

Abstract
Lipophagy is a type of selective autophagy that targets lipid droplets for degradation. Since the discovery of lipophagy in 2009, research has uncovered a central role for this process in cellular lipid metabolism, including in atherogenic foam cells. Therefore, increasing lipophagy might be a therapeutic target to reverse lipid build-up in atherosclerosis.
Subject terms: Atherosclerosis, Lipids
Autophagy is a ubiquitous cellular mechanism that allows the orderly digestion of cell components. Although autophagy always results in the breakdown of cytoplasmic constituents in lysosomes, four forms of autophagy have been identified: macroautophagy, microautophagy, chaperone-mediated autophagy (CMA) and crinophagy. Of these forms of autophagy, both macroautophagy and microautophagy deliver cytoplasmic constituents such as organelles to lysosomes, whereas CMA and crinophagy degrade and recycle cellular proteins and secretory granules, respectively. Additionally, autophagy can occur selectively or in bulk. Bulk autophagy occurs in response to nutrient starvation, whereas selective autophagy targets specific cargo for degradation. Selective autophagy often relies on specific cargo ‘tags’ (such as ubiquitin) and ubiquitin-dependent selective autophagy receptors (SARs) that bind ubiquitin and cargo simultaneously to activate autophagy. During macroautophagy, cytoplasmic constituents are sequestered into autophagosomes that subsequently fuse with lysosomes, whereas during microautophagy, cytoplasmic constituents are engulfed directly by lysosomes.
Lipophagy, the latest subtype of autophagy to be described, refers to the selective degradation of lipid droplets by autophagy. In this type of selective autophagy, lipid droplets can be targeted to lysosomes by ubiquitination and the recruitment of lipophagy selectivity factors and autophagy receptors1–3. The lipophagy selectivity factors and autophagy receptors bridge lipid droplets to the autophagy machinery that facilitates the translocation of whole or parts of lipid droplets into lysosomes via macroautophagy or microautophagy, respectively (Fig. 1). Subsequently, neutral lipids in the lipid droplet core — cholesterol esters and triglycerides — are hydrolysed in the lysosome lumen by the action of lysosomal acid lipase (LAL). The resulting free cholesterol and fatty acids are exported from cells or directed to mitochondria for fatty acid oxidation to fuel ATP production, respectively.
Fig. 1. Macrolipophagy and microlipophagy pathways.
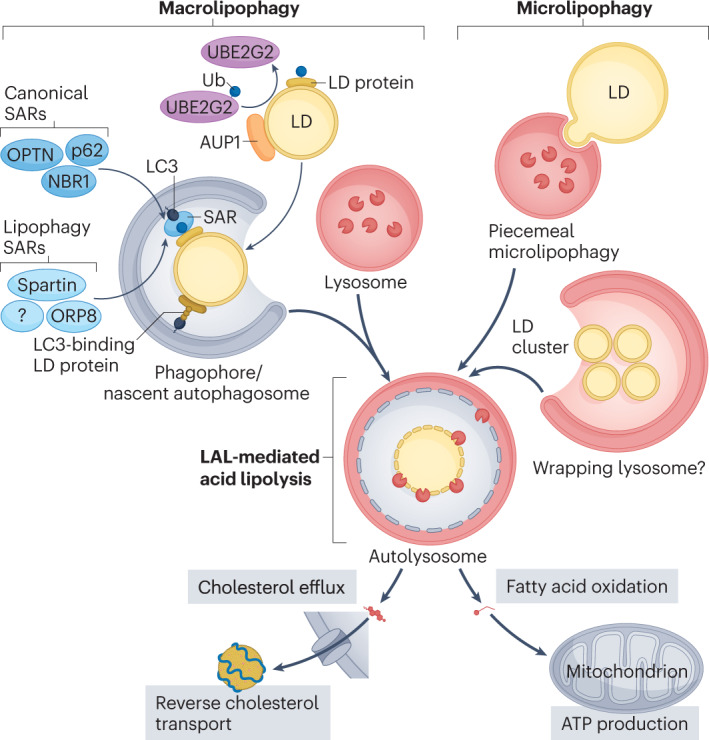
Lipid droplets (LDs) can be targeted to lysosomes by macrolipophagy or microlipophagy. In macrolipophagy, autophagosomes are recruited to LDs by LC3-binding domains present on LD surface proteins or on selective autophagy receptors (SARs) that recognize a ubiquitin (Ub) tag. Ancient ubiquitous protein 1 (AUP1) is presumed to facilitate the ubiquitination of lipid droplet proteins by recruiting the ubiquitin conjugating enzyme E2 G2 (UBE2G2). Fusion of LD-containing autophagosomes with lysosomes enables the breakdown of LDs by the lysosomal acid lipase (LAL). Alternatively, LDs can be directly engulfed into lysosomes by piecemeal microlipophagy and possibly via the wrapping of LD clusters by lysosomal compartments. Free cholesterol and fatty acids resulting from LD breakdown are channelled to the reverse cholesterol transport pathway and to mitochondria for ATP production, respectively. NBR1, autophagy cargo receptor; OPTN, optineurin; ORP8, oxysterol-binding protein-related protein 8; p62, sequestome 1.
Atherosclerosis, an inflammatory disease caused by the build-up of cholesterol-rich plaque in the arterial wall, can be mitigated by egress of cholesterol from plaques and/or by the resolution of inflammation. Autophagy has been shown to regulate lipid droplet clearance and efflux of stored cholesterol from macrophage foam cells for excretion from the body, an atheroprotective process called reverse cholesterol transport4. This discovery laid the foundation for subsequent studies that established a protective role for macrophage-mediated autophagy in atherosclerosis, identifying autophagy as a potential therapeutic target for the treatment of atherosclerosis.
Regulation of lipophagy in atherosclerosis
In macrophages, lipophagy is selectively activated in response to pro-atherogenic lipoproteins (oxidized low-density lipoprotein (LDL) and aggregated LDL), as shown by lipid droplet tagging with ubiquitin and the recruitment of the autophagy marker LC3 and several canonical SARs (sequestome 1 (also known as p62), NBR1 and optineurin) at the lipid droplet surface1,4. In these cells, inhibition of LAL or genetic ablation of autophagy by deletion of Atg5 reduces lipid droplet catabolism and ABCA1-mediated cholesterol efflux by ~50% (ref. 4), calling into question the central dogma that cytoplasmic neutral lipases are solely responsible for macrophage lipolysis and reverse cholesterol transport. Indeed, reverse cholesterol transport from Atg5–/– macrophages is markedly reduced in vivo4. Consistent with this finding, macrophage-specific deletion of Atg5 in Ldlr–/– or Apoe–/– hypercholesterolaemic mice promotes atherosclerosis and results in increased accumulation of lipids in plaques, apoptosis and necrosis, impaired plaque efferocytosis and inflammasome hyperactivation5,6. Conversely, increasing macrophage autophagy and lysosome biogenesis in mice reduces atherogenesis7.
Autophagic markers (such as LC3 and p62) are ubiquitously expressed in the vascular wall. Autophagy flux is active in nascent atheroma but becomes progressively blunted during the progression of atherosclerosis5. Although lipophagy has been known for over a decade to operate in several cell types, including macrophage foam cells, the mechanisms underlying lipophagy and its physiological functions are still poorly understood. Even less is known about the lipophagic capacity of vascular smooth muscle cell (VSMC) foam cells, which comprise the majority of foam cells in atherosclerotic plaques. First, the capacity and extent to which plaque VSMC foam cells undergo lipid droplet biogenesis is unclear, given their intrinsically low LAL activity and consequent inability to process lipoprotein-derived cholesterol esters in lysosomes8. However, increased expression of the lipid droplet marker perilipin 2 in modulated VSMCs during atherosclerosis progression suggests some degree of lipid droplet accumulation in VSMCs at later stages of atherosclerosis9. Interestingly, direct comparisons between macrophage and VSMC foam cells in vitro and in vivo revealed that VSMC foam cells cannot initiate autophagy and induce autophagic lipid droplet catabolism, unlike macrophage foam cells9. The autophagy activator metformin helps to overcome this deficiency and increases ABCG1-mediated cholesterol efflux to high-density lipoprotein9, suggesting that targeting VSMC foam cell lipophagy is an important therapeutic intervention in atherosclerosis.
In an effort to identify the protein machinery that targets lipid droplets for lipophagy, one preliminary study investigated spartin as a potential lipophagy receptor. Through its ubiquitin-binding region, lipid droplet-localized spartin recruited LC3 to lipid droplets to promote lysosomal lipid droplet degradation in SUM159 cells3. A second study identified hepatic ORP8 as an AMPK-regulated lipophagy receptor that mediates autophagic recognition and degradation of lipid droplets by recruiting either LC3 or GABA receptor-associated proteins2. A third study in macrophage foam cells identified 37 proteins that were enriched on lipid droplets after inhibition of autophagic lipid droplet degradation, several of which contained predicted LC3-interacting region motifs and predicted ubiquitin-associated motifs that are typical of SARs1. Yeast lipophagy assays revealed a genetic requirement for several candidate lipophagy factors, including HSPA5, UBE2G2 and AUP1 (ref. 1). Intriguingly, several lipophagy factors are dysregulated in atherosclerosis1, suggesting that alterations in lipophagy flux contribute to atherogenesis. Together, these findings have important biological and therapeutic implications. First, targeting lipophagy receptors and factors to selectively inhibit lipophagy will allow the role of lipophagy in the development of atherosclerosis to be directly tested. Conversely, increasing lipophagy in arterial foam cells could promote reverse cholesterol transport, slow atherogenesis and even drive the regression of pre-existing atherosclerotic plaques.
Targeting lipophagy in atherosclerosis
Mounting evidence points to various selective autophagy pathways, including lipophagy, as emerging therapeutic targets in cardiometabolic disorders. Currently, tools to selectively regulate and track lipophagy (and not simply bulk autophagy) in atherosclerosis and other metabolic disorders are lacking. In the past 3 years, lipophagy has been monitored in real-time in cell lines expressing lipid droplet-localized proteins fused to pH biosensors2,3. Bringing these lipophagy biosensors from cell culture into mouse models would substantially advance our understanding of the relative contribution of lipophagy to lipid homeostasis in distinct cell types and tissues that drive the development of atherosclerosis.
Excitingly, the first specific lipophagy-inducing compounds were generated in 2021 by ingenuously linking lipid droplet-interacting and LC3-interacting molecules together10. Promoting LC3 recruitment to lipid droplets, lipid droplet-targeting autophagy-tethering compounds (LD-ATTECs) markedly reduced lipid droplet accumulation in vitro in mouse embryonic fibroblasts and in 3T3-L1 adipocytes, and in mouse livers in vivo10. In atherosclerosis, the systemic administration of LD-ATTECs has the potential to regulate foam cell lipid metabolism and inflammation in the vascular wall in situ; in addition, they are expected to regulate these processes in other atherosclerosis-relevant tissues such as the liver and adipose tissue in the setting of hypercholesterolaemia.
Finally, a better understanding of lipophagy mechanisms is necessary to inform future lipophagy-targeted therapies. For example, in macrophage foam cells, both single lipid droplets and clusters of lipid droplets are tagged for autophagic degradation1. While a surprisingly large proportion of LC3-tagged lipid droplets (~45%) localize to cytosolic lipid droplet clusters in human THP1 macrophage foam cells1, whether single lipid droplets or clusters of lipid droplets are similarly degraded by macroautophagy and microautophagy mechanisms is unknown. Moreover, the specific contributions of macrolipophagy versus microlipophagy to overall lipophagy and cellular lipid metabolism in distinct cell types and tissues remain to be determined.
In conclusion, studies suggest that lipophagy contributes substantially to the atheroprotective effects of macrophage autophagy. Better tools are needed to selectively activate and track lipophagy in vitro and in vivo, to determine the relative contributions of macrolipophagy and microlipophagy to lipid metabolism, and to test the therapeutic potential of lipophagy activation for the treatment of atherosclerosis. Nonetheless, activation of lipophagy by lipophagy-inducing compounds such as LD-ATTECs in arterial foam cells holds tremendous potential to promote reverse cholesterol transport and regression of atherosclerosis.
Acknowledgements
The authors were supported by the Canadian Institutes for Health Research (PJT-391187 and Canada Research Chair to M.O.), the Natural Sciences and Engineering Research Council of Canada (Discovery Grant to M.O.), and a University of Ottawa Cardiac Endowment Fellowship (to T.L.).
Competing interests
The authors declare no competing interests.
References
- 1.Robichaud S, et al. Identification of novel lipid droplet factors that regulate lipophagy and cholesterol efflux in macrophage foam cells. Autophagy. 2021;17:3671–3689. doi: 10.1080/15548627.2021.1886839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pu M, et al. ORP8 acts as a lipophagy receptor to mediate lipid droplet turnover. Protein Cell. 2022 doi: 10.1093/procel/pwac063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung J, et al. The Troyer syndrome protein spartin mediates selective autophagy of lipid droplets. Preprint at. bioRxiv. 2021 doi: 10.1101/2021.08.18.456894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouimet M, et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razani B, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao X, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sergin I, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017;8:15750. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubland JA, et al. Low LAL (lysosomal acid lipase) expression by smooth muscle cells relative to macrophages as a mechanism for arterial foam cell formation. Arterioscler. Thromb. Vasc. Biol. 2021;41:e354–e368. doi: 10.1161/ATVBAHA.120.316063. [DOI] [PubMed] [Google Scholar]
- 9.Robichaud S, et al. Autophagy is differentially regulated in leukocyte and nonleukocyte foam cells during atherosclerosis. Circ. Res. 2022;130:831–847. doi: 10.1161/CIRCRESAHA.121.320047. [DOI] [PubMed] [Google Scholar]
- 10.Fu Y, et al. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 2021;31:965–979. doi: 10.1038/s41422-021-00532-7. [DOI] [PMC free article] [PubMed] [Google Scholar]