffold.
In a microcentrifuge tube, mix 0.4 μM scaffold, 0.4 μM DNA handle, 0.4 μM DNA template, 400 units T4 DNA ligase in T4 DNA ligase buffer to a total reaction volume of 50 μl. Incubate the mixture 16 h at 4 °C.
Prepare a 0.8% agarose gel in TAE buffer. In consecutive wells, load (i)1 Kb plus DNA ladder, (ii) 5 μl ligation product (from step 3), (iii) 22.5 μl ligation product (from step 3), (iv) 22.5 μl ligation product (from step 3). Run the gel at 100 V for 1.5 h.
Excise lanes (i) and (ii). Stain the excised lanes in SYBR Gold solution (following manufacturer’s staining protocol), and identify the 7500 bp DNA band. Use this stained gel fragment as a reference to excise the 7500 bp bands from the unstained gel fragment.
Extract the 7500 bp band from the agarose gel using the Qiaquick Gel Extraction Kit. Elute the band in 30 μl EB following manufacturer’s protocol.
3.2.4. Reconstitution of transcription elongation complex
In a microcentrifuge tube, mix 30 μl gel extracted ligated product (from subheading 3.2.3), 8 μl 5X YEB, and 1 μl of 2 μM RNA. Incubate 45 min at 30 °C.
Add 1 μl 300 nM RNAPII. Mix well and incubate 15 min at 30 °C.
Add 1 μl ACG mix and incubate 30 min at 30 °C.
Prepare 2 μl aliquots of the RNAPII TEC and freeze the aliquots in liquid nitrogen. Store at −80 °C.
3.2.5. Single-molecule optical trapping assay
Prepare anti-digoxigenin-coated polystyrene beads and avidin-coated polystyrene beads as described in subheadings 3.1.3 and 3.1.4.
Prepare flow cells as described in subheading 3.1.5.
Add 2.0 μl anti-digoxigenin-coated polystyrene beads and 2.0 μl avidin-coated polystyrene beads in two separate 0.5 ml microcentrifuge tubes. Resuspend the beads in each tube in 60 μl S. cerevisiae transcription buffer supplemented with 3 mg/ml BSA. (see Note 4)
Centrifuge the beads at 8500 × g. Remove the supernatant and resuspend each bead pellet in 2 μl S. cerevisiae transcription buffer. Sonicate on ice water for 5 min at 50% intensity. After sonication, place the beads on ice.
Add 2 μl RNAPII TEC (from subheading 3.2.4) to the avidin-coated beads. Gently mix with a pipette, and incubate at room temperature for 40 min. (see Note 14)
Centrifuge the RNAPII-bead mixture at 8500 × g for 30 s. Remove the supernatant and resuspend the beads in 1 μl S. cerevisiae transcription buffer. Sonicate the tube on ice water for 2 min at 50% intensity.
Add the anti-digoxigenin-coated beads from step 2. Mix gently with a pipette. Incubate at room temperature for 30 min and then place the tube on ice. At this stage, the “dumbbell” (bead-RNAPII-DNA-bead complex) is formed (see Fig. 2b) and is ready for optical trapping.
In a 0.5 ml microcentrifuge tube, add 1 μl of the bead-RNAPII-DNA-bead complex and 30 μl oxygen-scavenging system. Mix gently with a pipette and keep on ice.
Fill a flow cell channel with 7 μl suspension from step 7.
Prepare 30 μl 1 mM NTP solution in S. cerevisiae transcription buffer. Place on ice. (see Note 18)
Mount the flow cell on the microscope stage. Adjust the objective and condenser to bring the surface of the cover glass into focus.
Use a customized LabView program to maneuver the optical trap to pick up a free-floating 0.6 μm bead, and perform position calibration. In brief, a second, low-power detection laser is deployed to perform laser-based position detection. (see Note 19)
Flow 7 μl 1 mM NTP solution into the sample chamber.
Perform steps 10–13 in subheading 3.1.6.
4. Notes
The plasmid pALB3 encodes a bacterial template DNA containing the following modules in succession: (i) a 2640 bp upstream DNA sequence, (ii) a T7A1 promoter DNA, (iii) a 1693 bp downstream DNA sequence from the E. coli rpoB gene.
The forward primer and the reverse primer are used amplify a 4408 bp DNA fragment.
Biotin-X-cadaverine is reconstituted in DMSO.
BSA concentration in this buffer is critical for achieving optimal stickiness between the bead and the surface. With too high BSA concentration, most of the beads would not adhere to the surface. Conversely, with low BSA concentration, beads may adhere too strongly to surafce, making it very difficult to levitate the beads off of the surface using optical traps. If optical trapping is performed in presence in of other proteins and/or other buffer conditions, the optimal BSA concentration at this step may have to be reassessed.
Optical trapping can produce free radicals. This buffer contains a glucose/glucose oxidase/catalase mediated oxygen scavenging system to prevent the oxidative damage. Tyically the oxygen scavenging components can last for 1.5 h on ice, after which the pH of the solution may decrease. Thus, the buffer needs to be made fresh every 1.5 h.
Plasmid pPM172 contains a 4750 bp transcription template which consists of a fragment from the human PLR2A gene encoding the UTR and several exons and introns.
The forward primer and the reverse primer are used to amplify a 4750 bp DNA fragment.
Restriction digestion with DraIII creates a 5’ sticky end, capable of ligating to the DNA scaffold.
The forward primer and the reverse primer are used amplify a 2700 bp DNA fragment
Restriction digestion with StyI creates a 3’ sticky end, capable of ligating to the DNA scaffold.
Annealing of the template oligo produces a 70 bp scaffold which contains: (i) a 5’ DraIII sticky end, (ii) an artificial transcription bubble, (iii) a 3’ StyI sticky end.
At this stage, the beads can stay on the rotator at 4 °C for at least 6 months.
At this stage, the beads are ready for use and can last for at least one month on rotator at 4 °C.
When mixing, avoid introducing bubbles to the solution.
At this stage, the beads can stay on the rotator and last for at least 6 months.
In preparation to assemble the “dumbbell” tether complex, home-made polystyrene beads derivatized with avidin or anti-digoxigenin linkages need to be washed. This step required to suspend the beads in transcription buffer, and to coat the beads with BSA.
This step allows RNAP to form biotin-avidin linkage with the beads.
The solution should be used within 1.5 h. After 1.5 h on ice, the oxygen scavenging system is exhausted.
In brief, a low-power detection laser is deployed to perform position calibration which is achieved by performing back focal plane detection of the signal from interference of detection laser beam [15]. The interference is generated between forward-scattered detection laser light from the bead and unscattered light. To capture the signal, position sensitive detectors (PSD) are mounted at a plane that is conjugate to the back focal plane of the condenser. The main advantage of back focal plane detection scheme is that it is sensitive to the bead displacement from laser beam axis and insensitive to absolute bead position.
In brief, the customized LabView force clamp program actively measures tether extension, adjusts the steerable trap’s position through a feedback loop to maintain a constant tension on the “dumbbell” tether. The program records the change in tether extension in real time. This permits the monitoring of RNAP translocation as a continuous increase in DNA extension.
In order to identify the sequence-dependent kinetics of transcriptional elongation, it is necessary to convert the change in tether extension in nanometers to translocational position of RNAP in base pairs. This conversion requires the fitting of a DNA to a WLC model and subsequent estimation of the rise per base pair corrsponding to the assisting load (i.e. the force used in the force clamp program).
Acknowledgements:
This work was supported by grant number R37 GM057035-22 from the National Institute of General Medical Sciences to S.M.B.
References
- 1.Decker KB, Hinton DM (2013) Transcription regulation at the core: similarities among bacterial, archaeal, and eukaryotic RNA polymerases. Annu Rev Microbiol 67:113–139. doi: 10.1146/annurev-micro-092412-155756 [DOI] [PubMed] [Google Scholar]
- 2.Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C (2009) Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science 325 (5940):626–628. doi: 10.1126/science.1172926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horn AE, Goodrich JA, Kugel JF (2014) Single molecule studies of RNA polymerase II transcription in vitro. Transcription 5 (1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larson MH, Zhou J, Kaplan CD, Palangat M, Kornberg RD, Landick R, Block SM (2012) Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II. Proc Natl Acad Sci U S A 109 (17):6555–6560. doi: 10.1073/pnas.1200939109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Schweikhard V, Block SM (2013) Single-molecule studies of RNAPII elongation. Biochim Biophys Acta 1829 (1):29–38. doi: 10.1016/j.bbagrm.2012.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galburt EA, Grill SW, Wiedmann A, Lubkowska L, Choy J, Nogales E, Kashlev M, Bustamante C (2007) Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature 446 (7137):820–823. doi: 10.1038/nature05701 [DOI] [PubMed] [Google Scholar]
- 7.Galburt EA, Grill SW, Bustamante C (2009) Single molecule transcription elongation. Methods 48 (4):323–332. doi: 10.1016/j.ymeth.2009.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbert KM, Greenleaf WJ, Block SM (2008) Single-molecule studies of RNA polymerase: motoring along. Annu Rev Biochem 77:149–176. doi: 10.1146/annurev.biochem.77.073106.100741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herbert KM, La Porta A, Wong BJ, Mooney RA, Neuman KC, Landick R, Block SM (2006) Sequence-resolved detection of pausing by single RNA polymerase molecules. Cell 125 (6):1083–1094. doi: 10.1016/j.cell.2006.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuman KC, Abbondanzieri EA, Landick R, Gelles J, Block SM (2003) Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell 115 (4):437–447 [DOI] [PubMed] [Google Scholar]
- 11.Shaevitz JW, Abbondanzieri EA, Landick R, Block SM (2003) Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature 426 (6967):684–687. doi: 10.1038/nature02191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM (2005) Direct observation of base-pair stepping by RNA polymerase. Nature 438 (7067):460–465. doi: 10.1038/nature04268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang MD, Schnitzer MJ, Yin H, Landick R, Gelles J, Block SM (1998) Force and velocity measured for single molecules of RNA polymerase. Science 282 (5390):902–907 [DOI] [PubMed] [Google Scholar]
- 14.Yin H, Wang MD, Svoboda K, Landick R, Block SM, Gelles J (1995) Transcription against an applied force. Science 270 (5242):1653–1657 [DOI] [PubMed] [Google Scholar]
- 15.Neuman KC, Block SM (2004) Optical trapping. Rev Sci Instrum 75 (9):2787–2809. doi: 10.1063/1.1785844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perkins TT (2014) Angstrom-precision optical traps and applications. Annu Rev Biophys 43:279–302. doi: 10.1146/annurev-biophys-042910-155223 [DOI] [PubMed] [Google Scholar]
- 17.Schafer DA, Gelles J, Sheetz MP, Landick R (1991) Transcription by single molecules of RNA polymerase observed by light microscopy. Nature 352 (6334):444–448. doi: 10.1038/352444a0 [DOI] [PubMed] [Google Scholar]
- 18.Greenleaf WJ, Woodside MT, Abbondanzieri EA, Block SM (2005) Passive all-optical force clamp for high-resolution laser trapping. Phys Rev Lett 95 (20):208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visscher K, Gross SP, Block SM (1996) Construction of multiple-beam optical traps with nanometer-resolution position sensing. Ieee J Sel Top Quant 2 (4):1066–1076. doi:Doi 10.1109/2944.577338 [DOI] [Google Scholar]
- 20.Fazal FM, Meng CA, Murakami K, Kornberg RD, Block SM (2015) Real-time observation of the initiation of RNA polymerase II transcription. Nature 525 (7568):274–277. doi: 10.1038/nature14882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palangat M, Larson MH, Hu X, Gnatt A, Block SM, Landick R (2012) Efficient reconstitution of transcription elongation complexes for single-molecule studies of eukaryotic RNA polymerase II. Transcription 3 (3):146–153. doi: 10.4161/trns.20269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komissarova N, Kireeva ML, Becker J, Sidorenkov I, Kashlev M (2003) Engineering of elongation complexes of bacterial and yeast RNA polymerases. Methods Enzymol 371:233–251. doi: 10.1016/S0076-6879(03)71017-9 [DOI] [PubMed] [Google Scholar]
- 23.Kettenberger H, Armache KJ, Cramer P (2004) Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell 16 (6):955–965. doi:DOI 10.1016/j.molcel.2004.11.040 [DOI] [PubMed] [Google Scholar]
- 24.Bustamante C, Marko JF, Siggia ED, Smith S (1994) Entropic elasticity of lambda-phage DNA. Science 265 (5178):1599–1600 [DOI] [PubMed] [Google Scholar]